
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMolecular tweezers with a rotationally restricted linker and freely rotating porphyrin moieties†
Rhys B.
Murphy‡
 a,
Duc-Truc
Pham
b,
Jonathan M.
White
c,
Stephen F.
Lincoln
b and
Martin R.
Johnston
*a
a,
Duc-Truc
Pham
b,
Jonathan M.
White
c,
Stephen F.
Lincoln
b and
Martin R.
Johnston
*a
aFlinders Centre for NanoScale Science and Technology, College of Science and Engineering, Flinders University, Bedford Park, Adelaide, Australia. E-mail: martin.johnston@flinders.edu.au
bDepartment of Chemistry, The University of Adelaide, Adelaide, Australia
cSchool of Chemistry, The University of Melbourne, Melbourne, Australia
First published on 14th August 2018
Abstract
The effect of the degree of conformational rigidity and/or flexibility on preorganisation in artificial molecular receptors continues to be actively explored by supramolecular chemists. This work describes a bis-porphyrin architecture, linked via a rigid polycyclic backbone, in which a sterically bulky 2,3,5,6-tetramethylphenyl diimide core restricts rotation to afford two non-interconvertible tweezer conformations; syn- and anti-. After separation, the host–guest chemistry of each conformation was studied independently. The difference in host geometry allows only the syn-conformation to form a strong 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 bis-porphyrin complex with the diamino ligand 1,4-diazabicyclo[2.2.2]octane (DABCO) (K11 = 1.25 × 108 M−1), with the anti-conformation adopting a 2:2 sandwich complex with DABCO (K22 = 5.57 × 1017 M−3).
:1 bis-porphyrin complex with the diamino ligand 1,4-diazabicyclo[2.2.2]octane (DABCO) (K11 = 1.25 × 108 M−1), with the anti-conformation adopting a 2:2 sandwich complex with DABCO (K22 = 5.57 × 1017 M−3).
Introduction
The guest complexation behavior of molecular tweezers is heavily dependent on the crucial role played by the linker in providing preorganisation.1–3 In general terms, it is reported that conformationally rigid linkers afford larger association constants than systems with conformationally flexible linkers,4,5 as well as offering substrate selectivity provided that the dimensions of the tweezer cavity are suited to the substrate.4 Conformationally flexible linkers allow complexation of substrates of different sizes and shapes,4,6 and these systems may suffer from poor selectivity.7 However, it can be advantageous to balance the degree of host preorganisation and rigidity with conformational flexibility, as demonstrated by several notable examples:● a metalloporphyrin host which increases the rate of a hetero-Diels–Alder reaction,8
● disulfide-linked bis(cyclopeptides) which display high affinity and selectivity for the sulfate anion in aqueous solution,9
● the comparison of the effective molarities (EM) for a large number of porphyrin-pyridine complexes with different torsional degrees of freedom,10
● and the use of flexible cyclodextrins to template porphyrin nanorings.11
In a series of bis-porphyrin hosts each containing different linker structures,12–14 it has been observed that large association constants can be obtained for architectures containing either conformationally flexible, restricted, or more rigidly constrained linkers, and that the linker need only confer the system with moderate preorganization.§ Additional evidence for this is provided by both our recent work15 and a variety of porphyrin host–guest systems with varying degrees of preorganisation,6–8,12,13,16–27 including those which allow defined changes in interporphyrin distance. Further, there continues to be an active interest to explore linker conformational rigidity/flexibility in porphyrin tweezers.28,29
Biological receptors, which are dynamic and far from rigid, possess comparatively larger association constants compared to those of most artificial receptors.5,9 This has led to discussion that adopting a rigid-only approach to host design which relies solely on covalent linkages to establish geometry could limit the scope of artificial receptors.5,9
We have previously described the host–guest chemistry of bis-porphyrin tweezer 1,15 which forms a strong 1·DABCO bis-porphyrin:DABCO complex with the aliphatic diamino ligand 1,4-diazabicyclo[2.2.2]octane (DABCO), and more recently reported the synthesis of a tetra-porphyrin tweezer 2 with two cooperative binding sites30 (Fig. 1). In these systems, host preorganisation was achieved by using a bridged polycyclic linker. The favourable properties of fused [n]polynorbornyl scaffolds in molecular receptors, particularly with respect to host preorganisation, are reported in the literature by a number of research groups including those of Warrener and Butler,7,31–39 Paddon-Row,40–43 Johnston,19,44–46 Pfeffer,47–53 Clever,47,50,54 and Margetić.55–58
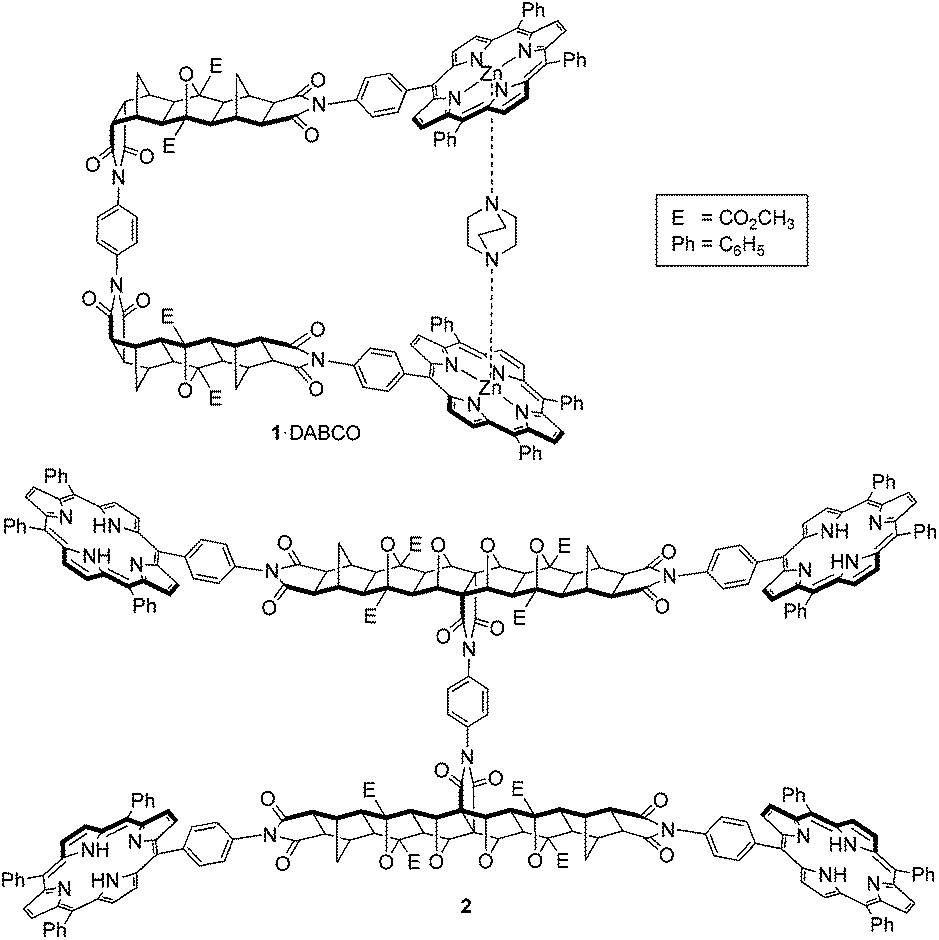 | ||
| Fig. 1 Previously described tweezer systems with a freely rotating phenyl diimide core; single binding site tweezer 115 (shown as complex 1·DABCO), and two binding site tweezer 2.30 | ||
Importantly, tweezers 1 and 2 share a freely rotating phenyl diimide core inserted within the otherwise rigid polycyclic linker, a feature which allows unhindered adjustment of the interporphyrin distance, to potentially allow the binding of guests of different lengths. However, because all conformations are readily accessible, the possibility of 1 assuming an anti-conformation arises, thereby decreasing the proportion of the syn-conformation and the formation of syn-1 bis-porphyrin:guest complexes. This possibility is also inherent in the design of other molecular tweezers in the literature (for several examples see ref. 24 and 59–63).
To investigate the role of the phenyl core on the host–guest behaviour of molecular architectures 1 and 2, we designed restricted rotation tweezer 3 (Fig. 2). Tweezer 3 maintains the rigid bridged polycyclic scaffold but contains a phenyl diimide core with sterically bulky methyl substituents (dotted box, Fig. 2). These methyl substituents drastically increase the energy barrier to rotation about the phenyl diimide core and afford non-interconvertible syn-3a and anti-3b conformations, providing the opportunity to study the host–guest complexation of each conformation independently.
 | ||
| Fig. 2 Restricted rotation single binding site tweezer 3, where non-interconvertible syn-3a and anti-3b conformations arise due to a sterically bulky 2,3,5,6-tetramethylphenyl diimide core. | ||
This manuscript reports on the synthesis, isolation and characterisation of the syn-3a and anti-3b conformers. Further, their complexation with DABCO was studied by UV-Vis and NMR spectroscopy and the resulting data fitted to various models of stoichiometry using non-linear least squares analysis.
Results and discussion
Synthesis
The synthesis of tweezer 3 was undertaken in a similar manner to tweezer 115 involving the condensation of endo-Mitsudo anhydride 415,55 with 2,3,5,6-tetramethyl-p-phenylenediamine 5, followed by ring closing to form imides 6a and 6b (Scheme 1).
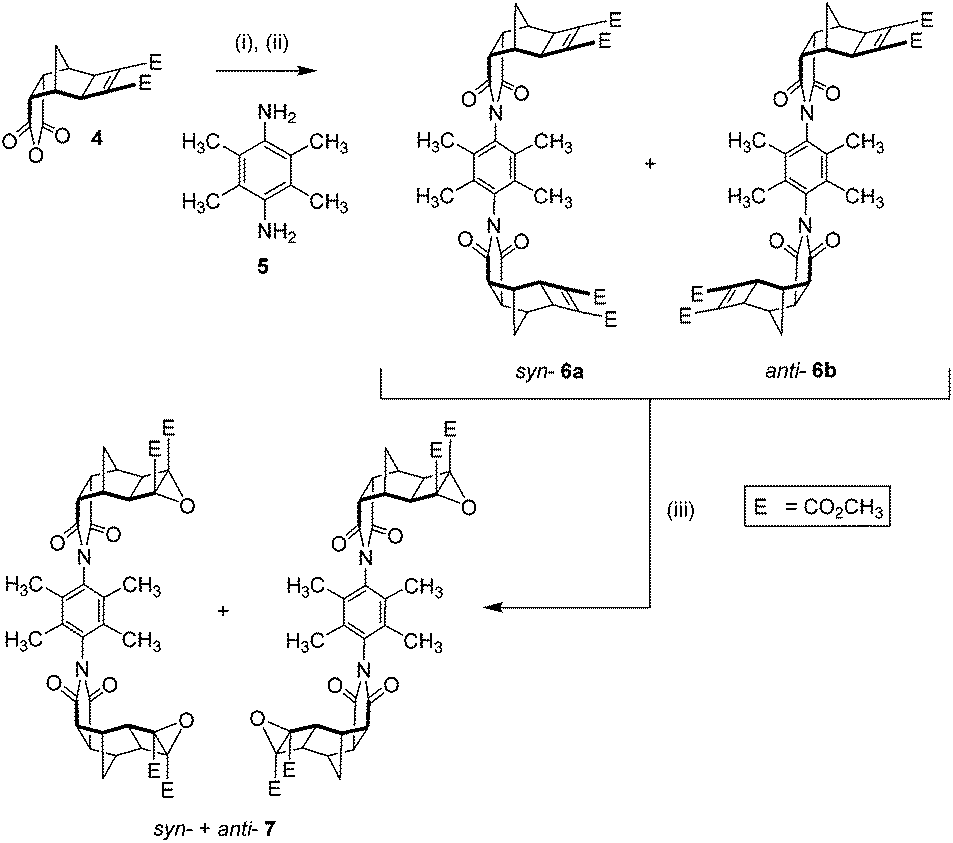 | ||
| Scheme 1 Synthesis of the restricted rotation linker. (i) 2,3,5,6-Tetramethyl-p-phenylenediamine 5 (0.5 eq.), dry DMSO, Ar deoxygenated, 80 °C, 1 day; (ii) NaOAc/Ac2O, 80 °C, 1 day, 72% (as a mixture of syn-6a + anti-6b); (iii) anhydrous tBuOOH in toluene (3.3 M, 2.5–5 eq.), dry CH2Cl2, 0 °C, 10 min, tBuOK (1–2 eq.), room temp., 3.5–7 h, 51%. | ||
The 1H NMR spectrum of the reaction mixture (not shown) revealed two pairs of signals for the methyl phenyl substituent proton resonances. This was attributed to the two different linker conformations of syn-6a and anti-6b. The mixture showed a slight deviation from statistical, in an approximately 60:40 ratio based on the relative integration between the two pairs of signals within the mixture.¶
To provide experimental evidence for separate syn-6a and anti-6b conformers, a small sample was separated by using a combination of column chromatography and selective recrystallisation. The partial 1H NMR spectra highlighting the methyl resonances are shown in Fig. 3 (full 1H NMR spectra S1 and S3†). In either conformation, the two methyl groups are non-equivalent resulting in two resonances. Additionally, the syn-6a and anti-6b conformations have different 13C NMR spectra (S2 and S4†); with pairs of non-equivalent methyl and phenyl carbon resonances evident.
