Facile synthesis and fundamental properties of an N-methylguanidine-bridged nucleic acid (GuNA[NMe])†
Received
2nd June 2018
, Accepted 27th June 2018
First published on 30th August 2018
Abstract
An N-methylguanidine-bridged nucleic acid (GuNA[NMe]), a guanidine-bridged nucleic acid (GuNA) bearing a methyl substituent at the bridge, was successfully synthesised and incorporated into oligonucleotides. By employing an acetyl protecting group, GuNA[NMe]-modified oligonucleotides bearing acid-sensitive purine nucleobases were successfully prepared. The obtained GuNA[NMe]-modified oligonucleotides exhibit excellent binding affinity towards the complementary single-stranded RNA and DNA. Furthermore, even a single GuNA[NMe] modification provides robust enzymatic stability, similar to that achieved by the well-established phosphorothioate backbone modification. These data indicate that such a GuNA[NMe] represents a valuable modification for the development of therapeutic oligonucleotides.
Introduction
The clinical application of antisense oligonucleotides (ASOs) is becoming a reality as several ASO drugs have been approved by the U.S. Food and Drug Administration (FDA).1 The ASOs used in clinical applications and/or in in vitro/in vivo experiments are generally chemically modified DNA/RNA oligos, where the modifications play an important role in enhancing the biophysical and pharmacokinetic properties of the ASOs (e.g. the enzymatic stability against nuclease-mediated degradation, duplex-forming ability towards target RNAs, and cellular/tissue distribution).2 One of the most widely used chemical modifications is the phosphorothioate (PS) backbone modification, which greatly increases the enzymatic stability and cellular/tissue distribution. Other important modifications are sugar modifications, such as 2′-fluoro (2′-F), 2′-O-methoxyethyl (MOE), and 2′,4′-bridged nucleic acid/locked nucleic acid (2′,4′-BNA/LNA, Fig. 1). These three modifications impart strong binding affinity towards target RNAs and a certain degree of enzymatic stability. The covalent attachment of cationic functional groups, such as guanidino and amino groups, is also a reliable modification to enhance the enzymatic stability, duplex-forming ability, and cellular uptake.3
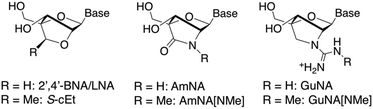 |
| | Fig. 1 Structures of 2′,4′-BNA/LNA, S-cEt, AmNA, AmNA[NMe], GuNA and GuNA[NMe]. The GuNA[NMe] motif was designed in the present study. Base = nucleobase. | |
We have recently designed and synthesised guanidine-bridged nucleic acid (GuNA, Fig. 1), bearing a positively charged 2′–4′ bridge structure.4 The GuNA-modified oligonucleotides (ONs) were found to exhibit extremely high binding affinity towards the complementary single-stranded RNA (ssRNA) and DNA (ssDNA) and show far superior enzymatic stability compared to their 2′,4′-BNA/LNA-modified counterparts. These data clearly demonstrate that GuNA is a promising approach for the modification of therapeutic ASOs.
A wide variety of 2′,4′-BNA/LNA analogues currently exist, and ONs/ASOs modified with such analogues have been rigorously evaluated in vitro and in vivo.2,5 Interestingly, recent studies showed that even a simple methyl substitution at the 2′–4′ bridge has a large impact on the pharmacokinetics as well as biophysical properties. For example, ONs/ASOs modified with an amido-bridged nucleic acid bearing a methyl group at the 2′–4′ amide bridge (AmNA[NMe]) exhibit higher enzymatic stability and in vitro antisense potency than those modified with AmNA.5a,6 In another example, ASOs modified with S-2′,4′-constrained-2′-O-ethyl (S-cEt, i.e. methyl-substituted 2′,4′-BNA/LNA) were found to show improved hepatotoxicity in animal models compared to the 2′,4′-BNA/LNA-modified ASOs.5b,7 Such positive data prompted us to design a novel methyl-substituted GuNA analogue, termed N-methylguanidine-bridged nucleic acid (GuNA[NMe], Fig. 1). In this report, we describe the facile synthesis of GuNA[NMe]-thymidine (GuNA[NMe]-T) phosphoramidite and the properties of GuNA[NMe]-modified ONs.
