
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of N-[(dialkylamino)methyl]acrylamides and N-[(dialkylamino)methyl]methacrylamides from Schiff base salts: useful building blocks for smart polymers†
Abdullah
Alzahrani
 ,
Styliana I.
Mirallai
*,
Benjamin A.
Chalmers
,
Patrick
McArdle
and
Fawaz
Aldabbagh‡
*
,
Styliana I.
Mirallai
*,
Benjamin A.
Chalmers
,
Patrick
McArdle
and
Fawaz
Aldabbagh‡
*
School of Chemistry, National University of Ireland Galway, University Road, Galway, Ireland. E-mail: Styliana.Mirallai@nuigalway.ie
First published on 9th May 2018
Abstract
The traditional thermal Mannich reaction is unsuitable for preparing polymerizable N-methylene amino substituted acrylamides and methacrylamides. Herein we provide a facile multi-gram high yield synthesis of these monomeric precursors to stimuli-responsive polymers by the addition of acrylamides and methacrylamides onto in situ generated or freshly isolated methylene Schiff base (iminium) salts. The X-ray crystal structure of the hydrated iminium salt, 1-(hydroxymethyl)azocan-1-ium chloride and monomer·HCl salt (N-[(azocan-1-yl)methyl]prop-2-enamide hydrochloride) is described.
Introduction
The three-component Mannich reaction is fundamental, allowing access to β-amino methylated carbonyl compounds.1,2N-[(Dialkylamino)methyl]acrylamide and methacrylamide analogues are valuable monomeric precursors to smart polymers, with dual functionalities of temperature and pH-responsiveness (Fig. 1), however potential applications have not been realised due to problems with their synthesis.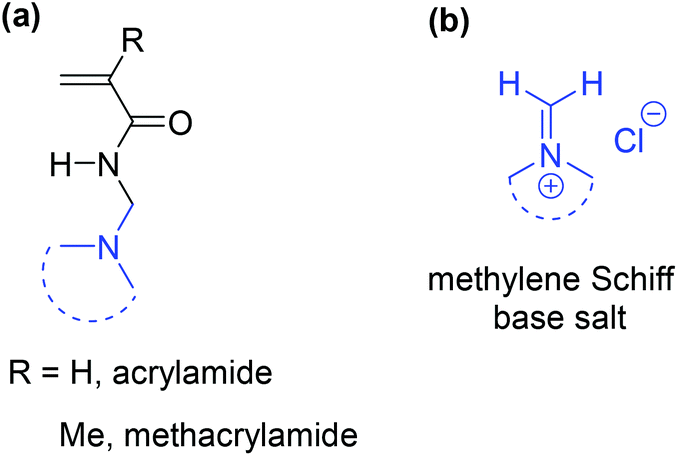 | ||
| Fig. 1 (a) Synthetic targets and (b) precursors. | ||
Literature routes have used one-pot Mannich condensation of (meth)acrylamide with formaldehyde to generate the Schiff base followed by secondary amine addition (Scheme 1).3–6 The reaction operates thermally (at ∼80 °C), and is inefficient in forming the Schiff base in situ, with the elevated temperature resulting in premature polymerization of the monomer and intermediates. The reaction has the added difficulty of monomer isolation, which requires vacuum distillation from the aqueous reaction mixture.
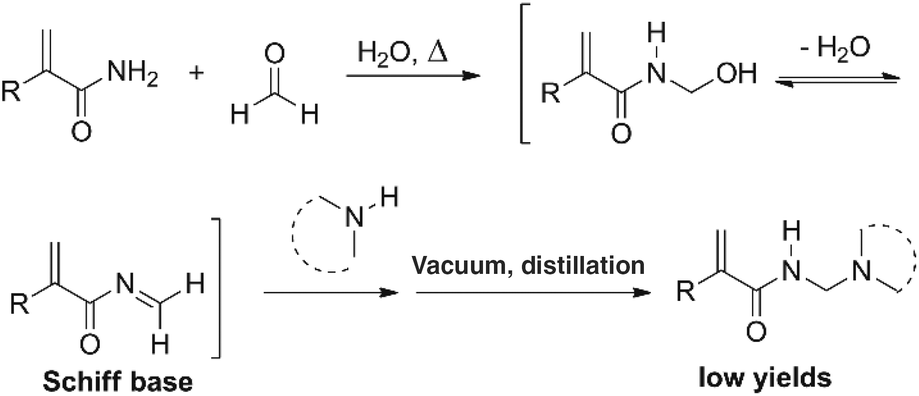 | ||
| Scheme 1 One-pot thermal Mannich reaction route. | ||
The most widely studied temperature-responsive polymers are those with lower critical solution temperature (LCST) close to physiological temperature, such as poly(N-isopropyl-acrylamide) and poly(N,N-diethylacrylamide) with LCST of 32–34 °C in water.7–9 The N-dialkyl amino (including saturated nitrogen heterocycle) of substituted acrylamides and methacrylamides can be reversibly ionized allowing for a pH-response that alters polymer hydrophobicity.9–15 Amphiphilic block copolymers comprising such monomers can self-assemble into a variety of nano-objects for use as stimuli-responsive polymersomes for targeted delivery of therapeutics.11–13 In a recent communication, the synthesis of the selected acrylamides containing N-methylene saturated nitrogen heterocycles, and their incorporation into well-defined water-soluble block copolymer polyacrylamides was realised.14 In this full paper, we expand on the monomer synthesis by providing efficient multi-gram routes to acrylamides and methacrylamides, including those with dialkyl acyclic and large saturated nitrogen heterocyclic rings. The synthesis involves efficient generation of the methylene Schiff base salt, which was characterized in the hydrated form.
Results and discussion
In contrast to the one-pot thermal Mannich condensation reaction in Scheme 1, our synthesis uses readily accessible aminals made from the condensation of formaldehyde with secondary amines at 0 °C (Scheme 2).16,17 Böhme pioneered the quaternization of the aminal to generate the Schiff base (iminium) salt, and this procedure using acetyl chloride was followed.17,18 There are numerous accounts of alkylation and nucleophilic addition onto methylene Schiff base salts,16–28 including by non-vinylic amides.27,28 The most utilised is commercial N,N-dimethylmethyleneiminium iodide or Eschenmoser's salt.20,29 Inspired by the simplicity and low temperatures, acrylamides and methacrylamides were added onto in situ generated methylene Schiff salts to give the monomer hydrochloride salts of morpholine, pyrrolidine and piperidine.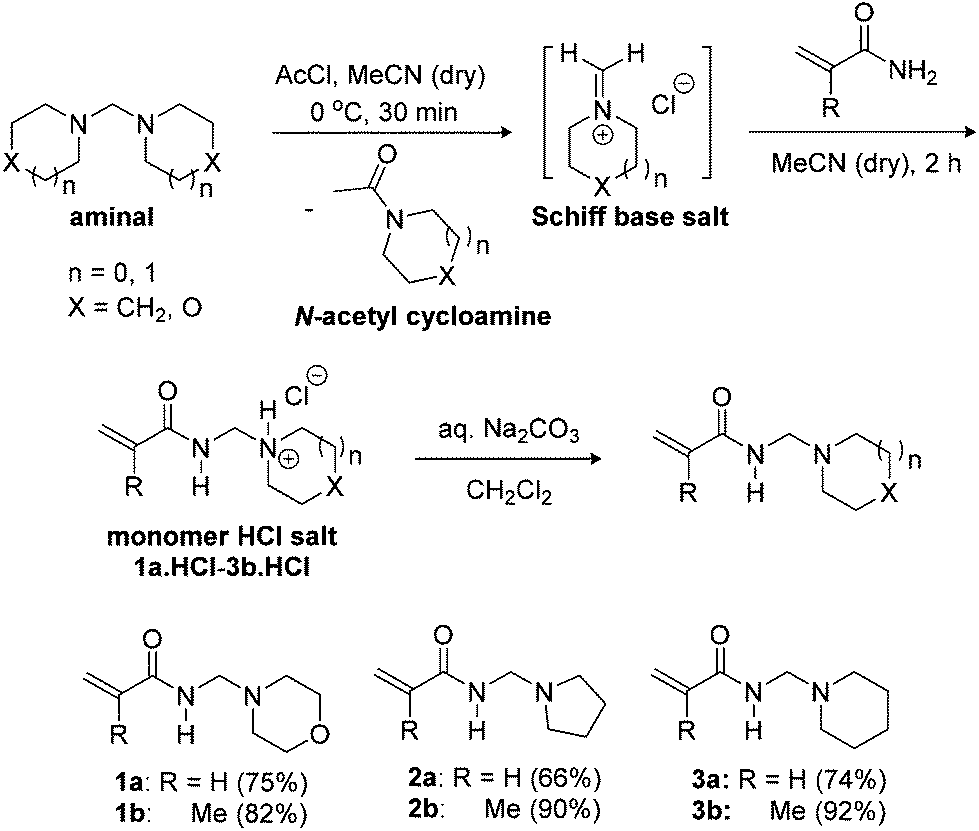 | ||
| Scheme 2 Synthesis of N-[(cycloalkylamino)methyl]acrylamides 1a–3a,14 and N-[(cycloalkylamino)methyl]methacrylamides 1b–3b using the in situ Schiff base salt approach. | ||
The monomer hydrochloride salts 1a·HCl–3b·HCl were precipitated upon the addition of diethyl ether to the reaction in acetonitrile, which allowed the separation of the soluble N-acetyl cycloamines (Scheme 2). The isolable 1a·HCl–3b·HCl salts are themselves useful as monomeric building blocks in aqueous solution polymerizations.14 Basification of the latter allowed the free N-[(cycloalkylamino)methyl]acrylamides 1a–3a to be isolated on a 20–25 g scale in yields of 66–75% with the N-[(cycloalkylamino)methyl]methacrylamides 1b–3b isolated in higher yields of 82–92% and on a ≥30 g scale.