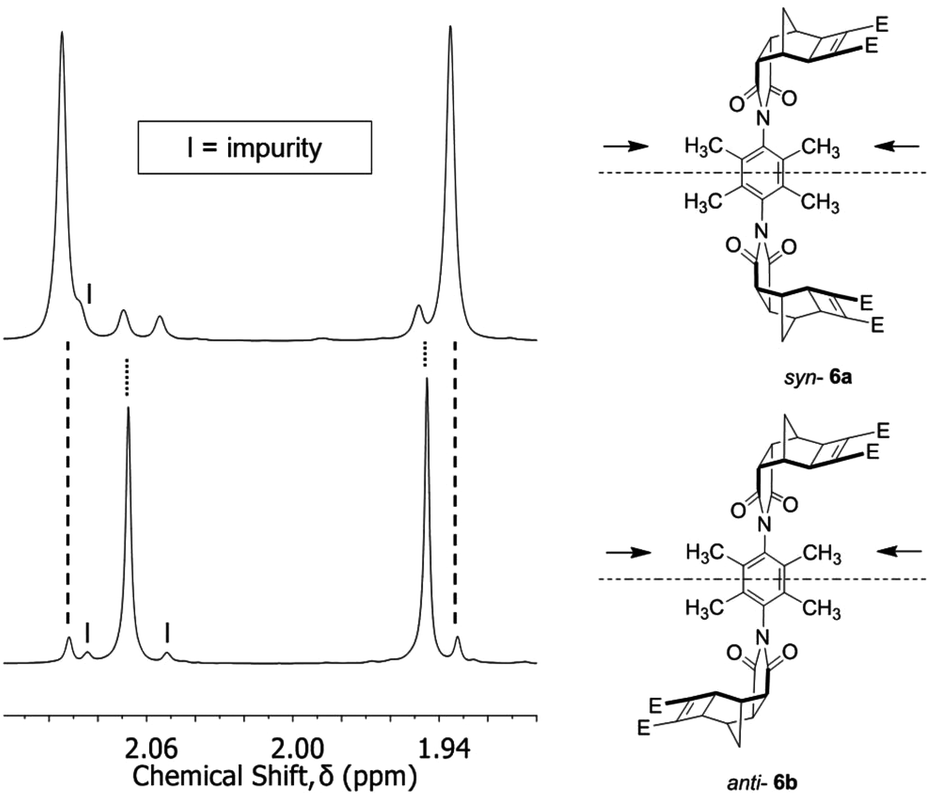 | ||
| Fig. 3 Selected region of the 1H NMR spectrum of the syn-6a and anti-6b restricted rotation linker (absolute conformation unassigned), showing the different chemical environments of the methyl groups. Each sample contained traces of the other conformation and these are shown by the dotted lines. Impurity is marked by the letter I. Spectra recorded at 600 MHz in CDCl3. | ||
The mixture of syn-6a and anti-6b was subsequently epoxidised under standard conditions for electron deficient alkenes (tBuOOH/tBuOK),33,35 to afford bis-epoxide 7 (Scheme 1). The bis-epoxide mixture 7 was subsequently appended with exo-porphyrin receptors 815via the alkene plus cyclobutane epoxide (ACE) reaction (Scheme 2) in a sealed tube.64 Overall, the reaction proceeded in a 23% yield, which upon NMR analysis was shown to exist as 10% syn-9a and 13% anti-9b conformations. The syn- and anti-assignment was achieved using 1H NMR of the zinc metallated porphyrin derivatives, 3a and 3b respectively (vide infra).
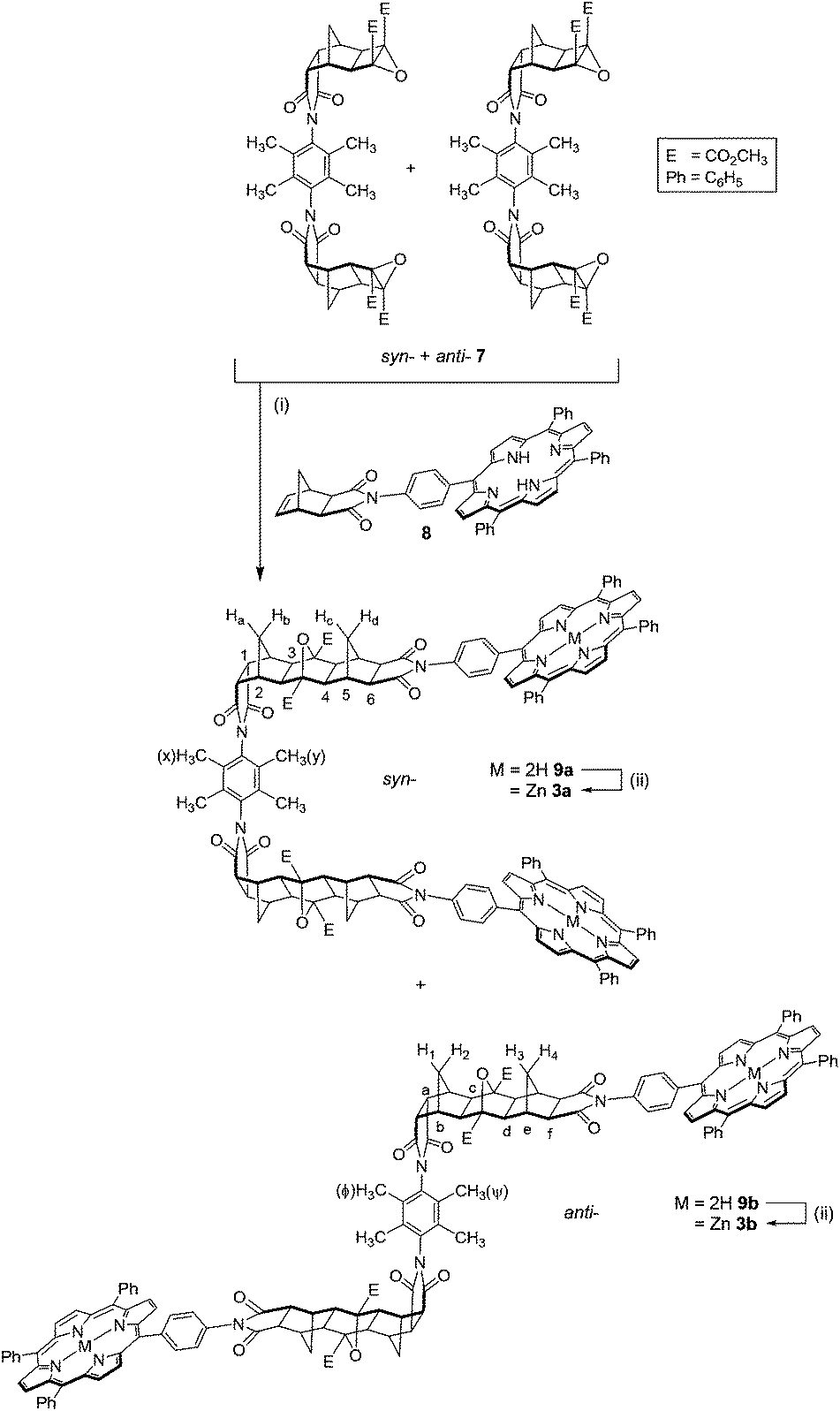 | ||
| Scheme 2 Synthesis of tweezer 3, syn-3a and anti-3b. (i) exo-Porphyrin receptor 8 (2 eq.), dry THF, sealed tube, 160 °C, 24 h, 23% (10% syn-3a + 13% anti-3b); (ii) Zn(OAc)2/MeOH/CHCl3, reflux, 1–2 h, 79–89%. | ||
The 1H NMR spectra of the syn-9a and anti-9b mixture contained several features characteristic of ACE-coupled reactions, including a small downfield shift for the methyl ester resonance compared to that of the epoxide 7,65 along with the disappearance of the norbornyl proton resonance from the exo-porphyrin receptor 8 at 6.45 ppm. The 13C NMR spectrum contained a resonance at 90 ppm, similar to other polycyclic systems,38 which is assigned to the bridgehead carbon atoms in the newly formed 7-oxanorbornane.38
Relative integration of the two inner pyrrole proton resonances in the 1H NMR spectrum of the crude mixture suggested the ratio of syn-:anti- was approximately 43:57. While separation by column chromatography yielded the anti-9b conformation completely free of syn-9a, the syn-9a conformation contained approximately 25% residual anti-9b. In an effort to conserve limited product, further purification to remove anti-9b from syn-9a was carried out post-zinc(II) metallation, as syn-3a. The identity of the partially purified freebase syn-9a and anti-9b were confirmed by using accurate MS (S9),† ESI-TOF, for two samples containing different ratios of syn-9a and anti-9b (sample 1 [M + 2H]2+ found: 1162.4267, calc: 1162.4260; sample 2 [M + 2Na]2+ found: 1184.4110, calc: 1184.4079).
The freebase syn-9a and anti-9b mixture and pure anti-9b were metallated with zinc(II) under standard conditions,66 giving syn-3a and anti-3b respectively (Scheme 2). Zinc(II) metallation was characterised by loss of the porphyrin inner pyrrole proton resonance at −2.80 (anti-) and −2.84 (syn-) in the 1H NMR spectrum. Several slow recrystallisations enabled the zinc(II) syn-3a conformation to be purified completely free of anti-3b. HRMS (MALDI-TOF) of syn-3a and anti-3b further confirmed zinc(II) metallation. MALDI-TOF of porphyrin compounds is reported to afford the radical molecular ion, [M]˙+, without the uptake of H+ or Na+.67 The monoisotopic mass for [M]˙+ was found to be 2446.694 for syn-3a and 2446.6770 for anti-3b, which compares well with the calculated value of 2446.6644 (5,10,15,20-tetraphenylporphin, H2TPP, was used for internal calibration). In addition, the profiles of the experimental and theoretical MS spectra are in excellent agreement (S15†).
In the 1H NMR spectrum for each adduct (syn-3a, S10,†anti-3b, S13†), ten polycyclic resonances occur as expected (excluding the methyl ester and two methyl phenyl substituent resonances); six singlets and four doublets. The doublets arise from two pairs of non-equivalent methylene bridge protons, Ha/Hb and Hc/Hd in the case of syn-3a, and H1/H2 and H3/H4 in the case of anti-3b (Scheme 2). These pairs (e.g. Ha/b and Hc/d, or H1/2 and H3/4) appear at significantly different chemical shifts, characteristic of steric compression by oxygen in these systems,55 and confirms the formation of a linear ACE product.55 These resonances occur at chemical shifts of 2.77/1.43 ppm and 2.49/1.13 ppm for syn-3a and 2.77/1.42 ppm and 2.54/1.22 ppm for anti-3b. The six signals correspond with the number of unique proton chemical environments along the polycyclic scaffold (marked 1–6 for syn-3a and a–f for anti-3b in Scheme 2). In addition, a pair of methyl phenyl substituent resonances for the linker core are observed at chemical shifts of 2.31 and 2.00 ppm for syn-3a and 2.28 and 2.04 ppm for anti-3b (marked x and y for syn-3a, Φ and Ψ for anti-3b in Scheme 2, absolute assignment not determined). This is in line with similar observations for the syn-6a and anti-6b linkers described in Fig. 3.
The different aromatic regions of the 1H NMR spectra of syn-3a and anti-3b are shown in Fig. 4. The splitting of the porphyrin resonances, in particular the β-pyrrole resonances, and the differences in integration of the meso-phenyl resonances between the two conformations indicates facial differentiation of the porphyrin macrocycles as a result of the close proximity of the opposing ring in the syn-conformation. For the anti-conformation, no major splitting of the porphyrin resonances was observed and the integrations are typical of compounds of the type mono-porphyrin 1015 (structure given in Table 1), presumably as the porphyrins cannot be positioned cofacially (unless intermolecular aggregation occurs). Additionally, the syn-conformation porphyrin resonances are shielded relative to the anti-, further demonstrating the effect of the adjacent π systems in the syn-conformation.
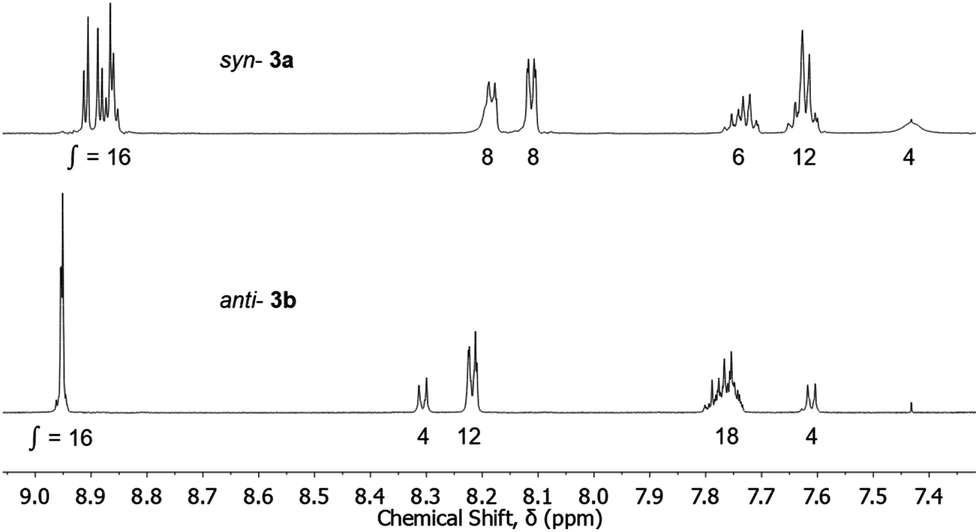 | ||
| Fig. 4 Partial 1H NMR (aromatic region) of the zinc(II) metallated syn-3a and anti-3b restricted rotation tweezers. Spectra recorded at 600 MHz in CDCl3. | ||
For the syn-3a tweezer, several of the porphyrin meso-phenyl 1H NMR resonances are broadened while others appear sharp. Furthermore, the 1H NMR spectrum was highly concentration dependent (S12(a)†), with significant resonance shifts observed for porphyrin and polycyclic resonances (Δδ = 0.3–0.7 ppm for samples more and less concentrated than 3.5 mM). Additional resonance shifts are observed between +20 °C and −50 °C (S12(b)†) Taken together, these data suggest that the syn-tweezer is undergoing dynamic conformational changes (but not to anti-) on the NMR timescale, and/or changes to intermolecular aggregation influenced by concentration and temperature, however this has not been investigated further.
