
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis and structural elucidation of a dehydrochloromethyltestosterone metabolite†
Nicolas
Kratena
 a,
Sarah M.
Pilz
a,
Matthias
Weil
b,
Günter
Gmeiner
c,
Valentin S.
Enev
*a and
Peter
Gärtner
*a
a,
Sarah M.
Pilz
a,
Matthias
Weil
b,
Günter
Gmeiner
c,
Valentin S.
Enev
*a and
Peter
Gärtner
*a
aInstitute of Applied Synthetic Chemistry, Vienna University of Technology, Getreidemarkt 9, 1060 Vienna, Austria. E-mail: valentin.enev@tuwien.ac.at; peter.gaertner@tuwien.ac.at
bInstitute for Chemical Technologies and Analytics, Vienna University of Technology, Getreidemarkt 9, 1060 Vienna, Austria
cDoping Control Laboratory, Seibersdorf Labor GmbH, 2444 Seibersdorf, Austria
First published on 6th March 2018
Abstract
The human urinary long-term metabolite “M3” (4-chloro-17β-hydroxymethyl-17α-methyl-18-norandrost-13-en-3-ol) of the common doping agent DHCMT has thus far been detected via GC/MS-MS, creating ambiguities concerning its absolute configuration. Its structure was elucidated via the synthesis of all eight possible stereoisomers with 17β-hydroxymethyl configuration. The highlights of the synthesis consist of a novel first generation approach to 4β-chloro-5β compounds as well as a divergent route which allows easy access to the remaining A-ring chlorohydrins.
Introduction
In doping control analysis, a tool with still increasing importance is high sophisticated mass spectrometry MS (HRMS or tandem MS) coupled either to an HPLC or a GC separation system.1 Due to a lack of structural information, especially in complex organic substances, it is often not possible to completely assign the configuration of in vivo formed stereogenic centers using these methods. In order to unambiguously identify the compound or its metabolites in an athlete's sample, the chemical synthesis of the target compound followed by analytical characterization is a suitable method.A novel long-term metabolite, adapted by the World Anti-Doping Agency (WADA) for testing in 2013 extended the window of detection for dehydrochloromethyltestosterone (DHCMT, 2) significantly to an estimated time frame of 40–50 days. This resulted in a significant increase in the number of adverse analytical findings for DHCMT in 2013 (1 to 87).2 The responsible metabolite M3 (1) (4-chloro-17β-hydroxymethyl-17α-methyl-18-norandrost-13-en-3-ol) was first detected and described by Sobolevsky and Rodchenkov in 2012.3 The metabolic transformations leading to this metabolite are full A-ring hydrogenation, Wagner–Meerwein-rearrangement and hydroxylation (or vice versa). It was assumed that the metabolite would be reduced in vivo forming a 5β-androstane, and the authors also argued that the C-3 hydroxyl should be α since 3β-steroids tend to be excreted as sulphate conjugates.4
The stereochemistry at C-4 was left uncommented on, but the literature suggests the formation of an equatorial 4β-chloride.5 The identity of the compound is not ensured and a reference material provided by organic synthesis would be beneficial for method development and substantially facilitate assay characterization efforts.
In order to prove the identity of the metabolite and elucidate the unknown stereochemistry we planned the synthesis of the most probable target, 4β-chloro-17β-hydroxymethyl-17α-methyl-18-nor-5β-androst-13-en-3α-ol (3a). In continuation of previous work on long-term metabolites with the 17-hydroxymethyl-17-methyl-18-nor-13-en fragment6 we herein report the synthesis of eight possible stereoisomers of compound 1 concerning configurations at C-3, C-4 and C-5 (see Fig. 1).
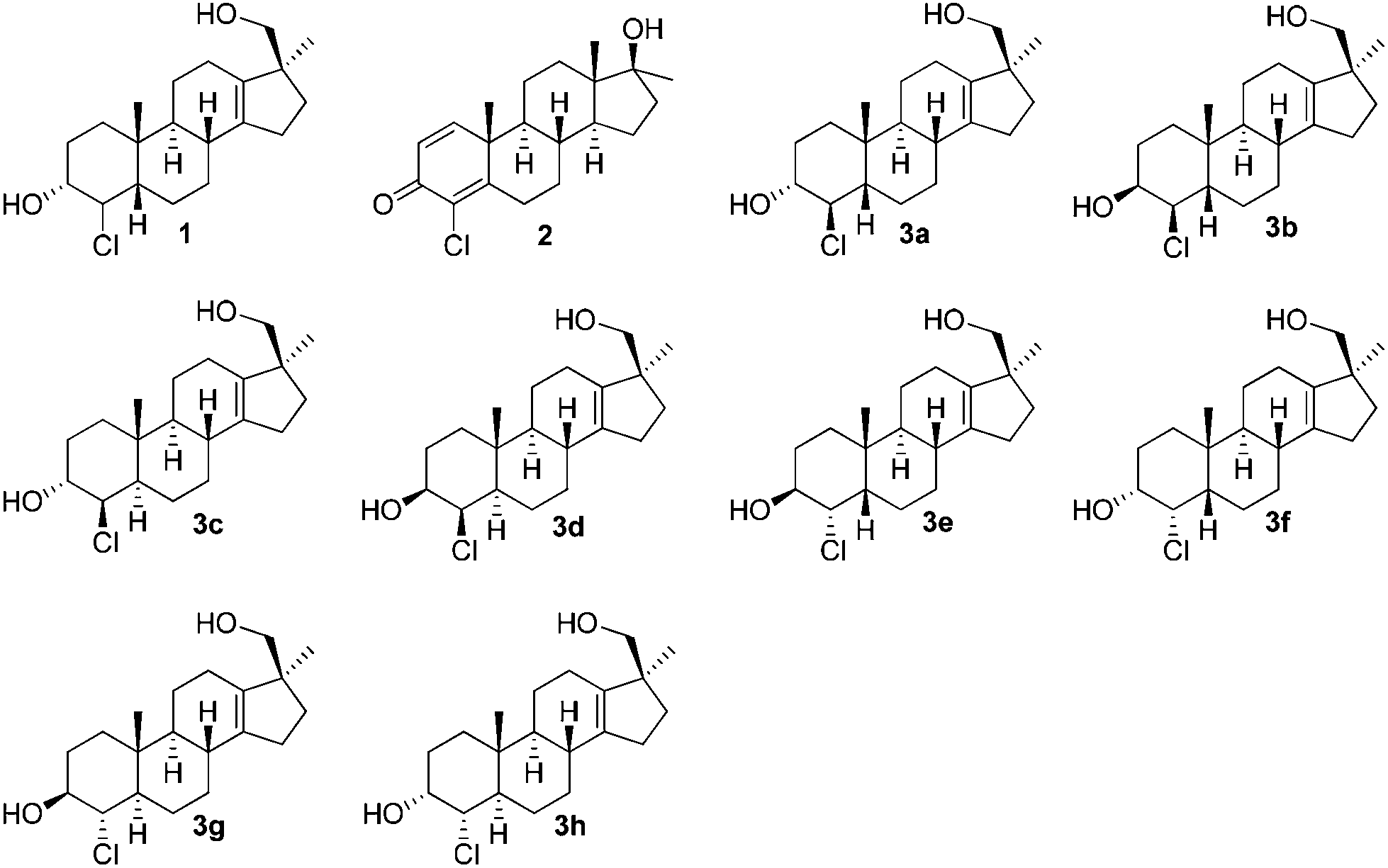 | ||
| Fig. 1 Relevant structures. | ||
Retrosynthetic analysis
The retrosynthetic analysis of 3a is depicted in Scheme 1 and starts with a Wagner–Meerwein-rearrangement to install the functionalities at the D-ring. Epoxide 11 can conceivably be formed from the corresponding C-17 exo-methylene, which might be obtained by the reduction of chloroketone 6. It is known that 3-keto-5β androstanes are kinetically deprotonated at C-4 regioselectively,7 so ketone 5 would be suited for chlorine introduction. The 3-keto-5β structural motif might come from α,β-unsaturated ketone which is a known intermediate from another metabolite synthesis.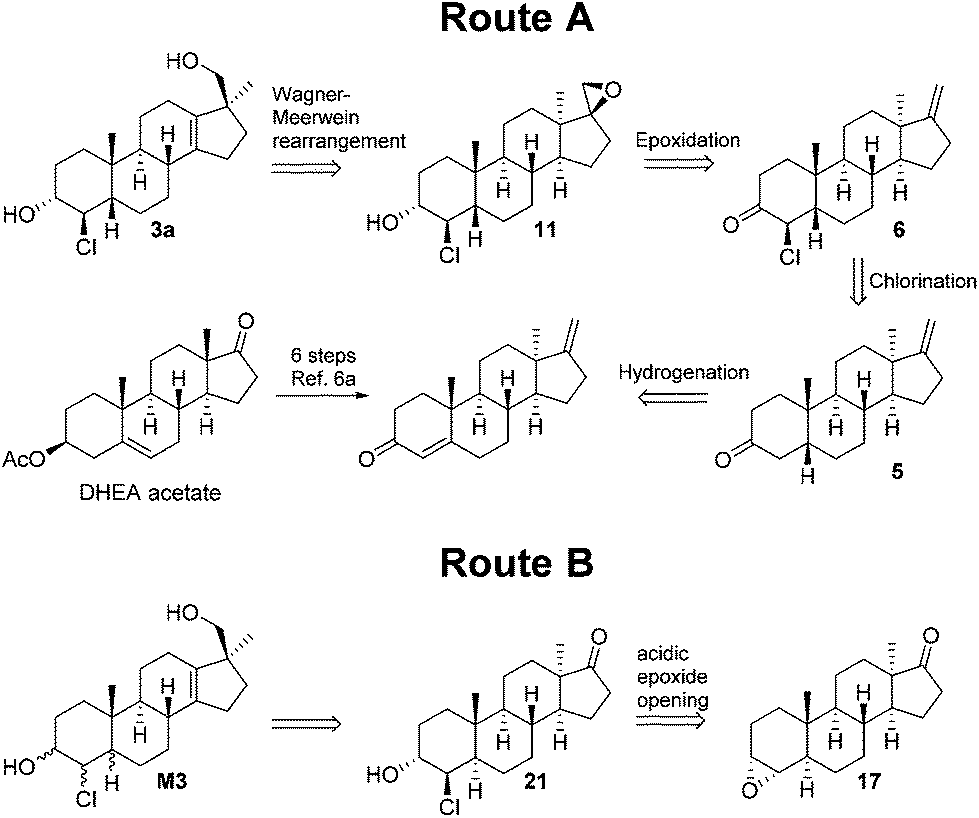 | ||
| Scheme 1 Retrosynthetic analysis of compound 3a and divergent route. | ||
For Route B, the retrosynthesis of the D-ring is as described above. The trans/trans-chlorohydrins could be constructed from the C-3-axial epoxides. If epoxide 17 can be accessed, a trans addition of the nucleophile8 would deliver the desired product, given the reaction proceeds under the right regioselectivity. The epoxides could be synthesized from the corresponding olefins. The literature precedence indicated straightforward access to these olefins via the Clemmensen reduction of an α,β-unsaturated ketone which can be derived from dehydroepiandrosterone acetate (DHEA acetate), a commercially available starting material. Inversion of the C-3 can be achieved by oxidation/reduction or a Mitsunobu reaction. To reach the trans/cis and cis/cis isomers of the 5α series a suitable 3-keto-4β-chloro intermediate needs to be epimerised.
Results and discussion
1st generation approach (Route A)
The starting material for this sequence, enone 4, was synthesized in 6 steps with an overall yield of 40% from DHEA acetate (Scheme 2).6a The chemoselective hydrogenation of the double bond in conjugation was achieved by organocatalytic transfer hydrogenation.9 The exocyclic double bond was not touched and ketone 5 was isolated as a 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 diastereomeric mixture favouring the desired 5β-product. Next, introducing the chloride through enolate chemistry was investigated and it was found that treating the lithium enolate of 5 with N-chlorosuccinimide (NCS) yields the 4β-chloro product 6 as a single diastereomer in 61% yield.
