
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of fused tricyclic systems by thermal Cope rearrangement of furan-substituted vinyl cyclopropanes†
Verena
Klaus
,
Stéphane
Wittmann
,
Hans M.
Senn
 and
J. Stephen
Clark
*
and
J. Stephen
Clark
*
WestCHEM, School of Chemistry, Joseph Black Building, University of Glasgow, University Avenue, Glasgow G12 8QQ, UK. E-mail: stephen.clark@glasgow.ac.uk; Tel: +44 (0)141 330 6296
First published on 15th May 2018
Abstract
A novel method for the stereoselective construction of hexahydroazuleno[4,5-b]furans from simple precursors has been developed. The route involves the use of our recently developed Brønsted acid catalysed cyclisation reaction of acyclic ynenones to prepare fused 1-furanyl-2-alkenylcyclopropanes that undergo highly stereoselective thermal Cope rearrangement to produce fused tricyclic products. Substrates possessing an E-alkene undergo smooth Cope rearrangement at 40 °C, whereas the corresponding Z-isomers do not react at this temperature. Computational studies have been performed to explain the difference in behaviour of the E- and Z-isomers in the Cope rearrangement reaction. The hexahydroazuleno[4,5-b]furans produced by Cope rearrangement have potential as advanced intermediates for the synthesis of members of the guaianolide family of natural products.
Introduction
Furans and benzofurans occur frequently as sub-units in pharmaceuticals and bioactive natural products.1,2 In many natural products, a furan or benzofuran is embedded in a polycyclic array or a macrocycle and is fused to one or more rings. An interesting and synthetically alluring group of furan-containing natural products comprises compounds that possess a furan or benzofuran fused to a seven-membered ring. Examples include myrrhterpenoid H,3 frondosin B4 and liphagal,5 which have been popular targets in recent years and have been shown to possess biological activities such as neuroprotective activity,3 binding to interlukin-8 receptors4 and inhibition of various kinases4,5 (Fig. 1).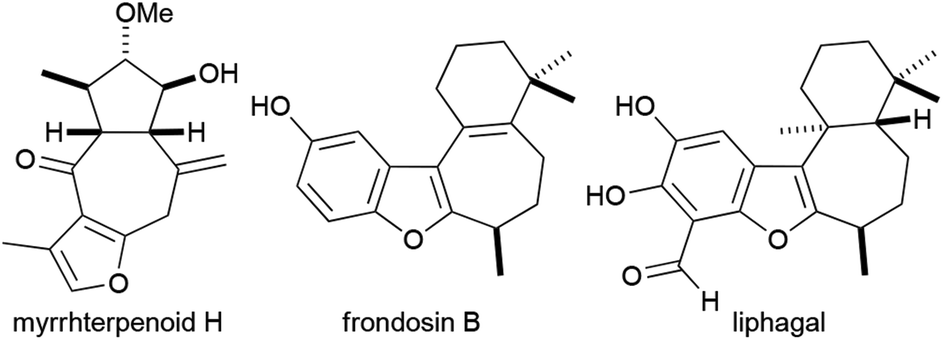 | ||
| Fig. 1 Examples of some bioactive natural products that possess a furan or benzofuran fused to a seven-membered ring. | ||
We have recently embarked on a programme directed toward the development of new methods for the concise and efficient synthesis of highly functionalised furans. As part of this programme, we wished to explore whether polycyclic systems containing the cyclohepta[b]furan unit could be synthesised from the cyclopropyl-substituted furans prepared by use of our recently-discovered stereoselective Brønsted acid catalysed cascade reaction (Scheme 1).6 In previous work, we have shown that treatment of the structurally diverse ynenediones 1 with chloroacetic acid results in intramolecular cyclisation to produce the tricyclic products 2 in which a trisubstituted furan is connected to a cyclopropyl substituent at the α position. The reaction is proposed to occur by carbonyl group protonation, and nucleophilic attack of the oxygen onto the central carbon of the putative allenyl intermediate to produce a carbene that reacts with the pendent alkene to form a cyclopropane.6
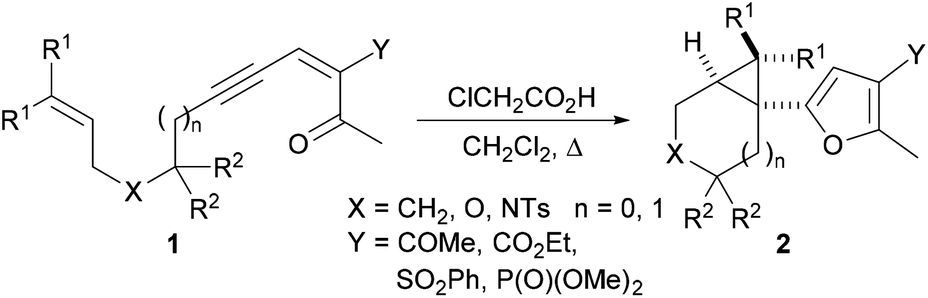 | ||
| Scheme 1 Synthesis of cyclopropyl-substituted furans by Brønsted acid mediated intramolecular cascade reactions. | ||
Cyclohepta[b]furans can be prepared from simple furfuryl alcohols by formal [4 + 3] cycloadditions reactions (Scheme 2). Pattenden and Winne have shown that the furan-containing diene 3 undergoes an intramolecular acid mediated reaction to produce the fused lactone 4, a tetracyclic compound that comprises most of the core structure of the marine diterpene rameswaralide.7 Winne and co-workers have developed an intermolecular variant of the reaction in which a Lewis acid catalysed reaction of a furfuryl alcohol with a simple diene delivers a cyclohepta[b]furan.8 For example, the reaction was used to convert the furfuryl alcohol 5 into the cyclohepta[b]furan 6 in reasonable yield (Scheme 2).
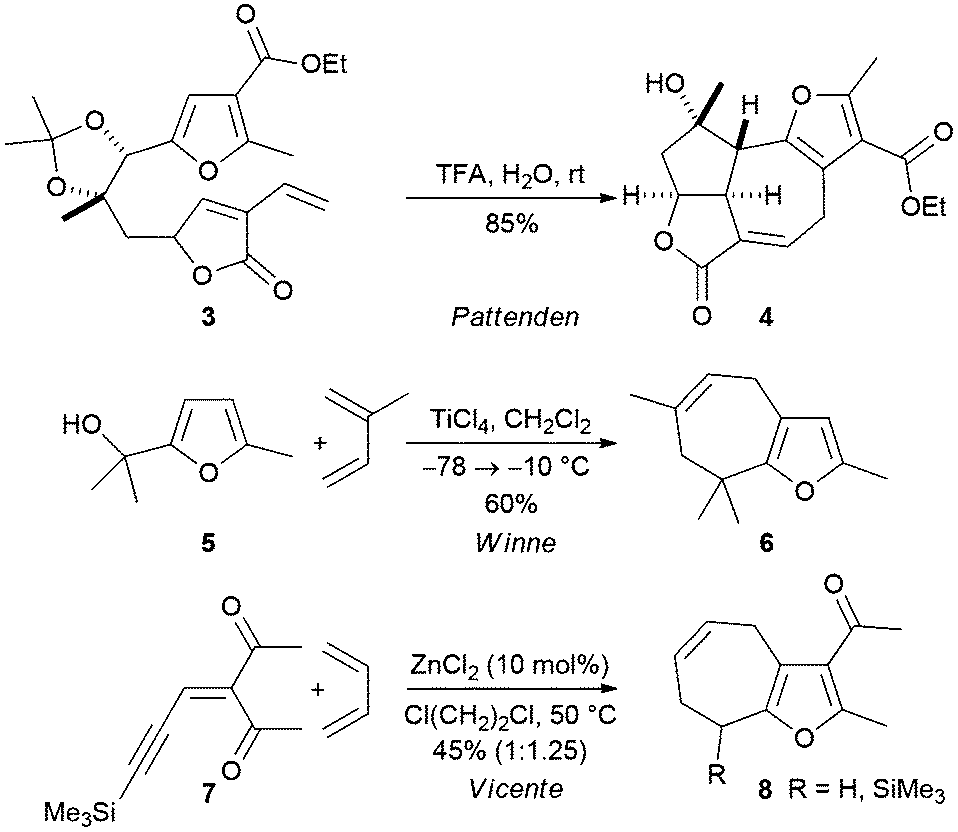 | ||
| Scheme 2 Preparation of partially reduced cyclohepta[b]furans by use of [4 + 3] cycloaddition reactions. | ||
The groups of Liang and Vicente have reported a one-pot method for the synthesis of fused cyclohepta[b]furans from acyclic precursors by Lewis acid mediated cyclisation of an ynenedione in the presence of a diene (Scheme 2).9,10 For example, treatment of the substrate 7 with zinc(II) chloride in the presence of butadiene, was found to deliver a mixture of the silylated and desilylated products 8.10 It was proposed that the products result from a formal [4 + 3] cycloaddition reaction, but cyclopropane formation and subsequent Cope rearrangement cannot be ruled out in these cases.
The successful preparation of highly functionalised cyclopropyl-substituted furans by use of our Brønsted acid promoted cyclisation reaction (Scheme 1) prompted us to explore whether these compounds could be rearranged to give fused tricyclic compounds containing an embedded furan. Our general approach to the synthesis of fused tricyclic compounds is depicted in Scheme 3. In the proposed reaction sequence, Brønsted acid promoted cyclisation of an acyclic ynenone 9, which possesses a tethered diene, would be used to produce a vinyl cyclopropane 10. Subsequent Cope rearrangement should deliver the fused tricyclic triene 11 and re-aromatization would result in formation of the furan-containing tricyclic product 12. It was anticipated that, under appropriate reaction conditions, the acyclic precursor 9 could be transformed directly into the fused tricyclic product 12 in a one-pot fashion with the creation of three rings, three bonds and three stereogenic centres in a single operation.
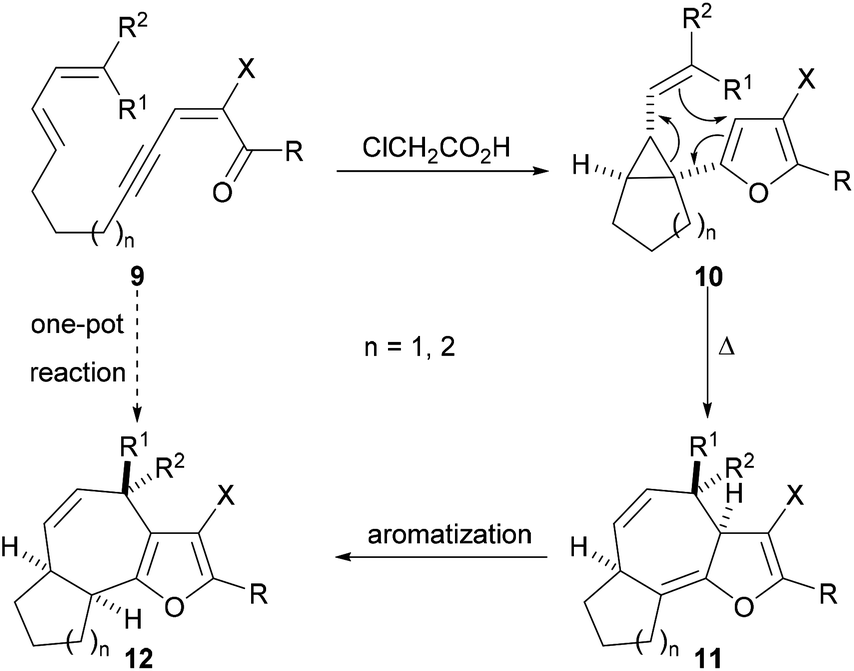 | ||
| Scheme 3 Proposed conversion of acyclic diene-tethered ynenones into highly functionalised fused tricyclic systems. | ||
There are just three examples of the thermal Cope rearrangement of 1-furanyl-2-vinylcyclopropanes.11,12 In 1980, Maas and Hummel described the rearrangement of the very simple substrates 13a and 13b to give the 3a,7-dihydro-4H-cyclohepta[b]furans 14a and 14b in low yield by heating them in toluene or deuterated benzene (Scheme 4).11 Aromatized 7,8-dihydro-4H-cyclohepta[b]furans 15a and 15b were obtained when toluene solutions of the substrates were heated at higher temperatures in a sealed tube. The only other example of the reaction is an unusual one published by Barluenga and co-workers, in which the unstable fused tricyclic lactone 17 was prepared by thermal rearrangement of the lactone-containing vinyl cyclopropane 16 at 85 °C.12 A systematic study of the scope of the thermal Cope rearrangement of 1-furanyl-2-vinylcyclopropanes has not been published and there are no examples of the use of the reaction to prepare more complex fused polycyclic systems from cyclopropyl-substituted furans that possess additional rings.
