
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthetic antibody protein mimics of infliximab by molecular scaffolding on novel CycloTriVeratrilene (CTV) derivatives†
Ondřej
Longin
,
Mohammed
Hezwani
,
Helmus
van de Langemheen
and
Rob M. J.
Liskamp
 *
*
School of Chemistry, Joseph Black Building, University of Glasgow, University Avenue, Glasgow G12 8QQ, UK and Department of Pharmaceutical Sciences, Faculty of Science, Utrecht University, P.O. Box 80082, NL-3508 TB Utrecht, The Netherlands. E-mail: Robert.liskamp@glasgow.ac.uk
First published on 5th June 2018
Abstract
Syntheses of novel semi-orthogonally protected CycloTriVeratrilene (CTV) analogues with enhanced water solubility, that is 3 and 4, derived from the previously described CTV scaffold derivative 2 are described here. These scaffolds 2–4 enabled a sequential introduction of three different complementarity determining region (CDR) mimics via Cu(I)-catalysed azide–alkyne cycloaddition towards medium-sized protein mimics denoted as “synthetic antibodies”. The highly optimised sequential introduction enabled selective attachment of three different CDR mimics in a one-pot fashion. This approach of obtaining synthetic antibodies, demonstrated by the synthesis of paratope mimics of monoclonal antibody infliximab (Remicade®), provided a facile access to a range of (highly) pre-organised molecules bearing three different (cyclic) peptide segments and may find a wide range of applications in the field of protein–protein interaction disruptors as well as in the development of synthetic vaccines or lectin mimics. The prepared synthetic antibodies were tested for their affinity towards tumour necrosis factor alpha using surface plasmon resonance and synthetic antibodies with micromolar affinities were uncovered.
Introduction
For several years we have been interested in the molecular construction and further development of protein mimics toward medium-sized molecule alternatives of biologics such as antibodies and vaccines.1–4 In this area, we are facing several important challenges, which may determine a successful outcome of this quest. One challenge of our continuous attention is the development of pre-organized molecular scaffolds as the core molecular unit for attaching complementarity determining region (CDR)-loops mimics. This goes hand-in-hand with our desire to develop scalable syntheses. In these endeavours we are increasingly faced with a limited or even poor solubility of our molecular constructs. With respect to this, it is of course realised that an important condition for the use of any bio-active compound is an adequate solubility in an aqueous environment. In fact a (very)poor water solubility or insolubility is responsible for the largest attrition rate of molecule candidates in the drug discovery process.5 If it turns out to be impossible to improve water solubility by chemical modification, then depending on the drug candidate, more sophisticated drug delivery approaches are called for. Thus, the solubility issue is a crucial challenge in our approaches and if it can be dealt with in an early stage of molecular construction of our protein mimics, this is a preferred route. Concomitantly, convenience and scalability of synthesis remain very important issues in the assembly of these medium-size molecules.Although paratope mimics consisting of a single CDR loop have recently been reported to be successful as synthetic antibody mimics,6 molecular construction of mimics having more than one cyclic peptide mimicking the individual CDR loops may still be very beneficial for their affinity and selectivity.1,2
Recently, we described4 the synthesis of the first semi-orthogonally protected derivative of a highly pre-organized CTV scaffold 1 (Fig. 1), the CTV scaffold derivative 2 (Fig. 2) onto which three different peptide segments representing CDR loops can be sequentially introduced. Furthermore, a convenient “click and cleavage” approach was presented for the sequential introduction of the three different CDR loop mimics onto this scaffold derivative towards the synthesis of synthetic antibodies.
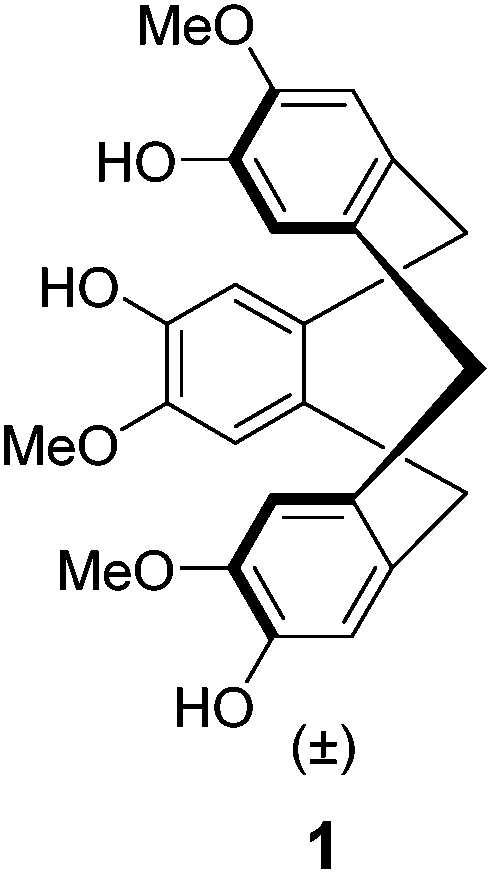 | ||
| Fig. 1 CTV scaffold enantiomers. | ||
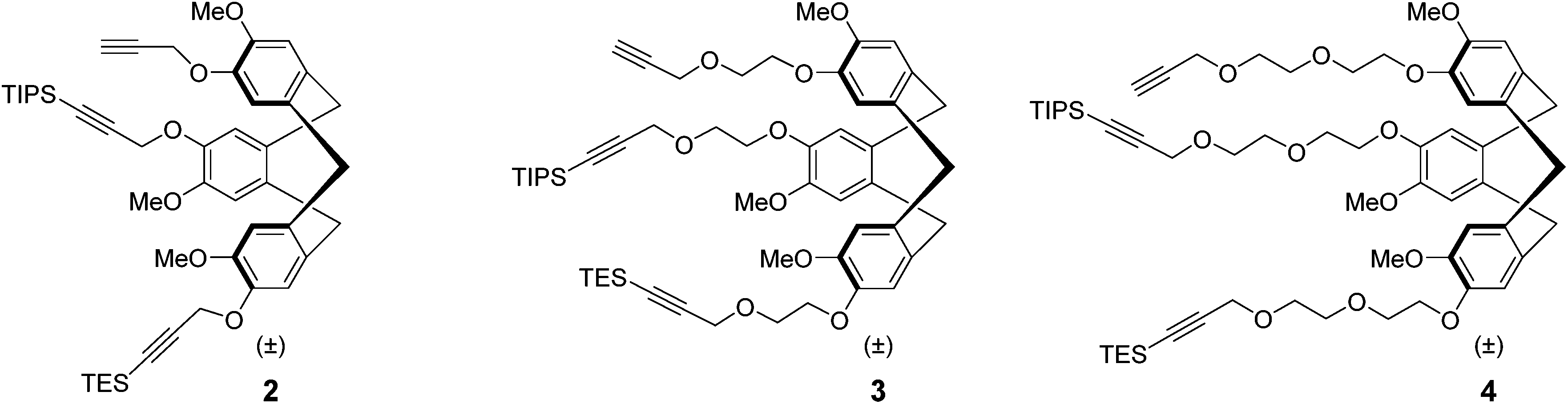 | ||
| Fig. 2 Previously prepared semi-orthogonally protected CTV scaffold derivative 2 and the newly synthesised mono and diethylene glycol spacer containing analogues 3 and 4. | ||
Although synthesis of relatively complex synthetic antibodies could be achieved quite conveniently, the water solubility of the resulting protein mimic was rather low. Hence, in order to improve water solubility and thus facilitate subsequent evaluation of synthetic antibodies, the semi-orthogonally protected CTV scaffold derivatives 3 and 4 (Fig. 2) with improved water solubility were developed. In addition, the incorporation of a mono or diethylene glycol spacer (MEG or DEG) will indicate whether it is necessary to allow more space for accommodation of the CDR loop mimics, since they are significantly larger than the CTV scaffold itself and an elongated spacer may provide additional room. To obtain some insight into the structure of these CTV derivatives, molecular models were constructed using the Maestro (http://www.schrödinger.com) modelling suite in which MacroModel software was incorporated (Fig. 3). These molecular models (without silyl protecting groups) clearly show the pre-organized bowl shape of all three CTV derivatives. In addition the mono ethylene glycol moieties containing CTV derivative 3 may, next to increasing water solubility, provide some room for the relatively large peptide loops to be attached. In the model of the diethylene glycol moieties containing CTV derivative 4, likely increased water solubility due to the diethylene glycol moieties, may reduce the pre-organization of the molecular construct, because of their increased flexibility.
 | ||
| Fig. 3 Molecular models of CTV derivatives 2–4 (without TES and TIPS protecting groups) showing their pre-organization and possible influence of the mono or diethylene glycol moieties. | ||
Furthermore, the previously described “click and cleavage” protocol for synthesis of synthetic antibodies, which required purification and lyophilisation after each step of synthesis, has been optimised in such degree that purification and lyophilisation is only required at the very end of the synthesis. Moreover, the optimised one-pot protocol includes splitting of the reaction mixture towards the end of synthesis allowing for two different synthetic antibodies to be prepared simultaneously. These changes significantly reduced time required for synthesis of synthetic antibodies and greatly improved the overall yield of synthesis. Thus, two synthetic antibodies have now been prepared within only 28 hours with overall yield of 12% to 21%, corresponding to 65% to 73% per step of the five-step synthesis.
This optimised protocol was applied in the synthesis of synthetic antibodies, which mimic the paratope of monoclonal antibody (mAb) infliximab (Remicade®). Infliximab is a clinically used biologic for treatment of human tumour necrosis factor alpha (hTNFα) mediated autoimmune diseases and directly binds to hTNFα. Cyclised versions of CDRs of the paratope, i.e. CDR mimics 28–31, were introduced on the CTV scaffold derivatives 2, 3 and 4 to yield a collection of synthetic antibodies comprising of 12 compounds. The ability of the prepared synthetic antibodies to bind to hTNFα was tested by Surface Plasmon Resonance (SPR) using expressed hTNFα immobilised on a CM5 chip. The best hTNFα-binder, antibody mimic 36, exhibited a KD of 11 μM.
Results and discussion
The recent development of a semi-orthogonally protected CycloTriVeratrylene (CTV) scaffold derivative 2 opened up the way towards design and synthesis of other semi-orthogonally protected CTV scaffold derivatives with altered physico-chemical properties; that is mono and diethylene glycol spacer containing CTV scaffold derivatives 3 and 4.The required (oligo)ethylene glycol linkers for alkylation of the CTV scaffold, i.e. linkers of a and b series, were prepared by similar synthetic pathways (Schemes 1 and 2). Firstly, mono ethylene glycol 6a and diethylene glycol 6b were treated with sodium hydride and potassium tert-butoxide, respectively, followed by a treatment with propargyl bromide to yield the corresponding propargyl ether derivatives 7a and 7b. Next, the hydroxyl group of propargyl containing derivatives 7a,b was protected with dihydropyran to provide tetrahydropyranyl (THP) protected derivatives 8a,b. Then, the free alkyne was silylated with triethylsilyl (TES) chloride or triisopropylsilyl (TIPS) chloride using n-butyl lithium as a base to yield TES and TIPS protected derivatives 9a,b and 10a,b, respectively. Finally, the THP protected hydroxyl groups of the derivatives 9a,b and 10a,b were converted into bromides using bromine and triphenylphospine to give the desired TES 11a,b and TIPS 12a,b protected alkylation linkers. The linker derivatives 13a and 13b bearing unprotected alkyne moieties were prepared from derivatives 7a and 8b by conversion of free or the THP protected hydroxyl groups into iodide and bromide, respectively.