For the zinc(II) anti-3b adduct, there are only minor (<0.1 ppm) shifts in the spectrum at −50 °C compared to room temperature. Concentration dependence of the 1H NMR could not be established for anti-3b due to poor solubility at concentrations greater than 0.85 mM.
The UV/Vis spectra of the zinc(II) metallated syn-3a and anti-3b adducts supported our conformational assignment. Zinc(II) anti-3b and zinc(II) mono-porphyrin 1015|| display very similar Soret maxima and peak bandwidths at half height (Table 1). The absence of exciton coupling interactions supports the assignment of the anti-conformation,59 where the bis-porphyrins are not positioned cofacially. Conversely, the Soret maxima of the zinc(II) syn-3a tweezer is blue shifted by 1 nm relative to the zinc(II) mono-porphyrin 10, and has a slightly larger peak bandwidth at half height than the zinc(II) freely rotating tweezer 1 (Table 1). Although the blue shift and broadening of the Soret band are both small, this could indicate weak exciton coupling interactions19,22,59,68–72 between the porphyrins in the syn-3a adduct. The weakness of the exciton coupling in the syn-3a adduct suggests that the bis-porphyrins are not fixed in a cofacial orientation, and that the porphyrin units are able to undergo rotation either about the meso-phenyl linking the porphyrin to the scaffold, and/or the two porphyrin arms can undergo restricted rotation about the diimide core.
2,3,5,6-Tetramethylphenyl diimide as the linker
Restricted rotation has been reported for N-phenyl imide derivatives with ortho-substituents other than hydrogen.73–83,94 As the size of this substituent increases, the energy barrier to rotation increases due to steric repulsion with the oxygen atom of the imide.84–86 In addition, the angle between the substituted phenyl and imide rings increases towards perpendicular to minimise this steric interaction.84,85,87,88 This comes at the expense of resonance delocalisation and conjugation favoured in the planar conformation89.**NMR studies for mono-ortho-methyl substituted N-phenyl imide derivatives73,74 report a mixture of rotational isomers at ambient conditions and which undergo thermally activated bond rotation. No interconversion between syn-3a and anti-3b was observed for our fully substituted 2,3,5,6-tetramethylphenyl 1,4-diimide under the conditions examined; the systems are both stable to interconversion at room temperature over the duration of NMR experiments, while the 1H NMR spectrum of the pure anti-3b adduct after microwave irradiation at 95 °C for 15 minutes (300 W, 50 psi), or sonication at 30 °C for 15 minutes, showed no detectable interconversion from anti-3b to syn-3a (higher temperatures and longer times were not explored). However, a range of intermediate conformations are envisaged to be energetically accessible within each of syn-3a and anti-3b, and would allow for some adjustment of the interporphyrin distance in the presence of guest.
Furthermore, in the course of this work, we examined several variations of linker 6,90 most notably the exo-analogues 11 (Fig. 5). During synthesis of 11, the crude filtrate and crude precipitate of the reaction mixture contained mainly the opposite conformational isomer respectively (different solubility). Conveniently, slow evaporation of acetonitrile solutions of the precipitate and filtrate afforded crystals suitable for X-ray diffraction measurements. The crystal structures are shown in Fig. 6.
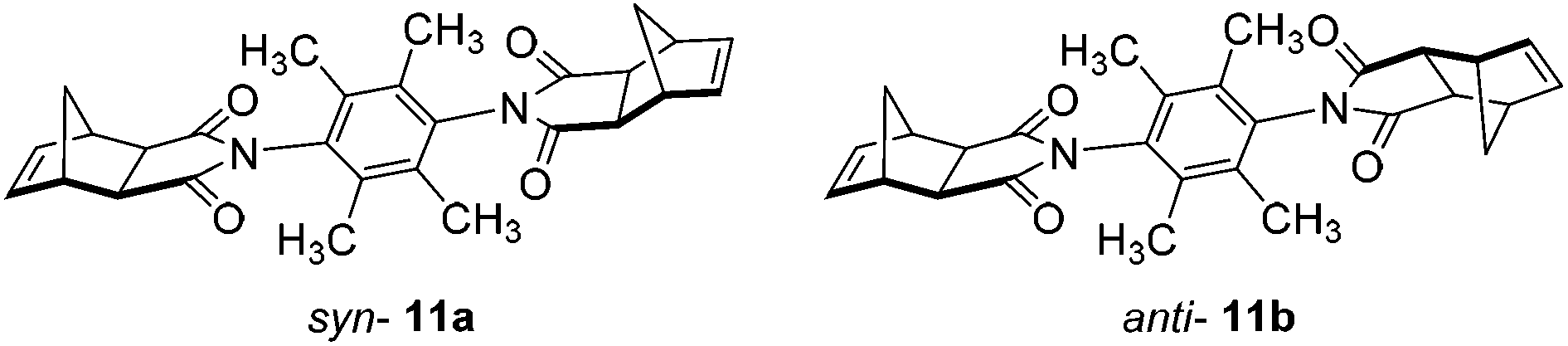 | ||
| Fig. 5 exo-Analogues syn-11a and anti-11b. | ||
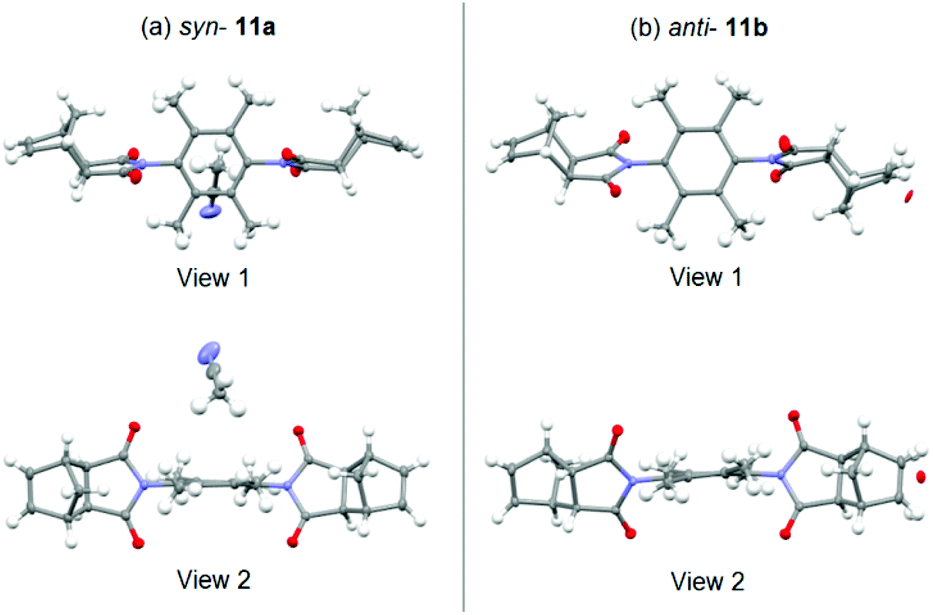 | ||
| Fig. 6 X-ray crystal structures of the exo-analogues; (a) syn-11a (crystallised with a molecule of acetonitrile), (b) anti-11b (co-crystallised with approximately 6% mono-epoxide). Images rendered in Mercury (CCDC).91 | ||
While crystal packing forces must be considered, these crystal structures provide unequivocal evidence supporting the formation of distinct syn-11a and anti-11b conformations (Fig. 6(a) and (b) respectively). Key parameters for the X-ray crystallographic measurements are provided in S18 and S21.†
An interesting structural feature revealed in Fig. 6 is the noticeable deviation of the 2,3,5,6-tetramethylphenyl core from planarity (view 2). The syn-11a conformation is co-crystallised with acetonitrile solvent, while the anti-11b conformation is co-crystallised with approximately 6% of a mono-epoxide impurity.
The presence of co-crystallised epoxide for anti-11b was unexpected, and re-examination of the high resolution mass spectrometry for samples prior to crystallisation revealed a second signal in the mass spectra of both compounds within 37 ppm of the theoretical mass for molecular formula with an additional oxygen atom (i.e. epoxide, [M + O + Na]+). There is a literature report of partial epoxide conversion of similar polycyclic derivatives during recrystallisation,92 in which epoxidation is also undertaken chemically using an initiator in the presence of molecular oxygen, however it is conceivable that uninitiated epoxidation could occur at a rate too low for convenient measurement.92
Host–guest complexations of tweezer 3 with DABCO
The interaction of the zinc(II) metallated tweezer 3 with the diamino ligand DABCO was studied independently for syn-3a and anti-3b, using UV/Vis and NMR spectroscopic titrations. Complexation models, association constants and speciation diagrams were calculated from the UV/Vis titration data similarly to other published approaches,14,17,28,95–103 using HypSpec and HySS2009 (Protonic software).104,105:1 (3a + DABCO) ⇌ 3a·DABCO; K11 = [3a·DABCO]/([3a][DABCO]), 1:2 (3a + 2·DABCO) ⇌ 3a·(DABCO)2; K12 = [3a·(DABCO)2]/([3a][DABCO]2), and 2:1 (2·3a + DABCO) ⇌ (3a)2·DABCO; K21 = [(3a)2·DABCO]/([3a]2[DABCO]) stoichiometries.
 | ||
| Fig. 7 Schematic representation of the various equilibria between syn-3a and DABCO. | ||
UV/Vis titration of a solution of DABCO into a solution of syn-3a resulted in a two-stage red shift of the Soret maximum (Fig. 8), indicating the stepwise formation of two dominant complexes between syn-3a and DABCO. The first red shift, from 418.3 nm (red line) to 423.4 nm (blue line) is characteristic of a bis-porphyrin DABCO sandwich complex formation.106,107 A second gradual red shift occurs to 428.6 nm (green line) by 400000 equivalents of DABCO, although this transition appears to only be partially complete. This is characteristic of simple mono-porphyrin DABCO complexes.106,107
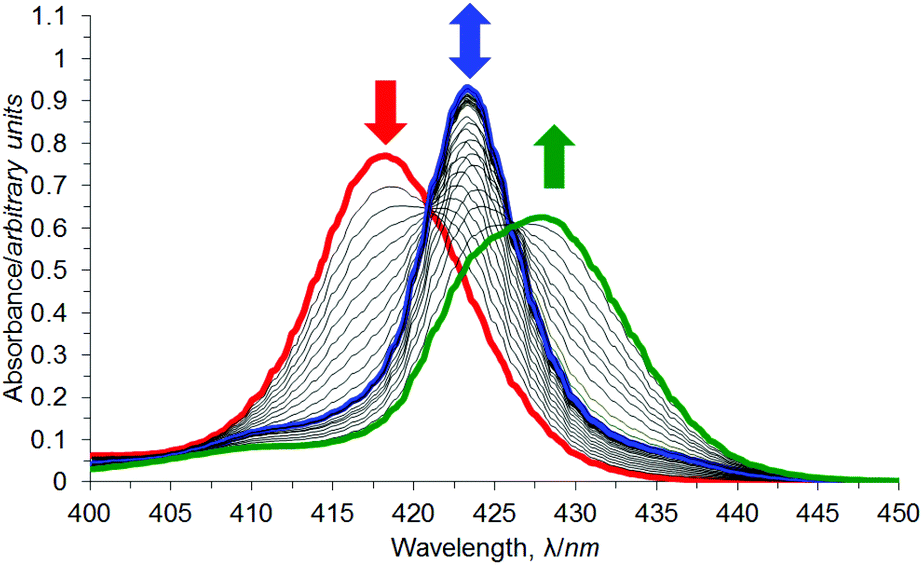 | ||
| Fig. 8 UV/Vis titration of syn-3a (red line) with DABCO in chloroform. | ||
Fitting of the UV/Vis titration data using HypSpec104,105 resulted in reasonable fits (visual inspection) to two different complexation models, including either the formation of 3a·DABCO and 3a·(DABCO)2 or the formation of (3a)2·(DABCO)2 and 3a·(DABCO)2 (Fig. 7).
Fitting of an algorithm (Fig. 9 using the HypSpec protocol104,105) for the formation of the 1:1 complex 3a·DABCO and the 1:2 complex 3a·(DABCO)2 (Fig. 7) to absorbance data at 419 nm obtained over a range of 200000 equivalents of added DABCO yielded K11 = 1.25 × 108 M−1 and K12 = 1.32 × 109 M−2 in CHCl3 at 20 °C. This corresponds to a stepwise K′12 = 10.6 M−1 for the complexation of DABCO by 3a·DABCO to give 3a·(DABCO)2. The derived speciations for the complexation in the 0–2 and 0–200000 equivalents of added DABCO ranges are show in Fig. 9(a) and (b), and the corresponding data fits at 419 nm are shown in Fig. 9(c) and (d), respectively. Similar data fits were obtained at 423 and 430 nm (not shown). The K11 = 1.25 × 108 M−1 characterising 3a·DABCO is much larger than K11 = 2.53 × 105 M−1 (not statistically corrected) for mono-porphyrin·quinuclidine,15 and is consistent with 3a·DABCO involving bis-porphyrin complexation in the sandwich structure shown in Fig. 7.13,17,18
 | ||
| Fig. 9 Derived speciation plots (a) and (b) for formation of 3a·DABCO (blue line) and 3a·(DABCO)2 (green line) in the 0–2 equivalent and 0–200000 equivalent ranges of added DABCO, respectively, in CHCl3 at 20 °C. The corresponding best fits, (c) and (d), of an algorithm for the formation of the 1:1 complex 3a·DABCO and the 1:2 complex 3a·(DABCO)2 (black lines) for the UV/Vis absorbance data obtained at 419 nm (black circles). | ||
The UV/Vis titration data for syn-3a/DABCO could also be fitted to a complexation model involving the formation of (3a)2:(DABCO)2 (K22 = 1.29 × 1022 M−3) and 3a:(DABCO)2 (K12 = 1.90 × 109 M−2) (S23†). However, the geometric constraints imposed by restricted phenyl core rotation in syn-3a limits the opportunity to form bis-porphyrin interactions with DABCO in (3a)2(DABCO)2.