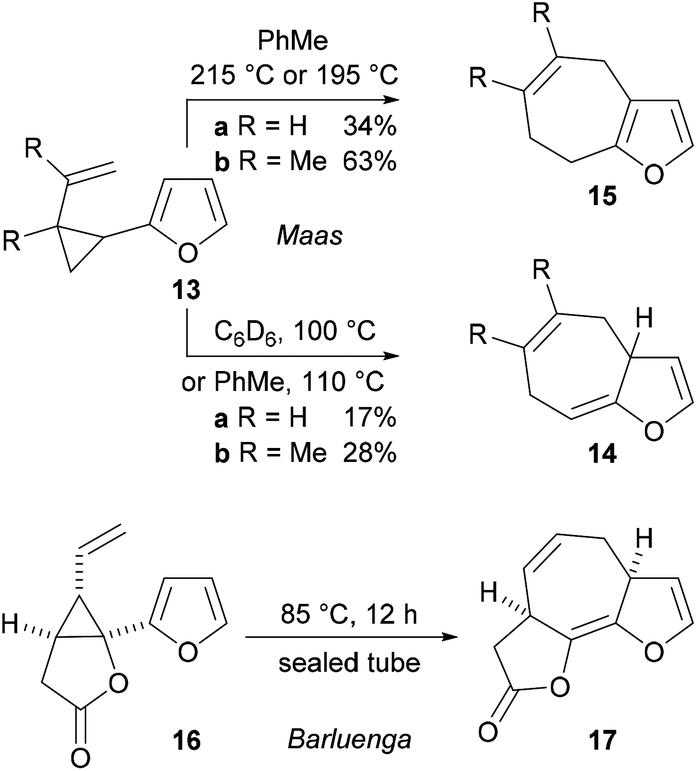 | ||
| Scheme 4 Previous examples of Cope rearrangement reactions of vinylcyclopropane-substituted furans. | ||
Results and discussion
Exploration of the Cope rearrangement reactions of vinylcyclopropane-substituted furans
Our studies commenced with the synthesis of the aldehyde 23, the key intermediate required for the preparation of the Cope rearrangement precursors (Scheme 5). The known alcohol 1813 was O-protected and the trimethylsilyl group was removed. The resulting terminal alkyne was then formylated to give the propargylic aldehyde 19. Knoevenagel condensation of the aldehyde 19 with acetylacetone produced the ynenedione 20 and this compound was then subjected to an acid-catalysed cascade cyclisation reaction to give the furan 21 in excellent yield and with high diastereoselectivity (>98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2). Acid-mediated cleavage of the t-butyldimethysilyl ether and oxidation of the resulting alcohol 22 with the Dess–Martin periodinane afforded the required aldehyde 23 in excellent yield.
:2). Acid-mediated cleavage of the t-butyldimethysilyl ether and oxidation of the resulting alcohol 22 with the Dess–Martin periodinane afforded the required aldehyde 23 in excellent yield.
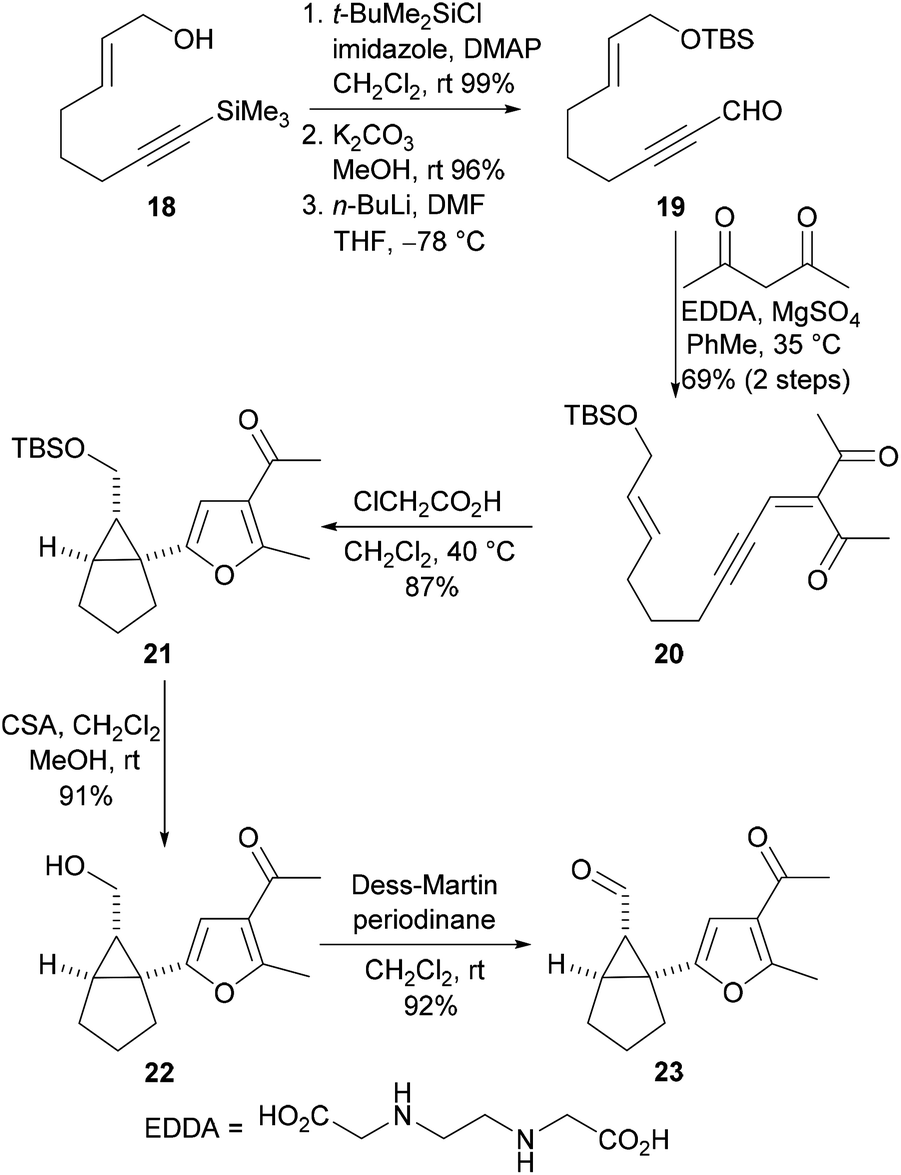 | ||
| Scheme 5 Synthesis of the tricyclic aldehyde 23. | ||
The substrates used in our study of the Cope rearrangement reaction were obtained by alkylidenation of the aldehyde 23 using a Wittig reaction (entries 1–4, Table 1) or a Julia–Kocienski reaction (entries 5–7, Table 1). The alkenes 24–27 were obtained with variable yields and with E:Z ratios that were dependent on the alkylidenation reaction that was employed.
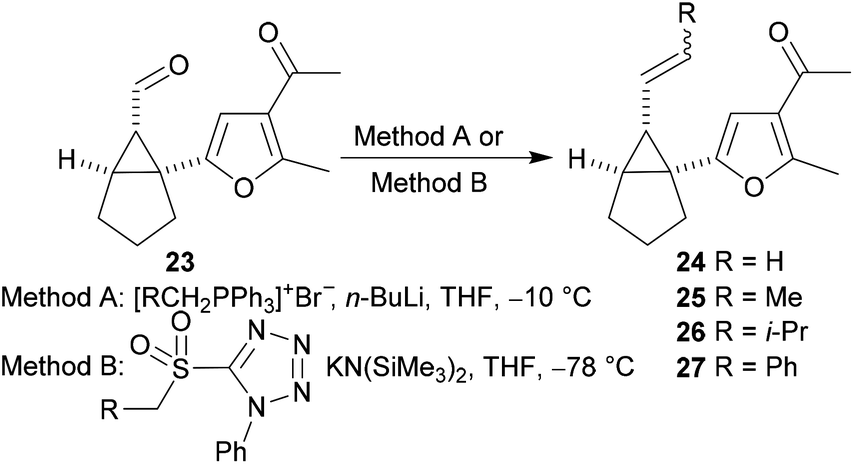
Rearrangement of the 1-furanyl-2-alkenylcyclopropane 24 was explored first. When a solution of the substrate was heated in toluene at reflux for 1 hour, the fused tricyclic product 28 was obtained as single diastereoisomer in 36% yield (entry 1, Table 2). Subsequent reactions were performed at lower temperatures, which resulted in reduced reaction rates and extended reaction times, but delivered higher yields of the product. The highest yield (63%) of the tricyclic product 28 was obtained when the reaction was performed in toluene at 40 °C (entry 2, Table 2). Inferior yields of this compound were obtained when rearrangement reactions performed in either THF or dichloromethane at 40 °C. Aromatisation to form the furan system did not occur at this reaction temperature.
|
|
|||||
|---|---|---|---|---|---|
| Entry | Substrate (E:Z ratio)a |
Time (h) | Product | Yieldb (%) | Recovered 24–27 (%) |
|
a Ratio determined by 1H NMR analysis.
b Yields of isolated compounds.
c Reaction performed at 110 °C.
d Only the Z isomer was recovered.
e 74:26 E:Z mixture of 25 recovered.
f 85:15 E:Z mixture of 26 recovered.
g 12:88 E:Z mixture of 27 recovered.
