
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis of oligosaccharides related to galactomannans from Aspergillus fumigatus and their NMR spectral data†
V. B.
Krylov
a,
D. A.
Argunov
a,
A. S.
Solovev
a,
M. I.
Petruk
a,
A. G.
Gerbst
a,
A. S.
Dmitrenok
a,
A. S.
Shashkov
a,
J.-P.
Latgé
b and
N. E.
Nifantiev
 *a
*a
aLaboratory of Glycoconjugate Chemistry, N.D. Zelinsky Institute of Organic Chemistry, Russian Academy of Sciences, Leninsky Prospect 47, 119991 Moscow, Russia. E-mail: nen@ioc.ac.ru; Fax: (+)7-499-135-87-84; Tel: (+)7-499-135-87-84
bUnité des Aspergillus, Institut Pasteur, 25 rue du Docteur Roux, 75724 Paris Cedex 15, France
First published on 29th January 2018
Abstract
The synthesis of model oligosaccharides related to antigenic galactomannans of the dangerous fungal pathogen Aspergillus fumigatus has been performed employing pyranoside-into-furanoside (PIF) rearrangement and controlled O(5) → O(6) benzoyl migration as key synthetic methods. The prepared compounds along with some previously synthesized oligosaccharides were studied by NMR spectroscopy with the full assignment of 1H and 13C signals and the determination of 13C NMR glycosylation effects. The obtained NMR database on 13C NMR chemical shifts for oligosaccharides representing galactomannan fragments forms the basis for further structural analysis of galactomannan related polysaccharides by a non-destructive approach based on the calculation of the 13C NMR spectra of polysaccharides by additive schemes.
Introduction
Aspergillus fumigatus is an opportunistic fungal pathogen that can cause allergic bronchopulmonary aspergillosis, chronic aspergillosis and especially invasive aspergillosis (IA) – a life threatening disease with high morbidity and mortality rates among immunocompromised patients.1–3 A major carbohydrate antigen produced by A. fumigatus is the polysaccharide galactomannan.4 Its identification in a patient's serum employing a commercial sandwich enzyme immunoassay is used for the diagnosis of IA.5Galactomannan is a complex heteropolysaccharide whose samples can be highly structurally diverse depending on the conditions of A. fumigatus cultivation. Despite numerous structural studies, some inconsistencies can be seen among the published data. Latgé et al. proposed a structure for two forms4,6 of galactomannan representing