
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of the proposed structure of talarolide A†
Shengping
Zhang
a,
Luis M.
De Leon Rodriguez
 b,
Renjie
Huang
a,
Ivanhoe K. H.
Leung
a,
Paul W. R.
Harris
abc and
Margaret A.
Brimble
*abc
b,
Renjie
Huang
a,
Ivanhoe K. H.
Leung
a,
Paul W. R.
Harris
abc and
Margaret A.
Brimble
*abc
aSchool of Chemical Sciences, The University of Auckland, 23 Symonds St, Auckland, 1142, New Zealand. E-mail: m.brimble@auckland.ac.nz; Fax: +649 3737422; Tel: +64 9 3737599
bMaurice Wilkins Centre for Molecular Biodiscovery, The University of Auckland, Auckland, 1142, New Zealand
cSchool of Biological Sciences, The University of Auckland, 3A Symonds Street, Auckland 1010, New Zealand
First published on 29th June 2018
Abstract
The proposed structure of talarolide A, a cycloheptapeptide featuring a hydroxamate moiety within the peptide backbone, was successfully synthesized. An initial attempt to synthesize a linear peptide precursor containing a C-terminal N-benzyloxy glycine residue was problematic due to an unreported on-resin reduction of N-benzyloxy glycine to glycine. After repositioning the peptide cyclization point, a new linear peptide sequence was successfully prepared using Fmoc-solid-phase peptide synthesis. Subsequent solution-phase cyclization and removal of protecting groups furnished the synthetic talarolide A in good yield. Despite the mismatch of the NMR data between the synthetic talarolide A and the natural product, a detailed structural analysis using 2D NMR spectroscopy, together with re-synthesis of the same synthetic material using two additional cyclization sites, confirmed that our synthetic product has the reported structure of talarolide A.
Introduction
The N-hydroxylation of peptide backbones is an important strategy for peptide post-translational modification which has mainly been found in the metabolites of microorganisms.1–4 Naturally-occurring peptides containing the N-hydroxy amide moiety have been reported as potential antibacterial and antitumor agents.2,3,5,6 Additionally, they can act as siderophores which chelate and transport metal ions essential for cell growth and proliferation.1 Synthetic peptides containing the N-hydroxy amide unit, also exhibit a wide spectrum of bioactivities including inhibition of metalloproteases and HIV protease, as well as immune suppression.7–10 It is proposed that the N-hydroxy amide functionality not only serves as a strong proton donor participating in hydrogen bonding and metal chelation but also confers enhanced stability to enzymatic degradation compared to its cognate amide counterpart.11,12Talarolide A (1) is a cyclic heptapeptide isolated from an Australian marine tunicate-associated fungus, Talaromyces sp. (CMB TU011) (Fig. 1).13 Its overall structural elucidation was recently reported by Capon et al. using de novo spectroscopic analysis and a combination of C3, C18, and 2D C3 Marfey's analyses.13 The relevant structural features of 1 are the presence of a characteristic N-hydroxy amide moiety within the peptide backbone, along with multiple N-methyl amino acids. Reports on naturally derived cyclic peptides containing both N-hydroxy and N-methyl residues are scarce and some of these peptides exhibit potent inhibitory activity against Gram-positive bacteria,2Mycobacterium tuberculosis (TB)14 and the oxytocin receptor.4 More importantly, only one recent account of their synthesis has been published to date, which involved the use of corrosive coupling reagents, orthogonal protecting groups and a multi-step synthesis of the N-hydroxylated amino acid.15 We therefore embarked on the first total synthesis of talarolide A (1) in order to confirm the proposed structure and establish a robust synthetic route towards this unusual family of peptides.