:1 diastereomeric mixture favouring the desired 5β-product. Next, introducing the chloride through enolate chemistry was investigated and it was found that treating the lithium enolate of 5 with N-chlorosuccinimide (NCS) yields the 4β-chloro product 6 as a single diastereomer in 61% yield.
 | ||
| Scheme 2 Synthesis of 3a and 3b with 1st generation approach: (a) D-CSA, Hantzsch ester, (S)-(2-pyrrolidinylmethyl)pyrrolidine, CH3CN, 70 °C; (b) LiHMDS, THF, NCS, −70 °C; (c) NaBH4, MeOH, rt; (d) m-CPBA, CH2Cl2, rt; (e) H3PO4, THF/H2O 2/1, rt. | ||
All other approaches (silyl enol ethers or other sources of electrophilic chlorine) failed or gave complex mixtures. At this stage, the structure of compound 6 was confirmed via single crystal X-ray diffraction to ascertain C-4, C-5 configuration.
Next, the reduction of 6 (sodium borohydride) gave a 1:1 mixture of alcohols which after separation yielded 7 and 8 in a ratio of 1:1.3. The epoxidation of these olefins with m-CPBA gives 17α-epoxy as major (10, 12, not shown in the scheme) and 17β-epoxy as minor (9, 11) products, and the latter were isolated in 25–30% yield. The following rearrangement was carried out with the 17β-epoxides only, since they possess the right stereoelectronic prerequisites.6b Accordingly, epoxides 9 and 11 were treated with phosphoric acid to give the first two putative metabolites 3a (trans product: JH3–H4 = 9.6 Hz) and 3b (cis product: JH3–H4 = 2.7 Hz) in 43% and 28% isolated yield, respectively. Additionally, 3β configuration in 7 which yielded 3b was confirmed by a reduction experiment of 6 with L-Selectride® which is known to give exclusively cyclohexanols with an axial hydroxyl group by the equatorial delivery of the hydride.
After the first two putative metabolites showed some differences in compound 1 found in excretion studies, the synthesis of the remaining stereoisomers was attempted. A number of approaches for the synthesis of 4α-chloro-5β compounds were unsuccessful (epimerisation, inversion via substitution, hydrogenation of a chloroalkenyl). Ultimately, the divergent synthesis outlined in Scheme 1 (Route B) was pursued.
2nd generation approach (Route B)
As depicted in Scheme 3, the divergent synthesis started with already known alcohol 13 (2 steps, 80%, from DHEA acetate)6a which was oxidized under Oppenauer conditions to give diketone 14 in 91% yield. Subjecting this diketone to excess zinc in acetic acid rapidly reduces the A-ring enone to an olefin10 giving rise to a new stereocenter at C-5 in a 1.3:1 ratio favouring the 5α isomer. The separation of the diastereomers 15 and 16 was not practical at this stage, instead the mixture of olefins was epoxidized with m-CPBA to give compounds 17 and 18 as the only two diastereomers. The epoxides were then treated with concentrated hydrochloric acid and gave a mixture of isomeric chlorohydrins 19, 20 and 21. While for the 5α-isomer the opening proceeded with distinct regioselection, for the 5β-isomer both possible regioisomers were observed. The separation of this three-component mixture was achieved with column chromatography and chlorohydrins 19, 20 and 21 were isolated in 14%, 19% and 42% yield (over three steps). The configuration and stereochemistry of diastereomer 19 were proved by single crystal X-ray determination. The undesired chlorohydrin 20 can be recycled to pure 18 by treatment with a strong base.11 Likewise chlorohydrin 21 was used to obtain a pure analytical sample of 17.
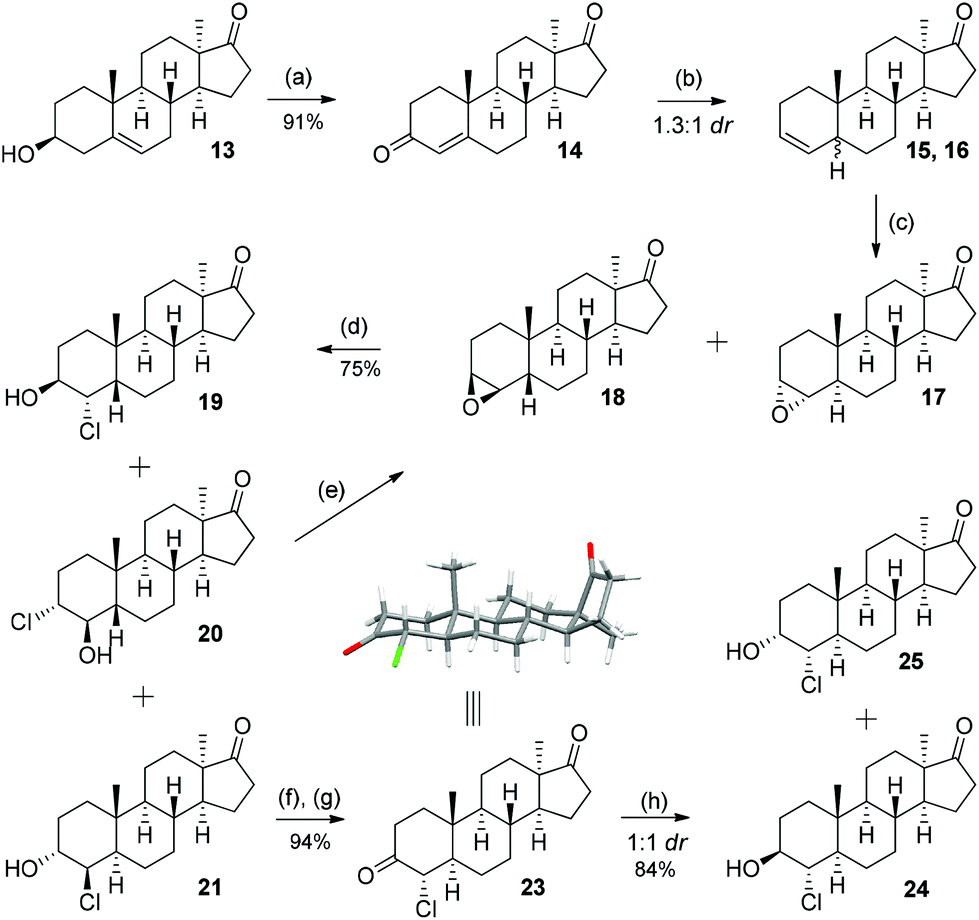 | ||
| Scheme 3 Synthesis of the 17-keto-chlorohydrins: (a) Al(O-iPr)3, cyclohexanone, toluene, reflux; (b) Zn, AcOH, reflux; (c) m-CPBA, CH2Cl2, rt; (d) conc. HCl, CHCl3, rt; (e) KOH, MeOH, reflux; (f) DMP, CH2Cl2, rt; (g) KOH, MeOH, rt; (h) Al(O-iPr)3, iPrOH, toluene, 70 °C. | ||
Chlorohydrins 19 and 21, if further elaborated, can lead to four new putative metabolites (vide infraSchemes 4 and 5). The last two of the eight possible diastereomers in this series are the 4α-chloro-5α compounds. These were obtained by the oxidation of 21 with DMP (Dess–Martin periodinane) and treatment of the resulting diketone 22 with potassium hydroxide, which effected complete C-4 epimerisation over two steps to give 23 in 94% yield.
 | ||
| Scheme 4 D-ring modifications of chlorohydrin 21: (a) TBSCl, imidazole, DMF, rt; (b) Nysted reagent, TiCl4, THF, 0 °C to rt; (c) m-CPBA, CH2Cl2, rt; (d) TMSOTf, 2,6-lutidine, toluene, −70 °C; (e) 2 M HCl, MeOH, rt; (f) DMP, CH2Cl2, rt; (g) NaBH4, MeOH, 0 °C. | ||
 | ||
| Scheme 5 D-ring modifications of chlorohydrin 19: (a) TBSCl, imidazole, DMF, rt; (b) Nysted reagent, TiCl4, THF, 0 °C to rt; (c) m-CPBA, CH2Cl2, rt; (d) TMSOTf, 2,6-lutidine, CH2Cl2, −70 °C; (e) 2 M HCl, MeOH, rt; (f) DMP, CH2Cl2, rt; (g) NaBH4, MeOH, 0 °C. | ||
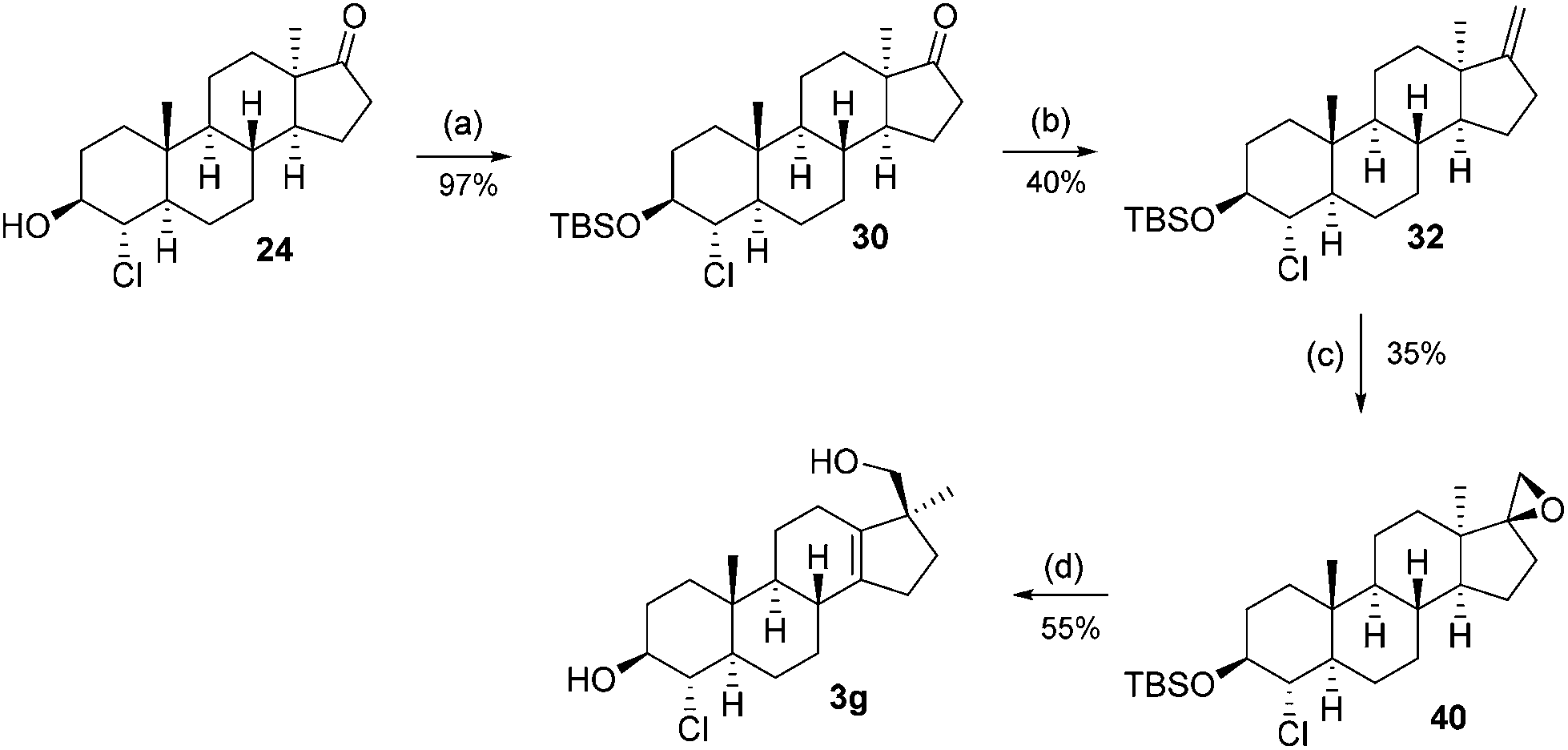
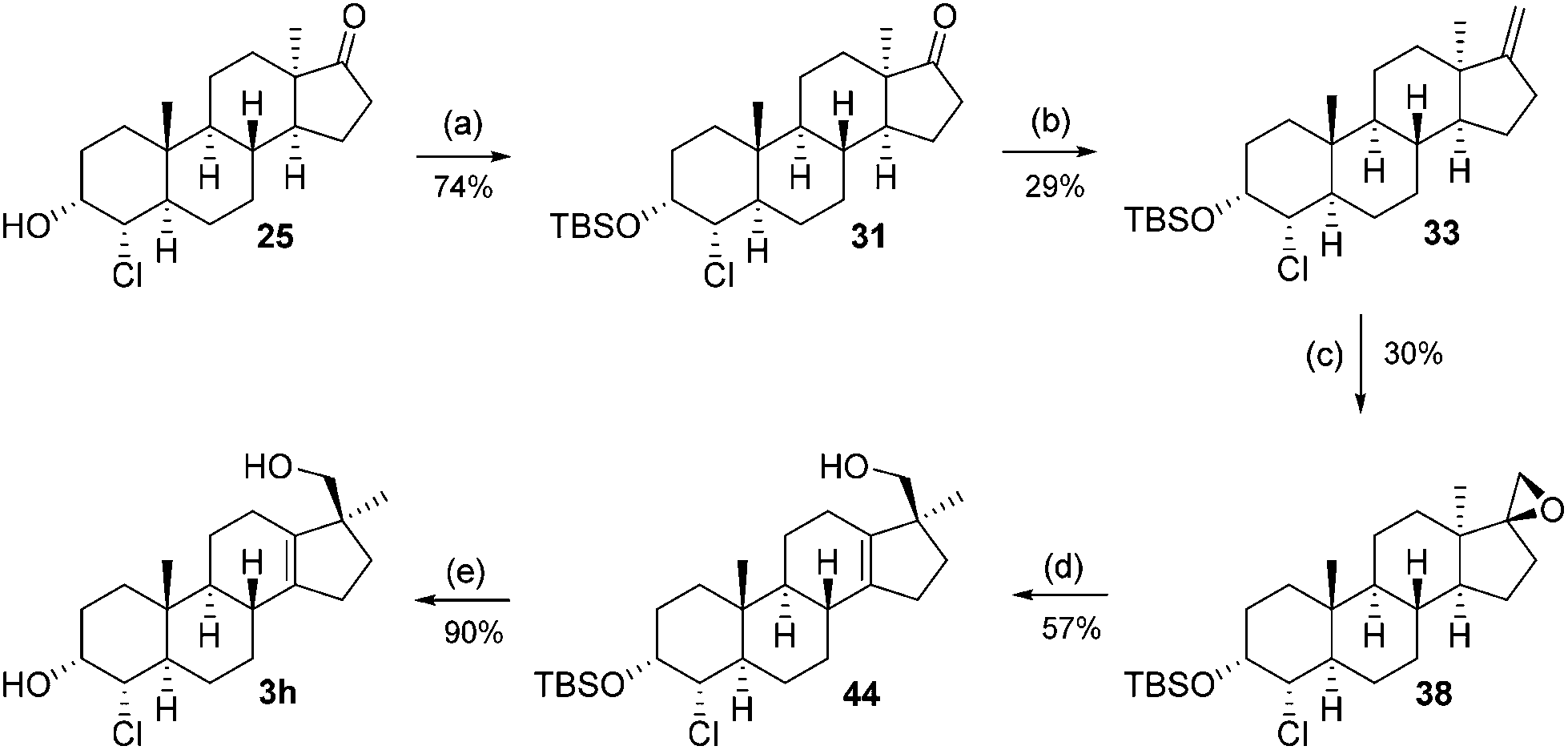
Its structure was also confirmed by single-crystal XRD. A reduction affecting only the C-3 ketone was needed and it was found that Meerwein–Ponndorf–Verley reduction results in a 1:1 mixture of C-3 epimeric alcohols 24 and 25 in 84% yield, which were separated and provides the starting point for the synthesis of the last two potential metabolites.