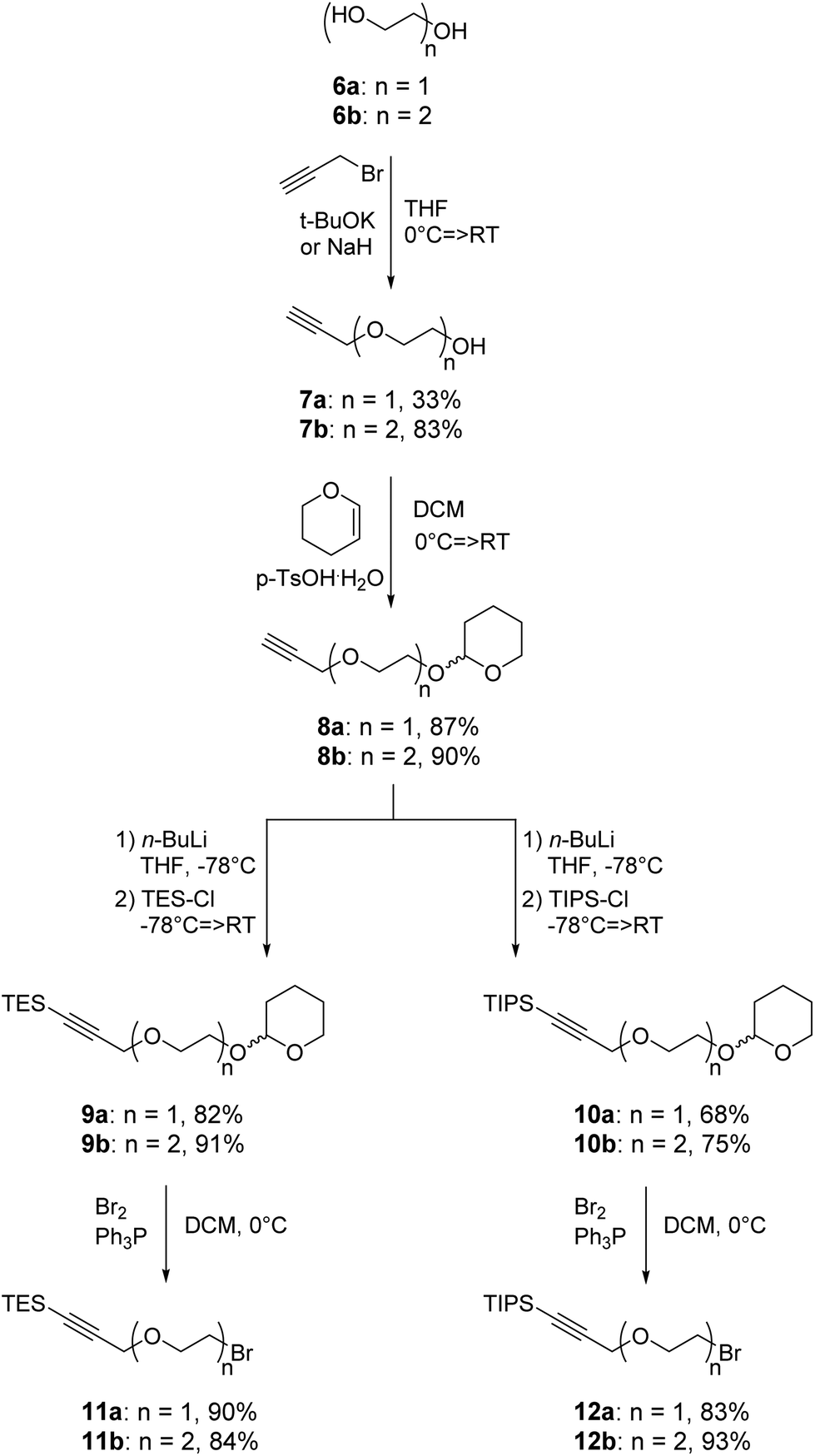 | ||
| Scheme 1 Synthesis of silyl protected alkylation linkers 11a,b and 12a,b. | ||
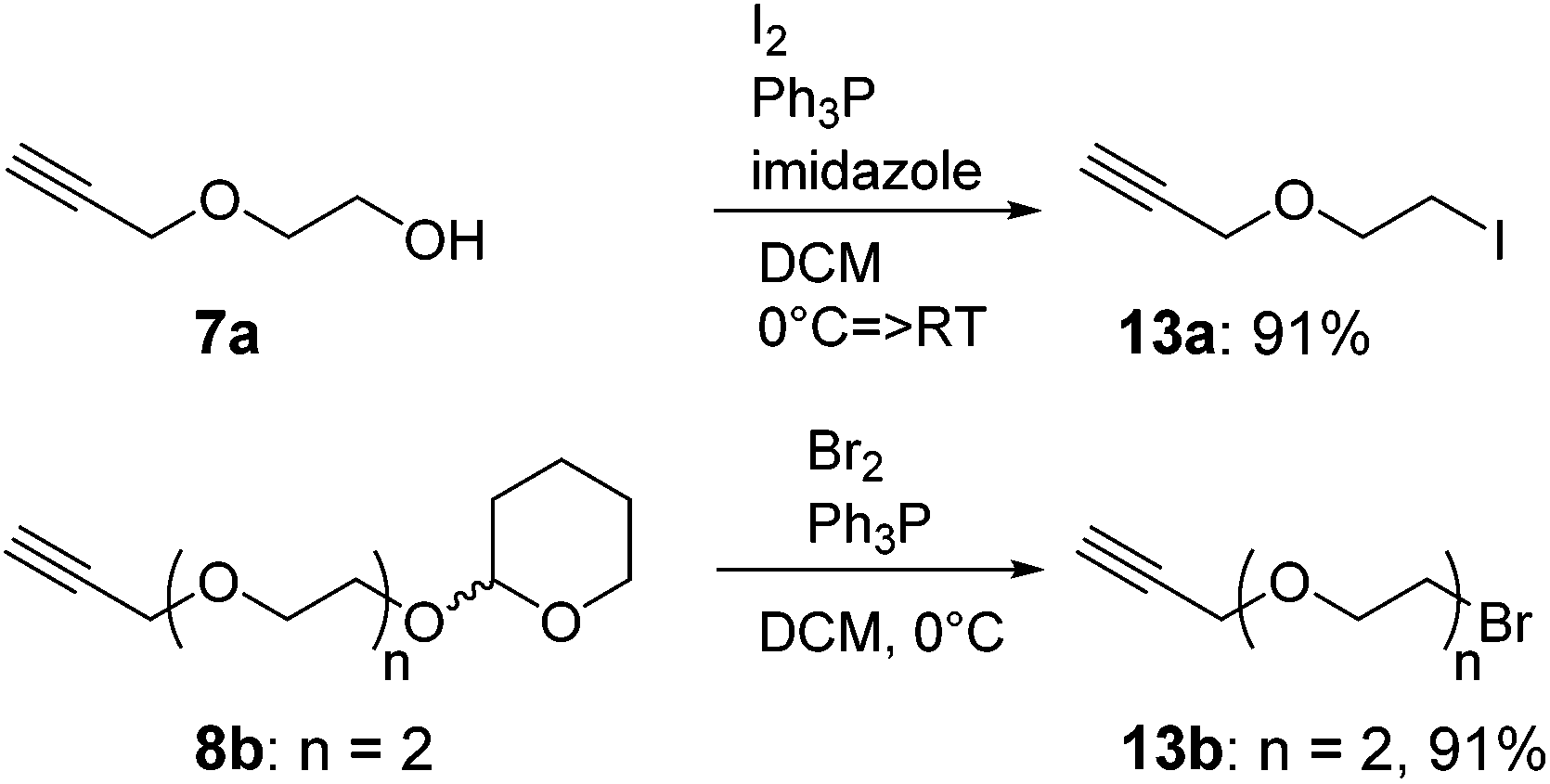 | ||
| Scheme 2 Synthesis of free alkyne bearing alkylation linkers 13a and 13b. | ||
After this CTV scaffold 1 was prepared according to the procedure of Canceill et al.7 Protection of CTV scaffold 1 as a ditetrahydropyranyl (diTHP) derivative 14 as well as the recovery of CTV scaffold 1 from side products of the protection was performed as previously described.4 Alkylation of diTHP protected scaffold 14 with (oligo)ethylene glycol linkers 13a,b was performed in acetonitrile in the presence of Cs2CO3 to yield monoalkylated diTHP protected CTV scaffold derivative 15a,b (Scheme 3) in high yield. Following the removal of THP protecting groups with 1 M HCl in methanol (Scheme 3), the dihydroxyl CTV scaffold derivatives 16a,b were alkylated with TIPS protected linkers 12a,b (Scheme 4) providing rather modest yields of the desired monoTIPS derivatives 17a,b along with unwanted diTIPS side products 18a,b (Scheme 4) while leaving some of the dihydroxyl CTV scaffold 16a,b unreacted. Finally, the TES protected linkers 11a,b were introduced to monoTIPS CTV scaffold derivatives 17a,b in DMF using Cs2CO3 as a base to yield the semi-orthogonally protected CTV scaffold derivatives 3 and 4 (Scheme 4). In case of the CTV scaffold derivative 3 a hydrolysis of TES protecting group and therefore formation of a side product 5 lacking the TES protecting group was observed.
