
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Identification of a novel ligand for the ATAD2 bromodomain with selectivity over BRD4 through a fragment growing approach†
Duncan C.
Miller‡
 a,
Mathew P.
Martin‡
b,
Santosh
Adhikari‡
a,
Alfie
Brennan
a,
Jane A.
Endicott
b,
Bernard T.
Golding
a,
Ian R.
Hardcastle
a,
Amy
Heptinstall
a,
Stephen
Hobson
a,
Claire
Jennings
b,
Lauren
Molyneux
a,
Yvonne
Ng
b,
Stephen R.
Wedge
b,
Martin E. M.
Noble
*b and
Celine
Cano
*a
a,
Mathew P.
Martin‡
b,
Santosh
Adhikari‡
a,
Alfie
Brennan
a,
Jane A.
Endicott
b,
Bernard T.
Golding
a,
Ian R.
Hardcastle
a,
Amy
Heptinstall
a,
Stephen
Hobson
a,
Claire
Jennings
b,
Lauren
Molyneux
a,
Yvonne
Ng
b,
Stephen R.
Wedge
b,
Martin E. M.
Noble
*b and
Celine
Cano
*a
aNewcastle Drug Discovery, Northern Institute for Cancer Research, School of Chemistry, Newcastle University, Newcastle upon Tyne, NE1 7RU, UK. E-mail: celine.cano@ncl.ac.uk; Tel: +44 (0)191 208 7060
bNewcastle Drug Discovery, Northern Institute for Cancer Research, Paul O'Gorman Building, Medical School, Framlington Place, Newcastle upon Tyne NE2 4HH, UK. E-mail: martin.noble@ncl.ac.uk; Tel: +44 (0)191 208 4466
First published on 15th February 2018
Abstract
ATAD2 is an ATPase that is overexpressed in a variety of cancers and associated with a poor patient prognosis. This protein has been suggested to function as a cofactor for a range of transcription factors, including the proto-oncogene MYC and the androgen receptor. ATAD2 comprises an ATPase domain, implicated in chromatin remodelling, and a bromodomain which allows it to interact with acetylated histone tails. Dissection of the functional roles of these two domains would benefit from the availability of selective, cell-permeable pharmacological probes. An in silico evaluation of the 3D structures of various bromodomains suggested that developing small molecule ligands for the bromodomain of ATAD2 is likely to be challenging, although recent reports have shown that ATAD2 bromodomain ligands can be identified. We report a structure-guided fragment-based approach to identify lead compounds for ATAD2 bromodomain inhibitor development. Our findings indicate that the ATAD2 bromodomain can accommodate fragment hits (Mr < 200) that yield productive structure–activity relationships, and structure-guided design enabled the introduction of selectivity over BRD4.
Introduction
Histone proteins undergo several post translational modifications (PTMs) that affect DNA transcription, replication and repair, and genomic architecture.1 Histone PTMs are regulated by three groups of proteins known as writers, erasers and readers. Bromodomains are a class of protein interaction modules, conserved across evolution, which recognise ε-N-acetyl lysine PTMs.2 A total of 61 bromodomains in 46 diverse proteins have been identified in the human proteome, and these are divided into eight structural classes. Within subfamily IV in the structure-based classification of bromodomain-containing proteins is ATAD2 (ATPase Family, AAA Domain Containing 2). ATAD2 is overexpressed in a wide range of human cancers, including breast, lung, and prostate carcinomas, and it is present in low levels in normal non-tumour cells.3–5 The overexpression of ATAD2 has been associated with a poor patient outcome to treatment in breast,6 lung,7 ovarian,8 hepatocellular,9 endometrial10 and gastric11 cancers. Functionally, ATAD2 has been shown to act as a coactivator of various transcription factors including the androgen receptor (AR),4 and the proto-oncogene MYC,5 and studies examining gene silencing of ATAD2, using siRNA or shRNA, report it to have a role in tumour cell proliferation and survival.3,12 These studies suggest that ATAD2 is a potential target for cancer drug discovery, and small-molecule inhibitors would provide insight into the phenotypic response to the inhibition of ATAD2.ATAD2 comprises a four helical bundle (αZ, αA, αB, αC) and two loops (ZA and BC). The acetyllysine binding pocket, created by helices αB, αC and the ZA loop, is polar and shallow compared to several other bromodomains.13 The ZA loop of ATAD2, which forms a major part of the binding site is polar, whereas, the binding site in the bromodomain BRD4 is mostly hydrophobic.14 The flexibility of the ZA loop coupled to the shallow and polar nature of the binding site resulted in the druggability of the ATAD2 bromodomain being classified as ‘difficult’.13,14
Compounds 1 and 2a–b were recently disclosed as relatively potent inhibitors of the bromodomain of ATAD2.15–17 Compound 1 was not selective for ATAD2 over BRD4. Enhanced ATAD2 potency and selectivity over BRD4 was achieved in this series through introduction of a cyclic sulfone (2a) or a difluorocyclohexane moiety (2b). These groups form favourable interactions with the sidechains of Arg1007 and Arg1077 in the ATAD2 binding site, and achieve selectivity due to unfavourable interactions with lipophilic Trp81 and Met149 sidechains in BRD4. Compounds 3–5 were reported as ATAD2 inhibitors arising from a fragment screen,18 but the low potency of these fragments makes them unsuitable as chemical probes of ATAD2 function. More recently BAY-850 (6) has been reported as a potent ATAD2 inhibitor arising from screening of a DNA-encoded library with an unusual dimer-inducing mode of action, although not BRD4 selectivity data was reported.19
This work describes the development of ATAD2 inhibitors employing structure-guided optimization of a fragment hit, with potential for development into selective chemical tools to investigate ATAD2 bromodomain function in biological systems.
