
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Campestarenes: new building blocks with 5-fold symmetry†
Seong
Nam
 a,
David C.
Ware
a and
Penelope J.
Brothers
*ab
a,
David C.
Ware
a and
Penelope J.
Brothers
*ab
aSchool of Chemical Sciences, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand. E-mail: p.brothers@auckland.ac.nz
bMacDiarmid Institute for Advanced Materials and Nanotechnology, New Zealand
First published on 21st August 2018
Abstract
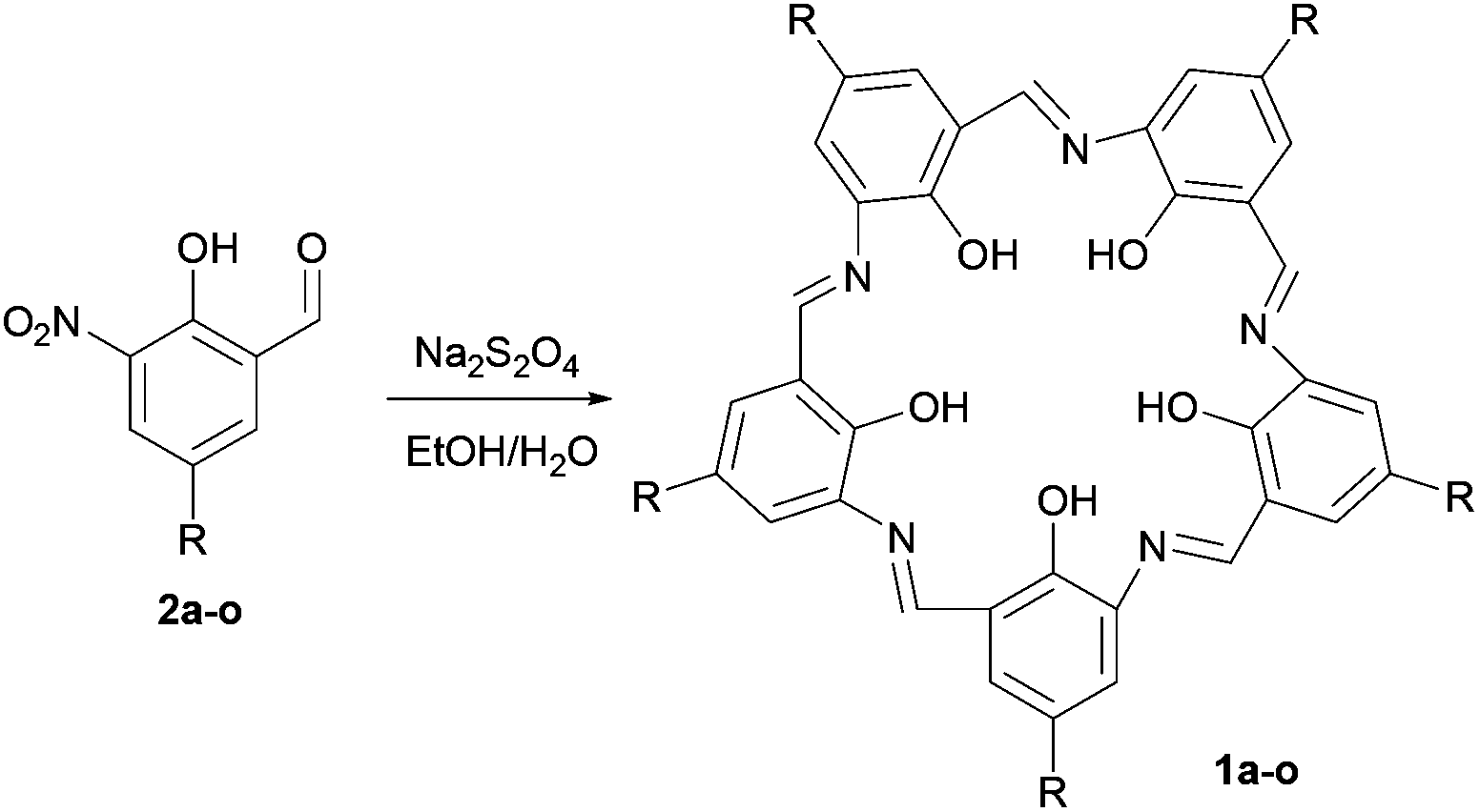
Campestarene is a planar, shape-persistent macrocycle with 5-fold symmetry. A range of derivatives bearing peripheral functional groups suitable for generating supramolecular interactions has been designed and synthesised for potential applications in creating 2D quasicrystal molecular assemblies. The new campestarene derivatives bear ester, carboxylic acid, methoxy, bromo, 4-pyridyl, 4-cyanophenyl and 4-phenyl carboxylic acid groups, including further derivatives of the latter two bearing alkyl chains on the phenyl groups to improve solubility. The campestarene derivatives were prepared by reductive condensation of phenol precursors bearing nitro and formyl groups using Na2S2O4. The target functional groups were installed either by pre-cyclisation derivatisation or by synthesis of methoxy-substituted campestarene and subsequent derivatisation. The cyclisation reaction is tolerant of the functional groups introduced. The ten new campestarene derivatives were characterised by NMR spectroscopy and MALDI-TOF MS, although the poor solubility of some examples precluded their detailed characterisation.
Introduction
Architects and builders designing and constructing houses need supply yards full of building materials of the right size, shape and material properties to create buildings with the desired dimensions, features and functions. The same is true for nano-architects and builders whose supplies of suitable molecular building blocks need to develop to keep pace with the increasing sophistication of their supramolecular nanoarchitectures. Just as the shape of an individual brick has a relationship with the symmetry and properties of the wall it is used to build, individual molecular building blocks also determine the symmetry and properties of supramolecular nanoarchitectures. For this reason, shape-persistent molecular building blocks have proved extremely useful for the design and construction of ordered 2D and 3D materials on the nanoscale. Amongst these, macrocycles with full or partial conjugation have proved especially useful. The macrocycles themselves often contain aryl units as integral components linked by amide, ethynyl or imine bridges which have well-defined spatial configurations and organise the overall shape of the macrocycles.1–6 Porphyrins, with their well-defined 4-fold symmetry and planar geometry, are quintessential examples.7,8 Shape-persistent building blocks with 2-, 3-, 4- and 6-fold symmetry are common, with many synthetically accessible examples available, and have been extensively studied for 2D and 3D assemblies which typically replicate the symmetry of their components.1,7,8 However, extending this principle to the use of building blocks with 5-fold symmetry to generate assemblies which demonstrate 5-fold symmetry in extended arrays has proved much more challenging, primarily because the expression of 5-fold symmetry in 2D and 3D assemblies is inherently more complex. The building blocks cannot pack regularly, as evidenced by the particular properties of Penrose tiling patterns in 2D and quasicrystal packing in 3D.This challenge has received growing interest in recent times, prompted in part by the observation that 3D quasicrystalline metal alloys show unusual properties in a range of applications.9–13 However, the rational design and assembly of 2D quasicrystal packing using molecular pentagons as building blocks remains an elusive goal. It requires an understanding of the unique symmetry properties of 2D crystal tiling patterns based on a pentagonal tile, which are ordered but translationally aperiodic.14–18 Amongst the conceivable experimental approaches, the most obvious is the deposition of planar molecular pentagons on a surface. Attempts to do this have shown that the hexagonal symmetry of the underlying surface rather than the pentagonal shape of the molecule determines the packing arrangement.19 In a serendipitous discovery, regions of 2D quasicrystalline, Penrose tile ordering of ferrocene carboxylic acid molecules on a surface were observed in which the ordering was directed by supramolecular interactions between the ferrocene carboxylic acid groups.20,21 This points to the need for inclusion of functional groups suitable for generating supramolecular interactions on the periphery of the building blocks. This approach has been shown to play a significant role in the packing orientation in 2D self-assembly.22
A further barrier to the exploration of quasicrystalline packing in 2D is the paucity of synthetically available, shape-persistent macrocyclic building blocks with 5-fold symmetry.23–32 Examples from the recent literature are the family of macrocyclic pentamers from Zeng's group,31,33,34 cyanostar reported by Flood et al.,32 and MacLachlan's campestarene.30,35 Although these molecules are planar and rigorously 5-fold symmetric, all of them bear alkyl groups as the peripheral substituents and so are not ideal as building blocks for 2D supramolecular assemblies.19,30,32,34 Suitably functionalised macrocyclic pentamers could be useful for this purpose, and also for dendrimer design and as building blocks for metal- or covalent-organic frameworks (MOFs or COFs).36 The goal of this study was to elaborate the synthesis of the campestarene framework to allow the inclusion of a range of different functional groups on the periphery which could serve as a supply of 5-fold symmetric building blocks for supramolecular assemblies.