 | ||
| Scheme 3 Indirect monoalkylation of CTV scaffold. | ||
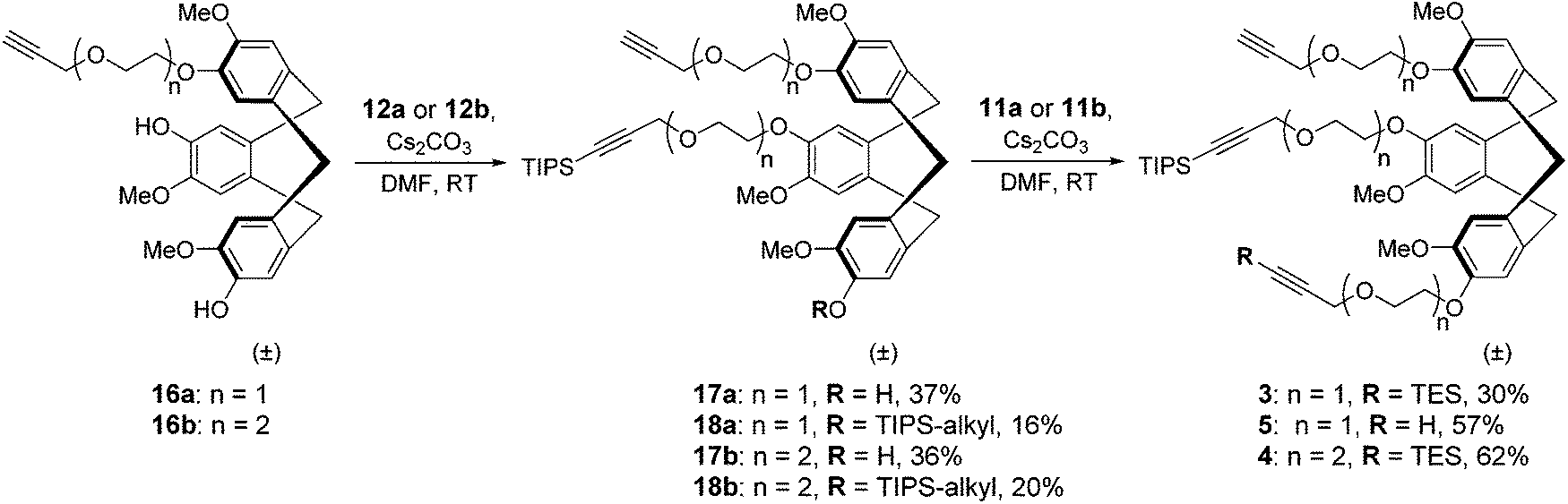 | ||
| Scheme 4 Introduction of TIPS/TES protected linkers onto monoalkylated CTV scaffold. | ||
Improvement of aqueous solubility by incorporation of ethylene glycol spacers
Since silyl groups are not present in the final products, i.e. synthetic antibodies, aqueous solubility of the desilylated analogues 20, 21, and 22 (Scheme 5) of CTV scaffold derivatives 2, 3, and 4, respectively, were determined, by measurement of dissolved quantities using reverse phase HPLC.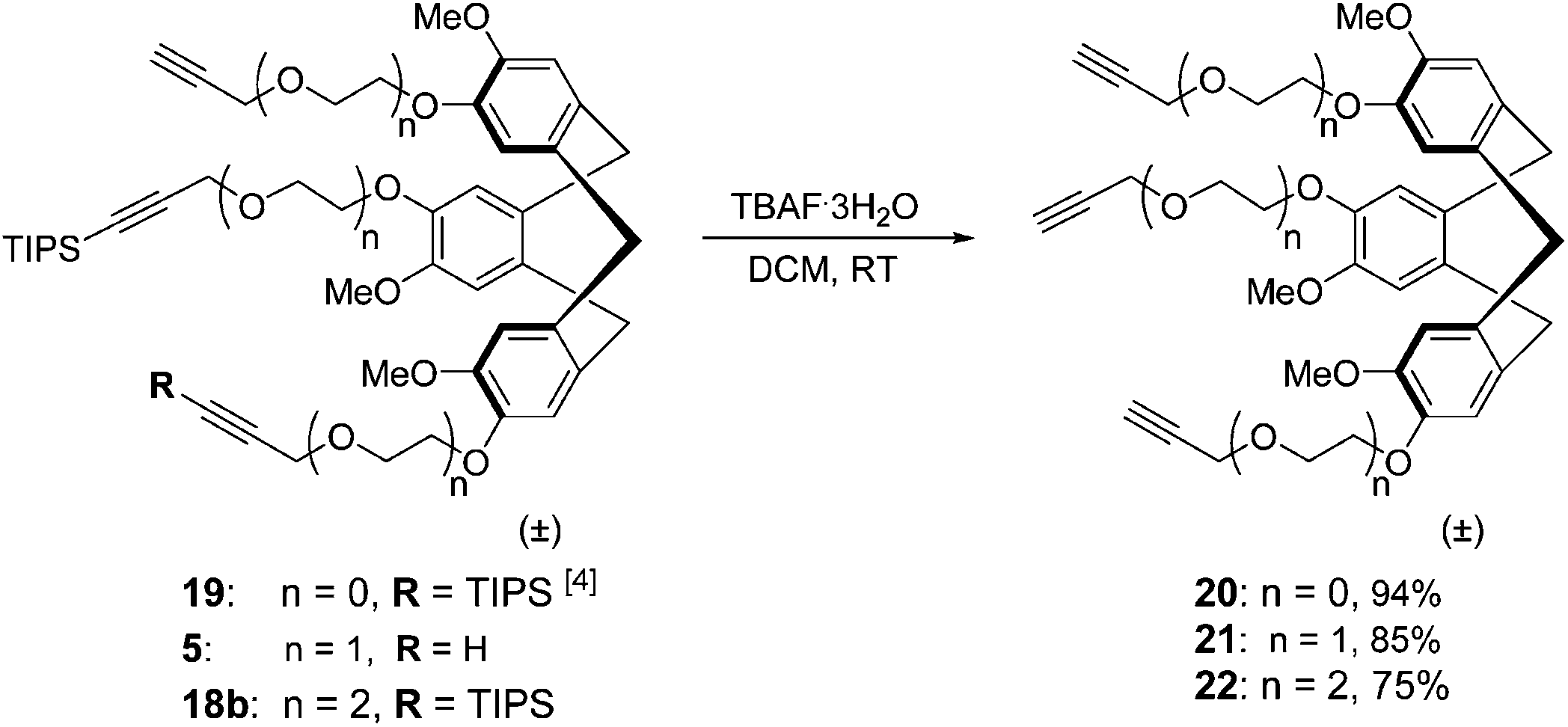 | ||
| Scheme 5 Synthesis of desilylated analogues 20, 21, 22 of CTV scaffold derivatives 2, 3, and 4. | ||
Indeed, incorporation of the (oligo)ethylene glycol linkers improved water solubility as the mono-ethylene glycol derivative 21 and diethylene glycol derivative 22 were found to be 5 and 14 times more soluble in deionised water (supplemented with 2% of ethanol) than CTV scaffold derivative 20 without a solubilizing linker.
Unfortunately, improvement of the solubility of the synthetic antibodies containing these CTV derivatives in PBS buffer pH 7.4 was not sufficient to allow for their biological activity evaluation on cell lines. The prepared synthetic antibodies were reasonably soluble in 2-(N-morpholino)ethanesulfonic acid hydrate (MES) buffer pH 6.0 containing 5% DMSO.‡
Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/h3_char_2009.gif) P determination
P determination
Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P values of the desilylated analogues 20, 21, and 22 were also determined using an Organisation for Economic Co-operation and Development (OECD) approved reverse phase HPLC method.8 LogP values of 4.1, 4.3, and 4.6 were determined for compounds 20, 22 and 21, respectively. The similar logP values indicate that the resulting protein mimics will have similar cell permeability properties and ensuing biological activity properties, which are not influenced by variation of the scaffold.
P values of the desilylated analogues 20, 21, and 22 were also determined using an Organisation for Economic Co-operation and Development (OECD) approved reverse phase HPLC method.8 LogP values of 4.1, 4.3, and 4.6 were determined for compounds 20, 22 and 21, respectively. The similar logP values indicate that the resulting protein mimics will have similar cell permeability properties and ensuing biological activity properties, which are not influenced by variation of the scaffold.
CDRs selection and peptide cyclisation
As was described previously4 selection of peptide sequences mimicking CDRs of the paratope of mAb infliximab (Remicade®) was based on the X-ray crystal structure9 of a complex of infliximab with hTNFα trimer. In this research, selection of CDR mimics was extended by including peptide segment 23 (Scheme 6) as its cyclised derivative 28. This particular sequence has been shown to be crucial for binding to human hTNFα as was demonstrated by a mutation study.9 Thus, a total of four peptide segments which mimic the selected CDRs were prepared. Three sequences, 23, 24, 26, correspond to the sequences present in the entire peptide segment of the CDR loops of infliximab. Peptide sequence 25 mainly consists of distinct amino acid residues present in the antibody which are important for binding to hTNFα as is apparent from the X-ray structure of the complex and which are in close spatial proximity to target amino acids in hTNFα. These peptide segments of the CDRs of infliximab are shown in colour (Fig. 4), which corresponds to the colours of the CDR loop mimics 28–31 used in the construction of the synthetic antibodies.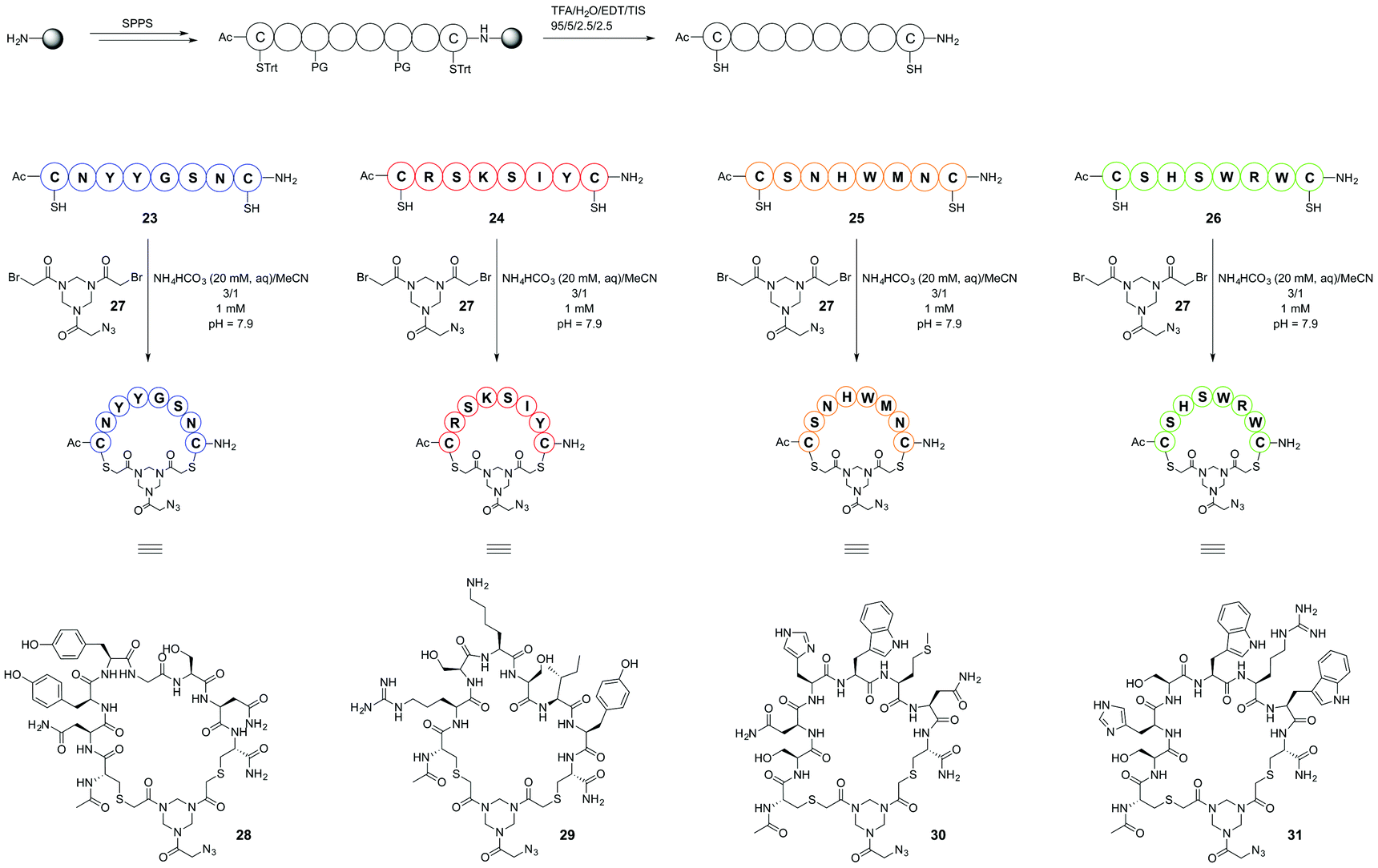 | ||
| Scheme 6 Synthesis and cyclisation of linear CDR mimics. | ||
 | ||
| Fig. 4 CDR loops in infliximab based on reported interactions between hTNFα and infliximab Fab.9 The colour coding corresponds to the peptides in the CDR mimics and toward the molecular synthetic antibody constructs. A top view (interaction site with hTNFα) and a side view are shown. The residue (Arg-52, Heavy chain), which was part of the red and green loop, is salmon-coloured. | ||
The selected peptide sequences were furnished with both an N and a C terminal cysteine residue to allow their chemoselective cyclisation using our recently developed polar hinge 2710 (Scheme 5) which should enhance water solubility of the cyclised peptides. The resulting azide handle containing cyclized peptides 28–31 were subsequently introduced via Cu(I)-catalyzed azide alkyne cycloaddition (CuAAC) onto the CTV scaffold derivatives 2, 3 and 4 as will be described below.
