
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The α-hydroxyphosphonate-phosphate rearrangement of a noncyclic substrate – some new observations†
Susanne
Prechelmacher
a,
Kurt
Mereiter
 b and
Friedrich
Hammerschmidt
*a
b and
Friedrich
Hammerschmidt
*a
aInstitute of Organic Chemistry, University of Vienna, Währingerstrasse 38, A-1090 Vienna, Austria. E-mail: friedrich.hammerschmidt@univie.ac.at
bInstitute of Chemical Technologies and Analytics, Vienna University of Technology, Getreidemarkt 9/164, A-1060 Vienna, Austria. E-mail: kurt.mereiter@tuwien.ac.at
First published on 2nd May 2018
Abstract
Racemic ethyl hydrogen (1-hydroxy-2-methylsulfanyl-1-phenylethyl)phosphonate was resolved with (R)-1-phenylethylamine. The (R)-configuration of the (−)-enantiomer was determined by chemical correlation. Esterification of the (−)-enantiomer with a substituted diazomethane derived from 3-hydroxy-1,3,5(10)-estratrien-17-one delivered two epimeric phosphonates separated by HPLC. Methylation with methyl fluorosulfate at the sulfur atom and treatment with a strong base induced an α-hydroxyphosphonate-phosphate rearrangement with formation of dimethyl sulphide and two enantiomerically pure enol phosphates. Their oily nature interfered with a single crystal X-ray structure analysis to determine the stereochemistry at the phosphorus atom.
Introduction
When phosphoric acid derivatives (±)-1 are treated with strong bases in stoichiometric amounts such as alkyl lithiums or lithium amides at low temperatures, they are deprotonated to give short-lived organolithiums (±)-2 containing dipole-stabilised1 carbanions (Scheme 1). These undergo rearrangements via (±)-3 to lithiated α-substituted phosphonates (±)-4 and on work up to α-hydroxy-, α-sulfanyl- and α-aminophosphonates (±)-5. This isomerisation discovered for X = O by Sturtz and Corbel2 is called phosphate–phosphonate or more specifically phosphate-α-hydroxyphosphonate rearrangement. This3–5 and the versions for X = S6,7 and N8 have extensively been studied by Hammerschmidt's group. The reverse process with many examples9–17 for X = O, the α-hydroxyphosphonate-phosphate rearrangement, also termed [1,2]-phospha-Brook rearrangement, has been found by Pudovik and Konovalova17 before the phosphate–phosphonate rearrangement. This isomerisation is normally catalysed by a variety of catalytic bases such as e.g. NaOH, NaOEt and DBU. While the transformation of (±)-1 into (±)-5 for X = O is feasible even for R2 = alkyl and R3 = H, the reverse process not. At least one of the substituents, R2 or R3, should stabilise the developing negative charge on the carbon atom in (±)-3 upon cleavage of the C–P bond. An aromatic substituent suffices to stabilise the intermediate carbanion. The driving force for the phosphate–phosphonate rearrangements is the stronger Li–O than Li–C bond. The reverse process (O–H + P–C → C–H + P–O) is dominated by the much higher P–O than P–C bond energy. These isomerisations are related to the Brook and retro-Brook rearrangements in silicon chemistry.18 | ||
| Scheme 1 Phosphate–phosphonate rearrangements and reverse processes. | ||
The phosphate–phosphonate rearrangement for X = O,3,5 S7 and N8 and the reverse process for X = O11,15,16 follow a retentive course at the respective carbon atoms. The stereochemistry at the phosphorus atom upon the a-hydroxyphosphonate-phosphate rearrangement follows a retentive course too, proven only for α-hydroxyphosphonates with the phosphorus atom as part of a six-membered ring.11,16 It was found that diastereomeric α-hydroxyphosphonates (R,SP)- and (R,RP)-7 obtained by esterification of enantiomer (R)-6 and fractional crystallisation rearrange stereospecifically (Scheme 2).10 Here the phosphorus atom was not part of a ring system and the developing negative charge on the α-carbon atom upon cleavage of the P–C bond eliminated a β-chloride, resulting in enantiomerically pure enol phosphates (+)- and (−)-8. As they were oils, their absolute configuration could not be determined by X-ray structure analysis and the stereochemistry at the phosphorus atom had to remain unanswered.
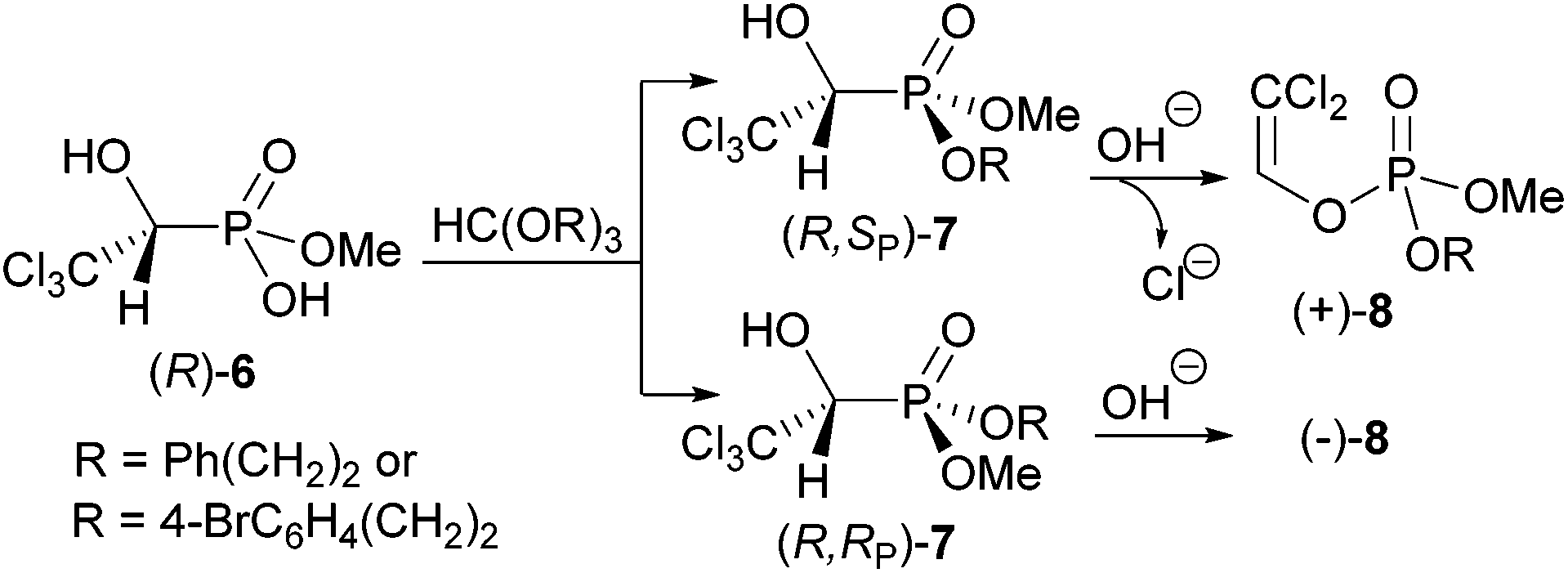 | ||
| Scheme 2 α-Hydroxyphosphonate-phosphate rearrangement of diastereomeric α-hydroxyphosphonates 7 to enantiomerically pure enol phosphates 8. | ||
Results and discussion
The highly enantioselective synthesis of acyclic phosphate triesters is difficult and challenging.19,20 While Hall and Inch19 built their syntheses on 5- and 6-membered cyclic phosphorus compounds derived from (−)-ephedrine and D-glucose, Nakayama and Thompson20 applied (S)-proline derivatives. We reasoned that the α-hydroxyphosphonate-phosphate rearrangement of acyclic substrates with a stereogenic P-atom of known configuration would give chiral, nonracemic phosphate triester and alkenyl dialkyl ester. In order to assign the configuration to the P-chiral product, the stereochemistry of the rearrangement at the phosphorus atom has to be known. Here we start another approach to unravel it.At first, an enantiomerically pure alkyl hydrogen α-hydroxyphosphonate was prepared and resolved (Scheme 3).