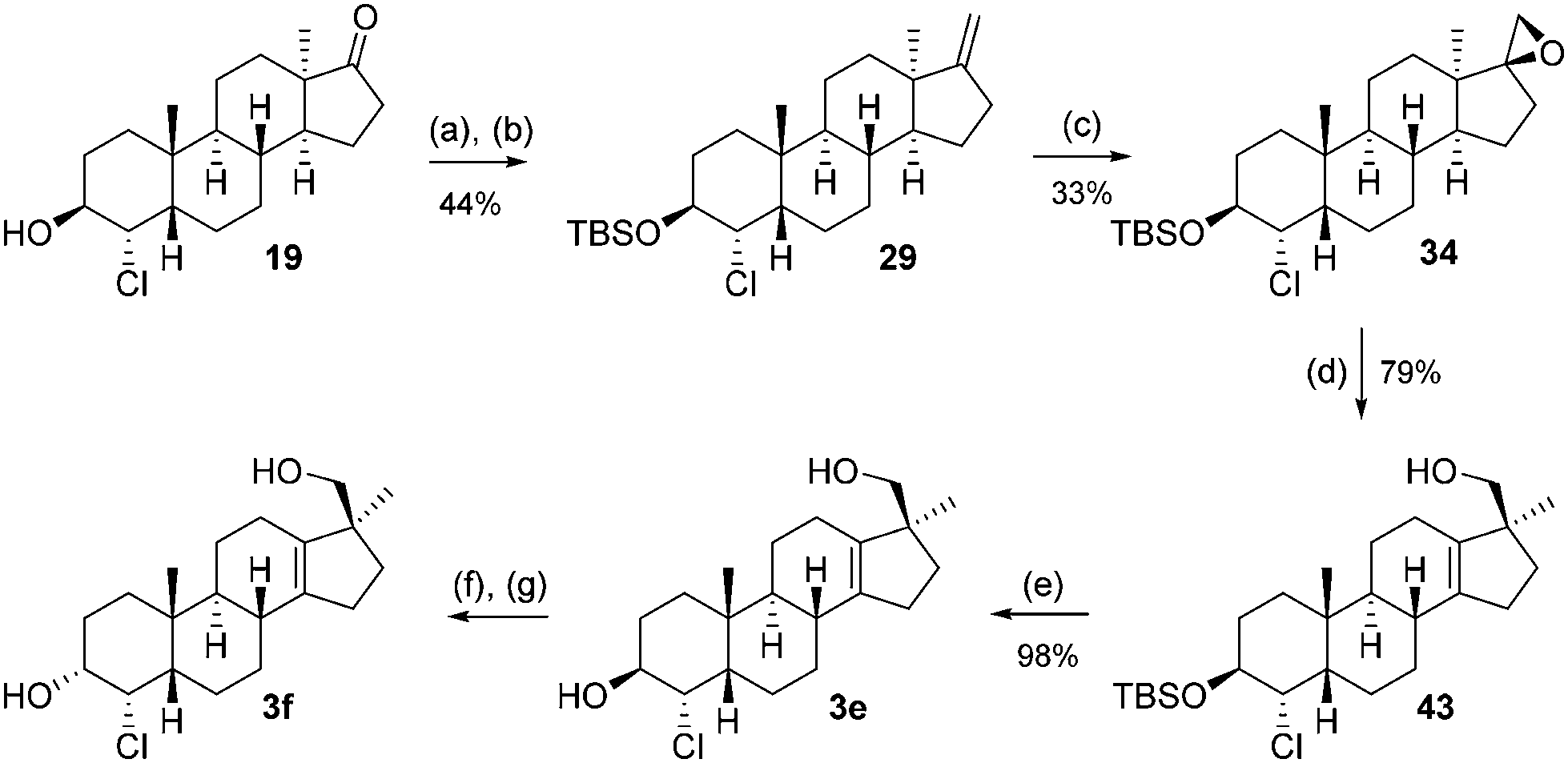
The synthesis from this point forward is the same for all four chlorohydrins and is discussed in detail for 21. First, the alcohol is protected as a TBS ether (26) and subsequently treated with the Nysted reagent12 and titanium tetrachloride to give exo-methylene product 28. The double-bond is epoxidized with m-CPBA to give a 2:1 mixture of diastereomers (major product 37: vide infraFig. 2) of which again only the minor product 36 was used. The Wagner–Meerwein-rearrangement was initiated by trimethylsilyl triflate/2,6-lutidine in dichloromethane or toluene at low temperatures.13 This method offers a slight increase in isolated yield compared to the aqueous acidic rearrangement protocol used before (typically 45–55%). Finally, the TBS ether in 42 is cleaved with hydrochloric acid in methanol to furnish the final trans (JH3–H4 = 2.61 Hz) product 3c. To arrive at the C-3 epimer global oxidation with DMP, followed by NaBH4 reduction, furnished the equatorial cis (JH3–H4 = 2.78 Hz) alcohol 3d.
 | ||
| Fig. 2 Molecular structures of relevant intermediates determined from single crystal X-ray diffraction data. | ||
This sequence was also carried out for the previously mentioned chlorohydrins: 19, 24 (JH3–H4 = 9.2 Hz) and 25, giving the isomers 3e–h and completing the synthesis of eight possible isomeric 4-chloro-17β-hydroxymethyl-17α-methyl-18-norandrost-13-en-3-ols (see Schemes 5–7; compounds 27, 35, 39, 41, and 45 not depicted). Single crystal X-ray analysis of 25 and 37 was performed to secure structural information on the stereochemistry of the A-ring substituents and the 17-spiroepoxides. The molecular structures resulting from the previously mentioned XRD experiments are depicted in Fig. 2.
 | ||
| Scheme 6 D-ring modifications of chlorohydrin 24: (a) TBSCl, imidazole, DMF, rt; (b) Nysted reagent, TiCl4, THF, 0 °C to rt; (c) m-CPBA, CH2Cl2, rt; (d) TMSOTf, 2,6-lutidine, CH2Cl2, −70 °C, then 2 M HCl. | ||
 | ||
| Scheme 7 D-ring modifications of chlorohydrin 25: (a) TBSCl, imidazole, DMF, rt; (b) Nysted reagent, TiCl4, THF, 0 °C to rt; (c) m-CPBA, CH2Cl2, rt; (d) TMSOTf, 2,6-lutidine, CH2Cl2, −70 °C; (e) 2 M HCl, MeOH, rt. | ||
Conclusions
To summarise, we have accomplished the first synthesis of the reported long-term metabolite “M3” of dehydrochloromethyltestosterone. All possible eight stereoisomers 3a–3h of this material have been synthesized using two different synthetic routes. A straightforward, short approach and a highly divergent second approach, which can deliver all eight stereoisomers from a common precursor, were developed. GC-MS/MS analysis of the products and their comparison with excretion studies of DHCMT have been performed in the laboratories of our collaborators and the identity of 3h was proved (4α-chloro-17β-hydroxymethyl-17α-methyl-18-nor-5α-androst-13-en-3α-ol) with the in vivo metabolite of DHCMT. Details of these studies have been submitted for publication elsewhere.14Experimental section
General
Dehydroepiandrosterone was purchased from FluoroChem. Dry toluene, dichloromethane, dimethylformamide and m-CPBA are from Acros Organics. HPLC grade solvents (acetonitrile, methanol, isopropanol) were from VWR. All other non-specified chemicals were from Sigma-Aldrich. Anhydrous tetrahydrofuran was pre-dried using an Innovative Technologies PureSolv system, degassed and stored under 3 Å molecular sieves. NMR spectra were recorded on a Bruker AC400 and AC600 using TMS as an internal standard. IR spectra were recorded on a PerkinElmer Spectrum 65 using thin films (ATR FT-IR). TLC-analysis was performed with pre-coated aluminium-backed plates (Silica gel 60 F254, Merck). Compounds were visualized by submerging in an acidic phosphomolybdic acid/cerium sulphate solution and heating. Melting points were determined with a Kofler hot-stage apparatus. HR-MS analysis was carried out from acetonitrile solutions (concentration: 10 ppm) by using an HTC PAL system autosampler, an Agilent 1100/1200 HPLC with binary pumps, a degasser and a column thermostat and an Agilent 6230 AJS ESI-TOF mass spectrometer.For single crystal X-ray diffraction analysis, crystals were embedded in perfluorinated polyether and mounted on MITGEN™ loops. X-ray diffraction data were obtained in a cold stream of nitrogen at T = 100 K on a Bruker APEX-II diffractometer with Mo-Kα radiation. The collection strategy for the measurement was optimized with APEX-2 using ω- and φ-scans. After the integration of the data with SAINT, a semi-empirical absorption correction was performed with SADABS. The crystal structures were solved by direct methods and refined using the SHELXTL program package. All H atoms were placed geometrically and refined in a riding model approximation. The H atoms of hydroxyl groups were taken from difference maps and refined freely. Crystallographic data for the structure(s) reported in this paper have been deposited with the Cambridge Crystallographic Data Centre as supplementary publication no. CCDC 1586619–1586622 and 1825768.†
General procedure A: epoxidation with m-CPBA
The olefin is dissolved in CH2Cl2 (0.05–0.1 M), and KHCO3 (3 eq.) and m-CPBA (1.25 eq.) are added in this order. The reaction mixture is stirred at room temperature for 1–3 h and quenched by the addition of water and saturated Na2S2O3 solution. The biphasic mixture is transferred to a separatory funnel and is shaken vigorously. The aqueous phase is extracted 3 times with CH2Cl2 and the pooled organic phases are washed with brine and dried over MgSO4. The crude product after evaporation is purified by column chromatography (60:1 silica:product) using LP/EtOAc mixtures as the eluent system. The 17α-epoxy compound is obtained as the major and the desired 17β-epoxy is obtained as the minor product in all instances.