Campestarenes are cyclic pentamers comprised of imine-linked phenol groups. Several tautomers can be envisaged, with enol–imine and keto-enamine forms as well as a zwitterionic structure. Overall, the regular, planar shape is reinforced by the 3-centered hydrogen bonds between the imines and hydroxy groups (Fig. 1).30 Campestarenes are prepared by sequential formylation and nitration of the corresponding phenols, followed by cyclisation via a Schiff base amine-aldehyde condensation which gives a homogeneous product in high yield. The high selectivity for the pentameric structure from the one-pot cyclisation is accounted for based on ab initio DFT calculations, which for both tautomers of the pentamer were in accord with the experimentally observed planar structure, whereas the hexamer was calculated to adopt a twisted confirmation.30 The planar structure favours intermolecular π–π stacking leading to aggregation in solution and in the gas phase.37,38 Substitution with bulky organosilyl groups improved their solubility in both polar and non-polar solvents allowing full characterisation, including a molecular structure determination.35 Experimental studies on the tautomerisation behaviour of campestarenes concluded that the location of the interior protons was on nitrogen (keto-enamine form) in polar solvents and on oxygen (enol–imine form) in non-polar solvents, in agreement with DFT calculations. Campestarene derivatives reported to date bear tert-butyl,30 1,1-dimethyl-propyl,30 1,1,3,3-tetramethylbutyl,30 triphenylsilyl35 and triisopropylsilyl groups35 on the periphery 1a–1e (Table 1). The current study extends the synthetic routes to campestarene derivatives containing ester, carboxylic acid, methoxy, bromo, 4-pyridyl, 4-cyanophenyl and 4-phenyl carboxylic acid groups, 1f–1o, chosen for their potential utility as supramolecular recognition groups for the construction of molecular assemblies.
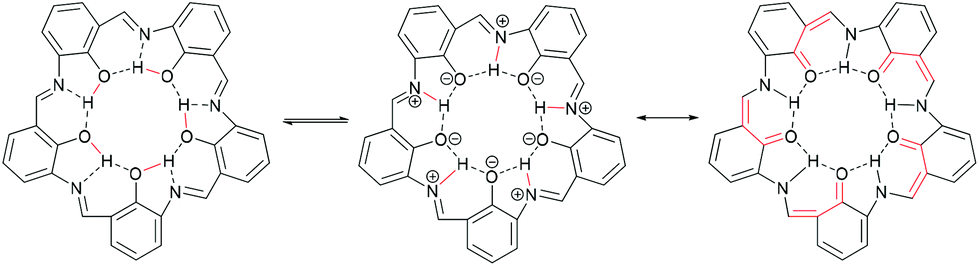 | ||
| Fig. 1 The enol–imine form with a shared hydrogen bond (left), the zwitterionic structure (centre) and the keto-enamine form (right). The major change of hydrogen bonding is highlighted in red. | ||
|
|
|||
|---|---|---|---|
| Compound | R | Compound | R |
| 1a, 2a 30 | tert-Butyl | 1h, 2h | OCH(CH3)COOH |
| 1b, 2b 30 | Isoamyl | 1i, 2i | OCH3 |
| 1c, 2c 30 | 1,1,3,3-Tetramethylbutyl | 1j, 2j | Br |
| 1d, 2d 35 | Triphenylsilyl | 1k, 2k | 4-C5H4N |
| 1e, 2e 35 | Triisopropylsilyl | 1l, 2l | 4-C6H4CN |
| 1f, 2f | OCH(CH3)COOEt | 1m, 2m | 4-C6H4COOH |
| 1g, 2g | OCH2COOH | 1n, 2n | C6H3-3-C4H9-4-COOH |
| 1o, 2o | C6H3-3-C7H15-4-COOH | ||
Results and discussion
The first approach to functionalising campestarenes is pre-cyclisation derivatisation where the target functional group is installed in the para-position of the monomeric phenol before the cyclisation. Scheme 1 shows three synthetic routes to the mono-substituted hydroquinones (5g-Et, 5g-tBu and 5f) required to prepare the precursors 2f–2h to the ester- and carboxylic acid-substituted campestarenes 1f–1h. Route 1 is a single substitution on hydroquinone using bromoacetate t-butyl ester to yield 5g-tBu. Surprisingly, if the bromoacetate ethyl ester was used then a mixture of the di-substituted product and unreacted hydroquinone resulted even when the reaction time, temperature, stoichiometry and solvent were varied. In routes 2 and 3 one hydroquinone hydroxyl group is protected by benzyl and sulfate groups, respectively, resulting in 5g-Et, 5g-tBu and 5f after deprotection. The next steps, formylation to give 6g-Et, 6g-tBu and 6f, followed by nitration, yielded the target ester-substituted campestarene precursors, 2g-Et, 2g-tBu and 2f, of which only 2f was taken on directly to the cyclisation step.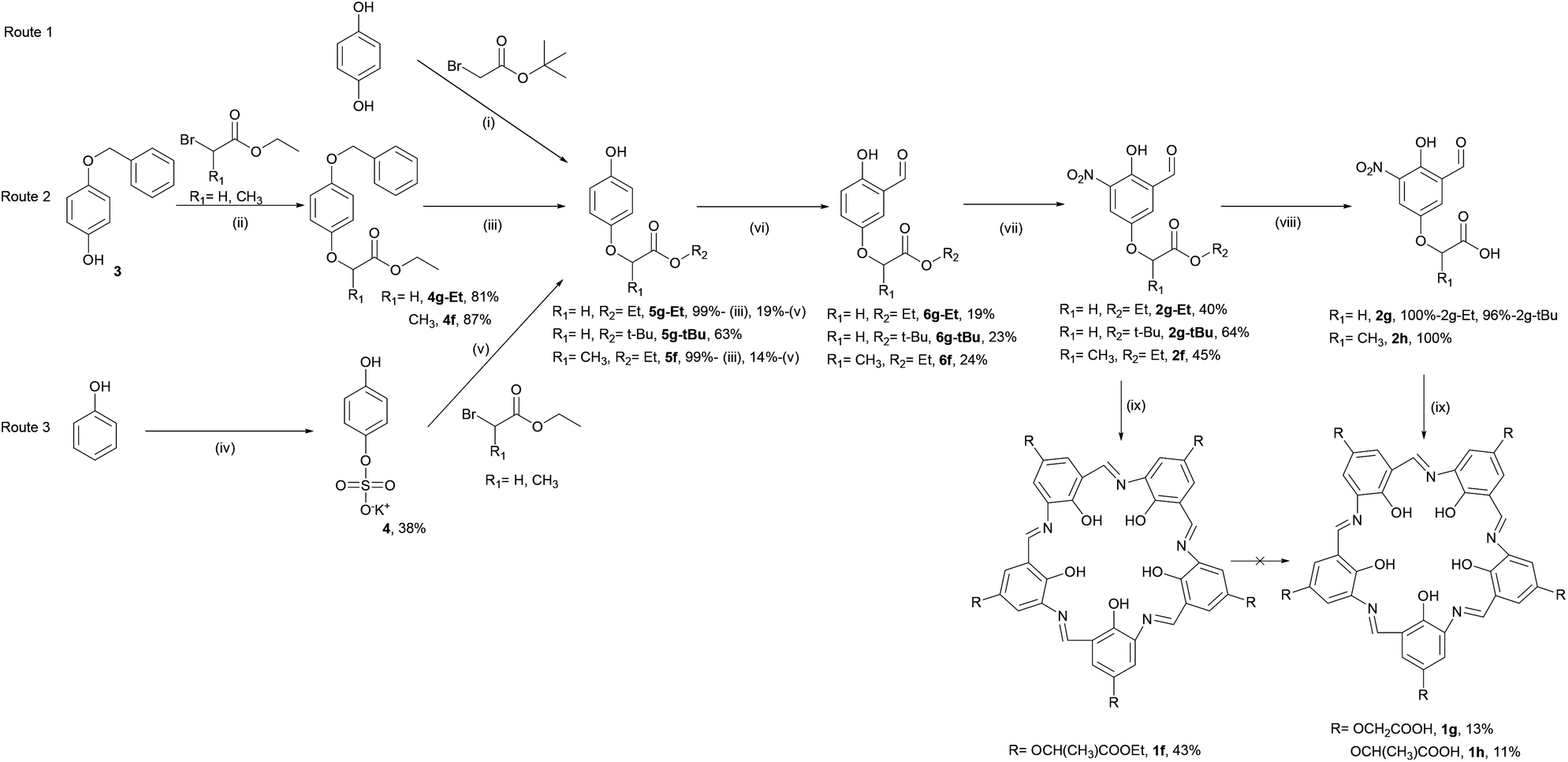 | ||
| Scheme 1 Syntheses of ester/carboxylic acid campestarenes (1f–1h): (i) NaOH, dioxane/H2O, 3 h, then HCl; (ii) K2CO3, acetone, reflux, 3 h, then HCl; (iii) Pd/C, H2, EtOH, 12 h; (iv) K2S2O8, NaOH, H2O, 24 h, then HCl; (v) K2CO3, EtOH, reflux 6 h, then AcOH, reflux, 2 h; (vi) Et3N, MgCl2, CH2O, MeCN, reflux, 24 h, then HCl; (vii) HNO3, AcOH, 2 h; (viii) R2 = Et, NaOH, MeOH/H2O, 12 h, then HCl or R2 = t-Bu, TFA/DCM, 12 h; (ix) Na2S2O4, EtOH/H2O, reflux, 2 h. | ||
Cyclisation of 2f using sodium dithionite in refluxing ethanol/water gave the ester-substituted campestarene 1f. De-esterification of 1f to afford the penta-carboxylic acid campestarene 1h using 1 or 2 M NaOH at 50 °C was unsuccessful. In the presence of strong base and heat the 3-centered hydrogen bonds in the macrocycle core were disrupted. Under milder conditions, <1 M NaOH with or without heating, the ester could not be converted into the carboxylic acid. However, the ester precursors 2g-Et and 2g-tBu could be de-esterified to form the carboxylic acid precursors 2g and 2h which were then successfully cyclised to produce the carboxylic acid campestarenes 1g and 1h.
Compound 1f was purified by flash column chromatography on alumina using dichloromethane/methanol as eluent to give a pure purple solid product after solvent removal. Both silica and alumina column chromatography decomposed 1g and 1h. Presumably, the five polar carboxylic acid groups on the macrocycles were excessively adsorbed onto silica and alumina. However, washing 1g and 1h with 0.1 M HCl followed by 0.1 M NaOH removed most of the organic by-products and chromatography on Sephadex G-10 gave solid purple products, 1g and 1h.
