
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Total synthesis of incargranine A†
Patrick D.
Brown
and
Andrew L.
Lawrence
 *
*
EaStCHEM School of Chemistry, University of Edinburgh, Joseph Black Building, David Brewster Road, Edinburgh, EH9 3FJ, UK. E-mail: a.lawrence@ed.ac.uk
First published on 4th April 2018
Abstract
Synthetic studies into the origins of the alkaloid incargranine A have resulted in the development of a four-step (longest linear sequence) total synthesis. This synthesis has been scaled-up to provide gram-scale quantities of material, which would alternatively require extraction of several metric-tons of dried-whole Chinese Trumpet-Creeper plants (Incarvillea mairei var. grandiflora).
In 2009 Zhang and co-workers isolated the alkaloid incargranine A (1) from Incarvillea mairei var. grandiflora, a Bignonia plant more commonly known as the Chinese Trumpet-Creeper plant (Scheme 1).1 Incargranine A (1) has not yet succumbed to total synthesis and represents a particularly scarce natural product, constituting just 0.0000002% by weight of the dried whole plant. Therefore, a practical – i.e., efficient and scalable – chemical synthesis of incargranine A (1) might advance a better understanding of its biological function. The novel framework of incargranine A (1) contains a synthetically daunting bridged-cyclohexane ring, in which all six-carbon atoms are stereogenic. Nevertheless, we were hopeful that if we could gain insight into how nature synthesizes this alkaloid a step-economical biomimetic strategy could be developed.
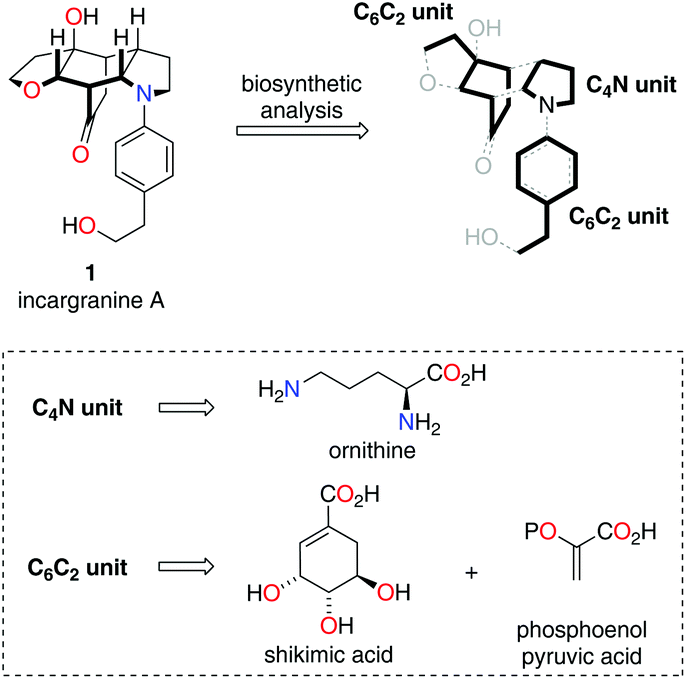 | ||
| Scheme 1 Structure and biosynthetic analysis of incargranine A. | ||
Our biosynthetic analysis, shown in Scheme 1, reveals incargranine A (1) is likely constructed from two shikimate-derived C6C2 units linked together by an ornithine-derived C4N unit. Our previous biomimetic studies on related phenylethanoid alkaloids provide important clues as to the potential origins of incargranine A (1).2 We recently proposed that a network of pathways, all originating from a simple biosynthetic precursor, diamine 2, could account for the formation of several structurally distinct phenylethanoid natural products (Scheme 2).2d In our proposal, diamine 2 can participate in a pair of divergent oxidative pathways (Scheme 2; pathways 1 and 2). As shown in Scheme 2, pathway 1 terminates in the formation of incarviditone (3)3 and incarvilleatone (4),4via the intermediacy of cornoside (5)5 and rengyolone (6),6 whereas pathway 2 results in the production of incargranine B (7).2a–c,7 It was proposed that these two divergent pathways could re-converge to give millingtonine (8),8via a crossed-dimerization of cornoside 5, from pathway 1, and a PLP (pyridoxal phosphate) derived enamine 9, from pathway 2 (Scheme 2; pathway 3).2d The chemical feasibility of this re-convergent pathway was demonstrated in our seven-step biomimetic total synthesis of millingtonine (8).2d Herein, we propose that an additional re-convergent pathway could give rise to incargranine A (1) (Scheme 2; pathway 4). Thus, a Michael reaction between PLP-enamine 9 and rengyolone (6) would give an intermediate imine 11, which would ring-close through a condensation/Mannich reaction sequence to give incargranine A (1).9 To investigate the feasibility of this second re-convergent pathway, and in the hope of establishing a practical solution to the supply problem associated with incargranine A (1),1 we decided to pursue the development of a biomimetic synthetic strategy.