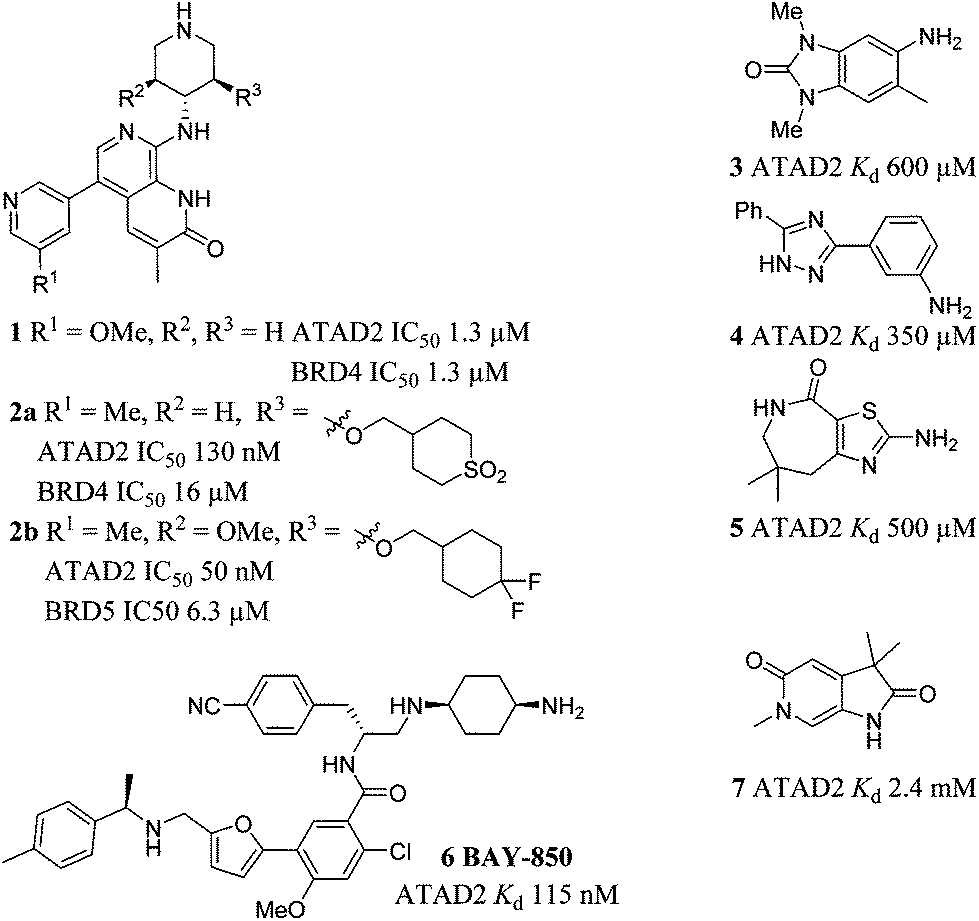
Synthesis
Screening of a small targeted fragment library identified 1,6-dihydro-2H-pyrrolo[2,3-c]pyridine-2,5(3H)-dione 7 as a promising scaffold for the design of inhibitors of the ATAD2 bromodomain. Synthesis of the bicyclic template started with protection of commercially available 6-methoxy-4-methylpyridin-3-amine 8 as its tert-butyl carbamate to give 9 (Scheme 1). Deprotonation followed by quenching with carbon dioxide gave 10. Acetic anhydride mediated ring closure in the presence of catalytic tetra-N-butylammonium acetate followed by bis-alkylation with iodomethane or iodoethane gave 12 and 13, respectively. Deprotection and N-alkylpyridone formation was achieved in a single step by heating 12 with iodomethane to give compound 7. Targets varying the N1 substituent (14a–f) were synthesised by alkylation of 7. 14f was deprotected using TBAF to provide 14g.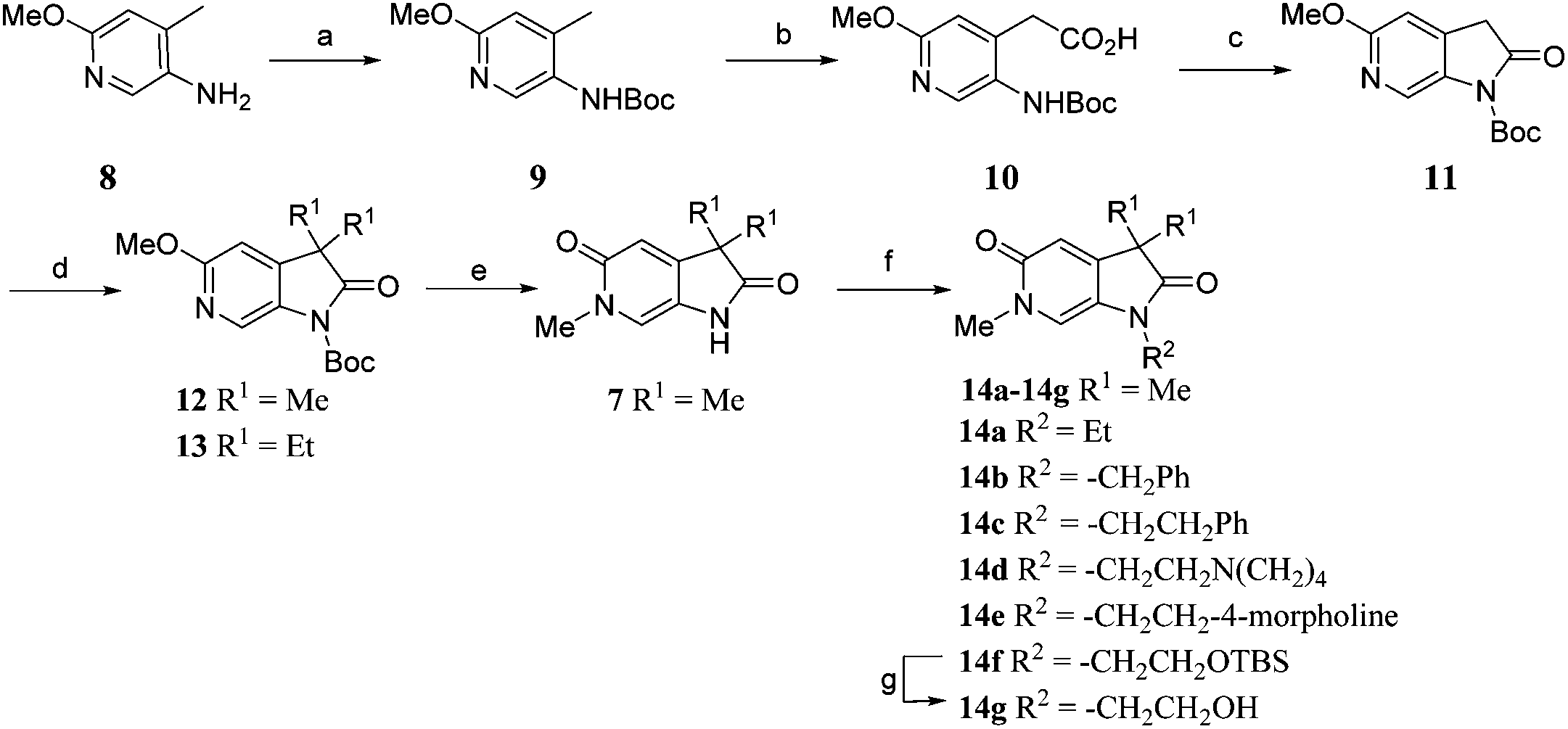 | ||
| Scheme 1 Reagents and conditions: (a) Boc2O, THF, Na2CO3, THF, r.t., 42 h, 95%; (b) (i) s-BuLi, THF, −78 °C, 15 min; (ii) CO2 (dry ice), −78 °C to r.t., 45 min, 74%; (c) Ac2O, tetrabutylammonium acetate, 65 °C, 1 h, 81%; (d) R1 = Me: MeI, Cs2CO3, MeCN, 60 °C, 3 h, 78%; R1 = Et: EtI, Cs2CO3, MeCN, 60 °C, 3 h, 51% (e) MeI, MeCN, 170 °C, μW, 1 h, quant; (f) for compounds 14a–c (i) NaH, DMF, r.t., 15 min; (ii) ethyl iodide or benzyl bromide or (2-bromoethyl)benzene, r.t., 3 h, 61% (14a), 61%; (14b), 30% (14c); For compounds 14d–f: Cs2CO3, DMF, 100 °C, μW, 30 min: 1-(2-chloroethyl)pyrrolidine hydrochloride 40% (14d), or 4-(2-chloroethyl)morpholine hydrochloride, 82% (14e), or (2-bromoethoxy)(tert-butyl)dimethylsilane, 55%; (14f); (g) TBAF, THF, r.t., 18 h, 90%. | ||
N 6-Alkylated target compounds were synthesised from 12 or 13 under microwave irradiation using an array of alkyl halides to give 15a–p and 16a–b, respectively. Subsequent N1-methylation gave compounds 17a–q and 18a–b (Scheme 2).