One-pot synthesis of synthetic antibodies
From the very start of this research, it was our desire to ultimately develop synthesis protocols for these medium-size molecules, which could be conveniently reproduced and applied also by researchers not directly working in the peptide field. Our recent CTV scaffold based protein mimic was assembled in a “click and cleavage” approach with several intermediate purifications.4 Here, this approach was extended to a one-pot procedure where the CDR loop mimics were subsequently introduced on each of the CTV scaffolds 2, 3 or 4. For clarity: only one CTV scaffold derivative was used in each reaction and not a mixture of 2–4. After introduction of the first and second CDR loop mimic the silyl protecting groups were removed and only after complete assembly the antibody mimics were purified.Thus, one equivalent of CDR loop mimic 31 was introduced via CuAAC onto one equivalent of either CTV scaffold derivative 2, 3, or 4 (Scheme 7) to give a one CDR loop containing protein mimic. Immediately thereafter, silver nitrate was added to the reaction mixture to selectively remove the TES protecting group11 and without intermediate purification the next CDR-mimicking peptide loop 30 was introduced via CuAAC leading to the two CDR loop mimic containing CTV derivatives. During this reaction aminoguanidine hydrochloride was added as a scavenger of the formed dehydroascorbic acid i.e.12 to prevent formation of ascorbic acid adducts with arginine residues in the peptide loops. Next, TBAF·3H2O was added to remove the TIPS protecting group giving two loop containing protein mimic intermediates 32, 33, 34 and the reaction mixture was split into two equal portions. To one portion, 1.2 equivalents of CDR loop mimic 29 were added while to the other, 1.2 equivalents of CDR loop mimic 28 were added. Thereby in a convenient manner two different synthetic antibodies were prepared. Upon completion of this one-pot procedure, the reaction mixtures were concentrated and the antibody mimics were purified by semi-preparative HPLC.
 | ||
| Scheme 7 One-pot synthetic approach towards synthetic antibodies 35–40. | ||
By using this same one-pot procedure albeit changing the sequences of addition of the various CDR-mimicking peptide loops to CTV scaffold derivative 2, 3, or 4 the other antibody mimics 44–49 were prepared (Scheme 8). Thus, CDR loop mimic 29 was introduced onto either scaffold 2, 3, or 4via CuAAC, followed by removal of TES group with silver nitrate and subsequent introduction of the CDR mimic 28 before TIPS protecting group was removed with TBAF·3H2O to give intermediates 41, 42, 43. After splitting of the reaction mixture in two equal parts, CDR loop mimic 30 was added to one part of the mixture while CDR loop mimic 31 was added to the other part.
 | ||
| Scheme 8 One-pot synthetic approach towards synthetic antibodies 44–49. | ||
Hence, the described approach led to a collection of twelve protein mimics 35–40 and 44–49 designated as “synthetic antibodies” (Fig. 5). This collection comprised all possible combinations of the four CDR loop mimics 28–31 onto each of the CTV scaffold derivatives 2, 3 or 4.
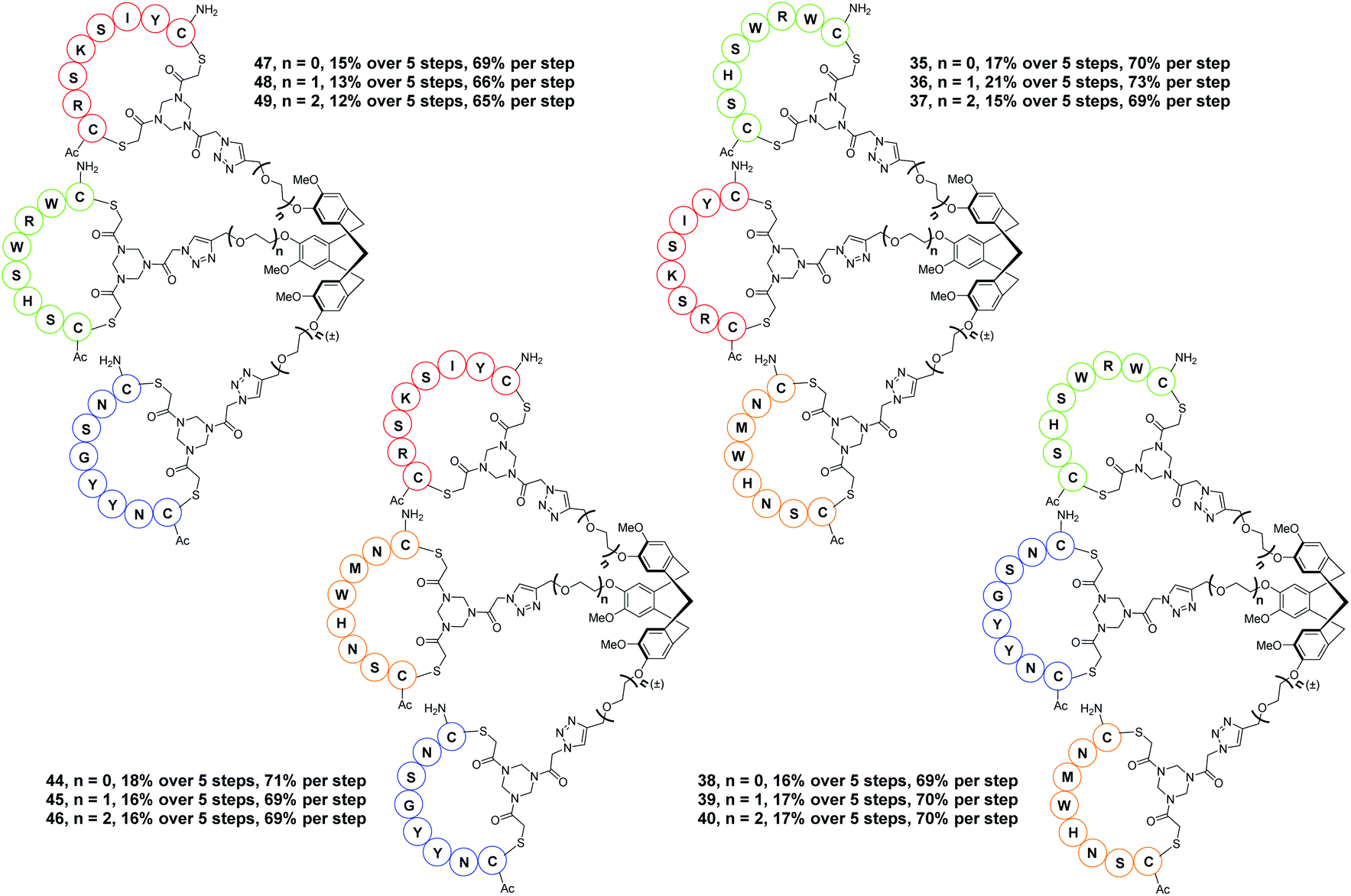 | ||
| Fig. 5 Prepared collection of antibody mimics toward synthetic antibodies. | ||
Progress of conversion of the CTV derivatives by subsequent peptide loop introduction was conveniently monitored by analytical reversed phase HPLC as the retention times of the intermediate products were sufficiently different (Fig. 6). As was apparent from the chromatograms, the reaction mixture remained very clean until the introduction of the third CDR loop mimic when side products, undesired combinations of previously unreacted CDR mimics on CTV scaffold derivative, with retention times close to that of the desired synthetic antibody mimic started to appear. Nevertheless, these side products could be largely separated with an optimised gradient on a preparative C8 reversed phase column. Thus, the desired protein mimics were obtained in purities of 91%–96%.