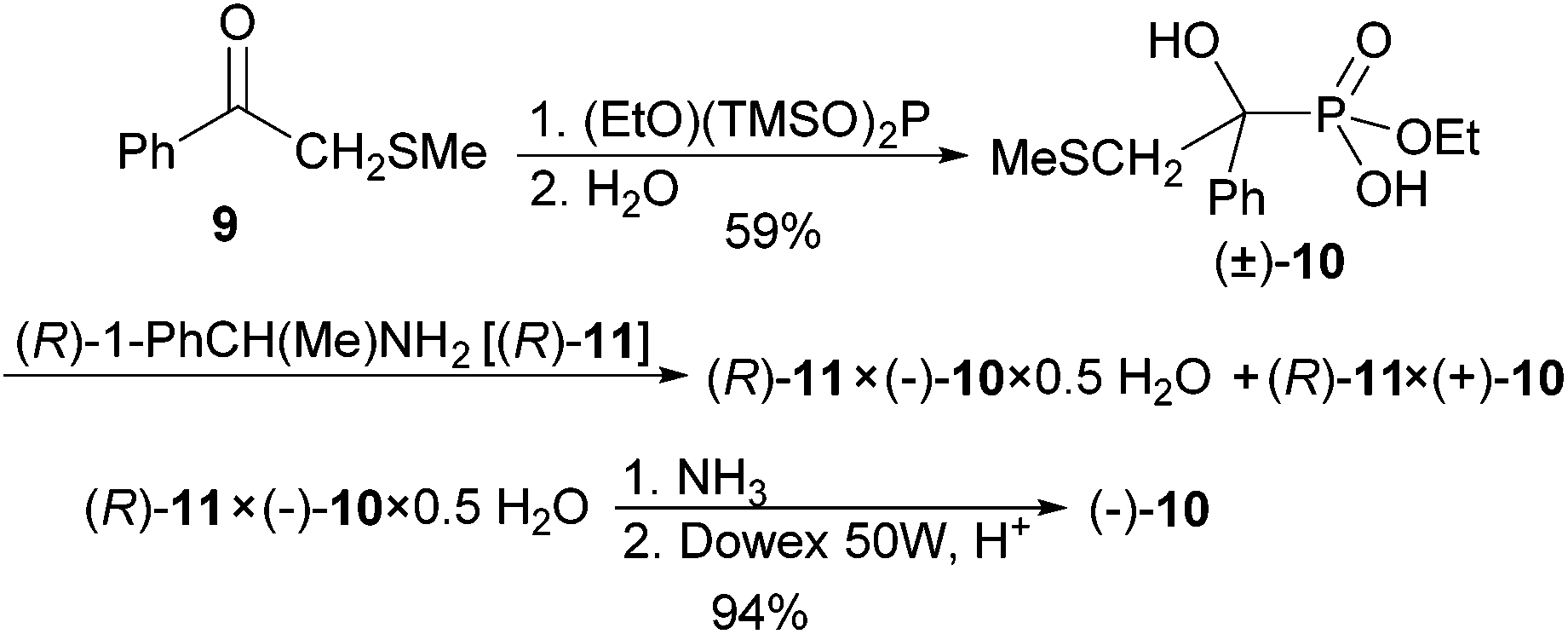 | ||
| Scheme 3 Preparation and resolution of ethyl hydrogen phosphonate (±)-10. | ||
Ethyl bis(trimethylsilyl) phosphite3 was added to ketone 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 21 to give a protected α-hydroxyphosphonate as intermediate, that was hydrolysed to phosphonic acid monoethyl ester (±)-10 upon aqueous workup and isolated in 59% yield. The phenyl ketone was selected, because the phenyl substituent with its anion-stabilising effect will ascertain that the α-hydroxyphosphonate-phosphate rearrangement at the end of the sequence will be feasible. The methylsulfanyl substituent can be methylated and give the good leaving group dimethyl sulfide. (R)-(+)-1-Phenylethylamine [(R)-11] was found to be a better resolving agent for phosphonic acid (±)-10 than brucine. The crystals obtained from Et2O/CH2Cl2 contained Et2O (by 1H NMR, salt/Et2O, 2.6:1.0) and had a de of already 86% (by 1H NMR, the two methylsulfanyl groups of the two diastereomers resonate as two singlets at δ 1.77 and 1.79). Two crystallisations from CHCl3 delivered crystals of hemihydrate (R)-11 × (−)-10 × 0.5H2O of de > 98% in 56% yield. When this salt was dissolved in aqueous ammonia (25%) and extracted with CH2Cl2, the (R)-1-phenylethylamine was recovered. The free acid (−)-10 was isolated from the aqueous phase by passage through Dowex 50 W, H+ and removal of water under reduced pressure.
21 to give a protected α-hydroxyphosphonate as intermediate, that was hydrolysed to phosphonic acid monoethyl ester (±)-10 upon aqueous workup and isolated in 59% yield. The phenyl ketone was selected, because the phenyl substituent with its anion-stabilising effect will ascertain that the α-hydroxyphosphonate-phosphate rearrangement at the end of the sequence will be feasible. The methylsulfanyl substituent can be methylated and give the good leaving group dimethyl sulfide. (R)-(+)-1-Phenylethylamine [(R)-11] was found to be a better resolving agent for phosphonic acid (±)-10 than brucine. The crystals obtained from Et2O/CH2Cl2 contained Et2O (by 1H NMR, salt/Et2O, 2.6:1.0) and had a de of already 86% (by 1H NMR, the two methylsulfanyl groups of the two diastereomers resonate as two singlets at δ 1.77 and 1.79). Two crystallisations from CHCl3 delivered crystals of hemihydrate (R)-11 × (−)-10 × 0.5H2O of de > 98% in 56% yield. When this salt was dissolved in aqueous ammonia (25%) and extracted with CH2Cl2, the (R)-1-phenylethylamine was recovered. The free acid (−)-10 was isolated from the aqueous phase by passage through Dowex 50 W, H+ and removal of water under reduced pressure.
To determine the absolute configuration of (−)-10, it was transformed into the known α-hydroxyphosphonate (S)-(−)-12 (Scheme 4). This was achieved by desulfurisation of the respective potassium salt with aged RANEY-nickel,22 followed by passage through Dowex 50 W, H+ to get the free acid. Esterification with diazoethane23 furnished phosphonic acid diethyl ester (−)-12 (in 22% overall yield), which has (S)-configuration based on the comparison of the specific optical rotation with the literature value.3 When freshly prepared RANEY-nickel was used, the CH3S and OH groups were both reductively removed. This experiment proved that phosphonic acid (−)-103 has (R)-configuration. The change of the descriptor is caused by the change in the priority for the substituents according to the CIP rules [for (S)-(−)-12: P > Ph > CH3; for (R)-(−)-10: P > CH2SCH3 > Ph.
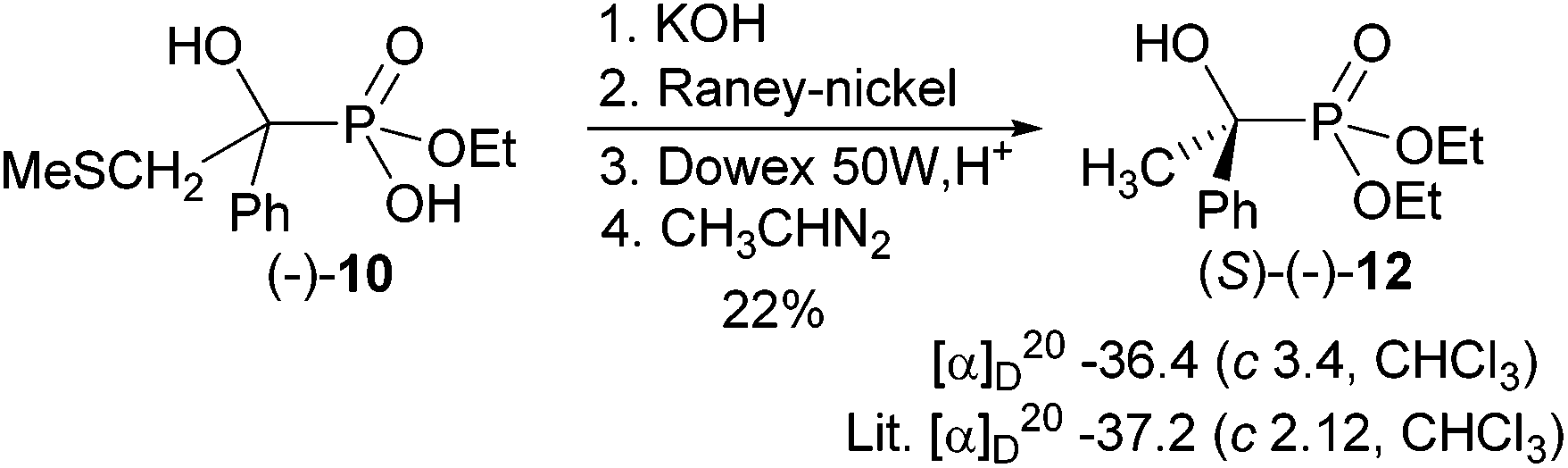 | ||
| Scheme 4 Determination of absolute configuration of (−)-10. | ||
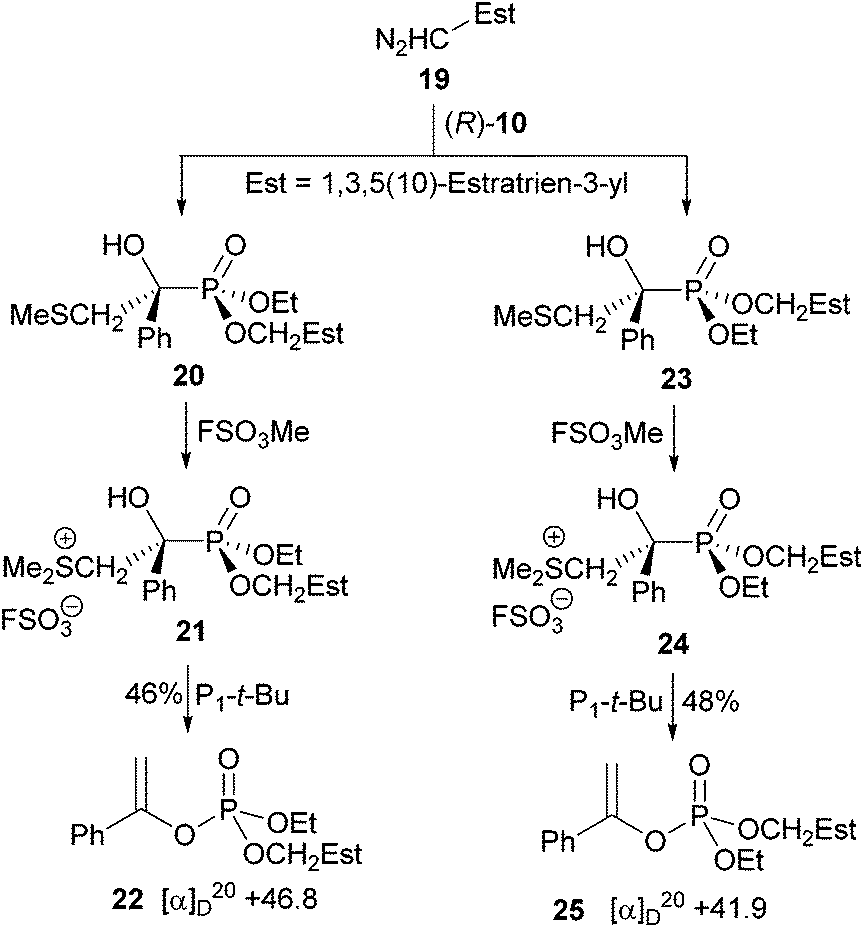
The next step was the esterification of phosphonic acid (R)-(−)-10 with a diazoalkane under mild conditions, which should give (1) separable diastereomeric α-hydroxyphosphonates and (2) at least one crystalline phosphate upon α-hydoxyphosphonate-phosphate rearrangement. Previously, a variety of bromoaryldiazomethanes were tested, but they delivered inseparable mixtures of α-hydroxyphosphonates and oily phosphates unfortunately.24 We reasoned that a steroid such as the fairly easily available 1,3,5(10)-estratrien-3-yldiazomethane (19) could fulfil the outlined requirements (Scheme 5). The centres of chirality of the steroid are too far away from the phosphorus atom to have an influence on the rearrangement. 1,3,5(10)-Estratrien-3-ol (13) prepared by a literature procedure25 from 3-hydroxy-1,3,5(10)-estratrien-17-one was esterified with triflic anhydride in the presence of 2,4,6-collidine and DMAP at −30 °C to give triflate 14 in 85% yield.26 Alkoxycarbonylation27 catalysed by Pd(OAc)2-1,3-bis(diphenylphosphino)propane of the phenolic triflate with CO/MeOH/Et3N at 70 °C delivered benzoate 15 in 80% yield, which was quantitatively reduced to benzyl alcohol 16 with LiAlH4. Swern oxidation of 16 to the aldehyde 17 was less effective (54% yield) than PCC oxidation (91%) in the presence of 3 Å molecular sieves,28 which facilitated a smooth reaction and workup. Heating a mixture of aldehyde 17 with tosyl hydrazine in MeOH29 at 40 °C, furnished in 95% yield tosyl hydrazone 17, the starting material for the preparation of the substituted diazomethane. Refluxing a mixture of the hydrazone and sodium bis(trimethylsiyl)amide in dry THF for 90 min gave the substituted diazomethane 19.29,30