 | ||
| Scheme 2 Reagents and conditions: (a) RX, MeCN, 170 °C, μW, 45 min, 22–93%; (b) MeI, Cs2CO3, DMF, 100 °C, μW, 30 min, 74–94%; (c) NaOH, EtOH, H2O, 100 °C, 23 h, 32%; Structures of 17a–q and 18a–b are given in Tables 2 and 3. | ||
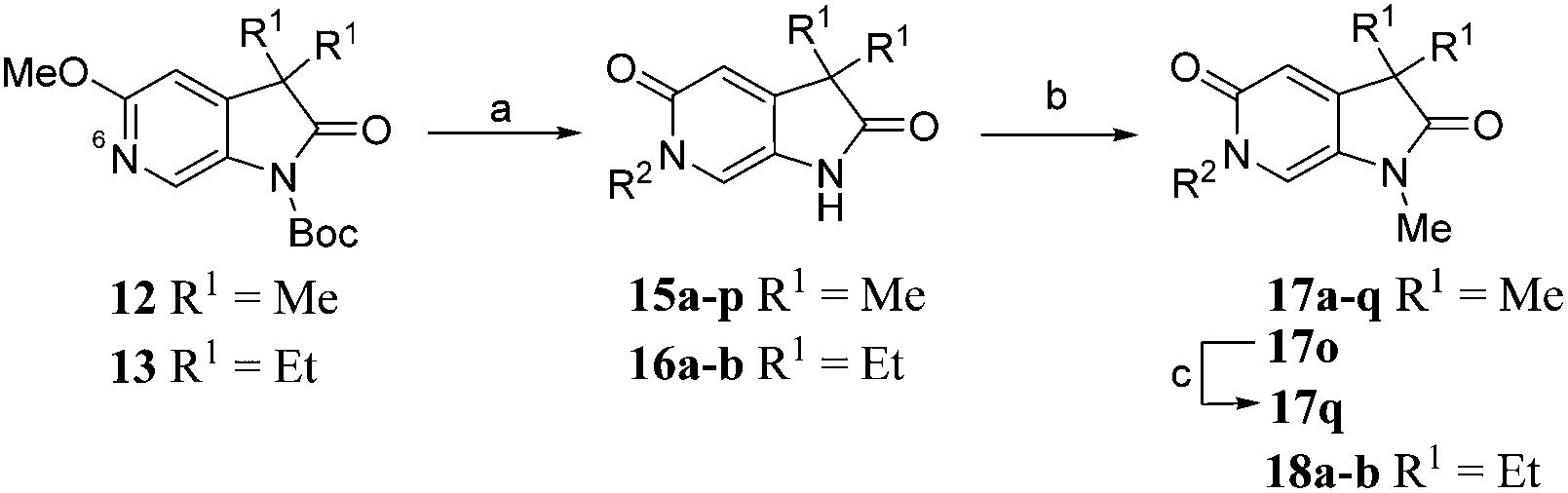
Diversification of the C3-position could be achieved using Knoevenagel condensation of aldehydes with 11 to provide 19a–j in high yield (Scheme 3). Subsequent alkene reduction, followed by methylation provided 21a–f. Treatment of methoxypyridines 21a–f with benzyl bromides at high temperature provided the target pyridones (22a–f) in moderate yields.
 | ||
| Scheme 3 Reagents and conditions: (a) RCHO, piperidine, THF, 100 °C, 30 min, 50–88%; (b) H2, 10% Pd/C, THF, MeOH, r.t., 2 h; (c) MeI, Cs2CO3, DMF, 50 °C, 1.5 h, 55–56% over 2 steps; (d) 4-chlorobenzyl bromide, MeCN, 170 °C μW, 45 min, 50–64%. Structures of 22a–f are given in Table 4. | ||
Results and discussion
A crystallographic screen of a small targeted library identified pyrrolidinopyridone 7 as a fragment which bound in the N-acetyllysine histone binding site of the bromodomain of ATAD2 (Fig. 1A and B). Isothermal titration calorimetry (ITC) was used to infer the binding affinity of 7 for the bromodomain of ATAD2, with an apparent Kd of 2.4 mM (Fig. 1C). The pyridone carbonyl forms a hydrogen bond with the carboxamide sidechain of Asn1064, and another through a water-mediated hydrogen bond to Tyr1021, with the N6-methyl group projecting towards a pocket containing four conserved water molecules. This interaction network mimics the acetylated lysine of the histone, and is consistent with small hydrophobic groups that occupy the equivalent pocket in other bromodomain family ligands such as the BRD4 inhibitors JQ120 and I-BET762.21 Selectivity over other bromodomain family members is important in the development of a tool compound to investigate the role of the ATAD2 bromodomain in biological systems.22 Minimizing the BET activity of an ATAD2 chemical probe is particularly necessary due to the known effects of BET inhibitors. Thus BRD4 was selected as representative BET family member to assess compound selectivity. The binding orientation of 7 in ATAD2 was designated as binding mode 1 for this template. The residues that line the acetylated lysine binding pocket of BRD4 and ATAD2 are highly conserved (Fig. 1D), with greater divergence in the more solvent exposed residues including the shelf region of the protein. Achieving selectivity for ATAD2 over BRD4 would thus require identification of fragments that extend away from the conserved core and towards the less conserved areas.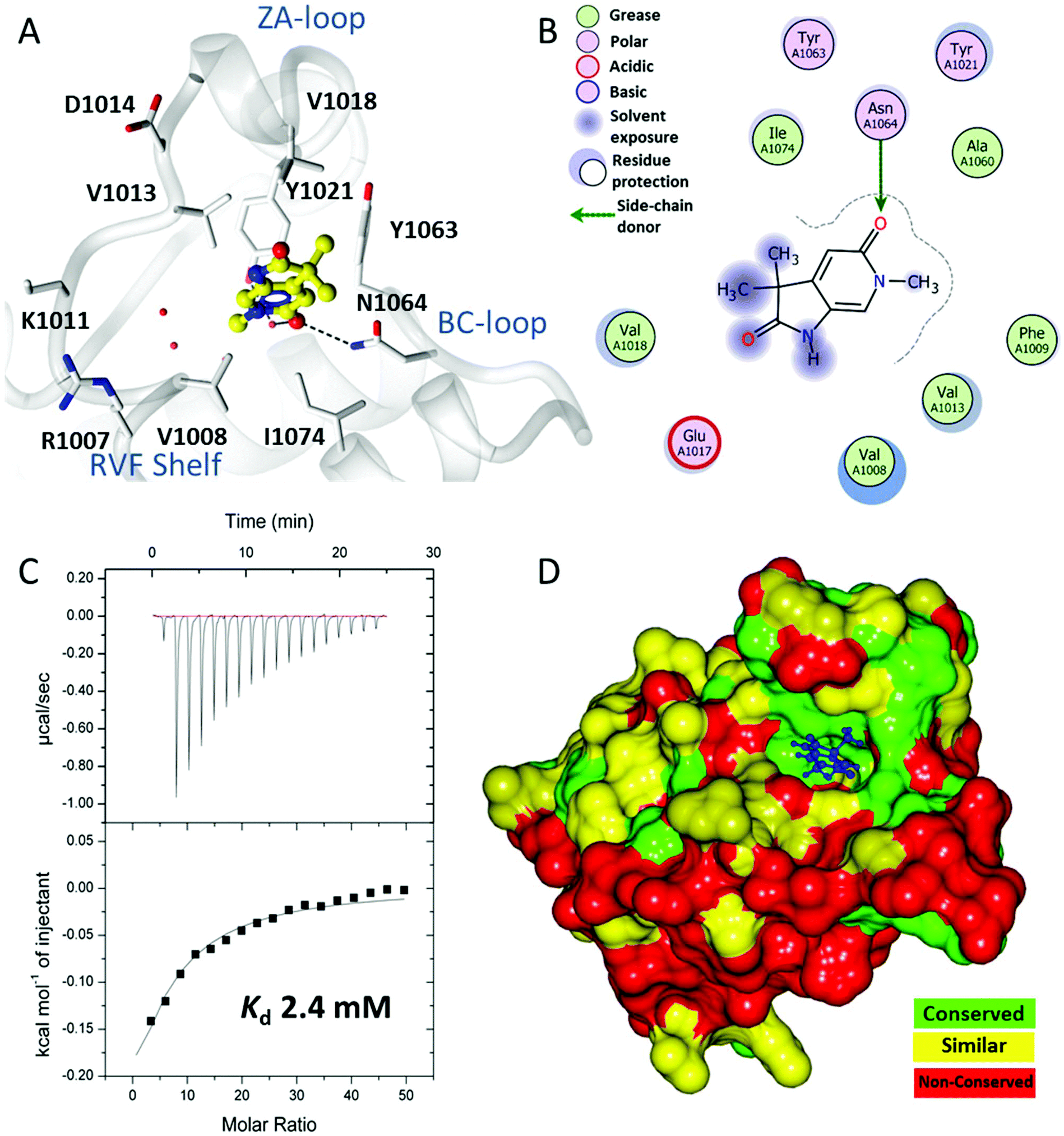 | ||
