
Synthesis of the [7-5-5] tricyclic core of Daphniphyllum alkaloids†
Yusuke
Kitabayashi
,
Tohru
Fukuyama
* and
Satoshi
Yokoshima
 *
*
Graduate School of Pharmaceutical Sciences, Nagoya University, Furo-cho, Chikusa-ku, Nagoya, 464-8601, Japan. E-mail: yokosima@ps.nagoya-u.ac.jp
First published on 18th April 2018
Abstract
The [7-5-5] tricyclic core of the Daphniphyllum alkaloids was constructed, featuring a Claisen-Ireland rearrangement to install the two contiguous stereogenic centers, E1cB elimination to form the tetrasubstituted C–C double bond, and a 2,3-Wittig rearrangement to construct the quaternary carbon. Ring-closing metathesis and an intramolecular carbonyl ene reaction were employed for construction of the requisite ring system.
Alkaloids with a vast range of diversity have been isolated from plants of the genus Daphniphyllum.1 According to their structure, these alkaloids can be classified into several groups, including yuzurimine-, yuzurine-, calyciphylline A-, calyciphylline C- and daphmanidin A-type (Fig. 1). Each group is composed of alkaloids that have the same carbon skeleton but different oxidation states and substituents. A common core structure is present across these groups, that is, a [7-5-5] tricyclic core 7 that features a quaternary carbon and two contiguous stereogenic centers, adjacent to which is a tetrasubstituted C–C double bond. Although a variety of syntheses of Daphniphyllum alkaloids have been reported to date,2 Smith's synthesis of calyciphylline N is the only example that has succeeded in constructing the above common core.2k,l
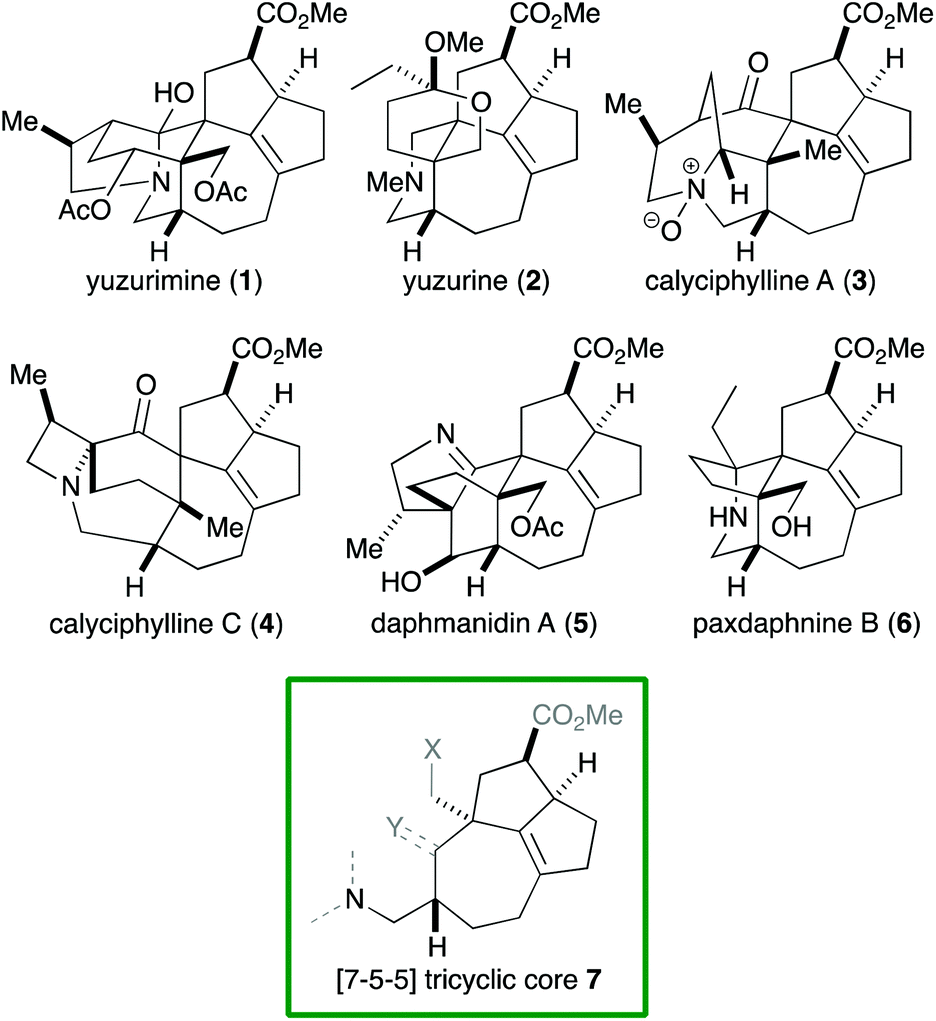 | ||
| Fig. 1 Daphniphyllum alkaloids. | ||