in both cases an α-(1→2)- and α-(1→6)-linked poly-D-mannoside backbone bearing β-(1→5)-linked oligogalactofuranoside side chains attached to some of the mannose units via either β-(1→3)- or β-(1→6)-bonds (Fig. 1A). Recently, Shibata et al.7 also revealed the presence of the β-(1→6)-linkage within the galactofuranoside chain8 and the β-(1→2)- but not the β-(1→3)-attachment of the galactofuranoside side chain to the mannan backbone (Fig. 1B). Significant variations in the polysaccharide structure have been reported under different culture growth conditions.7
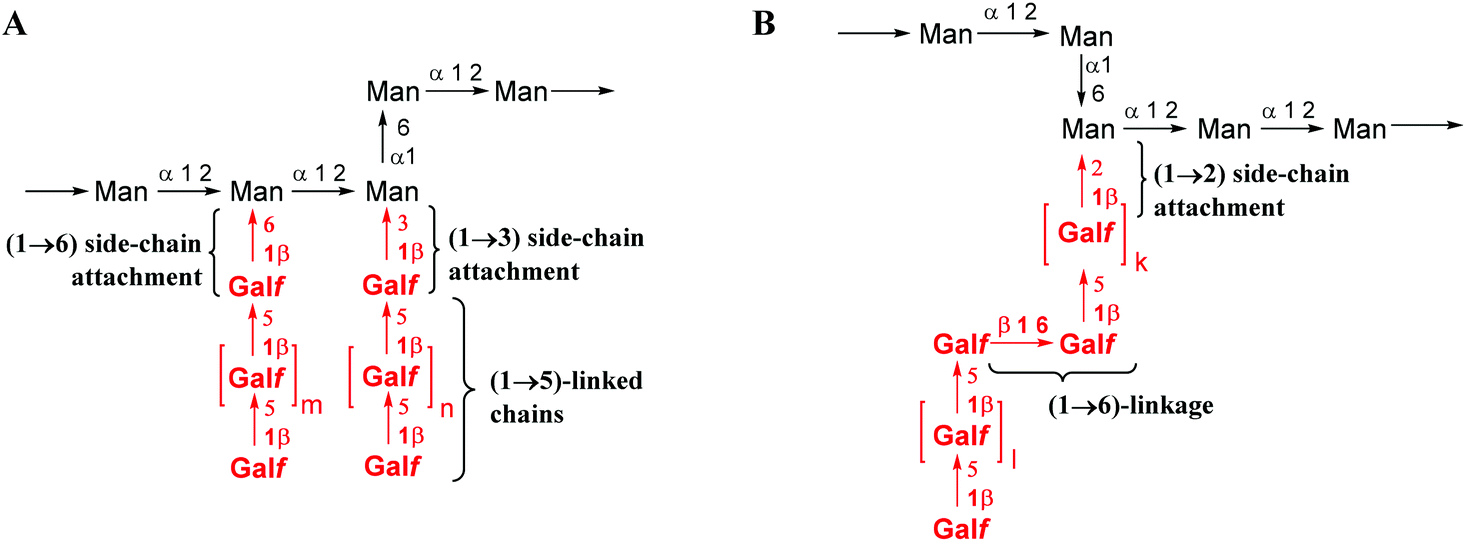 | ||
| Fig. 1 Proposed structure of galactomannan (A) reported by Latgé et al. and4 (B) additional structural elements reported by Shibata et al.7 | ||
Polysaccharides structurally related to galactomannans from A. fumigatus have been reported in other fungal species in particular Aspergillus ochraceus,9Malassezia furfur,10Malassezia pachydermatis,10Trichophyton rubrum,11Trichophyton mentagrophytes,11Paracoccidioides brasiliensis12 and others.13 This stimulates the development of sensitive methods for the structural analysis of galactomannans. NMR spectroscopy is the most potent non-destructive method used to accomplish these tasks.
The chemical syntheses of galactofuranoside-containing oligosaccharides, related to mycobacterial arabinogalactan14–16 or Aspergillus galactomannan17–19 and others,20 were previously reported. Surprisingly, there are well visible contradictions among the published 13C NMR data for very similar Galf-residues present in these oligosaccharides. For example, the 13C NMR chemical shifts of Galf-residues at the non-reducing end linked via (1→5)-bonds to the next Galf-ring reported by Lowary et al.,14 Reynolds et al.15 and Gallo-Rodriguez et al.17 (see compounds A–C in Table 1) are quite different. Similar deviations are found in data published for the terminal unit in Galf-(1→6)-Galf-fragments in oligosaccharides D and E. These examples of spectral discrepancy cannot be attributed to the differences of the used spectra recording conditions (temperature and concentration) or the remote influence of structural fragments along the oligosaccharide chains and probably are the results of the tentative assignment of NMR signals. Thus, a careful revision of the previous data is required for their further use in the NMR analysis of fungal galactomannans.