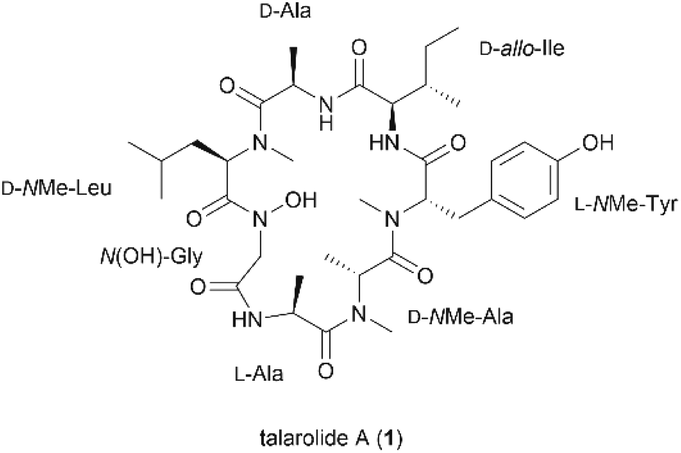 | ||
| Fig. 1 Structure of talarolide A (1). | ||
Results and discussion
We envisaged that 1 could be constructed employing Fmoc-solid-phase peptide synthesis (SPPS) to access the linear peptide precursor followed by a solution-phase head-to-tail cyclization. Specifically, a side-chain-protected linear peptide could be initially assembled on a hyperacid-labile resin and the N-hydroxy amide moiety could be introduced by incorporating an N-benzyloxy glycine (3) building block into the sequence; after releasing the linear peptide precursor from the resin, 1 can be obtained through a solution-phase macrolactamization followed by subsequent final deprotection (Scheme 1). Moreover, the presence of multiple N-methyl amino acids in the peptide sequence could improve the efficiency of the peptide cyclization step by inducing a β-turn conformation that reduces the average distance between the C- and N-terminal residues.16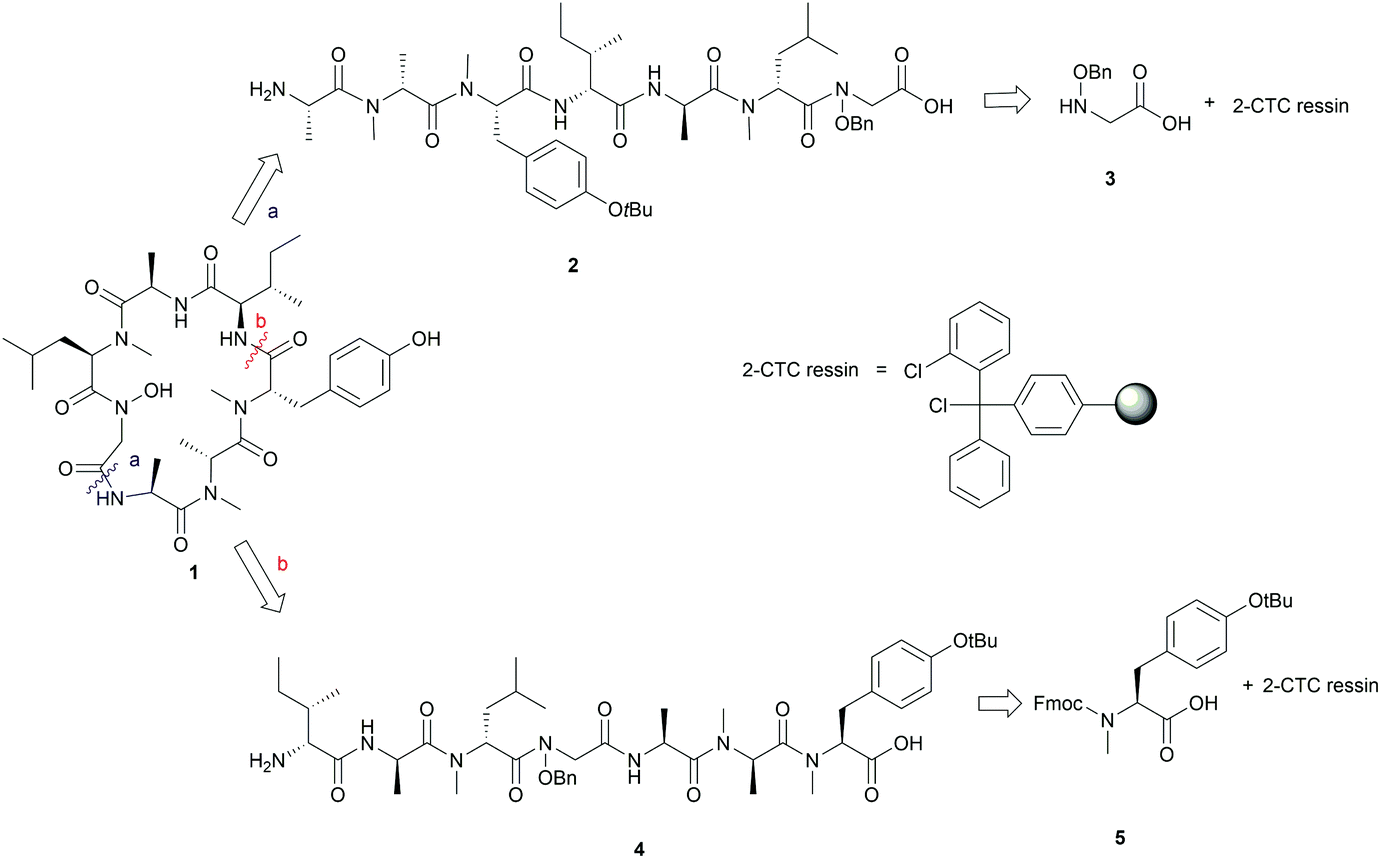 | ||
| Scheme 1 First (a) and second (b) retrosynthesis of talarolide A (1). | ||
Careful selection of the appropriate cyclization point is the key decision in order to successfully synthesize cyclic peptides. In this case, disconnections of 1 at sites that involved N-hydroxy and N-methyl amide bonds were excluded as it is known that cyclization at those sites is undesirable due to severe C-terminal epimerization and low reaction efficiency caused by the steric hindrance and decreased nucleophilicity of the terminal amine.16–19 Among the three remaining cyclization sites, the junction between N-OH-Gly and L-Ala was initially chosen, as the derived linear precursor 2 contains an achiral N-OH-Gly residue at the C-terminus, thus reducing the risk of epimerization during peptide cyclization (see disconnection a in Scheme 1).
The synthesis of 2 commenced with the attachment of the N-benzyloxy glycine (3) building block onto a 2-chlorotrityl chloride (2-CTC) resin (Scheme 2). The building block 3 was readily prepared from bromoacetic acid tert-butyl ester and O-benzylhydroxylamine according to reported methods.20,21 The absence of a protecting group on the benzyloxy amine of 3 did not affect the esterification of the first amino acid with 2-CTC resin due to its modified nucleophilicity and steric hindrance compared to unsubstituted amines.22 However, these intrinsic properties of the benzyloxy amine also posed a challenge during the following acylation step. Only strong acylating reagents, such as an acid chloride, a mixed anhydride, or O-(7-azabenzotriazol-1-yl)-N,N,N′,N′-tetramethyluronium hexafluorophosphate (HATU), were reported to be successful for the formation of an N-hydroxy amide bond.10,21,22 In this work, the coupling of Fmoc-D-NMe-Leu to the resin bound benzyloxy glycine was accomplished using HATU as the coupling reagent for 24 h. However, a second coupling was necessary to drive the reaction to completion and thus obtain 7 (Scheme 2). Pleasingly, the long reaction time did not give rise to significant epimerization which has been frequently observed in the acylation of sterically bulky amino acids.19,23
 | ||