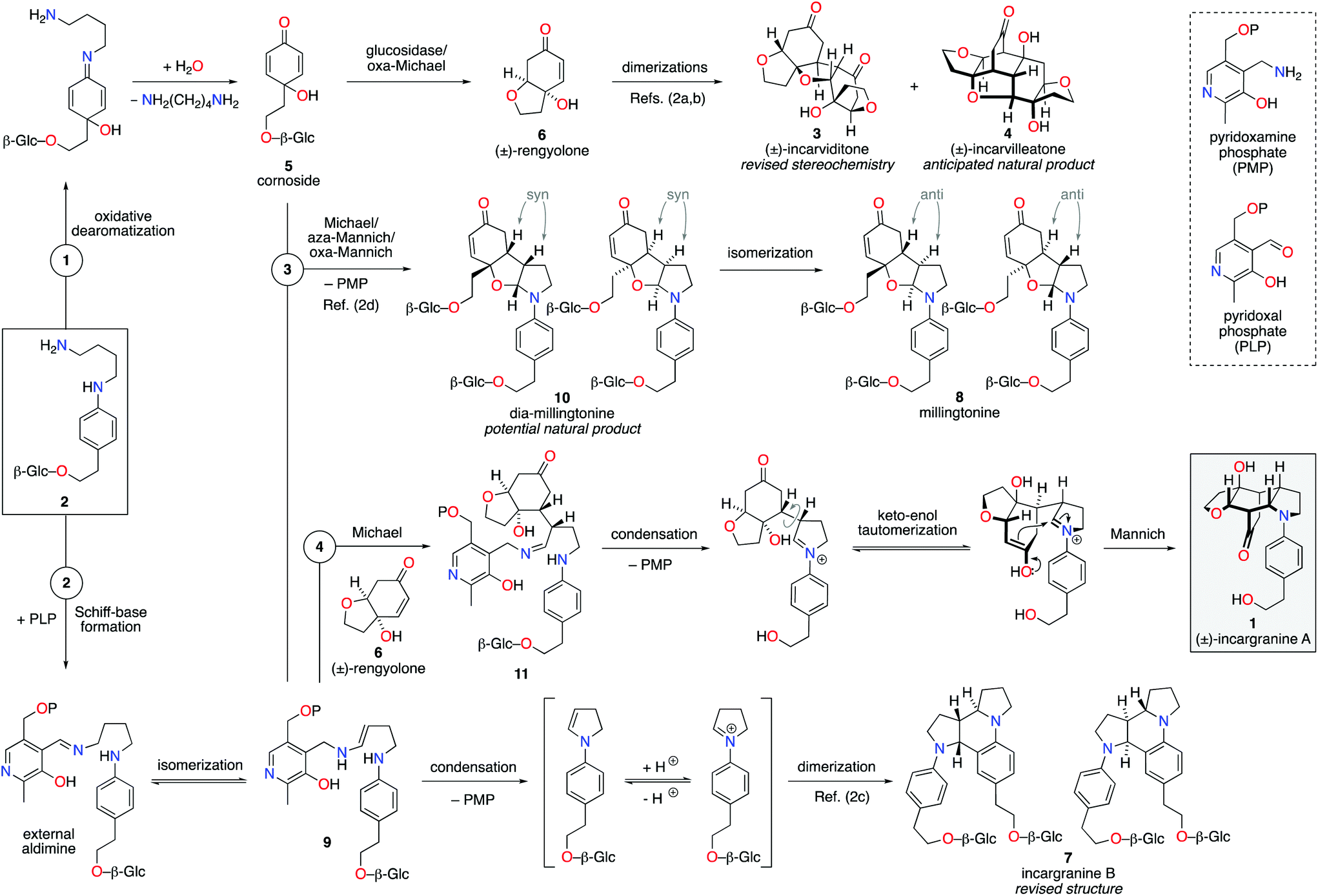 | ||
| Scheme 2 Proposed network of biosynthetic pathways towards a family of plant-derived phenylethanoid natural products, including incargranine A. | ||
Condensation of 4-aminophenethyl alcohol 12 with (Z)-1,4-dichlorobut-2-ene gave N-aryl-2,5-dihydropyrrole 13 in 87% yield (Scheme 3).10 The primary alcohol functional group was then protected under standard conditions as a tert-butyldimethylsilyl ether, to give alkene 14 in 84% yield. Exposure of alkene 14 to our previously developed RhHCO(PPh3)3 and pyrrolidine reaction conditions gave the expected aminal intermediate 15.2d,11 Due to the instability of aminal 15, and in the interests of practicality and efficiency, rengyolone (6), which can be readily prepared from tyrosol in 3 steps,2a was added directly to this crude reaction mixture. Monitoring the reaction by 1H NMR spectroscopy revealed it took 10 days at ambient temperature for aminal 15 to be consumed. Purification of the resulting crude reaction mixture by column chromatography resulted in a 12% isolated yield of an unwanted crossed-dimer 16, with no detectable formation of the desired product 17. Hemi-aminal 16 is presumably formed via a domino Michael/aza-Mannich/oxa-Mannich reaction sequence. In contrast, a final Mannich reaction between C5 and C1′′ would be required for formation of the incargranine A framework 17 (Scheme 3). Although this result demonstrates the viability of a crossed-dimerization between aminal 15 and rengyolone (6), several issues presented themselves with respect to using this strategy to access