Our in situ Schiff base salt approach was not applicable for larger cycloamines (azepane and azocane) and acyclic analogues. Isolation of the methylene Schiff base salts was deemed necessary in these cases due to the poor solubility of their aminals in acetonitrile. Seven and eight-membered heterocyclic base-containing monomers are useful for increased hydrophobicity in the ionisable block segment of amphiphilic copolymers promoting sharper pH-sensitivity of micelles.11,12 In contrast, linear dialkyl amine-containing polyacrylamides generally have greater water solubility in comparison to heterocyclic amine-containing analogues affording higher LCSTs.10 Treatment of the aminals with acetyl chloride in diethyl ether at 0 °C allowed access to both larger heterocyclic and acyclic methylene Schiff base salts 4a–4f, which were more conveniently characterised as N-hydroxymethyl hydrochloride salt derivatives 5a–5f (Scheme 3).
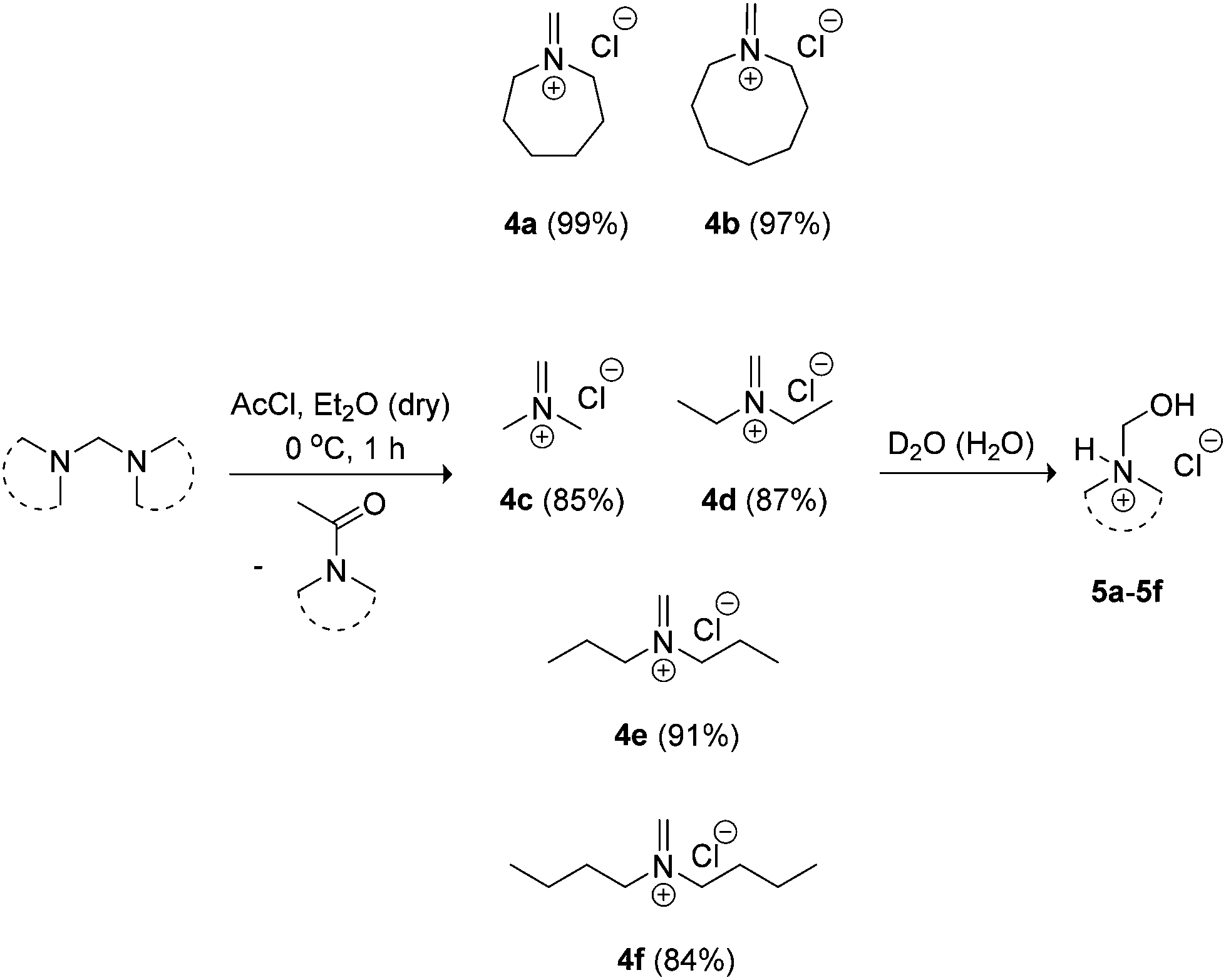 | ||
| Scheme 3 Synthesis of methylene Schiff base salts 4a–4f characterised by 1H NMR as hydrated derivatives 5a–5f. | ||
NMR spectra of iminium salts 4a–4f showed mixtures with their respective hydrated derivatives 5a–5f. For example, the 1H-NMR spectrum in CD2Cl2 gave similar intensity signals for the exo-methylene of N-methylideneazocan-1-ium chloride (4b) at 8.87 ppm and its N-hydroxymethyl derivative 5bexo-methylene at 4.74 ppm (Fig. 2a). Upon recrystallization from acetonitrile, the more stable N-(hydroxymethyl)azocan-1-ium chloride 5b was obtained (Fig. 2b). It was thus more convenient to characterize the moisture sensitive methylene Schiff base salts 4a–4f using NMR in D2O, as 5a–5f (Fig. 2c). An exception was N,N-dibutylmethaniminium chloride (4f), which appeared less hygroscopic. The NMR spectrum in CD2Cl2 contained only trace amounts of hydrated derivative 5f (Fig. 3a). The exo-methylene at 8.58 ppm in CD2Cl2 for the methylene Schiff base was replaced by the exo-methylene at 4.83 ppm in D2O for N-butyl-N-(hydroxymethyl)butan-1-aminium chloride (5f in Fig. 3b).