Scheme 2 Synthesis of the initial linear peptide precursor 2. Reagents and conditions: (i) 3 (2 equiv.), DIPEA (5 equiv.), CH2Cl2, RT, 2 h; (ii) Fmoc-NMe-D-Leu (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), RT, 2 × 1 day; (iii) iterative Fmoc-SPPS ((a) 20% piperidine in DMF, RT, 2 × 5 min; (b) Fmoc-AA-OH (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), DMF, RT, 1 h); (iv) 20% piperidine in DMF, RT, 2 × 5 min; (v) HFIP/CH2Cl2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :4), RT, 1 h. :4), RT, 1 h. | ||
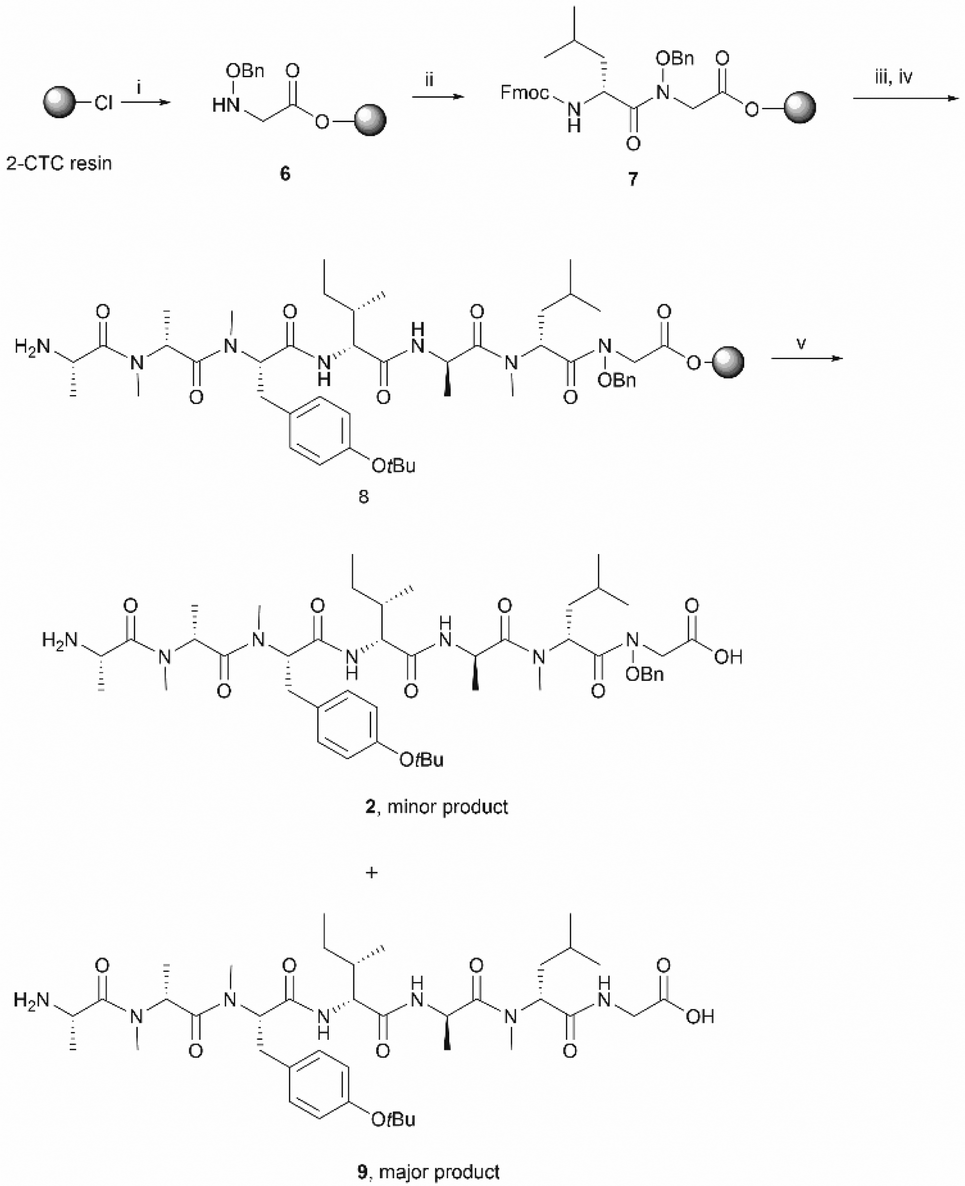
Subsequent peptide elongation with the remaining amino acids was then performed using HATU as the coupling reagent and 20% piperidine in DMF for Nα-Fmoc-deprotection, which led to the resin bound peptide 8. However, LC-MS analysis of a crude sample resulting from the cleavage of 2 from the resin showed an unexpected loss of the benzyloxyl fragment of the N-hydroxy glycine residue which resulted in the corresponding C-terminal glycine derivative 9 as the major product (Scheme 2). This side reaction was attributed to the repetitive treatment with piperidine, given that accumulation of the corresponding C-terminal reduced product was observed after each amino acid coupling cycle. For instance, while approximately 50% of the reduced tetrapeptide was observed after cleavage of the NH2-D-allo-Ile-D-Ala-D-NMe-Leu-(NOBn)Gly-OH peptide from the resin, the reduced peptide 9 (found 776.4 [M + H]+, calcd 776.5) accounted for ∼80% of the final product (Fig. S1, S2,†Scheme 2). The loss of the benzyloxy fragment was attributed to the thermal reduction of the N-benzyloxy glycine residue to glycine, which has not been reported in previous synthesis of hydroxamate-containing peptides.10,21 However, it is important to note that in those previous syntheses the N-hydroxy residues were located in the middle of the peptide sequence rather than in the C-terminal region, as is the case presented in Scheme 2. We therefore postulated that the ester bond linkage between the first amino acid and the resin rendered the N-acyl-N-benzyloxy glycine moiety prone to a base-induced elimination to afford the corresponding α-acylimine intermediate 10.22,24–26 After attack by a “soft nucleophile” such as piperidine, an N-substituted glycine derivative 11 is generated, which then undergoes further thermal reduction to give the corresponding glycine derivative 12 (Scheme 3).22,27
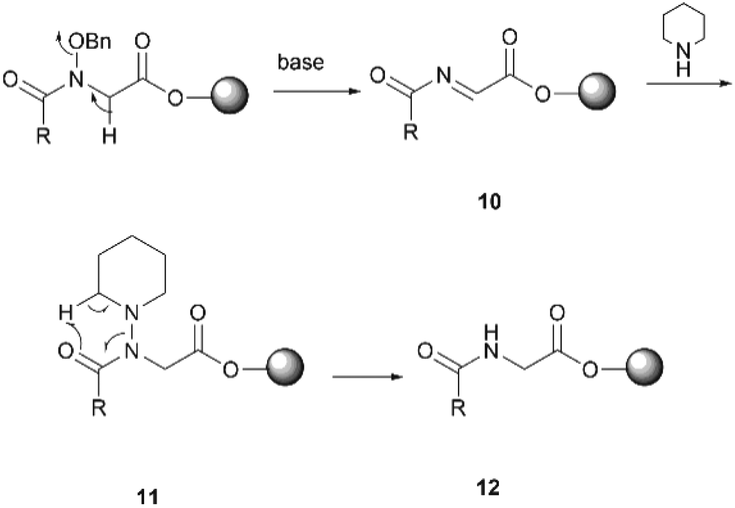 | ||
| Scheme 3 The proposed mechanism for the reduction of the C-terminal N-acyl-N-benzyloxyl glycine residue. | ||
Having established that the C-terminal N-acyl-N-benzyloxy glycine ester was not stable to repetitive treatment with bases such as DIPEA and piperidine, we decided to change the cyclization site to the junction between NMe-Tyr and D-allo-Ile so that the building block 3 would be repositioned in the middle of peptide sequence (Scheme 1, path b). The resulting linear precursor 4 was then assembled on a 2-CTC resin as shown in Scheme 4. The coupling of 3 to the resin-bound tripeptide 15 was performed using a mixture of 3, N,N′-diisopropylcarbodiimide (DIC) and 6-chloro-1-hydroxybenzotriazole (6-Cl-HOBt) in DMF for 24 h as powerful coupling reagents, such as HATU, might result in self–condensation of 3 during the long reaction time. For the Fmoc deprotection of 18 we encountered significant diketopiperazine formation which could not be alleviated using milder basic conditions (50% morpholine in DMF and 5% piperizine in DMF containing 0.1 M 6-Cl-HOBt). Fortunately, this undesirable side reaction was minimized using 20% piperidine in DMF and a short deprotection time (2 × 30 seconds), thereby affording 19 in almost quantitative yield (Scheme 4).