General procedure B: epoxide formation with base
The chlorohydrin starting material is dissolved in methanol (0.1 M) and solid KOH (5 eq.) is added. The base is dissolved by stirring and after a clear solution has been formed the mixture is heated to reflux for 10 hours. After cooling of the reaction mixture half the volume of saturated NH4Cl solution and half the volume water are added, and the solution is transferred to a separatory funnel. This is extracted twice with CH2Cl2 and the organic phases are combined, washed with brine and dried over MgSO4. The crude product can be purified by recrystallization from n-heptane.General procedure C: TBS protection
The chlorohydrin starting material is dissolved in dry DMF (0.3 M) at room temperature and imidazole (2.5 eq.) and TBSCl (1.3 eq.) are added. The reaction mixture is stirred at room temperature for 5–96 h and is then evaporated to dryness in vacuo. The solid residue is taken up in EtOAc and 0.2 M HCl. The phases are shaken in a separatory funnel and the aqueous phase is extracted twice with small portions of EtOAc. The pooled organic phases are washed with saturated NaHCO3 solution and brine and dried over MgSO4. After evaporating the solvent the crude product is purified via flash chromatography (10:1 silica:product).
General procedure D: Nysted methylenation
A Schlenk flask under an argon atmosphere was charged with Nysted reagent suspension (20 wt%, 5 eq.), which is diluted with half the volume of dry THF. The milky suspension is chilled to 0 °C in an ice-bath and TiCl4 (neat, 2.2 eq.) is added dropwise. The suspension is stirred for 10 minutes before removing the ice-bath and allowed to warm to room temperature. After 5 minutes the starting material dissolved in dry THF is added dropwise to the darkening suspension. After the addition is complete the flask is sealed and stirred for 14–20 hours (overnight). The reaction mixture is then poured onto a 1/1 mixture ice/2 M HCl and diluted by adding the same volume of Et2O. This is transferred to a separatory funnel and the mixture is shaken vigorously. After the separation of the phases the aqueous phase is extracted three times with Et2O and the combined organic phases are washed with saturated NaHCO3 solution, followed by brine and drying over MgSO4. After evaporation the crude product is purified via flash column chromatography on silica (15:1) using 10/1 → 3/1 LP/CH2Cl2 as the eluent.
General procedure E: Wagner–Meerwein rearrangement
A Schlenk flask under an argon atmosphere was charged with dry CH2Cl2 or toluene (0.05 M) and the solvent is cooled to −78 °C. Then 2,6-lutidine (2.5 eq.) and TMSOTf (2 eq.) are added at that temperature. After five minutes, the starting material is dissolved in CH2Cl2 or toluene and added dropwise to the chilled reaction mixture. The reaction mixture is stirred at −78 °C for 1 hour and then quenched by the addition of the same volume of methanol followed by 2 M HCl (10 eq.). The reaction vessel is taken out of the cooling bath and stirred at room temperature for 30 minutes. The mixture is then transferred to a separatory funnel and diluted with water and CH2Cl2. After the separation of the phases the aqueous phase is extracted twice with CH2Cl2 and the pooled organic phases are washed with saturated NaHCO3 solution and dried over MgSO4. The crude product obtained after evaporation is purified by column chromatography on silica (70:1) using between 10/1 and 5/1 LP/EtOAc as the eluent.
Cleavage of the TBS protecting group was observed under these conditions in some cases.
General procedure F: TBS deprotection
The starting material was fully dissolved in the smallest possible amount of methanol (ca. 0.1 M) and 2 M HCl (5 eq.) was added. The resulting solution was stirred at room temperature for 20–96 h. After the reaction is complete saturated NaHCO3 solution is added and the methanol is evaporated in vacuo. The reaction mixture is then extracted with CH2Cl2, and the extracts are combined and dried over MgSO4 and filtered over a small pad of silica (ca. 1 g). Evaporation of the solvent affords the product.4β-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5β-androst-13-en-3α-ol (3a)
Epoxide 11 (30 mg, 0.088 mmol, 1 eq.) was dissolved in a mixture of 2/1 THF/water (4 ml) and 0.1 ml concentrated H3PO4 and stirred at room temperature until TLC showed full conversion. The solution was then diluted with water, and extracted three times with small portions of CH2Cl2. These extracts were washed with brine and dried over MgSO4. The crude product (45 mg) was purified over a silica gel column with 8/1 LP/EtOAc as the eluent to give 13 mg (43%) of diol 3a as foam.1H-NMR (400 MHz, CDCl3): δH = 4.16 (2H, dd, J = 9.6 Hz, 11.8 Hz), 3.59 (1H, td, J = 9.71 Hz, 16.31 Hz), 3.35 (2H, dd, J = 63 Hz, 10.6 Hz), 2.06–2.33 (3H, m), 1.81–2.06 (5H, m), 1.65–1.81 (3H, m), 1.43–1.58 (4H, m), 1.04–1.27 (5H, m), 0.95 (3H, s), 0.93 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 141.11, 136.63, 76.55, 69.74, 69.09, 51.72, 49.51, 39.59, 38.33, 37.01, 34.17, 34.16, 30.62, 27.72, 25.16, 23.40, 23.26, 22.61, 22.48, 21.91. IR [cm−1]: 3336, 2929, 1450, 1071, 736. [α]20D = 14.0 (c 0.9, CH2Cl2). HRMS (M − OH): calcd for C20H30ClO: 321.1980, found: 321.1972 (Δ = 2.5 ppm).
4β-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5β-androst-13-en-3β-ol (3b)
Epoxide 9 (32 mg, 0.094 mmol, 1 eq.) was dissolved in a mixture of 2/1 THF/water (3 ml) and 0.1 ml concentrated H3PO4 and stirred at room temperature until TLC showed full conversion. The solution was then diluted with water, and extracted three times with small portions of CH2Cl2. These extracts were washed with brine and dried over MgSO4. The crude product was purified over a silica gel column with 8/1 LP/EtOAc as the eluent to give 9 mg (28%) of diol 3b as a colorless oil.1H-NMR (400 MHz, CDCl3): δH = 4.46 (1H, dd, J = 2.75 Hz, 11.88 Hz), 4.04 (1H, m), 3.37 (1H, dd, J = 63.27 Hz, 10.47 Hz), 1.90–2.02 (3H, m), 1.83–1.90 (3H, m), 1.71–1.79 (4H, m), 1.39–1.66 (5H, m), 1.08–1.24 (5H, m), 1.01 (1H, s), 0.95 (3H, s), 0.92 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 141.11, 136.63, 76.55, 69.74, 69.09, 51.72, 49.51, 39.59, 38.33, 37.01, 34.17, 34.16, 30.62, 27.72, 25.16, 23.40, 23.26, 22.61, 22.48, 21.91. IR [cm−1]: 3419, 2927, 1051, 907, 728. [α]20D = 22.84 (c 0.8, CH2Cl2). HRMS (M − OH): calcd for C20H30ClO: 321.1980, found: 321.1976 (Δ = 1.2 ppm).
4β-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5α-androst-13-en-3α-ol (3c)
The reaction was carried out following general procedure F. 30 mg (90%) of alcohol 3c was obtained as a white solid (barely soluble in CH2Cl2 or other conventional solvents for steroids).1H-NMR (600 MHz, CD3OD): δH = 3.98 (1H, dd, J = 2.61 Hz, 5.19 Hz), 3.90 (1H, m), 3.38 (1H, d, J = 10.91 Hz), 3.30 (1H, d, J = 10.91 Hz), 2.28 (1H, m), 2.06–2.17 (3H, m), 1.89–2.06 (4H, m), 1.76–1.88 (3H, m), 1.52–1.63 (2H, m), 1.39–1.50 (2H, m), 1.35 (1H, m), 1.21 (1H, qd, J = 11.88 Hz, 4.75 Hz), 1.08 (1H, m), 1.03 (3H, s), 0.94 (3H, s), 0.89–0.95 (1H, m). 13C-NMR (150 MHz, CD3OD): δC = 140.21, 138.95, 71.90, 68.96, 66.85, 54.73, 52.24, 44.50, 37.72, 37.49, 35.00, 32.59, 32.28, 30.90, 28.20, 24.24, 23.73, 22.68, 22.17, 14.39. IR [cm−1]: 3280, 2923, 1021. M.p.: 188–190 °C. [α]20D = −22.60 (c 0.2, CH2Cl2). HRMS (M + H): calcd for C20H32ClO2: 339.2086, found: 339.2054 (Δ = 9.4 ppm).
4β-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5α-androst-13-en-3β-ol (3d)
To a solution of aldehyde 46 (23 mg, 0.069 mmol, 1 eq.) in 4 ml dry methanol NaBH4 (10 mg, 0.274 mmol, 4 eq.) was added and the reaction mixture was stirred at room temperature. After 90 minutes the reaction mixture was quenched by the addition of saturated NH4Cl solution and was extracted with EtOAc (4×) and the extracts were washed with NaHCO3 solution once and dried over MgSO4. The crude product after evaporation was purified on silica gel (2.9 g) using 5/1 LP/EtOAc as the eluent giving 22 mg (95%) of alcohol 3d as a white solid.1H-NMR (400 MHz, CDCl3): δH = 4.29 (1H, dd, J = 2.78 Hz), 3.70 (1H, m), 3.46 (1H, d, J = 10.49 Hz), 3.30 (1H, d, J = 10.49 Hz), 2.29 (1H, m), 2.14 (2H, m), 1.92–2.05 (3H, m), 1.70–1.91 (5H, m), 1.40–1.61 (5H, m), 1.21 (2H, m), 1.08 (2H, m), 1.01 (3H, s), 0.94 (3H, s), 0.87 (1H, m). 13C-NMR (100 MHz, CDCl3): δC = 140.88, 136.70, 72.00, 70.77, 69.10, 53.38, 51.71, 49.28, 36.92, 36.52, 36.35, 31.32, 30.66, 27.36, 26.73, 22.53, 22.15, 21.93, 14.78. IR [cm−1]: 3288, 2925, 1443, 1043. [α]20D = −16.62 (c 0.85, CH2Cl2). M.p.: 178 °C. HRMS (M + H): calcd for C20H32ClO2: 339.2086, found: 339.2072 (Δ = 4.1 ppm).
4α-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5β-androst-13-en-3β-ol (3e)
The reaction was carried out following general procedure F. 38 mg (98%) of alcohol 3e was obtained as white foam.1H-NMR (400 MHz, CDCl3): δH = 4.25 (1H, dd, J = 10.66 Hz, 5.53 Hz), 3.58 (1H, td, J = 10.53 Hz, 4.74 Hz), 3.46 (1H, d, J = 10.30 Hz,), 3.30 (1H, d, J = 10.30 Hz,), 2.47 (1H, br s), 2.26 (1H, m), 1.77–2.13 (9H, m), 1.62–1.76 (2H, m), 1.41–1.61 (3H, m), 1.08–1.35 (5H, m), 0.93 (6H, s). 13C-NMR (100 MHz, CDCl3): δC = 141.08, 136.44, 71.35, 71.15, 68.84, 51.46, 48.17, 46.24, 37.75, 34.19, 32.92, 31.16, 30.59, 29.13, 28.30, 24.12, 22.32, 21.91, 19.35, 18.23. IR [cm−1]: 3376, 2924, 1036, 731. [α]20D = −102.77 (c 0.76, CH2Cl2). HRMS (M + H): calcd for C20H32ClO2: 339.2086, found: 339.2037 (Δ = 14.4 ppm).