Results and discussion
Approach using a Boc-protecting group for the synthesis of GuNA[NMe]-modified ONs
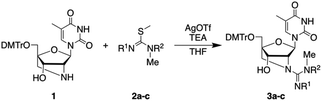
The preparation of GuNA[NMe]-T phosphoramidite, a building block for ON synthesis, started from 2′-amino-LNA 1,8 which was recently established as a key precursor for GuNA phosphoramidites9 (Table 1). At first, the guanidination of 1 was performed using N,N′-bis(tert-butoxycarbonyl)-N,S-dimethylisothiourea (2a)10 in the presence of silver(I) triflate11 and triethylamine to yield 3a, but this resulted in no reaction even at 60 °C (entries 1 and 2). In contrast, N′-Boc-N-methylisothiourea 2b successfully afforded the guanidinated product 3b in 88% yield (entry 3). The trace amounts of 3c obtained from N-Boc-N-methylisothiourea 2c (entry 4) indicated that this guanidination reaction proceeded via a carbodiimide mechanism.12 The 3′-hydroxyl group of product 3b was then phosphitylated to afford N-Boc-protected GuNA[NMe]-T phosphoramidite 4 (Scheme 1).
 |
| | Scheme 1 Reagents and conditions: (i) (i-Pr)2NP(Cl)O(CH2)2CN, DIPEA, CH2Cl2, rt, 76%. | |
Table 1 Guanidination of 1
|
|
| Entry |
Isothiourea |
R1 |
R2 |
Temperature |
Result |
| 1 |
2a
|
Boc |
Boc |
rt |
No reaction |
| 2 |
2a
|
Boc |
Boc |
60 °C |
No reaction |
| 3 |
2b
|
Boc |
H |
rt |
3b (88%) |
| 4 |
2c
|
H |
Boc |
rt |
3c (Trace) |
The ON synthesis was performed in an automated DNA synthesiser following the standard phosphoramidite protocol, except for a prolonged coupling time (16 min) for the incorporation of 4 into the ONs. After oligomer elongation, the Boc protecting groups were removed by treatment with TMSOTf and 2,6-lutidine in CH2Cl2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 9 (rt, 24 h), and the resulting ONs were cleaved from the solid support using aqueous ammonia (rt, 4 h; then 60 °C, 8 h). Finally, the GuNA[NMe]-modified T10-mers (ON1–ON5) were successfully isolated, as shown in Table 2. However, the preparation of ON6 containing acid-sensitive purine nucleobases13 was unsuccessful. These results show a sequence limitation for ON synthesis with N-Boc-protected phosphoramidite 4.
9 (rt, 24 h), and the resulting ONs were cleaved from the solid support using aqueous ammonia (rt, 4 h; then 60 °C, 8 h). Finally, the GuNA[NMe]-modified T10-mers (ON1–ON5) were successfully isolated, as shown in Table 2. However, the preparation of ON6 containing acid-sensitive purine nucleobases13 was unsuccessful. These results show a sequence limitation for ON synthesis with N-Boc-protected phosphoramidite 4.
Table 2 ONs prepared using N-Boc-protected phosphoramidite 4
| Oligonucleotide sequencea |
Yield (%) |
MALDI-TOF MS |
| Calcd [M − H]− |
Found [M − H]− |
![[T with combining low line]](https://www.rsc.org/images/entities/char_0054_0332.gif) = GuNA[NMe].
Complex mixture. = GuNA[NMe].
Complex mixture.
|
| 5′-d(TTTTTTTTT)-3′ (ON1) |
28 |
3060.5452 |
3060.5216 |
| 5′-d(TTTTTTTT)-3′ (ON2) |
20 |
3143.5935 |
3143.5313 |
| 5′-d(TTTTTTT)-3′ (ON3) |
34 |
3226.6419 |
3226.5907 |
| 5′-d(TTTTT)-3′ (ON4) |
35 |
3392.7385 |
3392.6876 |
| 5′-d(TTTTTTT)-3′ (ON5) |
27 |
3226.6419 |
3226.6913 |
| 5′-d(GCGTTTTTGCT)-3′ (ON6) |
—b |
3713.6574 |
— |
Approach using an Ac-protecting group for the synthesis of GuNA[NMe]-modified ONs
To overcome the above limitation, we decided to synthesise an N-Ac-protected GuNA[NMe]-T phosphoramidite, expecting that the alternative Ac group could be removed under aqueous ammonia conditions. The N-Ac-protected phosphoramidite 7 was prepared in the same way as N-Boc-protected phosphoramidite 4, as shown in Scheme 2. Briefly, 2′-amino-LNA 1 was guanidinated with N′-acetyl-N-methylisothiourea 5 to afford 6, which was then converted into the desired phosphoramidite 7.