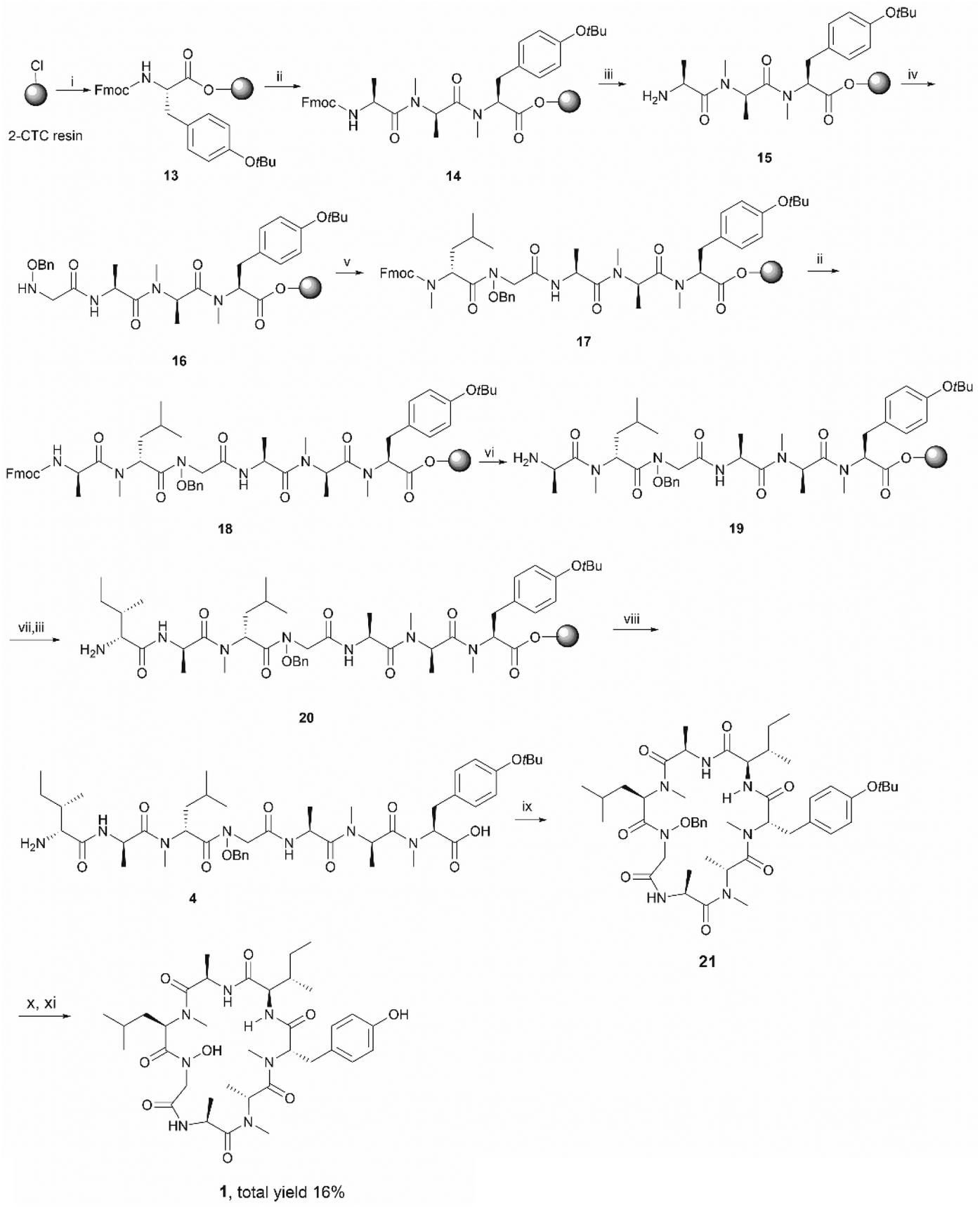 | ||
| Scheme 4 The second synthetic route towards talarodlide A (1). Reagents and conditions: (i) Fmoc-NMe-Tyr(OtBu)-OH (2 equiv.), DIPEA (5 equiv.), CH2Cl2, RT, 1 h; (ii) iterative Fmoc-SPPS ((a) 20% piperidine in DMF, RT, 2 × 5 min; (b) Fmoc-AA-OH (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), DMF, RT, 1 h); (iii) 20% piperidine in DMF, RT, 2 × 5 min; (iv) 3 (4 equiv.), DIC (4 equiv.), 6-Cl-HOBt (4 equiv.), DMF, RT, 24 h; (v) Fmoc-NMe-D-Leu (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), RT, 2 × 1 day; (vi) 20% piperidine in DMF, RT, 2 × 30 s; (vii) Fmoc-D-allo-Ile (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), DMF, RT, 1 h; (viii) HFIP/CH2Cl2 (1:4), RT, 1 h; (ix) PyBOP (3 equiv.), DIPEA (5 equiv.), DMF, RT, 1 day; (x) 90% formic acid in water, RT, 40 min; (xi) Pd–C, H2, MeOH, RT, 90 min. | ||
The protected linear precursor 4 (found 882.5 [M + H]+, calcd 882.5) was released from the resin upon treatment with 20% hexafluoroisopropanol (HFIP) in CH2Cl2 and the crude peptide was taken to the next step without further purification. The linear peptide 4 was then subjected to peptide cyclization in solution using (benzotriazol-1-yloxy)tripyrrolidinophosphonium hexafluorophosphate (PyBOP) as the cyclizing reagent under high dilution conditions (0.75 mM).28,29 LC-MS analysis of the crude reaction mixture indicated the rapid formation of a single product which exhibited the correct mass of the protected cyclic peptide 21. It is important to note that C-terminal epimerization was not detected during the synthesis, which echoed our hypothesis that a ring-closing conformation would be favored in a peptide sequence encompassing multiple N-methyl amino acids.
Subsequent deprotection of the t-butyl and benzyl group of 21, using 90% formic acid aqueous solution and palladium-catalyzed hydrogenation respectively, furnished the desired final product 1 in good yield (16% overall yield based on the determined loading of 2-CTC resin (0.48 mmol g−1)) (Scheme 4). It is also worth noting that cyclic peptide 21 was unstable in typical TFA-mediated t-butyl removal conditions, under which conditions it underwent a ring opening reaction to generate the corresponding linear peptide 2.
The synthetic talarolide A (1) was then characterized by 1H and 13C NMR spectroscopy and the obtained data were compared to those reported for the natural product. Unfortunately, the 1H and 13C NMR spectra of 1 did not match those of the natural product. More importantly, two sets of signals with an approximate 1:1 ratio were observed in the 1H-NMR spectrum of 1, which were attributed to the existence of potential conformers as the N-methyl or N-hydroxy amide moieties in talarolide A (1) are capable of assuming either a cis or trans conformation (Fig. 2).30–32