Compared to t-butyl campestarene (1a) which is observed to aggregate and exists as a dimer in the gas phase and in solution,30 the 1H NMR spectra for 1f–1h in DMSO-d6 show no evidence for aggregation (Fig. 2). The signals near 17 and 9 ppm can be assigned to the core hydrogens and the imine protons (N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH), respectively. The two aromatic protons can be observed near 7.2 and 7.7 ppm. Both 1g and 1h are soluble in methanol, although 1h is the more soluble of the two. The ester campestarene 1f is soluble in both methanol and dichloromethane indicating that the presence of the ester groups confers better solubility in organic solvents than the carboxylic acids. Compounds 1f–1h show limited solubility in methanol and dichloromethane. DMSO dissolved 1f–1h best among organic solvents. Due to the poor solubility of 1f, 1g and 1h, 13C NMR, HSQC and HMBC spectra could not be obtained even after more than 100
CH), respectively. The two aromatic protons can be observed near 7.2 and 7.7 ppm. Both 1g and 1h are soluble in methanol, although 1h is the more soluble of the two. The ester campestarene 1f is soluble in both methanol and dichloromethane indicating that the presence of the ester groups confers better solubility in organic solvents than the carboxylic acids. Compounds 1f–1h show limited solubility in methanol and dichloromethane. DMSO dissolved 1f–1h best among organic solvents. Due to the poor solubility of 1f, 1g and 1h, 13C NMR, HSQC and HMBC spectra could not be obtained even after more than 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 scans. The assignments of the 1H NMR spectra for 1f–1h are based on comparison with t-butyl campestarene (1a).30
000 scans. The assignments of the 1H NMR spectra for 1f–1h are based on comparison with t-butyl campestarene (1a).30
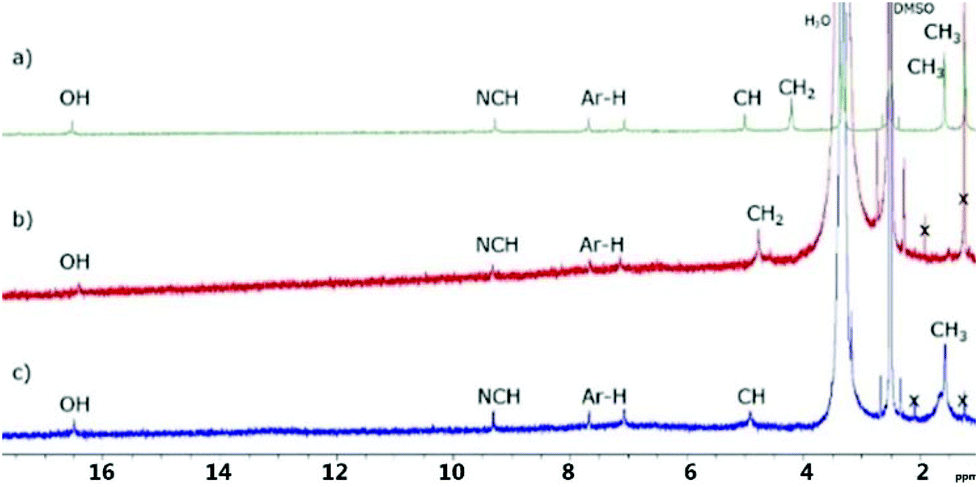 | ||
| Fig. 2 1H NMR spectra in DMSO-d6 of (a) 1f, (b) 1g and (c) 1h. X denotes acetic acid and grease. | ||
An alternative approach to functionalisation of campestarene was conceived via the synthesis of methoxy campestarene, 1i, with a plan to subsequently substitute the methoxy groups. 4-Methoxyphenol was formylated to form 7i and nitrated to prepare the precursor 2i which was cyclised to give 1i (Scheme 2). This purple solid was insoluble in most organic solvents except DMSO and DMF. The crude product 1i was purified by Soxhlet extraction with multiple solvents to remove impurities. Although aggregation of 1i with its very flat geometry was expected, no evidence of aggregation was observed by 1H NMR spectroscopy in DMSO-d6 (Fig. S44†) or MALDI-TOF MS. It is noted that ESI mass spectra could not be recorded for the campestarene derivatives. The attempted de-methylation of 1i to form hydroxy campestarene using boron tribromide did not proceed to completion, even after addition of excess BBr3. As shown in Fig. S46(b) and (c),† the signals assigned to the methoxy protons at 3.9 ppm could still be observed although with diminished intensities. The same difficulty in achieving complete de-methylation has also been reported for another 5-fold symmetric macrocycle.39
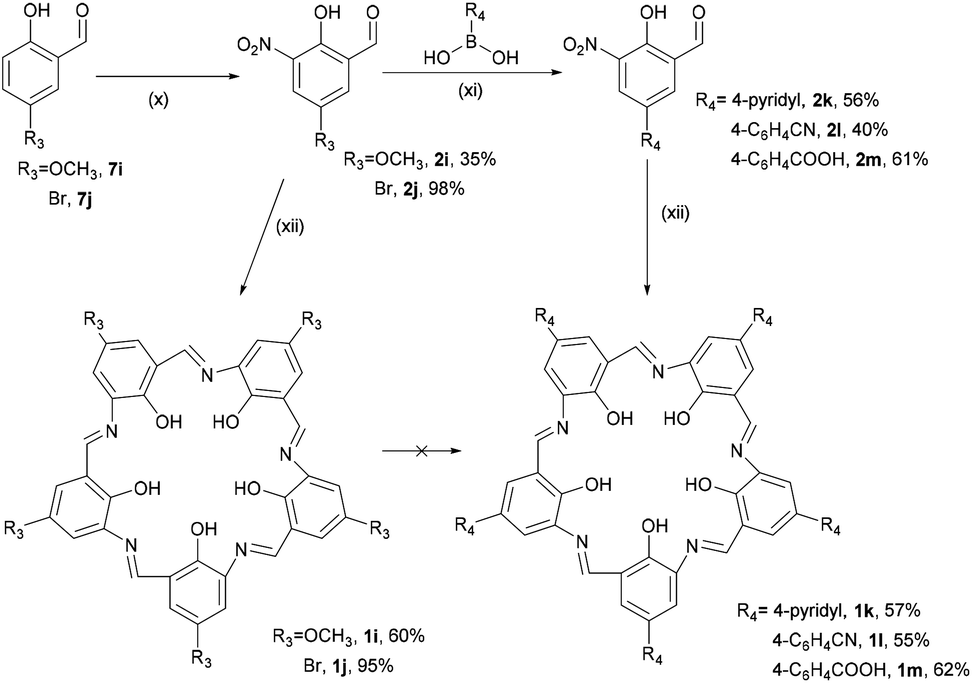 | ||
| Scheme 2 Syntheses of campestarenes (1i–1m): (x) HNO3, AcOH, 2 h; (xi) Na2CO3, Pd(PPh3)4, DMF/H2O, 105 °C, 6 h, then HCl; (xii) Na2S2O4, EtOH/H2O, reflux, 2 h. | ||
The brominated reagent 7j was used because halogens are useful synthons for coupling reactions. Commercially available reagent 7j was nitrated using fuming HNO3 to yield 2j which was then cyclised to form bromo-campestarene 1j. Post-cyclisation substitution of bromo-campestarene, 1j, has not yet been achieved due its poor solubility, being only very sparingly soluble in DMSO. In addition to the poor solubility issue, the 3-centered hydrogen bonds in the macrocycle core limits the use of strong regents that might cleave the imine bridges. Although the MALDI-TOF mass spectrum of 1j shows peaks for [M + H]+, [M + Na]+ and [M + K]+ with the correct isotope pattern for five bromine atoms, its poor solubility did not allow successful purification and consequently no further reactions were undertaken. Surprisingly, dimerised or trimerised 1j (aggregated) species were not found in the MALDI-TOF mass spectrum (see ESI†).
The third approach to preparing peripherally substituted campestarenes was to begin with boronic acid reagents and employ Suzuki cross-coupling to install the substituents on the precursors 2k–2m (Scheme 2). The boronic acid reagents bearing 4-pyridyl, 4-cyanophenyl and 4-carboxyphenyl groups were coupled to 2j, catalysed by tetrakis(triphenylphosphine)palladium(0), to give 2k–2m. Cyclisation of 2k–2m to synthesise the corresponding campestarenes was successfully achieved to yield the distinctive purple solid products 1k–1m, indicating that the condensation reaction is also tolerant to cyano and pyridyl groups. MALDI-TOF MS confirmed the presence of 1k–1m.
Unfortunately the solubilities of 1k–1m were too poor to complete their purification and characterisation beyond MALDI-TOF MS measurements. Even after multiple purification attempts with washing using the Soxhlet technique for 1k and 1l and acid–base washing followed by Sephadex G-10 column chromatography for 1m, the 1H NMR spectra of 1k–1m in DMSO-d6 showed broad signals at 7–8 ppm. Based on a report that the solubility of campestarenes could be improved by appending n-alkyl groups,30n-butyl and n-heptyl groups were attached to the boronic acid reagents, 10n and 10o, which were coupled to the intermediate 2j to synthesise the precursors 2n and 2o (which are alkyl-substituted derivatives of 2m). Cyclisations of 2n and 2o to synthesise campestarenes 1n and 1o were carried out under the same conditions (Scheme 3) and their presence confirmed by MALDI-TOF MS. The solubility of the resulting purple solid products was improved: the n-butyl campestarene, 1n, is soluble in methanol and the n-heptyl campestarene, 1o, can even be dissolved in dichloromethane. However, even with improved solubility, broadening of the signals in their 1H NMR spectra in DMSO-d6 (Fig. S44 and S45†) is still observed even after various attempts at further purification via silica or alumina flash column chromatography, washing using the Soxhlet technique and acid–base washing followed by Sephadex G-10 column chromatography. In addition to the broadening, the NMR spectra show additional structure in the region of the imine NCH and aryl CH peaks, as reported for 1a–1c and interpreted as evidence for aggregation.30 Aggregation probably also causes the poor solubility of 1k–1m and for all of 1k–1o most likely arises from the presence of 10 aromatic rings in each campestarene derivative.