incargranine A (1). Firstly, rengyolone (6) proved to be relatively unreactive in the crossed-dimerization, taking over a week to give full consumption of starting material 15, while comparable reactions with para-quinols were generally complete in 24 h.2d Furthermore, the low yield of crossed-dimer 16, even after these prolonged reaction times, was not a promising start to the development of an efficient synthesis. Finally, and most importantly, our attempts to rearrange hemi-aminal 16 to give the incargranine A framework 17, via a retro-oxa-Mannich/Mannich reaction sequence, were unsuccessful.12 This prompted us to reconsider our biosynthetic proposal and synthetic strategy.
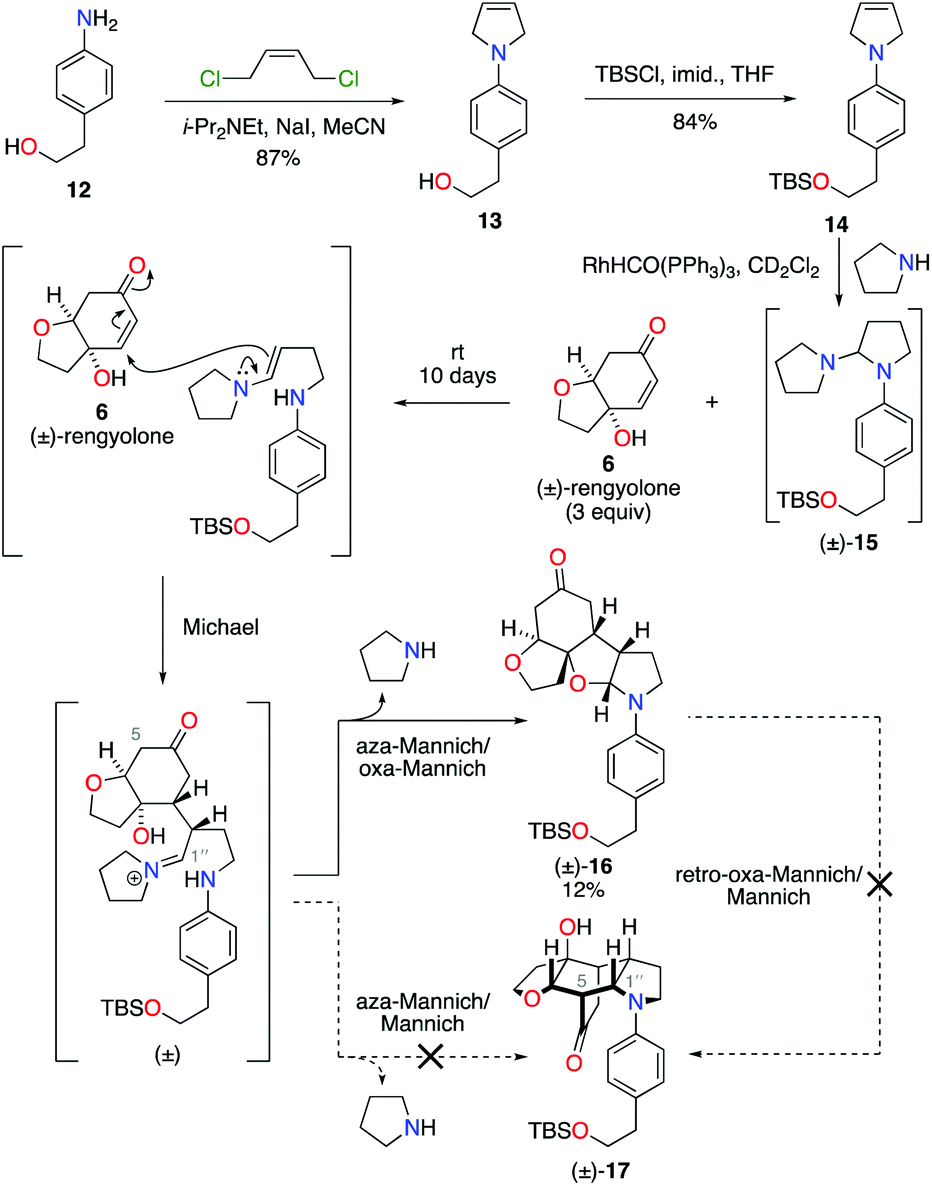 | ||
| Scheme 3 Failed approach to synthesize incargranine A. | ||
Upon further evaluation of the incargranine A (1) framework it became apparent that it might instead be derived from the syn-diastereomer of millingtonine, dia-millingtonine (10), which we had previously identified as a potential natural product and direct biosynthetic precursor to millingtonine (8) (Scheme 2; pathway 3).2d Specifically, the putative aglycone of dia-millingtonine, diol 18, could undergo a domino retro-oxa-Mannich/oxa-Michael/Mannich reaction sequence to give incargranine A (1) (Scheme 4).13 If this pathway could be shown to be chemically feasible it would lend further support to our proposal that dia-millingtonine (1) represents an as-yet-undiscovered natural product.2d
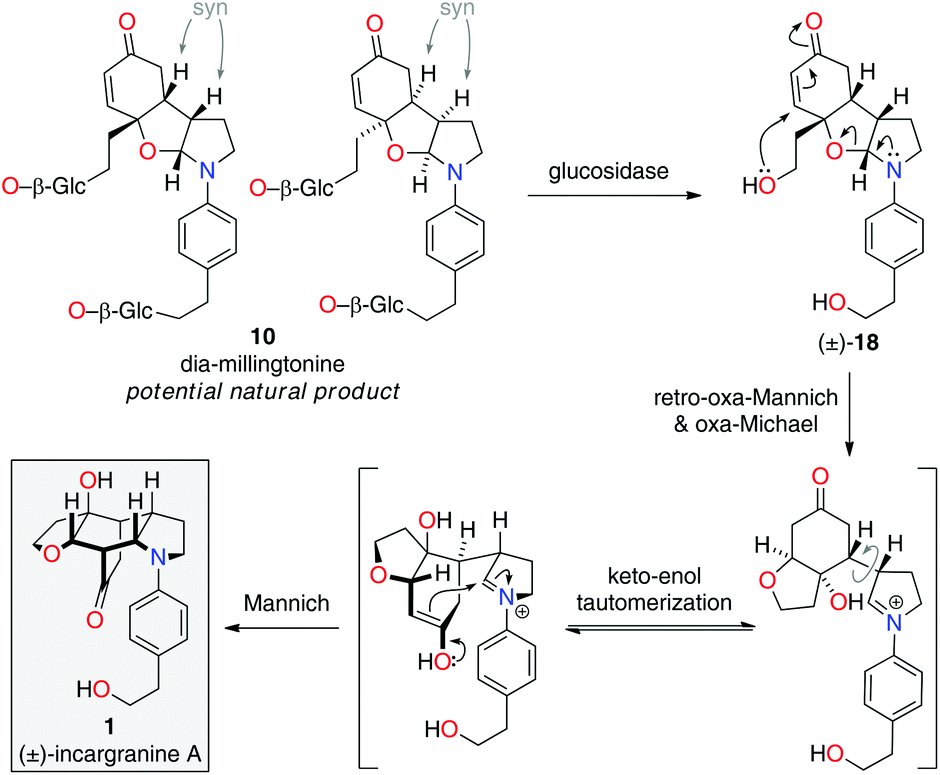 | ||
| Scheme 4 Revised biosynthetic hypothesis for incargranine A. | ||