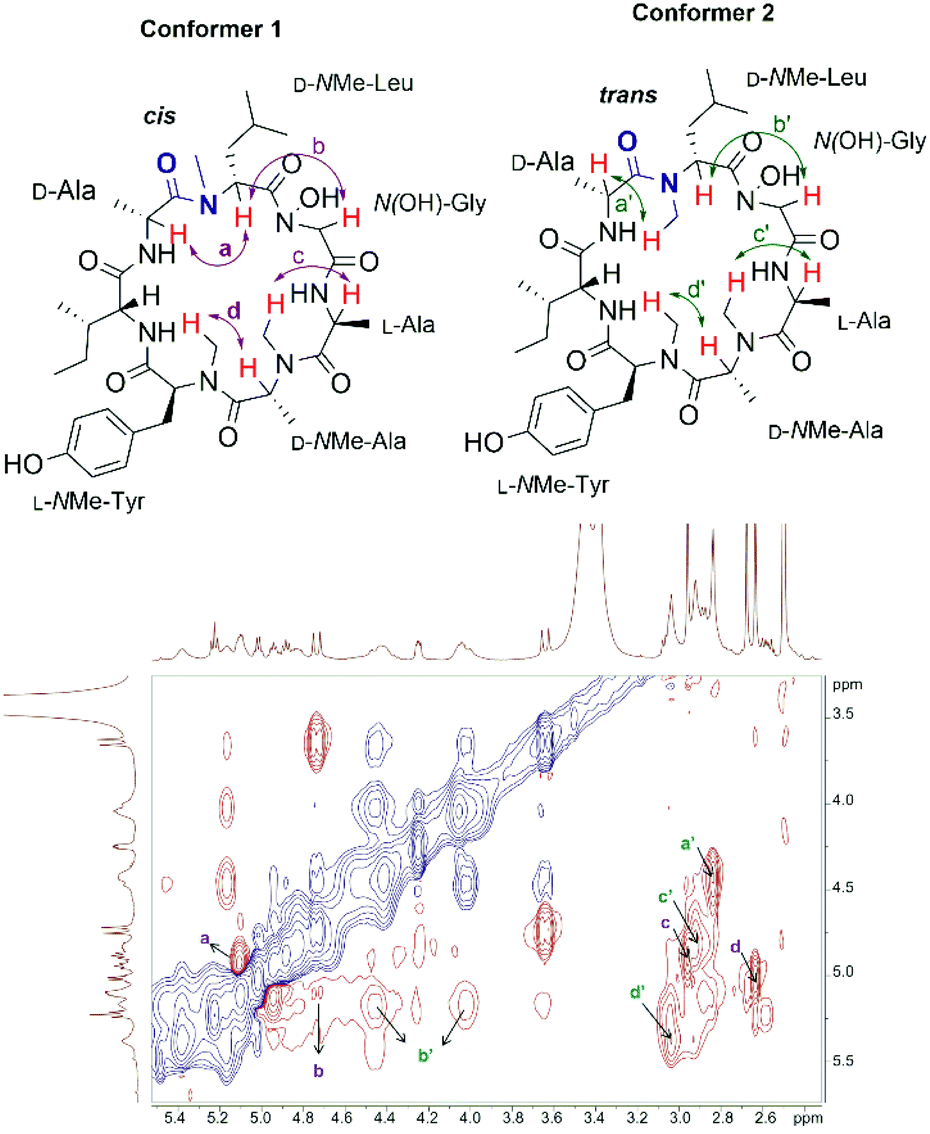 | ||
| Fig. 2 Expanded view of the ROESY spectrum of synthetic talarolide A (1). | ||
In order to support this hypothesis and unambiguously assign all the proton signals in the NMR spectrum to each individual conformer, full NMR characterization of the synthetic talarolide A (1) was carried out. The correct peptide sequences were confirmed for both conformers by HMBC. Moreover, the ROESY spectrum of 1 rendered a comprehensive profile of the spatial relationship between all the protons, from which the isomerization state for each N-methyl and N-hydroxy amide bonds can be deduced. As shown in Fig. 2, a correlation between the Hα of N(OH)-Gly and D-NMe-Leu residue (b and b′ in Fig. 2) was found in both conformers, thus indicating a cis conformation for the N-hydroxy amide moiety. In addition, all the amide bonds that involved N-methyl substitution assumed the trans conformation in both conformers, except the one between the D-NMe-Leu and D-Ala residues. The correlation observed between the NCH3 proton of D-NMe-Leu and the Hα of D-Ala in conformer 2 (a′ in Fig. 2) suggested the presence of a trans conformation while a cis conformation was identified in conformer 1, which was evidenced by the correlation between the Hα of D-NMe-Leu and D-Ala (a in Fig. 2). This finding pinpointed the position of amide bond where the cis/trans isomerization occurred in the synthetic talarolide A (1), thus providing deep insight into the structural difference between the two conformers observed in the NMR experiment. Unfortunately, the published ROESY spectrum of the natural product did not provide sufficient information regarding the isomerization state of each amide bond and only a trans conformation was determined for the amide bond between D-NMe-Leu and D-Ala residue.13
Variable temperature NMR experiments were then carried out. As shown in Fig. 3, the proton resonances of these two conformers were well resolved at 27 °C, possibly due to the slow interconversion between the cis and trans conformations (see below). As the temperature increased, the rate of interconversion increased, leading to peak broadening and signal coalescence, as demonstrated by the ratio between the two conformers, which went from 1:1 at RT to 1:0.5 at 60 °C (the peaks in the Cα–H region were integrated). However, a complete fusion of these two conformers had not been achieved even when the temperature reached 60 °C, thus indicating a relatively high energy barrier between them. A concentration-dependent NMR experiment was performed to examine the effect of peptide concentration on its 1H-NMR spectrum. The obtained 1H-NMR spectra of 1 at three different concentrations did not exhibit significant varation, hence it is concluded that peptide concentration was not a factor that could explain the differences observed in the NMR spectra of the synthetic and natural talarolide A peptides (Fig. S12†).