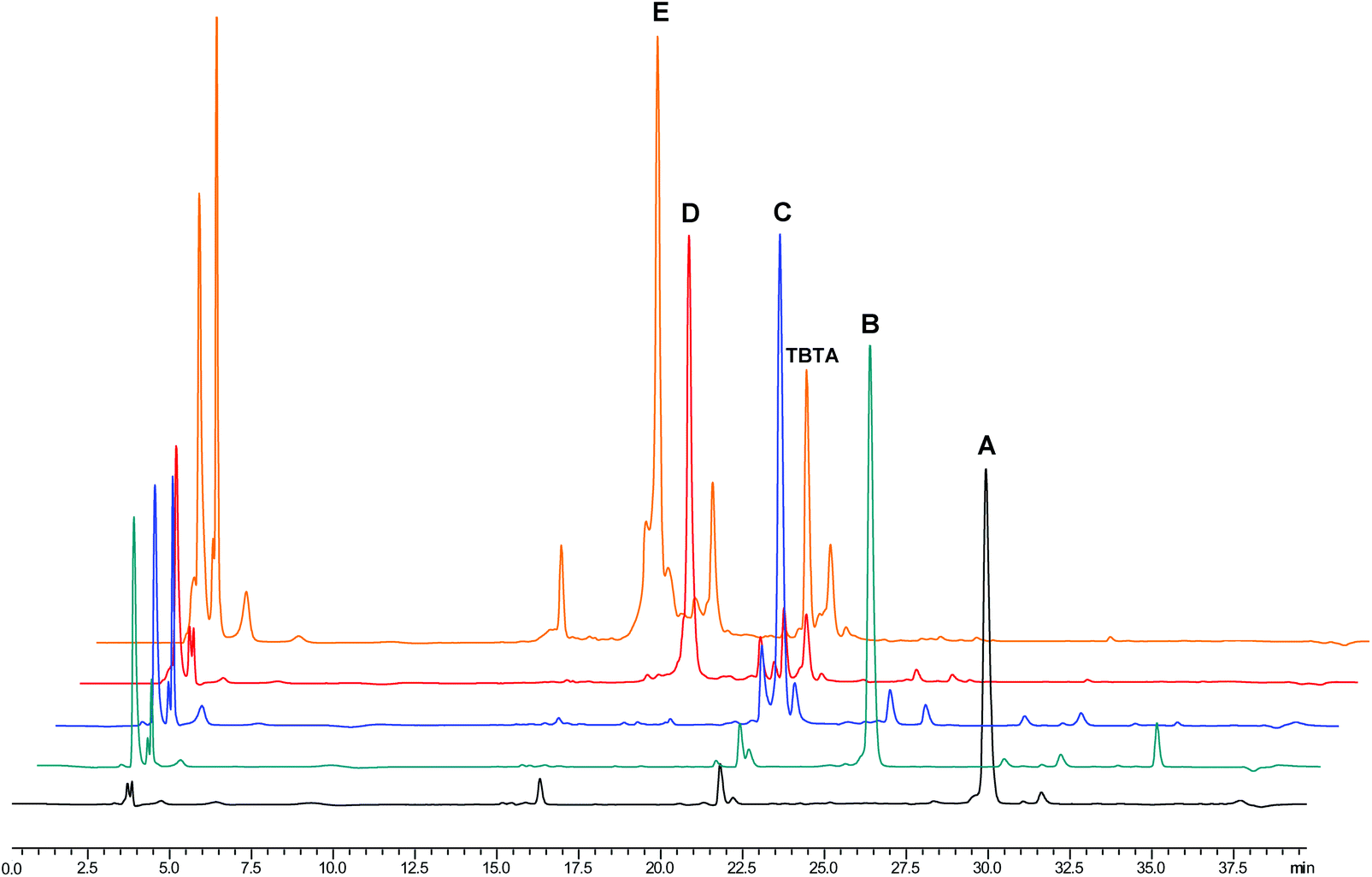 | ||
| Fig. 6 Representative analytical HPLC chromatogram of the one-pot synthesis of synthetic antibodies – overlay of the entire reaction sequence for synthetic antibody 37. Black (A) – product after introduction of CDR mimic 31 onto CTV scaffold derivative 4. Green (B) – product after TES protecting group removal. Blue (C) – product after introduction of CDR mimic 30. Red (D) – product after TIPS protecting group removal. Orange (E) – product after introduction of CDR mimic 29. Peak at tR = 21.8 min represents TBTA. | ||
Surface plasmon resonance
Initially, binding studies of protein mimics were attempted using Isothermal Titration Calorimetry (ITC). However, these were unsuccessful because of the need to dialyse hTNFα and synthetic antibodies to exactly match DMSO concentration (2% of final volume of appropriate buffer) in separately prepared samples and thus to avoid an artificial DMSO signal. Unfortunately, it was found that the synthetic antibodies stick to the cellulose dialysis membrane and as a consequence disappear from solution. Therefore binding studies of the prepared synthetic antibodies 35–40 and 44–49 were carried out by Surface Plasmon Resonance (SPR) on immobilised hTNFα. The integrity of immobilised hTNFα was first verified by flowing an anti-hTNFα specific monoclonal antibody (Life Technologies, Thermo Fisher Scientific, clone 68B 2B3) over the chip surface and indeed was capable of interacting with immobilised hTNFα (KD = 32 nM). After an initial screening of the prepared protein mimics at 2 μM (runs in duplicate) positive hits, i.e. compounds exhibiting binding, were selected and their binding characteristics were further determined. A representative sensogram of the best binding synthetic antibody 36 (Fig. 7) as well as kinetic parameters of synthetic antibodies 35, 36, 44, 47, and 48 (Table 1) are shown below.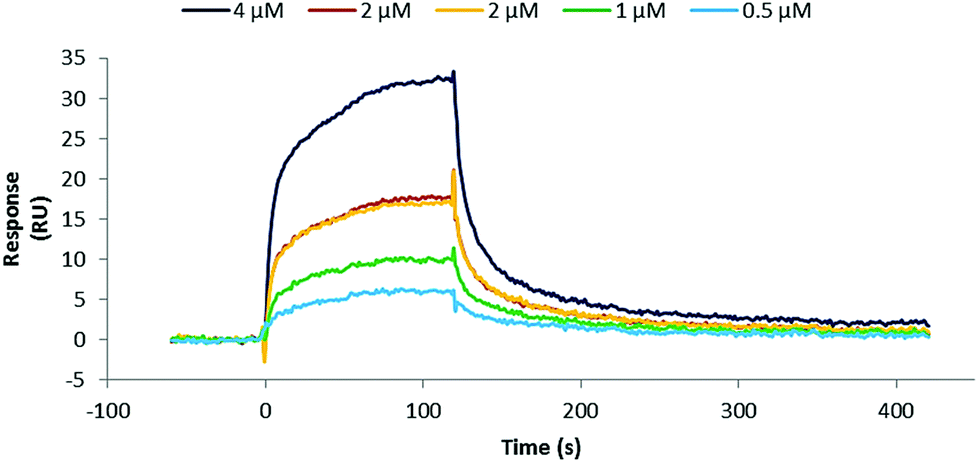 | ||
| Fig. 7 Sensogram representing binding of synthetic antibody 36 to immobilised hTNFα. Raw data presented.§ | ||
| Average values of 2 experiments performed in duplicate (n = 2) reported. |
|---|

|
The common crucial feature of synthetic antibodies 35, 36, 37, 44, 47, 48 and 49, capable of binding hTNFα, was the presence of CDR (red loop) mimic 29. CDR (green loop) mimic 31 proved to be important too as it was present in all synthetic antibodies capable of successfully binding to hTNFα except for synthetic antibody 44 which contained CDR (orange loop) 30 instead next to (red loop) mimic 29. In general, incorporation of combinations of CDR mimics (red loop) 29 and (green loop) 31 together with either CDR mimic (blue loop) 28 or (orange loop) 30 in a synthetic antibody led to antibody mimics with micromolar range affinity for hTNFα. Based on the obtained KD values (Table 1), the combination of CDR mimic (red loop) 29, (orange loop) 30, (green loop) 31 has slightly better affinity for hTNFα than combination of CDR mimics (blue loop) 28, (red loop) 29, (green loop) 31. Incorporation of the combination of CDR mimics (blue loop) 28 and (orange loop) 30 into a synthetic antibody resulted generally in lack of binding of the corresponding synthetic antibodies to hTNFα, i.e.38, 39, 40, 45 and 46, with the exception of construct 44, which still displayed a decent affinity (KD = 66 μM). Upon closer examination of this phenomenon supported by modelling (Fig. 8) it was concluded that CDR mimic (blue loop) 28 and (orange loop) 30 originate from an overlapping region of infliximab and thus may interfere with each other resulting in the lack of binding for hTNFα. This however is not the case for synthetic antibody 44 where the absence of an ethylene glycol linker might have prevented this interference.
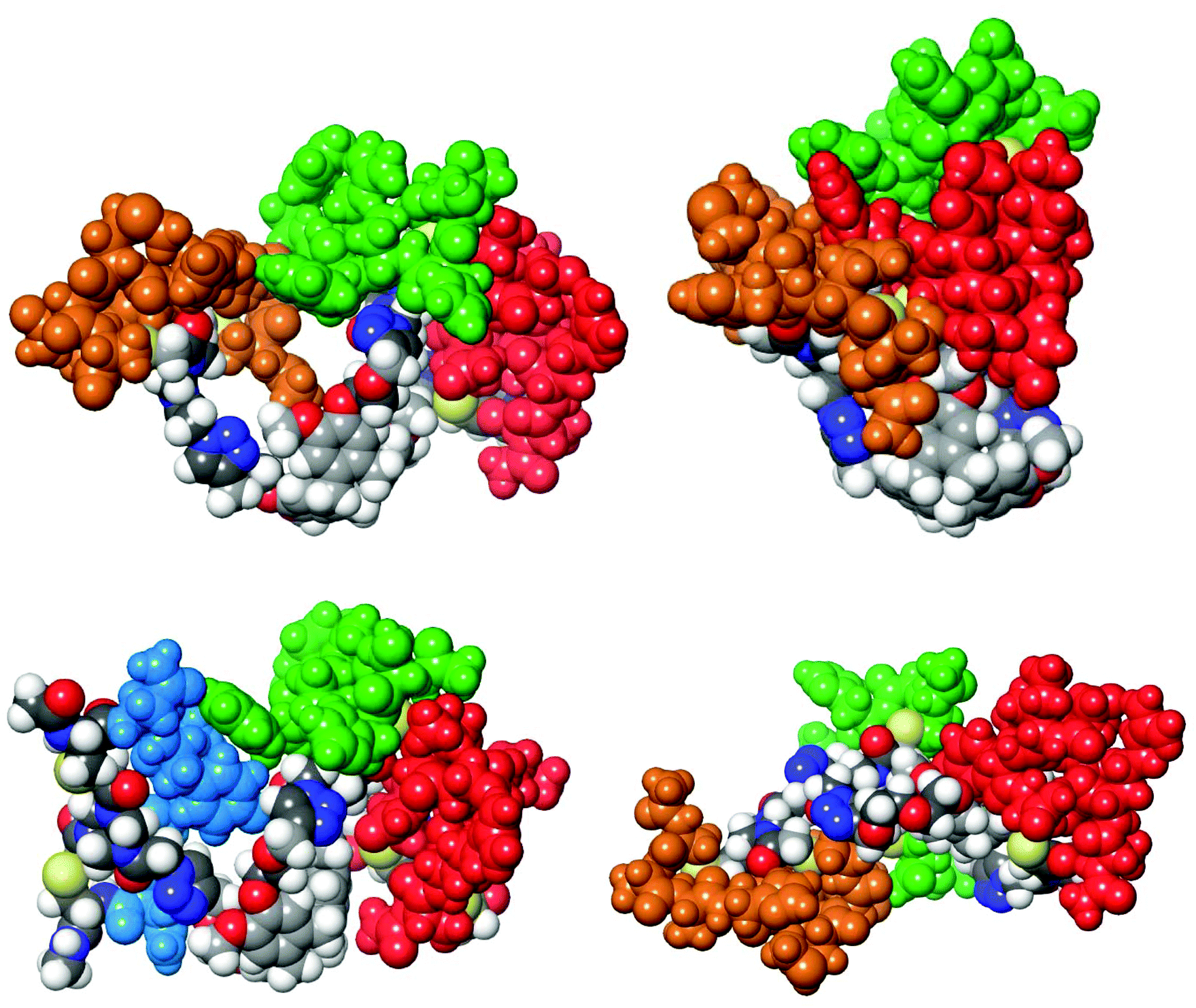 | ||
| Fig. 8 Molecular models of the synthetic antibody protein mimics 36 (top-left), 35 (top-right), 48 (bottom-left) and 37 (bottom-right). The first three are the best binding candidates. Molecular construct 37 is included to compare the influence of the diethylene glycol moiety with the constructs 36 and 35 having a mono ethylene glycol moiety or none, respectively. The weak affinity for immobilized hTNFα by 37 might be explained by the increased flexibility of the diethylene glycol moieties not allowing an optimal multivalent interaction. | ||
Synthetic antibodies derived from CTV scaffolds 2 or 3 capable of binding hTNFα, had a kinetic profile suitable for fitting of the measured curves. Synthetic antibodies derived from CTV scaffold derivative 4 had too fast kinetics (koff greater than 1 × 10−1 s−1) to reliably determine their dissociation constant. This might be due to a higher flexibility, of the diethylene glycol spacer, resulting in an increase of flexibility, possibly leading to a decrease in binding affinity. This is also apparent from Fig. 3 showing that the diethylene glycol spacer may lead to less directionality, so that the loops are gathered more around the CTV scaffold, rather than pointing in the same direction.