|
|||||
| 1c | 24 | 1 | 28 | 36 | — |
| 2 | 24 | 16 | 28 | 63 | — |
| 3 |
25 (27:73) |
18 | 29 | 23 | 40d |
| 4 |
25 (87:13) |
24 | 29 | 50 | 50e |
| 5 |
26 (12:88) |
36 | 30 | — | — |
| 6 |
26 (87:13) |
36 | 30 | 20 | 80f |
| 7 |
27 (47:53) |
18 | 31 | 43 | 23d |
| 8 |
27 (70:30) |
24 | 31 | 48 | 28g |
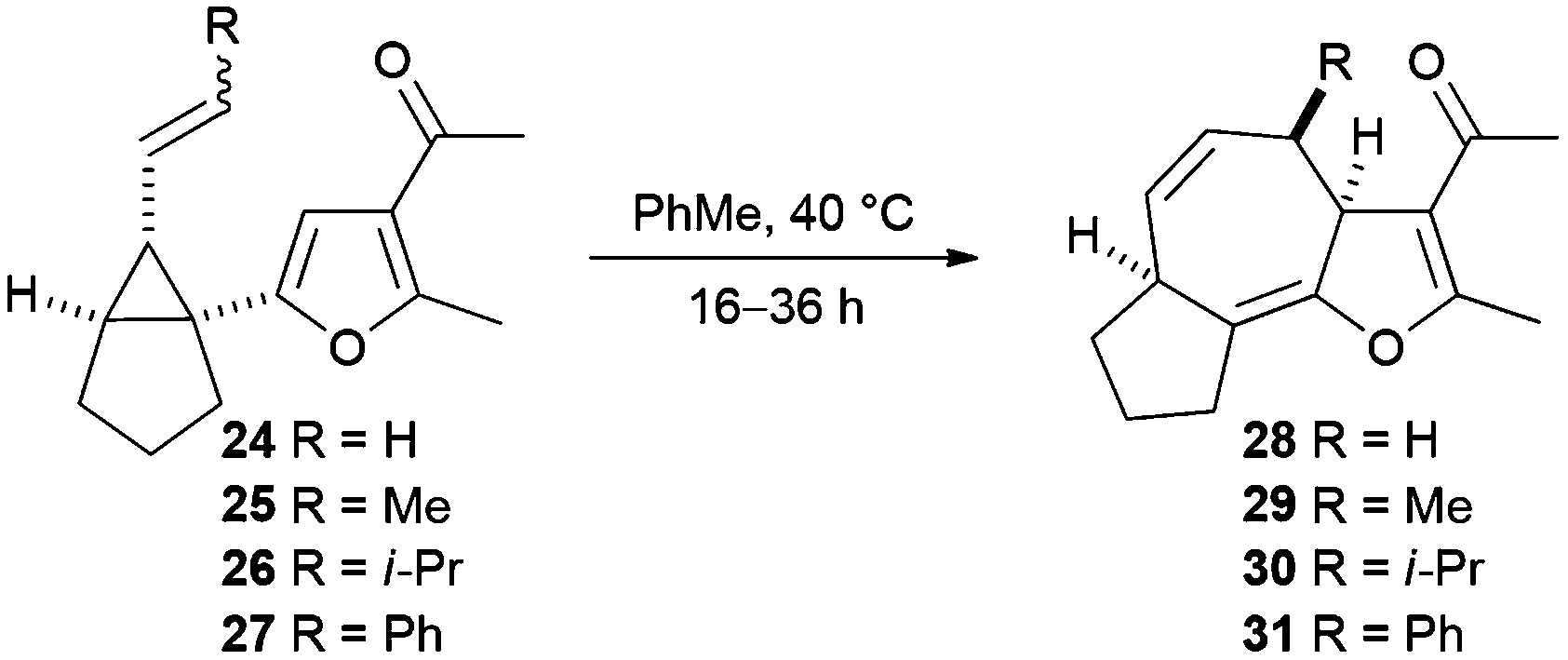
The Cope rearrangement reactions of substrates 25–27, which contain a non-terminal alkene, were then investigated (Table 2). The substrates – prepared and used as E/Z-mixtures – were dissolved in toluene and heated at 40 °C. Cope rearrangement of substrate 25 (27:73, E:Z), prepared by Wittig olefination, resulted in complete consumption of E-25 in 18 h to give the tricyclic product 29 in 23% yield;‡ unreacted Z-25 was recovered (40% mass recovery) and the product arising from rearrangement of this isomer was not observed (entry 3, Table 2). The substrate 25 (87:13, E:Z) prepared by Julia–Kocienski olefination underwent rearrangement to give the tricyclic product 29 in 50% yield and recovered starting material, enriched in Z-25 (72:28, E:Z), was recovered (50% mass recovery) (entry 4, Table 2). Rearrangement of the substrate 26 bearing a bulky isopropyl group was low yielding. Attempted rearrangement of a sample of the substrate 26 in which the Z isomer predominated (12:88, E:Z) failed to deliver the expected product and only starting material was recovered (entry 5, Table 2). The tricyclic rearrangement product 30 was obtained in 20% yield when substrate enriched in the E isomer (87:13, E:Z) was employed as the substrate and all of the unreacted starting material 26 (85:15, E:Z) was recovered after 36 h (entry 6, Table 2). The phenyl-substituted substrate 27 also underwent Cope rearrangement at 40 °C. A sample of the substrate 27 containing approximately equal amounts of both alkene isomers (47:53, E:Z) rearranged to give the cycloheptadiene 31 in 43% yield and unreacted isomer Z-27 was recovered without any evidence of Cope rearrangement of this isomer (entry 7, Table 2). When a sample of the substrate 27 enriched in the E isomer (70:30, E:Z) was subjected to the rearrangement conditions, the yield of the fused tricyclic product 31 increased to 48% and unreacted starting material (12:88, E:Z) was recovered (28% mass recovery) (entry 8, Table 2).
The stereochemical relationship between the C-3a and C-4 stereocentres was confirmed by analysis of 1H NMR data for the tricyclic ketone 31. Molecular modelling of the compounds 31 and epi-31 revealed an expected dihedral angle (for H–C–C–H) of 53° at C-3a and C-4 in the case of 31 and 175° in the case of epi-31. This would imply a coupling constant of 4 Hz between the protons for ketone 31 and 11–12 Hz for C-4 diastereomer (epi-31).14 The observed coupling between the protons is 3.7 Hz, an observation which confirms that we obtained the expected diastereomer from the reaction of the substrate E-27.
The results given in Table 2 reveal that increasing the size of the substituent (R) results in a lower rate of reaction; in the case of isopropyl-substituted substrate E-26, the reaction was incomplete even after 36 hours. The second important observation is that the Z isomers of substrates 25–27 are markedly less reactive than the corresponding E isomers; products arising from Cope rearrangement of the Z isomers were never isolated. This finding can be explained by consideration of the conformations leading to the respective transition states (Scheme 6). Cope rearrangement of cis-1,2-divinylcyclopropane is known to proceed through a boat-like transition state, resulting from an ‘endo/endo’ orientation of the alkenes in the diene precursor, to give (Z,Z)-1,4-cycloheptadiene.15 For the E isomers in our study, the alkyl group lies over the furan in the transition state (TS-E) and in the substrate conformation leading to that transition state. In contrast, for the Z isomers there is a highly unfavourable interaction between the alkene substituent (R) and the ring-junction hydrogen (Ha) as well as an eclipsing interaction between the alkene substituent (R) and the furan hydrogen (Hb) in both the transition state (TS-Z) and the conformation leading to that transition state. This means that TS-Z is higher in energy than TS-E and so rearrangement reactions of Z-25–27 to give the products 29–31 will only occur at significantly higher temperatures than the rearrangement reactions of E-25–27.
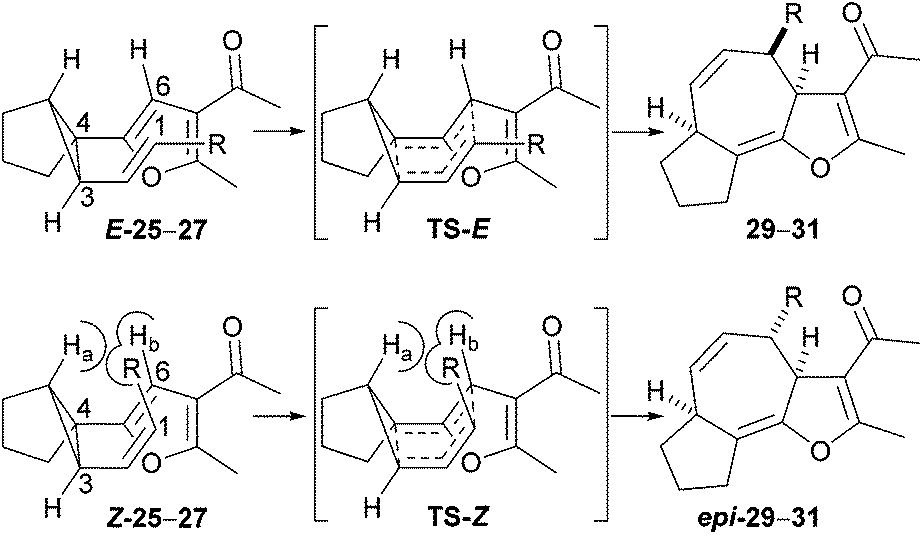 | ||
| Scheme 6 Transition states for Cope rearrangement of E and Z alkenyl cyclopropane isomers. | ||
A complicating feature of the reaction is that Cope rearrangement is potentially reversible. Normally, Cope rearrangement of a divinylcyclopropane to produce a 1,4-cycloheptadiene would favour formation of the larger ring because of relief of ring-strain upon opening of the cyclopropane. However, in the case of our reactions, the aromaticity of the furan is sacrificed during rearrangement and fused tricyclic products that possess a high degree of conformational rigidity are generated. Consequently, it is possible that unfavourable enthalpic and entropic factors in the products 29–31 could counter-balance the energy gain from relief of ring-strain in the substrates 25–27. It is even conceivable that reactions of the Z isomers are disfavoured thermodynamically, which would account for our failure to isolate the products expected from Cope rearrangement of these substrates.
In all of the cases studied, the 3a,4,6a,7,8,9-hexahydroazuleno[4,5-b]furans 29–31 were obtained rather than the aromatized 4,6a,7,8,9,9a-hexahydroazuleno[4,5-b]furans (cf. compound 12 in Scheme 3). This is unsurprising, given that Maas and Hummel found that reaction temperatures of approximately 200 °C are required to aromatize the 3a,7-dihydro-4H-cyclohepta[b]furans obtained from their Cope rearrangement reactions (Scheme 4).11
We attempted to prepare the 3a,4,6a,7,8,9-hexahydroazuleno-[4,5-b]furans 28–31 directly from the acyclic precursors in a one-pot fashion, as outlined in Scheme 3 (conversion of 9 into 12). In a preliminary study, the diene-containing cyclisation precursor 35 was prepared from the known α,β-unsaturated aldehyde 3213 as shown in Scheme 7. The diene 33 was prepared by alkylidenation of the aldehyde 32 with isopropylidene triphenylphosphorane. Alkyne desilylation followed by formylation then afforded the propargylic aldehyde 34. Knoevenagel reaction of the aldehyde 34 with acetylacetone produced the ynenedione 35 and subsequent acid-mediated cyclisation in dichloromethane at 40 °C afforded the 1-furanyl-2-vinylcyclopropane 36 in 59% yield. This Cope rearrangement precursor was also obtained by alkylidenation of the aldehyde 23 that we had prepared previously (Scheme 5).