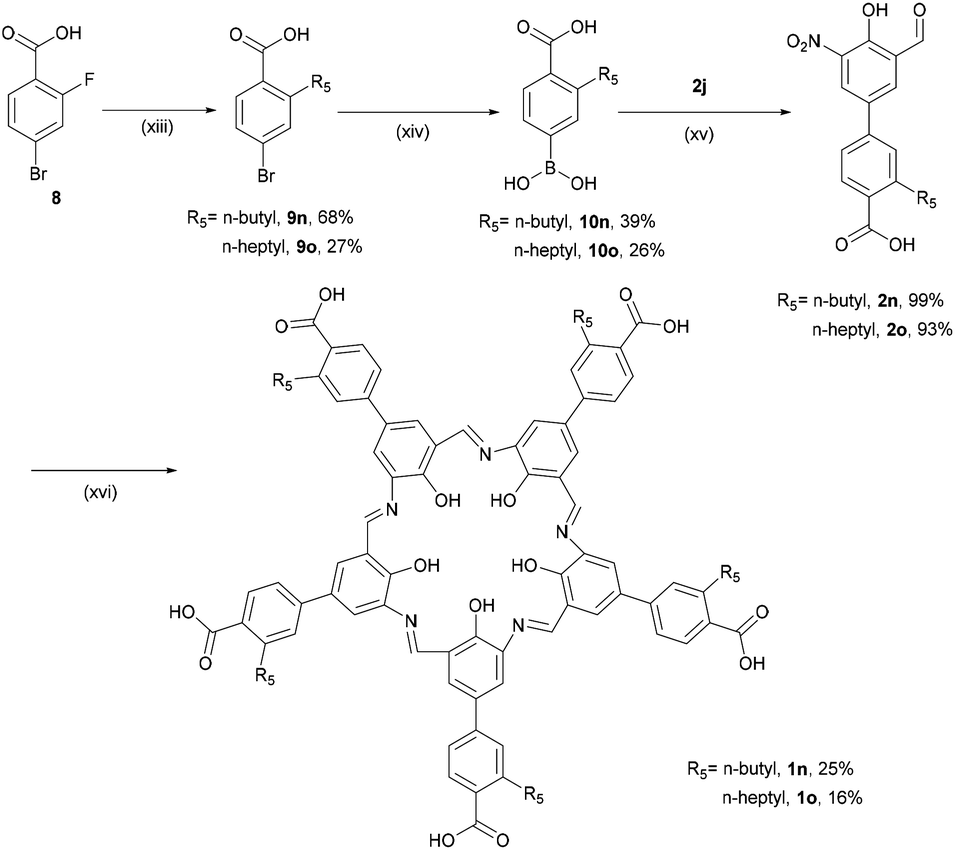 | ||
| Scheme 3 Syntheses of campestarenes (1n and 1o): (xiii) R5MgBr, THF, N2, 17 h, then HCl; (xiv) n-BuLi, B(O-i-Pr)3, THF, −78 °C, 3 h, then HCl; (xv) Na2CO3, Pd(PPh3)4, DMF/H2O, 105 °C, 6 h; (xvi) Na2S2O4, EtOH/H2O, reflux, 2 h. | ||
Conclusions
Campestarenes substituted with methoxy, alkyl ester and alkyl carboxylic acid can be prepared via sequential formylation and nitration of appropriately substituted precursor monomers followed by cyclisation. The cyclisation method is tolerant of several functional groups on the monomers. The products could be purified by Soxhlet extraction or acid–base washing followed by Sephadex G-10 column chromatography, allowing characterisation by 1H NMR spectroscopy and MALDI-TOF MS. Difficulties with postcyclisation substitution on some of the campestarenes may be attributed to the lower reactivity of campestarenes in comparison with the monomer molecules, for example in the de-methylation reaction. Disruption of the 3-centered hydrogen bonds in the macrocycle core by deprotonation in strongly basic media means that such conditions need to be avoided. The 4-bromophenyl-substituted campestarenes are potentially a useful synthon but suffer, as do other derviatives, from poor solubility.The syntheses of monomers substituted with various aryl functional groups were carried out via Suzuki coupling, followed by cyclisation to the long-chain alkyl-substituted campestarenes and the resulting products were characterised by MALDI-TOF MS. Even so, all the derivatives bearing substituted aryl rings were hampered by difficulties in purification of the sparingly soluble products. Presumably, the additional five aryl groups result in some aggregation with evidence of peak-broadening in the 1H NMR spectra of these campestarenes in DMSO-d6.
Overall, the new compounds reported here show that the rational synthesis of campestarenes bearing a range of functional groups can be achieved, expanding the potential utility of this 5-fold symmetric building block.
Experimental section
General information
All reagents and solvents were obtained from commercial suppliers and used as received unless otherwise noted. All dry solvents were collected from a solvent purifier manufactured by LC Technology Solutions Inc. (http://www.ictechinc.com). Sephadex G-10 gel was sourced from Amersham Bioscience. “MilliQ” water was used in all synthetic procedures and in the preparation of Sephadex G-10 columns.High resolution mass spectra were recorded on a Bruker microHTOFQ (Hybrid Quadrupole Time of Flight) mass spectrometer in electrospray ionisation (ESI) mode. MALDI-TOF MS analyses were performed using saturated α-cyano-4-hydroxycinnamic acid in 30% water in methanol as matrix on a Voyager-DE™ PRO MALDI-TOF mass spectrometer (Applied Biosystems, Inc., Foster City, CA). Prior to data collection, a linear external calibration was performed using the mass calibrants: bovine insulin (Mr = 5734), Escherichia coli thioredoxin (Mr = 11674) and equine apomyoglobin (Mr = 16952). For presentation, acquired ESI and MALDI mass spectra underwent smoothing and baseline subtraction using mMass (Version 5.5.0). The UV/Vis absorption measurements were obtained using a Shimadzu UV-Vis-NIR Spectrophotometer UV-3600 Plus and the software package UVProbe 2.50. Infrared spectra were recorded on a PerkinElmer Spectrum 100 infrared spectrometer.
1H, 11B, 13C, COSY, HSQC, HMBC and NOESY NMR spectra were recorded on a Bruker Avance III 300, 400 or HD 500 spectrometers. Spectra recorded in CDCl3, D2O, CD3OD, and DMSO-d6 were referenced to TSP-d4 for D2O, or the respective residual solvent peaks. Alkylation,40 boronation,40 formylation,30,41 nitration,30 Suzuki coupling42 and cyclisation30 were performed according to literature. Analytical grades of precursors 3, 7j, 8, hydroquinone, phenol, reagents and solvents were purchased and used without further purification. 2i,432j,442m,424,454g-Et,464f,465g-Et,45,465g-tBu,475f45,46 and 7i48 were synthesised using either reported or modified procedures. Their characterisation data matched literature values. 6g-Et, 6g-tBu, 6f, 2g-Et, 2g-tBu, 2f, 2g, 2h, 2k, 2l, 9n, 9o, 10n, 10o, 2n, 2o, and campestarenes 1f–1o were synthesised in this research. Compounds 6g-Et and 6g-tBu were prepared using Method A, 2g-Et, 2g-tBu and 2f using Method B, 1f–1o using Method C and 2k, 2l, 2n and 2o using Method D.
Method A (6g-Et and 6g-tBu)30
Two equivalents of Et3N were added dropwise to a mixture of 1 equivalent of the corresponding phenol (5g-Et or 5g-tBu), 2 equivalents of MgCl2 and 2.2 equivalents of paraformaldehyde in dry THF. The reaction mixture was refluxed for 24 h, cooled to r.t. and dilute HCl was added until the remaining solid was completely dissolved. The organic phase was removed by rotary evaporation and then the aqueous phase was extracted with CH2Cl2. The combined extracts were dried over MgSO4, filtered and the solvent removed under vacuum. The crude products were purified by silica gel flash column chromatography.Method B (2g-Et, 2g-tBu and 2f)30
1.1 Equivalents of fuming HNO3 were added dropwise to the corresponding hydroxybenzaldehyde (6g-Et, 6g-tBu or 6f) in glacial acetic acid. After stirring at r.t. for 2 h, water was added to the reaction mixture and a white precipitate formed. The crude product was collected by filtration and recrystallized from hot EtOH by addition of cold water.Method C (1f–1o)30
Six equivalents of sodium dithionite (Na2S2O4) were added to the corresponding 2-hydroxy-3-nitrobenzaldehyde (2f–2o) in EtOH and water. The reaction mixture was refluxed for 2 h, cooled to r.t. and the solvent removed under vacuum.Method D (2k, 2l and 2o)41
One equivalent of 2j, 1.2 equivalents of the corresponding boronic acid, 6 equivalents of sodium carbonate and 0.05 equivalents of tetrakis(triphenylphosphine)palladium(0) were dissolved/suspended in DMF/water (1:1). The reaction mixture was heated at 105 °C under N2 for 6 h. After cooling to r.t., 1 M NaOH was added to the reaction mixture which was then washed with CH2Cl2. The aqueous phase was acidified with 6 M HCl to give an orange/yellow precipitate which was washed with water and diethyl ether and then dried under vacuum.