Among the structural features of the [7-5-5] tricyclic core 7, the installation of the tetrasubstituted C–C double bond has been explored only to a limited extent. In Smith's synthesis,2k,l this feature was realized by selective hydrogenation of a diene moiety in 8 using the BArF analog of the Crabtree catalyst (Scheme 1).3 Dixon and coworkers reported isomerization of 10 into 11 under basic conditions,4 which was employed by Zhai and coworkers in their synthesis of daphnilongeranin B.2p Very recently, Li and coworkers also reported the synthesis of daphnilongeranin B by employing the Dixon's strategy.2r In this case, the carbonyl group of the resulting enone, which fixes the position of the C–C double bond, is the constituent of the target alkaloid. Embedding a planar C–C double bond in a bowl-like [5-5] bicyclic system induces a sizable strain; therefore, the double bond tends to move away from the ring juncture. In fact, Sakai and coworkers reported that an intramolecular aldol reaction of 14 under thermal and acidic conditions produced β,γ-unsaturated ketone 16 as the major product.5 Hayakawa and coworkers also reported that calculations predicted the same stability profile in a trisubstituted olefin and a tetrasubstituted olefin.6 Herein, we disclose our efforts on constructing the [7-5-5] tricyclic core of the Daphniphyllum alkaloids.
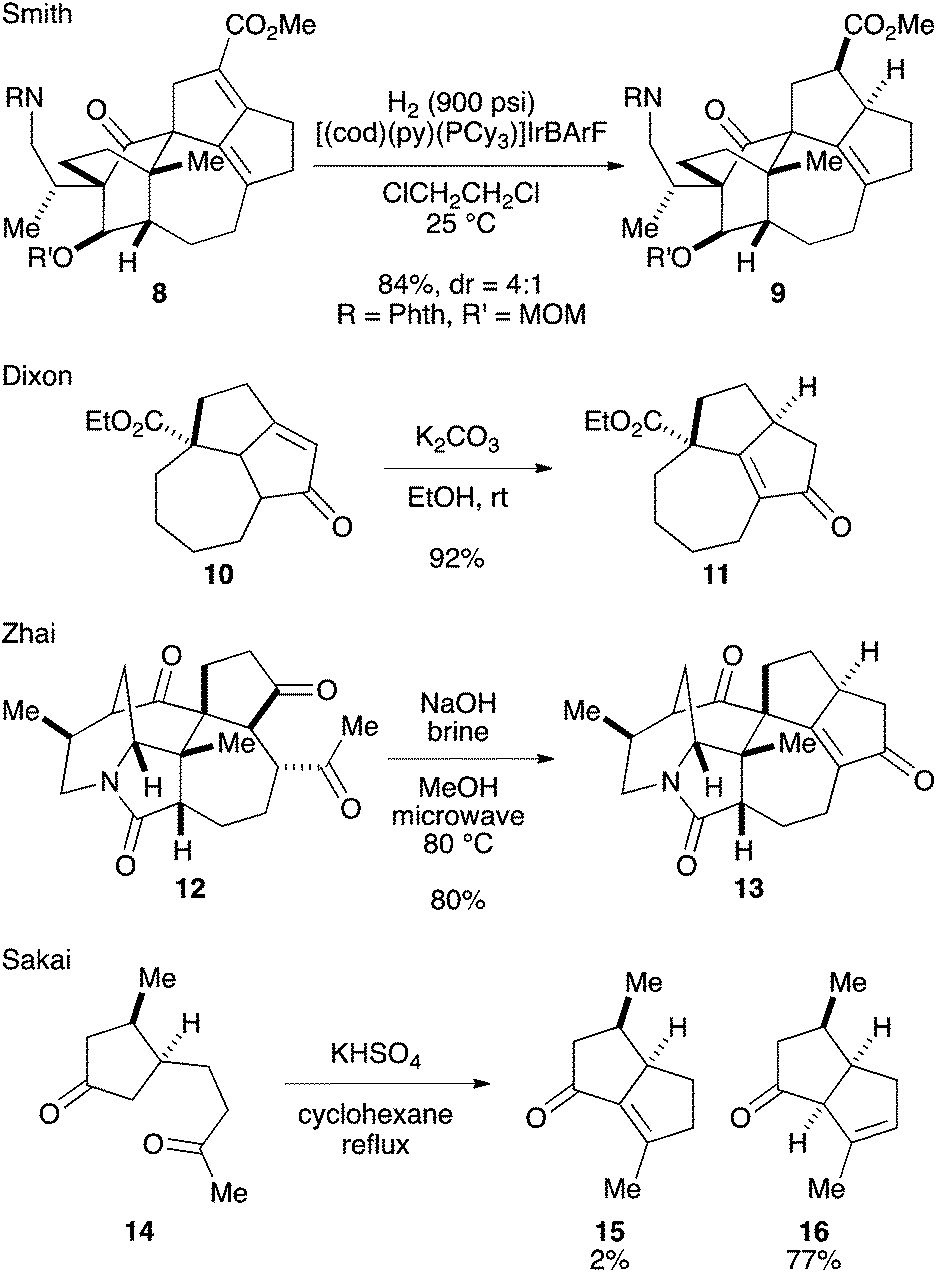 | ||
| Scheme 1 Installation of tetrasubstituted C–C double bond. | ||
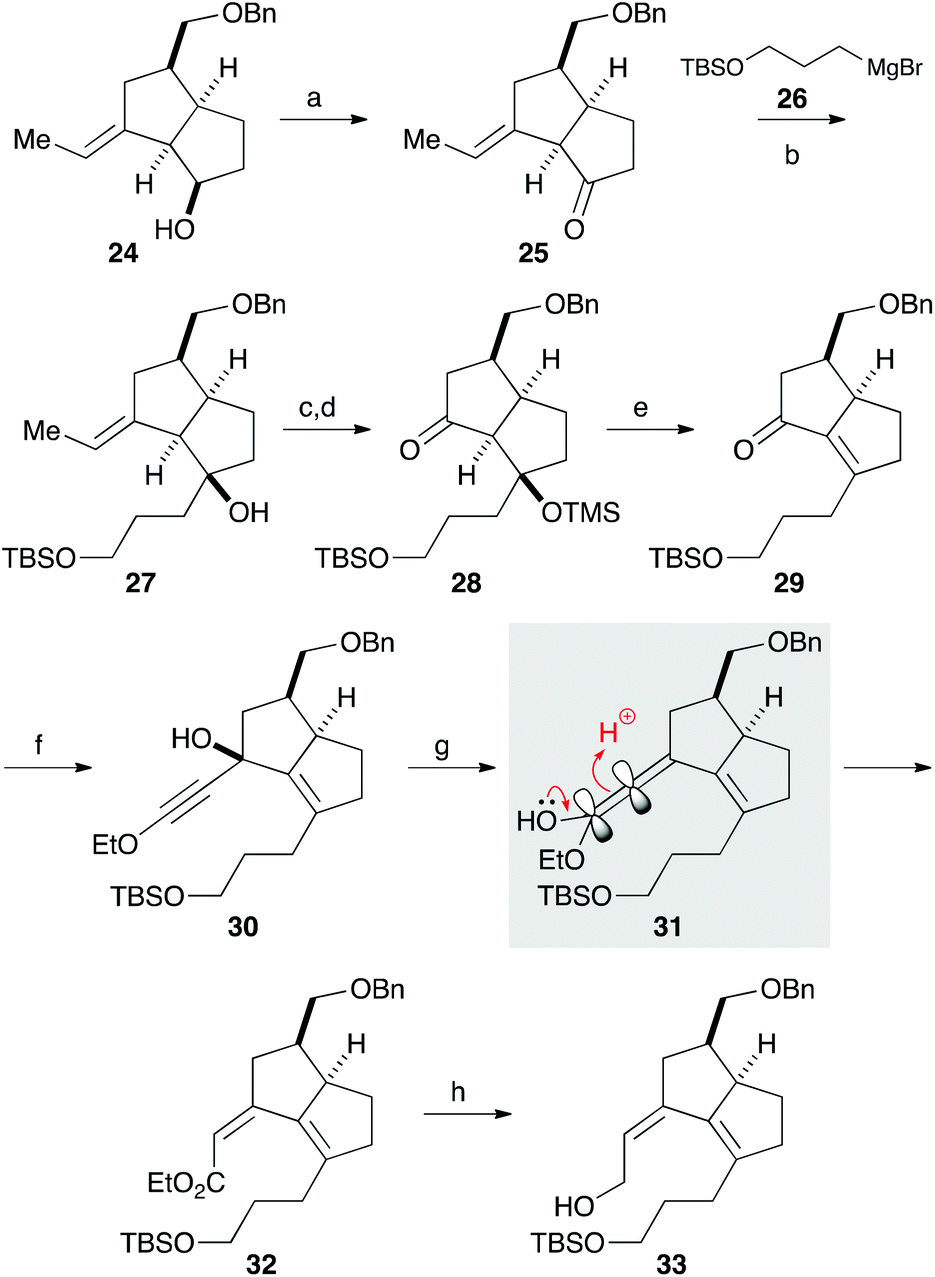