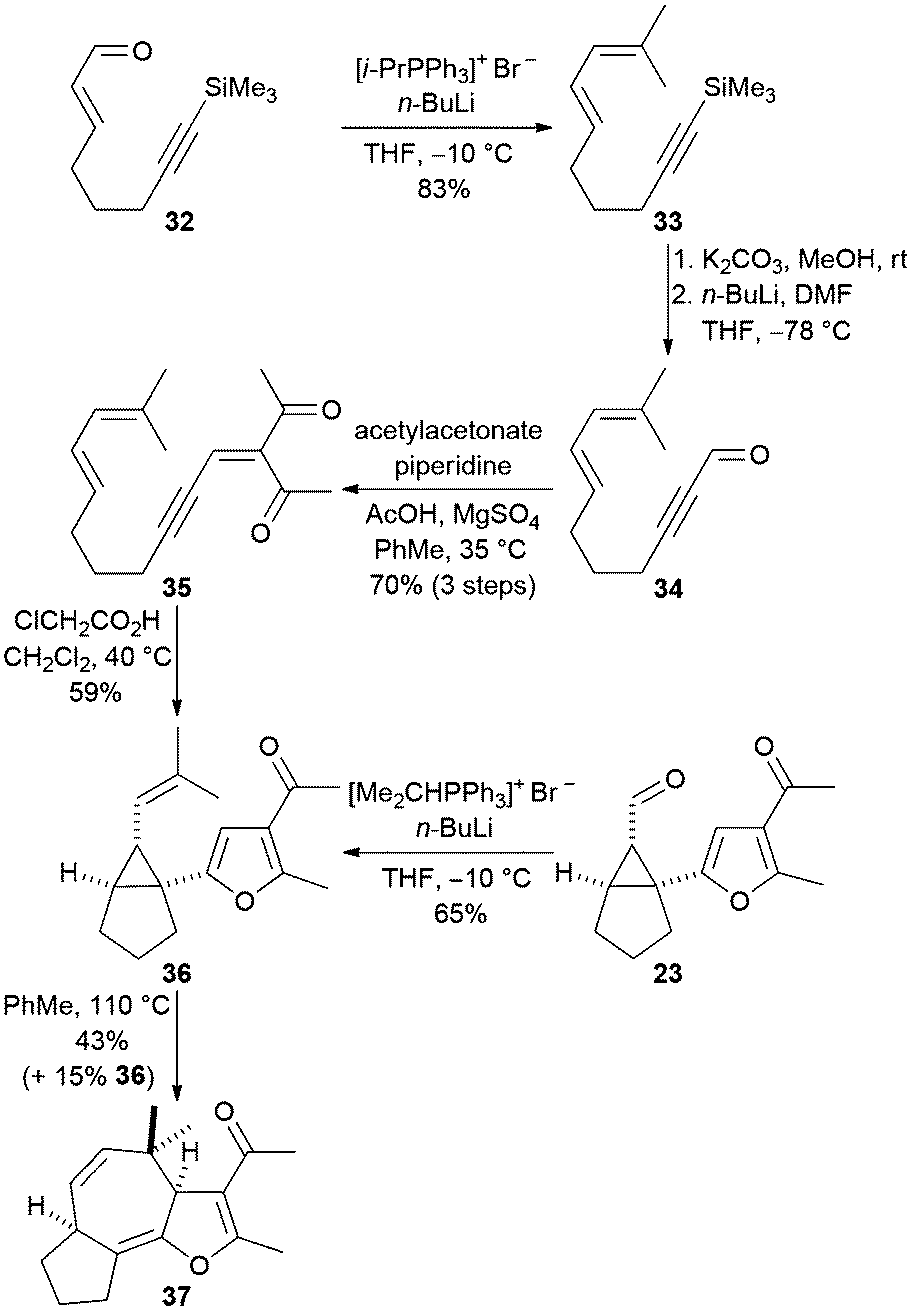 | ||
| Scheme 7 Direct acid-catalysed synthesis of the rearrangement precursor 27 from an acyclic diene-tethered ynenone. | ||
The trisubstituted alkene 36 did not undergo Cope rearrangement when heated at 40 °C as a solution in toluene and so the reaction temperature was increased to 110 °C. After a reaction time of three days, 15% of the starting material was recovered and the fused tricyclic product 37 was isolated in 43% yield (51% yield when recovered starting material is considered). Reducing the reaction time to 24 hours resulted in a lower conversion (32%) but a higher yield (68%) based on the amount of recovered starting material, a finding which suggests that either the substrate or product decomposes when heated at 110 °C for several days. Unfortunately, it was not possible to perform Knoevenagel condensation, acid-catalysed cyclisation and Cope rearrangement in a one-pot fashion to give the tricyclic ketone 37 directly from the aldehyde 34 because of the relatively high reaction temperature (110 °C) required to accomplish Cope rearrangement of the furan 36.
Attempts to perform acid-catalysed cyclisation reactions with other diene-containing ynenediones, analogous to the substrate 35, were hampered by the fact that the propargylic aldehydes bearing tethered dienes, required for the preceding Knoevenagel condensation reaction, underwent competitive intramolecular Diels–Alder cycloaddition and complex mixtures of products were obtained instead of the required ynenediones.
Computational study of the Cope rearrangement of the isomers of the vinylcyclopropane-substituted furan 25
DFT calculations were performed to explain the outcome of the reactions of substrates 25–27 and, specifically, to quantify the effects of the configuration of the 1-propenyl double bond of the substrate (E-25vs.Z-25) on the Cope rearrangement reaction to give the product 29 (see Scheme 6). The calculated energetic and structural parameters for reactants, transition states, and products of the Cope rearrangement are collated in Table 3. The rearrangement reaction proceeds as expected for a [3,3] sigmatropic process and is largely unaffected by the configuration of the alkene. The distinct double bonds (1–2, 5–6) and single bonds (2–3, 4–5) of the reactant equalise in the transition state and localise again into distinct single and double bonds, respectively, in the product. Concomitant cleavage of the cyclopropane bond 3–4 and formation of the new bond between C1 and C6, creates the cycloheptadiene.| ΔE | −TΔS | ΔG | d/Å | ||||||
|---|---|---|---|---|---|---|---|---|---|
| (kJ mol−1) | (kJ mol−1) | (kJ mol−1) | 1–2 | 2–3 | 3–4 | 4–5 | 5–6 | 6–1 | |
| E-25 | 0 | 0 | 0 | 1.33 | 1.48 | 1.53 | 1.46 | 1.35 | 3.55 |
| TS-E | 108 | 20 | 124 | 1.39 | 1.38 | 2.02 | 1.37 | 1.41 | 2.18 |
| 29 | −23 | 19 | −1 | 1.51 | 1.33 | 2.55 | 1.32 | 1.50 | 1.54 |
| Z-25 | 5 | 5 | 10 | 1.33 | 1.49 | 1.51 | 1.47 | 1.35 | 4.49 |
| TS-Z | 126 | 20 | 142 | 1.39 | 1.39 | 1.99 | 1.37 | 1.40 | 2.23 |
| epi-29 | −3 | 20 | 20 | 1.51 | 1.33 | 2.46 | 1.32 | 1.51 | 1.57 |
The configuration of the alkene has a minor effect on the relative stability of the reactants E/Z-25, disfavouring the Z isomer both enthalpically and entropically by 5 kJ mol−1 each. In Z-25, the 1-propenyl arm of the cyclopropane is rotated outwards about the single bond 2–3 so that unfavourable steric interactions of the terminal methyl group with the rest of the molecule are avoided, which results in the long 6–1 distance (Fig. 2). However, in the transition state, as the bond 2–3 acquires increasing double-bond character and the propenyl arm approaches the furan, the configuration of TS-Z results in steric clashes of the methyl group with the hydrogens Ha and Hb (see Scheme 6 and Fig. 2). In contrast, TS-E is sterically less encumbered, which is reflected in a lower enthalpic barrier for rearrangement of E-25 (Δ‡E of 108 vs. 121 kJ mol−1; barriers calculated relative to the energy of the respective starting compound). The entropic barrier is much smaller and similar for the two isomers (−TΔ‡S of 20 vs. 15 kJ mol−1). The more facile approach of C1 and C6 in TS-E is also reflected in a somewhat later transition state: the incipient C1–C6 bond is 0.05 Å shorter in TS-E than in TS-Z and the breaking C3–C4 bond is 0.03 Å longer.
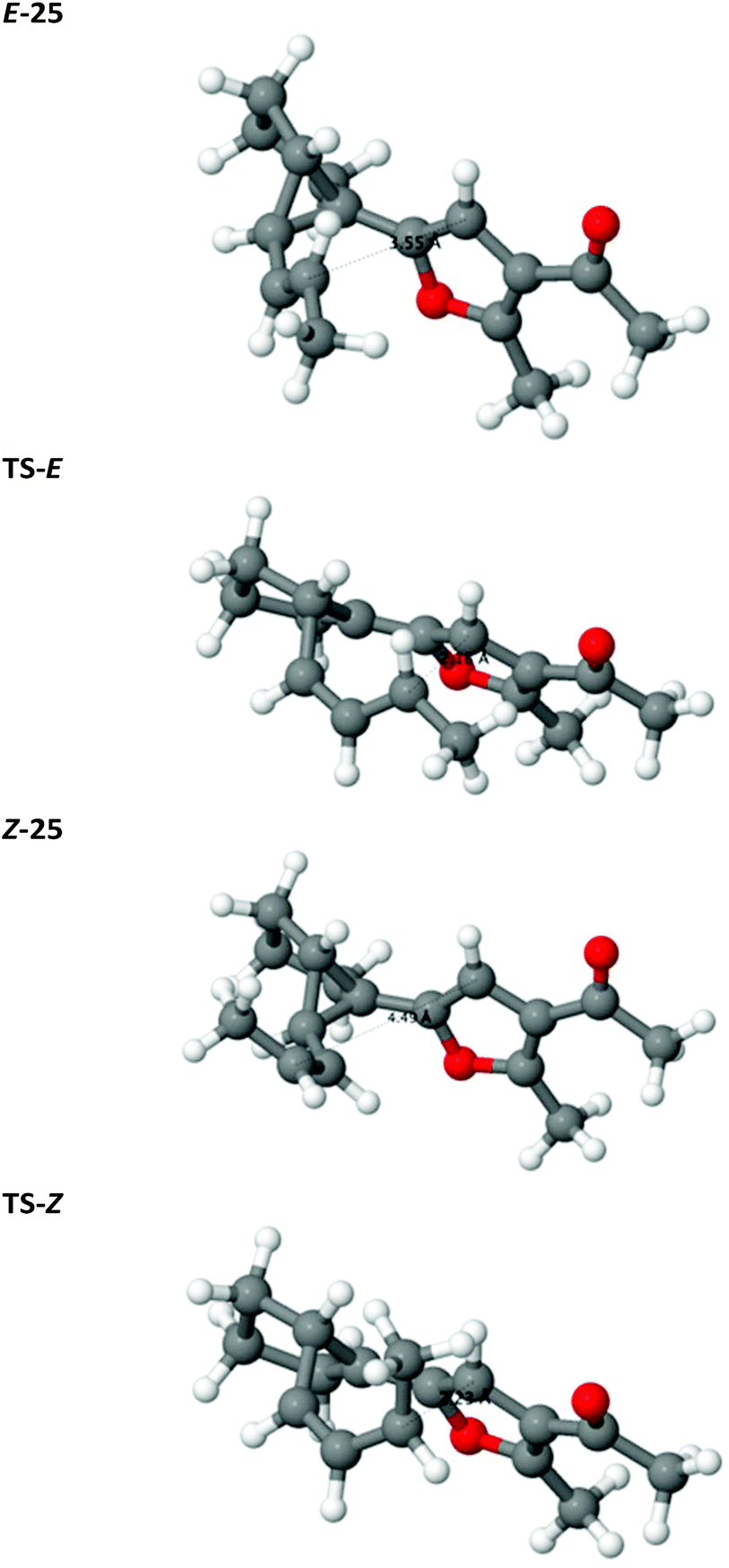 | ||
| Fig. 2 Optimised structures of the reactants and transition states of the Cope rearrangement reaction. | ||
The Gibbs free energy barrier is 8 kJ mol−1 lower for TS-E than TS-Z and so rearrangement of the E-25 is kinetically favoured. In terms of reaction rates, this difference in barrier corresponds to a 20-fold faster rate (at 40 °C) for the rearrangement of the E-25 relative to Z-25. The barrier is largely enthalpic and the entropic contribution is similar in both cases, so increasing the reaction temperature will not affect the kinetic selectivity significantly.
The tricyclic product 29, resulting from the rearrangement of E-25, is also favoured on thermodynamic grounds. The formation of this compound is thermoneutral at 40 °C, whereas the formation of epi-29 from Z-25 is endergonic by 10 kJ mol−1. The difference stems exclusively from the enthalpic part of the reaction free energy, which is exothermic. Because the product is conformationally rigid, the entropic penalty incurred during formation of the transition state remains ‘locked into’ the product. The entropic part of the reaction free energy is therefore unfavourable, of similar (or even larger) magnitude than the enthalpic part, and similar for the two isomers. A higher reaction temperature will thus shift the equilibrium further to the left, equally so for both isomers.
The near-zero, or even positive, reaction free energy means that when the temperature is sufficiently high for the forward reaction to proceed at a significant rate, so will the backward reaction; that is, reactant and product are in equilibrium. At 40 °C, the reaction free energies of −1 and +10 kJ mol−1 for 29 and epi-29, respectively, correspond to equilibrium constants of 2.6 and 0.02.