4α-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5β-androst-13-en-3α-ol (3f)
The crude aldehyde 45 (0.062 mmol, 1 eq.) was dissolved in 5 ml dry methanol and NaBH4 (12 mg, 0.32 mmol, 5 eq.) was added as a solid in one portion and the reaction mixture was stirred for 1 hour. After quenching with NH4Cl solution the reaction mixture was extracted with CH2Cl2 (3×), and the extracts were washed with water and dried over MgSO4. The crude product after evaporation was purified on silica gel (2.5 g) using 5/1 LP/EtOAc as the eluent to give 16 mg (76%) of 3f as a colorless oil.1H-NMR (600 MHz, CDCl3): δH = 4.46 (1H, m), 3.79 (1H, m), 3.46 (1H, d, J = 10.66 Hz), 3.29 (1H, d, J = 10.66 Hz), 2.29 (1H, m), 2.02–2.21 (3H, m), 1.98 (2H, m), 1.68–1.93 (8H, m), 1.51–1.67 (4H, m), 1.09–1.25 (2H, m), 0.95 (3H, s), 0.92 (3H, s), 0.88–1.01 (1H, m). 13C-NMR (150 MHz, C6D6): δC = 140.94, 137.01, 71.79, 70.71 (br s), 68.95, 51.7, 45.13 (2C), 36.67 (br s), 34.6, 33.81 (br s), 30.71, 30.22 (br s), 27.92 (br s), 26.89 (br s), 23.85 (br s), 23.70, 22.87, 21.96, 21.60 (br s). IR [cm−1]: 3397, 2930, 1038, 909. [α]20D = −33.97 (c 0.8, CH2Cl2). HRMS (M − OH): calcd for C20H30ClO: 321.1980, found: 321.1955 (Δ = 7.8 ppm).
4α-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5α-androst-13-en-3β-ol (3g)
The reaction was carried out following general procedure E. 27 mg (55%) of alcohol 3g was obtained as colourless foam.1H-NMR (400 MHz, CDCl3): δH = 3.77 (1H, m), 3.55 (1H, m), 3.46 (1H, d, J = 10.40 Hz), 3.30 (1H, d, J = 10.40 Hz), 2.29 (1H, m), 2.05–2.19 (3H, m), 1.79–2.05 (7H, m), 1.49–1.68 (3H m), 1.07–1.38 (6H, m), 0.89–1.02 (1H, m), 0.94 (3H, s), 0.83 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 140.81, 136.62, 76.57, 72.05, 69.08, 51.90, 51.67, 51.49, 38.88, 36.22, 35.57, 34.11, 30.79, 30.59, 28.72, 25.15, 22.63 (2C), 21.90, 12.79. IR [cm−1]: 3396, 2933, 1037, 750. [α]20D = −59.20 (c 1.2, CH2Cl2). HRMS (M + H): calcd for C20H32ClO2: 339.2086, found: 339.2008 (Δ = 22.7 ppm).
4α-Chloro-17β-hydroxymethyl-17α-methyl-18-nor-5α-androst-13-en-3α-ol (3h)
The reaction was carried out following general procedure F. 24 mg (90%) of alcohol 3h was obtained as white foam.1H-NMR (600 MHz, CDCl3): δH = 4.02 (1H, dd, J = 12.26 Hz, 2.58 Hz), 3.96 (1H, m), 3.39 (1H, d, J = 10.54 Hz), 3.23 (1H, d, J = 10.54 Hz), 2.19–2.34 (2H, m), 2.06 (2H, m), 1.67–1.95 (9H, m), 1.38–1.52 (3H, m), 1.04–1.21 (3H, m), 0.92–1.01 (2H, m), 0.88 (3H, s), 0.76 (3H, s). 13C-NMR (150 MHz, CDCl3): δC = 141.05, 136.46, 70.07, 69.79, 69.08, 51.77, 51.67, 45.93, 39.37, 36.40, 34.06, 31.13, 30.79, 30.56, 27.56, 25.00, 22.62, 22.39, 21.90, 12.03. IR [cm−1]: 3403, 2925, 1049, 1031. [α]20D = −66.70 (c 1.17, CH2Cl2). HRMS (M + H): calcd for C20H32ClO2: 339.2086, found: 339.2065 (Δ = 6.2 ppm).
17-Methylene-5β,13α-androstan-3-one (5)
To (S)-(2-pyrrolidinylmethyl)pyrrolidine (73.21 mg, 474 μmol, 0.25 eq.) dissolved in 20 mL acetonitrile was added (1S)-(+)-10-camphorsulfonic acid (110.25 mg, 474 μmol, 0.25 eq.) and the mixture was then stirred at room temperature for 5 min. Ketone 4 (540 mg, 1.9 mmol, 1 eq.) was added to the solution and it was stirred for 15 minutes before adding the Hantzsch ester (diethyl 1,4-dihydro-2,6-dimethyl-3,5-pyridinedicarboxylate) (961.75 mg, 3.8 mmol, 2 eq.). The solution was heated to 70 °C for 24 h and then evaporated. The crude product was purified via column chromatography on 22 g silica gel with 12/1 LP/EtOAc as the eluent. 440 mg of ketone 5 was obtained as a white solid (81%), dr = 10:1 (determined via1H-NMR).
1H-NMR (400 MHz, CDCl3): δH = 4.81 (1H, dd), 4.68 (1H, dd), 2.66 (1H, m), 2.47 (1H, dddd), 2.29–2.41 (2H, m), 2.14 (1H, dddd, J = 14.63 Hz, 4.28 Hz, 3.16 Hz, 2.31 Hz), 1.89–2.09 (3H, m), 1.87–1.69 (4H, m), 1.56 (1H, ddd, J = 13.24 Hz, 8.95 Hz, 2.02 Hz), 1.29–1.44 (4H, m), 1.28–1.20 (2H, m), 0.97–1.11 (2H, m), 0.94 (4H, s), 0.86 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 213.55, 157.11, 103.13, 54.04, 46.18, 44.10, 42.23, 39.51, 37.44, 37.06, 36.78, 35.25, 34.69, 31.48, 29.43, 26.86, 25.52, 24.54, 22.83, 21.4. IR [cm−1]: 2946, 1720, 1652, 1472, 1266, 875. [α]20D = −41.33 (c 1.5, CH2Cl2). HRMS (M + H): calcd for C20H31O: 287.2370, found: 287.2344 (Δ = 9 ppm).
4β-Chloro-17-methylene-5β,13α-androstan-3-one (6)
To ketone 5 (800 mg, 2.79 mmol, 1 eq.) dissolved in 10 mL dry THF and cooled to −78 °C, 1 M LiHMDS (584.16 mg, 3.49 mmol, 1.25 eq.) was added dropwise and the resulting mixture was stirred for 1 hour. Then, the solution was taken out of the cooling bath and stirred at 0 °C for 30 min. The mixture was then again chilled to −70 °C and solid N-chlorosuccinimide (466.16 mg, 3.49 mmol, 1.25 eq.) was added. After 2 hours, the reaction was quenched with saturated sodium bicarbonate solution and was extracted three times with 40 mL Et2O. The pooled organic phases were washed with brine, dried over Mg2SO4 and evaporated to dryness. The crude product was purified over 45 g silica gel with 1/1 LP/CH2Cl2 as the eluent to give 548 mg of chloroketone 6 (61%) as colorless crystals.1H-NMR (400 MHz, CDCl3): δH = 4.84 (1H, dd, J = 2.10 Hz), 4.80 (1H, d, J = 6.07 Hz), 4.70 (1H, dd), 2.32–2.62 (4H, m), 2.10–2.19 (1H, m), 1.94–2.09 (2H, m), 1.69–1.93 (4H, m), 1.51–1.68 (3H, m), 1.37–1.49 (3H, m), 1.28–1.37 (1H, m), 1.06–1.17 (1H, m), 0.99–1.06 (1H, m), 0.97 (3H, s), 0.93 (2H, s), 0.91 (1H, s). 13C-NMR (100 MHz, CDCl3): δC = 203.42, 156.80, 103.25, 65.16, 53.93, 53.77, 46.11, 40.68, 38.23, 37.09, 36.84, 36.66, 34.57, 31.31, 29.28, 25.76, 24.34, 23.96, 23.28, 21.50. IR [cm−1]: 3066, 2950, 1735, 1648, 1471, 1359, 872, 861. [α]20D = −8.48 (c 1, CH2Cl2). M.p.: 133–135 °C. HRMS (M − Cl): calcd for C20H29O: 285.2213 found: 285.2201 (Δ = 4.2 ppm).
Crystal data (CCDC 1586619†). C20H29ClO, M = 320.88, monoclinic, a = 8.5588(6). b = 6.9137(5), c = 15.0132(11) Å, β = 99.591(2)°, Z = 4, T = 100 K, space group P21 (no. 4), 27999 reflections measured, 6622 unique (Rint = 0.036), which were used in all calculations. The Flack parameter using 2651 quotients is −0.021(17), the final wR(F2) is 0.0829.
4β-Chloro-17-methylene-5β,13α-androstan-3-ol (7, 8)
Chloroketone 6 (339 mg, 1.06 mmol, 1 eq.) was dissolved in a 50 mL round bottom flask in a 1/1 mixture of THF/MeOH (10 mL) and stirred at 0 °C. To this solution was added NaBH4 (46 mg, 1.22 mmol, 1.15 eq.) as a solid in small portions over the course of 5 minutes. The reaction was complete in 1 hour after complete addition and was quenched by adding solid NH4Cl and concentrating the mixture in vacuo. The residue was taken up in Et2O and water and extracted 3 times with Et2O. The pooled organic phases were washed with saturated NaHCO3 solution and dried over MgSO4. The crude product after evaporating was purified on 20 g silica gel using 10/1 LP/EtOAc giving 102 mg (29%) of alcohol 7 and 131 mg (38%) of alcohol 8.(5β,13α)-4β-Chlorospiro[androstane-17ξ,2′-oxiran]-3β-ol (9, 10)
The reaction was carried out following general procedure A. 32 mg 17β-epoxy product 9 (28%) was obtained as a grey foam and 57 mg 17α-epoxy product 10 (49%) was obtained as a white solid.(5β,13α)-4β-Chlorospiro[androstane-17ξ,2′-oxiran]-3α-ol (11, 12)
The reaction was carried out following general procedure A. 30 mg 17β-epoxy product 11 (29%) was obtained as a white solid and 46 mg 17α-epoxy product 12 (44%) was obtained as off-white crystals.13α-Androst-4-en-3,17-dione (14)
A 500 mL round bottom flask was charged with 1.5 g 3β-hydroxy-13α-androst-5-en-17-one7 (13) (5.2 mmol, 1 eq.), 250 ml dry toluene and 10.75 ml cyclohexanone (104 mmol, 20 eq.). The solution was heated to reflux and after 15 minutes 2.65 g aluminium isopropoxide (13 mmol, 2.5 eq.) were added, upon which the solution turned yellow. After refluxing for 1 hour, the reaction was complete and was allowed to cool to room temperature. The mixture was then washed with 50 ml 0.1 M H2SO4 twice and once with saturated NaHCO3 solution and brine. After drying over MgSO4 and evaporating the volatiles the product was left to crystallize at 4 °C overnight. The crystals (390 mg) were filtered, and washed with small amounts of LP. The mother liquor was concentrated and purified on 55 g silica gel using 2/1 LP/EtOAc as the eluent. In total 1.36 g of diketone 14 (91%) were obtained as colorless needles.Analytical data were in accordance with the literature.15
13α,5ξ-Androst-3-en-17-one (15, 16)
A 250 ml round bottom flask was charged with 50 ml acetic acid and 2.85 g diketone 14 and heated to reflux. Zinc (20.15 g, 0.3 mol, 31 eq.) was added in portions over 25 minutes. The reaction was complete after the addition and was allowed to cool. The solids were filtered and washed thoroughly with ethyl acetate (100 ml). The filtrate was evaporated to dryness and taken up in CH2Cl2 and washed with water (25 ml). The aqueous phase was extracted once with 25 ml CH2Cl2 and the combined organic phases were dried over MgSO4 and evaporated to dryness. The crude mixture of ketones 15 and 16 (1.3:1 dr, 2.61 g, 96%, determined via1H-NMR) was used directly in the next step.