In view of the above results it was not entirely surprising that CDR (green loop) mimic 31 by itself showed affinity for hTNFα nevertheless the measured kinetics was too fast to reliably obtain a dissociation constant. However, the other three CDR mimics including the CDR (red loop) 29, had no affinity for hTNFα whatsoever. As anticipated, a multivalent effect induced by attachment of the appropriate CDR mimics on suitable CTV scaffold derivatives, resulting in increased affinities was observed. This also clearly demonstrated the crucial role of CTV scaffold derivatives in the molecular construction of antibody mimics by covalent assembly of individual CDR loop mimics.
Conclusion
A new one-pot approach allowing for rapid synthesis of synthetic antibodies has been described. The approach was applied in the construction of a collection of synthetic antibody candidates mimicking the paratope of mAb infliximab (Remicade®). The ability of the synthetic antibodies to mimic the original monoclonal antibody infliximab was determined by measuring their binding to hTNFα, using SPR. SPR measurements showed that 7 out of 12 prepared synthetic antibody candidates were capable of binding hTNFα with 5 of them displaying dissociation constants ranging from 11 μM to 66 μM. The relatively strong binding with a KD of 11 μM clearly supported the validity of the concept of molecular scaffolding of CDR mimics towards possible synthetic antibodies.Since the synthetic antibodies are prepared from racemic CTV, diastereomers of the synthetic antibodies have been obtained. However, it is likely that the size the attached CDR loop mimics will mask the difference between the enantiomeric CTV moieties.
Moreover, in this approach the rather low water solubility of the previously reported4 semi-orthogonally protected CTV scaffold derivative 2 was addressed by synthesising its monoethylene and diethylene glycol spacer containing analogues 3 and 4 respectively. Although incorporation of the glycol spacers did improve water solubility 5 times and 14 times respectively, these improvements were not sufficient for dissolution of these synthetic antibodies in PBS buffer pH 7.4 to allow bio-activity evaluation on cell lines. Therefore, under present investigation is further improvement of the water solubility of CTV derivatives.
In conclusion, the concept of replacing the majority of the structure of a monoclonal antibody by a molecular scaffold capable of performing a similar structural functional role as in the parent antibody leading to a significantly smaller protein mimic has proven to be valid. The obtained protein mimics denoted as a “synthetic antibodies” have less than 4% of the molecular mass of the mimicked monoclonal antibody infliximab. Nevertheless, the affinity of the prepared protein mimics is still considerably lower than that of the original monoclonal antibody, but the obtained micromolar range affinity might indicate that in addition to room for improvement, that this is a promising approach. It is realised, however that synthesis of these fairly complex molecular constructs has to be continuously improved if not simplified. In this work, considerable strides have been made toward significantly improving the synthesis of these protein mimics in terms of time and efficiency.
Two synthetic antibodies have been conveniently prepared in less than 30 hours while only requiring semi-preparative reverse phase HPLC purification at the very end of the synthesis. Thus, this synthetic protocol is a great improvement compared to our previously described methods for CTV scaffold derivatives as well as other TAC and related scaffolds,3,4,13 which required semi-preparative HPLC purification and isolation after practically each step of the synthesis of corresponding protein mimics, in total 4 times or 5 times. Furthermore, this protocol enabled attachment of three different ligands/substituents to one single molecule via Cu(I)-mediated azide–alkyne cycloaddition in a one pot reaction. Application of this protocol can however go beyond the introduction of peptide loops leading to synthetic antibodies as virtually any azide handle containing small or larger ligand can be introduced onto the CTV scaffold derivatives 2, 3, and 4 for example carbohydrates, nucleic acids and small molecules or combinations of these, giving rise to synthetic vaccines, molecular constructs interacting with carbohydrate binding proteins (lectins), covalent multi-enzyme complexes, enzyme mimics etc.
Experimental part
General information
All analytical or HPLC grade chemicals and solvents were purchased from commercial sources and were used as received unless stated otherwise. 1H NMR and the 13C NMR spectra were recorded on a Bruker AVIII 400 MHz or 500 MHz Spectrospin spectrometer in CDCl3. Chemical shifts (δ) are reported in parts per million (ppm) relative to trimethylsilane (TMS, 0.00 ppm) or CDCl3 (7.26 ppm). TLC was carried out on silica gel plates (Merck 60F254) and visualization was carried out by both UV detection (254 nm) and staining (cerium molybdenate, potassium permanganate) followed by heating. For column chromatography, silica gel Geduran® Si 60 (40–63 μm) was used. Dry solvents (THF, CH2Cl2) were dispensed from Pure Solv™ 500 Solvent Purification System and other dry solvents (acetonitrile, DMF) were obtained from freshly opened commercially available HPLC grade solvents by removal of residual water with activated 4 Å molecular sieves overnight. HRMS-ESI was recorded on Bruker microTOFq High Resolution Mass Spectrometer in the positive mode. EI-MS was recorded on Jeol MSTATION JMS-700 in the positive mode. Commercial solutions of n-butyl lithium were titrated using diphenyl acetic acid to determine their actual concentration prior to use. Degassed solvents for synthesis of synthetic antibodies were obtained upon sonication of the solvents in degas mode for 30 minutes while purging the headspace with nitrogen stream trough septum. Petroleum ether used refers to a mixture with boiling point of 40 °C–60 °C.Fmoc-amino acids were obtained from Activotec (Cambridge, United Kingdom) and N,N,N′,N′-tetramethyl-O-(6-chloro-1H-benzotriazol-1-yl)uranium hexafluorophosphate (HCTU) was obtained from Matrix Innovation (Quebec, Canada). Tentagel S RAM resin (particle size 90 μm, capacity 0.25 mmol g−1) was obtained from IRIS Biotech (Marktredwitz, Germany). Methyl tert-butyl ether (MTBE), n-hexane (HPLC grade) and TFA were obtained from Aldrich (Milwaukee, USA). DMF (Peptide grade) was obtained from VWR (Lutterworth, United Kingdom). Piperidine and DiPEA were obtained from AGTC Bioproducts (Hessle, United Kingdom), and 1,2-ethanedithiol (EDT) was obtained from Merck (Darmstadt, Germany). HPLC grade CH2Cl2 and acetonitrile were obtained from Fischer Scientific (Loughborough, United Kingdom). Solid phase peptide synthesis was performed on a PTI Tribute-UV peptide synthesizer. Lyophilisations were performed on a Christ Alpha 2–4 LDplus apparatus. Analytical high pressure liquid chromatography (HPLC) was carried out on a Shimadzu instrument comprising a communication module (CBM-20A), autosampler (SIL-20HT), pump modules (LC-20AT), UV/Vis detector (SPD-20A) and system controller (Labsolutions V5.54 SP), with a Phenomenex Gemini C18 column (110 Å, 5 μm, 250 × 4.60 mm). UV measurements were recorded at 214 and 254 nm, using a standard protocol: 100% buffer A (acetonitrile/H2O 5:95 with 0.1% TFA) for 2 min followed by a linear gradient of buffer B (acetonitrile/H2O 95:5 with 0.1% TFA) into buffer A (0–100%) over 30 min at a flow rate of 1.0 mL min−1. Purification of synthetic antibodies was performed on an Agilent Technologies 1260 infinity preparative system using UV detector with a Dr Maisch C8 column (110 Å, 10 μm, 250 × 20 mm) starting with 100% buffer A for 10 min followed by a linear gradient of buffer B into buffer A (27–37%) over 40 min at a flow rate of 10.0 mL min−1. CDR mimics were purified on the same system using Phenomenex Gemini C18 column (110 Å, 10 μm, 250 × 20 mm) starting with 100% buffer A for 5 min followed by and linear gradient of buffer B into buffer A (gradient specified for each compound) over 60 min at a flow rate of 12.5 mL min−1. The buffers had same composition as described for analytical HPLC. Auto-collection of fractions was based on the UV measurements at 214 nm. One-pot reaction mixtures were analysed by taking 10 μL of the reaction mixture, removing solvent under stream of nitrogen, dissolving the dried residue in buffer A/buffer B (1/1 v/v, 500 μL) and injecting of 50 μL of the obtained solution on analytical HPLC. Liquid chromatography mass spectrometry (LCMS) was carried out on a Thermo Scientific LCQ Fleet quadrupole mass spectrometer with a Dionex Ultimate 3000 LC using a Dr Maisch Reprosil Gold 120 C18 column (110 Å, 3 μm, 150 × 4.0 mm), using a 0–100% linear gradient of buffer B into buffer A and the same flow rate and buffers as described for analytical HPLC. Deionised water was obtained on Milli-Q® station (Merck Millipore).
Alkylation linker synthesis and CTV scaffold derivatisation
Peptide synthesis and Cu(I)catalyzed azide–alkyne cycloaddition chemistry
Linear peptide 23 , Ac-CNYYGSNC-NH2, tR = 13.9 min. LCMS-ESI: average mass calcd [M]+: 964.0; found: 964.3.
