
Cu-Catalyzed/mediated synthesis of N-fluoroalkylanilines from arylboronic acids: fluorine effect on the reactivity of fluoroalkylamines†
Xiang-Guo
Hu  *a
*a
*a
Abstract
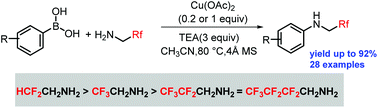
An oxidative coupling reaction of fluoroalkylamines with arylboronic acids has been achieved for the first time. Fluorine has profound influence on the reactivity and fluoroalkylated amines have the following reactivity trend: difluoroethylamine > trifluoroethylamine > pentafluoropropylamine ≈ heptafluorobutylamine.
 Please wait while we load your content...
Please wait while we load your content...