Our synthesis commenced with the construction of two contiguous stereogenic centers via a Claisen-Ireland rearrangement. Protection of 4-pentyn-1-ol (17) with a TBS group, followed by hydroxymethylation of the terminal alkyne, afforded alcohol 18 (Scheme 2). After partial reduction of the alkyne moiety, the resulting cis-allyl alcohol was condensed with carboxylic acid A to furnish ester 19.7 Upon treatment with LHMDS and TMSCl, 19 underwent a smooth Claisen-Ireland rearrangement,8 giving carboxylic acid 20 in 73% yield with a diastereomeric ratio of 6.3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. Sodium borohydride reduction of the mixed anhydride derived from the carboxylic acid afforded an alcohol, which was then protected as its benzyl ether. Ring closing metathesis of 21 was performed with the Zhan catalyst-1B (B) to give trisubstituted olefin 22. After a 2-step conversion of the silyl ether moiety in 22 to aldehyde 23, a carbonyl ene reaction was conducted by treatment with boron trifluoride to give bicyclic compound 24 in 93% yield.9
:1. Sodium borohydride reduction of the mixed anhydride derived from the carboxylic acid afforded an alcohol, which was then protected as its benzyl ether. Ring closing metathesis of 21 was performed with the Zhan catalyst-1B (B) to give trisubstituted olefin 22. After a 2-step conversion of the silyl ether moiety in 22 to aldehyde 23, a carbonyl ene reaction was conducted by treatment with boron trifluoride to give bicyclic compound 24 in 93% yield.9
 | ||
| Scheme 2 Stereoselective construction of the bicyclo[3.3.0]octane system. Reagents and conditions: (a) TBSCl, imidazole, CH2Cl2, rt, 99%; (b) n-BuLi, THF, −78 °C; (HCHO)n, −78 °C, 70%; (c) H2, Lindlar catalyst, quinoline, EtOAc, rt, quant; (d) A, EDCI, DMAP, CH2Cl2, rt, 89%; (e) LHMDS, TMSCl, Et2O, −78 to 40 °C, 73%, dr = 6.3:1; (f) Boc2O, pyridine, THF, rt; NaBH4, H2O, 0 °C, 98%; (g) BnBr, NaH, THF, 0 °C to rt, 83%; (h) B, toluene, 50 °C, 95%; (i) TBAF, THF, rt, 90%; (j) Dess–Martin periodinane, CH2Cl2, 0 °C to rt, 90%; (k) BF3·OEt2, CH2Cl2, −78 °C, 93%. | ||