Ethyl 2-(3-formyl-4-hydroxyphenoxy) acetate, 6g-Et
Method A. 5g-Et (1.80 g, 9.17 mmol), THF (80 mL) and 5% HCl (150 mL). Purified by flash silica column chromatography (eluent: 10% EtOAc in CH2Cl2) to give a yellow oil product. The first band was the product. Yield: 0.380 g, 18.5%; HRMS (ESI) [M + Na]+ = calcd 247.0582 m/z, found 247.0573 m/z; 1H NMR (300 MHz, CDCl3): δ = 10.62 (s, 1 H), 9.77 (s, 1 H), 7.16 (dd, 1 H, J = 9.1, 3.0 Hz), 7.00 (d, 1 H, J = 3.0 Hz), 6.89 (d, 1 H, J = 9.1 Hz), 4.56 (s, 2 H), 4.25 (q, 2 H, J = 7.1 Hz), 1.27 (t, 3 H, J = 7.1 Hz); 13C NMR (75 MHz, CDCl3): δ = 195.99, 168.70, 156.70, 151.00, 125.87, 120.06, 118.83, 117.22, 66.37, 61.44, 14.13.tert-Butyl 2-(3-formyl-4-hydroxyphenoxy) acetate, 6g-tBu
Method A. 5g-tBu (0.280 g, 1.25 mmol), THF (30 mL) and 3 M HCl (2 mL). Purified by flash silica column chromatography (eluent: 10% EtOAc in CH2Cl2) to give the product as a yellow oil. The first band was the product. Yield: 0.073 g, 23.2%; HRMS (ESI) [M + Na]+ = calcd 275.0895 m/z, found 275.0885 m/z; 1H NMR (500 MHz, CDCl3): δ = 10.60 (s, 1 H), 9.76 (s, 1 H), 7.13 (dd, 1 H, J = 9.0, 3.1 Hz), 6.97 (d, 1 H, J = 3.1 Hz), 6.88 (d, 1 H, J = 9.0 Hz), 4.46 (s, 2 H), 1.43 (s, 9 H); 13C NMR (125 MHz, CDCl3): δ = 195.99, 167.79, 156.55, 151.07, 125.74, 120.03, 118.76, 117.20, 82.52, 66.60, 28.01.Ethyl 2-(3-formyl-4-hydroxyphenoxy) propanoate, 6f
A mixture of 5f (2.52 g, 12.0 mmol), MgCl2 (1.71 g, 18.0 mmol), Et3N (6.36 mL, 45.6 mmol) and 5 equivalents of paraformaldehyde (1.80 g, 60.0 mmol) in MeCN (100 mL) was refluxed for 24 h, cooled to r.t. and poured into 5% HCl (150 mL). The crude product was extracted with diethyl ether, dried over MgSO4 and the solvent removed under vacuum. The yellow oil residue was purified by silica gel flash column chromatography (eluent: 10% EtOAc in n-hexane). The first band was the yellow oil product. Yield: 0.670 g, 23.5%; HRMS (ESI) [M + Na]+ = calcd 261.0739 m/z, found 261.0737 m/z; 1H NMR (400 MHz, CDCl3): δ = 10.53 (s, 1 H), 9.69 (s, 1 H), 7.06 (dd, 1 H, J = 8.9, 3.0 Hz), 6.92 (d, 1 H, J = 3.0 Hz), 6.79 (d, 1 H, J = 8.9 Hz), 4.62 (q, 1 H, J = 6.7 Hz), 4.13 (q, 2 H, J = 7.0 Hz), 1.51 (d, 3 H, J = 6.7 Hz), 1.15 (t, 3 H, J = 7.0 Hz); 13C NMR (100 MHz, CDCl3): δ = 195.916, 171.64, 156.43, 150.51, 126.23, 119.96, 118.54, 117.88, 73.66, 61.16, 18.29, 13.94.Ethyl 2-(3-formyl-4-hydroxy-5-nitrophenoxy) acetate, 2g-Et
Method B. 6g-Et (0.455 g, 2.03 mmol), acetic acid (2 mL) and water (100 mL). Purified by recrystallization from chloroform to give a yellow solid. Yield: 0.184 g, 40.4%; HRMS (ESI) [M + Na]+ = calcd 292.0433 m/z, found 292.0439 m/z; 1H NMR (300 MHz, CDCl3): δ = 10.91 (s, 1 H), 10.42 (s, 1 H), 7.90 (d, 1 H, J = 3.1 Hz), 7.72 (d, 1 H, J = 3.2 Hz), 4.67 (s, 2 H), 4.31 (q, 2 H, J = 7.1 Hz), 1.33 (t, 3 H, J = 7.1 Hz); 13C NMR (75 MHz, CDCl3): δ = 187.97, 167.92, 151.85, 150.39, 134.75 (found in HMBC), 126.38, 123.56, 116.97, 66.22, 61.97, 14.28.tert-Butyl 2-(3-formyl-4-hydroxy-5-nitrophenoxy) acetate, 2g-tBu
Method B. 6g-tBu (0.073 g, 0.089 mmol), acetic acid (1 mL) and water (20 mL). Purified by flash silica column chromatography (eluent: 33% EtOAc in CH2Cl2) to give a yellow/orange oil. The 2nd band was the product. Yield: 0.027 g, 63.5%; HRMS (ESI) [M + Na]+ = calcd 320.0746 m/z, found 320.0736 m/z; 1H NMR (400 MHz, CDCl3): δ = 10.90 (s, 1 H), 10.43 (s, 1 H), 7.88 (d, 1 H, J = 3.2 Hz), 7.71 (d, 1 H, J = 3.2 Hz), 4.57 (s, 2 H), 1.50 (s, 9 H).Ethyl 2-(3-formyl-4-hydroxy-5-nitrophenoxy) propanoate, 2f
Method B. 6f (0.467 g, 1.96 mmol), acetic acid (2 mL) and water (100 mL). An orange oil product. Yield: 0.251 g, 45.2%; HRMS (ESI) [M + Na]+ = calcd 306.0590 m/z, found 306.0573 m/z; 1H NMR (300 MHz, CDCl3): δ = 10.90 (s, 1 H), 10.41 (s, 1 H), 7.87 (d, 1 H, J = 3.1 Hz), 7.69 (d, 1 H, J = 3.1 Hz), 4.79 (q, 1 H, J = 6.8 Hz), 4.27 (q, 2 H, J = 7.1 Hz), 1.66 (d, 3 H, J = 6.8 Hz), 1.30 (t, 3 H, J = 7.1 Hz); 13C NMR (75 MHz, CDCl3): δ = 188.04, 171.02, 151.75, 150.17, 135.31 (found in HMBC), 126.32, 124.22, 117.44, 73.93, 61.93, 29.84, 18.44.2-(3-Formyl-4-hydroxy-5-nitrophenoxy) acetic acid, 2g
1 M NaOH (40 mL) was added to a solution of 2g-Et (0.184 g, 0.683 mmol) in MeOH (50 mL). The yellow/orange solution turned to dark red in colour immediately after the addition of 1 M NaOH. The reaction mixture was stirred at r.t. overnight. MeOH was removed under reduced pressure. 1 M HCl (25 mL) was added to the remaining solution to form a yellow solution (pH 1) which was extracted with CH2Cl2, dried over Na2SO4 and the solvent removed under vacuum to give yellow oily solid. Yield: 0.164 g, 99.6%.2g-tBu (0.027 g, 0.091 mmol) was dissolved in CH2Cl2 (5 mL) and TFA (0.02 mL, 0.272 mmol) was added at r.t. The reaction mixture was then stirred at r.t. overnight. The solvent was removed under vacuum and the remaining acid was co-evaporated with dioxane twice to give a yellow oil. Yield: 0.021 g, 95.8%; HRMS (ESI) [M − H]− = calcd 240.0233 m/z, found 240.0168 m/z; 1H NMR (500 MHz, CDCl3): δ = 11.85 (s, 1 H), 10.95 (s, 1 H), 10.45 (s, 1 H), 7.94 (d, 1 H, J = 3.4 Hz), 7.77 (d, 1 H, J = 3.4 Hz), 4.76 (s, 2 H); 13C NMR (125 MHz, CDCl3): δ = 187.96, 172.38, 151.94, 150.19, 126.43, 124.29, 123.48, 117.70, 65.67.