 | ||
| Scheme 5 Preparation of substituted diazomethane 19. | ||
Crude 19 was not purified, but immediately used for the esterification of phosphonic acid (R)-(−)-10 in CH2Cl2 at room temperature (Scheme 6). Flash chromatography of the crude product provided a 1:1 mixture of epimers 20 and 23 (by 1H NMR; epimers displayed the same polarity) in 90% yield. Separation by preparative HPLC delivered the less polar 20 of 88% de and the more polar 23 of 96% de. Crystallisation of epimer 20 from hexanes or cyclohexane furnished crystals of >98% de, which contained solvent (20/hexanes, 3.13:1; 20/cyclohexane, 2:1, by 1H NMR). The more polar epimer 23 was crystallised from hexanes/i-PrOH to give crystals of also de >98%, which were suitable for an X-ray crystal structure. It allowed to assign (R,RP)-configuration (Fig. 1) to the phosphonic acid part of 23 and consequently (R,SP)-configuration to that of 20 (the P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond is considered a single bond when the sequence rule is used!). Both epimers were methylated at the sulfur atom with methyl fluorosulfate at −35 °C. The respective sulfonium salts 21 and 24 were deprotonated at the hydroxyl groups with phosphazene base P1-t-Bu,31 a stronger base than DBU, to induce α-hydroxyphosphonate-phosphate rearrangements as detailed for 24 in Scheme 7.
O bond is considered a single bond when the sequence rule is used!). Both epimers were methylated at the sulfur atom with methyl fluorosulfate at −35 °C. The respective sulfonium salts 21 and 24 were deprotonated at the hydroxyl groups with phosphazene base P1-t-Bu,31 a stronger base than DBU, to induce α-hydroxyphosphonate-phosphate rearrangements as detailed for 24 in Scheme 7.
 | ||
| Scheme 6 Esterification of (R)-10 to give epimeric α-hydroxyphosphonates 20 and 23 for the rearrangement to phosphates 22 and 25. | ||
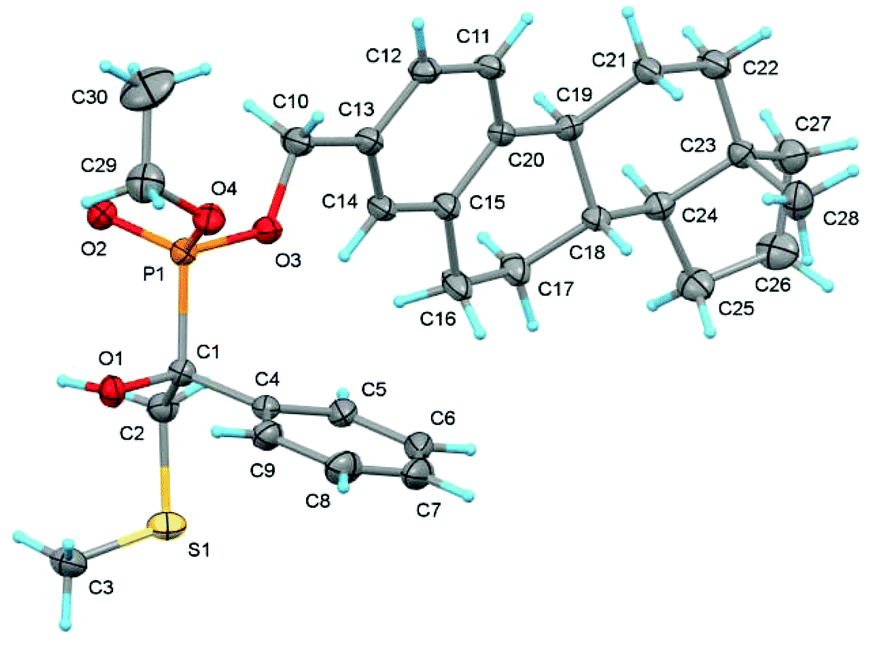 | ||
| Fig. 1 The molecular structure of 23 in solid state showing displacement ellipsoids at 20% probability. | ||
 | ||
| Scheme 7 Reaction pathways for alkoxide 26. | ||
The alkoxide 26 has two options. Firstly (pathway A), it can disintegrate (retro-Abramov reaction32) into phosphite anion 29 and sulfonium salt 28, which in turn react with each other to sulfonium ylide 30 and H-phosphonate 31. The carbonyl group of the ylide is not electrophilic enough to allow addition of the phosphite anion, which would lead to epimeric α-hydroxyphosphonates. Additionally, sulfur ylide 30 is not basic enough to deprotonate 31 to give 29. The H-phosphonate 31 was detected in the crude reaction mixture by 1H NMR spectroscopy [P(O)H: δH = 6.84, d, JHP = 698.0 Hz]. Secondly (pathway B), alkoxide 26 can undergo the rearrangement to enol phosphate 25via cyclic species 27, which might be either an intermediate or a transition state.33 We assume that 27 has a trigonal bipyramidal structure formed by an apical attack of the alkoxide anion on the electrophilc phosphorus atom from the less hindered side opposite to the EstCH2O substituent. The P–C bond will be equatorially orientated. The negative charge building up on the α-carbon atom in 25 upon cleavage of the P–C bond eliminates dimethyl sulphide. The two enol phosphates 22 and 25 were obtained in yields of 46% and 48%, respectively. Their specific optical rotations were [α]20D + 46.8 and + 41.9, respectively. These compounds contain beside the stereogenic phosphorus atom some stereogenic carbon ones in the steroidal substituent. Therefore the specific optical rotations cannot have the same absolute values with opposite signs. NMR spectroscopically, they are virtually identical (1H, 13C, 31P) except for the resonances of EstCH2OP group (AB parts of ABP systems) in the 1H NMR spectrum (Fig. 2). Inspection of the three segments of the relevant 1H NMR spectra reveal that the two enol phosphates 21 and 25 are enantiomerically pure. Unfortunately, none of the two oils could be induced to crystallise and the absolute configuration of the stereogenic phosphorus atom could not be determined by single X-ray structure analysis. The stereochemical course of the α-hydroxyphosphonate-phosphate rearrangement of a non-cyclic α-hydroxyphosphonates remains to be determined. However, it must be a stereospecific reaction yielding enantiomerically pure enol phosphates.