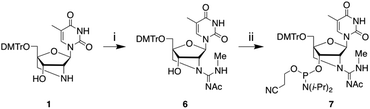 |
| | Scheme 2 Reagents and conditions: (i) N′-Acetyl-S,N-dimethylisothiourea (5), AgOTf, DIPEA, THF, rt, 85%; (ii) (i-Pr)2NP(Cl)O(CH2)2CN, DIPEA, CH2Cl2, rt, 84%. | |
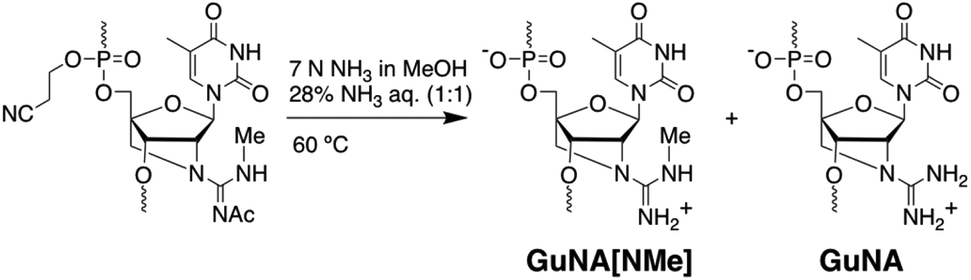
Phosphoramidite 7 was then incorporated into ONs, and the deprotection of the Ac group was subsequently conducted in a 7 N ammonia methanol solution/28% aqueous ammonia (1:1, 60 °C, 8 h). As expected, the cleavage of ONs from the solid support followed by Ac deprotection was successfully achieved in a one-pot process. However, the above reaction was slow and produced a considerable amount of by-products, arising from the replacement of the methylamino group with an amino group, i.e. GuNA (Fig. 2, ca. GuNA[NMe]/GuNA = 3:1). We found that switching the reagent to 7 N ammonia methanol solution/40% aqueous methylamine (1:1, rt, 4 h; then 60 °C, 10 h)14 efficiently minimised the conversion to GuNA and furnished the desired unprotected ONs in good yield (Table 3). Notably, ON6 containing purine nucleobases was successfully obtained by this method.
 |
| | Fig. 2 Deprotection in aqueous ammonia affords both the desired GuNA[NMe]-modified ONs and the undesired GuNA-modified ONs. | |
Table 3 ONs prepared using N-Ac-protected phosphoramidite 7
| Oligonucleotide sequencea |
Yield (%) |
MALDI-TOF MS |
| Calcd [M − H]− |
Found [M − H]− |
|
= GuNA[NMe].
|
| 5′-d(TTTTTTTTT)-3′ (ON1) |
39 |
3060.5452 |
3060.5549 |
| 5′-d(GCGTTTTTGCT)-3′ (ON6) |
23 |
3713.6574 |
3713.5872 |
| 5′-d(TTTTTTTTT)-3′ (ON7) |
16 |
3060.5452 |
3060.5425 |
Duplex-forming ability of GuNA[NMe]-modified ONs
The thermal stability of the duplexes formed by the GuNA[NMe]-modified ONs (ON1–ON6) and the complementary ssRNA or ssDNA was then evaluated by UV melting experiments, and the obtained melting temperatures (Tm) are summarised in Table 4. Compared to natural ONs (ON8 and ON9), the GuNA[NMe]-modified ONs showed extremely high binding affinity towards ssRNA (ON1–ON6: ΔTm = +5.0 to +8.8 °C per modification), with Tm values comparable to those of GuNA-modified ONs. As for the ssDNA complements, both GuNA[NMe] and GuNA greatly increased the Tm values depending on the number of modifications. The little or no difference in the Tm values between ON1–ON5 and ON10–ON144 indicates that methyl substitution at the guanidine bridge has no practical impact on the duplex-forming ability. Therefore, further functionalisation of GuNAs with no loss of their duplex-forming ability may be possible by introducing various substituents (e.g. fluorescent groups and tissue-targeting groups) at the guanidine bridge.
Table 4 Duplex-forming ability of GuNA[NMe]-modified ONs (ON1–ON6) towards the complementary ssRNA and ssDNAa
| Oligonucleotide sequence |
T
m (ΔTm/mod.) (°C) |
| ssRNA |
ssDNA |
|
Conditions: 10 mM sodium phosphate buffer (pH 7.2), 100 mM NaCl, and 4 μM oligonucleotide. The Tm values reflect the average of three measurements. ΔTm/mod.: the change in Tm value (ΔTm) per modification compared to the unmodified standard ONs (ON8 and ON9). Target strands: 5′-r(AAAAAAAAAA)-3′, 5′-r(AGCAAAAAACGC)-3′, 5′-d(AAAAAAAAAA)-3′ and 5′-d(AGCAAAAAACGC)-3′. T = GuNA. = GuNA[NMe].