2-(3-Formyl-4-hydroxy-5-nitrophenoxy) propanoic acid, 2h
1 M NaOH (80 mL) was added to a solution of 2f (0.356 g, 1.26 mmol) in MeOH (100 mL). The yellow/orange solution turned to dark red in colour immediately after the addition of 1 M NaOH. The reaction mixture was stirred at r.t. overnight. MeOH was removed under reduced pressure. 1 M HCl (50 mL) was added to the remaining solution to form a yellow solution (pH 1) which was extracted with CH2Cl2, dried over Na2SO4 and the solvent removed under vacuum to give yellow oily solid. Yield: 0.320 g, 99.6%; HRMS (ESI) [M + Na]+ = calcd 278.0277 m/z, found 278.0273 m/z; 1H NMR (500 MHz, CDCl3): δ = 10.91 (s, 1 H), 10.41 (s, 1 H), 7.89 (d, 1 H, J = 3.0 Hz), 7.70 (d, 1 H, J = 3.0 Hz), 4.85 (q, 1 H, J = 7.0 Hz), 1.70 (d, 3 H, J = 7.0 Hz); 13C NMR (125 MHz, CDCl3): δ = 188.12, 175.72, 151.94, 149.87, 135.06, 126.36, 124.05, 117.70, 73.33, 18.37.2-Hydroxy-3-nitro-5-(pyridin-4-yl)benzaldehyde, 2k
Method D. 4-Pyridinylboronic acid (0.148 g, 1.2 mmol), DMF (20 mL)/water (20 mL), 6 M HCl (40 mL), CH2Cl2 (3 × 10 mL), 1 M NaOH (60 mL) and diethyl ether (3 × 5 mL). Yield: 0.134 g, 55.7%; HRMS (ESI) [M + H]+ = calcd 245.0484 m/z, found 245.0559 m/z, [M + Na]+ = calcd 267.0382 m/z, found 267.0377 m/z; 1H NMR (500 MHz, DMSO-d6): δ = 10.25 (s, 1 H), 8.47 (d, 2 H, J = 4.8 Hz), 8.30 (d, 1 H, J = 4.8 Hz), 7.95 (d, 1 H, J = 2.6 Hz), 7.55 (d, 4 H, J = 2.6 Hz); 13C NMR (125 MHz, DMSO-d6): δ = 190.56, 168.71, 149.97, 146.00, 143.11, 130.98, 130.05, 130.00, 118.96, 112.98.3′-Formyl-4′-hydroxy-5′-nitro-[1,1′-biphenyl]-4-carbonitrile, 2l
Method D. 4-Cyanophenylboronic acid (0.176 g, 1.20 mmol), DMF (20 mL)/water (20 mL), 1 M NaOH (20 mL), CH2Cl2 (3 × 10 mL), 6 M HCl (20 mL) and diethyl ether (3 × 5 mL). Yield: 0.108 g, 40.3%; HRMS (ESI) [M − H]− = calcd 267.0484 m/z, found 267.0414 m/z; 1H NMR (300 MHz, DMSO-d6): δ = 10.25 (s, 1 H), 8.25 (d, 1 H, J = 2.9 Hz), 7.90 (d, 1 H, J = 2.9 Hz), 7.76 (d, 4 H, J = 2.0 Hz); 13C NMR (125 MHz, DMSO-d6): δ = 190.65, 168.46, 143.78, 142.87, 132.71, 131.77, 130.43, 130.11, 125.27, 119.19, 114.63, 107.49.4-Bromo-2-butylbenzoic acid, 9n
1-Bromobutane (0.740 mL, 6.85 mmol) was added to magnesium turnings (0.333 g, 13.7 mmol) in dry THF (7 mL) and the mixture was refluxed for 30 min. After cooling to r.t., the mixture was transferred to a solution of 8 (0.500 g, 2.28 mmol) in dry THF (5 mL) at 0 °C. The mixture was warmed to r.t. and stirred for 17 h under N2. Cold water (40 mL) was slowly added to the mixture in an ice bath. The mixture was then acidified with 6 M HCl until pH 1–2 and extracted with EtOAc (2 × 30 mL). The combined organic layers were washed with brine, dried over Na2SO4 and the solvent removed under vacuum to give a white solid. The crude product was purified by flash silica chromatography (eluent: CHCl3 to 30% of MeOH in CHCl3) to give a yellow solid. Yield: 0.396 g, 68.0%; HRMS (ESI) [M − H]− = calcd 255.0099 m/z, found 255.0026 m/z; 1H NMR (500 MHz, CDCl3): δ = 7.74 (t, 1 H, J = 8.0 Hz), 7.34 (m, 2 H), 2.94 (td, 2 H, J = 7.3, 3.1 Hz), 1.67 (p, 2 H, J = 7.6 Hz), 1.41–1.32 (m, 2 H), 0.94 (t, 3 H, J = 7.3 Hz); 13C NMR (125 MHz, CDCl3): δ = 197.88, 162.60, 160.55, 131.93, 131.90, 128.15, 128.12, 124.94, 124.83, 120.45, 120.24, 43.44, 43.39, 26.12, 22.44, 14.00.4-Bromo-2-heptylbenzoic acid, 9o
1-Bromoheptane (11 mL, 68.5 mmol) was added to magnesium turnings (3.33 g, 137 mmol) in dry THF (50 mL) and the mixture was refluxed for 30 min. After cooling to r.t., the mixture was transferred to a solution of 8 (5.00 g, 22.8 mmol) in dry THF (50 mL) at 0 °C. The mixture was warmed to r.t. and stirred for 24 h under N2. Cold water (400 mL) was slowly added to the mixture in an ice bath. The mixture was then acidified with 6 M HCl until pH 1–2 and extracted with EtOAc (2 × 200 mL). The combined organic layers were washed with brine, dried over Na2SO4 and the solvent removed under vacuum to give a white solid. The crude product was purified by flash silica chromatography (eluent: CHCl3 to 30% of MeOH in CHCl3) to give a yellow solid. Yield: 1.82 g, 26.7%; 1H NMR (400 MHz, CDCl3): δ = 11.59 (s, 1 H), 7.93 (t, 1 H, J = 7.8 Hz), 7.04 (m, 2 H), 2.98 (t, 2 H, J = 7.7 Hz), 1.64–1.59 (m, 2 H), 1.36–1.24 (m, 6 H), 0.90 (t, 3 H, J = 7.0 Hz); 13C NMR (100 MHz, CDCl3): δ = 170.07, 162.72, 161.53, 152.42, 134.13, 124.29, 116.97, 35.90, 31.84, 31.64, 30.76, 29.17, 22.76, 14.16.4-Borono-2-butylbenzoic acid, 10n
2.5 M n-butyllithium in n-hexane (21.5 mL, 53.9 mmol) was added dropwise to a solution of 9n (3.96 g, 15.4 mmol) in dry THF (200 mL) at −78 °C and the mixture was stirred at −78 °C for 10 min. Triisopropyl borate (12.5 mL, 53.9 mmol) was then added dropwise at −78 °C and the mixture was then stirred at −78 °C for 3 h. After warmed up to 0 °C, the reaction mixture was quenched with 2 M HCl (60.0 mL) and extracted with EtOAc (2 × 300 mL). The combined organic layers were stirred with 2.5 M NaOH (160 mL) for 10 min. The collected aqueous layer was acidified to pH 3 with 6 M HCl, extracted with EtOAc, dried over Na2SO4 and concentrated to give a white precipitate which was collected by filtration, washed with CH2Cl2 and dried under vacuum. Yield: 1.33 g, 39.0%; HRMS (ESI) [M − H]− = calcd 221.0993 m/z, found 221.0988 m/z; 1H NMR (500 MHz, MeOD): δ = 7.87–7.46 (m, 3 H), 2.97 (t, 2 H, J = 8.0 Hz), 1.60–1.54 (m, 2 H), 1.41–1.35 (m, 4 H), 0.95 (t, 3 H, J = 7.3 Hz); 13C NMR (125 MHz, MeOD): δ = 171.81, 144.02, 137.43, 133.07, 131.98, 130.39, 125.46, 35.39, 35.07, 23.81, 14.28; 11B NMR (160 MHz, MeOD): δ = 18.54 (s, 1 B).4-Borono-2-heptylbenzoic acid, 10o
2.5 M n-butyllithium in n-hexane (7.31 mL, 18.3 mmol) was added dropwise to a solution of 9o (1.82 g, 6.10 mmol) in dry THF (100 mL) at −78 °C and the mixture was stirred at −78 °C for 10 min. Triisopropyl borate (4.22 mL, 18.3 mmol) was then added dropwise at −78 °C and the mixture was then stirred at −78 °C for 3 h. After warming to 0 °C, the reaction mixture was quenched with 2 M HCl (25 mL) and extracted with EtOAc (2 × 30 mL). The combined organic layers were stirred with 2.5 M NaOH (25 mL) for 10 min. The collected aqueous layer was acidified to pH 3 with 6 M HCl, extracted with ethyl acetate, dried over Na2SO4 and concentrated to give a white precipitate which collected by filtration, was washed with CH2Cl2 and dried under vacuum. Yield: 0.413 g, 25.7%; HRMS (ESI) [M − H]− = calcd 263.1463 m/z, found 263.1466 m/z; 1H NMR (400 MHz, MeOD): δ = 7.76–7.46 (m, 3 H), 2.96 (t, 2 H, J = 7.9 Hz), 1.60–1.53 (m, 2 H), 1.35–1.29 (m, 8 H), 0.91 (t, 3 H, J = 7.3 Hz); 13C NMR (100 MHz, MeOD): δ = 162.72, 144.04, 137.09, 131.94, 131.56, 130.41, 35.34, 33.17, 33.01, 30.76, 30.26, 23.69, 14.41; 11B NMR (128 MHz, MeOD): δ = 18.76 (s, 1 B).3-Butyl-3′-formyl-4′-hydroxy-5′-nitro-[1,1′-biphenyl]-4-carboxylic acid, 2n
Method D. 10n (0.109 g, 0.491 mmol), DMF (10 mL)/water (10 mL), 1 M NaOH (10 mL), CH2Cl2 (3 × 5 mL), 6 M HCl (10 mL) and washing with water only. Yield: 0.167 g, 99.0%; HRMS (ESI) [M − H]− = calcd 342.0983 m/z, found 342.0994 m/z; 1H NMR (300 MHz, DMSO-d6): δ = 12.87 (s, 1 H), 10.33 (s, 1 H), 8.54 (d, 1 H, J = 2.5 Hz), 8.34 (d, 1 H, J = 2.5 Hz), 7.88 (d, 1 H, J = 8.2 Hz), 7.68 (d, 1 H, J = 2.1 Hz), 7.65 (dd, 1 H, J = 8.2, 2.1 Hz), 3.03 (t, 2 H, J = 8.0 Hz), 1.60 (p, 2 H, J = 7.5 Hz), 1.38 (s, 2 H, J = 7.5 Hz), 0.93 (t, 3 H, J = 7.2 Hz).3′-Formyl-3-heptyl-4′-hydroxy-5′-nitro-[1,1′-biphenyl]-4-carboxylic acid, 2o