 | ||
| Fig. 2 EstCH2O segments of 1H NMR spectra of enol phosphates 22 (left) and 25 (middle) and of 1:1 mixture of 22 and 25 (right). | ||
Conclusions
In summary, we prepared a racemic ethyl hydrogen α-hydroxyphosphonate, resolved it with (R)-1-phenylethylamine and esterified it with a diazomethane derived from 3-hydroxy-1,3,5(10)-estratrien-17-one. Each epimer obtained by HPLC separation was methylated at the methylsulfanyl substitutent and treated with base to induce α-hydroxyphosphonate-phosphate rearrangements. We found that the rearrangement is stereospecific. However, the stereochemistry could not be determined as the obtained phosphate was not crystalline to perform a single crystal X-ray structure analysis. The sequence allows to prepare enantiomerically pure enol phosphates.Experimental
General
1H, 13C (J-modulated) and 31P NMR spectra were recorded in CDCl3 on Bruker Avance AV 400 (1H: 400.13 MHz, 13C: 100.61 MHz, 31P: 161.97 MHz) and AV III 600 (1H: 600.25 MHz, 13C: 150.93 MHz, 31P: 242.94 MHz) spectrometers at 25 °C. Chemical shifts (δ) are reported in parts per million (ppm) relative to CHCl3/CDCl3 (δH 7.24, δC 77.00) and external H3PO4 (85%; δP 0.00) and coupling constants (J) in Hz. Data for 1H NMR spectra are reported as follows: chemical shift, multiplicity (s = singlet, d = doublet, t = triplet, q = quartet, sept = septet, m = multiplet), coupling constants, and integration. IR spectra were run as films between NaCl plates or on a silicon disc34 using a PerkinElmer 1600 FT-IR spectrometer. Optical rotations were measured on a PerkinElmer 351 polarimeter in a 1 dm cell. Analytical HPLC was performed on a Jasco System (PU-980 pump, UV 975 and RI 930) using a Nucleosil 50-4 column (Macherey-Nagel), Ø 0.4 cm × 25 cm. Preparative HPLC was performed on a Rainin System (Dynamix Model SD-1 pump, Model UV-1 UV detector, 254 nm) using a Nucleosil 50-7 column, Ø 6.3 cm × 28.8 cm. Melting points were measured on a Leica Galen III Thermovar instrument and are uncorrected. Flash (column) chromatography was performed with silica gel 60 (230–400 mesh) and monitored by TLC conducted on glass-backed 0.25 mm thick silica gel 60 F254. Spots were visualised by UV and/or dipping the plate into a solution of (NH4)6Mo7O24 × 4H2O (23.0 g) and Ce(SO4)2 × 4H2O (1.0 g) in 10% aqueous H2SO4 (500 mL), followed by heating with a heat gun.(±)-Ethyl hydrogen (1-hydroxy-2-methylsulfanyl-1-phenyl)-phosphonate [(±)-10]
A solution of ethyl bis(trimethylsilyl) phosphite3 (36.13 g, 142 mmol) and methylsulfanylmethyl phenyl ketone (9)21 (23.61 g, 142 mmol) in dry toluene (100 mL) was heated for 18 h at 70 °C under exclusion of moisture, cooled and then diluted with water (200 mL). After stirring vigorously for 30 min the mixture was neutralised with NaOH (2 M, phenolphthalein). The organic phase was separated and discarded. The aqueous one was continuously extracted with Et2O for 2 h and the extract was discarded. The aqueous layer was acidified with diluted H2SO4 (10 mL conc. H2SO4 and 30 mL H2O) and again continuously extracted with Et2O for 1 h. This extract was concentrated under reduced pressure and dried to yield crystalline phosphonic acid (±)-10 (28.0 g). Continuous extraction for another 2 h gave another 1 g phosphonic acid. The combined products were crystallised from Et2O (with cooling at −20 °C) to yield phosphonic acid (±)-10 (23.0 g, 59%) as colourless crystals. The analytical sample was recrystallised from EtOAc/Et2O; mp 95–98 °C.IR (nujol): ν 3409, 3100–2000, 1332, 1162, 1035 cm−1; 1H NMR (400.13 MHz, CDCl3): δ 1.16 (t, J = 7.1 Hz, 3H), 1.80 (s, 3H), 3.33 (AB part of ABP system, JAB = 14.1 Hz, J = 8.2, 6.6 Hz, 2H), 3.86–4.00 (m, 2H), 7.25–7.29 (m, 1H), 7.34 (t, J = 7.7 Hz, 2H), 7.40 (br. s, 2H), 7.54–7.60 (m, 2H); when excess (R)-(+)-1-phenylethylamine was added to the NMR sample, two diastereomeric salts formed with the methylsulfanyl groups resonating at 1.77 and 1.79 ppm. The singlet at lower field corresponds to the CH3S of the salt of the dextrorotary acid. 13C NMR (150.93 MHz, CDCl3): δ 16.2 (d, J = 5.9 Hz), 17.1, 43.1 (d, J = 6.8 Hz), 63.8 (d, J = 8.6 Hz), 74.2 (d, J = 165.4 Hz), 126.4 (d, J = 4.2 Hz), 127.8 (d, J = 2.8 Hz), 128.1 (d, J = 2.5 Hz), 138.6; 31P NMR (242.99 MHz, CDCl3): δ 23.4. Anal. calcd for C11H17O4PS: C, 47.82; H, 6.20; P, 11.21. Found: C, 47.95; H, 6.00; P, 11.58.
Optical resolution of (±)-ethyl hydrogen (1-hydroxy-2-methylsulfanyl-1-phenylethyl)phosphonate with (R)-1-phenylethylamine [(R)-11]
Racemic phosphonic acid (±)-10 (16.56 g, 60 mmol) was dissolved in CH2Cl2 (30 mL) and (R)-(+)-1-phenylethylamine (7.27 g, 60 mmol, 7.68 mL) was dropwise added with cooling. After the addition of Et2O (300 mL) and seeding crystals obtained by slow evaporation of solvent from a CHCl3 solution of this salt, the solution was left for 24 h at 4 °C. The formed crystals were collected, washed with Et2O/CH2Cl2 (10/1) and dried at 0.5 mm/20 °C for 30 min to give 9.1 g of salt, de 86% (by 1H NMR, salt/Et2O, 2.6:1.0). The crystals were dissolved in hot CHCl3 (136.5 mL). The flask with the solution was placed into a Dewar with warm water (50–55 °C). The Dewar topped with a Styropor plate was allowed to slowly cool in the fridge until the water had 4 °C. The colourless crystals not containing Et2O were collected, washed with cold CHCl3 and dried; 8.03 g, de 98%. The crystals were recrystallised from CHCl3 as before and furnished phosphonic acid salt (R)-11 × (−)-10 × 0.5H2O (6.8 g, 56%) as colourless crystals; mp 120–123 °C; [α]20D–7.0 (c. 1.53, CH2Cl2).
IR (nujol): ν = 3410, 3100–2000, 1620, 1550, 1300, 1190, 1170, 1160, 1050 cm−1; 1H NMR (600.25 MHz, CDCl3): δ = 0.99 (t, J = 6.9 Hz, 3H), 1.42 (d, J = 6.6 Hz, 3H), 1.76 (s, 3H), 2.11 (br. s), 3.26 (AB part of ABP system, JAB = 13.7 Hz, J = 3.1, 6.1 Hz, 2H), 3.50–3.70 (m, 2H), 3.98 (q, J = 6.9 Hz, 1H), 7.17–7.37 (m, 4H), 7.57 (d, J = 7.4 Hz, 1H), 8.32 (br. s); 13C NMR (150.93 MHz, CDCl3): δ 16.7 (d, J = 6.4 Hz), 17.1, 20.9, 44.5, 50.7, 62.0 (d, J = 6.6 Hz), 76.2 (d, J = 149.4 Hz), 126.5 (d, J = 3.1 Hz, 3C), 126.9 (2C), 127.6 (d, J = 1.6 Hz, 2C), 128.4, 128.8 (2C), 139.4, 142.1; 31P NMR (242.99 MHz, CDCl3): δ 17.4. Anal. calcd for C19H28NO4PS: C, 57.42; H, 7.10; N, 3.52; calcd for C19H28NO4PS × 0.5H2O: C, 56.14; H, 7.19; N, 3.44. Found: C, 56.12; H, 6.80; N, 3.39.