During the development of this new strategy, it was discovered that protection of the primary alcohol in N-aryl-2,5-dihydropyrrole 13 was not necessary for the subsequent alkene-isomerization/hydroamination reaction. Thus, exposure of free alcohol 13 to RhHCO(PPh3)3 and pyrrolidine gave the expected aminal intermediate 19 (Scheme 5).2d,11 TBS-protected para-quinol 20, which was prepared in 2 steps from tyrosol,2a was then added directly to this crude reaction mixture resulting in a kinetically-controlled crossed-dimerization to give syn-dimer 21 in 77% yield.2d
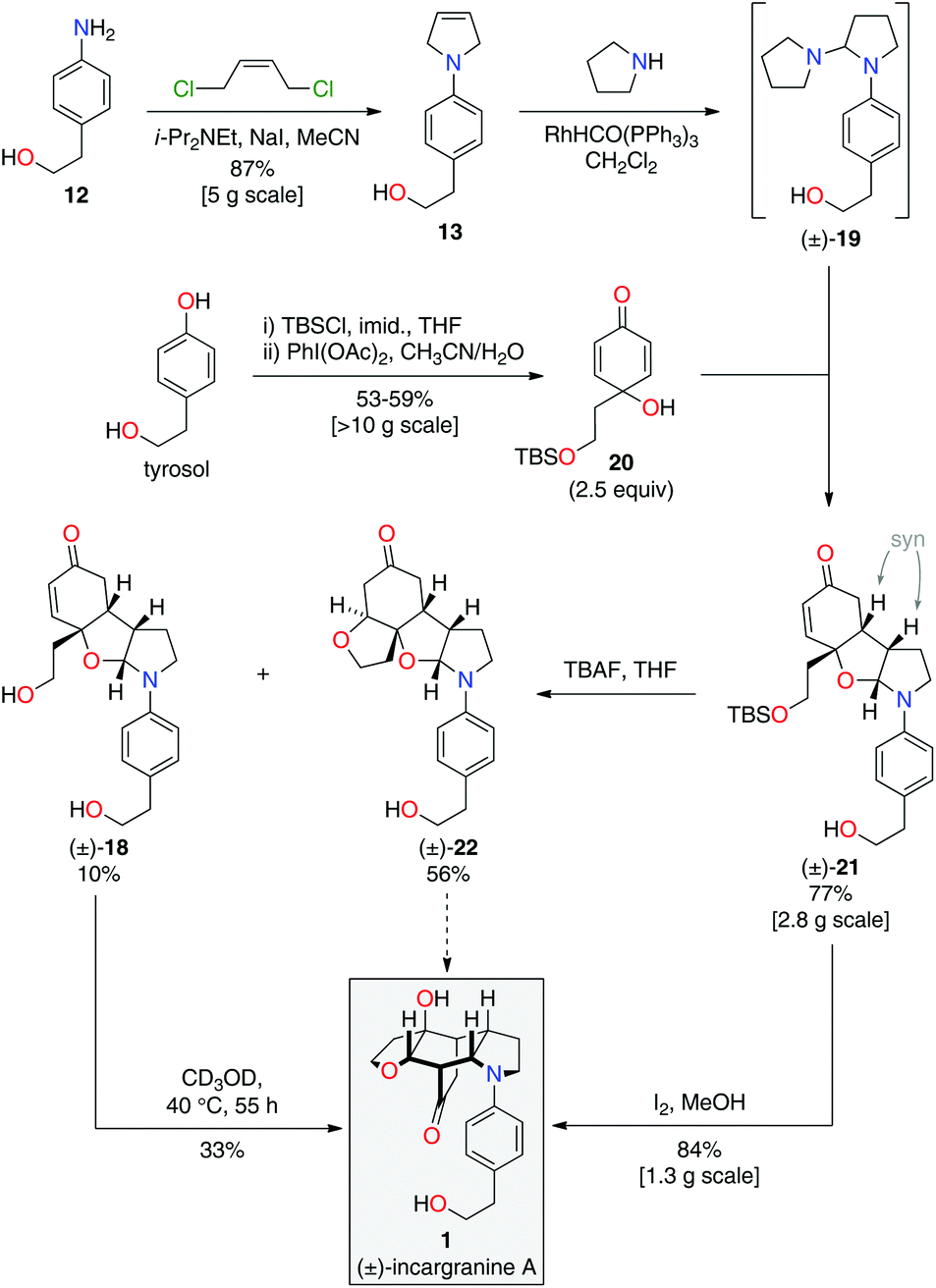 | ||
| Scheme 5 Total synthesis of incargranine A. | ||
Attention could now turn to the de-protection of crossed-dimer 21, a synthetic equivalent of dia-millingtonine (10), and its subsequent conversion to incargranine A (1). Cleavage of the tert-butyldimethylsilyl ether using standard TBAF (tetra-n-butylammonium fluoride) conditions gave the expected diol-aglycone 18 in just 10% yield, alongside a cyclized-aglycone 22 in 56% yield (Scheme 5). Remarkably, it was observed that diol-aglycone 18 spontaneously rearranges to give (±)-incargranine A (1) when dissolved in methanol at ambient temperature, albeit very slowly. Ultimately, a 33% isolated yield of (±)-incargranine A (1) was achieved when a CD3OD solution of diol-aglycone 18 was warmed to 40 °C for 2 days. The chemical feasibility of our proposed biosynthetic pathway between dia-millingtonine (10) and incargranine A (1) had thus been established. All efforts, however, to rearrange the cyclized-aglycone 22 to give incargranine A (1) were unsuccessful, akin to our failure to rearrange hemi-aminal 16 (Scheme 3).12