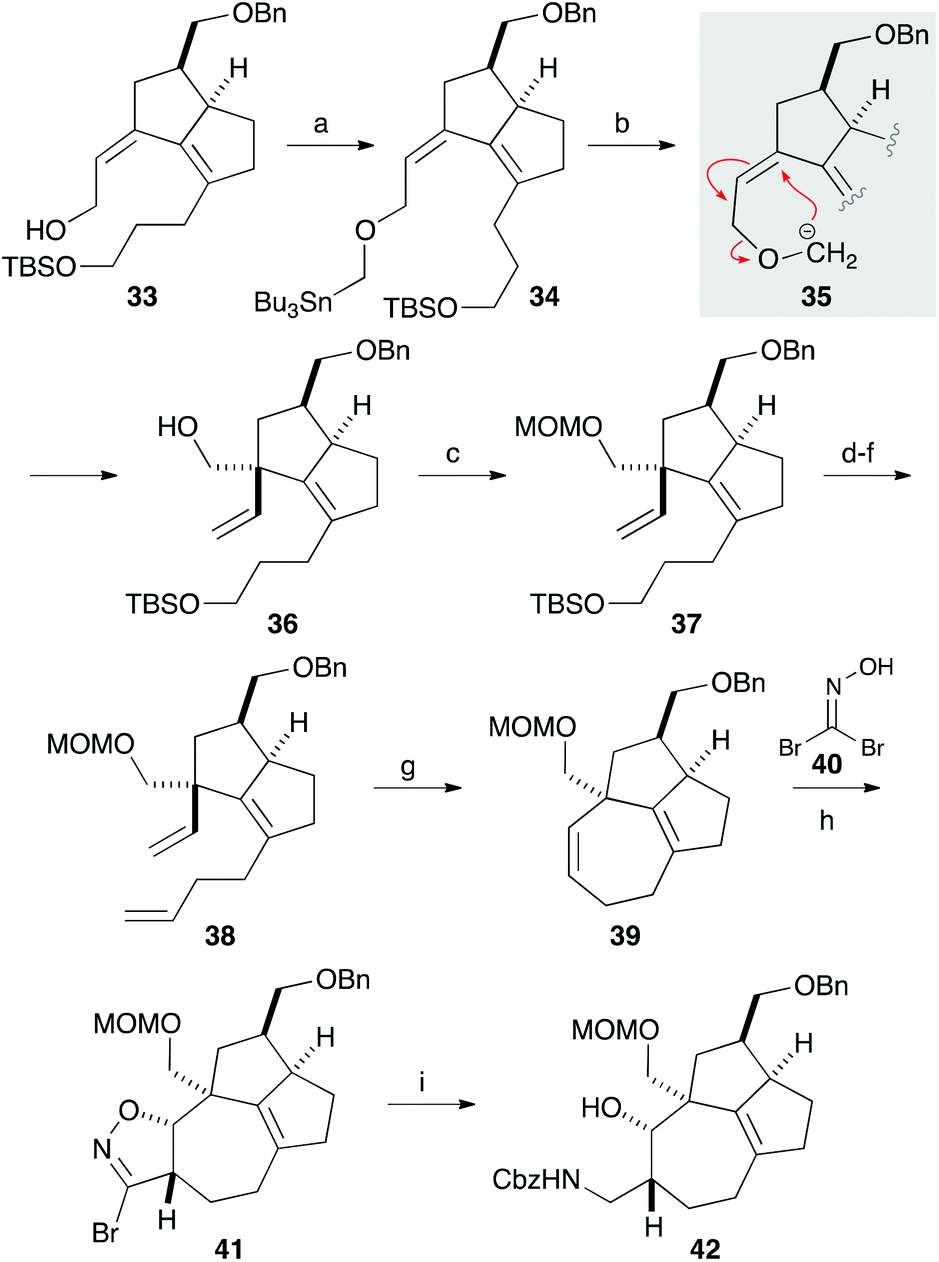
We next focused on introducing the tetrasubstituted C–C double bond. TPAP oxidation of the secondary alcohol moiety in 24,10 followed by the addition of three-carbon unit 26 in the presence of cerium chloride,11 afforded 27 (Scheme 3). Ozonolysis of the C–C double bond and protection of the tertiary alcohol moiety with a TMS group produced 28. We next conducted the E1cB-elimination of 28 to furnish α,β-unsaturated ketone 29.12 During the course of the investigations, we realized that the order of mixing the reagent was important. While addition of potassium tert-butoxide to a solution of the substrate induced isomerization of the α,β-unsaturated ketone to the corresponding β,γ-unsaturated ketone, addition of a solution of 28 in THF to a solution containing an excess amount of potassium tert-butoxide (10 equiv.) at 0 °C gave 29 in 78% yield. To the carbonyl group in enone 29 was added an ethoxyacetylene moiety, and the resulting product 30 was subjected to the Meyer–Schuster rearrangement by treatment with Sc(OTf)3.13,14 The reaction occurred via protonation from the less hindered side of the allenyl intermediate 31, giving unsaturated ester 32, which was reduced with DIBAL to afford alcohol 33.
 | ||
| Scheme 3 Formation of the tetrasubstituted olefin moiety. Reagents and conditions: (a) TPAP, NMO, MS4A, CH2Cl2, 0 °C, 84%; (b) TBSO(CH2)3MgBr (26), CeCl3, THF, 0 °C, 81%; (c) O3, CH2Cl2–MeOH, −78 °C; Ph3P, −78 °C to rt, 75%; (d) TMSCl, imidazole, DMF, rt, 92%; (e) KOt-Bu, THF, 0 °C, 78%; (f) EtMgBr, ethoxyacetylene, Et2O, 0 °C, 78%; (g) Sc(OTf)3, EtOH, CH2Cl2, 0 °C, 93%; (h) DIBAL, CH2Cl2, −78 to 0 °C, 77%. | ||