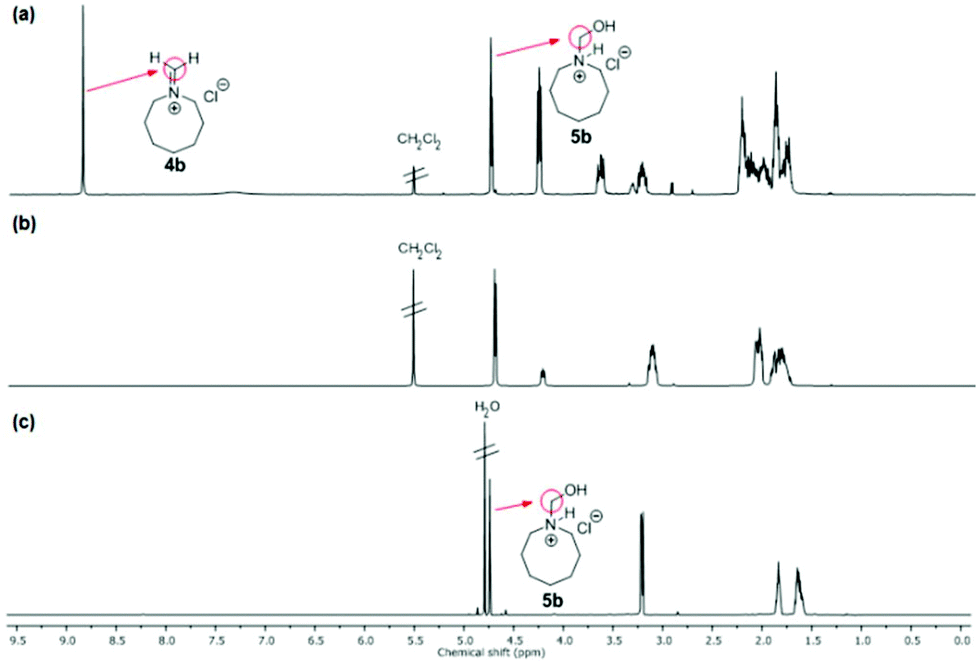 | ||
| Fig. 2 1H-NMR spectrum: (a) of the initially isolated mixture containing N-methylideneazocan-1-ium chloride (4b) and N-(hydroxymethyl)azocan-1-ium chloride (5b) in CD2Cl2 and spectra after recrystallization from MeCN in (b) CD2Cl2, and (c) D2O. | ||
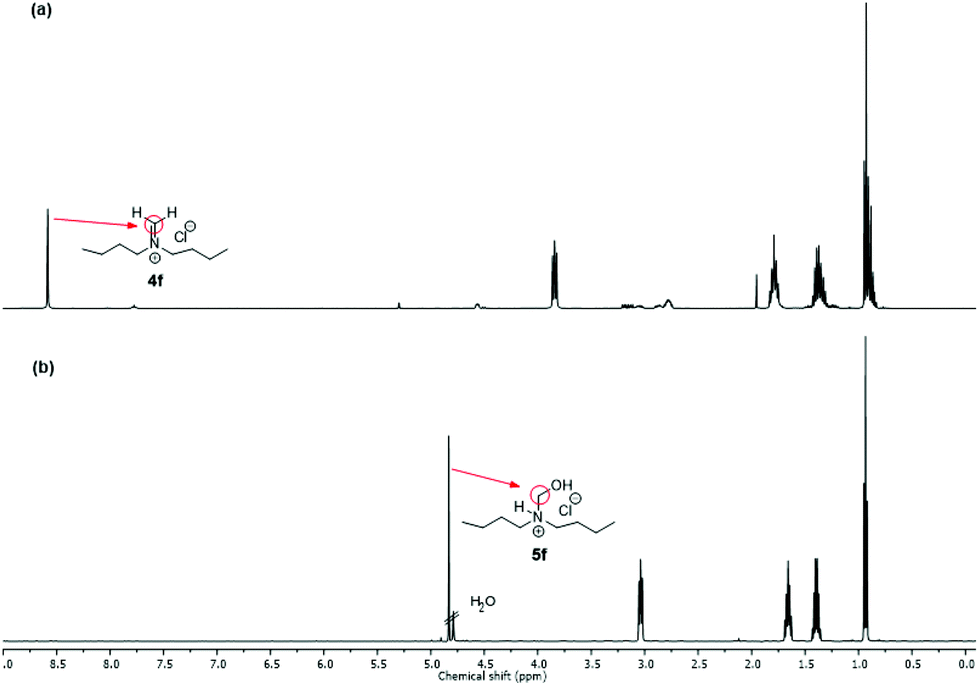 | ||
| Fig. 3 1H-NMR spectrum: (a) of N,N-dibutylmethaniminium chloride (4f) in CD2Cl2 and (b) of N-butyl-N-(hydroxymethyl)butan-1-aminium chloride (5f) in D2O. | ||
The X-ray crystal structure of N-(hydroxymethyl)azocan-1-ium chloride (5b) was obtained (Fig. 4a, Table S1†). The large eight membered ring of 5b was found to be disordered over two equally populated sites with both the N–H and O–H bonds found to be involved in H-bonding to the chloride counter ion (Fig. 4b). Interestingly, a search of the CCDC database for the R2NH–CH2–OH moiety gave only one hit, CSD code DIVDET, which was for a pyrimidine salt of tris(hydroxymethyl)ammonium chloride.30 The hydroxymethyl hydrochlorides 5a–5f were however difficult to isolate cleanly due to their susceptibility to decompose to formaldehyde.
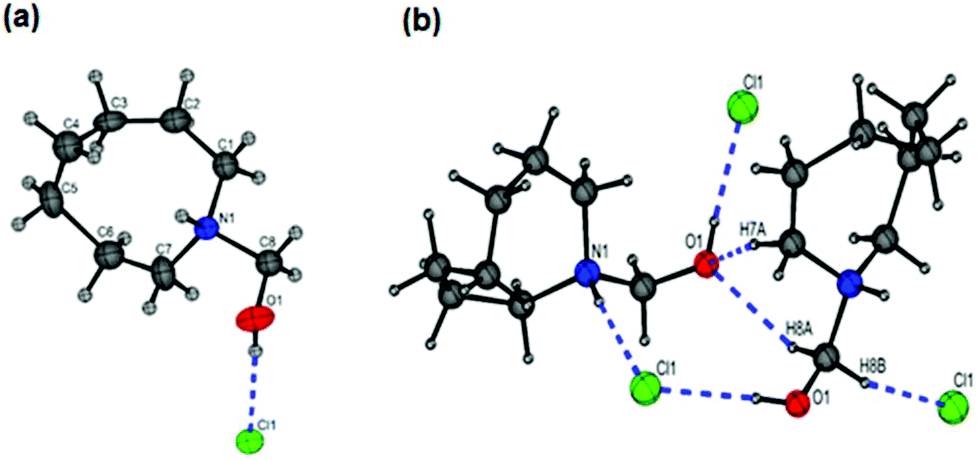 | ||
| Fig. 4 The X-ray crystal structure of N-(hydroxymethyl)azocan-1-ium chloride (5b): (a) only one component of the ring disorder shown for clarity and (b) H-bonding interactions (Table S1†). | ||
Nucleophilic addition of acrylamides or methacrylamides onto the freshly prepared methylene Schiff base salts of azepane and azocane 4a and 4b gave the hydrochloride monomer salts (6a·HCl, 6b·HCl, 7a·HCl and 7b·HCl) after precipitation from diethyl ether (Scheme 4). The monomer HCl salts were suspended in dichloromethane and basified to give the monomers 6a–6b and 7a–7b in high yields of 84–91% (from 4a–4b). Attempts to react acrylamide and methacrylamide with an analytically pure sample of N-(hydroxymethyl)azocan-1-ium chloride (5b) in dried acetonitrile resulted in the isolation of unreacted 5b and some degradation with the release of formaldehyde. It follows that yields of the monomer from addition onto methylene Schiff base salts were determined by the extent of hydration of the latter substrate.