The K11 = 1.25 × 108 M−1 characterising syn-3a·DABCO is approximately double that determined for freely rotating 1·DABCO (K11 = 8.1 × 107 M−1).15 This fits with the arc through which the two porphyrin moieties are able to move; 360° in 1 and 180° for syn-3a. This equates to syn-3a being approximately twice as pre-organised as freely rotating 1, and which is reflected in a nearly doubling of K11.
An assessment of the validity of the complexation model derived from the UV/Vis titration (carried out at micromolar concentration) may be made by using the derived complexation constants to predict the behaviour of the system during a 1H NMR titration (carried out at millimolar concentration).59,106–108 Thus, 1H NMR titrations of syn-3a/DABCO were undertaken in CDCl3.
The resonances of sandwich syn-3a porphyrin β-pyrrole and sandwich DABCO are diagnostic in the interpretation of the complexation occurring during 1H NMR titrations. At 20 °C, the β-pyrrole signals for free syn-3a occur at 8.92–8.75 ppm (Fig. 10(a)). The addition of up to 0.9 equivalents of DABCO (integration suggests 1 equivalent)†† results in the appearance of a second syn-3a β-pyrrole signal at 8.55 ppm. This upfield shift is typical of β-pyrrole protons in a bis-porphyrin DABCO sandwich complex as a consequence of shielding by opposing ring currents of two porphyrin aromatic systems in close proximity.96,106,107 The two syn-3a species are in slow exchange on the NMR chemical shift timescale, and the syn-3a complex resonance intensity increases as that of free syn-3a decreases. The relative integration of the syn-3a complex β-pyrrole resonance to that of the total syn-3a β-pyrrole resonance (free plus complex) is consistent with the formation of a 1:1 complex (within 10% error), such as 3a·DABCO or (3a)2·(DABCO)2, and is later shown to be the former. Addition of greater than 1 equivalent of DABCO causes the syn-3a β-pyrrole resonance to shift downfield as increasing proportions of the 3a·(DABCO)2 complex forms in fast exchange with 3a·DABCO on the NMR chemical shift timescale. However, the change in chemical shift of this syn-3a resonance after the addition of 5 equivalents of DABCO (Δδ = 0.033 ppm) is small compared to that of the freely rotating tweezer 1 (Δδ = 0.082 ppm (ref. 90)) under the same conditions, consistent with the 3a·DABCO complex being less amenable to the addition of a second DABCO than is the 1·DABCO complex.
 | ||
| Fig. 10 Selected 1H NMR spectra of syn-3a with various equivalents of DABCO at (a) 20 °C and (b) −50 °C. † identifies minor amounts of an additional sandwich complex. ● identifies the mono-porphyrin·DABCO signal broadened into the baseline due to chemical exchange. Spectra recorded at 600 MHz in CDCl3. | ||
The syn-3a porphyrin meso-phenyl proton resonances (not shown) are broadened in the presence of less than 1 equivalent of DABCO but sharpen in the presence of greater than 1 equivalent of DABCO and shift downfield, presumably as the sandwich complex forms and undergoes fast exchange with the open syn-3a complex with DABCO, 3a·(DABCO)2. Little information could be obtained from the polycyclic resonances.
Further understanding of the complexation of DABCO by syn-3a is gained from the DABCO methylene resonance (Fig. 10(a)). For the addition of up to 0.9 equivalents of DABCO (integration suggests 1 equivalent), a sharp singlet was observed at −4.84 ppm. This large upfield shift is typical of DABCO methylene protons in a bis-porphyrin DABCO sandwich complex, and again results from shielding by opposing ring currents of two porphyrin aromatic systems in close proximity.96,106,107,109–112 The relative integration of the DABCO sandwich resonance to the syn-3a ester signal‡‡ is consistent with the formation of a species with the empirical formula of 1:1 (within 10% error). At 1 equivalent and greater of DABCO, the sandwich DABCO resonance broadens, and migrates downfield towards the value for free DABCO. This is consistent with chemical exchange with another species occurring at a fast exchange rate on the NMR chemical shift timescale at 20 °C, most likely a combination of 3a·(DABCO)2 and free DABCO.
At less than 1 equivalent of DABCO, all resonances, including the syn-3a porphyrin and polycyclic resonances, are in slow exchange on the NMR chemical shift timescale, however, several of the free syn-3a host resonances undergo simultaneous resonance shifts as their signal intensity is depleted and the complex resonance increases (not shown). This was particularly obvious for the methylene bridgehead proton at 1.0–1.3 ppm and the porphyrin β-pyrrole resonance. This phenomenon appears to indicate a second process occurring in addition to formation of the sandwich complex. This could be attributed to the concentration dependence of the free host 1H NMR spectrum discussed earlier (S12(a)†). In this case, as the syn-3a complex forms, the free syn-3a concentration decreases and its concentration dependent chemical shift changes.
Slowing the exchange rate between the various syn-3a complexes in solution was achieved by lowering the sample temperature to −50 °C (Fig. 10(b)), and revealed several additional spectral features. Below 0.8 equivalents of DABCO (integration suggests 1 equivalent), a small amount of an additional sandwich syn-3a complex in addition to the main signal can be observed around −5 ppm (indicated by † in Fig. 10(b)). At greater than 0.8 equivalents of DABCO (spectral features suggest greater than 1 equivalent), the system moves into fast exchange on the NMR chemical shift timescale. A small amount of the open syn-3a complex can be observed at −3.06 ppm (indicated by ● in Fig. 10(b)) in addition to the main sandwich syn-3a complex resonance, now broadened due to chemical exchange. The resonance at −3.06 ppm is characteristic of the α methylene protons of DABCO bound to a single porphyrin,106,108,111 and most likely corresponds to conversion of the sandwich syn-3a complex to open species, in this case 3a·(DABCO)2.
The porphyrin meso-phenyl protons (not shown) are sharp and highly desymmetrised below 1 equivalent of DABCO, indicating facial differentiation of the porphyrins within the sandwich complex. For greater than 1 equivalent of DABCO, the porphyrin meso-phenyl protons broaden and resymmetrise, suggesting diminishing facial discrimination, then shift downfield, presumably as the sandwich complex undergoes fast exchange with open species on the NMR chemical shift timescale. Little information could be obtained from the polycyclic resonances.
To further investigate the composition of the complex formed between syn-3a and DABCO at NMR concentrations (millimolar), a simulated NMR speciation diagram was generated in HySS2009104,105 using the association constants for successfully fitted complexation models determined from the UV/Vis titrations (micromolar), and compared with experimental NMR titration data.106–108Fig. 11 shows this result for the 1:1 plus 1:2 complexation model, where the blue line represents the predicted NMR speciation for the growth and decay of 3a·DABCO, the green line represents the open species 3a·(DABCO)2, and the red line represents free host. The black circles and squares represent the experimental speciation for the slow and fast exchange NMR chemical shift timescale regions respectively, calculated as described in S22.†§§
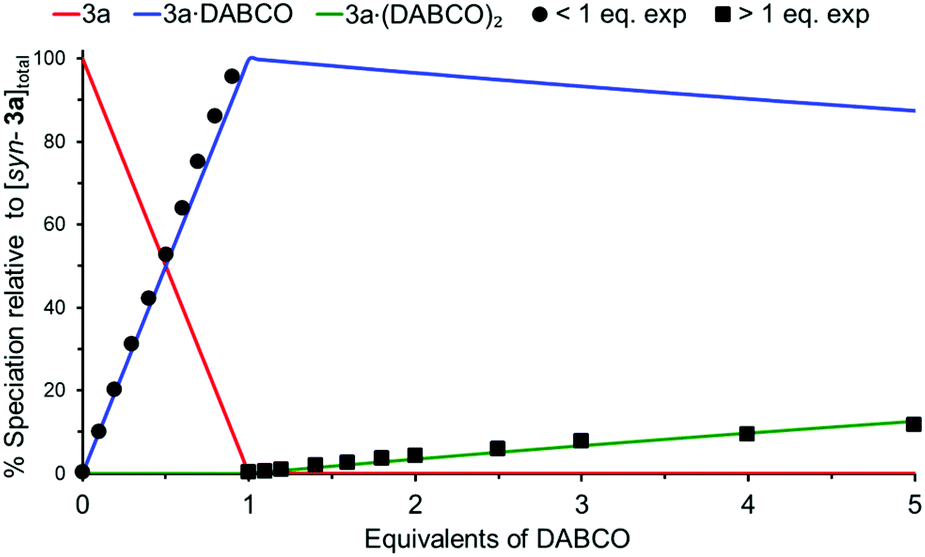 | ||
| Fig. 11 Simulated NMR speciation diagram (millimolar) generated from UV/Vis determined association constants K11 and K12 for syn-3a/DABCO (micromolar). Experimental NMR speciation has been overlayed for both the slow and fast exchange regions of the titration. | ||
As can be seen in Fig. 11, there is excellent agreement between the simulated and experimental speciation across both the slow and fast exchange NMR chemical shift timescale regions. This confirmed that the complexation model was the same at both UV/Vis and NMR concentrations for the syn-3a/DABCO system; formation of the sandwich complex 3a·DABCO and gradual decay of the sandwich complex into open complex 3a·(DABCO)2. The simulated NMR speciation for the alternative complexation model from the UV/Vis data involving the formation of (3a)2·(DABCO)2 and 3a·(DABCO)2 is shown in S23,† however, does not match the experimental data for the formation of the open species 3a·(DABCO)2.
:2 (2·3b + 2·DABCO) ⇌ (3b)2·(DABCO)2; K22 = [(3b)2·(DABCO)2]/([3b]2[DABCO]2), 1:2 (3b + 2·DABCO) ⇌ 3b·(DABCO)2; K12 = [3b·(DABCO)2]/([3b][DABCO]2), 2:1 (2·3b + DABCO) ⇌ (3b)2·DABCO; K21 = [(3b)2·DABCO]/([3b]2[DABCO]) stoichiometries, as well as a n:n polymeric assembly (n·3b + n·DABCO) ⇌ (3b)n·(DABCO)n; Knn = [(3b)n·(DABCO)n]/([3b]n[DABCO]n).
 | ||
| Fig. 12 Schematic representation of the various equilibria between anti-3b and DABCO. | ||
UV/Vis titration of a solution of DABCO into a solution of anti-3b again resulted in a two-stage redshift of the Soret maximum (Fig. 13), indicating the stepwise formation of two dominant complexes between anti-3b and DABCO. The first red shift, from 419.5 nm (red line) to 423.5 nm (blue line) is characteristic of a bis-porphyrin DABCO sandwich complex formation.106,107 However, this sandwich species is transient, and a second redshift occurs to 430.0 nm (green line) by 10000 equivalents of DABCO, after which this transition appears to be complete. This is characteristic of simple mono-porphyrin DABCO complexes.106,107
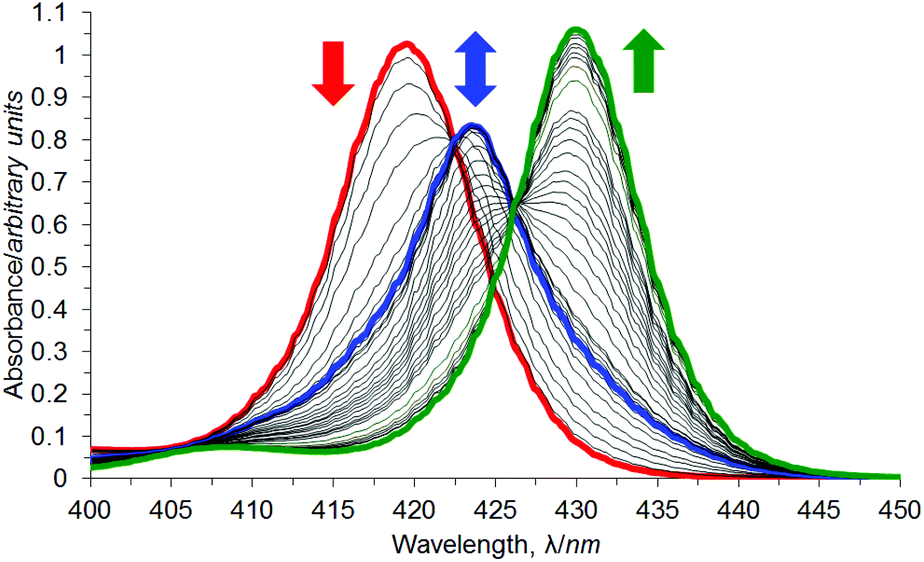 | ||
| Fig. 13 UV/Vis titration of anti-3b (red line) with DABCO in chloroform. | ||
Fitting of the UV/Vis titration data using HypSpec104,105 resulted in reasonable fits (visual inspection) to several different complexation models, including the formation of (3b)2·(DABCO)2 and 3b·(DABCO)2, and the formation of 3b·DABCO and 3b·(DABCO)2 (Fig. 12).