3ξ,4ξ-Epoxy-13α,5ξ-androst-17-one (17, 18)
The mixture of 15 and 16 (2.61 g, 9.6 mmol) from the previous step was dissolved in CH2Cl2 (100 ml) in a 250 ml round bottom flask. To this solution 2.02 g KHCO3 (19.2 mmol, 2 eq.) as well as 2.6 g meta-chloroperbenzoic acid (10.5 mmol, 1.1 eq.) were added over 5 minutes and the resulting suspension was stirred at room temperature for 1.5 hours. The reaction was flooded with NaHCO3 and Na2S2O3 solutions (25 ml each) and extracted with CH2Cl2. The combined organic phases were washed once with small portions of water and brine (10 ml) and dried over MgSO4. After evaporating and drying under high vacuum 2.83 g of crude epoxides 17 and 18 (102% yield, approx. 95% purity) were obtained. These were used in the next step directly.Analytical samples were obtained by the treatment of isolated chlorohydrins 20 and 21 according to general procedure B.
3ξ-Hydroxy-4ξ-chloro-5ξ,13α-androstan-17-one (19, 20, 21)
The isomeric mixture of epoxides 17 and 18 (2.83 g, 9.8 mmol, 1 eq.) was charged in a 250 ml round bottom flask and dissolved in 50 ml chloroform. 25 ml concentrated HCl (32%, 29 g, 254 mmol, 26 eq.) were added over the course of 5 minutes and the resulting biphasic mixture was stirred vigorously. After 30 minutes, the solution was transferred to a separatory funnel and the phases were separated. The aqueous phase was extracted with 25 ml CH2Cl2 and the pooled organic phases were washed with saturated NaHCO3 solution and brine. The extracts were dried over MgSO4 and evaporated. The residue was purified on silica gel (100:1) using 40/40/1 LP/CH2Cl2/CH3CN and subsequently recrystallized from n-heptane or diisopropylether to give 445 mg 5β-isomer 19 (14%), 600 mg chlorohydrin 20 (19%), and 1.33 g of 5α-isomer 21 (42%).
3β-Hydroxy-4α-chloro-5β,13α-androstan-17-one (19)
1H-NMR (400 MHz, CDCl3): δH = 4.12 (dd, 1H, J = 7.80, J = 5.12), 3.68 (m, 1H), 2.25–2.36 (m, 1H), 2.14–2.19 (m, 1H), 1.85–2.14 (m, 6H), 1.69–1.78 (m, 2H), 1.49–1.59 (m, 3H), 1.26–1.48 (m, 5H), 1.09–1.24 (m, 2H), 0.93 (s, 3H), 0.78 (s, 3H), 0.62–0.74 (m, 1H). 13C-NMR (100 MHz, CDCl3): δC = 222.50, 71.82, 69.85, 52.65, 50.34, 44.79 (2C), 37.41, 34.62, 34.15, 32.01, 31.81, 30.75, 27.09, 25.19, 24.23, 21.87, 21.61, 20.91. IR [cm−1]: 3482, 1721, 1178, 832. [α]20D = −114.34 (c 1.03, CH2Cl2). M.p.: 136–139 °C. HRMS (M + H): calcd for C19H30ClO2: 325.1929, found: 325.1925 (Δ = 1.2 ppm).Crystal data (CCDC 1586620†). C19H29ClO2, M = 324.87, monoclinic, a = 7.1254(5). b = 11.6665(8), c = 10.8645(8) Å, β = 103.497(2)°, Z = 2, T = 100 K, space group P21 (no. 4), 22481 reflections measured, 6706 unique (Rint = 0.040), which were used in all calculations. The Flack parameter using 2340 quotients is 0.00(3), and the final wR(F2) is 0.1013.
3α-Chloro-4β-hydroxy-5β,13α-androstan-17-one (20)
1H-NMR (400 MHz, CDCl3): δH = 3.73–3.87 (m, 2H), 2.40 (d, 1H, J = 2.34 Hz), 2.30–2.38 (m, 1H), 2.09–2.21 (m, 2H), 1.93–2.06 (m, 3H), 1.77–1.88 (m, 4H), 1.55–1.64 (m, 2H), 1.41–1.47 (m, 1H), 1.27–1.32 (m, 1H), 1.0–1.36 (m, 5H), 0.97 (s, 3H), 0.80 (s, 3H), 0.57–0.70 (m, 1H). 13C-NMR (100 MHz, CDCl3): δC = 222.23, 72.14, 69.34, 50.87, 50.39, 49.30, 40.23, 38.20, 37.16, 35.91, 33.90, 32.38, 30.23, 26.86, 25.41, 23.53, 22.86, 21.48, 21.42. IR [cm−1]: 3446, 1727, 1056, 878. [α]20D = −77.18 (c 1.41, CH2Cl2). M.p.: 148–151 °C. HRMS (M + H): calcd for C19H30ClO2: 325.1929, found: 325.1928 (Δ = 0.3 ppm).3α-Hydroxy-4β-chloro-5α,13α-androstan-17-one (21)
1H-NMR (400 MHz, CDCl3): δH = 4.09 (s, 1H), 3.91 (m, 1H), 2.28–2.38 (m, 1H), 1.97–2.21 (m, 5H), 1.78–1.87 (m, 2H), 1.63–1.77 (m, 2H), 1.40–1.61 (m, 5H), 1.23–1.35 (m, 2H), 0.98–1.17 (m, 2H), 0.94 (s, 3H), 0.88 (s, 3H), 0.64–0.73 (m, 2H). 13C-NMR (100 MHz, CDCl3): δC = 222.59, 71.33, 65.29, 53.21, 51.03, 50.29, 43.00, 37.76, 36.72, 34.12, 33.27, 32.18, 31.17, 26.61, 25.48, 23.81, 21.82, 21.63, 13.86. IR [cm−1]: 3353, 1719, 1251, 1133. [α]20D = −91.31 (c 1.02, CH2Cl2). M.p.: 191–193 °C. HRMS (M + H): calcd for C19H30ClO2: 325.1929, found: 325.1923 (Δ = 1.8 ppm).4β-Chloro-5α,13α-androstane-3,17-dione (22)
A 100 ml round bottom flask was charged with 1.8 g ketone 21 (5.54 mmol, 1 eq.) and 60 ml CH2Cl2. To this solution 3.17 g Dess–Martin periodinane (7.48 mmol, 1.35 eq.) was added in small portions over 15 minutes. After 1 hour, the reaction was quenched by adding aqueous Na2S2O3 solution and saturated NaHCO3. The biphasic system was separated and the aqueous phase was extracted three times with CH2Cl2. The pooled organic phases were washed with water and dried over MgSO4. The crude product after evaporation (1.9 g, 90% purity) was used in the next step without purification. An analytical sample of diketone 22 was obtained by recrystallizing from diisopropylether.1H-NMR (400 MHz, CDCl3): δH = 3.88 (dd, 1H, J = 3.48 Hz, J = 1.56 Hz), 2.81–2.93 (m, 1H), 2.19–2.23 (m, 1H), 1.92–2.19 (m, 7H), 1.63–1.78 (m, 3H), 1.17–1.45 (m, 5H), 1.02–1.03 (m, 1H), 0.98 (s, 3H), 0.84 (s, 3H), 0.54–0.73 (m, 2H). 13C-NMR (100 MHz, CDCl3): δC = 221.69, 204.79, 63.50, 52.45, 50.39, 49.97, 38.52, 37.34, 36.15, 33.71, 33.25, 32.66, 31.69, 25.61, 25.10, 22.08, 21.39, 13.41. IR [cm−1]: 2979, 1716, 1142, 744. [α]20D = −147.84 (c 1.06, CH2Cl2). M.p.: 165–168 °C. HRMS (M + H): calcd for C19H28ClO2: 323.1773, found: 323.17714 (Δ = 0.5 ppm).
4α-Chloro-5α,13α-androstane-3,17-dione (23)
140 ml methanol and 1.4 g of crude product 22 (4.34 mmol, 1 eq.) were charged in a 250 ml round bottom flask. At room temperature, 2.43 ml of a 10% solution of KOH was added in methanol (4.34 mmol, 1 eq.) dropwise. The solution turned yellow when the addition was complete and was stirred for 5 minutes, followed by 1 ml of acetic acid to neutralize. The reaction mixture was concentrated in vacuo and the resulting white precipitate was dissolved in water/CH2Cl2 and after shaking, the aqueous phase was again extracted twice with CH2Cl2. The pooled organic phases were washed with NaHCO3 solution and brine and dried over MgSO4. The crude product obtained after evaporating the solvent was purified on silica (60 g) using 10/1 to 4/1 LP/EtOAc as the eluent. 1.31 g (94%) of diketone 23 was obtained as a fine microcrystalline solid.1H-NMR (400 MHz, CDCl3): δH = 4.38 (d, 1H, J = 12.67 Hz), 2.44–2.60 (m, 2H), 2.32–2.41 (m, 1H), 2.02–2.23 (m, 6H), 1.79–1.89 (m, 1H), 1.50–1.62 (m, 4H), 1.34–1.43 (m, 1H), 1.22–1.33 (m, 1H), 1.13–1.21 (m, 1H), 0.97 (s, 3H), 0.93 (s, 3H), 0.74–0.84 (m, 3H). 13C-NMR (100 MHz, CDCl3): δC = 221.88, 202.19, 67.88, 54.76, 51.33, 50.40, 50.09, 38.64, 38.29, 37.36, 37.33, 33.74, 32.34, 31.89, 26.02, 25.14, 22.89, 21.32, 12.55. IR [cm−1]: 2955, 1717, 1255, 752. [α]20D = −109.54 (c 0.68, CH2Cl2). M.p.: 217–220 °C. HRMS (M + H): calcd for C19H28ClO2: 323.1773, found: 323.17721 (Δ = 0.3 ppm).
Crystal data (CCDC 1586621†). C19H27ClO2, M = 322.85, orthorhombic, a = 7.4333(17), b = 13.538(3), c = 16.411(4) Å, Z = 4, T = 100 K, space group P212121 (no. 19), 33711 reflections measured, 7201 unique (Rint = 0.037), which were used in all calculations. The Flack parameter using 2852 quotients is −0.038(14), and the final wR(F2) is 0.0731.
4α-Chloro-3ξ-hydroxy-5α,13α-androstan-17-one (24, 25)
In a 100 ml round bottom flask 860 mg (2.66 mmol, 1 eq.) diketone 23 were dissolved in 32 ml dry toluene under an argon atmosphere. Half the volume of isopropanol (213 mmol, 80 eq.) was added and the solution was warmed to 60 °C. At this temperature, 272 mg Al(O-iPr)3 (1.33 mmol, 0.5 eq.) was added. The reaction mixture was stirred at 60–70 °C for 4 hours and was subsequently quenched by adding 50 ml 0.1 M H2SO4 and shaking vigorously in a separatory funnel. After separation of the phases the aqueous phase was extracted twice with toluene. The pooled organic phases were washed with saturated NaHCO3 solution, dried over MgSO4 and evaporated to dryness. The crude residue was purified on silica gel (60:1) using 12/1 LP/EtOAc as the eluent giving 365 mg (42%) 3β-alcohol 24 and 361 mg (42%) 3α-alcohol 25 as white crystalline solids.
Crystal data (CCDC 1586622†). C19H29ClO2, M = 324.87, orthorhombic, a = 7.5312(3), b = 20.9084(8), c = 21.2244(9) Å, Z = 8 (Z′ = 2), T = 100 K, space group P212121 (no. 19), 31190 reflections measured, 10688 unique (Rint = 0.022), which were used in all calculations. The Flack parameter using 4448 quotients is −0.021(9), and the final wR(F2) is 0.0760.