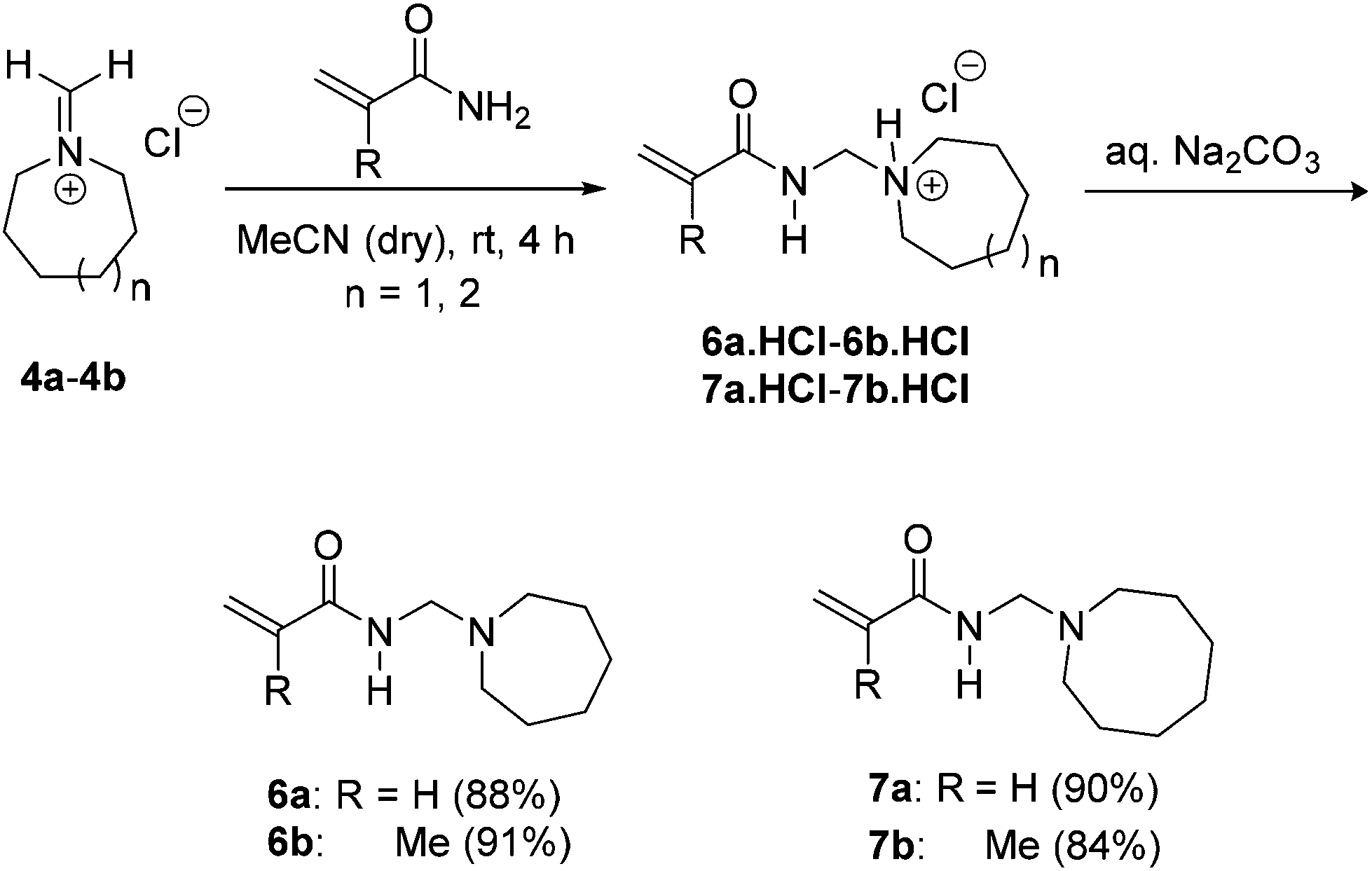 | ||
| Scheme 4 Synthesis of seven and eight-membered N-[(cycloalkylamino)methyl]acrylamides 6a–7a and N-[(cycloalkylamino)methyl]methacrylamides 6b–7b from the freshly prepared methylene Schiff base salts. | ||
The X-ray crystal structure of N-[(azocan-1-yl)methyl]prop-2-enamide hydrochloride (7a·HCl) was obtained with very small fitting errors suggesting that the ring was in the optimal conformation (Fig. 5). The crystal structure showed N–H bonds forming H-bonding interactions with the chloride anions and the oxygen atoms resulting in intermolecular packing with some weaker C–H⋯Cl and C–H⋯O (see Fig. S1, S2 and Table S2†).
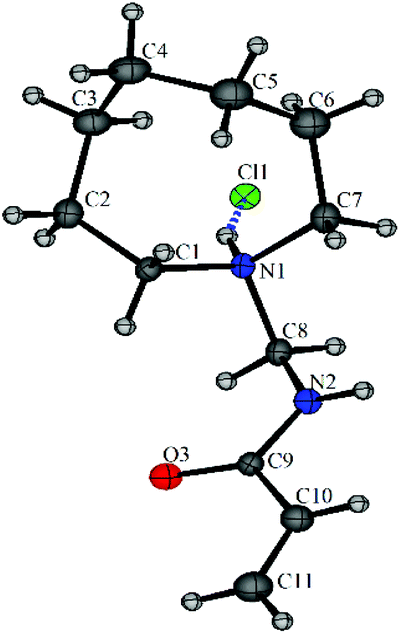 | ||
| Fig. 5 The X-ray crystal structure of N-[(azocan-1-yl)methyl]prop-2-enamide hydrochloride (7a·HCl) with one molecule from the asymmetric unit shown. | ||
For the preparation of N-dialkyl amino substituted monomers, N-[(dialkylamino)methyl]acrylamides 8a–11a and N-[(dialkylamino)methyl]methacrylamides 8b–11b, freshly prepared acyclic Schiff base salts 4c–4f were reacted with acrylamides and methacrylamides in acetonitrile at room temperature (Scheme 5). In this case, the isolation of the N-dialkyl amino substituted monomer hydrochloride salts proved difficult due to appreciable solubility in the reaction solvent and attempted precipitation solvents (including diethyl ether). Thus basification of the reaction mixture was preferred and the free monomer bases were isolated in multi-gram quantities (22–49 g) in yields of 69–90% without the isolation of the intermediate salts.
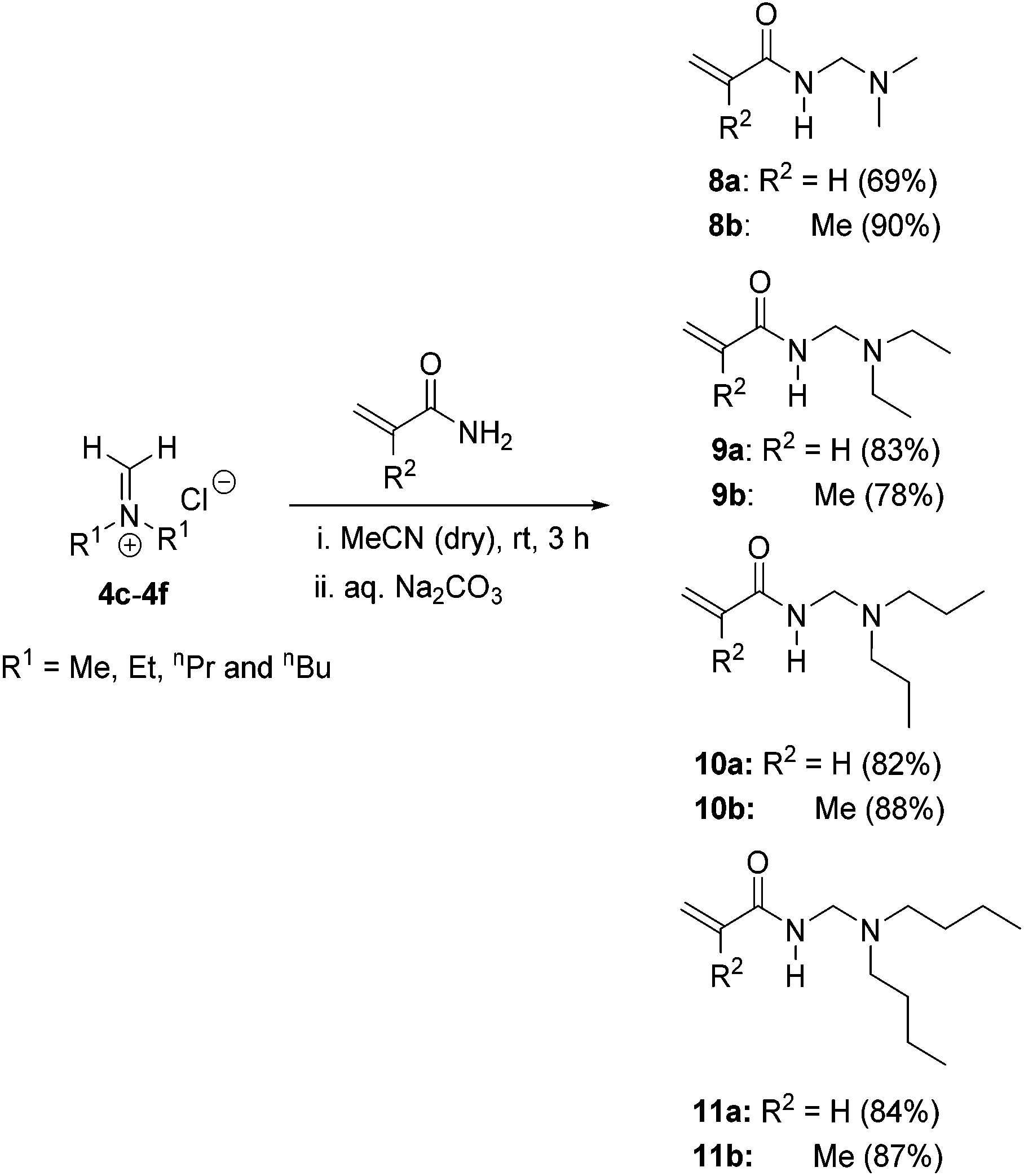 | ||
| Scheme 5 Synthesis of N-[(dialkylamino)methyl]acrylamides 8a–11a and N-[(dialkylamino)methyl]methacrylamides 8b–11b from the freshly prepared Schiff base salts. | ||
Conclusions
Readily accessible methylene Schiff base (iminium) salts have allowed the preparation of eighteen previously inaccessible acrylamides and methacrylamides containing methylene N-amino groups (both heterocyclic and acyclic). Heterocyclic substituted monomer syntheses have the added advantage of allowing the isolation of the monomer hydrochloride salt intermediate useful for polymerizations in water. For the preparation of monomers substituted with azepane, azocane, and acyclic derivatives, the iminium salts should be first isolated, prior to reactions with acrylamides and methacrylamides. Syntheses occur at low or ambient temperatures avoiding premature polymerization of vinyl compounds. Iminium salts are however hygroscopic and X-ray crystal structures of the hydrated eight-membered Schiff base salt, 1-(hydroxymethyl)azocan-1-ium chloride (5b), and the related monomer, N-[(azocan-1-yl)methyl]prop-2-enamide hydrochloride (7a·HCl) are described. Future research will involve controlled radical polymerizations of this new vinyl monomer class to give amphiphilic block copolymers for use as smart stimuli (temperature, pH, CO2)-responsive materials.Experimental
General information
Melting points were measured on a Stuart Scientific SMP1 melting point apparatus. Infrared spectra were recorded using a PerkinElmer Spec 1 with ATR attached. 1H NMR spectra were recorded at 400 or 500 MHz and 13C NMR were recorded at 101 or 125 MHz using a 400 MHz JEOL ECX and a 500 MHz Varian instrument respectively. The chemical shifts were recorded in ppm relative to Me4Si. NMR assignments were supported by DEPT. Deuterated solvents were used for homonuclear lock, and the signals were referenced to the deuterated solvent peaks. 1,4-Dioxane was used as a reference for 13C NMR in D2O. High resolution mass spectra (HRMS) were recorded using an ESI time-of-flight mass spectrometer (TOFMS) in positive mode. The precision of all accurate mass measurements was better than 5 ppm. All reactions were performed under inert conditions.Materials