Fitting of an algorithm (Fig. 14 using the HypSpec protocol104,105) for the formation of the 2:2 complex (3b)2·(DABCO)2 and the 1:2 complex 3b·(DABCO)2 (Fig. 12) to absorbance data at 419 nm obtained over a range of 10000 equivalents of added DABCO yielded K22 = 5.57 × 1017 M−3 and K12 = 1.94 × 1010 M−2 in CHCl3 at 20 °C. The value for K22 compares well to other similar porphyrin-DABCO sandwich complexes in the literature (K22 = 6.2 × 1016–6.3 × 1022 M−3).106–108 The derived speciation for the complexation with 0–10000 equivalents of added DABCO is shown in Fig. 14(a), and the corresponding data fit at 419 nm is shown in Fig. 14(b) and (c), for 0–100 and 0–10000 equivalents of added DABCO respectively. Similar data fits were obtained at 423 and 430 nm (not shown).
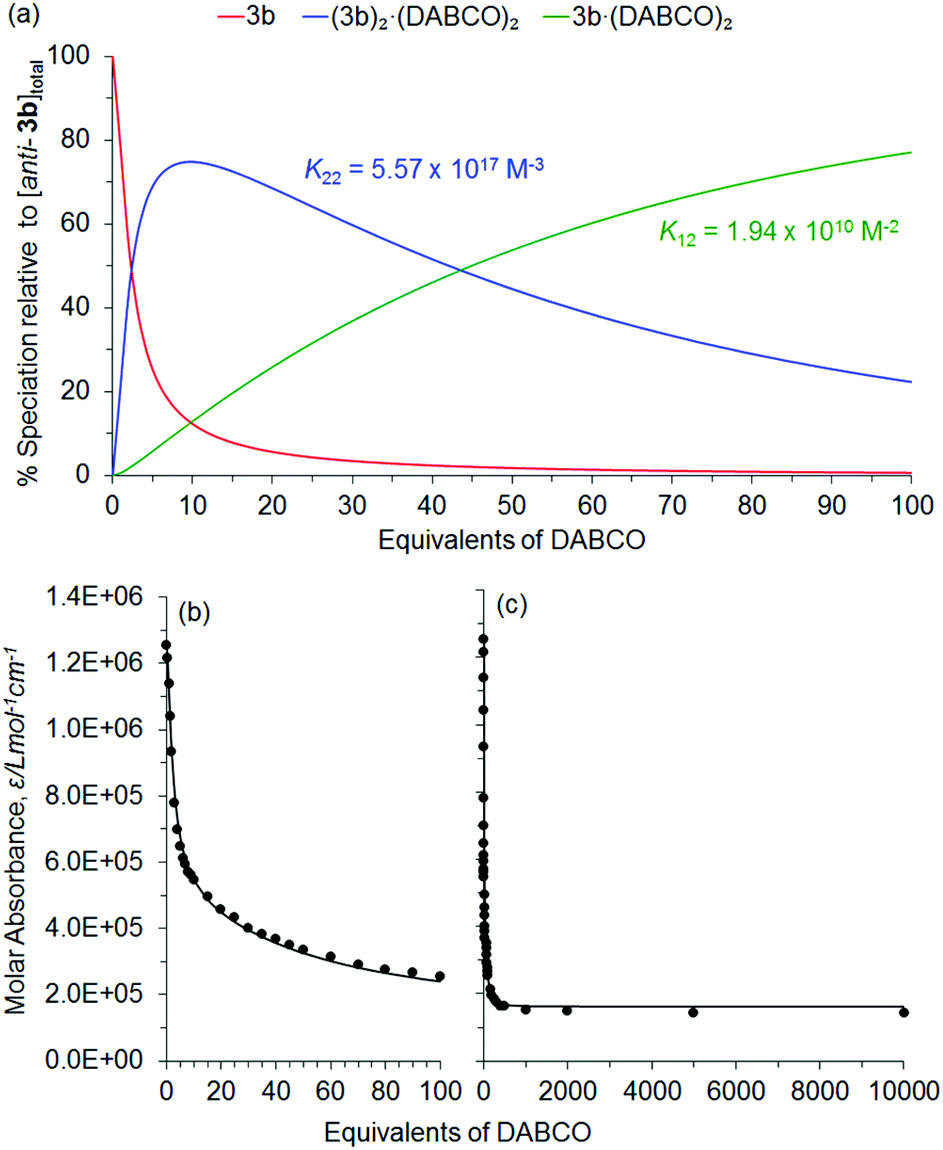 | ||
| Fig. 14 Derived speciation plot (a) for formation of (3b)2·(DABCO)2 (blue line) and 3b·(DABCO)2 (green line) for 0–10000 equivalents of added DABCO in CHCl3 at 20 °C. The corresponding best fits, (c) and (d), of an algorithm for the formation of the 2:2 complex (3b)2·(DABCO)2 and the 1:2 complex 3b·(DABCO)2 (black lines) for the UV/Vis absorbance data obtained at 419 nm (black circles) for 0–100 and 0–10000 equivalents of added DABCO respectively. | ||
The UV/Vis titration data for anti-3b/DABCO could also be fitted to a complexation model involving the formation of 3b·DABCO (K11 = 7.16 × 105 M−1) and 3b·(DABCO)2 (K12 = 2.17 × 1010 M−2) (S24†). However, the goodness of the fit alone cannot be used as the criterion to decide the correct complexation model,106 and all aspects of the fitting output should be examined (S22†). In this case, the calculated UV/Vis spectra in the HypSpec output (not shown) suggested the 3b·DABCO species had a wavelength maxima centred around 423 nm, which would experimentally suggest a bis-porphyrin DABCO sandwich complex. However, the geometric constraints imposed by restricted phenyl core rotation in anti-3b limit the opportunity to form bis-porphyrin interactions with DABCO in 3b·DABCO, and so this complexation model was excluded.
Subsequently, 1H NMR titrations of anti-3b/DABCO were undertaken in CDCl3. At 20 °C, the β-pyrrole signals for free anti-3b occur at 8.96–8.94 ppm (Fig. 15(a)). The addition of up 0.9 equivalents of DABCO (integration suggests 1 equivalent)†† results in the appearance of a second anti-3b β-pyrrole signal for the complex at 8.55 ppm. The two anti-3b species are in slow to medium exchange on the NMR chemical shift timescale (resonances are broad but distinct) and the anti-3b complex resonance intensity increases as that of free anti-3b decreases. The relative integration of the anti-3b complex β-pyrrole resonance to that of the total anti-3b β-pyrrole resonance (free plus complex) is consistent with the formation of a species with the empirical formula of 1:1 (within 10% error), such as (3b)2·(DABCO)2 or (3b)n·(DABCO)n. This chemical shift of this complex resonance (8.55 ppm) is typical of β-pyrrole protons in a bis-porphyrin DABCO sandwich,96,106,107 and so a complexation model involving 3b·DABCO can again be excluded due to geometric constraints as previously discussed. Addition of greater than 1 equivalent of DABCO causes the anti-3b β-pyrrole resonance to shift downfield as increasing proportions of 3b·(DABCO)2 forms in fast exchange with (3b)2·(DABCO)2 on the NMR chemical shift timescale. However, the change in chemical shift of this anti-3b resonance after the addition of 5 equivalents of DABCO (Δδ = 0.282 ppm) is larger than for the freely rotating 1 and syn-3a tweezer systems (Δδ = 0.082 (ref. 90) and 0.033 ppm respectively) under the same conditions. This demonstrates the increased formation of 3b·(DABCO)2 from (3b)2·(DABCO)2 for excess DABCO, compared to 1·(DABCO)2 from 1·DABCO, and 3a·(DABCO)2 from 3a·DABCO.
 | ||
| Fig. 15 Selected 1H NMR spectra of anti-3b with various equivalents of DABCO at (a) 20 °C and (b) −50 to −60 °C. † identifies additional sandwich complexes. * identifies DABCO sandwich signals broadened into the baseline due to chemical exchange. ● identifies the mono-porphyrin·DABCO signal broadened into the baseline due to chemical exchange. Spectra recorded at 600 MHz in CDCl3. | ||
The anti-3b porphyrin β-pyrrole (shown) and meso-phenyl signals (not shown) are broadened in the presence of less than 5 equivalents of DABCO, at which point they sharpen after the shift downfield, presumably as (3b)2·(DABCO)2 undergoes fast exchange with increasing proportions of 3b·(DABCO)2. Little information could be obtained from the polycyclic resonances.
Further understanding of the complexation of DABCO by anti-3b is gained from the DABCO methylene resonance (Fig. 15(a)). At 0.1 equivalents of DABCO (not shown), two signals can be observed; major at −4.826 ppm and minor at −4.846 ppm. This large upfield shift is typical of DABCO methylene protons in a bis-porphyrin DABCO sandwich complex,96,106,107,109–112 and indicates the formation of two different sandwich complexes. At 0.5 equivalents of DABCO, only the major signal can be observed, and is a sharp singlet for up to 0.9 equivalents of DABCO (integration suggests 1 equivalent). The relative integration of the DABCO sandwich resonance to the anti-3b ester signal‡‡ is consistent with the formation of a species with the empirical formula of 1:1 (within 10–15% error), such as (3b)2·(DABCO)2 or (3b)n·(DABCO)n. At 1 equivalent and greater of DABCO, the sandwich DABCO resonance broadens. This is consistent with chemical exchange with another species occurring at a fast exchange rate on the NMR chemical shift timescale at 20 °C, most likely a combination of 3b·(DABCO)2 and free DABCO. At 5 equivalents of DABCO, a broad signal was observed between 0.8–2.2 ppm under the polycyclic signals (not shown), shifting towards the resonance for free DABCO.
Slowing the exchange rate between the various anti-3b complexes in solution was achieved by lowering the sample temperature to −50 and −60 °C (Fig. 15(b)), and revealed several additional spectral features. Below 0.9 equivalents of DABCO (integration suggests 1 equivalent), two sandwich anti-3b complexes can be identified from the DABCO signals at −4.89 ppm and −4.90 ppm. The species at −4.90 ppm is dominant below 0.3 equivalents of DABCO after which the species at −4.89 ppm becomes dominant. A trace quantity of a third sandwich complex was observed at −4.98 ppm. These sandwich species are marked † in Fig. 15(b). Additional evidence for two sandwich anti-3b complexes at low temperature was observed in several of the polycyclic resonances (not shown), several of which are split into three; free anti-3b and two complexes. These are most likely to be different molecular orientations of (3b)2·(DABCO)2 (see molecular modelling section), or (3b)n·(DABCO)n. At greater than 1 equivalent of DABCO, the system remained in slow to intermediate exchange on the NMR chemical shift timescale, and allowed the resonances for the open anti-3b/DABCO complex, 3b·(DABCO)2, to be assigned. The open porphyrin β-pyrrole resonance for 3b·(DABCO)2 occurs at 8.88 ppm, while the DABCO α-methylene resonance in 3b·(DABCO)2 occurs at −3.04 ppm (marked ● in Fig. 15(b)). Both of these chemical shifts are consistent with DABCO interacting with a mono-porphyrin.106,108,111 These resonances increase in intensity at the expense of those for the sandwich anti-3b complex, (3b)2·(DABCO)2, with the conversion to 3b·(DABCO)2 almost complete at 2 equivalents of DABCO. This again demonstrates the increased formation of 3b·(DABCO)2 from (3b)2·(DABCO)2 for excess DABCO, compared to 1·(DABCO)2 from 1·DABCO, and 3a·(DABCO)2 from 3a·DABCO. At 3 equivalents of DABCO, the mono-porphyrin DABCO resonance for 3b·(DABCO)2 broadens, presumably due to exchange with free DABCO.
Interestingly, there is a broad signal at a chemical shift of 0.82 ppm (not shown) at −50 °C, which increases in intensity between 1–2 equivalents of DABCO. This was tentatively assigned to the β-methylene of DABCO interacting with a mono-porphyrin, such as 3b·(DABCO)2, in the absence of a literature value (quinuclidine β-methylene protons occur at −0.64 ppm for their complex with mono-porphyrins111).
To further investigate the composition of the complex formed between anti-3b and DABCO at NMR concentrations (millimolar), a simulated NMR speciation diagram was generated in HySS2009104,105 using the association constants for successfully fitted complexation models determined from the UV/Vis titrations (micromolar), and compared with experimental NMR titration data.106–108Fig. 16 shows this result for the 2:2 plus 1:2 complexation model, where the blue line represents the predicted NMR speciation for the growth and decay of (3b)2·(DABCO)2, the green line represents the open species 3b·(DABCO)2, and the red line represents free host. The black circles and squares represent the experimental speciation for the slow and fast exchange NMR chemical shift timescale regions respectively, calculated as described in S22.†¶¶§§
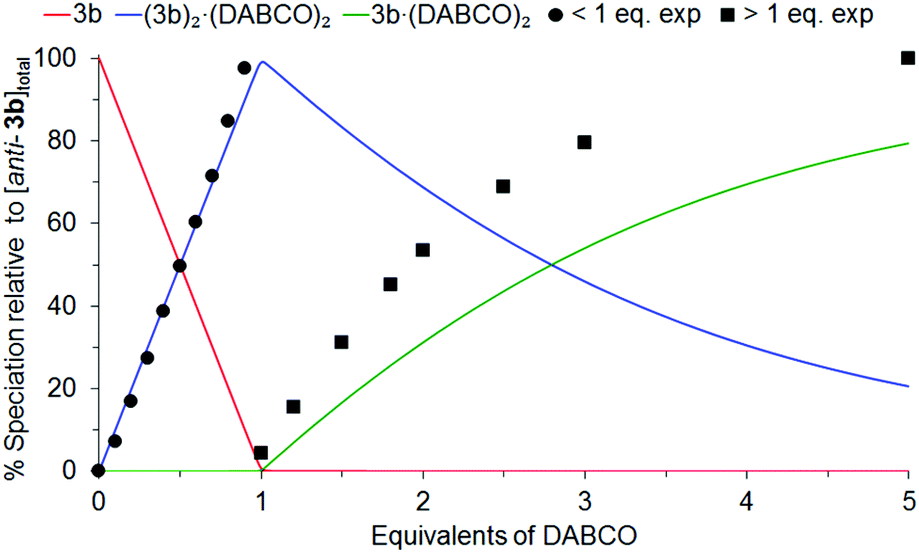 | ||
| Fig. 16 Simulated NMR speciation diagram (millimolar) generated from UV/Vis determined association constants K22 and K12 for anti-3b/DABCO (micromolar). Experimental NMR speciation has been overlayed for both the slow and fast exchange regions of the titration. | ||
As can be seen in Fig. 16, there is excellent agreement between the simulated and experimental speciation in the slow exchange NMR chemical shift timescale region (<1 eq. DABCO), while the simulation follows the same trend as the experimental data in the fast exchange NMR chemical shift timescale region (>1 eq. DABCO). This confirmed that the complexation model was the same at both UV/Vis and NMR concentrations for the anti-3b/DABCO system; formation of the sandwich complex (3b)2·(DABCO)2 and decay of the sandwich complex into open complex 3b·(DABCO)2 (assuming that the major and minor sandwich species observed in the NMR at low equivalents of DABCO are two different molecular orientations of (3b)2·(DABCO)2, and not (3b)n·(DABCO)n). The simulated NMR speciation for the alternative complexation model, 3b·DABCO and 3b·(DABCO)2, excluded during the analysis of both the UV/Vis and NMR data, is shown in S24,† however, does not match the experimental data for the formation of either 3b·DABCO or 3b·(DABCO)2.