| Fig. 1 Binding of initial fragment hit 7. (A) Binding mode of fragment hit 7 (yellow ball and stick) bound to ATAD2 (white). ATAD2 active site residues shown in cylinder and conserved water molecules shown as red spheres. Potential hydrogen bonding interactions shown in black dash. (B) 2D Interaction map of 7 with the active site residues of ATAD2 represented by Lidia within Coot. (C) Isothermal titration calorimetry of 7 binding with saturation to ATAD2. (D) ATAD2 represented in solid surface and coloured through conservation of sequence identity between ATAD2 and the first bromodomain of BRD4. | ||
Methylation of the N1 position (17a) resulted in a change in binding mode (Fig. 2A), whereby the fragment core rotates and the pyrrolidinone carbonyl now forms a hydrogen bond with the carboxamide sidechain of Asn1064, and a second through a water-mediated hydrogen bond to Tyr1021. In this binding mode the N1-methyl group projects towards the conserved water molecules. Rotation of the pyrrolidinone core allows the pyridone carbonyl to interact through a water molecule with the backbone NH of Asp1014 on the ZA loop. One of the C3-geminal methyl groups occupies a small hydrophobic pocket formed between the sidechains of ZA-loop residues Tyr1021, Val1018 and the Tyr1063 residue that reside next to the conserved asparagine. Additional hydrophobic interactions are formed with the base of the binding pocket, where Val1008 and Ile1074 interact with the second C3-geminal methyl group and pyrrolidinone core, respectively. This binding orientation was designated as binding mode 2. Intriguingly, when the N1 substituent was further elaborated to an ethyl group (14a: Fig. 2B) the compound reverted to binding mode 1, suggesting the conserved water-rich pocket was unable to accommodate the larger ethyl group.
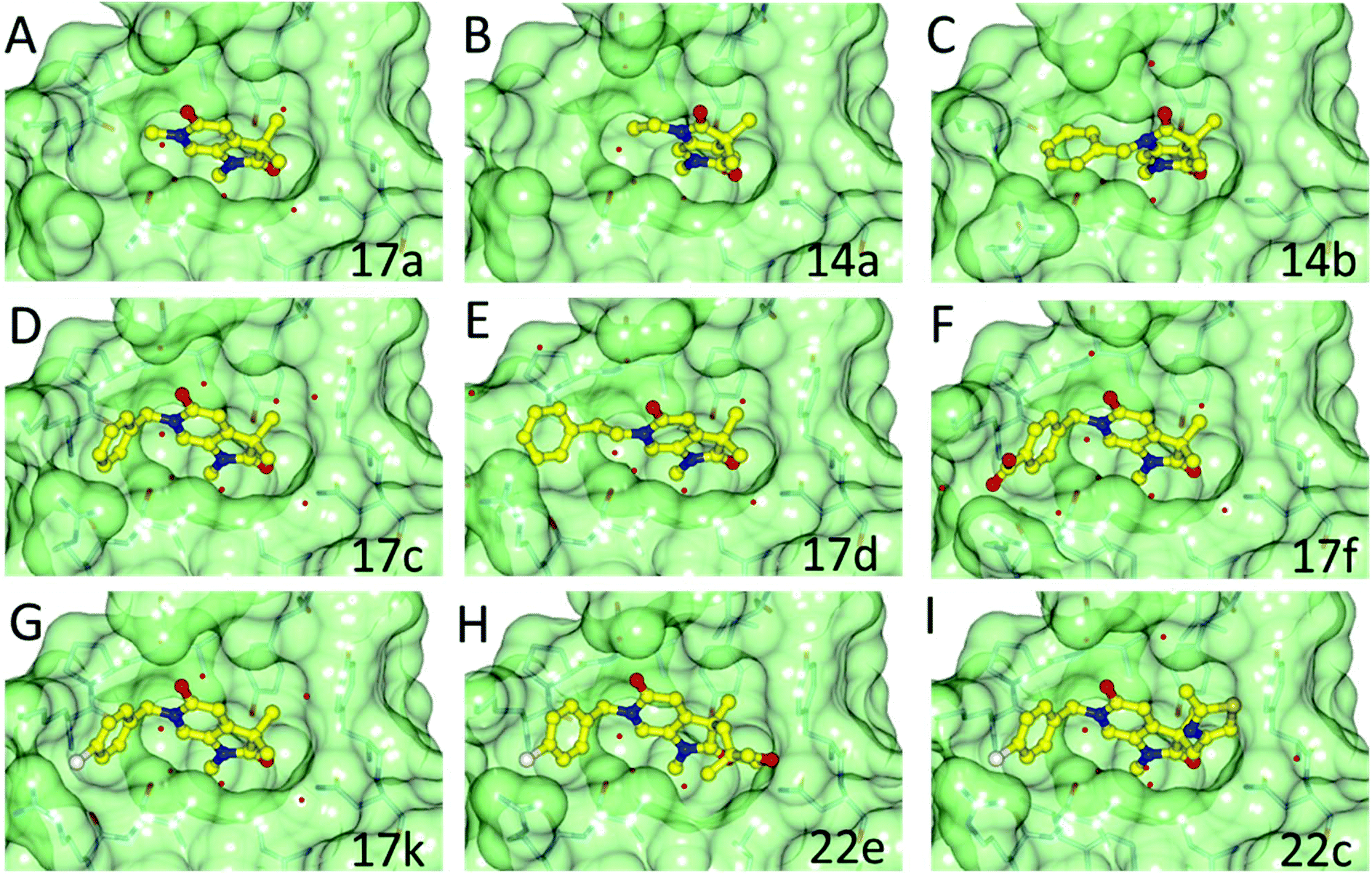 | ||
| Fig. 2 Overlay of the crystal structures of fragment 17a, 14a, 14b, 17c, 17d, 17f, 17k, 22e and 22c bound to ATAD2. Compounds represented in yellow as ball and stick. Binding pocket of ATAD2 shown in transparent surface, with neighbouring residues of compound shown in stick representation. Conserved waters are represented by red spheres. | ||
Fragments 7, 17a, and 14a bound very weakly and were unable to displace the acetylated histone ligand sufficiently in a homogeneous time-resolved fluorescence (HTRF) assay to enable a Kd to be determined, although some inhibition was observed at the highest concentration assayed (1000 μM; Table 1). Surface plasmon resonance (SPR) was used to assess the direct binding of the fragments and provide Kd values for binding to both ATAD2 and BRD4. SPR can yield artefactual data where the concentration of a small-molecular analyte exceeds ∼250 μM, and accordingly, this was the highest concentration used in the assays. Where the Kd exceeded this limit, projected Kds were determined by extrapolation, using the observed response units for compound binding (RU) relative to the molecular weight of a known control compound.23 Compounds 7 and 17a bound too weakly to ATAD2 to allow Kd values to be calculated. However, 17a did show detectable binding to BRD4. Compound 14a showed a remarkable increase in affinity towards BRD4, reporting a Kd of 12.7 μM and a binding constant 100-fold weaker towards ATAD2 (Table 1).