The quaternary carbon was next constructed using the allyl alcohol moiety in 33 (Scheme 4). After extensive investigations involving reactions related to Claisen rearrangement15 or Lewis acid-mediated rearrangement of epoxyalcohol derivatives,16 we found that 2,3-Wittig rearrangement was effective for achieving this transformation.17 Alkylation of the hydroxy group in 33 with n-Bu3SnCH2I in the presence of potassium hydride furnished 34, which was treated with methyllithium in diethyl ether.18 The requisite 2,3-Wittig rearrangement stereoselectively occurred on the less hindered, convex face of the bicyclic system to produce alcohol 36 having a vinyl group19 that was to be used for construction of the 7-membered ring. After protecting the hydroxy group with a MOM group, the three-carbon chain was elongated in a 3-step sequence comprising cleavage of the TBS ether, oxidation to aldehyde, and a Wittig reaction. The resulting diene 38 was subjected to ring-closing metathesis with the second-generation Grubbs catalyst to form a seven-membered ring in a quantitative yield.20 Thus, the [7-5-5] tricyclic core containing the quaternary carbon, two contiguous stereogenic centers, and the tetrasubstituted C–C double bond was successfully constructed. A preliminary investigation involving 1,3-dipolar cycloaddition of a nitrile oxide, generated from 40, to 39 afforded isoxazoline 41 in a 1.1:1 diastereomer ratio.21 Reduction of the isoxazoline ring with nickel boride, followed by protection of the resulting amine with a Cbz group, gave 42.