Conversion of (R)-1-phenylethylammonium salt of ethyl hydrogen (1-hydroxy-2-methylsulfanyl-1-phenylethyl)-phosphonate to free phosphonic acid (−)-10 (general procedure A)
The (R)-1-phenylethylammonium salt hemihydrate (R)-11 × (−)-10 × 0.5H2O (1.105 g, 2.72 mmol), CH2Cl2 (20 mL), water (20 mL) and ammonia solution (2 mL, 25%) were mixed. The organic phase was separated and the aqueous one was extracted with CH2Cl2 (2 × 15 mL). The organic phases containing the amine were discarded and the aqueous phase was concentrated under reduced pressure. The residue was dissolved in water and applied to a Dowex 50W × 8, H+ column and eluted with water until neutral. The eluate was concentrated under reduced pressure and dried (0.5 mbar/RT) to give phosphonic acid (−)-10 (0.710 g, 94%) as colourless gum, which crystallised; mp 61–63 °C (i-Pr2O/few drops of CH2Cl2); [α]18D–16.9 (c. 1.45, dry EtOH). Anal. calcd for C11H17O4PS: C, 47.82; H, 6.20; O, 23.16; S, 11.60. Found: C, 47.83; H, 6.20; O, 23.40; S, 11.71.Desulfurisation of potassium salt of (−)-ethyl hydrogen (1-hydroxy-2-methylsulfanyl-1-phenylethyl)phosphonate (−)-10
RANEY-nickel prepared by a literature procedure22 was washed with water (10 × 250 mL portions) and stored in water for 72 h at room temperature prior to use (it has to be handled quickly when moist as it is pyrophoric!).Diazoethane:23 To a solution of KOH (15 g) in water (45 mL) and Et2O (30 mL) cooled at −35 °C (bath temperature) N-nitroso-N-ethylurea35 (4.0 g) was added in portions within 5 min. The mixture was stirred until the urea had dissolved (20 min). The yellow ethereal solution of diazoethane was used directly for esterification.
The free phosphonic acid obtained by general procedure A from (R)-1-phenylethylammonium salt hemihydrate (R)-11 × (−)-10 × 0.5H2O (0.80 g, 1.97 mmol) was dissolved in a mixture of ethanol (12 mL) and water (8 mL) and neutralised with KOH (10%, phenolphthalein). After the addition of moist RANEY-nickel (5.3 g) the mixture was stirred for 15 h at room temperature and filtered. The RANEY® nickel was washed with a mixture of EtOH/water (the spent RANEY®-nickel was inactivated by storage under CH2Cl2). The filtrate was passed through Dowex 50 W × 8, (H+) and eluted with water until neutral. The eluate was concentrated under reduced pressure, dissolved in EtOH and esterified with diazoethane. The solution was concentrated under reduced pressure. The oily residue was flash chromatographed (CH2Cl2/EtOAc, 5:1, Rf 0.17 and 0.07). The less polar product (0.060 g) although evidently homogeneous by TLC was an inseparable mixture of diethyl 1-hydroxy-2-methylsulfanyl-1-phenylethylphosphonate and diethyl 1-phenylethylphosphonate (ratio by 1H NMR: 19:81). The more polar product was flash chromatographed a second time (CH2Cl2/EtOAc, 2:1, Rf 0.17) to give diethyl 1-hydroxy-1-phenylethylphosphonate (−)-12 (0.11 g, 22%) as a colourless oil; [α]20D–36.4 (c. 3.4, CHCl3), after distillation (115–120 °C/0.005 mm) [α]20D–35.67 (c. 1.8, CHCl3) {lit.3 [α]20D–37.2 (c. 2.12, CHCl3) for known 1-hydroxy-1-phenylethylphosphonate (S)-(−)-12}.
1,3,5(10)-Estratrien-3-yl trifluoromethanesulfonate (14)
1,3,5(10)-Estratrien-3-ol25 (13) (11.7 g, 45.6 mmol, crystalline product, freed from EtOH by dissolution in toluene and concentration under reduced pressure) was dissolved in dry CH2Cl2 (150 mL) under argon atmosphere. 2,4,6-Trimethylpyridine (9.53 g, 78.7 mmol, 10.4 mL, 1.73 equiv.) and 4-dimethylaminopyridine (1.30 g, 10.6 mmol, 0.23 equiv.) were added, followed by cooling at −30 °C and dropwise addition of triflic anhydride (19.4 g, 69 mmol, 11.33 mL, 1.5 equiv.).26 The mixture was stirred for 10 min at −30 °C and 2 h at room temperature. The mixture was washed with 2 M HCl, water and a saturated aqueous solution of NaHCO3 (each with 100 mL). The organic phase was dried (MgSO4), concentrated under reduced pressure. The residue was flash chromatographed (hexanes, Rf 0.27) to give triflate 14 (15.1 g, 85%) as colourless crystals; mp 51–52 °C (hexanes); [α]20D + 58.7 (c. 1.06, acetone).IR (Si): ν 2935, 2870, 1490, 1424, 1249, 1211, 1172, 1143 cm−1; 1H NMR (400.13 MHz, CDCl3): δ 0.73 (s, 3H), 1.09–1.80 (m, 11H), 1.85–1.97 (m, 2H), 2.18–2.30 (m, 2H), 2.83–2.93 (m, 2H), 6.94 (d, J = 2.6 Hz, 1H), 6.99 (dd, J = 8.6, 2.6 Hz, 1H), 7.32 (d, J = 8.6 Hz, 1H); 13C NMR (100.61 MHz, CDCl3): δ 17.4, 20.5, 25.2, 26.5, 27.6, 29.7, 38.5, 38.7, 40.4, 41.0, 44.2, 53.6, 118.0, 118.8 (q, JCF = 321.0 Hz, CF3), 121.1, 127.2, 139.6, 141.3, 147.4. Anal. calcd for C19H23F3O3S: C, 58.75; H, 5.97. Found: C, 58.65; H, 6.03.
Methyl 1,3,5(10)-estratriene-3-carboxylate (15)
This reaction was performed in a well vented hood (CO!). 1,3,5(10)-Estratrien-3-yl trifluoromethanesulfonate (14) (14.4 g, 37.1 mmol) were dissolved in a stirred mixture of dry methanol (74 mL) and dry DMSO (110 mL). Triethylamine (8.2 g, 81 mmol, 11.3 mL, 2.2 equiv.), Pd(OAc)2 (0.499 g, 2.22 mmol, 0.06 equiv.) and 1,3-bis(diphenylphosphino)propane (0.914 g, 2.22 mmol, 0.06 equiv.) were added.27 The apparatus was flushed with CO for 15 min and then the mixture was heated at 70 °C (oil bath temperature) under the CO atmosphere for 4 h. After cooling to room temperature water (380 mL) was added and the mixture was extracted with CH2Cl2 (3 × 120 mL). The combined organic layers were washed with HCl (2 M), water and a saturated aqueous solution of NaHCO3, dried (Na2SO4) and concentrated under reduced pressure. The residue was flash chromatographed (hexanes/CH2Cl2, 2:1, Rf 0.24) to give methyl ester 15 (8.83 g, 80%) as colourless crystals; mp 92–94 °C (hexanes); [α]20D + 78.6 (c. 1.58, acetone).
IR (Si): ν 2948, 2868, 1723, 1435, 1291, 1263, 1193 cm−1; 1H NMR (400.13 MHz, CDCl3): δ 0.73 (s, 3H), 1.08–1.82 (m, 11H), 1.84–1.91 (m, 2H), 2.23–2.36 (m, 2H), 2.85–2.95 (m, 2H), 3.87 (s, 3H), 7.34 (d, J = 8.2 Hz, 1H), 7.73 (d, J = 1.6 Hz, 1H), 7.77 (dd, J = 8.2, 1.6 Hz, 1H); 13C NMR (100.61 MHz, CDCl3): δ 17.5, 20.5, 25.2, 26.4, 27.8, 29.5, 38.6, 38.7, 40.4, 41.0, 44.8, 51.9, 53.7, 125.4, 126.6, 127.2, 130.1, 137.0, 146.3, 167.4. Anal. calcd for C20H26O2: C, 80.50; H, 8.78. Found: C, 80.28; H, 8.79.