Conclusions
In summary, a new route for the stereoselective construction of hexahydroazuleno[4,5-b]furans from simple acyclic precursors has been developed that involves an acid-catalysed cyclisation reaction to give 1-furanyl-2-vinylcyclopropanes and their subsequent Cope rearrangement. We have shown that E-configured substrates undergo smooth Cope rearrangement at 40 °C but the corresponding Z-isomers do not rearrange at this temperature. A cascade procedure for the direct formation of the tricyclic products from acyclic ynenone precursors was investigated, but in most cases intermediates underwent intramolecular Diels–Alder cycloaddition prior to, or during, the Knoevenagel condensation reaction used to prepare the precursors required for the furan-forming reaction. The Cope rearrangement product 28 maps on the core structure found in sesquiterpene natural products of the guaianolide family, such as the lactones 38 and 39 (Fig. 3).16,17 Studies are in progress to apply this reaction to the synthesis of these and structurally related guaianolides.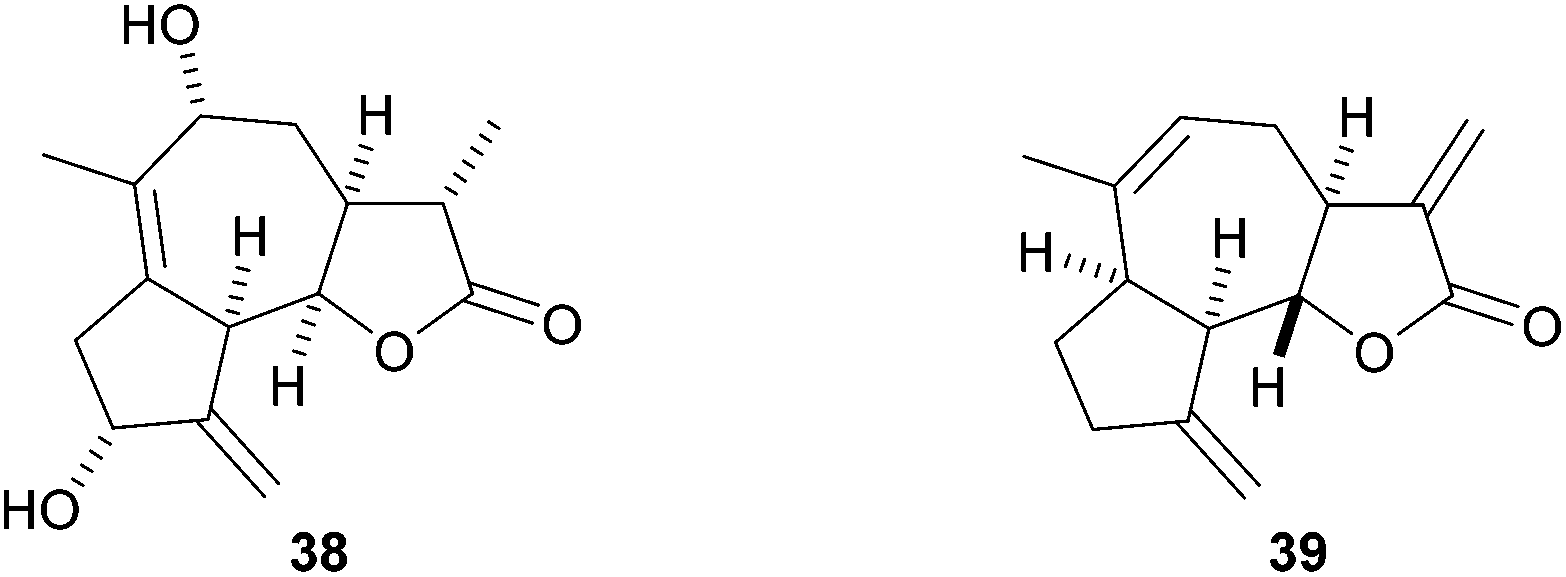 | ||
| Fig. 3 Potential guaianolide natural product targets 38 and eremanthine (39). | ||
Experimental section
Air or moisture-sensitive reactions were performed under an atmosphere of argon in flame-dried glassware.When required, tetrahydrofuran, toluene, dichloromethane and diethyl ether were dried using a Pure-Solv™ solvent purification system. Other dry organic solvents and reagents were purchased from commercial supplies and used without further purification unless otherwise specified.
Reactions were monitored by thin-layer chromatography (TLC) on Merck silica gel 60 plates. The TLC plates were visualised under UV light and stained with acidic ethanolic anisaldehyde solution or potassium permanganate solution.
Column chromatography was performed under forced flow with silica gel (Fluorochem LC60A, 35–70 micron or Merck Geduran Si 60, 40–63 micron). Petroleum ether used for column chromatography was the 40–60 °C fraction.
Melting points were recorded using an Electrothermal IA 9100 instrument.
IR spectra were recorded on a Shimadzu FT IR-8400S ATR instrument. The IR spectrum of each compound was acquired directly on a thin film (liquid) or powder (solid) at room temperature. 1H NMR and 13C NMR spectra were recorded using a Bruker Avance III 400 MHz or Bruker Avance III UltraShield 500 MHz spectrometer at ambient temperature. 13C NMR spectra were recorded at 101 MHz or 126 MHz.
High resolution mass spectrometry (HRMS) was performed by the analytical service of the University of Glasgow with an Jeol MStation JMS-700 instrument using positive chemical ionization (CI using isobutene) or a positive ion impact (EI) techniques, or on a Bruker micro TOFq High Resolution instrument using positive ion electrospray (ESI) techniques.
(E)-8-(Trimethylsilyl)-2-octen-7-yn-1-ol (18)13
To a stirred solution of the ethyl (E)-8-(trimethylsilyl)-2-octen-7-ynoate (10.03 g, 42.07 mmol) in dichloromethane (200 mL) was added diisobutylaluminium hydride (93 mL, 1.0 M solution in hexane, 93 mmol) dropwise at −78 °C. The reaction was stirred at −78 °C for 2 h and was then quenched by addition of methanol (50 mL) and of a saturated aqueous solution of Rochelle's salt (200 mL). The mixture was stirred for 3 h at room temperature and the phases were then separated. The aqueous phase was extracted with Et2O (2 × 150 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure to give alcohol 18 (8.24 g), which was used for the next step without further purification.(E)-1-(t-Butyldimethylsilyloxy)-8-(trimethylsilyl)-2-octen-7-yne
To a stirred solution of alcohol 18 (8.24 g, 42.0 mmol) in dichloromethane (130 mL) was added t-butyldimethylsilyl chloride (7.0 g, 46 mmol), imidazole (4.6 g, 68 mmol) and DMAP (50 mg, 0.41 mmol). The solution was stirred for one day at room temperature and the reaction was monitored by TLC. Further t-butyldimethylsilyl chloride (3 g, 0.02 mol), imidazole (2 g, 0.03 mol) and DMAP (10 mg, 0.082 mmol) were added and the solution was stirred for a further day. The reaction was quenched by addition of water (100 mL) and the phases were separated. The aqueous phase was extracted with dichloromethane (3 × 100 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The crude product was purified by flash column chromatography on silica gel (petroleum ether–Et2O, 99:1) to afford the title compound (13.0 g, 99% over 2 steps).
(E)-1-(t-Butyldimethylsilyloxy)-2-octen-7-yne
To a stirred solution of (E)-1-(t-butyldimethylsilyl-oxy)-8-(trimethylsilyl)-2-octen-7-yne (2.03 g, 6.54 mmol) in MeOH (65 mL) at rt was added K2CO3 (0.99 g, 7.2 mmol) in one portion. The mixture was stirred for 12 h and then the reaction was quenched by addition of water (70 mL). The mixture was diluted with Et2O (30 mL) and the phases were separated. The aqueous phase was extracted with Et2O (3 × 30 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 300:1) to afford (E)-1-(t-butyldimethylsilyloxy)-2-octen-7-yne (1.50 g, 96%) as a colourless oil. Rf = 0.04 (petroleum ether); 1H NMR (400 MHz, CDCl3) δ 5.66–5.53 (2H, m), 4.14–4.10 (2H, m), 2.20 (2H, td, J = 7.2, 2.7 Hz), 2.17–2.12 (2H, m), 1.95 (1H, t, J = 2.7 Hz), 1.61 (2H, tt, J = 7.2, 7.1 Hz), 0.90 (9H, s), 0.07 (6H, s); 13C NMR (101 MHz, CDCl3) δ 130.3, 130.0, 84.4, 68.5, 64.0, 31.2, 28.1, 26.1, 18.6, 18.0, −5.0; νmax (film) 3325, 2955, 2930, 2897, 1472, 833, 816, 773, 731, 667, 629 cm−1; HRMS (ESI) calcd for C14H26NaO3Si [M + Na]+ 261.1645, found 261.1640.
3-[(E)-9-(t-Butyldimethylsilyloxy)-7-nonen-2-yn-1-ylidene]-pentane-2,4-dione (20)
To a stirred solution of (E)-1-(t-butyl-dimethylsilyloxy)-2-octen-7-yne (2.30 g, 9.64 mmol) in THF (96 mL) at −78 °C was added n-BuLi (6.6 mL of a 2.2 M solution in hexanes, 15 mmol) over a period of 10 min. The mixture was stirred at −78 °C for 15 min and then anhydrous DMF (1.5 mL, 19 mmol) was added. The mixture was stirred at −78 °C for a further 30 min and then the reaction was quenched by addition of 10% aqueous KH2PO4 (100 mL). The mixture was diluted with EtOAc (50 mL), stirred for 10 min and the phases were separated. The aqueous phase was extracted with EtOAc (3 × 50 mL) and the combined organic extracts were washed with brine (80 mL), dried over MgSO4, filtered and concentrated under reduced pressure to afford crude propargylic aldehyde 19 as a yellow oil. The aldehyde was used directly in the next step without further purification.To a stirred solution of crude propargylic aldehyde 19 and acetylacetone (0.99 mL, 9.6 mmol) in toluene (10 mL) at rt were added MgSO4 (0.23 g, 1.9 mmol) and ethylenediamine-N,N′-diacetic acid (0.17 g, 0.96 mmol). The mixture was stirred at 35 °C for 1 h and then the reaction was quenched by addition of saturated aqueous NH4Cl (30 mL). The mixture was diluted with Et2O (20 mL) and the phases were separated. The aqueous phase was extracted with Et2O (2 × 10 mL) and the combined organic extracts were washed with brine (20 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford ynenedione 20 (2.33 g, 69% over 2 steps) as a pale yellow oil. Rf = 0.09 (petroleum ether–EtOAc, 10:1); 1H NMR (500 MHz, CDCl3) δ 6.68 (1H, t, J = 2.5 Hz), 5.63–5.52 (2H, m), 4.12–4.10 (2H, m), 2.45 (3H, s), 2.43 (2H, td, J = 7.2, 2.4 Hz), 2.30 (3H, s), 2.15–2.09 (2H, m), 1.64 (2H, tt, J = 7.2, 7.1 Hz), 0.89 (9H, s), 0.05 (6H, s); 13C NMR (126 MHz, CDCl3) δ 201.4, 195.9, 149.7, 130.7, 129.4, 123.3, 110.1, 77.2, 63.9, 31.3, 31.1, 27.7, 27.3, 26.1, 19.7, 18.6, −5.0, −5.0; νmax (film) 2930, 2833, 2208, 1718, 1692, 1667, 970, 835, 775 cm−1; HMRS (ESI) calcd for C20H32NaO3Si [M + Na]+ 371.2013, found 371.1997.