 | ||
| Scheme 4 Construction of the [7-5-5] tricyclic core. Reagents and conditions: (a) KH, Bu3SnCH2I, THF, rt; (b) MeLi, Et2O, 0 °C; (c) MOMCl, i-Pr2NEt, CH2Cl2, rt, 64% (3 steps); (d) TBAF, THF, rt, 92%; (e) TPAP, NMO, MS4A, CH2Cl2, 0 °C; (f) Ph3PCH3Br, n-BuLi, THF, 0 °C, 53% (2 steps); (g) Grubbs II, ClCH2CH2Cl, 50 °C, quant; (h) 40, NaHCO3, CH2Cl2, rt, 70%, dr = 1.1:1; (i) NiCl2, NaBH4, MeOH, −78 °C; CbzCl, aq NaHCO3, CH2Cl2, 0 °C, 39%. | ||
In conclusion, we have constructed the [7-5-5] tricyclic core of the Daphniphyllum alkaloids. The present synthesis features a Claisen-Ireland rearrangement to install the two contiguous stereogenic centers, E1cB elimination to form the tetrasubstituted C–C double bond, and a 2,3-Wittig rearrangement to construct the quaternary carbon. Ring-closing metathesis and an intramolecular carbonyl ene reaction formed the ring system. These findings provide a basis for the total synthesis of the Daphniphyllum alkaloids that have the [7-5-5] tricyclic core.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by JSPS KAKENHI (Grant Numbers 16H01141 and 17H01523), by the Takeda Science Foundation, and by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research; BINDS) from the Japan Agency for Medical Research and Development (AMED) under Grant Number JP17am0101099.Notes and references
- (a) J. Kobayashi and T. Kubota, Nat. Prod. Rep., 2009, 26, 936 RSC; (b) B. Kang, P. Jakubec and D. J. Dixon, Nat. Prod. Rep., 2014, 31, 550 RSC; (c) A. K. Chattopadhyay and S. Hanessian, Chem. Rev., 2017, 117, 4104 CrossRef CAS PubMed.
- (a) C. H. Heathcock, S. K. Davidsen, S. Mills and M. A. Sanner, J. Am. Chem. Soc., 1986, 108, 5650 CrossRef CAS; (b) R. B. Ruggeri, M. M. Hansen and C. H. Heathcock, J. Am. Chem. Soc., 1988, 110, 8734 CrossRef CAS; (c) R. B. Ruggeri, K. F. McClure and C. H. Heathcock, J. Am. Chem. Soc., 1989, 111, 1530 CrossRef CAS; (d) S. Piettre and C. H. Heathcock, Science, 1990, 248, 1532 CAS; (e) R. B. Ruggeri and C. H. Heathcock, J. Org. Chem., 1990, 55, 3714 CrossRef CAS; (f) J. A. Stafford and C. H. Heathcock, J. Org. Chem., 1990, 55, 5433 CrossRef CAS; (g) C. H. Heathcock, J. A. Stafford and D. L. Clark, J. Org. Chem., 1992, 57, 2575 CrossRef CAS; (h) C. H. Heathcock, J. C. Kath and R. B. Ruggeri, J. Org. Chem., 1995, 60, 1120 CrossRef CAS; (i) M. E. Weiss and E. M. Carreira, Angew. Chem., Int. Ed., 2011, 50, 11501 CrossRef CAS PubMed; (j) Z. Lu, Y. Li, J. Deng and A. Li, Nat. Chem., 2013, 5, 679 CrossRef CAS PubMed; (k) A. Shvartsbart and A. B. Smith III, J. Am. Chem. Soc., 2014, 136, 870 CrossRef CAS PubMed; (l) A. Shvartsbart and A. B. Smith III, J. Am. Chem. Soc., 2015, 137, 3510 CrossRef CAS PubMed; (m) R. Yamada, Y. Adachi, S. Yokoshima and T. Fukuyama, Angew. Chem., Int. Ed., 2016, 55, 6067 CrossRef CAS PubMed; (n) J. Li, W. Zhang, F. Zhang, Y. Chen and A. Li, J. Am. Chem. Soc., 2017, 139, 14893 CrossRef CAS PubMed; (o) H. Shi, I. N. Michaelides, B. Darses, P. Jakubec, Q. N. N. Nguyen, R. S. Paton and D. J. Dixon, J. Am. Chem. Soc., 2017, 139, 17755 CrossRef CAS PubMed; (p) X. Chen, H.-J. Zhang, X. Yang, H. Lv, X. Shao, C. Tao, H. Wang, B. Cheng, Y. Li, J. Guo, J. Zhang and H. Zhai, Angew. Chem., Int. Ed., 2018, 57, 947 CrossRef CAS PubMed; (q) Y. Chen, W. Zhang, L. Ren, J. Li and A. Li, Angew. Chem., Int. Ed., 2018, 57, 952 CrossRef CAS PubMed; (r) W. Zhang, M. Ding, J. Li, Z. Guo, M. Lu, Y. Chen, L. Liu, Y.-H. Shen and A. Li, J. Am. Chem. Soc., 2018, 140, 4227 CrossRef CAS PubMed.