All chemicals were obtained from commercial sources. Aminals, 1,1′-methylenedipyrrolidine,16 1,1′-methylenedipiperidine,17 1,1′-methylenebis(azepane),31N,N,N′,N’-tetramethylmethanediamine,16N,N,N′,N’-tetraethylmethanediamine,16 and N,N,N′,N’-tetrapropyl-methanediamine31 were readily prepared in high yields from the reaction of formaldehyde (Sigma-Aldrich, 37 wt% in H2O) with the appropriate secondary amine according to the literature procedures. Distilled aminals were stored under vacuum and dry atmospheres in desiccators at room temperature. Heptamethyleneimine (Sigma-Aldrich, 98%), acetyl chloride (AcCl, Sigma-Aldrich, 98%), acrylamide (Sigma-Aldrich, 97%), and methacrylamide (Sigma-Aldrich, 98%) were used as received. CH2Cl2 (Sigma-Aldrich, ≥99%), CDCl3 (Sigma-Aldrich, 99.8 atom%), D2O (Sigma-Aldrich, 99.9 atom%), KOH pellets (Sigma-Aldrich, ≥85%), Na2CO3 (Sigma-Aldrich, ≥99%), and MgSO4 (Sigma-Aldrich, ≥99.99%) were used as received. The synthesis of N-[(morpholin-4-yl)methyl]prop-2-enamide 1a, 2-methyl-N-[(morpholin-4-yl)methyl]prop-2-enamide 1b, N-[(pyrrolidin-1-yl)methyl]prop-2-enamide 2a and N-(piperidin-1-ylmethyl)prop-2-enamide 3a is included in our recent communication.14 For Schiff base salt and monomer synthesis all solvents were freshly distilled, and the reactions were carried out using anhydrous solvents using an inert nitrogen atmosphere. Acetonitrile (MeCN, Sigma-Aldrich, ≥99.9%) was freshly distilled over 3 Å molecular sieves and then CaH2 (Sigma-Aldrich, 95%) and Et2O (Et2O, Sigma-Aldrich, ≥99.5%) were freshly distilled over Na wire and benzophenone (Sigma-Aldrich, 95%).Synthesis of N-[(cycloalkylamino)methyl]methacrylamides using the in situ Schiff base salt approach
AcCl (14.30 mL, 0.20 mol) was added over 30 min to aminal (0.20 mol) in MeCN (40 mL) at ca. 0 °C. Methacrylamide (17.02 g, 0.20 mol) in MeCN (40 mL) was added, and stirred at ca. 20 °C for 2 h. Et2O (200 mL) was added and the hydrochloride salt of the monomer was precipitated, filtered, and dried under vacuum. The hydrochloride salts (1b·HCl–3b·HCl) were recrystallized, dried, and characterized. An aqueous solution of Na2CO3 (100 mL, 3 M) was added to a suspension of the hydrochloride salt in CH2Cl2 (100 mL) and stirred for 30 min. The organic layer was separated, and the aqueous layer was washed with CH2Cl2 (4 × 250 mL). The combined organic extracts were dried (MgSO4), filtered, and evaporated to dryness to give the monomer, which was recrystallized.![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O).
O), 1450, 1362, 1356, 1299, 1208, 1134, 1046; 1H NMR (400 MHz, CDCl3) δ 1.75–1.79 (m, 4H), 1.96 (s, 3H), 2.62 (t, J 6.4 Hz, 4H), 4.24 (dd, J 0.7, 6.2 Hz, 2H), 5.33–5.34 (m, 1H), 5.69–5.70 (m, 1H), 6.17–6.31 (brs, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 18.8 (Me), 23.7, 51.0, 58.6, 119.8 (all CH2), 140.1 (C), 168.7 (CO); HRMS (ESI) m/z [M + H]+, C9H17N2O calcd 169.1341 observed 169.1334.
O).
O), 1655, 1615, 1453, 1453, 1440, 1368, 1333, 1306, 1158, 1110, 1034; 1H NMR (400 MHz, CDCl3) δ 1.38–1.44 (m, 2H), 1.54–1.59 (m, 4H), 1.95 (s, 3H), 2.45–2.59 (m, 4H), 4.10 (d, J 6.4 Hz, 2H), 5.34 (s, 1H), 5.71 (s, 1H), 6.29–6.38 (brs, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 18.8 (Me), 24.2, 25.9, 51.6, 62.4, 119.8 (all CH2), 140.2 (C), 168.9 (CO); HRMS (ESI) m/z [M + H]+, C10H19N2O calcd 183.1497 observed 183.1493.
O).
O), 1450, 1362, 1356, 1299, 1208, 1134, 1046; 1H NMR (400 MHz, CDCl3) δ 1.75–1.79 (m, 4H), 1.96 (s, 3H), 2.62 (t, J 6.4 Hz, 4H), 4.24 (dd, J 0.7, 6.2 Hz, 2H), 5.33–5.34 (m, 1H), 5.69–5.70 (m, 1H), 6.17–6.31 (brs, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 18.8 (Me), 23.7, 51.0, 58.6, 119.8 (all CH2), 140.1 (C), 168.7 (CO); HRMS (ESI) m/z [M + H]+, C9H17N2O calcd 169.1341 observed 169.1334.
O).
O), 1655, 1615, 1453, 1453, 1440, 1368, 1333, 1306, 1158, 1110, 1034; 1H NMR (400 MHz, CDCl3) δ 1.38–1.44 (m, 2H), 1.54–1.59 (m, 4H), 1.95 (s, 3H), 2.45–2.59 (m, 4H), 4.10 (d, J 6.4 Hz, 2H), 5.34 (s, 1H), 5.71 (s, 1H), 6.29–6.38 (brs, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 18.8 (Me), 24.2, 25.9, 51.6, 62.4, 119.8 (all CH2), 140.2 (C), 168.9 (CO); HRMS (ESI) m/z [M + H]+, C10H19N2O calcd 183.1497 observed 183.1493.