The low yields and lack of selectivity achieved in the final de-protection and rearrangement steps rendered this synthesis unsuitable for scale-up. Alternative deprotection conditions were therefore screened in the hope of favoring production of diol 18, whilst avoiding formation of the seemingly intractable ring-closed aglycone 22. Vaino and Szarek have reported iodine in methanol as mild reaction conditions for the cleavage of tert-butyldimethylsilyl ethers.14 Unexpectedly, however, exposure of syn-dimer 21 to iodine in methanol did not result in the formation of diol 18, nor ring-closed aglycone 22, but instead gave (±)-incargranine A (1) directly. Thus, in a single step, 2 new bonds, 2 new rings and 3 new stereogenic centres are formed in an impressive 84% yield. This synthetic sequence was readily scaled-up to provide gram-scale quantities of (±)-incargranine A (1), which compares very favorably to the effort required to obtain this material from the natural source; over four metric-tons of dried Incarvillea mairei var. grandiflora would need to be extracted to isolate one gram of natural incargranine A (1).1
Zhang and co-workers reported an optical rotation for natural incargranine A (1), [α]22D = +2 (c = 0.175, CHCl3).1 However, given our biosynthetic speculation and the small magnitude of the reported optical rotation value, we consider it likely that natural incargranine A (1) exists as a racemic mixture. Unfortunately, no authentic sample was available to validate