Method D. 10o (0.100 g, 0.379 mmol), DMF (8 mL)/water (8 mL), 1 M NaOH (8 mL), CH2Cl2 (3 × 4 mL), 6 M HCl (8 mL) and washing with water alone. Yield: 0.113 g, 92.6%; HRMS (ESI) [M + Na]+ = calcd 408.1418 m/z, found 408.1404 m/z; 1H NMR (300 MHz, CDCl3): δ = 11.41 (s, 1 H), 10.50 (s, 1 H), 8.61 (d, 1 H, J = 2.5 Hz), 8.39 (d, 1 H, J = 2.5 Hz), 8.16 (d, 1 H, J = 8.8 Hz), 7.52 (d, 1 H, J = 1.9 Hz), 7.49 (m, 1 H), 3.13 (t, 2 H, J = 7.6 Hz), 1.72 (p, 2 H, J = 6.7 Hz), 1.45–1.30 (m, 8 H), 0.91 (t, 3 H, J = 6.6 Hz); 13C NMR (75 MHz, CDCl3): δ = 188.99, 170.92, 156.27, 147.49, 141.11, 135.37, 132.89, 135.66, 132.45, 129.52, 129.33, 128.08, 126.09, 124.13, 34.98, 32.11, 31.98, 29.92, 29.26, 22.81, 14.25.Penta-ethylpropanoate(oxy)-campestarene, 1f
Method C. 2f (0.251 g, 0.886 mmol) in EtOH (20 mL) and water (3 mL). Purified twice by flash alumina column chromatography (eluent: 3–10% MeOH in CH2Cl2). On column chromatography impurities were removed with 3% MeOH in CH2Cl2 and the pure purple product was collected with 4–10% MeOH in CH2Cl2. Yield: 90.0 mg, 43.2%; MALDI-TOF-MS [M + H]+ = calcd 1176.4223 m/z, found 1176.9572 m/z, [M + Na]+ = calcd 1198.4121 m/z, found 1198.9564 m/z, [M + K]+ = calcd 1214.3860 m/z, found 1214.9379 m/z; 1H NMR (500 MHz, DMSO-d6): δ = 16.51 (s, 5 H, OH), 9.27 (s, 5 H, HCN), 7.67 (s, 5 H, Ar–H), 7.06 (s, 5 H, Ar–H), 5.01 (q, 5 H, J = 6.7 Hz, CH), 4.22 (q, 10 H, J = 6.7 Hz, CH2), 1.57 (d, 15 H, J = 6.7 Hz, CH3), 1.23 (t, 15 H, J = 6.7 Hz, CH3). UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 555 (1334), 440 (3016), 304 (3408).
Pentaaceto-campestarene, 1g
Method C. 2g (0.020 g, 0.083 mmol), EtOH (2 mL) and water (0.3 mL). Purification: after addition of water (10 mL), the mixture was acidified with 0.1 M HCl to give purple precipitate which was re-dissolved in 0.1 M NaOH (1 mL) and purified by Sephadex G-10 column chromatography. The collected purple solution was acidified with 0.1 M HCl to give purple precipitate which was collected by centrifuge, washed with minimum amount of water and dried under vacuum to give purple solid products. Yield: 2.00 mg, 12.5%; MALDI-TOF-MS [M + H]+ = calcd 966.1875 m/z, found 966.3094 m/z, [M + Na]+ = calcd 988.1773 m/z, found 988.2966 m/z, [M + K]+ = calcd 1004.1512 m/z, found 1004.3279 m/z; 1H NMR (300 MHz, DMSO-d6): δ = 16.39 (s, 5 H, OH), 9.31 (s, 5 H, HCN), 7.66 (s, 5 H, Ar–H), 7.12 (s, 5 H, Ar–H), 4.75 (s, 10 H, CH2). UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 545 (1337), 321 (3692).
Pentapropionoxy-campestarene, 1h
Method C. 2h (0.320 g, 1.26 mmol) in EtOH (35 mL) and water (4 mL). Purification: after addition of water (20 mL), the mixture was acidified with 0.1 M HCl to give purple precipitate which was re-dissolved in 0.1 M NaOH (1 mL) and purified by Sephadex G-10 column chromatography. The collected purple solution was acidified with 0.1 M HCl to give purple precipitate which was collected by centrifuge, washed with minimum amount of water and dried under vacuum to give purple solid products. Yield: 28.0 mg, 10.8%; MALDI-TOF-MS [M + H]+ = calcd 1036.2658 m/z, found 1036.4210 m/z, [M + Na]+ = calcd 1058.2556 m/z, found 1058.4048 m/z, [M + K]+ = calcd 1074.2295 m/z, found 1074.4120 m/z; 1H NMR (400 MHz, DMSO-d6): δ = 16.49 (s, 5 H, OH), 9.30 (s, 5 H, HCN), 7.66 (s, 5 H, Ar–H), 7.06 (s, 5 H, Ar–H), 4.90 (d, 5 H, J = 7.0 Hz, CH), 1.57 (d, 15 H, J = 7.0 Hz, CH3). UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 551 (5303), 450 (4807), 319 (6429).
Pentamethoxy-campestarene, 1i
Method C. 2i (0.182 g, 0.923 mmol) in EtOH (20 mL) and water (3 mL). The purple crude product was purified twice by Soxhlet technique using CH2Cl2 and MeOH as solvents for 24 h each and multiple washing with water using an ultra-sonic bath. Yield: 0.082 g, 59.4%; MALDI-TOF-MS [M + H]+ = calcd 746.2384 m/z, found 746.3896 m/z, [M + Na]+ = calcd 768.2282 m/z, found 768.3630 m/z, [M + K]+ = calcd 784.2021 m/z, found 784.3323 m/z; 1H NMR (400 MHz, DMSO-d6): δ = 16.34 (s, 5 H, OH), 9.26 (s, 5 H, HCN), 7.53 (s, 5 H, Ar–H), 7.51 (s, 5 H, Ar–H), 3.84 (s, 15 H, CH3). UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 442 (966), 303 (1148).
Pentabromo-campestarene, 1j
Method C. 2j (1 g, 4.06 mmol) in EtOH (20 mL) and water (3 mL). The purple crude product was purified twice by Soxhlet technique using CH2Cl2 and MeOH as solvents for 24 h each and multiple washing with water using an ultra-sonic bath. Yield: 0.764 g, 94.9%; MALDI-TOF-MS [M + H]+ = calcd 989.7340 m/z, found 989.7634 m/z, [M + Na]+ = calcd 1011.7238 m/z, found 1011.7281 m/z, [M + K]+ = calcd 1027.6977 m/z, found 1027.7227 m/z. UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 534 (3001), 304 (2826).Penta(pyridyl)-campestarene, 1k
Method C. 2k (0.134 g, 0.549 mmol) in EtOH (20 mL) and water (3 mL). The purple crude product was purified twice by Soxhlet technique using CH2Cl2 and MeOH as solvents for 24 h each and multiple washing with water using an ultra-sonic bath. Yield: 0.062 g, 57.4%; MALDI-TOF-MS [M + H]+ = calcd 981.3183 m/z, found 981.4041 m/z, [M + Na]+ = calcd 1003.3081 m/z, found 1003.3911 m/z, [M + K]+ = calcd 1019.2820 m/z, found 1019.3751 m/z. UV-vis (DMSO) λmax/nm: 538, 317.Penta(cyanoaryl)-campestarene, 1l
Method C. 2l (0.108 g, 0.402 mmol) in EtOH (20 mL) and water (3 mL). The purple crude product was purified twice by Soxhlet technique using CH2Cl2 and MeOH as solvents for 24 h each and multiple washing with water using an ultra-sonic bath. Yield: 0.049 g, 55.1%; MALDI-TOF-MS [M + H]+ = calcd 1101.3183 m/z, found 1101.5008 m/z, [M + Na]+ = calcd 1123.3081 m/z, found 1123.4918 m/z, [M + K]+ = calcd 1139.2820 m/z, found 1139.4703 m/z. UV-vis (DMSO) λmax/nm: 537, 319.Penta(arylcarboxylic acid)-campestarene, 1m
Method C. 2m (0.176 g, 0.612 mmol) in EtOH (20 mL) and water (3 mL). The purple crude product was purified twice by Soxhlet technique using CH2Cl2 and MeOH as solvents for 24 h each and multiple washing with water using an ultra-sonic bath. Yield: 0.091 g, 62.3%; MALDI-TOF-MS [M + Na]+ = calcd 1218.2810 m/z, found 1218.5125 m/z. UV-vis (DMSO) λmax/nm: 538.Penta(n-butylarylcarboxylic acid)-campestarene, 1n
Method C. 2n (0.167 g, 0.486 mmol) in EtOH (17 mL) and water (2 mL). Purification: after addition of water (10 mL), the mixture was acidified with 0.1 M HCl to give purple precipitate which was re-dissolved in 0.1 M NaOH (0.5 mL) and purified by Sephadex G-10 column chromatography. The collected purple solution was acidified with 0.1 M HCl to give purple precipitate which was collected by centrifuge, washed with minimum amount of water and dried under vacuum to give purple solid products. Yield: 35.1 mg, 24.5%; MALDI-TOF-MS [M + H]+ = calcd 1476.6042 m/z, found 1476.7339 m/z, [M + Na]+ = calcd 1498.5940 m/z, found 1498.8278 m/z, [M + K]+ = calcd 1514.5679 m/z, found 1514.8114 m/z. UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 544 (4342), 314 (9788).Penta(n-heptylarylcarboxylic acid)-campestarene, 1o
Method C. 2o (0.113 g, 0.293 mmol) in EtOH (17 mL) and water (2 mL). Purification: after addition of water (10 mL), the mixture was acidified with 0.1 M HCl to give purple precipitate which was re-dissolved in 0.1 M NaOH (1 mL) and purified by Sephadex G-10 column chromatography. The collected purple solution was acidified with 0.1 M HCl to give purple precipitate which was collected by centrifuge, washed with minimum amount of water and dried under vacuum to give purple solid products. Yield: 15.7 mg, 15.9%; MALDI-TOF-MS [M + H]+ = calcd 1686.8390 m/z, found 1686.9863 m/z, [M + Na]+ = calcd 1780.8287 m/z, found 1780.9654 m/z, [M + K]+ = calcd 1724.8027 m/z, found 1724.9409 m/z. UV-vis (λmax/nm (ε/M−1 cm−1), DMSO): 547 (6644), 313 (12210).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
SJN is grateful to the University of Auckland, Faculty of Science for the award of a PhD fellowship.Notes and references
- Y. Jin, Q. Wang, P. Taynton and W. Zhang, Acc. Chem. Res., 2014, 47, 1575–1586 CrossRef PubMed.