Analytical data of linear peptides 24–26 were identical to those described previously.4
Cyclic peptide
28
,  purified using 0% to 40% of buffer B in buffer A. Overall yield: 14% (19 steps, average yield 90% per step). tR = 14.5 min. LCMS-ESI: average mass calcd [M]+: 1214.3; found: 1214.3.
purified using 0% to 40% of buffer B in buffer A. Overall yield: 14% (19 steps, average yield 90% per step). tR = 14.5 min. LCMS-ESI: average mass calcd [M]+: 1214.3; found: 1214.3.
Purification and analytical data of cyclic peptides (CDR mimics) 29–31 were described previously.4
Preparation of synthetic antibodies 35–40 and 44–49
000g for 6 min) and the product in the supernatant was purified by semi-preparative reverse phase HPLC using a gradient of 27% to 37% of buffer B in buffer A. Fractions containing the product were pooled and lyophilised. In the majority of the experiments, significant amounts (89% on average) of desired synthetic antibodies were obtained in purities of 91%–96% along with smaller portion of impure fractions which were re-purified using the same gradient.
Synthetic antibody 35 (2.8 mg, overall yield 17%, average yield 70% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.1 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1472.3; found: 1472.0. MALDI: exact mass calcd [M + H]+ 4411.7; found: 4411.1.
Synthetic antibody 36 (3.4 mg, overall yield 21%, average yield 73% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.2 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1516.4; found: 1516.0. MALDI: exact mass calcd [M + H]+ 4543.8; found: 4543.9.
Synthetic antibody 37 (2.6 mg, overall yield 15%, average yield 69% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.3 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1560.4; found: 1560.2. MALDI: exact mass calcd [M + H]+ 4675.9; found: 4675.6.
Synthetic antibody 38 (2.4 mg, overall yield 16%, average yield 69% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.7 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1460.2; found: 1460.0. MALDI: exact mass calcd [M + H]+ 4375.6; found: 4376.0.
Synthetic antibody 39 (2.6 mg, overall yield 17%, average yield 70% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.7 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1504.3; found: 1504.0. MALDI: exact mass calcd [M + H]+ 4507.6; found: 4507.5.
Synthetic antibody 40 (2.8 mg, overall yield 17%, average yield 70% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.8 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1548.4; found: 1548.0. MALDI: exact mass calcd [M + H]+ 4639.7; found: 4639.8.
Synthetic antibody 44 (2.7 mg, overall yield 18%, average yield 71% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.2 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1425.2; found: 1424.9. MALDI: exact mass calcd [M + H]+ 4270.6; found: 4270.9.
Synthetic antibody 45 (2.4 mg, overall yield 16%, average yield 69% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.3 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1469.3; found: 1469.0. MALDI: exact mass calcd [M + H]+ 4402.7; found: 4402.6.
Synthetic antibody 46 (2.5 mg, overall yield 16%, average yield 69% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.3 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1513.3; found: 1513.0. MALDI: exact mass calcd [M + H]+ 4534.7; found: 4534.5.
Synthetic antibody 47 (2.4 mg, overall yield 15%, average yield 69% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.2 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1448.6; found: 1448.3. MALDI: exact mass calcd [M + H]+ 4340.7; found: 4340.7.
Synthetic antibody 48 (2.0 mg, overall yield 13%, average yield 66% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.3 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1492.6; found: 1492.3. MALDI: exact mass calcd [M + H]+ 4472.8; found: 4472.8.
Synthetic antibody 49 (1.9 mg, overall yield 12%, average yield 65% per step, 5 steps) was obtained as a white fluffy solid. tR = 17.3 min. LCMS-ESI: average mass calcd [M + 3H]3+: 1536.7; found: 1536.4. MALDI: exact mass calcd [M + H]+ 4604.8; found: 4604.7.
1. Solvents had to be degassed prior to carrying out the reaction sequence otherwise the CuAAC introduction of the CDR mimics proceeded poorly. This was especially important for introduction of the third CDR mimic else almost no conversion was observed.
2. Using less than 5.5 equivalents of AgNO3 led, in some cases, to incomplete TES removal within 2 h. However, using more than 5.5 equivalents of AgNO3 (10 equivalents at the maximum) slowed down the subsequent introduction of the second CDR mimic although it did expedite TES removal.
3. Addition of the least soluble CDR mimic (that is compound 28 which did not fully dissolve in the chosen solvent mixture) to a scaffold already bearing another CDR mimic prevented the need for prolonged reaction times which was not the case when the least soluble CDR mimic 28 was attached as the first mimic to either scaffold 2, 3 or 4. In the latter case, the first CuAAC CDR mimic introduction required up to 8 h for completion and subsequent reactions usually required more time than described in the general procedure.
4. In cases when CDR mimics 28 and 29 were attached onto CTV scaffold derivatives 3 or 4, conversion of TIPS removal decreased to ca. 60%–75%. However, close to complete conversion could be achieved by increasing the amount of used TBAF·3H2O in degassed THF from 17.5 to 35 equivalents.
5. Fresh portions of CuSO4, sodium ascorbate and TBTA had to be added for every CuAAC introduction of CDR mimics to scaffolds 2, 3 or 4 as well as its CDR mimic(s) containing derivatives in order to secure good conversion. “Recycling” of copper from the first CuAAC or alternatively from the first and the second CuAAC by reducing the initially added CuSO4 with additional sodium ascorbate for the subsequent CuAAC did not lead to satisfactory conversions.
6. Extension of reaction times of CuAAC introduction of the third CDR mimic beyond 2.5 h hardly improved the conversion even when additional CuSO4, sodium ascorbate and TBTA were added during the reaction. The reaction tended to stop and a slow degradation of the product as well as formation of side products were observed after ca. 4 h.
7. Formation of dehydroascorbic acid adducts of arginine containing CDR mimics, which was mainly observed at the very end of the synthesis when reaction mixture was concentrated under stream of nitrogen, could be effectively prevented by addition of aminoguanidine hydrochloride as a scavenger of the formed dehydroascorbic acid.12
8. After evaluation of several gradients on C4, C8, and C18 reversed-phase columns, a gradient of 27% to 37% of buffer B into buffer A on a C8 column was selected as the best one for purification of all prepared synthetic antibodies.
Surface plasmon resonance experiments
The surface plasmon resonance experiments were performed using Biacore™ X100 SPR biosensor (Biacore AB, Uppsala, Sweden) equipped with CM5 sensor chip (Biacore AB, Uppsala, Sweden). hTNFα (ca. 51 kDa = trimer, >97% pure based on SDS-PAGE) was immobilised using amine-coupling chemistry.22 Surfaces of two flow cells were activated for 7 min with a 1:1 mixture of 0.1 M NHS (N-hydroxysuccineimide) and 0.4 M EDC [3-(N,N-dimethylamino)propyl-N-ethylcarbodiimide] at a flow rate of 5 μL min−1. hTNFα at a concentration of 30 μg mL−1 in 10 mM sodium acetate, pH 4.5, was immobilised at a density of 984 RU and 1024 RU, representing two different CM5 chips, on flow cell 2. Flow cell 1 was left blank to serve as a reference surface. Both surfaces were blocked with a 7 min injection of 1 M ethanolamine, pH 8.5, at a flow rate of 5 μL min−1. The immobilisation was performed at 21 °C with MES running buffer (10 mM MES, 150 mM NaCl, 3 mM EDTA, 0.05% P20, pH 6.0) using Biacore immobilisation wizard with target density of 1600 RU. Following the immobilisation, non-covalently bound oligomeric units of hTNFα were removed by injections of regeneration solution (5 mM NaOH, 150 mM NaCl, ca. 6 × 20 s).
The activity of immobilised hTNFα was verified with anti-hTNFα specific monoclonal antibody (Life Technologies, Thermo Fisher Scientific, clone 68B 2B3) in a single-cycle experiment with 3-fold dilutions of the mAb (highest concentration 100 nM) at a flow rate of 30 μL min−1 and a temperature of 21 °C. The experiment was performed using two different running buffers; the above mentioned MES buffer as well as its DMSO supplemented version, MES-DMSO, (10 mM MES, 150 mM NaCl, 3 mM EDTA, 0.05% P20, 5% DMSO, pH 6.0). Duration of the association/dissociation phase was set to 180 s and 420 s, respectively. The sensogram data were fitted according to 1:1 binding model using Biacore X100 Evaluation software version 2.0.1 (Biacore AB, Uppsala, Sweden). The anti-hTNFα specific mAb bound immobilised hTNFα with KD of 15 nM and 32 nM in MES and MES-DMSO buffer, respectively. Thus, a 2-fold decrease in affinity for experiments performed in MES-DMSO buffer was observed.
Solutions of synthetic antibodies were prepared at a concentration of 16 μM by dissolution of the synthetic antibody in DMSO (volume corresponding to 5% of the final volume) and subsequent dropwise dilution with 1.05 × MES-DMSO (10.5 mM MES, 157.5 mM NaCl, 3.15 mM EDTA, 0.0525% P20, pH 6.0) aided by vigorous stirring by vortex. The affinity of synthetic antibodies towards hTNFα was screened at 2 μM and the synthetic antibodies which showed binding were further tested in a multi-cycle kinetic experiment at 8 μM with 2-fold dilutions at a flow rate of 30 μL min−1 and a temperature of 21 °C in MES-DMSO buffer. Duration of the association/dissociation phase was set to 120 s and 300 s, respectively. Regeneration of the sensor surface was accomplished by a 12 s injection of a regeneration solution (5 mM NaOH, 150 mM NaCl) followed by a 300 s stabilisation period. Experiments were performed in duplicate in two independent experiments (n = 2) on chips with different immobilisation level of hTNFα (984 RU and 1024 RU). The medium concentration (2 μM) was injected twice as a control of stability of the experimental setup. During data processing the highest tested concentration, i.e. 8 μM was excluded from fitting as the obtained curves had irregular shape. The blank subtracted and injection peaks cleaned data were evaluated in Biacore X100 Evaluation software version 2.0.1 (Biacore AB, Uppsala, Sweden) using heterogeneous ligand fitting model. The heterogeneous fitting model was used as three out of six lysine residues of hTNFα, via which immobilisation happens, reside near the binding site of infliximab and thus synthetic antibodies. Following data evaluation, KD representing the binding event contributing the most to total Rmax was selected as the value best describing the strength of the observed binding.