3α-(t-Butyldimethylsilyloxy)-4β-chloro-5α,13α-androstan-17-one (26)
The reaction was carried out following general procedure C. 976 mg (97%) of TBS protected ketone 26 was obtained as a white solid.1H-NMR (400 MHz, CDCl3): δH = 3.97 (dd, 1H, J = 5.4 Hz, J = 2.7 Hz), 3.76 (s, 1H), 2.27–2.37 (m, 1H), 1.95–2.20 (m, 5H), 1.62–1.74 (m, 2H), 1.38–1.55 (m, 5H), 1.21–1.38 (m, 3H), 1.08–1.21 (m, 1H), 0.96–1.06 (m, 1H), 0.94 (s, 3H), 0.87 (s, 9H), 0.85 (s, 3H), 0.61–0.68 (m, 2H), 0.03 (d, 6H). 13C-NMR (100 MHz, CDCl3): δC = 222.59, 71.66, 66.17, 53.20, 50.89, 50.22, 42.96, 37.71, 36.55, 34.07, 33.32, 32.09, 31.23, 26.59, 25.93 (3C), 25.39, 24.19, 21.73, 21.58, 18.17, 13.89, −4.69, −4.71. IR [cm−1]: 2930, 1739, 1046, 832. [α]20D = −76.44 (c 0.74, CH2Cl2). M.p.: 159–160 °C. HRMS (M + H): calcd for C25H44ClO2Si: 439.2794, found: 439.2774 (Δ = 4.5 ppm).
3β-(t-Butyldimethylsilyloxy)-4α-chloro-5β,13α-androstan-17-one (27)
The reaction was carried out following general procedure C. 662 mg (83%) ketone 27 as white crystals as well as 68 mg (11%) recovered starting material 19 were obtained.1H-NMR (400 MHz, CDCl3): δH = 3.93–3.97 (m, 1H), 3.87 (dd, 1H, J = 6.24 Hz, J = 4.28 Hz), 2.26–2.35 (m, 1H), 2.07–2.21 (m, 2H), 1.91–2.06 (m, 4H), 1.76–1.86 (m, 3H), 1.63–1.71 (m, 1H), 1.43–1.61 (m, 6H), 1.13–1.32 (m, 3H), 0.95 (s, 3H), 0.85 (s, 9H), 0.76 (s, 3H), 0.03 (d, 6H). 13C-NMR (100 MHz, CDCl3): δC = 222.65, 72.71, 67.67, 51.64, 50.40, 40.19, 36.69, 35.94, 33.92, 32.36, 28.89, 28.37, 25.85, 25.28, 24.91, 24.10, 23.86, 23.62, 21.26, 18.11, −4.75, −4.77. IR [cm−1]: 2931, 1733, 1109, 835. [α]20D = −190.84 (c 1.03, CH2Cl2). M.p.: 165–168 °C. HRMS (M + H): calcd for C25H44ClO2Si: 439.2794, found: 439.2788 (Δ = 1.4 ppm).
3α-(t-Butyldimethylsilyloxy)-4β-chloro-17-methylene-5α,13α-androstane (28)
The reaction was carried out following general procedure D. 360 mg (45%) of olefin 28 as well as 445 mg (55%) recovered starting material 26 were obtained.1H-NMR (400 MHz, CDCl3): δH = 4.80 (s, 1H), 4.66 (s, 1H), 3.97 (dd, 1H, J = 5.46 Hz, J = 2.73 Hz), 3.76 (s, 1H), 2.28–2.49 (m, 2H), 1.94–2.09 (m, 2H), 1.72–1.93 (m, 4H), 1.58–1.71 (m, 1H), 1.47–1.57 (m, 2H), 1.39–1.45 (m, 2H), 1.27–1.38 (m, 3H), 1.12–1.26 (m, 4H), 0.92 (s, 3H), 0.88 (s, 9H), 0.05 (d, 6H). 13C-NMR (100 MHz, CDCl3): δC = 157.56, 102.93, 71.78, 66.43, 54.25, 54.21, 46.02, 43.09, 36.92, 36.57, 34.49, 33.28, 31.59, 31.35, 29.63, 26.84, 25.96, 24.73, 24.30, 20.30, 18.18, 14.04, −4.75, −4.78. IR [cm−1]: 2931, 1257, 1052, 833. [α]20D = −28.77 (c 0.88, CH2Cl2). M.p.: 110–112 °C. HRMS (M + Na): calcd for NaC26H45ClOSi: 459.2820, found: 459.2840 (Δ = 4.4 ppm).
3β-(t-Butyldimethylsilyloxy)-4α-chloro-17-methylene-5β,13α-androstane (29)
The reaction was carried out following general procedure D. 141 mg (47%) of olefin 29 as well as 145 mg (48%) recovered starting material 27 were obtained.1H-NMR (400 MHz, CDCl3): δH = 4.79 (s, 1H), 4.65 (s, 1H), 3.94–3.98 (m, 1H), 3.82–3.87 (dd, 1H, J = 7.02 Hz, J = 4.68 Hz), 1.94–2.03 (m, 2H), 1.83–1.92 (m, 2H), 1.74–1.80 (m, 1H), 1.68–1.72 (m, 1H), 1.58–1.67 (m, 2H), 1.48–1.57 (m, 3H), 1.37–1.43 (m, 3H), 1.31–1.36 (m, 1H), 1.22–1.30 (m, 4H), 0.92 (s, 3H), 0.86 (s, 9H), 0.80 (s, 3H), 0.03 (d, 6H). 13C-NMR (100 MHz, CDCl3): δC = 157.86, 102.81, 72.92, 67.99, 55.09, 46.28, 41.44, 40.92, 36.24, 35.61, 34.77, 31.50, 29.80, 29.61, 29.38, 28.61, 25.97, 24.80, 24.54, 23.88, 22.27, 18.23, −4.70, −4.72. IR [cm−1]: 2929, 1249, 1107, 836. [α]20D = −25.43 (c 0.68, CH2Cl2). M.p.: 125–127 °C. HRMS (M + Na): calcd for NaC26H45ClOSi: 459.2820, found: 459.2918 (Δ = 0.4 ppm).
3β-(t-Butyldimethylsilyloxy)-4α-chloro-5α,13α-androstan-17-one (30)
The reaction was carried out following general procedure C. 698 mg (97%) ketone 30 were obtained as white crystals.1H-NMR (400 MHz, CDCl3): δH = 3.66 (1H, dd, J = 11.26 Hz, 9.12 Hz), 3.51 (1H, m), 2.35 (1H, m), 2.01–2.22 (5H, m), 1.69–1.89 (3H, m), 1.45–1.61 (3H, m), 1.00–1.28 (4H, m), 0.97 (3H, s), 0.89 (9H, s), 0.84–0.92 (2H, m), 0.68 (3H, s), 0.61–0.76 (2H, m), 0.11 (3H, s), 0.07 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 222.30, 77.38, 70.15, 51.89, 51.85, 50.77, 50.19, 38.59, 37.50, 35.91, 33.91, 32.73, 32.21, 31.44, 26.00, 25.32, 24.96, 22.74, 21.40, 18.29, 13.13, −4.27, −4.36. IR [cm−1]: 2929, 1730, 1091. [α]20D = −76.74 (c 0.75, CH2Cl2). M.p.: 185–186 °C. HRMS (M + H): calcd for C25H44ClO2Si: 439.2794, found 439.28126 (Δ = 4.2 ppm).
3α-(t-Butyldimethylsilyloxy)-4α-chloro-5α,13α-androstan-17-one (31)
The reaction was carried out following general procedure C. 529 mg (74%) ketone 31 as a white crystalline solid as well as 135 mg (25%) recovered starting material 25 were obtained.1H-NMR (400 MHz, CDCl3): δH = 3.96 (1H, dd, J = 2.51 Hz), 3.83 (1H, dd, J = 11.87 Hz, 2.63 Hz), 2.23–2.33 (1H, m), 1.86–2.19 (5H, m), 1.79 (1H, dd, J = 13.28 Hz, 9.05 Hz), 1.69 (1H, td, J = 12.07 Hz, 2.8 Hz), 1.57–1.64 (2H, m), 1.45–1.53 (2H, m), 1.33–1.41 (2H, m), 1.14 (1H, td, J = 13.15 Hz, 4.05 Hz), 0.96–1.07 (1H, m), 0.92 (3H, s), 0.79–0.95 (2H, m), 0.88 (9H, s), 0.55–0.74 (2H, m), 0.61 (3H, s), 0.05 (3H, s), 0.01 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 222.11, 71.07, 67.15, 51.73, 50.64, 49.96, 45.45, 39.08, 37.50, 33.78, 32.54, 32.05, 31.34, 29.70, 25.98, 25.17, 24.38, 22.38, 21.24, 18.27, 12.38, −4.58, −4.70. IR [cm−1]: 2950, 1729, 1082. [α]20D = −124.81 (c 0.7, CH2Cl2). M.p.: 147–148 °C. HRMS (M + H): calcd for C25H44ClO2Si: 439.2794, found 439.27985 (Δ = 1 ppm).
3β-(t-Butyldimethylsilyloxy)-4α-chloro-17-methylene-5α,13α-androstane (32)
The reaction was carried out following general procedure D. 178 mg (40%) olefin 32 as colorless crystals as well as 250 mg (56%) recovered starting material 30 were obtained.1H-NMR (400 MHz, CDCl3): δH = 4.82 (1H, s), 4.68 (1H, s), 3.67 (1H, dd, J = 11.34 Hz, 9.10 Hz), 3.52 (1H, m), 2.24–2.53 (2H, m), 2.11 (1H, ddd, J = 13.13 Hz, 3.23 Hz), 1.95–2.02 (1H, m), 1.91 (1H, dt, J = 13.64 Hz, 3.16 Hz), 1.70–1.87 (3H, m), 1.48–1.62 (2H, m), 0.97–1.41 (6H, m), 0.94 (3H, s), 0.90 (9H, s), 0.61–0.88 (3H, m), 0.71 (3H, s), 0.12 (3H, s), 0.08 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 157.32, 102.99, 77.45, 70.37, 54.04, 52.88, 52.01, 45.99, 38.63, 36.63, 36.02, 34.57, 32.64, 31.57, 31.41, 29.48, 26.03, 25.17, 24.49, 21.28, 18.29, 13.32, −4.27, −4.34. IR [cm−1]: 2857, 1095, 841. [α]20D = −58.36 (c 0.5, CH2Cl2). M.p.: 137–138 °C. HRMS (M + H): calcd for C26H46ClOSi: 437.3001, found 437.2923 (Δ = 17.8 ppm).
3α-(t-Butyldimethylsilyloxy)-4α-chloro-17-methylene-5α,13α-androstane (33)
The reaction was carried out following general procedure D. 143 mg (29%) olefin 33 as white crystals as well as 340 mg (68%) recovered starting material 31 were obtained.1H-NMR (400 MHz, CDCl3): δH = 4.81 (1H, s), 4.68 (1H, s), 4.01 (1H, dd, J = 2.73 Hz), 3.90 (1H, dd, J = 11.90 Hz, 2.73 Hz), 2.25–2.53 (2H, m), 1.87–2.02 (3H, m), 1.62–1.86 (4H, m), 1.58 (1H, m), 1.30–1.47 (2H, m), 1.19–1.28 (2H, m), 0.97–1.11 (2H, m), 0.95 (3H, s), 0.93 (9H, s), 0.71–0.90 (4H, m), 0.69 (3H, s), 0.11 (3H, s), 0.07 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 157.62, 102.86, 71.32, 67.67, 54.09, 52.91, 45.95, 45.69, 39.31, 36.80, 34.57, 32.58, 31.56, 31.45, 29.95, 29.54, 26.10, 24.72, 24.49, 21.09, 18.42, 12.68, −4.45, −4.58. IR [cm−1]: 2950, 1381, 1089. [α]20D = −95.90 (c 0.65, CH2Cl2). M.p.: 128–129 °C.