|
|
|||||
|---|---|---|---|---|---|
| ID | R | ATAD2 IC50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) a (μM) a (μM) |
ATAD2 LE | ATAD2 Kdb (μM) |
BRD4 Kdb (μM) |
|
a HTRF format assay.
b SPR format assay.
c Isothermal titration calorimetry. Ligand efficiency (LE) = 1.4(−logIC50)/N, where N is number of non-hydrogen atoms.
|
|||||
| 7 | H | >1000 | N/A | >2000 | >2000 |
| 2400c | |||||
| 17a | Me | >1000 | N/A | >2000 | 1500 |
| 14a | Et | >1000 | N/A | 1300 | 12.7 |
| 14b | Bn | 810 ± 57 | 0.21 | >2000 | 1050 |
| 14c | –CH2CH2Ph | 868 ± 33 | 0.19 | 1400 | 800 |
| 14d |
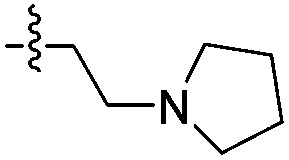
|
>1000 | N/A | >2000 | 1700 |
| 14e |
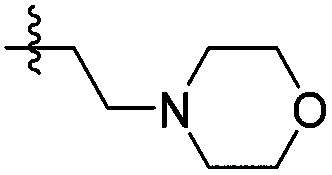
|
>1000 | N/A | >2000 | >2000 |
| 14g | –CH2CH2OH | >1000 | N/A | >2000 | >2000 |
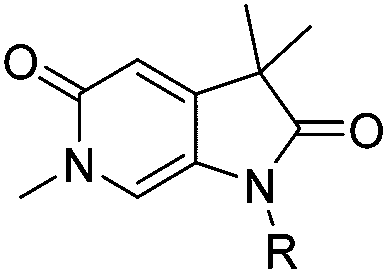
The two binding modes offered different vectors to explore structure activity relationships (SARs) of substituents directed towards the RVF shelf and the ZA loop. Thus a set of N1-alkylated pyrrolidinopyridones was prepared to attempt to access this region while maintaining binding mode 1 (Table 1). Lipophilic benzyl (14b) and phenethyl (14c) substituents exhibited sub-millimolar HTRF IC50 values, and showed the first detectable signs of direct binding to both ATAD2 and BRD4 when analysed by SPR. Co-crystallisation confirmed that 14b retained binding mode 1 (Fig. 2C). Hydrophilic substituents (14d–g) gave no measurable ATAD2 inhibition or binding in HTRF or SPR format experiments.
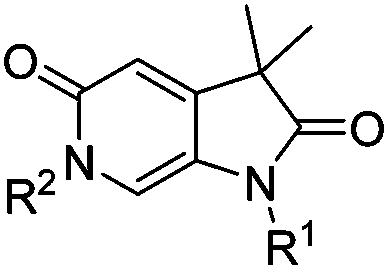
To attempt to exploit binding mode 2, a set of N6-alkylated pyrrolidinopyridones were also prepared (Table 2) with N6-benzyl (17c) and N6-phenethyl (17d) analogues giving sub-millimolar ATAD2 IC50 values. As expected, 17c and 17d retained binding mode 2 (Fig. 2D and E), with the benzyl group sitting above the RVF shelf, forming a hydrophobic interaction with the sidechain of Lys1011. Pairwise comparison with N1-des-methyl analogue 15c confirmed the importance of a methyl group in the conserved water pocket. 17c and 17d bound more tightly to BRD4 than to ATAD2 in the SPR experiments.
|
|
||||||
|---|---|---|---|---|---|---|
| ID | R1 | R2 | ATAD2 IC50a (μM) |
ATAD2 LE | ATAD2 Kdb (μM) |
BRD4 Kdb (μM) |
|
a HTRF format assay.
b SPR format assay. Ligand efficiency (LE) = 1.4(−logIC50)/N, where N is number of non-hydrogen atoms.