|
| 5′-d(TTTTTTTTTT)-3′ (ON8) |
18 |
|
21 |
|
| 5′-d(GCGTTTTTTGCT)-3′ (ON9) |
47 |
|
51 |
|
| 5′-d(TTTTTTTTTT)-3′ (ON10) |
25 |
(+7.0) |
25 |
(+4.0) |
| 5′-d(TTTTTTTTTT)-3′ (ON11) |
30 |
(+6.0) |
36 |
(+7.5) |
| 5′-d(TTTTTTTTTT)-3′ (ON12) |
41 |
(+7.3) |
50 |
(+9.7) |
| 5′-d(TTTTTTTTTT)-3′ (ON13) |
62 |
(+9.2) |
75 |
(+10.8) |
| 5′-d(TTTTTTTTTT)-3′ (ON14) |
39 |
(+7.0) |
50 |
(+9.7) |
| 5′-d(TTTTTTTTT)-3′ (ON1) |
26 |
(+8.0) |
25 |
(+4.0) |
| 5′-d(TTTTTTTT)-3′ (ON2) |
31 |
(+6.5) |
38 |
(+8.5) |
| 5′-d(TTTTTTT)-3′ (ON3) |
41 |
(+7.7) |
55 |
(+11.3) |
| 5′-d(TTTTT)-3′ (ON4) |
62 |
(+8.8) |
74 |
(+10.6) |
| 5′-d(TTTTTTT)-3′ (ON5) |
39 |
(+7.0) |
50 |
(+9.7) |
| 5′-d(GCGTTTTTGCT)-3′ (ON6) |
52 |
(+5.0) |
56 |
(+5.0) |
Enzymatic stability of GuNA[NMe]-modified ONs
The stability of the GuNA[NMe]-modified ONs against enzymatic degradation was then evaluated. T10-mers with the 3′-terminal thymidine replaced with GuNA[NMe]-T, GuNA-T, LNA, or 5′-PS-linked thymidine (5′-PS-T) were incubated with 3′-exonuclease (Crotalus adamanteus venom phosphodiesterase, CAVP) in Tris-HCl buffer (pH 8.0) at 37 °C, and the percentage of intact ONs was analysed by HPLC over time. As shown in Fig. 3, the natural T10-mer (ON8) was degraded within 5 min and the LNA-modified T10-mer (ON15) was degraded within 10 min. In contrast, the GuNA[NMe]- and GuNA-modified congeners (ON7 and ON16, respectively) showed dramatically improved enzymatic stability. ON17 with the well-established PS modification exhibited similar stability to that of ON7 and ON16, revealing that GuNA and GuNA[NMe] are both valuable modifications to enhance the enzymatic stability. The strong resistance of ON7 against nuclease activity possibly arises mainly from the positive charge of the guanidine moiety, since the methyl substitution seems to contribute little to the resistance (ON7vs.ON16).
 |
| | Fig. 3 Stability of ONs against CAVP. Conditions: 0.25 μg CAVP, 10 mM MgCl2, 50 mM Tris-HCl (pH 8.0), and 7.5 μM oligonucleotide at 37 °C (total volume: 100 μL). The sequence used was 5′-TTTTTTTTTX-3′. X = natural thymidine (orange closed diamond, ON8), LNA-T (purple closed square, ON15), GuNA[NMe]-T (red open circle, ON7), GuNA-T (blue open triangle, ON16), or 5′-PS-T (green cross, ON17). | |
Conclusions
GuNA[NMe]-T phosphoramidite was successfully synthesised and incorporated into ONs. By employing an acetyl protecting group, the facile preparation of GuNA[NMe]-modified ONs with/without purine nucleobases was successfully accomplished. The GuNA[NMe]-modified ONs were found to show excellent duplex-forming ability towards the complementary ssRNA and ssDNA. Moreover, T10-mers modified with GuNA[NMe] exhibited extremely high enzymatic stability similar to that of oligomers modified with a PS linkage. These results demonstrate that GuNA[NMe], as well as GuNA, may serve as valuable modifications for the development of therapeutic ONs/ASOs.
Experimental
All moisture-sensitive reactions were carried out in well-dried glassware under an N2 atmosphere. 1H spectra were recorded at 300, 400, or 500 MHz. 13C NMR spectra were recorded at 76, 100, or 126 MHz. 31P NMR spectra were recorded at 162 or 202 MHz. Chemical shifts (δ) are expressed in ppm relative to internal tetramethylsilane (0.00 ppm) or residual CHCl3 (7.26 ppm) in the 1H NMR spectra, internal tetramethylsilane (0.00 ppm) or chloroform-d1 (δ = 77.0 ppm) in the 13C NMR spectra, and 85% H3PO4 (δ = 0.00 ppm) in the 31P NMR spectra. MALDI-TOF mass spectra of all new compounds were obtained on a SpiralTOF JMS-S3000 instrument. For column chromatography, PSQ-60B or PSQ-100B silica gel was used. The progress of the reactions was monitored by analytical thin layer chromatography (TLC) on glass plates (TLC silica gel 60 F254) and the products were visualised by UV light.