1-(5-{(1R*,5S*,6S*)-6-[(t-Butyldimethylsilyloxy)methyl]bicyclo-[3.1.0]hex-1-yl}-2-methyl-3-furanyl)ethanone (21)
To a stirred solution of ynenedione 20 (2.03 g, 5.82 mmol) in CH2Cl2 (23 mL) at rt was added chloroacetic acid (0.55 g, 5.8 mmol) in one portion. The mixture was stirred at 40 °C for 4 d and then concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford furan 21 (1.77 g, 87%) as a pale yellow oil. Rf = 0.45 (petroleum ether–EtOAc, 10:1); 1H NMR (500 MHz, CDCl3) δ 6.27 (1H, s), 3.60 (1H, dd, J = 11.0, 6.4 Hz), 3.44 (1H, dd, J = 11.0, 7.8 Hz), 2.53 (3H, s), 2.36 (3H, s), 2.13 (1H, dd, J = 12.5, 8.0 Hz), 1.90–1.80 (3H, m), 1.75–1.67 (1H, m), 1.53 (1H, dd, J = 4.0, 4.0 Hz), 1.38–1.29 (2H, m); 0.84 (9H, s), −0.03 (3H, s), −0.04 (3H, s); 13C NMR (126 MHz, CDCl3) δ 194.5, 157.0, 154.6, 122.0, 106.4, 62.8, 33.0, 30.7, 29.3, 29.0, 27.8, 27.5, 26.0, 22.0, 18.4, 14.6, −5.1, −5.2; νmax (film) 2955, 2928, 2863, 1678, 1570, 947, 835, 775 cm−1; HRMS (EI) calcd for C20H32O3Si [M]+ 348.2121, found 348.2125; anal. calcd for C20H32O3Si: C, 68.92%; H, 9.25%. Found: C, 68.71%; H, 9.11%.
1-(5-{(1R*,5S*,6S*)-6-(Hydroxymethyl)bicyclo[3.1.0]hex-1-yl}-2-methyl-3-furanyl)ethanone (22)
To a stirred solution of protected alcohol 21 (247 mg, 0.709 mmol) in MeOH/CH2Cl2 (v/v 5:2, 7 mL) at rt was added 10-camphorsulfonic acid (32 mg, 0.14 mmol) in one portion. The mixture was stirred for 1 h and then the reaction was quenched by addition of water (20 mL) and saturated aqueous NaHCO3 (5 mL). The mixture was diluted with Et2O (10 mL) and the phases were separated. The aqueous phase was extracted with Et2O (3 × 5 mL) and the combined organic extracts were washed with brine (2 × 5 mL), dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 2:1) to afford alcohol 22 (151 mg, 89%) as a colourless oil. Rf = 0.08 (petroleum ether–EtOAc, 2:1); 1H NMR (500 MHz, CDCl3) δ 6.30 (1H, s), 3.69 (1H, ddd, J = 11.5, 6.5, 6.5 Hz), 3.36 (1H, ddd, J = 11.5, 8.8, 3.3 Hz), 2.55 (3H, s), 2.37 (3H, s), 2.16 (1H, dd, J = 12.4, 8.0 Hz), 1.90–1.81 (3H, m), 1.77–1.70 (1H, m), 1.59 (1H, dd, J = 4.0, 4.0 Hz), 1.44–1.31 (3H, m); 13C NMR (126 MHz, CDCl3) δ 194.3, 157.2, 154.5, 122.2, 106.9, 62.8, 33.1, 30.6, 29.3, 29.2, 27.8, 27.4, 22.1, 14.6; νmax (film) 3410 (br), 2953, 2928, 2862, 1663, 1566, 947, 802, 675, 630 cm−1; HRMS (EI) calcd for C14H18O3 [M]+ 234.1256, found 234.1257.
(1R*,5S*,6S*)-1-(4-Acetyl-5-methyl-2-furanyl)bicyclo[3.1.0]-hexane-6-carboxaldehyde (23)
To a stirred solution of alcohol 22 (0.15 g, 0.64 mmol) in CH2Cl2 (7 mL) at 0 °C was added Dess–Martin periodinane (0.45 g, 1.1 mmol) in small portions. The mixture was stirred at rt for 16 h and then the reaction was quenched by sequential addition of saturated aqueous Na2S2O3 (10 mL) and saturated aqueous NaHCO3 (10 mL). The mixture was diluted with Et2O (10 mL), stirred until two clear layers were obtained (ca. 30 min) and the phases were separated. The aqueous phase was extracted with Et2O (3 × 10 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:3) to afford aldehyde 23 (0.14 g, 94%) as a pale yellow oil. Rf = 0.16 (petroleum ether–EtOAc, 10:3); 1H NMR (400 MHz, CDCl3) δ 9.03 (1H, d, J = 6.5 Hz), 6.37 (1H, s), 2.55 (1H, dd, J = 4.0, 3.9 Hz), 2.53 (3H, s), 2.36 (3H, s), 2.26 (1H, dd, J = 13.0, 8.4 Hz), 2.10–1.97 (3H, m), 2.05 (1H, dd, J = 6.5, 4.0 Hz), 1.81 (1H, ddd, J = 13.7, 8.4, 8.4 Hz), 1.33 (1H, ddddd, J = 13.7, 11.3, 11.3, 8.4, 8.4 Hz); 13C NMR (101 MHz, CDCl3) δ 199.7, 194.0, 157.6, 151.3, 122.2, 107.9, 37.5, 36.6, 33.5, 32.4, 29.3, 27.3, 20.9, 14.6; νmax (film) 2959, 2942, 2867, 2743, 1701, 1674, 1568, 949, 810, 631 cm−1; HRMS (ESI) calcd for C14H16NaO3 [M + Na]+ 255.0992, found 255.0991.
1-(5-{(1R*,5S*,6S*)-6-Ethenylbicyclo[3.1.0]hex-1-yl}-2-methyl-3-furanyl)ethanone (24)
To a stirred solution of methyltriphenylphosphonium bromide (0.21 g, 0.59 mmol) in THF (8 mL) at −10 °C was added n-BuLi (0.16 mL of a 2.3 M solution in hexanes, 0.37 mmol). The mixture was stirred at −10 °C for 1 h and then added to a stirred solution of aldehyde 23 (71 mg, 0.31 mmol) in THF (10 mL) at −10 °C. The mixture was stirred at rt for 2 h and then the reaction was quenched by pouring the solution into a mixture of pH 7 buffer (50 mL) and Et2O (20 mL). The mixture was stirred for 10 min and the phases were separated. The aqueous phase was extracted with Et2O (3 × 20 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on aluminium oxide (activated, basic Brockmann I, petroleum ether–EtOAc, 10:1) to afford vinylcyclopropane 24 (57 mg, 81%) as a colourless oil. Rf = 0.13 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, CDCl3) δ 6.25 (1H, s), 5.39 (1H, ddd, J = 17.1, 10.2, 9.3 Hz), 5.08 (1H, dd, J = 17.1, 1.8 Hz), 4.88 (1H, dd, J = 10.2, 1.8 Hz), 2.54 (3H, s), 2.37 (3H, s), 2.16 (1H, dd, J = 12.6, 8.4 Hz), 1.96–1.87 (3H, m), 1.81–1.78 (1H, m), 1.77–1.71 (1H, m), 1.74 (1H, dd, J = 4.0, 9.1 Hz), 1.44–1.32 (1H, m); 13C NMR (126 MHz, CDCl3) δ 194.5, 157.1, 154.4, 137.1, 122.1, 113.9, 106.5, 33.4, 32.7, 32.4, 30.2, 29.3, 27.5, 21.7, 14.6; νmax (film) 2955, 2926, 2862, 1678, 1570, 949, 889, 633 cm−1; HMRS (ESI) calcd for C15H18NaO2 [M + Na]+ 253.1199, found 253.1187.
1-(5-{(1R*,5S*,6S*)-6-(1-Propenyl)bicyclo[3.1.0]hex-1-yl}-2-methyl-3-furanyl)ethanone (E-25 and Z-25)
:1) to afford an inseparable mixture of E-25 and Z-25 (49 mg, 79%; 27:73 E:Z) as a colourless oil.
:1) to afford an inseparable mixture of E-25 and Z-25 (85 mg, 51%; 87:13, E:Z) as a colourless oil. Rf = 0.16 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, C6D6) δ 6.18 (1H, s), 6.18 (1H, s), 5.50 (1H, dq, J = 15.2, 6.5 Hz), 5.44 (1H, dqd, J = 10.8, 6.8, 1.0 Hz), 5.25–5.21 (1H, m), 5.19 (1H, ddq, J = 10.8, 9.5, 1.7 Hz), 2.37 (3H, s), 2.37 (3H, s), 2.11 (1H, ddd, J = 12.5, 8.2, 0.6 Hz), 2.08 (1H, dd, J = 8.0, 4.5, 1.0 Hz), 1.94–1.84 (4H, m), 2.02 (3H, s), 2.02 (3H, s), 1.73–1.67 (6H, m), 1.64 (3H, dd, J = 6.8, 1.7 Hz), 1.54 (3H, dd, J = 6.5, 1.6 Hz), 1.51–1.43 (1H, m), 1.51–1.43 (1H, m), 1.22–1.07 (1H, m), 1.22–1.07 (1H, m); 13C NMR (126 MHz, C6D6) δ 192.6, 192.6, 156.7, 156.6, 154.7, 154.6, 129.8, 129.2, 125.0, 123.9, 122.6, 122.5, 106.9, 106.8, 33.6, 33.2, 33.1, 33.0, 33.0, 32.1, 29.5, 28.9, 27.8, 27.7, 24.7, 22.1, 22.0, 18.2, 14.3, 14.3, 13.5; νmax (film) 3023, 2955, 2933, 2862, 1676, 1569, 949, 730, 632 cm−1; HMRS (ESI) calcd for C16H20NaO2 [M + Na]+ 267.1356, found 267.1343.
1-(5-{(1R*,5S*,6S*)-6-(3-Methyl-1-buten-1-yl)bicyclo[3.1.0]hex-1-yl}-2-methyl-3-furanyl)-ethanone (E-26 and Z-26)
:4) to afford an inseparable mixture of E-26 and Z-26 (38 mg, 30%; 12:88 E:Z) as a colourless oil.
:3) to afford an inseparable mixture of E-26 and Z-26 (20 mg, 17%; 87:13, E:Z) as a colourless oil. E-261H NMR (400 MHz, C6D6) δ 6.18 (1H, s), 5.49 (1H, dd, J = 15.4, 6.8 Hz), 5.20 (1H, ddd, J = 15.4, 8.4, 1.0 Hz), 2.40 (3H, s), 2.22–2.11 (1H, m), 2.09–2.04 (1H, m), 2.04 (3H, s), 1.93–1.85 (1H, m), 1.75–1.64 (4H, m), 1.52–1.43 (1H, m), 1.19–1.06 (1H, m), 0.92 (3H, d, J = 6.7 Hz), 0.91 (3H, d, J = 6.7 Hz); Z-261H NMR (400 MHz, C6D6) δ 6.19 (1H, s), 5.27 (1H, ddd, J = 10.8, 9.2, 1.0 Hz), 5.07 (1H, ddd, J = 10.7, 9.3, 1.1 Hz), 2.73 (1H, dqqd, J = 9.2, 6.7, 6.6, 1.0 Hz), 2.37 (3H, s), 2.11 (1H, ddd, J = 12.6, 8.3, 0.9 Hz), 2.02 (3H, s), 1.93–1.85 (2H, m), 1.74–1.66 (3H, m), 1.53–1.45 (1H, m), 1.22–1.11 (1H, m), 0.99 (3H, d, J = 6.6 Hz), 0.97 (3H, d, J = 6.7 Hz); E-2613C NMR (101 MHz, C6D6) δ 192.6, 156.5, 154.6, 137.9, 125.8, 122.5, 107.0, 33.3, 32.9, 32.2, 31.6, 29.1, 28.8, 27.7, 23.0, 22.9, 22.0, 14.3; Z-2613C NMR (101 MHz, C6D6) δ 192.6, 156.7, 154.5, 137.8, 126.1, 122.5, 106.9, 33.6, 33.2, 33.0, 28.8, 27.7, 27.5, 25.0, 23.4, 22.1, 14.3; νmax (film) 2955, 2930, 2864, 1676, 1570, 964, 945, 891, 802, 673, 633 cm−1; HMRS (ESI) calcd for C18H24NaO2 [M + Na]+ 295.1669, found 295.1661.