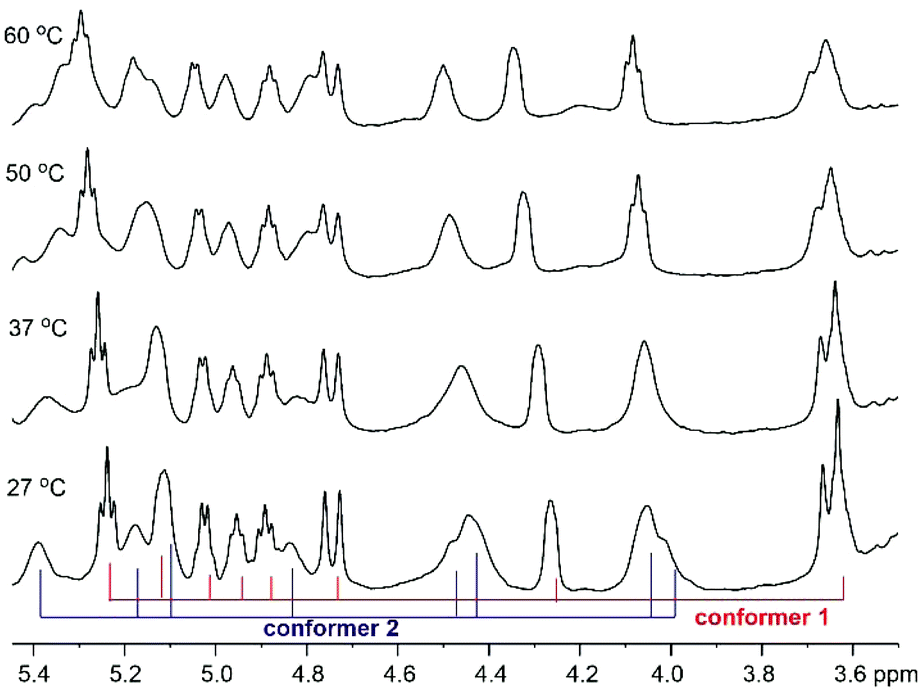 | ||
| Fig. 3 Cα–H region of the 1H NMR spectra (500 MHz, DMSO-d6) of synthetic talarolide A (1) at 27 °C, 37 °C, 50 °C and 60 °C. | ||
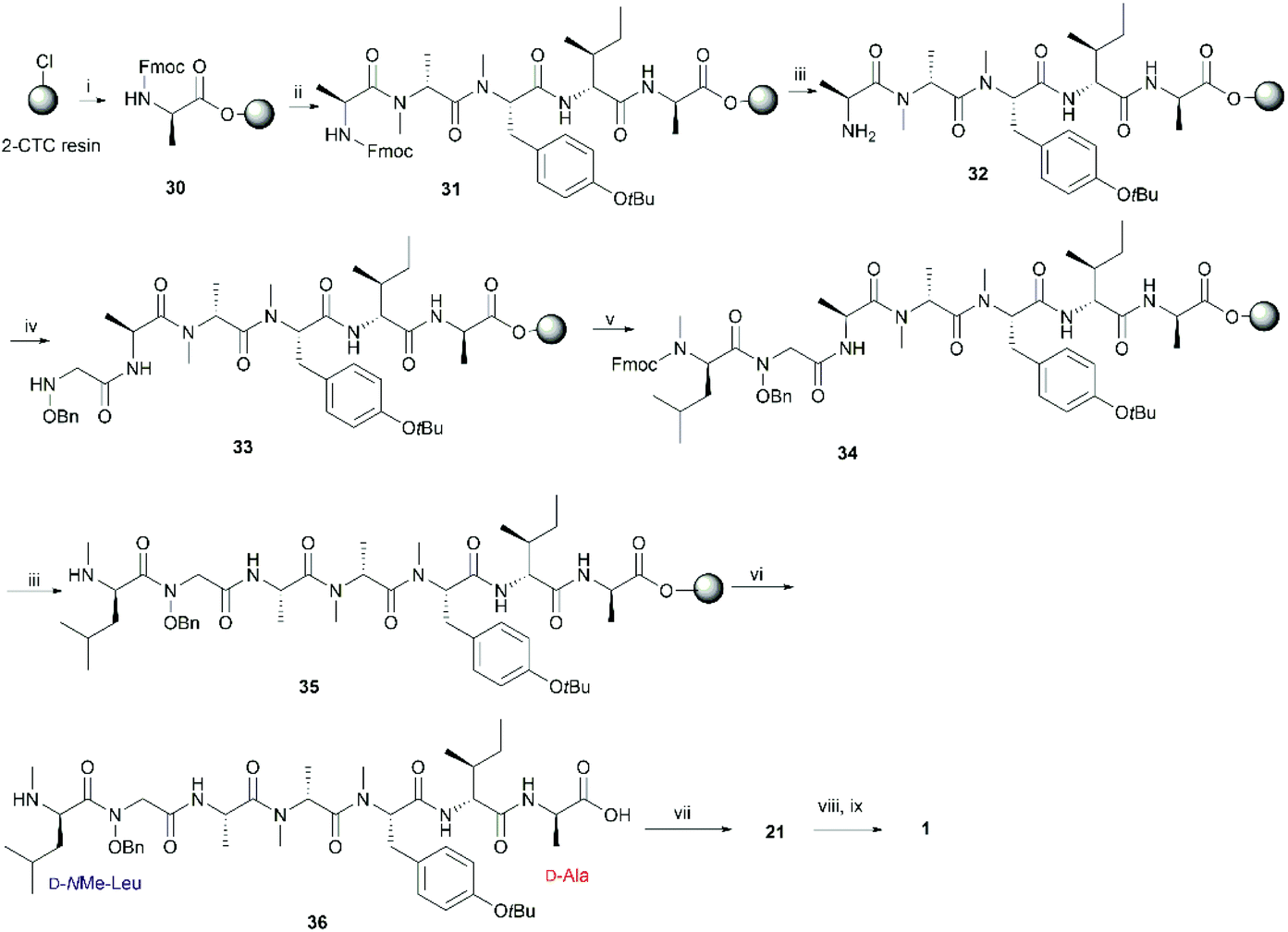
In order to further confirm the validity of our synthesis of 1 and to evaluate the impact of different cyclizing sites on peptide conformation, we also synthesized 1 using two different disconnecting points (Schemes 5 and 6). Both linear precursors 29 and 36 were successfully prepared on 2-CTC resin using the same method as described above for the synthesis of 4. Macrolactamization of 29 in DMF (0.75 mM) proceeded smoothly using PyBOP while cyclization of 36 using the same conditions failed to give the desired peptide 21 probably due to the steric hindrance of the N-methylated amine at N-terminus. Linear peptide 36 was successfully cyclized using HATU, but significant C-terminal epimerization was observed during this process. This phenomenon agreed well with previous reports where severe epimerization took place during the cyclization of peptides that contained an N-methyl amino acid at the N-terminus.17–19 After removal of protecting groups, the final product 1 derived from 29 and 36 was obtained in 7% and 2% overall yield respectively (based on the corresponding loading of 2-CTC resin (0.41 mmol g−1 and 0.55 mmol g−1)). Finally, the samples of synthetic talarolide A (1) obtained from these two different cyclizing points were characterized by 1H-NMR, both of which exhibited the same 1H-NMR spectrum as the one we obtained via the synthetic route outlined in Scheme 4, thereby ruling out the existence of possible epimers in the final product (Fig. 4). Moreover, given the fact that the two conformers were consistently observed in all the 1H-NMR spectra of 1 (Fig. 4), it was concluded that the selection of a different cyclization site for execution of the synthesis had a negligible effect on the conformation of the final peptide.
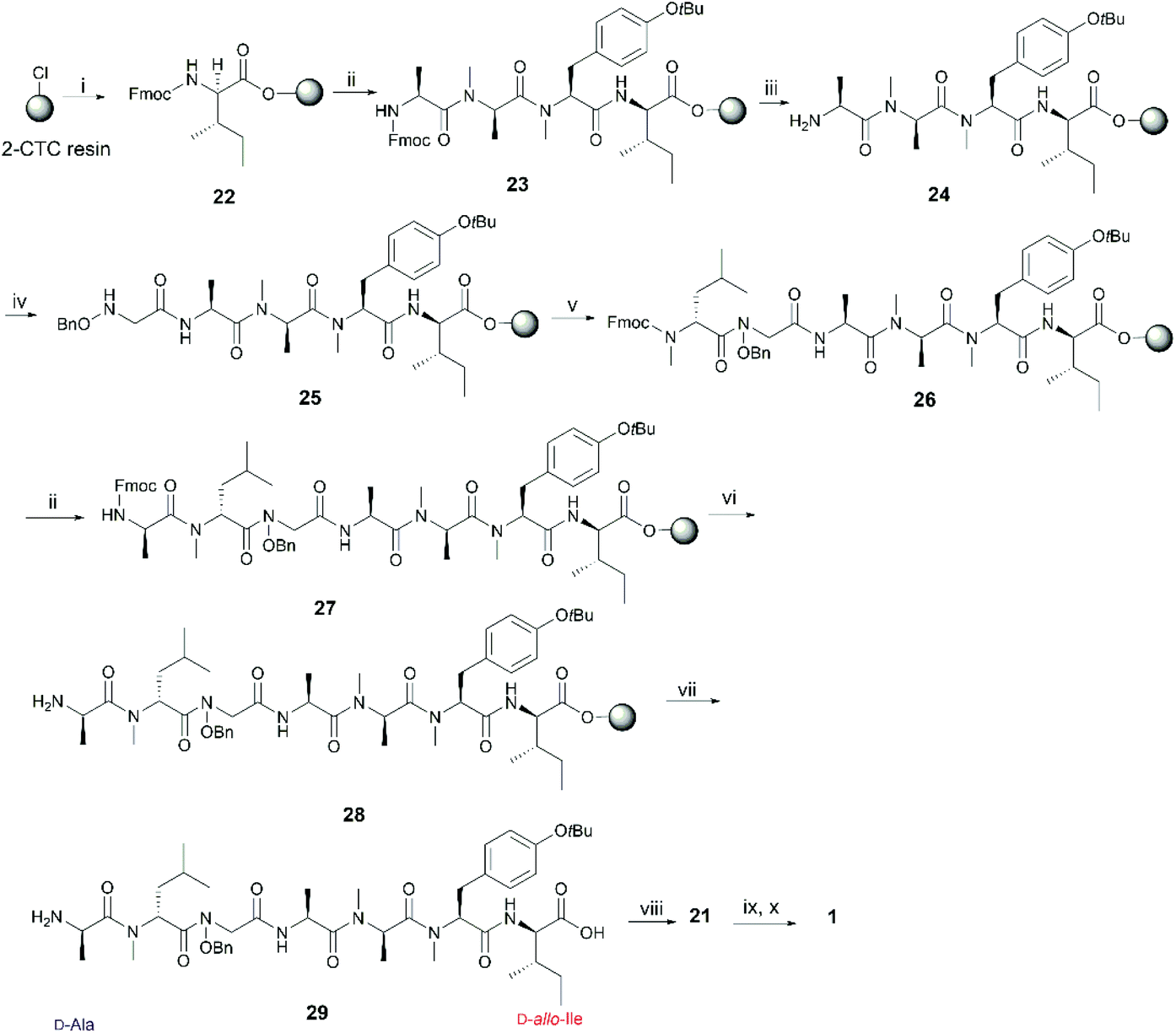 | ||
| Scheme 5 Re-synthesis of 1 from the junction between D-allo-Ile and D-Ala. Reagents and conditions: (i) Fmoc-D-allo-Ile-OH (2 equiv.), DIPEA (5 equiv.), CH2Cl2, RT, 1 h; (ii) iterative Fmoc-SPPS ((a) 20% piperidine in DMF, RT, 2 × 5 min; (b) Fmoc-AA-OH (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), DMF, RT, 1 h); (iii) 20% piperidine in DMF, RT, 2 × 5 min; (iv) 3 (4 equiv.), DIC (4 equiv.), 6-Cl-HOBt (4 equiv.), DMF, RT, 24 h; (v) Fmoc-NMe-D-Leu (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), RT, 2 × 1 day; (vi) 20% piperidine in DMF, RT, 2 × 30 s; (vii) HFIP/CH2Cl2 (1:4), RT, 1 h; (viii) PyBOP (3 equiv.), DIPEA (5 equiv.), DMF, RT, 1 day; (ix) 90% formic acid in water, RT, 40 min; (x) Pd-C, H2, MeOH, RT, 90 min. | ||