this hypothesis.15 In all other respects, however, the spectroscopic data for our synthetic material matched that reported for natural incargranine A (1).1,15 We propose that this successful synthesis provides new evidence in support of the proposal that dia-millingtonine (10) is a natural product.2d,16 In fact, it is possible that incargranine A (1) is only produced from dia-millingtonine (10) during the extraction and isolation process. This would not necessarily mean that incargranine A (1) is an unimportant artifact of human intervention.17 It is known, for example, that plants can use glycosidic-metabolites as chemical defense systems, wherein damage to the plant brings gycosidase enzymes into contact with the glycosides to release the active aglycones.18
Conclusions
In just three-linear steps from 4-aminophenethyl alcohol 12 we have selectively formed 2 new C–N bonds, 2 new C–C bonds, 2 new rings, and 6 new contiguous stereogenic centres, in 56% overall yield.19 Key to the development of this efficient synthetic strategy has been the probing and refinement of a biosynthetic proposal using chemical synthesis. Ultimately, this has led to new evidence in support of the notion that dia-millingtonine (10) is an as-yet-undiscovered natural product.16 Practical quantities of these metabolites are now available for interested parties to study their biological function.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Prof. Wei-Dong Zhang (School of Pharmacy, Second Military Medical University, Shanghai) for kindly providing copies of the processed NMR spectra for natural incargranine A. The Royal Society is thanked for the award of a Research Grant. P. D. B. thanks the University of Edinburgh for the provision of a studentship.Notes and references