Synthesis of 1,1′-methylenebis(azocane)
Heptamethyleneimine (15.0 g, 0.13 mol) was added over 30 min to formaldehyde (37 wt% in H2O, 6.2 mL, 0.07 mol) with stirring at ca. 0 °C. The solution was stirred overnight at ca. 20 °C, after which KOH pellets were added to form a saturated solution, and stirring was continued for 30 min. H2O (40 mL) was added and the mixture was extracted with Et2O (4 × 40 mL). The organic layers were combined and washed with H2O (3 × 20 mL), dried (MgSO4), and evaporated to dryness. Fractional distillation under reduced pressure gave the title compound as a colorless liquid (14.69 g, 88%); bp 138–140 °C (0.25 mmHg); vmax (neat, cm−1) 2915, 2846, 2779, 1657, 1472, 1448, 1358, 1250, 1158, 1094, 1046, 1017; 1H NMR (400 MHz, CDCl3) δ 1.51–1.68 (m, 20H), 2.53–2.60 (m, 8H), 3.02 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 26.1, 27.9, 28.2, 52.9, 83.8 (all CH2); HRMS (ESI) m/z [M + H]+, C15H31N2, calcd 239.2487, observed 239.2312.Synthesis of N,N,N′,N’-tetrabutylmethanediamine
Dibutylamine (68.00 mL, 0.40 mol) was added over 30 min to formaldehyde (37 wt% in H2O, 15.00 mL, 0.20 mol) with stirring at ca. 0 °C. The solution was stirred overnight at ca. 20 °C. KOH pellets were added to form a saturated solution, and the drying agent was removed by filtration. Fractional distillation under reduced pressure gave the title compound as a colorless liquid (45.98 g, containing about 10% of suspected (dibutylamino)methanol impurity by 1H NMR); bp 112–114 °C (0.25 mmHg); 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J 7.2 Hz, 12H), 1.28 (m, 8H), 1.33–1.41 (m, 8H), 2.42 (t, J 7.3 Hz, 8H), 2.98 (s, 2H); 13C NMR (101 MHz, CDCl3) δ 14.3 (Me), 20.9, 29.5, 52.0, 75.6 (all CH2). HRMS (ESI) m/z [M + H]+, C17H39N2 calcd 271.3113 observed 271.3143. The title compound was used without further purification to prepare N,N-dibutylmethaniminium chloride (4f).Synthesis of Schiff base salts
AcCl (21.33 mL, 0.3 mol) was added over 30 min to a stirred solution of aminal (0.3 mol) in Et2O (150 mL) at ca. 0 °C, and the resulting white precipitate was stirred for an additional 30 min. Et2O (200 mL) was added and the precipitate was filtered, and dried under vacuum to give the methylene Schiff-base salt (4a–4f). Iminium salts 4a–4f were immediately used in addition reactions with acrylamides and methacrylamides due to their hygroscopic nature.1H NMR (400 MHz, D2O) δ 1.63–1.66 (m, 4H), 1.78–1.85 (brs, 4H), 3.17–3.21 (m, 4H), 4.77 (d, J 0.8 Hz, 2H); 13C NMR (101 MHz, D2O, 1,4-dioxane added) δ 25.2, 26.6, 46.8, 82.4 (all CH2).
1H NMR (500 MHz, D2O) δ 1.57–1.67 (m, 6H), 1.81–1.86 (m, 4H), 3.21 (t, J 5.8 Hz, 4H), 4.74 (s, 2H); 13C NMR (125 MHz, D2O, 1,4-dioxane added) δ 23.9, 24.8, 25.2, 45.8, 82.4 (all CH2).
1H NMR (400 MHz, D2O) δ 2.79 (s, 6H), 4.56 (s, 2H); 13C NMR (101 MHz, D2O, 1,4-dioxane added) δ 35.2 (Me), 82.4 (CH2).
1H NMR (400 MHz, D2O) δ 1.23 (t, J 7.3 Hz, 6H), 3.00–3.07 (m, 4H), 4.78 (s, 2H); 13C NMR (101 MHz, D2O, 1,4-dioxane added) δ 11.2 (Me), 42.9, 82.4 (both CH2).
1H NMR (400 MHz, D2O) δ 0.92 (t, J 7.6 Hz, 6H), 1.65 (sext, J 7.6 Hz, 4H), 2.95 (t, J 7.6 Hz, 4H), 4.78 (s, 2H); 13C NMR (101 MHz, D2O, 1,4-dioxane added) δ 10.8 (Me), 19.7, 49.7, 82.4 (all CH2).
1H NMR (400 MHz, D2O) δ 0.93 (t, J 6.0 Hz, 6H), 1.39 (sext, J 6.0 Hz, 4H), 1.66 (quint, J 6.0 Hz, 4H), 3.04 (t, J 6.0 Hz, 4H), 4.83 (s, 2H); 13C NMR (101 MHz, D2O, 1,4-dioxane added) δ 13.4 (Me), 19.8, 28.2, 47.9, 82.4 (all CH2).
Synthesis of seven and eight-membered N-[(cycloalkylamino)-methyl]acrylamides and N-[(cycloalkylamino)methyl]methacrylamides
A solution of acrylamide or methacrylamide (0.03 mol) in MeCN (10 mL) was added to a solution of freshly prepared Schiff base salt 4a or 4b (0.04 mol) in MeCN (30 mL) and stirred at ca. 20 °C for 4 h. Et2O (200 mL) was added and the hydrochloride salt of the monomer was precipitated, filtered, and dried under vacuum. The hydrochloride salts (6a·HCl–6b·HCl and 7a·HCl–7b·HCl) were recrystallized, dried, and characterized. An aqueous solution of Na2CO3 (15 mL, 3 M) was added to a suspension of the hydrochloride salt in CH2Cl2 (20 mL) and left to stir for an additional 30 min and extracted with CH2Cl2 (4 × 50 mL). The organic layer was dried (MgSO4), filtered and evaporated to dryness to give the corresponding monomers 6a–6b and 7a–7b.O).
O), 1623, 1539, 1452, 1405, 1365, 1309, 1227, 1133, 1080; 1H NMR (400 MHz, CDCl3) δ 1.52–1.61 (m, 8H), 2.68 (t, J 5.6 Hz, 4H), 4.23 (d, J 6.2 Hz, 2H), 5.61 (dd, J 1.6, 10.2 Hz, 1H), 6.11 (dd, J 10.2, 17.0 Hz, 1H), 6.25 (dd, J 1.6, 17.0 Hz, 1H), 6.31–6.40 (brs, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 26.9, 28.6, 53.1, 62.6, 126.7 (all CH2), 131.1 (CH), 166.1 (CO); HRMS (ESI) m/z [M + H]+, C10H19N2O calcd 183.1497 observed 183.1516.
O).
O), 1526, 1373, 1313, 1201, 1133, 1088, 1020; 1H NMR (400 MHz, CDCl3) δ 1.56–1.68 (m, 8H), 1.96 (s, 3H), 2.72 (t, J 5.5 Hz, 4H), 4.24 (d, J 6.1 Hz, 2H), 5.33 (s, 1H), 5.69 (s, 1H), 6.12–6.22 (brs, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 18.8 (Me), 27.0, 28.6, 53.2, 62.7, 119.6 (all CH2), 140.3 (C), 168.8 (CO); HRMS (ESI) m/z [M + H]+, C11H21N2O calcd 197.1654 observed 197.1708.
O).
O), 1624, 1536, 1363, 1232, 1162, 1095, 1060; 1H NMR (400 MHz, CDCl3) δ 1.50–1.62 (m, 10H), 2.62–2.70 (m, 4H), 4.27 (d, J 6.0 Hz, 2H), 5.65 (d, J 10.2 Hz, 1H), 5.91–6.02 (brs, 1H), 6.10 (dd, J 10.2, 17.0 Hz, 1H), 6.28 (d, J 17.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 26.1, 27.7, 28.0, 51.6, 62.6, 126.7 (all CH2), 131.2 (CH), 166.0 (CO); HRMS (ESI) m/z [M + H]+, C11H21N2O, calcd 197.1656, observed 197.1654.
O).