Molecular modelling
Molecular modelling93 was undertaken to provide information on the equilibrium geometry of the restricted rotation tweezer in the absence of guest (semi-empirical, AM1), and is shown in Fig. 17. For the syn-3a conformation, rotation of the polycyclic arms can be observed about the central 2,3,5,6-tetramethylphenyl diimide group. Although the bis-porphyrins in the free host are not arranged in a cofacial orientation in the model, rotation about porphyrin meso-phenyl groups is well known,113–115 and this would enable the formation of the sandwich syn-3a·DABCO complex that is observed experimentally. For the anti-3b conformation, it is evident from the model that a sandwich anti-3b·DABCO complex cannot be accessed due to geometry constraints in the free host, but that the sandwich anti-(3b)2·(DABCO)2 complex that was observed experimentally is supported by the model. | ||
| Fig. 17 Equilibrium geometry of syn-3a and anti-3b in the absence of guest (semi-empirical, AM1). | ||
Finally, molecular modelling (semi-empirical, AM1)93 was undertaken to determine the equilibrium geometry of DABCO complexes of syn-3a and anti-3b (Fig. 18 and 19). The model of the syn-3a·DABCO complex (Fig. 18) appears similarly to our previously described freely rotating tweezer 1·DABCO.15 Rotation is observed about the porphyrin moieties as well as between the two polycyclic arms of the tweezer about the central 2,3,5,6-tetramethylphenyl diimide group. The core is partially distorted from planarity,|||| as was observed experimentally in the X-ray crystallographic structures of related compound 11 (Fig. 6). Overlay of the polycyclic arms in models with and without DABCO (not shown) revealed only minimal distortion, providing further evidence that the polycyclic arms are essentially rigid. The overall complexed structure does not appear to be significantly strained, and this supports the high association constant that has been determined experimentally.
 | ||
| Fig. 18 Equilibrium geometry (semi-empirical, AM1) of syn-3a·DABCO. Image rendered in GaussView.116 | ||
 | ||
| Fig. 19 Equilibrium geometry (semi-empirical, AM1) of (a)–(c) different orientations of anti-(3b)2·(DABCO)2; (d) n:n polymeric assembly anti-(3b)n·(DABCO)n (truncated). Image rendered in GaussView.116 | ||
The anti-(3b)2·(DABCO)2 complex could exist in several molecular orientations, for example where the polycyclic linkers are stacked horizontally, vertically, or adopt a four sided structure (Fig. 19(a)–(c) respectively). In addition, the n:n polymeric assembly, anti-(3b)n·(DABCO)n, is shown in Fig. 19(d), truncated to show a single repeating unit. No further investigation has been conducted into the identity of the several sandwich species observed during the NMR titration of anti-3b with DABCO.
Conclusions
The degree of rotation in our tweezer architecture was restricted from freely rotating by fully substituting the phenyl diimide linker core with methyl groups. Evidence of restricted rotation preventing interconversion between the syn- and anti-conformations was provided by UV/Vis and NMR spectroscopy, host–guest complexation models and speciation, and X-ray crystallography.UV/Vis and NMR titration confirmed that complexation between syn-3a and DABCO is best described by a 3a·DABCO and 3a·(DABCO)2 complexation model. The large association constant obtained for syn-3a·DABCO (K11 = 1.25 × 108 M−1) indicates the tweezer is sufficiently preorganised for DABCO. This is a similar outcome to freely rotating 1·DABCO (K11 = 8.1 × 107 M−1 (ref. 15)), which is also sufficiently preorganised for DABCO. The microscopic effective molarity,117–119 EM (divided by the statistical factor, Kσ), was calculated to be 9.76 × 10−4 M for syn-3a·DABCO (Kσ = 8), 6.33 × 10−4 M for freely rotating 1·DABCO (Kσ = 8), and 1.70 × 10−5 M for anti-(3b)2·(DABCO)2 (Kσ = 128) (S25†). Effective molarity values of other porphyrin host–guest systems in the literature usually range between 10−3–101 M,10,59,106,107,109,118*** although larger values have been reported.11,119 Further work is underway in our laboratory to examine the difference in preorganisation between syn-3a and freely rotating 1, which is expected to be manifested for ligands with length much greater than DABCO (2.64 Å) where the required interporphyrin distance for guest complexation requires significant rotation through the phenyl diimide linker core. Molecular modelling of syn-3a·DABCO suggests that the interporphyrin distance is modulated by partial rotation of the polycyclic scaffold about the 2,3,5,6-tetramethylphenyl diimide core, with the polycyclic scaffold remaining rigid. Nevertheless, the combined results of both this manuscript and our previous work15 are in agreement with the concept that the linker need only confer the system with moderate or sufficient preorganisation to obtain a large association constant that is enhanced compared to the reference monomeric complex.§
As expected, the UV/Vis and NMR titrations of anti-3b with DABCO confirmed its non-tweezer like behaviour compared to syn-3a, and was best described by a (3b)2·(DABCO)2 and 3b·(DABCO)2 complexation model. The NMR data indicated the formation of major and minor sandwich species, most likely different orientations of anti-(3b)2·(DABCO)2, or n:n polymeric anti-(3b)n·(DABCO)n, as represented by molecular modelling.
Experimental
Molecular modelling
Molecular modelling was undertaken using the Wavefunction Spartan ‘10 software package.93 Equilibrium geometry energy minimisation was calculated using a semi-empirical AM1 model.Characterisation
Melting points were measured using a Barloworld Scientific SMP10 melting point apparatus. Theoretical mass spectra were generated by using the mMass software package.120NMR spectra were recorded on a Bruker UltraShield Avance III 600 MHz NMR Spectrometer running the TopSpin software package at 293 K (20 °C). Spectra were calibrated to the residual solvent signal.121 CDCl3 was deacidified by passing through neutral activated aluminium oxide (Scharlau, activity degree 1, 70–290 mesh, grain size 0.05–0.2 mm) and stored under a nitrogen atmosphere over silver foil/molecular sieves in a brown glass bottle.
X-ray diffraction intensity data for compounds 11a and 11b was collected with an Oxford Diffraction SuperNova CCD diffractometer using Cu-Kα radiation. The temperature during data collection was maintained at 130.00(10) using an Oxford Cryosystems cooling device. The structure was solved by direct methods and difference Fourier synthesis.122 Thermal ellipsoid plots were generated using the program ORTEP-3123 integrated within the WINGX124 suite of programs. Additional information on the crystal structures of compounds 11a and 11b can be found in the ESI (see S18 and S21†). CCDC 1526930–1526931† contain the supplementary crystallographic data for this paper.
UV/Vis spectra were recorded on a Cary 50 instrument at 20 °C in a Starna Type 21 SX 1 cm2 quartz cuvette with the following parameters: average time 0.05 s, data interval 0.15 nm, scan rate 180 nm min−1, wavelength range 300–700 nm, baseline correction to blank solvent. Dry CHCl3 for recording spectra was prepared under a nitrogen atmosphere first by reflux over P2O5 then distillation,125 and deacidified and stored the same as above for CDCl3.
Reagents
Where necessary, solvents and reagents for synthesis were purified according to the methods published.125 Dry THF was freshly distilled from sodium/benzophenone, dry CH2Cl2 freshly distilled from CaH2, and dry DMF and DMSO were distilled under reduced pressure onto fresh molecular sieves after stirring on molecular sieves overnight. The following chemicals were purified by sublimation under high vacuum at 0.17 mmHg at the following temperatures, then stored under a nitrogen atmosphere, protected from light, and in a desiccator at room temperature unless otherwise specified: potassium tert-butoxide at 160 °C, DABCO twice at 75 °C and stored at −20 °C. 2,3,5,6-tetramethyl-p-phenylenediamine 5 (Sigma-Aldrich) was used as purchased.Silica gel (Davisil, 60 Å, 40–63 μm, Grace Davison Discovery Sciences) was used for column chromatography. Kieselgel silica gel 60 F254 aluminium sheets (Merck) was used for TLC. Colourless compounds were visualised using a UV lamp or permanganate dip stain.
Methodology for host–guest titrations
All samples for host–guest titrations were weighed using a five decimal point balance (Shimadzu AUW220D or AandD GR-202). Volumetric glassware (class A) was used for volumes >1 mL. Gas tight microlitre syringes (SGE, Hamilton) were used for volumes ≤1 mL. UV/Vis host–guest titrations were carried out at constant host concentrations of 10−6–10−7 M in CHCl3 (absorbance of free host <1 absorbance unit). Complexation models, association constants and speciation diagrams were calculated from the UV/Vis titration data using the HypSpec and HySS2009 software packages (Protonic software)104,105 over 400–450 nm. A more detailed discussion of examining the fitting can be found in S22.†1H NMR host–guest titrations were carried out in CDCl3 at non-constant host concentration, starting at 10−3–10−4 M, and gradually diluted by aliquots of guest solution (usually 5–10 μL per 0.1 equivalents of guest). Low temperature NMR was undertaken at −50 to −60 °C.General procedure for sealed tube ACE reactions
An 80 mL sealed tube (BSG Glassware, Tasmania, screw thread stopper with upper and lower Viton O-rings) was fitted with a stirrer bar and loaded with 0.8 g combined starting materials suspended in no more than 20 mL of solvent (less than one quarter total volume of flask). The tube was wrapped in alfoil, immersed in an oil bath relative to the solvent level, and heated to 160 °C with stirring for 24 hours behind a blast shield. O-rings (Viton) were replaced for each reaction.General procedure for porphyrin zinc(II) metallation
Using standard porphyrin zinc(II) metallation conditions,66 a solution of Zn(OAc)2·2H2O (excess, usually 5–10 eq. per porphyrin) in MeOH was added dropwise down the condenser to a refluxing solution of porphyrin in CHCl3 (or CH2Cl2)/MeOH (approx. 4:1), such that the combined volume was CHCl3 ≥ MeOH. The solution was refluxed for 1–2 hours, with further CHCl3 added during this time as necessary to prevent evaporation to dryness or precipitation of the porphyrin. The mixture was cooled, diluted with CHCl3, washed with H2O, dried with Na2SO4, filtered, and the solvent removed in vacuo. The material was purified by column chromatography (silica) and recrystallised from CDCl3/MeOH to afford purple crystals.
Synthesis of compounds and characterisation data
15,55 (3.73 g, 12.2 mmol) and 2,3,5,6-tetramethyl-p-phenylenediamine 5 (1.0 g, 6.1 mmol, 0.5 eq.) in degassed dry DMSO (60 mL) was heated at 80 °C under an argon atmosphere for 1 day. The DMSO was removed by distillation under reduced pressure, the mixture redissolved in Ac2O (80 mL), NaOAc (7.2 g, 53 mmol) added, and the solution heated at 80 °C under a nitrogen atmosphere for a further 1 day, after which a precipitate was observed. Preliminary 1H NMR of the reaction mixture suggested two isomers in an approximately 60:40 ratio (although may be due to different solubility), distinguished by outer and inner pairs of non-equivalent CH3 phenyl substituent resonances. The precipitate was removed by filtration and the solids redissolved in CHCl3 (200 mL), washed with H2O (2 × 200 mL), aqueous NaOH solution (2 M, 3 × 200 mL), aqueous HCl solution (2 M, 1 × 100 mL), H2O (1 × 100 mL), dried (Na2SO4), and the solvent removed in vacuo to afford a white powder (3.25 g, 72%, m.p. > 300 °C). The linker was used as a mixture due to difficulty in separation of the isomers.
A small sample of the isomers was separated by column chromatography, using either of the following methods or a combination of both. Method A: silica (10% THF/CHCl3) to partially resolve isomers, if bands co-elute then silica (5% MeOH/CHCl3), followed by recrystallisation from CHCl3/MeOH, solids washed with MeOH. Method B: silica plug (EtOAc), then silica (load CHCl3, elute 50% EtOAc/CHCl3, poor solubility and precipitation on column). Unfortunately the assignment of each isomer as either syn- or anti- could not be achieved by 1H NOE spectroscopy, and so are referred to by the inner and outer CH3 isomer nomenclature established above.