|
||||||
| 17b | H | Pr | >1000 | N/A | >2000 | 1000 |
| 15c | H | Bn | >1000 | N/A | 2000 | 1400 |
| 17c | Me | Bn | 681 ± 120 | 0.21 | 700 | 101 |
| 17d | Me | –CH2CH2Ph | 741 ± 74 | 0.20 | 1000 | 161 |
|
|
|||||
|---|---|---|---|---|---|
| ID | R | ATAD2 IC50a (μM) | ATAD2 LE | ATAD2 Kdb (μM) | BRD4 Kdb (μM) |
|
a HTRF format assay.
b SPR format assay. Ligand efficiency (LE) = 1.4(−logIC50)/N, where N is number of non-hydrogen atoms.
|
|||||
| 17e | 4-CO2Me | 651 ± 1 | 0.18 | 900 | 141 |
| 17f | 4-CO2H | 533 ± 40 | 0.19 | 600 | 1500 |
| 17g | 4-CN | 562 ± 67 | 0.20 | 900 | 142 |
| 17h | 4-CONH2 | 680 ± 66 | 0.18 | 700 | 300 |
| 17i | 4-SO2Me | 359 ± 37 | 0.19 | 600 | 200 |
| 17j | 4-Cl | 190 ± 63 | 0.24 | 200 | 40 |
| 17k | 4-Br | 236 ± 36 | 0.23 | 138 | 48 |
| 17l | 4-Me | 274 ± 75 | 0.23 | 400 | 58 |
| 17m | 4-CF3 | 179 ± 43 | 0.21 | 400 | 124 |
| 17n | 3,4-DiCl | 299 ± 28 | 0.21 | 159 | 65 |
| 17o | 2-CN | >1000 | N/A | >2000 | 300 |
| 17p | 3-CN | 668 ± 28 | 0.19 | 800 | 184 |
| 17q | 2-CO2H | >1000 | N/A | >2000 | 300 |
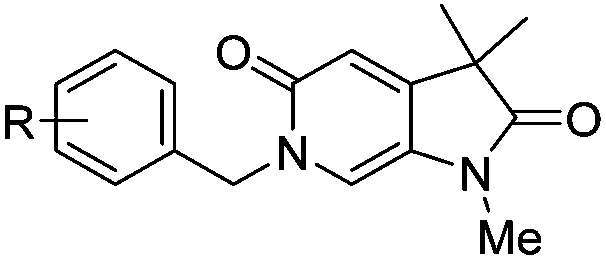
With the aim of improving binding affinity towards ATAD2 the sidechain of Arg1007 was targeted through substitution of the 4-position of the N6-benzyl group. Compound 17f showed similar ATAD2 affinity to 17c, and the co-crystal structure with ATAD2 (Fig. 2F) confirmed that the carboxylate did not form the projected charge–charge interaction with Arg1007. Other analogues bearing functionality with the potential to form hydrogen bonds with Arg1007 gave no further improvement in inhibition (17g–i). Lipophilic substituents at the 4-position of the benzyl group (17j–m, Fig. 2G) provided the most potent benzylic analogues, but continued to be selective for BRD4. Addition of a second chloro substituent at the 3-position (17n) resulted in a modest drop in potency. N6-Benzyl analogues with substituents at the 2- and 3-positions of the benzylic ring (17o–q) were prepared to attempt to introduce further interactions with the ZA loop, or to favourably influence the conformation of the ligand, but no further improvement in ATAD2 affinity was observed.
Attention turned to exploring substitution at the C3-geminal dimethyl group. The pro-S methyl group occupies a small lipophilic pocket in the ligand binding site of ATAD2, whereas the pro-R methyl provides opportunities for fragment growth. It was envisioned that larger substituents at the pro-R position may occupy the cleft between the ZA and BC loops where the substrate acetylated lysine sidechain enters the active site, and may also provide improved selectivity over the more sterically demanding BRD4. Synthetic chemistry to access chiral C3 analogues that retained one methyl but differed in the second C3 substituent proved challenging, with mono-alkylation strategies found to be low yielding. Knoevenagel condensation was successful when the aldehyde coupling partner did not contain an enolisable proton, allowing the synthesis of 22a–f (Table 4) as racemic mixtures. Symmetrical C3-diethyl analogues 18a and 18b were also prepared.
|
|
|||||||
|---|---|---|---|---|---|---|---|
| ID | R1 | R2 | X | ATAD2 IC50a (μM) | ATAD2 LE | ATAD2 Kdb (μM) | BRD4 Kdb (μM) |
|
a HTRF format assay.
b SPR format assay. Ligand efficiency (LE) = 1.4(−logIC50)/N, where N is number of non-hydrogen atoms.
|
|||||||
| 22a | Me | Bn | Cl | >1000 | N/A | 500 | >2000 |
| 22b | Me |
|
Cl | >1000 | N/A | 300 | 1700 |
| 22c | Me |
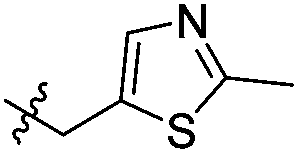
|
Cl | 352 | 0.17 | 110 | 1500 |
| 22d | Me | CH2C(CH3)3 | Cl | 489 ± 18 | 0.18 | 125 | >2000 |
| 22e | Me |
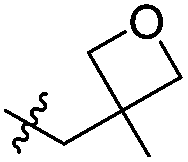
|
Cl | 214 | 0.19 | 500 | >2000 |
| 22f | Me |
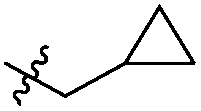
|
Cl | 304 ± 83 | 0.20 | 113 | 800 |
| 18a | Et | Et | Cl | 163 ± 23 | 0.22 | 107 | 1000 |
| 18b | Et | Et | CF3 | 163 ± 37 | 0.20 | 300 | >2000 |
The crystal structure of 18a bound to ATAD2 (Fig. 3A) showed that the diethyl analogues delve further into the hydrophobic pockets formed within the ZA-loop residues Tyr1021, Val1018 and with Ile1074 in the base of the binding site. Gratifyingly, these analogues resulted in a significant improvement in selectivity over BRD4 while retaining ATAD2 binding and inhibition (Table 4) (Fig. 3B).