O), 1523, 1452, 1363, 1201, 1162, 1095, 1060; 1H NMR (400 MHz, CDCl3) δ 1.49–1.59 (m, 10H), 1.93 (s, 3H), 2.61–2.66 (m, 4H), 4.21 (t, J 4.3 Hz, 2H), 5.28 (s, 1H), 5.64 (s, 1H), 6.12–6.26 (brs, 1H, NH); 13C NMR (101 MHz, CDCl3) δ 18.8 (Me), 26.0, 27.7, 28.0, 51.5, 62.7, 119.2 (all CH2), 140.4 (C), 168.9 (CO); HRMS (ESI) m/z [M + H]+, C12H23N2O, calcd 211.1838, observed 211.1810.
Synthesis of N-[(dialkylamino)methyl]acrylamides and N-[(dialkyl-amino)methyl]methacrylamides
A solution of acrylamide or methacrylamide (0.25 mol) in MeCN (50 mL) was added to a stirred solution of freshly prepared Schiff base salt 4c–4f (0.25 mol) in MeCN (50 mL) and stirred at ca. 20 °C for 3 h. An aqueous solution of Na2CO3 (150 mL, 3 M) was added and the solution was stirred for an additional 30 min and extracted with CH2CL2 (4 × 250 mL). The organic layer was dried (MgSO4), filtered and evaporated to give the corresponding acrylamides 8a–11a and methacrylamides 8b–11b.O), 1625, 1536, 1407, 1230, 1029; 1H NMR (400 MHz, CDCl3) δ 2.24 (s, 6H), 4.05 (d, J 6.4 Hz, 2H), 5.62 (dd, J 1.6, 10.2 Hz, 1H), 6.12 (dd, J 10.2, 17.0 Hz, 1H), 6.26 (dd, J 1.6, 17.0 Hz, 1H), 6.71–6.82 (brs, 1H); 13C NMR (101 MHz, CDCl3) δ 41.8 (Me), 61.5, 126.0 (both CH2), 130.7 (CH), 166.2 (CO); HRMS (ESI) m/z [M + H]+, C6H13N2O, calcd 129.1028, observed 129.1026.
O), 1619, 1523, 1453, 1311, 1196, 1049, 1033; 1H NMR (400 MHz, CDCl3) δ 1.92 (s, 3H), 2.23 (s, 6H), 4.03 (d, J 6.3 Hz, 2H), 5.30 (s, 1H), 5.66 (s, 1H), 6.31–6.45 (brs, 1H); 13C NMR (101 MHz, CDCl3) δ 18.7, 42.3 (both Me), 62.2, 119.5 (both CH2), 140.1 (C), 169.0 (CO); HRMS (ESI) m/z [M + H]+, C7H15N2O calcd 143.1184, observed 143.1180.
O), 1624, 1536, 1464, 1233, 1206, 1067; 1H NMR (400 MHz, CDCl3) δ 1.08 (t, J 7.2 Hz, 6H), 2.56 (q, J 7.2 Hz, 4H), 4.29 (d, J 6.1 Hz, 2H), 5.64 (dd, J 1.4, 10.2 Hz, 1H), 5.90–5.99 (brs, 1H), 6.09 (dd, J 10.2, 17.0 Hz, 1H), 6.28 (dd, J 1.4, 17.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 12.7 (Me), 45.4, 56.9, 126.6 (all CH2), 131.0 (CH), 166.1 (CO); HRMS (ESI) m/z [M + H]+, C8H17N2O, calcd 157.1341, observed 157.1337.
O), 1617, 1522, 1455, 1375, 1197, 1066, 1046; 1H NMR (400 MHz, CDCl3) δ 1.03 (t, J 7.2 Hz, 6H), 1.90 (s, 3H), 2.52 (q, J 7.2 Hz, 4H), 4.22 (d, J 6.0 Hz, 2H), 5.27 (s, 1H), 5.63 (s, 1H), 6.11–6.21 (brs, 1H); 13C NMR (101 MHz, CDCl3) δ 12.7, 18.8 (both Me), 45.4, 57.3, 119.5 (all CH2), 140.2 (C), 168.9 (CO); HRMS (ESI) m/z [M + H]+, C9H19N2O calcd 171.1497, observed 171.1789.
O), 1623, 1550, 1457, 1246, 1185, 1069; 1H NMR (500 MHz, CDCl3) δ 0.82 (t, J 7.4 Hz, 6H), 1.44 (sext, J 7.4 Hz, 4H), 2.39 (t, J 7.4 Hz, 4H), 4.23 (d, J 6.0 Hz, 2H), 5.58 (dd, J 1.6, 10.2 Hz, 1H), 6.11 (dd, J 10.2, 17.0 Hz, 1H), 6.22 (dd, J 1.6, 17.0 Hz, 1H), 6.28–6.39 (brs, 1H); 13C NMR (125 MHz, CDCl3) δ 11.9 (Me), 20.9, 54.0, 58.1, 126.5 (all CH2), 131.1 (CH), 166.1 (CO); HRMS (ESI) m/z [M + H]+, C10H21N2O calcd 185.1654, observed 185.1663.
O), 1619, 1524, 1456, 1374, 1183, 1075, 1052; 1H NMR (400 MHz, CDCl3) δ 0.85 (t, J 7.4 Hz, 6H), 1.46 (sext, J 7.4 Hz, 4H), 1.93 (s, 3H), 2.42 (t, J 7.4 Hz, 4H), 4.22 (d, J 6.0 Hz, 2H), 5.29 (s, 1H), 5.65 (s, 1H), 6.02–6.15 (brs, 1H); 13C NMR (101 MHz, CDCl3) δ 11.9, 18.8 (both Me), 20.9, 54.1, 58.5, 119.4 (all CH2), 140.3 (C), 168.9 (CO); HRMS (ESI) m/z [M + H]+, C11H23N2O calcd 199.1810, observed 199.1800.
O), 1625, 1542, 1456, 1459, 1366, 1180, 1071; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J 7.3 Hz, 6H), 1.24–1.33 (m, 4H), 1.40–1.47 (m, 4H), 2.45 (t, J 7.5 Hz, 4H), 4.26 (d, J 6.0 Hz, 2H), 5.63 (dd, J 1.5, 10.2 Hz, 1H), 5.88–6.00 (brs, 1H), 6.10 (dd, J 10.2, 17.0 Hz, 1H), 6.27 (dd, J 1.5, 17.0 Hz, 1H); 13C NMR (101 MHz, CDCl3) δ 14.1 (Me), 20.7, 30.0, 51.9, 58.3, 126.7 (all CH2), 131.0 (CH), 166.0 (CO); HRMS (ESI) m/z [M + H]+, C12H25N2O calcd 213.1967, observed 213.1952.
O), 1525, 1456, 1374, 1296, 1179, 1083, 1034; 1H NMR (400 MHz, CDCl3) δ 0.89 (t, J 7.4 Hz, 6H), 1.25–1.34 (m, 4H), 1.40–1.47 (m, 4H), 1.95 (s, 3H), 2.46 (t, J 7.4 Hz, 4H), 4.24 (d, J 5.9 Hz, 2H), 5.32 (s, 1H), 5.66 (s, 1H), 5.98–6.06 (brs, 1H); 13C NMR (101 MHz, CDCl3) δ 14.1, 18.8 (both Me), 20.7, 30.0, 51.9, 58.6, 119.4 (all CH2), 140.4 (C), 168.9 (CO); HRMS (ESI) m/z [M + H]+, C13H27N2O calcd 227.2123, observed 227.2129.
X-ray crystallographic studies
Single crystal X-ray diffraction data were collected using an Oxford Diffraction Xcalibur system operated using the CrysAlisPro software36 and the data collection temperature was controlled at 150 K using a Cryojet system from Rigaku Oxford Diffraction. The crystals were hygroscopic and were first coated in cold paraffin oil before being transferred to the cold stream on the diffractometer. The crystal structures were solved using ShelxT version 2014/5,37 and refined using ShelxL version 2017/138 both of which were operated within the Oscail software package.39![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) , a = 10.1121(7), b = 10.4420(6), c = 12.0205(14) Å, α = 95.695(8), β = 91.086(8), γ = 97.671(5)°, V = 1251.02(19) Å3, Z = 4, T = 150.0(1) K, ρcalcd = 1.236 g cm−3, refinement of 271 parameters on 4487 independent reflections out of 7456 measured reflections (Rint = 0.0638) led to R1 = 0.0684 (I > 2σ(I)), wR2 = 0.2128 (all data), and S = 0.979 with the largest difference peak and hole of 0.852 and −0.705 e Å−3.