(1R,3R,4R,7S)-5-(N′-tert-Butoxycarbonyl-N-methylcarbamimidoyl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2,2,1]heptane (3b)
To a mixture of 1 (451 mg, 0.79 mmol) and N′-tert-butoxycarbonyl-N,S-dimethylisothiourea (2b) (258 mg, 1.3 mmol) in anhydrous THF (8.0 mL), silver triflate (410 mg, 1.6 mmol) and triethylamine (0.45 mL, 3.2 mmol) were added at 0 °C. The resulting mixture was then stirred overnight at room temperature. After the completion of the reaction, water was added and the product was extracted with CHCl3. The organic phase was washed with sat. aq. NH4Cl and brine, dried over Na2SO4, and concentrated under reduced pressure. The product was purified by column chromatography (CHCl3/MeOH = 18:1) to afford 3b (508 mg, 88%) as a white solid. [α]24D: 16.7 (c 1.0, CHCl3); IR (KBr): 3215, 3067, 2977, 2836, 1692, 1606, 1509, 1447, 1390, 1365, 1273, 1254, 1176, 1149, 1038 cm−1; 1H NMR (CDCl3): δ 1.40 (9H, s), 1.69 (3H, s), 2.91 (3H, s), 3.37, 3.62 (2H, AB, J = 10.0 Hz), 3.43, 3.51 (2H, AB, J = 11.0 Hz), 3.77 (6H, s), 4.21 (1H, s), 4.50 (1H, s), 5.51 (1H, s), 6.82 (4H, d, J = 8.9 Hz), 7.18–7.45 (9H, m), 7.53 (1H, s), 9.78 (1H, s) ppm; 13C NMR (CDCl3): δ 12.6, 28.2, 30.2, 54.6, 55.2, 59.3, 63.5, 70.1, 78.5, 86.3, 86.5, 88.8, 110.8, 113.2, 127.0, 128.0, 130.0, 130.0, 134.3, 135.2, 135.5, 144.3, 150.5, 158.6, 161.2, 161.4, 164.1, 185.3 ppm; HRMS (MALDI) calcd for C39H45N5O9Na [M + Na]+ 750.3110, found 750.3107.
(1R,3R,4R,7S)-5-(N′-tert-Butoxycarbonyl-N-methylcarbamimidoyl)-7-[2-cyanoethoxy(diisopropylamino)phosphanyloxyl]-1-(4,4′-dimethoxytrityloxymethyl)-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2,2,1]heptane (4)
To a solution of 3b (423 mg, 0.58 mmol) in anhydrous CH2Cl2 (6.0 mL), N,N-diisopropylethylamine (240 μL, 1.4 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (200 μL, 0.87 mmol) were added, and the resulting mixture was stirred for 6 h at room temperature. After the completion of the reaction, sat. aq. NaHCO3 was added and the product was extracted with CH2Cl2. The organic phase was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The product was purified by column chromatography (n-hexane/AcOEt = 1:3) to afford 4b (411 mg, 76%) as a white solid. 1H NMR (CDCl3): δ 0.96 (3H, d, J = 6.9 Hz), 1.02 (3H, d, J = 6.9 Hz), 1.09 (3H, d, J = 6.9 Hz), 1.12 (3H, d, J = 7.9 Hz), 1.44 (9H, s), 1.59 (3/2H, d, J = 1.2 Hz), 1.60 (3/2H, d, J = 0.7 Hz), 2.38–2.63 (2H, m), 3.05 (3/2H, d, J = 6.5 Hz), 3.07 (3/2H, d, J = 5.2 Hz), 3.30–3.38 (2H, m), 3.41–3.70 (4H, m), 3.75–3.91 (2H, m), 3.79 (3H, s), 3.80 (3H, s), 4.32 (1/2H, d, J = 6.5 Hz), 4.41 (1/2H, d, J = 9.3 Hz), 4.66 (1/2H, s), 4.69 (1/2H, s), 5.65 (1/2H, s), 5.66 (1/2H, s), 6.81–6.87 (4H, m), 7.24–7.36 (7H, m), 7.41–7.47 (2H, m), 7.71 (1/2H, d, J = 1.4 Hz), 7.74 (1/2H, d, J = 1.4 Hz), 8.13 (1/2H, br), 8.28 (1/2H, br), 8.95 (1H, br) ppm; 31P NMR (CDCl3): δ 148.2, 148.5 ppm; HRMS (MALDI) calcd for C48H62N7O10NaP [M + Na]+ 950.4188, found 950.4200.