 | ||
| Scheme 6 Re-synthesis of 1 from the junction between D-Ala and NMe-D-Leu. Reagents and conditions: (i) Fmoc-D-Ala (2 equiv.), DIPEA (5 equiv.), CH2Cl2, RT, 1 h; (ii) iterative Fmoc-SPPS ((a) 20% piperidine in DMF, RT, 2 × 5 min; (b) Fmoc-AA-OH (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), DMF, RT, 1 h); (iii) 20% piperidine in DMF, RT, 2 × 5 min; (iv) 3 (4 equiv.), DIC (4 equiv.), 6-Cl-HOBt (8 equiv.), DMF, RT, 24 h; (v) Fmoc-NMe-D-Leu (4 equiv.), HATU (4 equiv.), DIPEA (8 equiv.), RT, 2 × 1 day; (vi) HFIP/CH2Cl2 (1:4), RT, 1 h; (vii) HATU (3 equiv.), HOAt (3 equiv.), DIPEA (5 equiv.), DMF, RT, 1 day; (viii) 90% formic acid in water, RT, 40 min; (ix) Pd–C, H2, MeOH, RT, 90 min. | ||
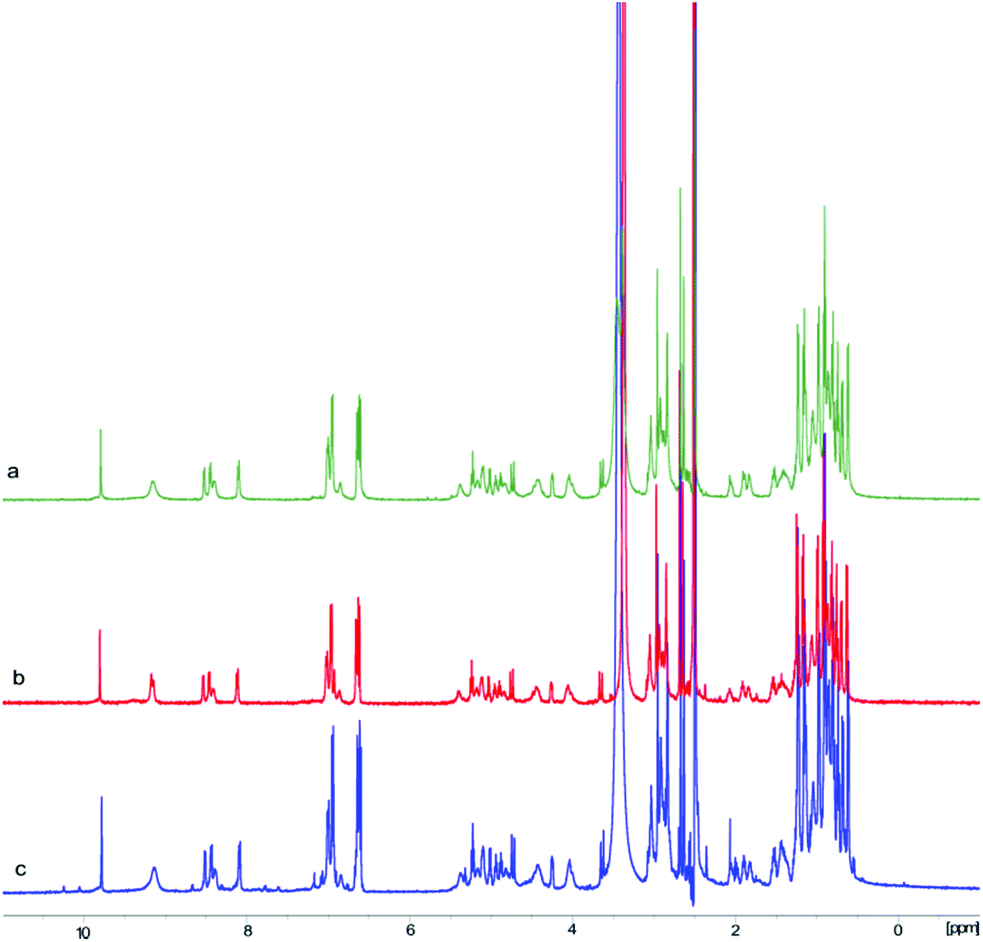 | ||
| Fig. 4 Stacked 1H-NMR (500 MHz, DMSO-d6) spectra of 1 prepared from three different cyclization sites. (a) 1H-NMR spectrum of 1 synthesized via linear peptide 4; (b) 1H-NMR spectrum of 1 synthesized via linear peptide 29; (c) 1H-NMR spectrum of 1 synthesized via linear peptide 36. The slight differences in the three spectra are likely due to impurities from the purification steps. | ||
Conclusions
In conclusion, the proposed structure of talarolide A (1) was successfully synthesized using a combination of Fmoc-SPPS and solution-phase macrolactamization. Interestingly, a novel on-resin reduction of the N-benzyloxy glycine residue was identified during the synthesis, which led to a low-yielding synthesis of the initial linear peptide. This side reaction was circumvented by repositioning the N-benzyloxy glycine residue to the middle of the linear peptide sequence. In this case, after cyclization of the linear peptide and removal of protecting groups, 1 was successfully obtained in good yield. Unfortunately, the 1H and 13C NMR spectra of the synthetic talarolide A (1) did not match those of the natural product and revealed the existence of two different conformers. Subsequent 2D-NMR analysis of 1, together with the re-synthesis of 1 using two different cyclization points, fully supported 1 as the structure of our synthetic talarolide A (1) and also pinpointed the structural difference between the two coexisting conformers. The work reported herein suggests that further studies are required to establish the structural difference between synthetic and natural talarolide A.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful for financial support from the Maurice Wilkins Centre for Molecular Biodiscovery and a PhD scholarship from the China Scholarship Council (S. Z.).Notes and references
- H. Drechsel and G. Jung, J. Pept. Sci., 1998, 4, 147–181 CrossRef PubMed.