, a = 10.1121(7), b = 10.4420(6), c = 12.0205(14) Å, α = 95.695(8), β = 91.086(8), γ = 97.671(5)°, V = 1251.02(19) Å3, Z = 4, T = 150.0(1) K, ρcalcd = 1.236 g cm−3, refinement of 271 parameters on 4487 independent reflections out of 7456 measured reflections (Rint = 0.0638) led to R1 = 0.0684 (I > 2σ(I)), wR2 = 0.2128 (all data), and S = 0.979 with the largest difference peak and hole of 0.852 and −0.705 e Å−3.
Crystallographic data for compounds 5b and 7a·HCl have been deposited with the Cambridge Crystallographic Data Centre with deposition numbers CCDC 1819145 and 1819144 respectively.†
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Minstry of Education of the Kingdom of Saudi Arabia for supporting the PhD of Abdullah Alzahrani, and the Irish Research Council (IRC) for Goverment of Ireland Postdoctoral Fellowships for Styliana I. Mirallai and Benjamin A. Chalmers.Notes and references
- V. C. Mannich and W. Krösche, Arch. Pharm., 1912, 250, 647–667, DOI:10.1002/ardp.19122500151.
- F. F. Blicke, The Mannich Reaction, Org. React., 2011, 1(10), 303–341, DOI:10.1002/0471264180.or001.10.
- V. E. Müller, K. Dinges and W. Graulich, Die Makromol. Chem., 1962, 57, 27–51, DOI:10.1002/macp.1962.020570103.
- V. E. Müller and H. Thomas, Angew. Makromol. Chem., 1973, 34, 111–133, DOI:10.1002/apmc.1973.050340108.
- R. C. Baltieri, L. H. Innocentini-Mei, W. M. S. C. Tamashiro, L. Peres and E. Bittencourt, Eur. Polym. J., 2002, 38, 57–62, DOI:10.1016/S0014-3057(01)00177-X.
- M. L. Eritsyan, Z. B. Barsegyan, R. A. Karamyan, S. M. Manukyan, T. D. Karapetyan and K. A. Martirosyan, Russ. J. Appl. Chem., 2011, 84, 1257–1260, DOI:10.1134/S1070427211070238.
- E. C. Cho, J. Lee and K. Cho, Macromolecules, 2003, 36, 9929–9934, DOI:10.1021/ma034851d.
- D. Roy, W. L. A. Brooks and B. S. Sumerlin, Chem. Soc. Rev., 2013, 42, 7214–7243, 10.1039/c3cs35499g.
- Z. Song, K. Wang, C. Gao, S. Wang and W. Zhang, Macromolecules, 2015, 49, 162–171, DOI:10.1021/acs.macromol.5b02458.
- K. Wang, Z. Song, C. Liu and W. Zhang, Polym. Chem., 2016, 7, 3423–3433, 10.1039/c6py00526h.
- K. Zhou, Y. Wang, X. Huang, K. Luby-Phelps, B. D. Sumer and J. Gao, Angew. Chem., Int. Ed., 2011, 50, 6109–6114, DOI:10.1002/anie.201100884.
- H.-J. Li, J.-Z. Du, J. Liu, X.-J. Du, S. Shen, Y.-H. Zhu, X. Wang, X. Ye, S. Nie and J. Wang, ACS Nano, 2016, 10, 6753–6761, DOI:10.1021/acsnano.6b02326.
- B. A. Chalmers, C. Magee, D. L. Cheung, P. B. Zetterlund and F. Aldabbagh, Eur. Polym. J., 2017, 97, 129–137, DOI:10.1016/j.eurpolymj.2017.10.004.
- B. A. Chalmers, A. Alzahrani, G. Hawkins and F. Aldabbagh, J. Polym. Sci., Part A: Polym. Chem., 2017, 55, 2123–2128, DOI:10.1002/pola.28607.
- X. Su, M. F. Cunningham and P. G. Jessop, Polym. Chem., 2014, 5, 940–944, 10.1039/c3py01382k.
- H. Heaney, G. Papageorgiou and R. F. Wilkins, Tetrahedron, 1997, 53, 2941–2958, DOI:10.1016/S0040-4020(96)01174-X.
- A. Porzelle and C. M. Williams, Synthesis, 2006, 3025–3030, DOI:10.1055/s-2006-942539.
- H. Böhme and K. Hartke, Chem. Ber., 1960, 93, 1305–1309, DOI:10.1002/cber.19600930610.
- H. Böhme and P. Backhaus, Liebigs Ann. Chem., 1975, 1790–1796, DOI:10.1002/jlac.197519751007.
- J. Schreiber, H. Maag, N. Hashimoto and A. Eschenmoser, Angew. Chem., Int. Ed. Engl., 1971, 10, 330–331, DOI:10.1002/anie.197103301.
- G. Kinast and L.-F. Tietze, Angew Chem., Int. Ed. Engl., 1976, 15, 239–240, DOI:10.1002/anie.197602391.
- N. Abe, F. Fujisaki and K. Sumoto, Chem. Pharm. Bull., 1998, 46, 142–144, DOI:10.1248/cpb.46.142.
- N. Pemberton, V. Åberg, H. Almstedt, A. Westermark and F. Almqvist, J. Org. Chem., 2004, 69, 7830–7835, DOI:10.1021/jo048554y.
- Y.-Y. Ku, T. Grieme, Y.-M. Pu, A. V. Bhatia and S. A. King, Tetrahedron Lett., 2005, 46, 1471–1474, DOI:10.1016/j.tetlet.2005.01.027.
- B. R. Buckley, P. C. B. Page, H. Heaney, E. P. Sampler, S. Carley, C. Brocke and M. A. Brimble, Tetrahedron, 2005, 61, 5876–5888, DOI:10.1016/j.tet.2005.03.130.
- V. Werner, M. Ellwart, A. J. Wagner and P. Knochel, Org. Lett., 2015, 17, 2026–2029, DOI:10.1021/acs.orglett.5b00801.
- D. Alker, L. M. Harwood and C. E. Williams, Tetrahedron, 1997, 53, 12671–12678, DOI:10.1016/S0040-4020(97)00788-6.
- H. Möhrle and G. Keller, Z. Naturforsch., B: J. Chem. Sci., 2003, 58, 885–902, DOI:10.1021/jo01291a032.
- T. A. Bryson, G. H. Bonitz, C. J. Reichel and R. E. Dardis, J. Org. Chem., 1980, 45, 524–525, DOI:10.1021/jo01291a032.
- C. A. M. A. Huq, S. Fouzia and M. NizamMohideen, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2013, 69, 1766, DOI:10.1107/S1600536813030559.
- A. R. Bhat, A. I. Bhat, F. Athar and A. Azam, Helv. Chim. Acta, 2009, 92, 1644–1656, DOI:10.1002/hlca.200800461.
- C. Karakus, L. H. Fischer, S. Schmeding, J. Hummel, N. Risch, M. Schäferling and Elisabeth Holder, Dalton Trans., 2012, 41, 9623–9632, 10.1039/C2DT30835E.
- K. A. Jensen and L. Henriksen, Acta Chem. Scand., Ser. B, 1975, 29, 877–883, DOI:10.3891/acta.chem.scand.29b-0877.
- C. Rochin, O. Babot, J. Dunoguès and F. Duboudin, Synthesis, 1986, 228–229, DOI:10.1055/s-1986-31627.
- H. Böhme and E. Raude, Chem. Ber., 1981, 114, 3421–3429, DOI:10.1002/cber.19811141023.
- CrysAlisPro, 1.171.37.38, Rigaku Corporation, Oxford, UK, 2015. http://journals.iucr.org/e/services/stdswrefs.html Search PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A: Found. Crystallogr., 2015, 71, 3–8, DOI:10.1107/S2053273314026370.
- G. Sheldrick, Acta Crystallogr., Sect. C: Cryst. Struct. Commun., 2015, 71, 3–8, DOI:10.1107/S2053229614024218.
- P. McArdle, J. Appl. Crystallogr., 2017, 50, 320–326, DOI:10.1107/S1600576716018446.
Footnotes |
| † Electronic supplementary information (ESI) available. CCDC 1819144 and 1819145. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob00811f |
| ‡ Present address: Department of Pharmacy, School of Life Sciences, Pharmacy and Chemistry, Kingston University, Penrhyn Road, Kingston upon Thames, KT1 2EE, UK. E-mail: F.Aldabbagh@kingston.ac.uk. |
| This journal is © The Royal Society of Chemistry 2018 |