(1R,3R,4R,7S)-5-(N′-Acetyl-N-methylcarbamimidoyl)-1-(4,4′-dimethoxytrityloxymethyl)-7-hydroxy-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2,2,1]heptane (6)
To a mixture of 1 (2.0 g, 3.5 mmol) and N′-acetyl-N,S-dimethylisothiourea (5) (560 mg, 3.9 mmol) in anhydrous THF (35 mL), silver triflate (1.4 g, 5.4 mmol) and N,N-diisopropylethylamine (0.80 mL, 5.2 mmol) were added at 0 °C. The resulting mixture was then stirred overnight at room temperature. After the completion of the reaction, AcOEt (40 mL) and sat. aq. NH4Cl (15 mL) were added, and the mixture was filtered. The filtrate was extracted with AcOEt, washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The product was purified by column chromatography (CHCl3/MeOH = 5:1) to afford 6 (2.0 g, 85%) as a white solid. [α]24D: 16.2 (c 1.0, CHCl3); IR (KBr): 2953, 2835, 2247, 1695, 1608, 1511, 1441, 1251, 1176, 1153, 1111, 1068, 1041 cm−1; 1H NMR (CDCl3): δ 1.65 (3H, s), 2.05 (3H, s), 2.87 (3H, s), 3.35–3.54 (4H, m), 3.77 (3H, s), 3.78 (3H, s), 4.30 (1H, s), 4.56 (1H, s), 5.53 (1H, s), 6.83 (2H, d, J = 9.2 Hz), 6.83 (2H, d, J = 9.2 Hz) 7.20–7.44 (9H, m), 7.57 (1H, s), 8.74 (1H, br) ppm; 13C NMR (CDCl3): δ 12.4, 25.9, 29.3, 53.7, 55.2, 59.1, 63.4, 69.8, 85.8, 86.7, 88.5, 111.0, 113.2, 127.0, 127.9, 128.2, 130.0, 130.1, 134.2, 135.1, 135.5, 144.1, 150.3, 158.6, 161.2, 163.9 ppm; HRMS (MALDI) calcd for C36H40N5O8 [M + H]+ 670.2871, found 670.2874.
(1R,3R,4R,7S)-5-(N′-Acetyl-N-methylcarbamimidoyl)-7-[2-cyanoethoxy(diisopropylamino)phosphanyloxyl]-1-(4,4′-dimethoxytrityloxymethyl)-3-(thymin-1-yl)-2-oxa-5-azabicyclo[2,2,1]heptane (7)
To a solution of 6 (2.0 g, 3.0 mmol) in anhydrous CH2Cl2 (37 mL), N,N-diisopropylethylamine (2.1 mL, 12 mmol) and 2-cyanoethyl-N,N-diisopropylchlorophosphoramidite (1.3 mL, 6.0 mmol) were added, and the resulting mixture was stirred for 6 h at room temperature. After the completion of the reaction, sat. aq. NaHCO3 was added and the product was extracted with CHCl3. The organic phase was washed with water and brine, dried over Na2SO4, and concentrated under reduced pressure. The product was purified by column chromatography (CHCl3/MeOH = 12:1) to afford 7 (2.2 g, 84%) as a white solid. 1H NMR (CDCl3): δ 0.97 (3H, d, J = 6.9 Hz), 1.03 (3H, d, J = 6.9 Hz), 1.09 (3H, d, J = 6.9 Hz), 1.13 (3H, d, J = 6.9 Hz), 1.58 (3H, s), 2.02 (3/2H, s), 2.03 (3/2H, s), 2.38–2.42 (1H, m), 2.50–2.64 (1H, m), 2.99 (3/2H, d, J = 4.6 Hz), 3.04 (3/2H, d, J = 4.1 Hz), 3.25–3.31 (1H, m), 3.36–3.40 (1H, m), 3.43–3.83 (6H, m), 3.79 (3/2H, s), 3.79 (3/2H, s), 3.80 (3/2H, s), 3.80 (3/2H, s), 4.39 (1/2H, d, J = 6.4 Hz), 4.47 (1/2H, d, J = 8.7 Hz), 4.77 (1/2H, s), 4.82 (1/2H, s), 5.68 (1H, s), 6.82–6.87 (4H, m), 7.23–7.36 (7H, m), 7.42–7.47 (2H, m), 7.73 (1/2H, s), 7.75 (1/2H, s), 8.76 (1H, br), 9.24 (1H, br) ppm; 31P NMR (CDCl3): δ 148.1, 148.5 ppm; HRMS (MALDI) calcd for C45H56N7O9NaP [M + Na]+ 892.3769, found 892.3755.
Oligonucleotide synthesis using N-Boc-protected GuNA[NMe]-T phosphoramidite
The synthesis at 0.2 μmol scale of oligonucleotides modified with GuNA[NMe] was performed using an nS-8 oligonucleotide synthesiser (GeneDesign, Inc.) according to the standard phosphoramidite protocol with 0.5 M 5-ethylthio-1H-tetrazole as the activator. Custom Primer Support™ T 40s (GE Healthcare Life Sciences) was used as the solid support. The amidite solutions were dehydrated by using Molecular Sieves Pack 3A (Wako). The standard synthesis cycle was used for the assembly of the reagents and synthesis of the oligonucleotides, except that the coupling time was extended to 16 min. The synthesis was carried out in “trityl on” mode. The Boc protecting groups were removed using 1.0 M TMSOTf and 1.25 M 2,6-lutidine in CH2Cl2 (rt, 24 h). The resulting oligonucleotides were cleaved from the solid support by treatment with concentrated ammonium hydroxide (rt, 4 h; then 60 °C, 8 h).