(5β,13α)-3β-(t-Butyldimethylsilyloxy)-4α-chlorospiro[androstane-17ξ,2′-oxirane] (34, 35)
The reaction was carried out following general procedure A. 67 mg (33%) of the desired 17β-epoxy product 34 and 133 mg (66%) of 17α-epoxy product 35 were obtained.(5α,13α)-3α-(t-Butyldimethylsilyloxy)-4β-chlorospiro[androstane-17ξ,2′-oxirane] (36, 37)
The reaction was carried out following general procedure A. 84 mg (26%) of the desired 17β-epoxy product 36 and 182 mg (57%) of 17α-epoxy product 37 were obtained.Crystal data (CCDC 1825768†). C26H45ClO2Si, M = 453.16, monoclinic, a = 13.2696(16), b = 7.0208(8), c = 28.171(3) Å, β = 97.170(2)°, Z = 4, T = 100 K, space group C2, 24646 reflections measured, 6247 unique (Rint = 0.0576), which were used in all calculations. The Flack parameter using 2512 quotients is −0.02(4), and the final wR(F2) is 0.1434.
(5α,13α)-3α-(t-Butyldimethylsilyloxy)-4α-chlorospiro[androstane-17ξ,2′-oxirane] (38, 39)
The reaction was carried out following general procedure A. 40 mg (30%) of the desired 17β-epoxy product 38 and 83 mg (62%) of 17α-epoxy product 39 were obtained.(5α,13α)-3β-(t-Butyldimethylsilyloxy)-4α-chlorospiro[androstane-17ξ,2′-oxirane] (40, 41)
The reaction was carried out following general procedure A. 66 mg (35%) of the desired 17β-epoxy product 40 and 111 mg (58%) of 17α-epoxy product 41 were obtained.3α-(t-Butyldimethylsilyloxy)-4β-chloro-17β-hydroxymethyl-17α-methyl-18-nor-5α-androst-13-ene (42)
The reaction was carried out following general procedure E. 17 mg (65%) of alcohol 42 were obtained as colourless crystals. 1H-NMR (400 MHz, CDCl3): δH = 4.01 (1H, m), 3.78 (1H, m), 3.46 (1H, d, J = 10.36 Hz), 3.30 (1H, br d, J = 10.36 Hz), 2.28 (1H, m), 2.04–2.18 (3H, m), 1.93–2.03 (2H, m), 1.84–1.93 (3H, m), 1.72–1.83 (2H, m), 1.50–1.61 (2H, m), 1.29–1.47 (3H, m), 1.03–1.21 (3H, m), 1.00 (3H, s), 0.95 (3H, s), 0.89 (9H, s), 0.85–0.93 (1H, m), 0.06 (3H, s), 0.06 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 141.38, 136.40, 71.75, 69.17, 66.39, 53.23, 51.72, 43.32, 36.56, 36.49, 34.18, 31.38, 31.25, 30.68, 27.05, 25.93, 24.35, 22.51, 21.92, 18.17, 14.00, −4.76, −4.81. IR [cm−1]: 3343, 2927, 1040, 774. M.p.: 168–170 °C. [α]20D = −25.12 (c 0.85, CH2Cl2). HRMS (M + H): calcd for C26H46ClO2Si: 453.2950, found 453.2947 (Δ = 0.7 ppm).3β-(t-Butyldimethylsilyloxy)-4α-chloro-17β-hydroxymethyl-17α-methyl-18-nor-5β-androst-13-ene (43)
The reaction was carried out following general procedure E. 53 mg (79%) of alcohol 43 was obtained as colourless oil.1H-NMR (400 MHz, CDCl3): δH = 4.10 (1H, dd, J = 8.40 Hz, 5.35 Hz), 3.66 (1H, td, J = 8.46 Hz, 3.92 Hz), 3.46 (1H, d, J = 10.03 Hz), 3.29 (1H, d, J = 10.03 Hz), 2.28 (1H, m), 2.02–2.17 (3H, m), 1.97 (1H, m), 1.77–1.93 (5H, m), 1.60–1.75 (2H, m), 1.49–1.59 (2H, m), 1.30–1.47 (4H, m), 1.10–1.28 (2H, m), 0.94 (3H, s), 0.92 (3H, s), 0.89 (9H, s), 0.09 (3H, s), 0.06 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 141.56, 136.24, 72.60, 69.01, 68.59, 51.59, 45.66 (br s), 44.59 (br s), 36.89, 34.25, 32.72 (br s), 31.79 (br s), 30.66, 29.20 (br s), 28.04, 25.96 (3C), 23.93, 22.49, 21.97, 20.81 (2C, br s), 18.29, −4.46, −4.55. IR [cm−1]: 3373, 2927, 1111, 733. [α]20D = −56.40 (c 1.0, CH2Cl2). HRMS (M + H): calcd for C26H46ClO2Si: 453.2950, found 453.2941 (Δ = 2 ppm).
3α-(t-Butyldimethylsilyloxy)-4α-chloro-17β-hydroxymethyl-17α-methyl-18-nor-5α-androst-13-ene (44)
The reaction was carried out following general procedure E. 38 mg (57%) of alcohol 44 was obtained as colourless oil.1H-NMR (400 MHz, CDCl3): δH = 4.04 (1H, m), 3.92 (1H, dd, J = 11.83 Hz, 2.66 Hz), 3.46 (1H, d, J = 10.50 Hz), 3.30 (1H, d, J = 10.50 Hz), 2.30 (1H, m), 2.05–2.19 (2H, m), 1.76–2.05 (5H, m), 1.67–1.75 (2H, m), 1.45–1.65 (6H, m), 1.01–1.23 (3H, m), 0.97 (3H, s), 0.92 (9H, s), 0.80 (3H, s), 0.89–0.95 (1H, m), 0.11 (3H, s), 0.07 (3H, s). 13C-NMR (100 MHz, CDCl3): δC = 141.45, 136.24, 71.37, 69.21, 67.36, 51.90, 51.73, 45.79, 39.21, 36.48, 34.21, 31.44, 30.91, 30.65, 30.02, 26.09, 24.93, 22.63, 22.49, 21.91, 18.42, 12.25, −4.47, −4.57. IR [cm−1]: 3350, 2927, 1079, 832. [α]20D = −55.72 (c 0.76, CH2Cl2). HRMS (M + H): calcd for C26H46ClO2Si: 453.2950, found 453.2946 (Δ = 0.9 ppm).
4α-Chloro-17α-methyl-18-nor-3-oxo-5β-androst-13-en-17β-carbaldehyde (45)
In a 50 ml round bottom flask alcohol 3e (21 mg, 0.062 mmol, 1 eq.) was dissolved in 6 ml CH2Cl2 at room temperature and DMP (C13H13IO8, 79 mg, 0.19 mmol, 3 eq.) was added in one portion and the solution was stirred for 20 minutes. The reaction mixture was then washed with NaHCO3 solution followed by Na2S2O3 solution and the organic phase was dried over MgSO4 and evaporated to dryness. The compound decomposes (epimerizes) on silica gel, so the crude product was instead used directly in the next step.4β-Chloro-17α-methyl-18-nor-3-oxo-5α-androst-13-en-17β-carbaldehyde (46)
In a 25 ml round bottom flask alcohol 3c (30 mg, 0.088 mmol, 1 eq.) was dissolved in 7 ml CH2Cl2 at room temperature and DMP (112 mg, 0.26 mmol, 3 eq.) was added in one portion and the solution was stirred for 60 minutes. The reaction mixture was then washed with NaHCO3 solution followed by Na2S2O3 solution and the organic phase was dried over MgSO4 and evaporated to dryness. The crude product was purified by chromatography on silica gel (2.5 g) using 25/1 LP/EtOAc as the eluent giving 21 mg (71%) aldehyde 46 as small crystals.1H-NMR (400 MHz, CDCl3): δH = 9.39 (1H, s), 4.02 (1H, dd, J = 3.89 Hz, 1.77 Hz), 3.06 (1H, td, J = 14.94 Hz, 5.90 Hz), 2.42 (1H, m), 2.32 (1H, m), 2.19–2.28 (2H, m), 2.03–2.18 (2H, m), 1.93 (1H, td, J = 12.80 Hz, 4.14 Hz), 1.73–1.86 (3H, m), 1.67 (1H,), 1.24–1.50 (4H, m), 1.22 (3H, s), 1.17 (1H, m), 1.12 (3H, s), 0.95 (1H, m), 0.87 (1H, m). 13C-NMR (100 MHz, CDCl3): δC = 205.17, 203.14, 143.49, 134.15, 63.82, 61.85, 52.45, 50.31, 38.73, 36.64, 36.28, 33.54, 32.59, 31.21, 30.76, 26.17, 23.16, 22.19, 18.04, 13.68. IR [cm−1]: 2930, 2855, 1717, 1455. [α]20D = −268.46 (c 0.7, CH2Cl2). M.p.: 148–151 °C. HRMS (M − CHO): calcd for C19H26ClO: 305.1667, found: 305.1663 (Δ = 1.3 ppm).
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We are thankful for financial support from the World Anti-Doping Agency (WADA). We thank Prof. Christian Hametner for NMR sample measurements. The X-ray Centre of TU Wien is acknowledged for providing access to the X-ray diffractometer.Notes and references
- (a) W. Schänzer, M. K. Parr, G. Fußhöller, M. Gütschow and C. Hess, Recent Advances in Doping Analysis, Sportverlag Strauß, 2010, vol. 18, p. 64 Search PubMed; (b) M. Thevis, A. Thomas and W. Schänzer, Anal. Bioanal. Chem., 2011, 401, 405 CrossRef CAS PubMed; (c) W. Schänzer and M. Thevis, Mass Spectrom. Rev., 2017, 36, 16 CrossRef PubMed; (d) M. Thevis, A. Thomas and W. Schänzer, Anal. Bioanal. Chem., 2013, 405, 9655 CrossRef CAS PubMed.
- W. Geyer, W. Schänzer and M. Thevis, Br. J. Sports Med., 2014, 48, 820 CrossRef PubMed.
- T. Sobolevsky and G. Rodchenkov, J. Steroid Biochem. Mol. Biol., 2012, 128, 121 CrossRef CAS PubMed.
- W. Schänzer, Clin. Chem., 1996, 42, 1001 Search PubMed.
- K. Schubert, J. Schlegel, C. Hörhold and H. Frischleder, J. Steroid Biochem., 1970, 1, 229 CrossRef CAS.
- (a) N. Kratena, V. S. Enev, G. Gmeiner and P. Gärtner, Steroids, 2016, 115, 75 CrossRef CAS PubMed; (b) N. Kratena, B. Stöger, M. Weil, V. S. Enev and P. Gärtner, Tetrahedron Lett., 2017, 58, 1316 CrossRef CAS.
- (a) M. Sobukawa, M. Nakajima and K. Koga, Tetrahedron: Asymmetry, 1990, 5, 295 CrossRef; (b) J. W. Huffman and W. H. Balke, J. Org. Chem., 1988, 53, 3828 CrossRef CAS.
- A. Fürst and P. A. Plattner, Helv. Chim. Acta, 1949, 32, 275 CrossRef.
- D. B. Ramachary, R. Sakthidevi and P. S. Reddy, RSC Adv., 2013, 3, 13497 RSC.
- (a) J. A. R. Salvador, M. L. Sá e Melo and A. S. Campos Neves, Tetrahedron Lett., 1993, 34, 357 CrossRef CAS; (b) J. McKenna, J. K. Norymberski and R. D. Stubbs, J. Chem. Soc., 1959, 2502 RSC; (c) E. J. Tavares da Silva, F. M. F. Roleira, M. L. Sá e Melo, A. S. C. Neves, J. A. Paixão, M. J. de Almeida, M. R. Silva and L. C. R. Andrade, Steroids, 2002, 67, 311 CrossRef CAS.
- C. A. Horiuchi and J. Y. Satoh, Bull. Chem. Soc. Jpn., 1987, 60, 426 CrossRef CAS.
- L. N. Nysted, US Patent, 3865848, 1975 Search PubMed.
- R. Noyori, S. Murata and M. Suzuki, Tetrahedron, 1981, 37, 3899 CrossRef CAS.
- G. Forsdahl, T. Geisendorfer, L. Göschl, S. Pfeffer, P. Gärtner, M. Thevis and G. Gmeiner, Drug Test. Anal., 2018 DOI:10.1002/dta.2385.
- (a) L. J. Chinn, J. Org. Chem., 1965, 30, 4165 CrossRef CAS; (b) J. R. Hanson, A. C. Hunter and S. Roquier, Collect. Czech. Chem. Commun., 1998, 63, 1646 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 1586619–1586622 and 1825768. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8ob00122g |
| This journal is © The Royal Society of Chemistry 2018 |