1,3,5(10)-Estratrien-3-ylmethanol (16)
A solution of methyl 1,3,5(10)-estratriene-3-carboxylate (15) (8.57 g, 28.7 mmol) in dry Et2O (40 mL) was dropwise added to a stirred suspension of LiAlH4 (0.82 g, 21.5 mmol, 1.5 equiv.) in dry Et2O (50 mL) at 0 °C. The mixture was refluxed for 2 h and cooled at 0 °C. Then water (12 mL, 0 °C) was dropwise added, followed by H2SO4 (120 mL, 2 M). The organic phase was separated and the aqueous one extracted with Et2O (2 × 120 mL). The combined organic layers were washed with brine (120 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (CH2Cl2, Rf 0.32) to give 1,3,5(10)-estratrien-3-ylmethanol (16) (7.51 g, 97%) as colourless crystals; mp 99–101 °C (methanol); [α]20D + 84.7 (c. 1.99, acetone).IR (Si): ν 3286, 2932, 2868, 1452, 1428, 1377, 1155, 1046, 1014, 1002 cm−1; 1H NMR (400.13 MHz, CDCl3): δ 0.73 (s, 3H), 1.09–1.54 (m, 8H), 1.55 (br s, 1H), 1.60–1.81 (m, 3H), 1.84–1.91 (m, 2H), 2.20–2.34 (m, 2H), 2.80–2.95 (m, 2H), 4.61 (s, 2H), 7.08 (s, 1H), 7.12 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.0 Hz, 1H); 13C NMR (100.61 MHz, CDCl3): δ 17.5, 20.5, 25.2, 26.6, 28.0, 29.6, 38.8, 39.0, 40.5, 41.0, 44.4, 53.6, 65.3, 124.3, 125.6, 127.7, 137.1, 138.0, 140.4. Anal. calcd for C19H26O: C, 84.39; H, 9.70. Found: C, 83.83; H; 9.80.
1,3,5(10)-Estratriene-3-carbaldehyde (17)
Pyridinium chlorochromate (10.91 g, 50.6 mmol) was portion wise added to a stirred mixture of 1,3,5(10)-estratrien-3-ylmethanol (16) (6.84 g, 25.3 mmol) und molecular sieves (25 g, 3 Å)28 in dry CH2Cl2 (125 mL) under cooling with cold water. The mixture was stirred for 1.5 h at room temperature. After addition of Et2O (380 mL), the mixture was filtered through silica 60 (50 g). The reaction flask was washed with Et2O (3 × 80 mL). The filtrate was concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/CH2Cl2, 2:1, Rf 0.17) to give aldehyde 17 (6.18 g, 91%) as colourless crystals; mp 95–97 °C (hexanes/CH2Cl2); [α]20D + 88.4 (c. 2.04, acetone).
IR (Si): ν 2946, 1691, 1606, 1568, 1453, 1378, 1281, 1226, 1153 cm−1; 1H NMR (400.13 MHz, CDCl3): δ 0.73 (s, 3H), 1.10–1.82 (m, 11H), 1.85–2.00 (m, 2H), 2.24–2.37 (m, 2H), 2.88–2.98 (m, 2H), 7.45 (d, J = 8.0 Hz, 1H), 7.57 (d, J = 1.2 Hz, 1H), 7.63 (dd, J = 8.0, 1.2 Hz, 1H), 9.92 (s, 1H); 13C NMR (100.61 MHz, CDCl3): δ 17.5, 20.5, 25.2, 26.4, 27.7, 29.4, 38.5, 38.7, 40.4, 40.9, 45.0, 53.7, 126.1, 127.0, 130.3, 134.0, 137.8, 148.3, 192.4. Anal. calcd for C19H24O: C, 85.03; H, 9.01. Found: C, 85.12; H, 9.07.
1,3,5(10)-Estratriene-3-carbaldehyde tosylhydrazone (18)
A solution of 1,3,5(10)-estratriene-3-carbaldehyde (17) (6.10 g, 22.7 mmol) and tosyl hydrazide (4.95 g, 26.6 mmol, 1.17 equiv.) in dry methanol (75 mL) was stirred for 1 h at room temperature and 1 h at 40 °C.29 The solution was concentrated under reduced pressure. The residue was purified by flash chromatography (CH2Cl2, Rf 0.29) to yield hydrazone 18 (9.40 g, 95%) as crystals; mp 189–192 °C (toluene/EtOH); [α]20D + 46.9 (c. 1.92, CHCl3).IR (Si): ν 3196, 2925, 2867, 1451, 1364, 1321, 1167, 1052 cm−1; 1H NMR (400.13 MHz, CDCl3): δ 0.71 (s, 3H), 1.07–1.80 (m, 11H), 1.82–1.95 (m, 2H), 2.18–2.31 (m, 2H), 2.38 (s, 3H), 2.79–2.87 (m, 2H), 7.24–7.34 (m, 5H), 7.68 (s, 1H), 7.74 (br s, 1H), 7.82–7.87 (m, 2H); 13C NMR (100.61 MHz, CDCl3): δ 17.5, 20.5, 21.5, 25.2, 26.4, 27.8, 29.5, 38.69, 38.74, 40.4, 41.0, 44.7, 53.6, 124.7, 125.7, 127.8, 127.9 (2C), 129.7 (2C), 130.3, 135.3, 137.3, 143.8, 144.1, 148.5. Anal. cald for C26H32N2O2S: C, 71.52; H, 7.39; N, 6.42. Found: C, 71.43; H, 7.29; N, 6.35.
(1R,Sp)- and (1R,Rp)-(1′,3′,5′(10′)-estratrien-3′-ylmethyl) ethyl (1-hydroxy-2-methylsulfanyl-1-phenylethyl)phosphonate (20 and 23)
Preparation of 1,3,5(10)-estratrien-3-yl-diazomethane (19) from 1,3,5(10)-estratrien-3-carbaldehyde tosyl hydrazone (18): A mixture of tosyl hydrazone 18 (1.51 g, 3.46 mmol) and NaHMDS (0.80 g, 4.15 mmol, 95%, 1.2 equiv.) in dry THF (45 mL) was refluxed for 90 min30 After cooling at room temperature the solvent was removed under reduced pressure. Water (60 mL) was added and the mixture was extracted with CH2Cl2 (3 × 40 mL). The combined organic layers were washed with water, dried (Na2SO4) and concentrated under reduced pressure. The residue was twice dissolved in toluene und concentrated each time under reduced pressure. The dark red residue was dried for 10 min (0.5 mbar/RT) and then immediately used for the next step.2. Esterification of phosphonic acid: A solution of (R)-(−)-ethyl hydrogen (1-hydroxy-2-methylsulfanyl-1-phenylethyl)-phosphonate [(R)-(−)-10] (0.577 g, 2.1 mmol, prepared from the (R)-1-phenylethylammonium salt by general procedure A) in dry CH2Cl2 (20 mL) was dropwise added to a stirred solution of the above prepared crude steroidal diazomethane in dry CH2Cl2 (25 mL) within 15 min at room temperature. While the reaction mixture was stirred for 40 min at room temperature, the colour changed from deep red to orange. Excess diazomethane was destroyed by dropwise addition of AcOH (colour changed to yellow). The solvent was removed under reduced pressure. The residue was purified by flash chromatography (CH2Cl2/EtOAc, 10:1, Rf 0.28) to give a mixture of epimers 20 and 23 (0.99 g, 90%; ratio 1:1, by 1H NMR) as a colourless oil. The epimers were separated by HPLC (analytical HPLC: Nucleosil 50–4 column, 0.46 × 25 cm, 5% i-PrOH in hexanes, 1 mL × min−1, tR = 10.7 and 11.4 min; preparative HPLC: Nucleosil 50–7 column, 6.3 × 28.8 cm, 2.5% i-PrOH in hexanes). The less polar epimer 20 had a de of 88% and the more polar 23 of 96%. Crystallisation increased the de of the former to >98% [from hexanes, crystals contained solvent; 20/hexanes, 3.13:1, by 1H NMR] and of the latter to also >98% (hexanes/i-PrOH). Crystals of 23 were unsolvated and used for the determination of the X-ray structure.
20: Less polar epimer; for crystals from hexanes: mp 52–54 °C; [α]20D + 18.24 (c. 1.03, CHCl3). Crystallisation from cyclohexane furnished crystals containing cyclohexane (20/cyclohexane, 2:1, by 1H NMR), mp 49–52 °C.