1-(5-{(1R*,5S*,6S*)-6-(2-Phenylethenyl)bicyclo[3.1.0]hex-1-yl}-2-methyl-3-furanyl)ethanone (E-27 and Z-27)
:1) to afford an inseparable mixture of E-27 and Z-27 (60 mg, 91%; 47:53, E:Z) as a colourless oil.
:5) to afford an inseparable mixture of E-27 and Z-27 (27 mg, 41%; 70:30, E:Z) as a colourless oil. Rf = 0.32 (petroleum ether–EtOAc); 1H NMR (400 MHz, C6D6) δ 7.43 (2H, d, J = 7.6 Hz), 7.23 (2H, d, J = 7.9), 7.22 (2H, dd, J = 7.6, 7.9 Hz), 7.10–7.07 (3H, m), 6.98 (1H, t, J = 7.4 Hz), 6.45 (1H, d, J = 15.8 Hz), 6.44 (1H, d, J = 11.4 Hz), 6.23 (1H, s), 6.22 (1H, s), 6.00 (1H, dd, J = 15.8, 9.2 Hz), 5.43 (1H, dd, J = 11.4, 9.3 Hz), 2.28 (3H, s), 2.36 (3H, s), 2.18 (1H, dd, J = 9.3, 4.2 Hz), 2.09 (1H, ddd, J = 12.6, 8.2, 5.6 Hz), 2.08 (1H, ddd, J = 12.6, 8.2, 5.6 Hz), 2.03 (3H, s), 1.99 (3H, s), 1.90 (1H, ddd, J = 12.6, 11.5, 8.6 Hz), 1.88 (1H, ddd, J = 12.6, 11.5, 8.6 Hz), 1.79–1.76 (2H, m), 1.75–1.71 (3H, m), 1.69–1.64 (2H, m), 1.52–1.46 (1H, m), 1.43–1.37 (1H, m), 1.21–1.12 (1H, m), 1.07–0.98 (1H, m); 13C NMR (101 MHz, C6D6) δ 192.6, 192.5, 156.8, 156.7, 154.3, 154.2, 138.2, 138.2, 130.9, 130.1, 129.6, 129.4, 129.2, 128.9, 128.6, 127.1, 127.0, 126.1, 122.6, 122.6, 107.2, 107.0, 34.5, 34.3, 34.1, 33.2, 33.1, 32.6, 30.2, 28.9, 28.8, 27.8, 27.6, 26.7, 21.9, 21.9, 14.3, 14.2; νmax (film) 2957, 2932, 2862, 1676, 1568, 945, 799, 767, 748, 694, 673, 633 cm−1; HMRS (ESI) calcd for C21H22NaO2 [M + Na]+ 329.1512, found 329.1502.
{(3aS*,7S*)-3a,4,6a,7,8,9-Hexahydro-2-methylazuleno[4,5-b]-furan-3-yl}ethanone (28)
A solution of vinylcyclopropane 24 (70 mg, 0.30 mmol) in toluene (6 mL) was stirred at 40 °C for 16 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford cycloheptadiene 28 (44 mg, 63%) as a pale yellow oil. Rf = 0.49 (petroleum ether–EtOAc, 10:3); 1H NMR (400 MHz, C6D6) δ 5.50–5.46 (2H, m), 4.18–4.14 (1H, m), 3.23–3.15 (1H, m), 2.77–2.68 (1H, m), 2.57–2.49 (1H, m), 2.44–2.33 (1H, m), 2.08–1.97 (1H, m), 1.89 (3H, d, J = 1.4 Hz), 1.85 (3H, s), 1.84–1.79 (1H, m), 1.60–1.53 (1H, m), 1.32–1.23 (2H, m); 13C NMR (126 MHz, C6D6) δ 192.0, 164.8, 150.4, 131.2, 126.4, 118.6, 117.1, 44.2, 42.0, 36.4, 32.0, 29.4, 29.1, 25.0, 14.7; νmax (film) 2953, 2935, 1744, 1668, 1618, 991, 949 cm−1; HMRS (ESI) calcd for C15H18NaO2 [M + Na]+ 253.1199, found 253.1187.
{(3aS*,4S*,7S*)-3a,4,6a,7,8,9-Hexahydro-2,4-dimethylazuleno[4,5-b]furan-3-yl}ethanone (29)
An isomeric mixture of the vinylcyclopropanes 25 (35 mg, 0.14 mmol; 27:73, E:Z) in toluene (4 mL) was stirred at 40 °C for 18 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford cycloheptadiene 29 (7.9 mg, 23%) as a colourless gum and recovered Z-25 (14 mg, 40%).
An isomeric mixture of the vinylcyclopropanes 25 (22 mg, 0.090 mmol; 87:13, E:Z) in toluene (2.6 mL) was stirred at 40 °C for 24 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford cycloheptadiene 29 (11 mg, 50%) as a colourless gum and recovered starting material 25 (11 mg, 50%; 74:26, E:Z). Rf = 0.45 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, C6D6) δ 5.58 (1H, ddd, J = 12.2, 6.8, 2.5 Hz), 5.43 (1H, dd, J = 12.2, 2.1 Hz), 4.38–4.33 (1H, m), 3.15–3.07 (1H, m), 2.86–2.80 (1H, m), 2.60–2.51 (1H, m), 2.46–2.35 (1H, m), 1.91 (3H, d, J = 1.4 Hz), 1.89 (3H, s), 1.83–1.76 (1H, m), 1.61–1.53 (1H, m), 1.29–1.19 (2H, m), 0.93 (3H, d, J = 7.0 Hz); 13C NMR (101 MHz, C6D6) δ 193.3, 164.0, 150.7, 134.3, 130.6, 118.7, 116.9, 49.8, 41.5, 39.3, 36.1, 29.4, 29.3, 25.2, 21.4, 14.3; νmax (film) 2955, 2928, 2860, 1744, 1670, 1651, 1610, 991, 951, 717 cm−1; HMRS (ESI) calcd for C16H20NaO2 [M + Na]+ 267.1356, found 267.1345.
{(3aS*,4S*,7S*)-3a,4,6a,7,8,9-Hexahydro-4-isopropyl-2-methylazuleno[4,5-b]furan-3-yl}-ethanone (30)
An isomeric mixture of the vinylcyclopropanes 26 (20 mg, 0.067 mmol; 87:13, E:Z) in toluene (2.1 mL) was stirred at 40 °C for 36 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 85:15) to afford cycloheptadiene 30 (4 mg, 20%) as a colourless gum and recovered starting material (16 mg, 80%; 85:15 E:Z). Rf = 0.25 (petroleum ether–EtOAc, 85:15); 1H NMR (400 MHz, C6D6) δ 5.65 (1H, dd, J = 12.7, 2.2 Hz), 5.57 (1H, ddd, J = 12.7, 6.3, 2.2 Hz), 4.47–4.41 (1H, m), 3.19–3.10 (1H, m), 3.02–2.97 (1H, m), 2.57–2.50 (1H, m), 2.35–2.23 (1H, m), 1.91 (3H, d, J = 1.3 Hz), 1.90 (3H, s), 1.89–1.80 (2H, m), 1.62–1.53 (1H, m), 1.28–1.20 (2H, m), 0.94 (3H, d, J = 7.1 Hz), 0.92 (3H, d, J = 6.8 Hz); 13C NMR (101 MHz, C6D6) δ 192.0, 164.8, 148.5, 131.9, 127.4, 117.2, 116.8, 47.6, 43.4, 42.3, 36.6, 29.4, 29.4, 29.3, 24.8, 23.6, 19.0, 14.7; νmax (film) 2955, 2928, 2870, 2857, 1745, 1666, 1645, 1620, 986, 974, 956, 943, 918, 885, 736 cm−1; HMRS (ESI) calcd for C18H24NaO2 [M + Na]+ 295.1669, found 295.1656.
{(3aS*,4S*,7S*)-3a,4,6a,7,8,9-Hexahydro-2-methyl-4-phenylazuleno[4,5-b]furan-3-yl}ethanone (31)
An isomeric mixture of the vinylcyclopropanes 27 (60 mg, 0.20 mmol; 47:53, E:Z) in toluene (6 mL) was stirred at 40 °C for 16 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford cycloheptadiene 31 (26 mg, 43%) as a colourless gum and recovered Z-27 (14 mg, 23%).
An isomeric mixture of the vinylcyclopropanes 27 (25 mg, 0.082 mmol; 70:30, E:Z) in toluene (6 mL) was stirred at 40 °C for 24 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford cycloheptadiene 31 (12 mg, 48%) as a colourless gum and recovered starting material 27 (7 mg, 28%; 12:88, E:Z). Rf = 0.45 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, C6D6) δ 7.29–7.25 (2H, m), 7.16–7.12 (2H, dd, m), 7.06 (1H, tt, J = 7.3, 1.3 Hz), 5.66 (1H, ddd, J = 12.2, 6.0, 2.3 Hz), 5.61 (1H, dd, J = 12.2, 1.6, 0.8 Hz), 4.67–4.64 (1H, m), 4.35 (1H, dt, J = 6.0, 3.7 Hz), 3.24–3.21 (1H, m), 2.57–2.50 (1H, m), 2.46–2.36 (1H, m), 1.93 (3H, s), 1.90–1.84 (1H, m), 1.64–1.57 (1H, m), 1.47 (3H, d, J = 1.4 Hz), 1.38–1.24 (2H, m); 13C NMR (126 MHz, C6D6) δ 191.9, 164.6, 147.8, 140.9, 131.0, 130.0, 129.8, 127.9, 126.9, 117.6, 117.3, 49.4, 44.3, 41.8, 36.5, 29.4, 29.4, 25.2, 14.4; νmax (film) 2955, 2924, 2855, 1745, 1620, 955, 760, 702 cm−1; HMRS (EI) calcd for C21H22O2 [M]+ 306.1620, found 306.1623.
[(E)-9-Methyl-6,8-decadien-1-yn-1-yl]trimethylsilane (33)
To a stirred solution of isopropyltriphenylphosphonium iodide (2.14 g, 4.95 mmol) in THF (20 mL) at −10 °C was added n-BuLi (1.6 mL of a 2.2 M solution in hexanes, 3.5 mmol). The mixture was stirred at −10 °C for 2 h and then added to a stirred solution of aldehyde (E)-8-trimethylsilyl-2-octen-7-ynal (32)13 (534 mg, 2.75 mmol) in THF (20 mL) at −10 °C. The mixture was stirred for 1 h and then the reaction was quenched by addition of brine (40 mL). The mixture was diluted with Et2O (30 mL) and the phases were separated. The aqueous phase was extracted with Et2O (3 × 20 mL). The combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 300:1) to afford diene 33 (501 mg, 83%) as a pale yellow oil. Rf = 0.43 (petroleum ether); 1H NMR (500 MHz, CDCl3) δ 6.24 (1H, ddt, J = 15.0, 10.8, 1.3 Hz), 5.78 (1H, d, J = 10.8 Hz), 5.50 (1H, dt, J = 15.0, 7.2 Hz), 2.22 (2H, t, J = 7.2 Hz), 2.18 (2H, br dt, J = 7.2, 7.1 Hz), 1.75 (3H, s), 1.73 (3H, s), 1.60 (2H, tt, J = 7.2, 7.1 Hz) 0.14 (9H, s); 13C NMR (126 MHz, CDCl3) δ 133.0, 130.5, 127.7, 125.1, 107.3, 84.7, 31.9, 28.5, 26.0, 19.3, 18.3, 0.3; νmax (film) 3025, 2965, 2863, 2174, 957, 839, 758, 698, 636 cm−1; HMRS (CI, isobutane) calcd for C14H25Si [M + H]+ 221.1726, found 221.1726.