Oligonucleotide synthesis using N-Ac-protected GuNA[NMe]-T phosphoramidite
The synthesis at 0.2 μmol scale of oligonucleotides modified with GuNA[NMe] was performed using an nS-8 oligonucleotide synthesiser (GeneDesign, Inc.) according to the standard phosphoramidite protocol with 0.5 M 5-ethylthio-1H-tetrazole as the activator. Custom Primer Support™ T 40s (GE Healthcare Life Sciences) or NittoPhase® UnyLinker™ 50 (Kinovate Life Sciences) was used as the solid support. The amidite solutions were dehydrated by using Molecular Sieves Pack 3A (Wako). The standard synthesis cycle was used for the assembly of the reagents and synthesis of the oligonucleotides, except that the coupling time was extended to 16 min. The synthesis was carried out in “trityl on” mode. The oligonucleotides were cleaved from the solid support by treatment with a mixture of 7 N ammonia in methanol and 40% aq. methylamine (50:50) (rt, 4 h; then 60 °C, 10 h).
Oligonucleotide purification and characterisation
The oligonucleotides were initially purified with a Waters Sep-Pak Plus C18 Cartridge. The oligonucleotides were further purified by reverse-phase HPLC with Waters XBridge™ C18 (4.6 × 50 mm analytical and 10 × 50 mm preparative) columns with a linear gradient of MeCN in 0.1 M triethylammonium acetate (pH 7.0). The oligonucleotides were analysed for purity by reverse-phase HPLC and characterised by MALDI-TOF mass spectrometry (SpiralTOF JMS-S3000).
UV melting experiments and melting profile
UV melting experiments were carried out on a Shimadzu UV-1650 spectrophotometer equipped with a Tm analysis accessory. Equimolecular amounts of the target RNA or DNA strand and oligonucleotide were dissolved in 10 mM sodium phosphate buffer at pH 7.2 containing 100 mM NaCl to give a final strand concentration of 4 μM. The samples were annealed by heating at 95 °C, followed by slow cooling to room temperature. The melting profile was recorded at 260 nm from 0 to 90 °C at a scan rate of 0.5 °C min−1. The Tm values, i.e. the temperatures of half-dissociation of the formed duplexes, were determined by the first derivative of the melting curve.
Nuclease resistance evaluation
The synthesis and purification of the LNA-, GuNA-, or 5′-PS-T-modified oligonucleotides (ON15–ON17) were conducted by Gene Design Inc. (Osaka, Japan). The 5′-PS-T-modified oligonucleotide was prepared as a stereoisomeric mixture. The sample solutions were prepared by dissolving 0.75 μmol of oligonucleotides in 50 mM Tris-HCl buffer (pH 8.0) containing 10 mM MgCl2. To each sample solution, 0.25 μg CAVP was added and the cleavage reaction was carried out at 37 °C. Aliquots of each reaction mixture were removed at timed intervals and heated to 90 °C for 2 min to deactivate the nuclease, which were then analysed by reverse-phase HPLC to evaluate the amount of intact oligonucleotides remaining. The percentage of intact oligonucleotides in each sample was calculated and plotted against the digestion time to obtain a degradation curve.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was supported in part by the JSPS KAKENHI Grant Number 16K1793, the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS)) from the AMED under Grant Number JP18am0101084, and the Basic Science and Platform Technology Program for Innovative Biological Medicine from the AMED under Grant Number JP18am0301004.
Notes and references
-
(a) N. Goyal and P. Narayanaswami, Muscle Nerve, 2018, 57, 356–370 CrossRef PubMed;
(b) J. C. Kaczmarek, P. S. Kowalski and D. G. Anderson, Genome Med., 2017, 9, 60–75 CrossRef PubMed;
(c) V. Sharma, R. Sharma and S. Singh, MedChemComm, 2014, 5, 1454–1471 RSC;
(d) T. Yamamoto, M. Nakatani, K. Narukawa and S. Obika, Future Med. Chem., 2011, 3, 339–365 CrossRef PubMed;
(e) C. F. Bennett and E. E. Swayze, Annu. Rev. Pharmacol. Toxicol., 2010, 50, 259–293 CrossRef PubMed.
-
(a) W. B. Wan and P. P. Seth, J. Med. Chem., 2016, 59, 9645–9667 CrossRef PubMed;
(b) G. F. Deleavey and M. J. Damha, Chem. Biol., 2012, 19, 937–954 CrossRef PubMed.