- Y.-Q. Su, Y.-H. Shen, S. Lin, J. Tang, J.-M. Tian, X.-H. Liu and W.-D. Zhang, Helv. Chim. Acta, 2009, 92, 165–170 CrossRef CAS.
- For previous biomimetic studies on related dimeric natural products, see: (a) P. D. Brown, A. C. Willis, M. S. Sherburn and A. L. Lawrence, Org. Lett., 2012, 14, 4537–4539 CrossRef CAS PubMed; (b) K. Zhao, G.-J. Cheng, H. Yang, H. Shang, X. Zhang, Y.-D. Wu and Y. Tang, Org. Lett., 2012, 14, 4878–4881 CrossRef CAS; (c) P. D. Brown, A. C. Willis, M. S. Sherburn and A. L. Lawrence, Angew. Chem., 2013, 125, 13515–13517 ( Angew. Chem. Int. Ed. , 2013 , 52 , 13273–13275 ) CrossRef; (d) P. D. Brown and A. L. Lawrence, Angew. Chem., 2016, 128, 8561–8565 ( Angew. Chem. Int. Ed. , 2016 , 55 , 8421–8425 ) CrossRef.
- Y. Q. Chen, Y. H. Shen, Y. Q. Su, L. Y. Kong and W. D. Zhang, Chem. Biodiversity, 2009, 6, 779–783 CrossRef CAS.
- Y. P. Gao, Y. H. Shen, S. D. Zhang, J. M. Tian, H. W. Zeng, J. Ye, H. L. Li, L. Shan and W. D. Zhang, Org. Lett., 2012, 14, 1954–1957 CrossRef CAS.
- J. S. Rosendal, A. Kjaer and N. B. Juhl, Acta Chem. Scand., 1973, 27, 367–369 CrossRef.
- (a) K. Endo and H. Hikino, Can. J. Chem., 1984, 62, 2011–2014 CrossRef CAS; (b) I. Messana, M. Sperandei, G. Multari, C. Galeffi and G. B. Marini Bettolo, Phytochemistry, 1984, 23, 2617–2619 CrossRef CAS; (c) J. Tian, Q. S. Zhao, H. J. Zhang, Z. W. Lin and H. D. Sun, J. Nat. Prod., 1997, 60, 766–769 CrossRef CAS.