Modelling of the molecular constructs
Molecular models of CTV-derivatives, peptide loops and synthetic antibody mimics were constructed using the Maestro software (http://www.Schrödinger.com) interfaced with MacroModel23 minimization and conformational analysis options using the MMFFs and OPLS-2005 force fields, PRCG minimization with water as a solvent. In short CDR loop mimics were generated by construction of the peptides in the grow mode, followed by introduction of the cyclization hinge 27 and minimization of obtained CDR peptide loops 28–31. Multi conformer analysis options allowed obtaining CDR mimics close to the global minimum energy conformations. The CDR mimics were connected subsequently to the appropriate CTV scaffold derivatives 2, 3 or 4 and minimized after each connection step. At this stage MD (Molecular Dynamics) studies were omitted, since the primary goal was to obtain a visual insight as a qualitative explanation of the observed results.Aqueous solubility of CTV scaffold derivatives determination
Excess of scaffold derivatives 20, 21, and 22 (ca. 1.0 mg) was suspended in EtOH (2% of final volume) and diluted with deionised water (to a final volume of 1 mL). The suspension was briefly sonicated (<10 s), stirred by vortex at 2500 rpm for 5 min, allowed to stand for 25 min and centrifuged (13000g for 10 min) before a sample was taken for analytical HPLC analysis (50 μL injected). HPLC runs were performed using isocratic gradient of acetonitrile/H2O 50:50 at a flow rate of 1 mL min−1 using a Phenomenex Gemini C18 column (110 Å, 5 μm, 250 × 4.60 mm). Calibration curves were constructed using 5 concentration points of 2-fold dilutions of an appropriate CTV scaffold derivative in acetonitrile/H2O 50/50, v/v. The results are a mean of three independent experiments performed in triplicate. The solubilisation had to be aided by addition of 2% of ethanol in order to break clusters of namely compound 21 which would otherwise barely interact with water. Even though such addition alters the final solubility, the ratio of solubility between the three scaffold derivatives should remain the same.
LogP determination
Following the OECD method,8 6 standards of known logP (anisole, toluene, bromobenzene, phenyl benzoate, diphenyl ether, bibenzyl) were used to construct a calibration curve. LogP values of compounds 20, 21, and 22 were determined from their retention time using regression equation obtained from the calibration curve of standards. Analytical HPLC runs were performed using isocratic gradient of acetonitrile/H2O 50/50, v/v at a flow rate of 1 mL min−1 using a Phenomenex Gemini C18 column (110 Å, 5 μm, 250 × 4.60 mm). Dead volume of the HPLC system was determined to be 2.89 min using uracil.
Recombinant hTNFα expression
Soluble recombinant hTNFα (Val77 – Leu233) was obtained by cloning its E. coli optimised gene sequence into a pDEST14™ expression vector (Thermo Fisher Scientific). The expression vector was transformed into competent E. coli BL21-AI™ One Shot® according to the manufacturer's protocol. A single colony of transformed cells was selected and allowed to expand in 2-YT bacterial growth media (16 g of tryptone, 10 g of yeast extract, 5 g of NaCl per 1 L supplemented with 50 μg mL−1 carbenicillin; 20 mL) at 37 °C overnight with 165 RPM shaking. The overnight bacterial culture was expanded into fresh 2-YT media at a ratio of 1/20 and the culture was incubated at 37 °C with 165 RPM shaking until the cells reached log phase (approximately 2 h). The cells were then induced for 5 h with the addition of L-(+)-arabinose (final concentration of 0.2% w/v) to initiate protein expression. After induction, the cells were pelleted by centrifugation at 12000g for 5 min at 4 °C. The pellet was suspended in cold lysis buffer (20 mM Tris-HCl, 1 mM EDTA, pH 8.0, supplemented with EDTA-free protease inhibitor cocktail; 10 mL) and the cells were lysed on ice by sonication (10 s with 25% amplitude followed by 30 s break, repeated 15 times). The lysate was centrifuged at 12000g for 15 min at 4 °C and the supernatant was transferred into a new tube. The remaining pellet was resuspended in fresh ice cold lysis buffer (10 mL) and the above procedure was repeated and the supernatant was pooled. The hTNFα was purified from the supernatant using an optimised procedure from Zhang et al.24 In short, the supernatant containing recombinant hTNFα was applied on to two HiTrap™ DEAE Fast Flow (GE Healthcare) columns with 10 mL of total column volume (CV) using an AKTA Start purification system (GE Healthcare) at a flow rate of 1 mL min−1. The columns were equilibrated with 8 CV of buffer A (20 mM Tris-HCl, pH 8.0) and hTNFα was eluted by applying 15% step elution gradient of buffer B (20 mM Tris-HCl with 1 M NaCl, pH 8.0). The second purification step was carried out on a HiTrap™ CM FF (GE Healthcare) column with 5 mL of CV (equilibrated with 5 CV of buffer C) after 1/5 dilution of hTNFα containing fractions from DEAE column with buffer C (20 mM Na2HPO4/KH2PO4, pH 6.0) to decrease conductivity below 5 mS cm−1. Elution of hTNFα was achieved by applying gradient (0%–30%) of buffer D (20 mM Na2HPO4/KH2PO4 with 1 M NaCl, pH 6.0). Fractions containing hTNFα were pooled and dialysed (benzoylated cellulose, MWCO 2 kDa) against PBS pH 7.4 overnight at 4 °C. The dialysed protein was sterile-filtered using a 0.2 μm filter and aliquots were stored at −78 °C. The activity and purity of hTNFα was assessed by an MTT assay utilising L929 cell line and SDS-PAGE respectively. Typically, 10 mg–14 mg of TNFα with a purity ≥97% and IC50 of 0.07 ng mL−1 was obtained from a 250 mL of bacterial culture.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. Arvind H. Patel of the MRC-University of Glasgow Centre for Virus Research for using the SPR equipment.References
- M. Hijnen, D. J. van Zoelen, C. Chamorro, P. van Gageldonk, F. R. Mooi, G. Berbers and R. M. Liskamp, Vaccine, 2007, 25, 6807 CrossRef PubMed.
- G. E. Mulder, H. L. Quarles van Ufford, J. van Ameijde, A. J. Brouwer, J. A. Kruijtzer and R. M. Liskamp, Org. Biomol. Chem., 2013, 11, 2676 RSC.
- P. R. Werkhoven, M. Elwakiel, T. J. Meuleman, H. C. Q. van Ufford, J. A. W. Kruijtzer and R. M. J. Liskamp, Org. Biomol. Chem., 2016, 14, 701 RSC.
- O. Longin, H. van de Langemheen and R. M. J. Liskamp, Bioorg. Med. Chem., 2017, 25, 5008 CrossRef PubMed.
- S. Kalepu and V. Nekkanti, Acta Pharm. Sin. B, 2015, 5, 442 CrossRef PubMed.
- R. U. Kadam, J. Juraszek, B. Brandenburg, C. Buyck, W. B. G. Schepens, B. Kesteleyn, B. Stoops, R. J. Vreeken, J. Vermond, W. Goutier, C. Tang, R. Vogels, R. H. E. Friesen, J. Goudsmit, M. J. P. van Dongen and I. A. Wilson, Science, 2017, 358, 496 CrossRef PubMed.
- J. Canceill, J. Gabard and A. Collet, J. Chem. Soc., Chem. Commun., 1983, 122 RSC.
- Partition Coefficient (n-octanol/water), High Performance Liquid Chromatography (HPLC) Method, OECD Guideline for Testing of Chemicals, adopted on 30.03.1989.
- S. Liang, J. Dai, S. Hou, L. Su, D. Zhang, H. Guo, S. Hu, H. Wang, Z. Rao, Y. Guo and Z. Lou, J. Biol. Chem., 2013, 288, 13799 CrossRef PubMed.
- H. van de Langemheen, V. Korotkovs, J. Bijl, C. Wilson, S. S. Kale, C. Heinis and R. M. J. Liskamp, ChemBioChem, 2017, 18, 387 CrossRef PubMed.
- I. E. Valverde, A. F. Delmas and V. Aucagne, Tetrahedron, 2009, 65, 7597 CrossRef.
- A. C. Conibear, K. Farbiarz, R. L. Mayer, M. Matveenko, H. Kahlig and C. F. W. Becker, Org. Biomol. Chem., 2016, 14, 6205 RSC.
- P. R. Werkhoven, H. van de Langemheen, S. van der Wal, J. A. Kruijtzer and R. M. Liskamp, J. Pept. Sci., 2014, 20, 235 CrossRef PubMed.
- J. A. Nieman, S. K. Nair, S. E. Heasley, B. L. Schultz, H. M. Zerth, R. A. Nugent, K. Chen, K. J. Stephanski, T. A. Hopkins, M. L. Knechtel, N. L. Oien, J. L. Wieber and M. W. Wathen, Bioorg. Med. Chem. Lett., 2010, 20, 3039 CrossRef PubMed.
- T. Mayer and M. E. Maier, Eur. J. Org. Chem., 2007, 4711 CrossRef.
- P. A. Allegretti and E. M. Ferreira, Org. Lett., 2011, 13, 5924 CrossRef PubMed.
- M. Inouye, K. Akamatsu and H. Nakazumi, J. Am. Chem. Soc., 1997, 119, 9160 CrossRef.
- D. Bom, D. P. Curran, S. Kruszewski, S. G. Zimmer, J. Thompson Strode, G. Kohlhagen, W. Du, A. J. Chavan, K. A. Fraley, A. L. Bingcang, L. J. Latus, Y. Pommier and T. G. Burke, J. Med. Chem., 2000, 43, 3970 CrossRef PubMed.
- F. Bellotta, M. V. D'Auria, V. Sepe and A. Zampella, Tetrahedron, 2009, 65, 3659 CrossRef.
- T. Harada, K. Muramatsu, K. Mizunashi, C. Kitano, D. Imaoka, T. Fujiwara and H. Kataoka, J. Org. Chem., 2008, 73, 249 CrossRef PubMed.
- V. Percec, P. Leowanawat, H. J. Sun, O. Kulikov, C. D. Nusbaum, T. M. Tran, A. Bertin, D. A. Wilson, M. Peterca, S. Zhang, N. P. Kamat, K. Vargo, D. Moock, E. D. Johnston, D. A. Hammer, D. J. Pochan, Y. Chen, Y. M. Chabre, T. C. Shiao, M. Bergeron-Brlek, S. Andre, R. Roy, H. J. Gabius and P. A. Heiney, J. Am. Chem. Soc., 2013, 135, 9055 CrossRef PubMed.
- C. Poiesi, A. Albertini, S. Ghielmi, G. Cassani and A. Corti, Cytokine, 1993, 5, 539 CrossRef PubMed.
- F. Mohamadi, N. G. J. Richards, W. C. Guida, R. Liskamp, M. Lipton, C. Caufield, G. Chang, T. Hendrickson and W. C. Still, J. Comput. Chem., 1990, 11, 440 CrossRef.
- C. Zhang, Y. D. Liu, D. W. Zhao, X. N. Li, R. Yu and Z. G. Su, Protein Expression Purif., 2014, 95, 195 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob01104d |
| ‡ A few of the synthetic antibody constructs derived from CTV scaffold derivative 2 tended to precipitate from the solution within several hours of solution preparation. Nevertheless, this time frame still allowed for carrying out the SPR experiments. |
| § Selected data, cleaned and fitted with heterogeneous ligand binding model, are included in the ESI.† |
| This journal is © The Royal Society of Chemistry 2018 |