-
(a) R. O. Dempcy, K. A. Browne and T. C. Bruice, J. Am. Chem. Soc., 1995, 117, 6140–6141 CrossRef;
(b) J. Robles, A. Grandas and E. Pedroso, Tetrahedron, 2001, 57, 179–194 CrossRef;
(c) C. J. Wilds, M. A. Maier, V. Tereshko, M. Manoharan and M. Egli, Angew. Chem., Int. Ed., 2002, 41, 115–117 CrossRef;
(d) V. Roig and U. Asseline, J. Am. Chem. Soc., 2003, 125, 4416–4417 CrossRef PubMed;
(e) S. R. Gerrard, M. M. Edrees, I. Bouamaied, K. R. Fox and T. Brown, Org. Biomol. Chem., 2010, 8, 5087–5096 RSC;
(f) T. P. Prakash, A. Püschl, E. Lesnik, V. Mohan, V. Tereshko, M. Egli and M. Manoharan, Org. Lett., 2004, 6, 1971–1974 CrossRef PubMed;
(g) G. Deglane, S. Abes, T. Michel, P. Prévot, E. Vives, F. Debart, I. Barvik, B. Lebleu and J.-J. Vasseur, ChemBioChem, 2006, 7, 684–692 CrossRef PubMed;
(h) J. Brzezinska, J. D'Onofrio, M. C. R. Buff, J. Hean, A. Ely, M. Marimani, P. Arbuthnot and J. W. Engels, Bioorg. Med. Chem., 2012, 20, 1594–1606 CrossRef PubMed;
(i)
J. Barman, D. Gurav, O. P. Oommen and O. P. Varghese, RSC Adv., 2015, 5, 12257–12260 RSC.
- A. R. Shrestha, Y. Kotobuki, Y. Hari and S. Obika, Chem. Commun., 2014, 50, 575–577 RSC.
-
(a) T. Yamamoto, A. Yahara, R. Waki, H. Yasuhara, F. Wada, M. Harada-Shiba and S. Obika, Org. Biomol. Chem., 2015, 13, 3757–3765 RSC;
(b) P. P. Seth, A. Siwkowski, C. R. Allerson, G. Vasquez, S. Lee, T. P. Prakash, E. V. Wancewicz, D. Witchell and E. E. Swayze, J. Med. Chem., 2009, 52, 10–13 CrossRef PubMed;
(c) T. Yamamoto, M. Harada-Shiba, M. Nakatani, S. Wada, H. Yasuhara, K. Narukawa, K. Sasaki, M. Shibata, H. Torigoe, T. Yamaoka, T. Imanishi and S. Obika, Mol. Ther.-Nucleic Acids, 2012, 1, e22 CrossRef PubMed;
(d) T. Yamamoto, H. Yasuhara, F. Wada, M. Harada-Shiba, T. Imanishi and S. Obika, J. Nucleic Acids, 2012, 2012, 707327–707333 Search PubMed;
(e) K. Setoguchi, L. Cui, N. Hachisuka, S. Obchoei, K. Shinkai, F. Hyodo, K. Kato, F. Wada, T. Yamamoto, M. Harada-Shiba, S. Obika and K. Nakano, Mol. Ther.-Nucleic Acids, 2017, 9, 170–181 CrossRef PubMed.
- A. Yahara, A. R. Shrestha, T. Yamamoto, Y. Hari, T. Osawa, M. Yamaguchi, M. Nishida, T. Kodama and S. Obika, ChemBioChem, 2012, 13, 2513–2251 CrossRef PubMed.
- P. P. Seth, G. Vasquez, C. A. Allerson, A. Berdeja, H. Gaus, G. A. Kinberger, T. P. Prakash, M. T. Migawa, B. Bhat and E. E. Swayze, J. Org. Chem., 2010, 75, 1569–1581 CrossRef PubMed.
-
(a) S. K. Singh, R. Kumar and J. Wengel, J. Org. Chem., 1998, 63, 10035–10039 CrossRef;
(b) M. D. Sørensen, M. Petersenb and J. Wengel, Chem. Commun., 2003, 2130–2131 RSC.
-
S. Obika, E. Kawanishi, H. Sawamoto, S. Yamakoshi, Y. Arai and S. Kumagai, WO2017047816, 2017 Search PubMed.
-
M. Nakatsuka, Y. Ueno, S. Okada and F. Nishikaku, US6100260, 2000 Search PubMed.
- Y. Fukahori, Y. Takayama, T. Imaoka, O. Iwamoto and K. Nagasawa, Chem. – Asian J., 2013, 8, 244–250 CrossRef PubMed.
- A. Williams and I. T. Ibrahim, Chem. Rev., 1981, 81, 589–636 CrossRef.
-
(a) J. A. Zoltewicz, D. F. Clark, T. W. Sharpless and G. Grahe, J. Am. Chem. Soc., 1970, 92, 1741–1750 CrossRef PubMed;
(b) T. Lindahl and B. Nyberg, Biochemistry, 1972, 11, 3610–3618 CrossRef PubMed.
- Similar conditions have been reported previously. See: M. P. Reddy, N. B. Hanna and F. Farooqui, Tetrahedron Lett., 1994, 35, 4311–4314 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob01307a |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence b,
Yutaro
Kotobuki
a,
Takao
Yamaguchi
b,
Yutaro
Kotobuki
a,
Takao
Yamaguchi