- The diene moiety was also constructed by Weiss and Carreira in their synthesis of daphmanidin E. See ref. 2i.
- B. Darses, I. N. Michaelides, F. Sladojevich, J. W. Ward, P. R. Rzepa and D. J. Dixon, Org. Lett., 2012, 14, 1684 CrossRef CAS PubMed.
- H. Suemune, K. Oda, S. Saeki and K. Sakai, Chem. Pharm. Bull., 1988, 36, 172 CrossRef CAS.
- I. Hayakawa, K. Niida and H. Kigoshi, Chem. Commun., 2015, 51, 11568 RSC.
- S. Yokoshima, T. Ueda, S. Kobayashi, A. Sato, T. Kuboyama, H. Tokuyama and T. Fukuyama, J. Am. Chem. Soc., 2002, 124, 2137 CrossRef CAS PubMed.
- (a) R. E. Ireland, R. H. Mueller and A. K. Willard, J. Am. Chem. Soc., 1976, 98, 2868 CrossRef CAS; (b) R. E. Ireland, P. Wipf and J. D. Armstrong, J. Org. Chem., 1991, 56, 650 CrossRef CAS; (c) R. E. Ireland, P. Wipf and J. N. Xiang, J. Org. Chem., 1991, 56, 3572 CrossRef CAS.
- (a) B. B. Snider, M. Karras, R. T. Price and D. J. Rodini, J. Org. Chem., 1982, 47, 4538 CrossRef CAS; (b) N. H. Andersen, S. W. Hadley, J. D. Kelly and E. R. Bacon, J. Org. Chem., 1985, 50, 4144 CrossRef CAS; (c) S. Sakane, K. Maruoka and H. Yamamoto, Tetrahedron, 1986, 42, 2203 CrossRef CAS; (d) J. A. Marshall and M. W. Andersen, J. Org. Chem., 1993, 58, 3912 CrossRef CAS; (e) L. A. Paquette, Y. Hu, A. Luxenburger and R. L. Bishop, J. Org. Chem., 2007, 72, 209 CrossRef CAS PubMed.
- (a) W. P. Griffith, S. V. Ley, G. P. Whitcombe and A. D. White, J. Chem. Soc., Chem. Commun., 1987, 1625 RSC; (b) S. V. Ley, J. Norman, W. P. Griffith and S. P. Marsden, Synthesis, 1994, 639 CrossRef.
- (a) T. Imamoto, T. Kusumoto, Y. Tawarayama, Y. Sugiura, T. Mita, Y. Hatanaka and M. Yokoyama, J. Org. Chem., 1984, 49, 3904 CrossRef CAS; (b) T. Imamoto, Y. Sugiura and N. Takiyama, Tetrahedron Lett., 1984, 25, 4233 CrossRef CAS.