Inner CH3 isomer. 1H NMR (600 MHz, 20 °C, CDCl3): 3.80 (s, 12H), 3.50–3.47 (m, 4H), 3.11 (s, 4H), 2.98–2.95 (m, 4H), 2.07 (s, 6H), 1.95 (s, 6H), 1.86 (d, J = 11.6 Hz, 2H), 1.59 (d, J = 11.6 Hz, 2H). 13C NMR (150 MHz, CDCl3): 175.72, 160.80, 141.45, 133.16, 132.62, 132.04, 52.27, 48.16, 43.15, 36.53, 34.83, 16.18, 15.51. HRMS (ESI-TOF-MS) for C40H40N2O12Na+ [M + Na]+: calc.: 763.2479. Found: 763.2494.
Outer CH3 isomer. 1H NMR (600 MHz, 20 °C, CDCl3): 3.80 (s, 12H), 3.50–3.46 (m, 4H), 3.12 (s, 4H), 2.98–2.96 (m, 4H), 2.09 (s, 6H), 1.94 (s, 6H), 1.86 (d, J = 11.5 Hz, 2H), 1.59 (d, J = 11.5 Hz, 2H). 13C NMR (150 MHz, CDCl3): 175.74, 160.79, 141.45, 133.11, 132.71, 132.03, 52.27, 48.18, 43.15, 36.55, 34.84, 16.17, 15.58. HRMS (ESI-TOF-MS) for C40H40N2O12Na+ [M + Na]+: calc.: 763.2479. Found: 763.2494.
anti-9b. 1H NMR (600 MHz, 20 °C, CDCl3): 8.85 (s, 16H), 8.30 (d, J = 8.1 Hz, 4H), 8.25–8.18 (m, 12H), 7.82–7.73 (m, 18H), 7.62 (d, J = 8.1 Hz, 4H), 3.98 (s, 12H), 3.34–3.30 (m, 4H), 2.92 (s, 4H), 2.90 (s, 4H), 2.85–2.82 (m, 4H), 2.76 (d, J = 10.9 Hz, 2H), 2.58–2.51 (m, 6H, two overlapping signals), 2.32 (s, 4H), 2.28 (s, 6H), 2.04 (s, 6H), 1.41 (d, J = 10.9 Hz, 2H), 1.22 (d, J = 11.9 Hz, 2H), −2.80 (s, 4H).
syn-9a. Characterised as the zinc(II) derivative. 1H NMR spectrum provided in S8† as mixture with anti-9b.
HRMS (ESI-TOF-MS) for two samples containing different ratios of syn-9a/anti-9b. Sample 1 [C146H116N12O18]2+ [M + 2H]2+: calc.: 1162.4260. Found: 1162.4267. Sample 2 [C146H114N12O18Na2]2+ [M + 2Na]2+: calc.: 1184.4079. Found: 1184.4110.
syn-11a (filtrate, outer CH3). M.p. > 300 °C (crystals crack, partial decomposition). 1H NMR (600 MHz, 20 °C, CDCl3): 6.34 (m, 4H), 3.42 (m, 4H), 2.91 (s, 4H), 2.051 (s, 6H), 1.967 (s, 6H), 1.68 (m, 4H). 13C NMR (150 MHz, CHCl3): 176.72, 138.03, 133.08, 132.67, 131.86, 48.47, 45.29, 43.80, 15.90, 15.63. HRMS (ESI-TOF-MS) for C28H28N2O4Na+ [M + Na]+: calc.: 479.1947. Found: 479.1957. Single crystal for X-ray diffraction analysis was grown from CH3CN by slow evaporation. CCDC 1526930† contains the supplementary crystallographic data for this paper.
anti-11b (precipitate, inner CH3). M.p. > 300 °C (crystals transition from clear to white 195–205 °C, partial decomposition by 300 °C). 1H NMR (600 MHz, 20 °C, CDCl3): 6.34 (m, 4H), 3.42 (m, 4H), 2.91 (s, 4H), 2.039 (s, 6H), 1.973 (s, 6H), 1.67 (m, 4H). 13C NMR (150 MHz, CHCl3): 176.70, 138.01, 133.14, 132.58, 131.84, 48.46, 45.28, 43.78, 15.99, 15.50. HRMS (ESI-TOF-MS) for C28H28N2O4Na+ [M + Na]+: calc.: 479.1947. Found: 479.1949. Single crystal for X-ray diffraction analysis was grown from CH3CN by slow evaporation, and solved as the structure co-crystallised with approx. 6% mono-epoxide (at the norbornene). CCDC 1526931† contains the supplementary crystallographic data for this paper.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
R. B. M wishes to thank Flinders University for the provision of an Australian Postgraduate Award and the Playford Memorial Trust for a PhD top-up scholarship. R. B. M also thanks the following for their efforts and expertise in accurate mass spectrometry: Dr Sally Duck (Monash University) porphyrin compound 9, Dr Craig Brinkworth (Defence Science and Technology Group, Australia) porphyrin compounds syn-3a and anti-3b, Dr Daniel Jardine (Flinders Analytical) all other compounds.Notes and references
- J. W. Steed and J. L. Atwood, Supramolecular Chemistry, John Wiley & Sons, Ltd, 1st edn, 2000 Search PubMed.
- D. Cram, Science, 1988, 240, 760–767 CrossRef PubMed.
- D. J. Cram, G. M. Lein, T. Kaneda, R. C. Helgeson, C. B. Knobler, E. Maverick and K. N. Trueblood, J. Am. Chem. Soc., 1981, 103, 6228–6232 CrossRef.
- V. Valderrey, G. Aragay and P. Ballester, Coord. Chem. Rev., 2014, 258–259, 137–156 CrossRef.
- S. Otto, Dalton Trans., 2006, 2861–2864 RSC.
- N. Voyer and F. Maltais, Adv. Mater., 1993, 5, 568–570 CrossRef.
- S. P. Gaynor, M. J. Gunter, M. R. Johnston and R. N. Warrener, Org. Biomol. Chem., 2006, 4, 2253–2266 RSC.
- M. Nakash and J. K. M. Sanders, J. Org. Chem., 2000, 65, 7266–7271 CrossRef PubMed.
- Z. Rodriguez-Docampo, E. Eugenieva-Ilieva, C. Reyheller, A. M. Belenguer, S. Kubik and S. Otto, Chem. Commun., 2011, 47, 9798–9800 RSC.
- H. Sun, C. A. Hunter and E. M. Llamas, Chem. Sci., 2015, 6, 1444–1453 RSC.
- L. Pengpeng, P. Neuhaus, D. V. Kondratuk, T. S. Balaban and H. L. Anderson, Angew. Chem., Int. Ed., 2014, 53, 7770–7773 CrossRef PubMed.
- N. Solladié, S. Bouatra, R. Rein and J. Roeser, J. Porphyrins Phthalocyanines, 2005, 9, 779–787 CrossRef.
- N. Solladié, F. Aziat, S. Bouatra and R. Rein, J. Porphyrins Phthalocyanines, 2008, 12, 1250–1260 CrossRef.
- S. Merkas, S. Bouatra, R. Rein, I. Piantanida, M. Zinic and N. Solladié, J. Porphyrins Phthalocyanines, 2015, 19, 535–546 CrossRef.
- R. B. Murphy, D.-T. Pham, S. F. Lincoln and M. R. Johnston, Eur. J. Org. Chem., 2013, 2985–2993 CrossRef.
- H. L. Anderson, S. Anderson and J. K. M. Sanders, J. Chem. Soc., Perkin Trans. 1, 1995, 2231–2245 RSC.
- J. Etxebarria, A. Vidal-Ferran and P. Ballester, Chem. Commun., 2008, 5939–5941 RSC.
- N. Solladié, N. Aubert, S. Bouatra, C. Bourgogne, F. Bregier, J. Brettar, J.-P. Gisselbrecht, M. Gross, R. Rein, C. Sooambar, V. Troiani and M. Walther, J. Porphyrins Phthalocyanines, 2003, 7, 270–281 CrossRef.
- M. R. Johnston and D. M. Lyons, Supramol. Chem., 2005, 17, 503–511 CrossRef.
- D. Kim, S. Lee, G. Gao, H. S. Kang and J. Ko, J. Organomet. Chem., 2009, 695, 111–119 CrossRef.
- Y. Kubo, Y. Murai, J.-i. Yamanaka, S. Tokita and Y. Ishimaru, Tetrahedron Lett., 1999, 40, 6019–6023 CrossRef.
- S. Yagi, M. Ezoe, I. Yonekura, T. Takagishi and H. Nakazumi, J. Am. Chem. Soc., 2003, 125, 4068–4069 CrossRef PubMed.
- L. H. Tong, J.-L. Wietor, W. Clegg, P. R. Raithby, S. I. Pascu and J. K. M. Sanders, Chem. – Eur. J., 2008, 14, 3035–3044 CrossRef PubMed.
- C.-H. Lee, H. Yoon and W.-D. Jang, Chem. – Eur. J., 2009, 15, 9972–9976 CrossRef PubMed.
- M. Dudič, P. Lhoták, H. Petříčková, I. Stibor, K. Lang and J. Sýkora, Tetrahedron, 2003, 59, 2409–2415 CrossRef.
- D. Jokic, Z. Asfari and J. Weiss, Org. Lett., 2002, 4, 2129–2132 CrossRef PubMed.
- D. Jokic, C. Boudon, G. Pognon, M. Bonin, K. J. Schenk, M. Gross and J. Weiss, Chem. – Eur. J., 2005, 11, 4199–4209 CrossRef PubMed.
- P. Mondal and S. P. Rath, Isr. J. Chem., 2016, 56, 144–155 CrossRef.
- M. Blom, S. Norrehed, C.-H. Andersson, H. Huang, M. Light, J. Bergquist, H. Grennberg and A. Gogoll, Molecules, 2016, 21, 16 CrossRef PubMed.
- R. B. Murphy, R. E. Norman, J. M. White, M. V. Perkins and M. R. Johnston, Org. Biomol. Chem., 2016, 14, 8707–8720 RSC.
- R. N. Warrener, D. N. Butler, W. Y. Liao, I. G. Pitt and R. A. Russell, Tetrahedron Lett., 1991, 32, 1889–1892 CrossRef.
- R. N. Warrener, I. G. Pitt and D. N. Butler, J. Chem. Soc., Chem. Commun., 1983, 1340–1342 RSC.
- R. N. Warrener, A. C. Schultz, D. N. Butler, S. Wang, I. B. Mahadevan and R. A. Russel, Chem. Commun., 1997, 1023–1024 RSC.
- R. N. Warrener, S. Wang and R. A. Russell, Tetrahedron, 1997, 53, 3975–3990 CrossRef.
- R. N. Warrener, D. N. Butler, D. Margetic, F. M. Pfeffer and R. A. Russell, Tetrahedron Lett., 2000, 41, 4671–4675 CrossRef.
- M. Golić, M. R. Johnston, D. Margetić, A. C. Schultz and R. N. Warrener, Aust. J. Chem., 2006, 59, 899–914 CrossRef.
- D. Margetic, M. R. Johnston, E. R. T. Tiekink and R. N. Warrener, Tetrahedron Lett., 1998, 39, 5277–5280 CrossRef.
- R. N. Warrener, D. N. Butler, L. Liu, D. Margetic and R. A. Russell, Chem. – Eur. J., 2001, 7, 3406–3414 CrossRef.
- M. J. Gunter, H. Tang and R. N. Warrener, J. Porphyrins Phthalocyanines, 2002, 6, 673–684 CrossRef.
- A. M. Napper, I. Read, N. J. Head, A. M. Oliver and M. N. Paddon-Row, J. Am. Chem. Soc., 2000, 122, 5220–5221 CrossRef.
- N. J. Head, A. M. Oliver, K. Look, N. R. Lokan, G. A. Jones and M. N. Paddon-Row, Angew. Chem., Int. Ed., 1999, 38, 3219–3222 CrossRef PubMed.
- M. J. Shephard and M. N. Paddon-Row, J. Phys. Chem. A, 2000, 104, 11628–11635 CrossRef.
- M. J. Shephard and M. N. Paddon-Row, J. Phys. Chem. A, 1999, 103, 3347–3350 CrossRef.
- M. R. Johnston, M. J. Latter and R. N. Warrener, Org. Lett., 2002, 4, 2165–2168 CrossRef PubMed.
- M. Johnston, Molecules, 2001, 6, 406–416 CrossRef.
- M. R. Johnston, M. J. Latter and R. N. Warrener, Aust. J. Chem., 2001, 54, 633–636 CrossRef.
- M. D. Johnstone, E. K. Schwarze, J. Ahrens, D. Schwarzer, J. J. Holstein, B. Dittrich, F. M. Pfeffer and G. H. Clever, Chem. – Eur. J., 2016, 22, 10791–10795 CrossRef PubMed.
- M. D. Johnstone, M. Frank, G. H. Clever and F. M. Pfeffer, Eur. J. Org. Chem., 2013, 5848–5853 CrossRef.
- R. N. Robson and F. M. Pfeffer, Chem. Commun., 2016, 52, 8719–8721 RSC.
- M. D. Johnstone, E. K. Schwarze, G. H. Clever and F. M. Pfeffer, Chem. – Eur. J., 2015, 21, 3948–3955 CrossRef PubMed.
- A. J. Lowe, B. M. Long and F. M. Pfeffer, Chem. Commun., 2013, 49, 3376–3388 RSC.
- B. M. Long and F. M. Pfeffer, Chem. – Asian J., 2014, 9, 1091–1098 CrossRef PubMed.
- R. N. Robson, B. P. Hay and F. M. Pfeffer, ChemistrySelect, 2017, 2, 4605–4608 CrossRef.
- G. H. Clever and P. Punt, Acc. Chem. Res., 2017, 50, 2233–2243 CrossRef PubMed.