IR (Si): ν 3280, 2932, 2867, 1449, 1376, 1220, 1100, 1014, 985, 972 cm−1. NMR spectra are given for cyclohexane-containing crystals. 1H NMR (400.27 MHz, CDCl3): δ 0.72 (s, 3H), 1.06 (td, J = 7.0, 0.4 Hz, 3H), 1.09–1.80 (m, 11H), 1.41 (s, 6H, cyclohexane), 1.81 (s, 3H), 1.83–1.95 (m, 2H), 2.18–2.33 (m, 2H), 2.77–2.91 (m, 2H), 3.39 (AB part of ABP system, JAB = 14.0 Hz, J = 7.8, 7.4 Hz, 2H), 3.47 (d, J = 17.6 Hz, 1H), 3.67–3.79 (m, 1H), 3.82–3.93 (m, 1H), 5.03 (AB part of ABP system, JAB = 11.6 Hz, J = 7.8, 6.9 Hz, 2H), 7.02 (br. s, 1H), 7.09 (dd, J = 8.0, 1.5 Hz, 1H), 7.25–7.37 (m, 4H), 7.60–7.65 (m, 2H); 13C NMR (100.65 MHz, CDCl3): δ 16.2 (d, J = 5.8 Hz), 17.1, 17.5, 20.6, 25.2, 26.6, 26.9 (cyclohexane), 28.0, 29.6, 38.8, 38.9, 40.5, 41.0, 43.8 (d, JPC = 6.5 Hz), 44.5, 53.7, 63.9 (d, J = 6.5 Hz), 68.6 (d, J = 7.5 Hz), 75.0 (d, J = 161.7 Hz), 125.3, 125.6, 126.4 (d, J = 4.2 Hz, 2C), 127.8 (d, J = 2.8 Hz), 128.2 (d, J = 2.7 Hz, 2C), 128.7, 133.3 (d, J = 6.7 Hz), 137.1, 138.9, 141.2. 31P NMR (162.03 MHz, CDCl3): δ 21.37. Anal. calcd for C30H41O4PS×0.5C6H12:C, 69.44; H, 8.30. Found: C, 69.06; H, 8.12.
23: More polar epimer; mp 108–112 °C (hexanes/i-PrOH); [α]20D + 31.7 (c. 0.99, CHCl3). IR (Si): ν 3280, 2922, 2866, 1449, 1377, 1222, 1102, 1047, 1037, 999, 985 cm−1. 1H NMR (400.13 MHz, CDCl3): δ 0.72 (s, 3H), 1.07–1.81 (m, 11H), 1.25 (t, J = 7.0 Hz, 3H), 1.83 (s, 3H), 1.85–1.96 (m, 2H), 2.16–2.33 (m, 2H), 2.73–2.90 (m, 2H), 3.40 (AB part of ABP system, JAB = 14.1 Hz, J = 7.8, 7.5 Hz, 2H), 3.61 (d, J = 17.4 Hz, 1H), 4.05–4.18 (m, 2H), 4.71 (AB part of ABP system, JAB = 11.5 Hz, J = 7.5, 6.5 Hz, 2H), 6.88 (s, 1H), 6.96 (d, J = 7.9 Hz, 1H), 7.22 (d, J = 7.9 Hz, 1H), 7.26–7.39 (m, 3H), 7.60–7.67 (m, 2H); 13C NMR (100.61 MHz, CDCl3): δ 16.3 (d, J = 5.8 Hz), 17.1, 17.5, 20.5, 25.2, 26.5, 27.9, 29.6, 38.77, 38.83, 40.4, 41.0, 43.7 (d, J = 6.6 Hz), 44.4, 53.6, 63.5 (d, J = 7.6 Hz), 68.9 (d, J = 7.3 Hz), 75.0 (d, J = 161.3 Hz), 125.1, 125.5, 126.4 (d, J = 4.1 Hz, 2C), 127.8 (d, J = 2.8 Hz), 128.1 (d, J = 2.3 Hz, 2C), 128.5, 133.2 (d, JPC = 6.5 Hz), 136.9, 138.9, 141.0; 31P NMR (161.98 MHz, CDCl3): δ 22.13. Anal. calcd for C30H41O4PS: C, 68.16; H, 7.82. Found: C, 68.36; H, 7.77.
[1′,3′,5′(10′)-Estratrien-3′-ylmethyl] ethyl 1-phenylethenyl phosphate [22, prepared from 20]
A solution of methyl fluorosulfate (0.32 g, 2.8 mmol, 0.22 mL, 2.0 equiv.) in dry CH2Cl2 (1.2 mL) was dropwise added to a stirred solution of α-hydroxyphosphonate 20 (0.741 g, 1.4 mmol) in dry CH2Cl2 (10 mL) at −35 °C. The mixture was stirred for 35 min at −35 °C and 2 h at room temperature. Then, the solvent was removed under reduced pressure. The residue was dried for 45 min and dissolved in dry DMSO (10 mL). Phosphazene base P1-t-Bu31 (0.656 g, 2.8 mmol, 0.71 mL, 2.0 equiv.) was added. After stirring for 30 min at room temperature, water (75 mL) was added and the mixture was twice extracted with Et2O (75 mL and 50 mL). To improve phase separation, the aqueous layer was saturated with NaCl. The combined organic layers were washed with water (3 × 50 mL), dried (MgSO4) and concentrated under reduced pressure. The residue was purified by flash chromatography (hexanes/EtOAc, 3:1; Rf 0.31) to give phosphate 22 (0.308 g, 46%) as a colourless oil; [α]20D + 46.8 (c. 2.01, ethanol).
IR (Si): ν 2933, 2868, 1635, 1449, 1377, 1270, 1158, 1103, 1014 cm−1. 1H NMR (400.13 MHz, CDCl3): δ 0.72 (s, 3H), 1.09–1.82 (m, 11H), 1.32 (td, J = 7.4, 0.8 Hz, 3H), 1.80–1.96 (m, 2H), 2.22–2.32 (m, 2H), 2.76–2.91 (m, 2H), 4.17 (quin, J = 7.4 Hz, 2H), 5.07 (AB part of ABP system, JAB = 11.5 Hz, J = 8.0, 7.9 Hz, 2H), 5.21 (≈ t, J = 2.6 Hz, 1H), 5.26 (≈ t, J = 2.5 Hz, 1H), 7.05 (br. s, 1H), 7.11 (br. d, J = 8.1 Hz, 1H), 7.26 (d, J = 8.1 Hz, 1H), 7.29–7.36 (m, 3H), 7.50–7.56 (m, 2H); 13C NMR (100.61 MHz, CDCl3): δ 16.1 (d, J = 6.9 Hz), 17.5, 20.5, 25.2, 26.5, 27.9, 29.5, 38.79, 38.83, 40.5, 41.0, 44.4, 53.6, 64.6 (d, J = 6.1 Hz), 69.8 (d, J = 5.7 Hz), 97.3 (d, J = 3.6 Hz), 125.21 (2C), 125.23, 125.6, 128.3 (2C), 128.6, 129.0, 132.6 (d, J = 6.9 Hz), 134.3 (d, J = 6.9 H), 137.1, 141.3, 152.3 (d, J = 7.9 Hz). Anal. cald for C29H37O4P: C, 72.48; H, 7.76. Found: C, 72.05; H, 7.71.
[1′,3′,5′(10′)-Estratrien-3′-ylmethyl] ethyl 1-phenylethenyl phosphate [25, prepared from 23]
The α-hydroxyphosphonate 23 (0.741 g, 1.4 mmol) was converted to 25 (0.322 g, 48%) as colourless oil by the procedure as used for the preparation of 22; [α]20D + 41.9 (c. 1.92, ethanol).The IR spectrum and the 13C and 31P NMR spectra are identical to those of 22. The 1H NMR spectrum is identical to that of 22 except for the resonances of the POCH2 group (see Fig. 2): δ 5.07 (AB part of ABP system, JAB = 11.6 Hz, JAP = JBP = 7.9 Hz, 2H). Anal. calcd for C29H37O4P: C, 72.48; H, 7.76. Found: C, 72.20; H, 7.66.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank S. Felsinger for recording NMR spectra, S. Schneider for the preparative HPLC separation, J. Theiner for combustion analyses, H. Völlenkle for assistance with X-ray diffraction, and Schering AG for a gift of 3-hydroxy-1,3,5(10)-estratrien-17-one. S. Prechelmacher thanks the Austrian Academy of Sciences for a DOC scholarship. This work was supported by the Austrian Science Fund (FWF, grant no. P19869-N19).References
- (a) P. Beak and D. B. Reitz, Chem. Rev., 1978, 78, 275 CrossRef CAS; (b) M. C. Whisler, S. McNeil, V. Snieckus and P. Beak, Angew. Chem., Int. Ed., 2004, 43, 2206 CrossRef CAS PubMed.
- (a) G. Sturtz and B. Corbel, C. R. Acad. Sci., Ser. C, 1973, 276, 1807 CAS; (b) G. Sturtz, J.-J. Yaouanc, F. Krausz and B. Labeeuw, Synthesis, 1980, 289 CrossRef CAS.
- F. Hammerschmidt and H. Völlenkle, Liebigs Ann. Chem., 1986, 2053 CrossRef CAS.
- F. Benayoud, D. J. deMendonca, C. A. Digits, G. A. Moniz, T. C. Sanders and G. B. Hammond, J. Org. Chem., 1996, 61, 5159 CrossRef CAS.
- F. Hammerschmidt and S. Schmidt, Eur. J. Org. Chem., 2000, 2239 CrossRef CAS.
- S. Masson, M. Saquet and P. Marchand, Tetrahedron, 1998, 54, 1523 CrossRef CAS.
- V. Philippitsch and F. Hammerschmidt, Org. Biomol. Chem., 2011, 9, 5220 CAS.
- (a) F. Hammerschmidt and M. Hanbauer, J. Org. Chem., 2000, 65, 6121 CrossRef CAS PubMed; (b) E. Kuliszewska, M. Hanbauer and F. Hammerschmidt, Chem. – Eur. J., 2008, 14, 8603 CrossRef CAS PubMed.