- M. Igarashi, R. Sawa, N. Kinoshita, H. Hashizume, N. Nakagawa, Y. Homma, Y. Nishimura and Y. Akamatsu, J. Antibiot., 2008, 61, 1881–1469 CrossRef PubMed.
- H. Hashizume, R. Sawa, K. Yamashita, Y. Nishimura and M. Igarashi, J. Antibiot., 2017, 70, 699 CrossRef PubMed.
- M. A. Goetz, C. D. Schwartz, L. R. Koupal, J. M. Liesch, O. D. Hensens, R. Freidinger, P. S. Anderson, D. J. Pettibone and B. H. Woodruff, Merck & Co Inc (US), EP0256847A2, 1988 Search PubMed.
- H. Hashizume, H. Adachi, M. Igarashi, Y. Nishimura and Y. Akamatsu, J. Antibiot., 2010, 63, 279 CrossRef PubMed.
- K. Umezawa, K. Nakazawa, T. Uemura, Y. Ikeda, S. Kondo, H. Naganawa, N. Kinoshita, H. Hashizume, M. Hamada and T. Takeuchi, Tetrahedron Lett., 1998, 39, 1389–1392 CrossRef.
- D. V. Patel, M. G. Young, S. P. Robinson, L. Hunihan, B. J. Dean and E. M. Gordon, J. Med. Chem., 1996, 39, 4197–4210 CrossRef PubMed.
- E. Bourdel, S. Doulut, G. Jarretou, C. Labbe-Jullie, J. A. Fehrentz, O. Doumbia, P. Kitabgi and J. Martinez, Int. J. Pept. Protein Res., 1996, 48, 148–155 CrossRef PubMed.
- M. Marastoni, M. Bazzaro, S. Salvadori, F. Bortolotti and R. Tomatis, Bioorg. Med. Chem., 2001, 9, 939–945 CrossRef PubMed.
- A. Bianco, C. Zabel, P. Walden and G. Jung, J. Pept. Sci., 1998, 4, 471–478 CrossRef PubMed.
- T. Kolasa, Tetrahedron, 1983, 39, 1753–1754 CrossRef.
- A. Bianco, D. Kaiser and G. Jung, Chem. Biol. Drug Des., 1999, 54, 544–548 Search PubMed.
- P. Dewapriya, P. Prasad, R. Damodar, A. A. Salim and R. J. Capon, Org. Lett., 2017, 19, 2046–2049 CrossRef PubMed.
- S. B. Singh, J. Odingo, M. A. Bailey, B. Sunde, A. Korkegian, T. O'Malley, Y. Ovechkina, T. R. Ioerger, J. C. Sacchettini and K. Young, bioRxiv, Microbiol., 2018, 279307 Search PubMed.
- Y. M. Elbatrawi, C. W. Kang and J. R. Del Valle, Org. Lett., 2018, 20, 2707–2710 CrossRef PubMed.
- C. J. White and A. K. Yudin, Nat. Chem., 2011, 3, 509 CrossRef PubMed.
- A. K. Ghosh and C.-X. Xu, Org. Lett., 2009, 11, 1963–1966 CrossRef PubMed.
- S. Zhang, L. M. De Leon Rodriguez, E. Lacey, A. M. Piggott, I. K. Leung and M. A. Brimble, Eur. J. Org. Chem., 2017, 149–158 CrossRef.
- M. Teixidó, F. Albericio and E. Giralt, J. Pept. Res., 2005, 65, 153–166 CrossRef PubMed.
- X. Hu, J. Zhu, S. Srivathsan and D. Pei, Bioorg. Med. Chem. Lett., 2004, 14, 77–79 CrossRef PubMed.
- Y. Ye, M. Liu, J. L. K. Kao and G. R. Marshall, Pept. Sci., 2003, 71, 489–515 CrossRef PubMed.
- H. C. J. Ottenheijm and J. D. M. Herscheid, Chem. Rev., 1986, 86, 697–707 CrossRef.
- A. Siow, G. Opiyo, I. Kavianinia, F. F. Li, D. P. Furkert, P. W. R. Harris and M. A. Brimble, Org. Lett., 2018, 20, 788–791 CrossRef PubMed.
- R. E. Steiger, J. Biol. Chem., 1944, 153, 691–692 Search PubMed.
- H. C. J. Ottenheijm, R. Plate, J. H. Noordik and J. D. M. Herscheid, J. Org. Chem., 1982, 47, 2147–2154 CrossRef.
- T. Kolasa, Synthesis, 1983, 539–539 CrossRef.
- J. D. M. Herscheid, R. J. F. Nivard, M. W. Tijhuis, H. P. H. Scholten and H. C. J. Ottenheijm, J. Org. Chem., 1980, 45, 1880–1885 CrossRef.
- H. Kaur, A. M. Heapy, R. Kowalczyk, Z. Amso, M. Watson, J. Cornish and M. A. Brimble, Tetrahedron, 2014, 70, 7788–7794 CrossRef.
- S. Zhang, Z. Amso, L. M. De Leon Rodriguez, H. Kaur and M. A. Brimble, J. Nat. Prod., 2016, 79, 1769–1774 CrossRef PubMed.
- Y. Takeuchi and G. R. Marshall, J. Am. Chem. Soc., 1998, 120, 5363–5372 CrossRef.
- V. Dupont, A. Lecoq, J. P. Mangeot, A. Aubry, G. Boussard and M. Marraud, J. Am. Chem. Soc., 1993, 115, 8898–8906 CrossRef.
- M. Akiyama, K. Lesaki, A. Katoh and K. Shimizu, J. Chem. Soc., Perkin Trans. 1, 1986, 851–855 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob01230j |
| This journal is © The Royal Society of Chemistry 2018 |