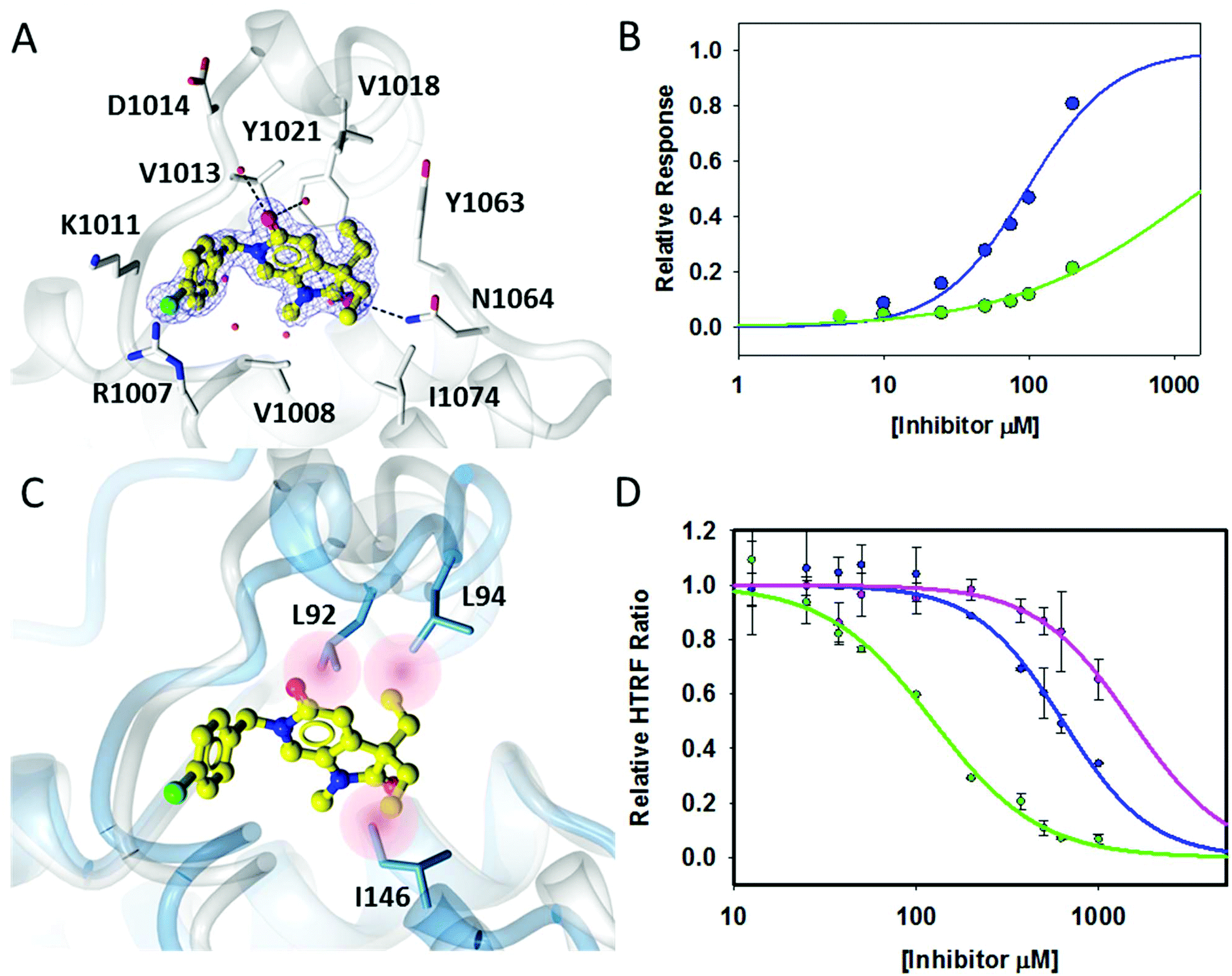 | ||
| Fig. 3 Binding interactions of 18a. (A) Crystal structure of 18a (yellow ball and stick) within the histone binding site of ATAD2 shown in white ribbon, active site residues shown in cylinder and conserved water molecules shown in red sphere. Key potential hydrogen bonding interactions shown in black dash. (B) Direct binding data using SPR of 18a with ATAD2 (blue) and BRD4 (green). (C) Superimposition of 18a bound to ATAD2 with BRD4 showing potential steric clashes of 18a within the binding site of BRD4 (red). (D) HTRF data of improved potency within the series 17a pink, 17c blue, 18a green. | ||
Superimposition of the crystal structure of 18a with the first bromodomain of BRD4 (PDB entry 2OSS) indicates that the selectivity for ATAD2 over BRD4 may be due to a steric clash of one of the ethyl groups with Leu94 and the other with Ile146 in the BRD4 structure (Fig. 3C).
Conclusions
Pyrrolidinopyridone 7 was identified through crystallographic fragment screening as a novel ligand for the ATAD2 bromodomain. Assessment in HTRF, ITC and SPR assays confirmed that this was a low affinity fragment. Structure-guided fragment growing at the N1, N6 and C3 substituents led to the identification of two binding modes for this template in ATAD2. For binding mode 1, methylation of N1, and incorporation of substituted benzyl groups at N6 led to compounds with ATAD2 binding affinities below 200 μM (Fig. 3D), but these compounds bound more tightly to BRD4 than to ATAD2. Increasing steric demand at the C3 position led to compounds 18a and 18b, retaining ATAD2 binding affinity and activity in the HTRF biochemical assay. These C3-substituents conferred selectivity over BRD4 through the introduction of unfavorable interactions with lipophilic amino acid sidechains in the more sterically demanding BRD4 N-acetyllysine binding site.Overall, we have demonstrated that structure-based optimisation of ATAD2 fragments was able to bring millimolar hits into a micromolar potency range. Although inhibition of ATAD2 in the pyrrolidinopyridone series reached a plateau at 100–200 μM, we have succeeded in identifying interactions that can enhance potency while introducing selectivity versus BRD4. Application of these lessons might contribute to the development of improved chemical probes of ATAD2's role and structure–function relationship, if applied to fragments with superior initial ligand efficiency.
Author contributions
Duncan C. Miller – Experimental design and interpretation, data analysis, synthesis and characterization of compounds and manuscript preparation.Mathew Martin – Protein expression, crystallisation and structure determination, assay development and manuscript preparation. †Santosh Adhikari – Synthesis and characterization of compounds.
Alfie Brennan – Synthesis and characterization of compounds.
Jane A. Endicott – Project conception, experimental design and interpretation.
Bernard T. Golding – Experimental design and interpretation, data analysis.
Ian R. Hardcastle – Experimental design and interpretation, data analysis.
Amy Heptinstall – Synthesis and characterization of compounds.
Stephen Hobson – Synthesis and characterization of compounds.
Claire Jennings – Protein expression, crystallisation and structure determination.
Lauren Molyneux – Synthesis and characterization of compounds.
Yvonne Ng – Assay development.
Stephen R. Wedge – Project conception, experimental design and interpretation.
Martin E. M. Noble – Project conception, experimental design and interpretation, data analysis and manuscript preparation.
Céline Cano – Project conception, experimental design and interpretation, data analysis and manuscript preparation (submitting author).
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
The authors thank Cancer Research UK (Grant Reference C2115/A21421) and Astex Pharmaceuticals for financial support. We thank Phil Day and Tom Heightman, Astex Pharmaceuticals for helpful discussions and advice on the crystallography of ATAD2. The use of the EPSRC Mass Spectrometry Service at the University of Wales (Swansea) is also gratefully acknowledged. We also thank beamline staff at The Diamond Light Source who provided excellent facilities for data collection.References
- A. J. Bannister and T. Kouzarides, Cell Res., 2011, 21, 381 CrossRef CAS PubMed.
- P. Filippakopoulos and S. Knapp, Nat. Rev. Drug Discovery, 2014, 13, 337 CrossRef CAS PubMed.