(E)-9-Methyl-6,8-decadien-1-yne
To a stirred solution of protected alkyne 33 (575 mg, 2.61 mmol) in MeOH (13 mL) at rt was added K2CO3 (361 mg, 2.61 mmol) in one portion. The mixture was stirred for 12 h and then the reaction was quenched by addition of water (20 mL). The mixture was diluted with Et2O (10 mL) and the phases were separated. The aqueous phase was extracted with Et2O (3 × 5 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was filtered through a small pad of silica gel (petroleum ether–EtOAc, 300:1) to afford (E)-9-methyldeca-6,8-dien-1-yne as a colourless oil. The volatile dienyne (355 mg) was used directly in the next step without further purification. Rf = 0.33 (petroleum ether); 1H NMR (400 MHz, CDCl3) δ 6.25 (1H, ddt, J = 15.0, 10.8, 1.2 Hz), 5.79 (1H, d, J = 10.8 Hz), 5.51 (1H, dt, J = 15.0, 7.1 Hz), 2.24–2.16 (2H, m), 2.20 (2H, m), 1.95 (1H, t, J = 2.7 Hz), 1.76 (3H, s), 1.74 (3H, s), 1.62 (2H, tt, J = 7.2, 7.1 Hz).
3-[(E)-10-Methyl-7,9-undecadien-2-yn-1-ylidene]pentane-2,4-dione (35)
To a stirred solution of (E)-9-methyl-6,8-decadien-1-yne (355 mg) in THF (24 mL) at −78 °C was added n-BuLi (1.6 mL of a 2.1 M solution in hexanes, 3.6 mmol) over a period of 10 min. The mixture was stirred at −78 °C for 15 min and then anhydrous DMF (0.37 mL, 4.8 mmol) was added. The mixture was stirred at −78 °C for a further 30 min and then the reaction was quenched by addition of 10% aqueous KH2PO4 solution (50 mL). The mixture was diluted with Et2O (20 mL), stirred for 10 min and the phases were separated. The aqueous phase was extracted with Et2O (3 × 20 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure to afford crude propargylic aldehyde 34 as a yellow oil. The reactive aldehyde was used directly in the next step without further purification. Rf = 0.39 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, CDCl3) δ 9.10 (1H, t, J = 0.9 Hz), 6.26 (1H, ddt, J = 15.0, 10.8, 1.2 Hz), 5.78 (1H, d, J = 10.8 Hz), 5.48 (1H, dt, J = 15.0, 7.1 Hz), 2.42 (2H, td, J = 7.2, 0.8 Hz), 2.21 (2H, dt, J = 7.2, 7.1 Hz), 1.76 (3H, s), 1.74 (3H, s), 1.69 (2H, tt, J = 7.2, 7.2 Hz).
To a stirred solution of crude (E)-10-methyl-7,9-undecadien-2-ynal (34) and acetylacetone (0.23 mL, 2.3 mmol) in toluene (24 mL) at rt were added MgSO4 (58 mg, 0.48 mmol), piperidine (21 μL, 0.21 mmol), acetic acid (0.10 mL, 1.7 mmol). The mixture was stirred at 35 °C for 1 h and then the reaction was quenched by addition of water (50 mL). The mixture was diluted with EtOAc (25 mL) and the phases were separated. The aqueous phase was extracted with EtOAc (3 × 20 mL) and the combined organic extracts were dried over MgSO4, filtered and concentrated under reduced pressure. The residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford ynenedione 35 (469 mg, 70% over 3 steps) as a pale yellow oil. Rf = 0.13 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, CDCl3) δ 6.69 (1H, t, J = 2.5 Hz), 6.24 (1H, ddt, J = 15.0, 10.8, 1.2 Hz), 5.77 (1H, d, J = 10.8 Hz), 5.48 (1H, dt, J = 15.0, 7.2 Hz), 2.47 (3H, s), 2.44 (2H, td, J = 7.1, 2.5 Hz), 2.31 (3H, s), 2.18 (2H, dtd, J = 7.2, 7.1, 1.2 Hz), 1.75 (3H, s), 1.73 (3H, s), 1.65 (2H, tt, J = 7.1, 7.1 Hz); 13C NMR (101 MHz, CDCl3) δ 201.4, 195.9, 149.7, 133.8, 129.8, 128.2, 124.9, 123.3, 110.2, 77.2, 32.0, 31.1, 28.1, 27.4, 26.0, 19.8, 18.4; νmax (film) 3023, 2928, 2865, 2211, 1715, 1690, 1665, 1576, 988, 959 cm−1; HMRS (ESI) calcd for C17H22NaO2 [M + Na]+ 281.1512, found 281.1501.
1-(5-{(1R*,5S*,6S*)-6-(2-Methyl-1-propen-1-yl)bicyclo[3.1.0]-hex-1-yl}-2-methyl-3-furanyl)-ethanone (36)
:1) to afford vinylcyclopropane 36 (139 mg, 65%) as a colourless oil.
:1) to afford vinylcyclopropane 36 (25 mg, 59%) as a colourless oil. Rf = 0.48 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, C6D6) δ 6.19 (1H, s), 4.98 (1H, dqq, J = 8.7, 1.3, 1.2 Hz), 2.38 (3H, s), 2.13 (1H, dd, J = 12.4, 8.4, 0.6 Hz), 2.01 (3H, s), 1.91 (1H, ddd, J = 12.4, 11.4, 8.4 Hz), 1.81 (1H, dd, J = 8.7, 4.1 Hz), 1.76–1.71 (2H, m), 1.69–1.65 (1H, m), 1.64 (3H, d, J = 1.2 Hz), 1.59 (3H, d, J = 1.3 Hz), 1.55–1.46 (1H, m), 1.27–1.12 (1H, m); 13C NMR (101 MHz, C6D6) δ 192.6, 156.6, 154.9, 132.1, 123.3, 122.6, 106.7, 33.3, 33.1, 33.0, 28.8, 27.9, 25.8, 25.7, 22.2, 18.5, 14.3; νmax (film) 3025, 2959, 2926, 2864, 1677, 1570, 949, 635 cm−1; HMRS (ESI) calcd for C17H22NaO2 [M + Na]+ 281.1512, found 281.1499.
{(3aS*,7S*)-3a,4,6a,7,8,9-Hexahydro-2,4,4-trimethylazuleno[4,5-b]furan-3-yl}ethanone (37)
A solution of vinylcyclopropane 36 (48 mg, 0.19 mmol) in toluene (4 mL) was stirred at 110 °C for 16 h. The mixture was concentrated under reduced pressure and the residue was purified by flash column chromatography on silica gel (petroleum ether–EtOAc, 10:1) to afford cycloheptadiene 37 (13 mg, 27%, 68% brsm) as a colourless gum. Rf = 0.45 (petroleum ether–EtOAc, 10:1); 1H NMR (400 MHz, C6D6) δ 5.36 (1H, dd, J = 12.3, 2.0 Hz), 5.27 (1H, dd, J = 12.3, 2.5 Hz), 4.20–4.18 (1H, br), 3.14–3.06 (1H, m), 2.59–2.50 (1H, m), 2.44–2.33 (1H, m), 1.95 (3H, d, J = 1.3 Hz), 1.94 (3H, s), 1.86–1.77 (1H, m), 1.61–1.54 (1H, m), 1.33–1.16 (2H, m), 1.01 (3H, s), 0.95 (3H, s); 13C NMR (101 MHz, C6D6) δ 193.6, 164.0, 149.3, 139.4, 128.6, 117.6, 117.1, 53.0, 41.1, 38.6, 36.2, 30.2, 29.3, 29.2, 25.3, 24.2, 14.1; νmax (film) 2955, 2928, 2863, 1744, 1668, 1652, 1609, 949, 729 cm−1; HMRS (ESI) calcd for C17H22NaO2 [M + Na]+ 281.1512, found 281.1501.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the University of Glasgow for studentship funding for VK and a postdoctoral fellowship for SW.Notes and references
- J. B. Sperry and D. L. Wright, Curr. Opin. Drug Discovery Dev., 2005, 8, 713 Search PubMed.
- (a) H. Hikino and C. Konno, Heterocycles, 1976, 4, 817 CrossRef; (b) R. Maurya and P. P. Yadav, Nat. Prod. Rep., 2005, 22, 400 RSC; (c) Y. Liu, S. Zhang and P. J. M. Abreu, Nat. Prod. Rep., 2006, 23, 630 RSC; (d) P. A. Roethle and D. Trauner, Nat. Prod. Rep., 2008, 25, 298 RSC; (e) S. O. Simonetti, E. L. Larghi, A. B. Bracca and T. S. Kaufman, Nat. Prod. Rep., 2013, 30, 941 RSC.
- J. Xu, Y. Guo, Y. Li, P. Zhao, C. Liu, Y. Ma, J. Gao, W. Hou and T. Zhang, Planta Med., 2011, 77, 2023 CrossRef PubMed.
- A. D. Patil, A. J. Freyer, L. Killmer, P. Offen, B. Carte, A. J. Jurewicz and R. K. Johnson, Tetrahedron, 1997, 53, 5047 CrossRef.
- F. Marion, D. E. Williams, B. O. Patrick, I. Hollander, R. Mallon, S. C. Kim, D. M. Roll, L. Feldberg, R. Van Soest and R. J. Andersen, Org. Lett., 2006, 8, 321 CrossRef PubMed.
- J. S. Clark, F. Romiti, K. F. Hogg, M. H. S. A. Hamid, S. C. Richter, A. Boyer, J. C. Redman and L. J. Farrugia, Angew. Chem., Int. Ed., 2015, 54, 5744 CrossRef PubMed.
- G. Pattenden and J. M. Winne, Tetrahedron Lett., 2009, 50, 7310 CrossRef.
- J. M. Winne, S. Catak, M. Waroquier and V. Van Speybroeck, Angew. Chem., Int. Ed., 2011, 50, 11990 CrossRef PubMed.
- B. Song, L.-H. Li, X.-R. Song, Y.-F. Qiu, M.-J. Zhong, P.-X. Zhou and Y.-M. Liang, Chem. – Eur. J., 2014, 20, 5910 CrossRef PubMed.
- (a) S. Mata, L. A. López and R. Vicente, Synlett, 2015, 2685–2689 Search PubMed; (b) S. Mata, J. González, R. Vicente and L. A. López, Eur. J. Org. Chem., 2016, 2681 CrossRef.
- G. Maas and C. Hummel, Chem. Ber., 1980, 113, 3679 CrossRef.
- J. Barluenga, F. Aznar, I. Gutiérrez and J. A. Martín, Org. Lett., 2002, 4, 2719 CrossRef PubMed.
- M. Goto, I. Miyoshi, Y. Ishii, Y. Ogasawara, Y.-I. Kakimoto, S. Nagumo, A. Nishida, N. Kawahara and M. Nishida, Tetrahedron, 2002, 58, 2339 CrossRef.
- (a) K. G. R. Pachler, J. Chem. Soc., Perkin Trans. 2, 1972, 1936 RSC; (b) C. A. G. Haasnoot, F. A. A. M. de Leeuw and C. Altona, Tetrahedron, 1980, 36, 2783 CrossRef.
- S. Krüger and T. Gaich, Beilstein J. Org. Chem., 2014, 10, 163 CrossRef PubMed.
- T. Shen, C. W. Weng, W. D. Xie and K. H. Row, J. Chem. Res., 2009, 33, 623 CrossRef.
- W. Vichnewski and B. Gilbert, Phytochemistry, 1972, 11, 2563 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra (1H and 13C) for compounds 20–33 and 35–37 plus details of computational studies. See DOI: 10.1039/c8ob00924d |
| ‡ The relative configuration of the 3a,4,6a,7,8,9-hexahydroazuleno[4,5-b]furans 29–31 was confirmed by the magnitude of the coupling between the protons at C3 and C4, which was very small and indicative of a dihedral angle of approximately 90°. If the C4 epimers of the products 29–31 had been obtained, this coupling would have been relatively large because the dihedral angle would have been close to 180°. |
| This journal is © The Royal Society of Chemistry 2018 |