- B. Gong and Z. Shao, Acc. Chem. Res., 2013, 46, 2856–2866 CrossRef PubMed.
- Z.-T. Li, J.-L. Hou, C. Li and H.-P. Yi, Chem. – Asian J., 2006, 1, 766–778 CrossRef PubMed.
- W. Zhang and J. S. Moore, Angew. Chem., Int. Ed., 2006, 45, 4416–4439 CrossRef PubMed.
- J. S. Moore, Acc. Chem. Res., 1997, 30, 402–413 CrossRef.
- N. E. Borisova, M. D. Reshetova and Y. A. Ustynyuk, Chem. Rev., 2007, 107, 46–79 CrossRef PubMed.
- C. Grave and A. D. Schlüter, Eur. J. Org. Chem., 2002, 3075–3098 CrossRef.
- M. Iyoda, J. Yamakawa and M. J. Rahman, Angew. Chem., Int. Ed., 2011, 50, 10522–10553 CrossRef PubMed.
- M. E. Zoorob, M. D. B. Charlton, G. J. Parker, J. J. Baumberg and M. C. Netti, Nature, 2000, 404, 740–743 CrossRef PubMed.
- A. P. Tsai and M. Yoshimura, Appl. Catal., A, 2001, 214, 237–241 CrossRef.
- J. Y. Park, D. F. Ogletree, M. Salmeron, R. A. Ribeiro, P. C. Canfield, C. J. Jenks and P. A. Thiel, Science, 2005, 309, 1354–1356 CrossRef PubMed.
- T. Matsui, A. Agrawal, A. Nahata and Z. V. Vardeny, Nature, 2007, 446, 517–521 CrossRef PubMed.
- A. I. Goldman, Sci. Technol. Adv. Mater., 2014, 15, 044801 CrossRef PubMed.
- T. Tarnai, Z. Gáspár and L. Szalai, Biophys. J., 1995, 69, 612–618 CrossRef PubMed.
- D. Levine and P. J. Steinhardt, Phys. Rev. B: Condens. Matter, 1986, 34, 596–616 CrossRef.
- G. Kuperberg and W. Kuperberg, Discrete Comput. Geom., 1990, 5, 389–397 CrossRef.
- D. L. D. Caspar and E. Fontano, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 14271–14278 CrossRef.
- A. J. Olson, Y. H. E. Hu and E. Keinan, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 20731–20736 CrossRef PubMed.
- T. Bauert, L. Merz, D. Bandera, M. Parschau, J. S. Siegel and K.-H. Ernst, J. Am. Chem. Soc., 2009, 131, 3460–3461 CrossRef PubMed.
- N. A. Wasio, R. C. Quardokus, R. P. Forrest, C. S. Lent, S. A. Corcelli, J. A. Christie, K. W. Henderson and S. A. Kandel, Nature, 2014, 507, 86–89 CrossRef PubMed.
- O. Guillermet, E. Niemi, S. Nagarajan, X. Bouju, D. Martrou, A. Gourdon and S. Gauthier, Angew. Chem., Int. Ed., 2009, 48, 1970–1973 CrossRef PubMed.
- S. Mohnani and D. Bonifazi, Coord. Chem. Rev., 2010, 254, 2342–2362 CrossRef.
- J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134–5175 CrossRef PubMed.
- H. W. Kroto, J. R. Heath, S. C. O'Brien, R. F. Curl and R. E. Smalley, Nature, 1985, 318, 162–163 CrossRef.
- R. Misra and T. K. Chandrashekar, Acc. Chem. Res., 2008, 41, 265–279 CrossRef PubMed.
- Z. Zhang, Y. Luo, B. Xia, C. Han, Y. Yu, X. Chen and F. Huang, Chem. Commun., 2011, 47, 2417–2419 RSC.
- Z. Zhang, B. Xia, C. Han, Y. Yu and F. Huang, Org. Lett., 2010, 12, 3285–3287 CrossRef PubMed.
- C. Han, F. Ma, Z. Zhang, B. Xia, Y. Yu and F. Huang, Org. Lett., 2010, 12, 4360–4363 CrossRef PubMed.
- J. Kim, I.-S. Jung, S.-Y. Kim, E. Lee, J.-K. Kang, S. Sakamoto, K. Yamaguchi and K. Kim, J. Am. Chem. Soc., 2000, 122, 540–541 CrossRef.
- S. Guieu, A. K. Crane and M. J. MacLachlan, Chem. Commun., 2011, 47, 1169–1171 RSC.
- B. Qin, X. Chen, X. Fang, Y. Shu, Y. K. Yip, Y. Yan, S. Pan, W. Q. Ong, C. Ren, H. Su and H. Zeng, Org. Lett., 2008, 10, 5127–5130 CrossRef PubMed.
- S. Lee, C.-H. Chen and A. H. Flood, Nat. Chem., 2013, 5, 704–710 CrossRef PubMed.
- Z. Du, C. Ren, R. Ye, J. Shen, V. Maurizot, Y. Lu, J. Wang and H. Zeng, Chem. Commun., 2011, 47, 12488–12490 RSC.
- C. Ren, F. Zhou, B. Qin, R. Ye, S. Shen, H. Su and H. Zeng, Angew. Chem., 2011, 123, 10800–10803 CrossRef.
- Z. Chen, S. Guieu, N. G. White, F. Lelj and M. J. MacLachlan, Chem. – Eur. J., 2016, 22, 17657–17672 CrossRef PubMed.
- J. Bacsa, R. J. Less, H. E. Skelton, Z. Soracevic, A. Steiner, T. C. Wilson, P. T. Wood and D. S. Wright, Angew. Chem., Int. Ed., 2011, 50, 8279–8282 CrossRef PubMed.
- D. Zhao and J. S. Moore, J. Org. Chem., 2002, 67, 3548–3554 CrossRef PubMed.
- D. J. Hill and J. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5053–5057 CrossRef PubMed.
- B. Qin, L. Jiang, S. Shen, C. Sun, W. Yuan, S. F. Y. Li and H. Zeng, Org. Lett., 2011, 13, 6212–6215 CrossRef PubMed.
- M. Hammond, D. G. Washburn, T. H. Hoang, S. Manns, J. S. Frazee, H. Nakamura, J. R. Patterson, W. Trizna, C. Wu, L. M. Azzarano, R. Nagilla, M. Nord, R. Trejo, M. S. Head, B. Zhao, A. M. Smallwood, K. Hightower, N. J. Laping, C. G. Schnackenberg and S. K. Thompson, Bioorg. Med. Chem. Lett., 2009, 19, 4441–4445 CrossRef PubMed.
- Z.-N. Sun, F.-Q. Liu, Y. Chen, P. K. H. Tam and D. Yang, Org. Lett., 2008, 10, 2171–2174 CrossRef PubMed.
- J. B. Patterson, D. G. Lonergan, G. A. Flynn, Q. Zeng and P. V. Pallai, US8614253B2, 2013 Search PubMed.
- R. Baker and J. L. Castro, J. Chem. Soc., Perkin Trans. 1, 1990, 47–65 RSC.
- M. R. Heinrich, W. Steglich, M. G. Banwell and Y. Kashman, Tetrahedron, 2003, 59, 9239–9247 CrossRef.
- K. Watson and A. Serban, Aust. J. Chem., 1995, 48, 1503–1509 CrossRef.
- D.-I. Kato, S. Mitsuda and H. Ohta, J. Org. Chem., 2003, 68, 7234–7242 CrossRef PubMed.
- Ł. Winiarski, J. Oleksyszyn and M. Sieńczyk, J. Med. Chem., 2012, 55, 6541–6553 CrossRef PubMed.
- D.-J. Barrios Antunez, M. D. Greenhalgh, C. Fallan, A. M. Z. Slawin and A. D. Smith, Org. Biomol. Chem., 2016, 14, 7268–7274 RSC.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob00957k |
| This journal is © The Royal Society of Chemistry 2018 |