- M. Shang, R. N. Warrener, D. N. Butler, Y. Murata and D. Margetić, Mol. Diversity, 2011, 15, 541–560 CrossRef PubMed.
- P. Trošelj, A. Briš, Y. Murata and D. Margetić, Struct. Chem., 2012, 23, 791–799 CrossRef.
- P. Trošelj, I. Đilović, D. Matković-Čalogović and D. Margetić, J. Heterocycl. Chem., 2013, 50, 83–90 CrossRef.
- H. Tang, Z. Dong, Z. Merican, D. Margetić, Ž. Marinić, M. J. Gunter, D. Officer, D. N. Butler and R. N. Warrener, Tetrahedron Lett., 2009, 50, 667–670 CrossRef.
- A. R. Mulholland, P. Thordarson, E. J. Mensforth and S. J. Langford, Org. Biomol. Chem., 2012, 10, 6045–6053 RSC.
- G. Giancane, V. Borovkov, Y. Inoue and L. Valli, J. Colloid Interface Sci., 2012, 385, 282–284 CrossRef PubMed.
- V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, Tetrahedron Lett., 1999, 40, 5051–5054 CrossRef.
- V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, J. Phys. Chem. B, 1999, 103, 5151–5156 CrossRef.
- V. V. Borovkov, J. M. Lintuluoto and Y. Inoue, J. Am. Chem. Soc., 2001, 123, 2979–2989 CrossRef PubMed.
- R. N. Warrener, M. R. Johnston and M. J. Gunter, Synlett, 1998, 593–595 CrossRef.
- R. C. Foitzik, A. Lowe and F. M. Pfeffer, Tetrahedron Lett., 2009, 50, 2583–2584 CrossRef.
- J.-H. Fuhrhop and K. M. Smith, Laboratory methods in porphyrin and metalloporphyrin research, Elsevier Scientific Pub. Co., 1975 Search PubMed.
- E. Stulz, C. C. Mak and J. K. M. Sanders, J. Chem. Soc., Dalton Trans., 2001, 604–613 RSC.
- M. Kasha, H. R. Rawls and M. A. El-Bayoumi, Pure Appl. Chem., 1965, 11, 371–392 Search PubMed.
- A. Osuka and K. Maruyama, J. Am. Chem. Soc., 1988, 110, 4454–4456 CrossRef.
- C. A. Hunter, J. K. M. Sanders and A. J. Stone, Chem. Phys., 1989, 133, 395–404 CrossRef.
- C. K. Chang and I. Abdalmuhdi, J. Org. Chem., 1983, 48, 5388–5390 CrossRef.
- H. A. Staab and T. Carell, Angew. Chem., Int. Ed. Engl., 1994, 33, 1466–1468 CrossRef.
- Y. Yu, L. Shi, D. Yang and L. Gan, Chem. Sci., 2013, 4, 814–818 RSC.
- S. Verma and N. Singh, Aust. J. Chem., 1976, 29, 295–300 CrossRef.
- D. P. Curran, S. Geib and N. DeMello, Tetrahedron, 1999, 55, 5681–5704 CrossRef.
- K. Tanaka, M. Okano, H. Toshino, H. Kita and K.-I. Okamoto, J. Polym. Sci., Part B: Polym. Phys., 1992, 30, 907–914 CrossRef.
- Y. Zhang, J. M. Lavin and K. D. Shimizu, J. Am. Chem. Soc., 2009, 131, 12062–12063 CrossRef PubMed.
- D.-S. Choi, Y. S. Chong, D. Whitehead and K. D. Shimizu, Org. Lett., 2001, 3, 3757–3760 CrossRef PubMed.
- Y. Chen, M. D. Smith and K. D. Shimizu, Tetrahedron Lett., 2001, 42, 7185–7187 CrossRef.
- R. D. Rasberry, X. Wu, B. N. Bullock, M. D. Smith and K. D. Shimizu, Org. Lett., 2009, 11, 2599–2602 CrossRef PubMed.
- C. F. Degenhardt, J. M. Lavin, M. D. Smith and K. D. Shimizu, Org. Lett., 2005, 7, 4079–4081 CrossRef PubMed.
- J. M. Lavin and K. D. Shimizu, Chem. Commun., 2007, 228–230 RSC.
- W. R. Carroll, P. Pellechia and K. D. Shimizu, Org. Lett., 2008, 10, 3547–3550 CrossRef PubMed.
- C. W. Miller, C. E. Hoyle, E. J. Valente, D. H. Magers and E. S. Jönsson, J. Phys. Chem. A, 1999, 103, 6406–6412 CrossRef.
- K. Kondo, H. Fujita, T. Suzuki and Y. Murakami, Tetrahedron Lett., 1999, 40, 5577–5580 CrossRef.
- M. Mao, J. England and S. R. Turner, Polymer, 2011, 52, 4498–4502 CrossRef.
- C. W. Miller, E. S. Jönsson, C. E. Hoyle, K. Viswanathan and E. J. Valente, J. Phys. Chem. B, 2001, 105, 2707–2717 CrossRef.
- C. Miller, C. Hoyle, E. Valente, J. Zubkowski and E. S. Jönsson, J. Chem. Crystallogr., 2000, 30, 563–571 CrossRef.
- D. P. Curran and N. C. DeMello, J. Chem. Soc., Chem. Commun., 1993, 1314–1317 RSC.
- R. B. Murphy, PhD thesis, Flinders University, Adelaide, South Australia, 2016.
- Mercury 3.8, CCDC, 2016, http://www.ccdc.cam.ac.uk/mercury/ Search PubMed.
- P. D. Bartlett, G. L. Combs, A. X. T. Le, W. H. Watson, J. Galloy and M. Kimura, J. Am. Chem. Soc., 1982, 104, 3131–3138 CrossRef.
- Spartan ‘10 for Windows, Wavefunction, Inc., 18401 Von Karman Avenue, Suite 370, Irvine, CA 92612 USA, http://www.wavefun.com/index.html Search PubMed.
- B. A. Langowski, R. Rothchild and A.-M. Sapse, Spectrosc. Lett., 2001, 34, 235–251 CrossRef.
- A. Camara-Campos, C. A. Hunter and S. Tomas, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 3034–3038 CrossRef PubMed.
- P. N. Taylor and H. L. Anderson, J. Am. Chem. Soc., 1999, 121, 11538–11545 CrossRef.
- H.-T. Nguyen, D.-T. Pham, C. J. Easton and S. F. Lincoln, Aust. J. Chem., 2013, 66, 1057–1064 CrossRef.
- E. A. Kataev and T. A. Shumilova, Molecules, 2015, 20, 3354–3370 CrossRef PubMed.
- R. Montis, M. C. Aragoni, M. Arca, C. Bazzicalupi, A. J. Blake, C. Caltagirone, G. De Filippo, A. Garau, P. Gratteri, F. Isaia, V. Lippolis and A. Pintus, Inorg. Chim. Acta, 2012, 381, 170–180 CrossRef.
- F. Ulatowski, K. Dąbrowa, T. Bałakier and J. Jurczak, J. Org. Chem., 2016, 81, 1746–1756 CrossRef PubMed.
- S. A. Ikbal, S. Brahma and S. P. Rath, Chem. Commun., 2015, 51, 895–898 RSC.
- A. Dhamija, S. A. Ikbal and S. P. Rath, Inorg. Chem., 2016, 55, 13014–13026 CrossRef PubMed.
- E. A. Kataev, N. Backmann, T. A. Shumilova, T. Rüffer and H. Lang, Supramol. Chem., 2016, 28, 53–61 CrossRef.
- Protonic Software, 2 Templegate Avenue, Leeds, LS150HD, UK. ( http://www.hyperquad.co.uk) Search PubMed.
- P. Gans, A. Sabatini and A. Vacca, Talanta, 1996, 43, 1739–1753 CrossRef PubMed.
- P. Ballester, A. Costa, A. M. Castilla, P. M. Deyá, A. Frontera, R. M. Gomilla and C. A. Hunter, Chem. – Eur. J., 2005, 11, 2196–2206 CrossRef PubMed.
- L. Baldini, P. Ballester, A. Casnati, R. M. Gomila, C. A. Hunter, F. Sansone and R. Ungaro, J. Am. Chem. Soc., 2003, 125, 14181–14189 CrossRef PubMed.
- P. Ballester, A. I. Oliva, A. Costa, P. M. Deyà, A. Frontera, R. M. Gomila and C. A. Hunter, J. Am. Chem. Soc., 2006, 128, 5560–5569 CrossRef PubMed.
- H. L. Anderson, Inorg. Chem., 1994, 33, 972–981 CrossRef.
- C. C. Mak, N. Bampos and J. M. K. Sanders, Angew. Chem., Int. Ed., 1998, 37, 3020–3023 CrossRef PubMed.
- C. A. Hunter, M. N. Meah and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5773–5780 CrossRef.
- H. L. Anderson, C. A. Hunter, M. N. Meah and J. K. M. Sanders, J. Am. Chem. Soc., 1990, 112, 5780–5789 CrossRef.
- S. S. Eaton and G. R. Eaton, J. Chem. Soc., Chem. Commun., 1974, 576–577 RSC.
- R. W. Wagner, T. E. Johnson and J. S. Lindsey, J. Am. Chem. Soc., 1996, 118, 11166–11180 CrossRef.
- S. S. Eaton and G. R. Eaton, J. Am. Chem. Soc., 1975, 97, 3660–3666 CrossRef PubMed.
- R. Dennington, T. Keith and J. Millam, GaussView, Version 5.0.9, Semichem Inc., Shawnee Mission, KS, 2009 Search PubMed.
- G. Ercolani and L. Schiaffino, Angew. Chem., Int. Ed., 2011, 50, 1762–1768 CrossRef PubMed.
- C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488–7499 CrossRef PubMed.
- H. J. Hogben, J. K. Sprafke, M. Hoffmann, M. Pawlicki and H. L. Anderson, J. Am. Chem. Soc., 2011, 133, 20962–20969 CrossRef PubMed.
- M. Strohalm, mMass 5.4.1, 2012http://www.mmass.org/ Search PubMed.
- H. E. Gottlieb, V. Kotlyar and A. Nudelman, J. Org. Chem., 1997, 62, 7512–7515 CrossRef PubMed.
- G. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2008, 64, 112–122 CrossRef PubMed.
- L. Farrugia, J. Appl. Crystallogr., 1997, 30, 565 CrossRef.
- L. Farrugia, J. Appl. Crystallogr., 1999, 32, 837–838 CrossRef.
- D. D. Perrin, L. Armarego and D. R. Perrin, Purification of Laboratory Chemicals, Pergamon Press, Ltd., Oxford, 1966 Search PubMed.
- J. G. Hill, B. E. Rossiter and K. B. Sharpless, J. Org. Chem., 1983, 48, 3607–3608 CrossRef.
- D. Craig, J. Am. Chem. Soc., 1951, 73, 4889–4892 CrossRef.
- G. Ercolani, C. Piguet, M. Borkovec and J. Hamacek, J. Phys. Chem. B, 2007, 111, 12195–12203 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: NMR, UV/Vis, MS, X-ray, complexation models, statistical analysis, effective molarity. CCDC 1526930 and 1526931. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob00944a |
| ‡ Present address: Australian Nuclear Science and Technology Organisation, Locked Bag 2001, Kirrawee DC, NSW 2232, Australia. |
| § Although there are examples where conformationally flexible linkers are insufficiently preorganised and small association constants are obtained.13,14 |
| ¶ This apparent non-statistical mixture could result from differential solubility of the syn-6a and anti-6b conformations in the precipitate and filtrate after the reaction, however this has not been investigated further. |
| || Compound 10 was used due to the poor solubility of the zinc(II) metallated adduct of exo-porphyrin receptor 8 in chloroform. |
| ** Ref. 89 relates to anilide derivatives rather than imides, however, the principle of balancing steric and electronic factors is the same here. |
| †† The error between the number of equivalents of DABCO titrated with tweezer 3, based on mass weighed, compared to the NMR signal integration was estimated to be 10% at 20 °C for both syn-3a and anti-3b, 20% at −50 °C for syn-3a, and 10–15% at −50 to −60 °C for anti-3b. |
| ‡‡ The ester signal was selected as a reference because it does not change chemical shift significantly on complexation, and its integration is the sum of free plus complexed species. Conveniently, the number of protons in the host ester resonance is equal to the number of protons in an isolated molecule of free DABCO, and so their relative integrations do not require any normalisation. |
| §§ A chemical shift of 8.548 ppm (syn-3a + 0.9 eq. DABCO) was selected for the value for fully complexed species. A chemical shift of 8.829 ppm (anti-3b + 5 eq. DABCO) was selected for the value for fully open species. |
| ¶¶ The major and minor signals for the sandwich species observed at low equivalents of DABCO were not baseline resolved and so were integrated together, which assumes that the two resonances arise from different molecular orientations of the same complex (3b)2·(DABCO)2. |
| |||| While the 2,3,5,6-tetramethylphenyl diimide group is distorted from planarity, no curvature was observed along the norbornyl diimide plane. A degree of curvature was observed in the analogous model of the freely rotating tweezer with guest, 1·DABCO.90 |
| *** Effective molarity is sometimes reported as the microscopic effective molarity multiplied by the statistical factor, Kσ.117–119 Our Kσ·EM values are 7.81 × 10−3 M for syn-3a·DABCO, 5.06 × 10−3 M for freely rotating 1·DABCO, and 2.18 × 10−3 M for anti-(3b)2·(DABCO)2. Additionally, the determination of Kσ sometimes varies,128 which can influence the statistically corrected microscopic effective molarity. |
| This journal is © The Royal Society of Chemistry 2018 |