- C. Caron, C. Lestrat, S. Marsal, E. Escoffier, S. Curtet, V. Virolle, P. Barbry, A. Debernardi, C. Brambilla, E. Brambilla, S. Rousseaux and S. Khochbin, Oncogene, 2010, 29, 5171 CrossRef CAS PubMed.
- J. X. Zou, L. Guo, A. S. Revenko, C. G. Tepper, A. T. Gemo, H. J. Kung and H. W. Chen, Cancer Res., 2009, 69, 3339 CrossRef CAS PubMed.
- M. Ciró, E. Prosperini, M. Quarto, U. Grazini, J. Walfridsson, F. McBlane, P. Nucifero, G. Pacchiana, M. Capra, J. Christensen and K. Helin, Cancer Res., 2009, 69, 8491 CrossRef PubMed.
- E. V. Kalashnikova, A. S. Revenko, A. T. Gemo, N. P. Andrews, C. G. Tepper, J. X. Zou, R. D. Cardiff, A. D. Borowsky and H. W. Chen, Cancer Res., 2010, 70, 9402 CrossRef CAS PubMed.
- Y. Zhang, Y. Sun, Y. Li, Z. Fang, R. Wang, Y. Pan, H. Hu, X. Luo, T. Ye, H. Li, L. Wang, H. Chen and H. Ji, Ann. Surg. Oncol., 2013, 20, 577 CrossRef PubMed.
- W.-N. Wan, Y.-X. Zhang, X.-M. Wang, Y.-J. Liu, Y.-Q. Zhang, Y.-H. Que and W.-J. Zhao, Asian Pac. J. Cancer Prev., 2014, 15, 2777 CrossRef PubMed.
- H. W. Hwang, S. Y. Ha, H. Bang and C.-K. Park, Cancer Res. Treat., 2015, 47, 853 CrossRef CAS PubMed.
- P. Shang, F. Meng, Y. Liu and X. Chen, Tumor Biol., 2015, 36, 4479 CrossRef CAS PubMed.
- M. J. Zhang, C. Z. Zhang, W. J. Du, X. Z. Yang and Z. P. Chen, Clin. Transl. Oncol., 2016, 18, 776 CrossRef CAS PubMed.
- M. B. Raeder, E. Birkeland, J. Trovik, C. Krakstad, S. Shehata, S. Schumacher, T. I. Zack, A. Krohn, H. M. J. Werner, S. E. Moody, E. Wik, I. M. Stefansson, F. Holst, A. M. Oyan, P. Tamayo, J. P. Mesirov, K. H. Kalland, L. A. Akslen, R. Simon, R. Beroukhim and H. B. Salvesen, PLoS One, 2013, 8, e54873 CAS.
- A. Chaikuad, A. M. Petros, O. Fedorov, J. Xu and S. Knapp, MedChemComm, 2014, 5, 1843 RSC.
- L. R. Vidler, N. Brown, S. Knapp and S. Hoelder, J. Med. Chem., 2012, 55, 7346 CrossRef CAS PubMed.
- E. H. Demont, C.-w. Chung, R. C. Furze, P. Grandi, A.-M. Michon, C. Wellaway, N. Barrett, A. M. Bridges, P. D. Craggs, H. Diallo, D. P. Dixon, C. Douault, A. J. Emmons, E. J. Jones, B. V. Karamshi, K. Locke, D. J. Mitchell, B. H. Mouzon, R. K. Prinjha, A. D. Roberts, R. J. Sheppard, R. J. Watson and P. Bamborough, J. Med. Chem., 2015, 58, 5649 CrossRef CAS PubMed.
- P. Bamborough, C.-w. Chung, R. C. Furze, P. Grandi, A.-M. Michon, R. J. Sheppard, H. Barnett, H. Diallo, D. P. Dixon, C. Douault, E. J. Jones, B. Karamshi, D. J. Mitchell, R. K. Prinjha, C. Rau, R. J. Watson, T. Werner and E. H. Demont, J. Med. Chem., 2015, 58, 6151 CrossRef CAS PubMed.
- P. Bamborough, C.-w. Chung, E. H. Demont, R. C. Furze, A. J. Bannister, K. H. Che, H. Diallo, C. Douault, P. Grandi, T. Kouzarides, A.-M. Michon, D. J. Mitchell, R. K. Prinjha, C. Rau, S. Robson, R. J. Sheppard, R. Upton and R. J. Watson, Angew. Chem., Int. Ed., 2016, 55, 11382 CrossRef CAS PubMed.
- M. J. Harner, B. A. Chauder, J. Phan and S. W. Fesik, J. Med. Chem., 2014, 57, 9687 CrossRef CAS PubMed.
- A. E. Fernández-Montalván, M. Berger, B. Kuropka, S. J. Koo, V. Badock, J. Weiske, V. Puetter, S. J. Holton, D. Stöckigt, A. ter Laak, P. A. Centrella, M. A. Clark, C. E. Dumelin, E. A. Sigel, H. H. Soutter, D. M. Troast, Y. Zhang, J. W. Cuozzo, A. D. Keefe, D. Roche, V. Rodeschini, A. Chaikuad, L. Díaz-Sáez, J. M. Bennett, O. Fedorov, K. V. M. Huber, J. Hübner, H. Weinmann, I. V. Hartung and M. Gorjánácz, ACS Chem. Biol., 2017, 12, 2730 CrossRef PubMed.
- P. Filippakopoulos, J. Qi, S. Picaud, Y. Shen, W. B. Smith, O. Fedorov, E. M. Morse, T. Keates, T. T. Hickman, I. Felletar, M. Philpott, S. Munro, M. R. McKeown, Y. Wang, A. L. Christie, N. West, M. J. Cameron, B. Schwartz, T. D. Heightman, N. La Thangue, C. A. French, O. Wiest, A. L. Kung, S. Knapp and J. E. Bradner, Nature, 2010, 468, 1067 CrossRef CAS PubMed.
- C.-w. Chung, H. Coste, J. H. White, O. Mirguet, J. Wilde, R. L. Gosmini, C. Delves, S. M. Magny, R. Woodward, S. A. Hughes, E. V. Boursier, H. Flynn, A. M. Bouillot, P. Bamborough, J.-M. G. Brusq, F. J. Gellibert, E. J. Jones, A. M. Riou, P. Homes, S. L. Martin, I. J. Uings, J. Toum, C. A. Clément, A.-B. Boullay, R. L. Grimley, F. M. Blandel, R. K. Prinjha, K. Lee, J. Kirilovsky and E. Nicodeme, J. Med. Chem., 2011, 54, 3827 CrossRef CAS PubMed.
- P. Workman and I. Collins, Chem. Biol., 2010, 17, 561 CrossRef CAS PubMed.
- A. M. Giannetti, in Methods Enzymol, ed. L. C. Kuo, Academic Press, Editon edn., 2011, vol. 493, pp. 169 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8ob00099a |
| ‡ These authors contributed equally to the work described in this paper. |
| This journal is © The Royal Society of Chemistry 2018 |