- Y.-H. Shen, Y.-Q. Su, J.-M. Tian, S. Lin, H.-L. Li, J. Tang and W. D. Zhang, Helv. Chim. Acta, 2010, 93, 2393–2396 CrossRef CAS.
- T. Hase, K. Ohtani, R. Kasai, K. Yamasaki and C. Picheansoonthon, Phytochemistry, 1996, 41, 317–321 CrossRef CAS.
- For our earliest biosynthetic proposals, see page 36 of the ESI for ref. 2c.
- J. M. Bobbitt, L. H. Amundsen and R. I. Steiner, J. Org. Chem., 1960, 25, 2230–2231 CrossRef CAS.
- (a) W. A. Herrmann and M. Prinz, Applied Homogeneous Catalysis with Organometallic Compounds, Wiley-VCH, Weinheim, 2nd edn, 2002 Search PubMed; (b) T. J. Donohoe, T. J. C. O'Riordan and C. P. Rosa, Angew. Chem., 2009, 121, 1032–1035 ( Angew. Chem. Int. Ed. , 2009 , 48 , 1014–1017 ) CrossRef; (c) A. Vasseur, J. Bruffaerts and I. Marek, Nat. Chem., 2016, 8, 209–219 CrossRef CAS.
- Attempts to rearrange hemi-aminals 16 and 22 failed. Heating solutions in MeOH, EtOH or MeCN returned starting material. Treatment with TFA appeared to give isomerization from the syn to the anti configuration, with no sign of further rearrangement. Treatment with LiOH in refluxing MeOH/H2O resulted in slow decomposition.
- Treatment of millingtonine with glucosidase enzymes has been shown to result in a retro-oxa-Mannich/oxa-Michael/Mannich reaction sequence to give a diastereomer of incargranine A, see ref. 8.
- A. R. Vaino and W. A. Szarek, Chem. Commun., 1996, 20, 2351–2352 RSC.
- Professor Zhang very kindly provided pdf files of the processed NMR spectra for natural incargranine A, see the ESI† for direct comparisons.
- For recent examples of natural product anticipation from biomimetic studies, see: (a) D. P. O'Malley, K. Li, M. Maue, A. L. Zografos and P. S. Baran, J. Am. Chem. Soc., 2007, 129, 4762–4775 CrossRef; (b) P. Sharma, B. Lygo, W. Lewis and J. E. Moses, J. Am. Chem. Soc., 2009, 131, 5966–5972 CrossRef CAS; (c) M. Gavagnin, E. Mollo and G. Cimino, Rev. Bras. Farmacogn., 2015, 25, 588–591 CrossRef CAS; (d) A. L. Lawrence, R. M. Adlington, J. E. Baldwin, V. Lee, J. A. Kershaw and A. L. Thompson, Org. Lett., 2010, 12, 1676–1679 CrossRef CAS; (e) V. Sofiyev, J.-P. Lumb, M. Volgraf and D. Trauner, Chem. – Eur. J., 2012, 18, 4999–5005 CrossRef CAS; (f) S. Strych, G. Journot, R. P. Pemberton, S. C. Wang, D. J. Tantillo and D. Trauner, Angew. Chem., 2015, 127, 5168–5172 ( Angew. Chem. Int. Ed. , 2015 , 54 , 5079–5083 ) CrossRef; (g) P. Ellerbrock, N. Armanino, M. K. Ilg, R. Webster and D. Trauner, Nat. Chem., 2015, 7, 879–882 CrossRef CAS.
- P. Champy, Artifacts and Natural Substances Formed Spontaneously, in Biomimetic Organic Synthesis, ed. E. Poupon and B. Nay, Wiley-VCH, Weinheim, 2011, vol. 2, pp. 849–934 Search PubMed.
- A. Mithofer and W. Boland, Annu. Rev. Plant Biol., 2012, 63, 431–450 CrossRef PubMed.
- N. J. Green and M. S. Sherburn, Aust. J. Chem., 2013, 66, 267–283 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob00702k |
| This journal is © The Royal Society of Chemistry 2019 |