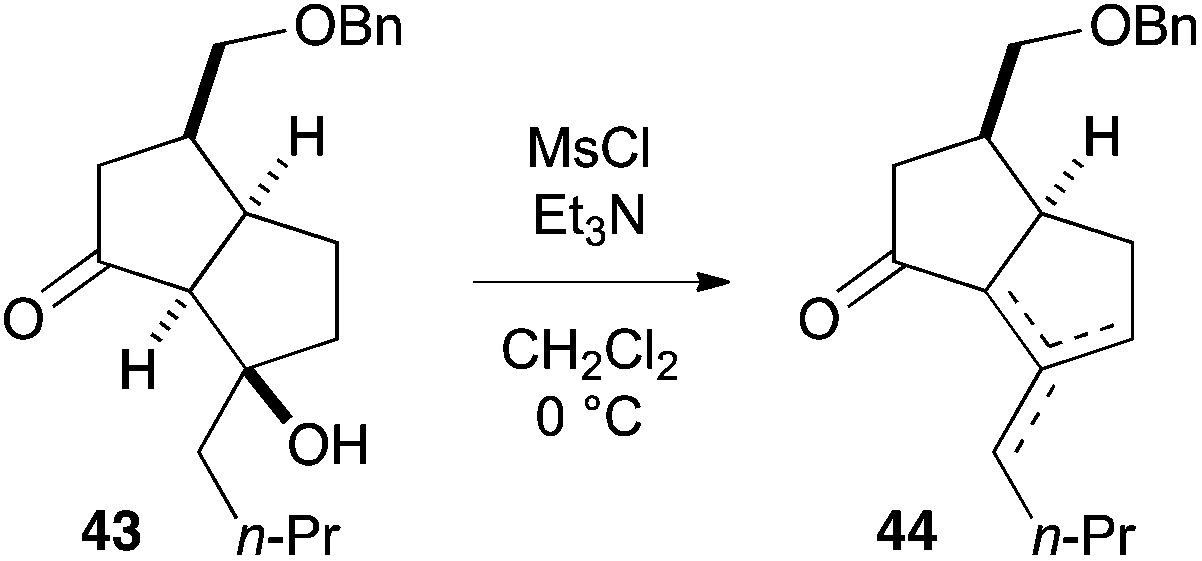
- Mesylation of β-hydroxyketone 43 induced elimination to furnish a mixture of olefinic isomers
. - (a) S. S. Lopez, D. A. Engel and G. B. Dudley, Synlett, 2007, 0949 CAS; (b) D. A. Engel, S. S. Lopez and G. B. Dudley, Tetrahedron, 2008, 64, 6988 CrossRef CAS.
- The attempted Horner–Wadsworth–Emmons reaction or Peterson reaction did not proceed.
- (a) A. Jefferson and F. Scheinmann, Q. Rev., Chem. Soc., 1968, 22, 391 RSC; (b) F. E. Ziegler, Chem. Rev., 1988, 88, 1423 CrossRef CAS; (c) A. M. Martín Castro, Chem. Rev., 2004, 104, 2939 CrossRef PubMed.
- (a) K. Maruoka, T. Ooi and H. Yamamoto, J. Am. Chem. Soc., 1989, 111, 6431 CrossRef CAS; (b) K. Maruoka, T. Ooi, S. Nagahara and H. Yamamoto, Tetrahedron, 1991, 47, 6983 CrossRef CAS.
- S. Hanessian, S. Dorich, A. K. Chattopadhyay and M. Büschleb, J. Org. Chem., 2013, 78, 8915 CrossRef CAS PubMed.
- P. Stanetty and M. D. Mihovilovic, J. Org. Chem., 1997, 62, 1514 CrossRef CAS.
- The stereochemistry of the quaternary carbon was confirmed by the NOESY spectrum of 37. For details, see the ESI.†.
- M. Scholl, S. Ding, C. W. Lee and R. H. Grubbs, Org. Lett., 1999, 1, 953 CrossRef CAS PubMed.
- For selected application of 40 in natural product synthesis, see: (a) D. M. Vyas, P. M. Skonezny, T. A. Jenks and T. W. Doyle, Tetrahedron Lett., 1986, 27, 3099 CrossRef CAS; (b) T. J. Maimone, J. Shi, S. Ashida and P. S. Baran, J. Am. Chem. Soc., 2009, 131, 17066 CrossRef CAS PubMed; (c) G. Cheng, X. Wang, R. Zhu, C. Shao, J. Xu and Y. Hu, J. Org. Chem., 2011, 76, 2694 CrossRef CAS PubMed; (d) J. Poulin, C. M. Grisé-Bard and L. Barriault, Angew. Chem., Int. Ed., 2012, 51, 2111 CrossRef CAS PubMed; (e) C. Ebner and E. M. Carreira, Angew. Chem., Int. Ed., 2015, 54, 11227 CrossRef CAS PubMed; (f) Y. Yoshii, H. Tokuyama and D. Y. K. Chen, Angew. Chem., Int. Ed., 2017, 56, 12277 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental protocols, characterization data and NMR spectra of all new compounds. See DOI: 10.1039/c8ob00859k |
| This journal is © The Royal Society of Chemistry 2018 |