- (a) W. F. Barthel, B. H. Alexander, P. A. Giang and S. A. Hall, J. Am. Chem. Soc., 1955, 77, 2424 CrossRef CAS; (b) W. Lorenz, A. Henglein and G. Schrader, J. Am. Chem. Soc., 1955, 77, 2554 CrossRef CAS; (c) M. S. Kharasch and I. S. Bengelsdorf, J. Org. Chem., 1955, 20, 1356 CrossRef CAS; (d) I. S. Bengelsdorf, J. Org. Chem., 1956, 21, 475 CrossRef CAS; (e) G. W. Fischer and P. Scheider, J. Prakt. Chem., 1977, 319, 399 CrossRef CAS.
- (a) M.-J. Brienne and M. J. Jacques, C. R. Acad. Sci., Ser. C, 1975, 280, 291 CAS; (b) M. J. Brienne, J. Jacques, M. C. Brianso and E. Surcouf, Nouv. J. Chim., 1978, 2, 19 CAS.
- K. Pallitsch, A. Roller and F. Hammerschmidt, Chem. – Eur. J., 2015, 21, 10200 CrossRef CAS PubMed.
- F. Hammerschmidt and E. Zbiral, Monatsh. Chem., 1980, 111, 1015 CrossRef CAS.
- A. E. Wroblewski and W. Karolczak, Pol. J. Chem., 1999, 73, 1191 CAS.
- A selection: (a) L. A. R. Hall, C. W. Stephens and J. J. Drydale, J. Am. Chem. Soc., 1957, 79, 1768 CrossRef CAS; (b) H. Timmler and J. Kurz, Chem. Ber., 1971, 104, 3740 CrossRef CAS; (c) F. Hammerschmidt, E. Schneyder and E. Zbiral, Chem. Ber., 1980, 113, 3891 CrossRef CAS; (d) M. Kuroboshi, T. Ishihara and T. Ando, J. Fluorine Chem., 1988, 39, 293 CrossRef CAS; (e) C. Meier, Angew. Chem., Int. Ed. Engl., 1993, 32, 1704 CrossRef; (f) C. Meier, L. W. Halbel, J. Balzarini and E. De Clercq, Liebigs Ann. Chem., 1995, 2195 CrossRef CAS; (g) R. Gancarz, I. Gancarz and A. Deron, Phosphorus, Sulfur Silicon, 2000, 161, 61 CrossRef CAS; (h) K. Pachamuthu and R. R. Schmidt, Chem. Commun., 2004, 1078 RSC; (i) C. C. Bausch and J. S. Johnson, Adv. Synth. Catal., 2005, 347, 1207 CrossRef CAS; (j) A. S. Demir, Ö. Reis, A. Òª. İğdir, İ. Esiringü and S. Eymur, J. Org. Chem., 2005, 70, 10584 CrossRef CAS PubMed; (k) L. E. Kaim, L. Gaultier, L. Grimaud and A. D. Santos, Synlett, 2005, 2335 CrossRef; (l) A. S. Demir, B. Reis, Ö. Reis, S. Eymür, M. Göllü, S. Tural and G. Saglam, J. Org. Chem., 2007, 72, 7439 CrossRef CAS PubMed; (m) R. Ruel, J.-P. Bouvier and R. N. Young, J. Org. Chem., 1995, 60, 5209 CrossRef CAS; (n) D. Coffinier, L. E. Kaim and L. Grimaud, Synlett, 2008, 1133 CAS; (o) M. Hayashi and S. Nakamura, Angew. Chem., Int. Ed., 2011, 50, 2249 CrossRef CAS PubMed; (p) M. T. Corbett, D. Uraguchi, T. Ooi and J. S. Johnson, Angew. Chem., Int. Ed., 2012, 51, 4685 CrossRef CAS PubMed; (q) A. Kondoh and M. Terada, Org. Lett., 2013, 15, 4568 CrossRef CAS PubMed; (r) A. Kondoh, T. Aoki and M. Terada, Org. Lett., 2014, 16, 3528 CrossRef CAS PubMed; (s) A. Kondoh and M. Terada, Org. Chem. Front., 2015, 2, 801 RSC; (t) A. Kondoh, T. Aoki and M. Terada, Chem. – Eur. J., 2015, 21, 12577 CrossRef CAS PubMed; (u) M. A. Horwitz, N. Tanaka, T. Yokosaka, D. Uraguchi, J. S. Johnson and T. Ooi, J. Chem. Sci., 2015, 6, 6086 RSC; (v) A. Kondo, A. Takai and M. Terada, Synlett, 2016, 1848 Search PubMed; (w) H. Yoneyama, K. Uemura, Y. Usami and S. Hurusawa, Tetrahedron, 2017, 73, 6109 CrossRef CAS; (x) for the rearrangement of a β-oxophosphonate to a phosphate see: M. Anitha, G. Gangadhararao and K. C. Kumara Swamy, Org. Biomol. Chem., 2016, 14, 3591 RSC.
- F. Hammerschmidt, Monatsh. Chem., 1993, 124, 1063 CrossRef CAS.
- S. Jankowski, J. Marczak, A. Olczak and M. L. Główka, Tetrahedron Lett., 2006, 47, 3341 CrossRef CAS.
- (a) A. N. Pudovik and I. V. Konovalova, Zh. Obshch. Khim., 1962, 32, 467 CAS; (b) A. N. Pudovik and M. G. Zimin, Pure Appl. Chem., 1980, 52, 989 CrossRef CAS.
- (a) C. M. Rojas, in From Name Reactions for Homologations-2, ed. J. J. Li, John Wiley & Sons, Inc., Hoboken, New Jersey, 2009, Pt. 2, p. 406 Search PubMed; (b) A. G. Brook, Acc. Chem. Res., 1974, 7, 77 CrossRef CAS.
- C. R. Hall and T. D. Inch, Tetrahedron, 1980, 36, 2059 CrossRef CAS.
- K. Nakayama and W. J. Thompson, J. Am. Chem. Soc., 1990, 112, 6936 CrossRef CAS.
- V. Prelog, V. Hahn, H. Brauchl and H. C. Beyerman, Helv. Chim. Acta, 1944, 47, 1209 CrossRef.
- L. F. Fieser and M. Fieser, Reagents for Organic Synthesis, John Wiley and Sons, Inc., 1967, p. 729 Search PubMed.
- (a) H. G. O. Becker, et al., Organikum (Organisch-chemisches Grundpraktikum), 21. Auflage, Wiley-VCH, Weinheim, New York, Chichester, Brisbane, Singapore, Toronto, 2001, p. 633 Search PubMed; (b) B. Eistert, M. Regitz, G. Heck and H. Schwall, Methoden der Organischen Chemie (Houben-Weyl, Hsg. E. Müller), 4. Auflage, Band X/4, Georg Thieme Verlag, Stuttgart, 1968, p. 539 Search PubMed.
- F. Hammerschmidt , unpublished results.
- Huang-Minlon, J. Am. Chem. Soc., 1949, 71, 3301 CrossRef CAS.
- P. J. Stang, M. Hanack and L. R. Subramanian, Synthesis, 1982, 85 CrossRef CAS.
- R. E. Dolle, S. J. Schmidt and L. I. Kruse, J. Chem. Soc., Chem. Commun., 1987, 904 RSC.
- J. Herscovici, M. J. Egron and K. Antonakis, J. Chem. Soc., Perkin Trans. 1, 1982, 1967 RSC.
- X. Creary, J. Org. Synth., Coll., 1990, 7, 438 Search PubMed.
- D. G. Farnum, J. Org. Chem., 1963, 28, 870 CrossRef CAS.
- R. Schwesinger, C. Hasenfratz, H. Schlemper, L. Walz, E.-M. Peters, K. Peters and H. G. von Schnering, Angew. Chem., Int. Ed. Engl., 1993, 32, 1361 CrossRef.
- (a) V. S. Abramov, Zh. Obshch. Khim, 1952, 22, 647 CAS; (b) A. N. Pudovik and I. V. Konovalova, Synthesis, 1979, 81 CrossRef CAS.
- (a) G.-V. Roeschenthaler, Organophosphorus Chem., 2008, 37, 247 CAS; (b) C. D. Hall, Organophosphorus Chem., 1986, 16, 51 CAS; (c) O. I. Kolodiazhnyi and A. Kolodiazhna, Tetrahedron: Asymmetry, 2017, 28, 1651 CrossRef CAS.
- W. Mikenda, Vib. Spectrosc., 1992, 3, 327–334 CrossRef CAS.
- (a) H. G. O. Becker et al. , Organikum (Organisch-chemisches Grundpraktikum), 21. Auflage, Wiley-VCH, Weinheim, New York, Chichester, Brisbane, Singapore, Toronto, 2001, p. 627 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra. Crystallographic data. CCDC 1818362. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob00419f |
| This journal is © The Royal Society of Chemistry 2018 |