| # | δ(C1) | δ(C2) | δ(C3) | δ(C4) | δ(C5) | δ(C6) |
|---|---|---|---|---|---|---|
| a Not described. A 15 β-D-Galf-(1→5)-β-D-Galf-OC8H17. B14 β-D-Galf-(1→5)-β-D-Galf-(1→6)-β-D-Galf-(1→5)-β-D-Galf-OC8H17. C17 β-D-Galf-(1→5)-β-D-Galf-(1→5)-β-D-Galf-STol. D15 β-D-Galf-(1→6)-β-D-Galf-OC8H17. E14 β-D-Galf-(1→6)-β-D-Galf-(1→5)-β-D-Galf-OC8H17. | ||||||
| A 15 | 107.71 | 81.58 | 76.74 | 82.81 | 70.77 | 62.07 |
| B 14 | 108.7 | 82.7 | 77.6 | 85.5 | 76.7 | 61.9 |
| C 17 | 109.0 | 83.6 | 78.7 | 84.6 | n.d.a | 64.2 |
| D 15 | 108.25 | 81.24 | 77.08 | 83.31 | 71.09 | 63.10 |
| E 14 | 107.9 | 81.2 | 77.6 | 83.9 | 70.4 | 61.9 |
Herein, we report an NMR spectral study of a series of synthetic galactomannan related oligosaccharides 1–13 (Fig. 2). This work is aimed towards the development of NMR database suitable for further structural analysis of Aspergillus galactomannan and structurally related oligo- and polysaccharides. The synthesis of compounds 1–6, 8 and 13 is also reported, while the preparation of oligosaccharides 7 and 9–12 was previously published.21,22
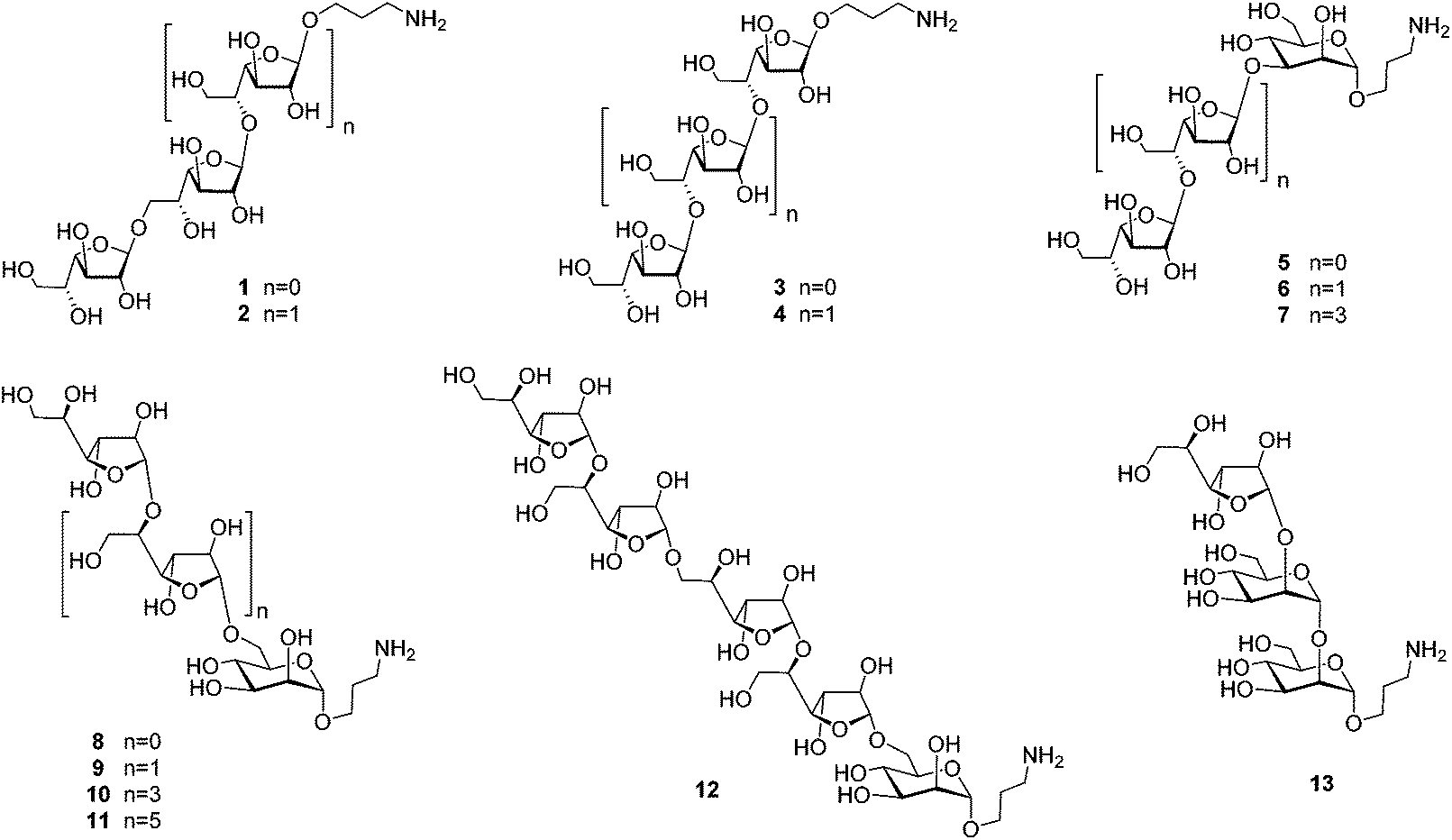 | ||
| Fig. 2 Studied library of oligosaccharides related to galactomannans of A. fumigatus. | ||
Results and discussion
Synthesis of oligosaccharides
The investigation of fine correlation between a galactomannan structure and NMR chemical shift values requires a representative series of model oligosaccharides with strictly defined composition containing different fragments of studied polysaccharide. Previously,21 we reported the synthesis of two galactomannan related pentasaccharides 7 and 10 and oligosaccharides 9, 11 and 12.22 In the present work, additionally required models such as homo-galactofuranosides 1–4 containing both β-(1→5)- and β-(1→6)-linked galactofuranosyl units and heterosaccharides 5, 6, 8 and 13 have been obtained (Fig. 2). For their synthesis, glycosyl donor 16![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 22 was chosen as a key galactofuranosyl building block. Its main feature was the presence of Fmoc-protection at O(6) which could be selectively removed by deblocking either a 6-OH or 5-OH group depending on the reaction conditions.22 This made building block 16 suitable for assembling both types of linkages in the oligogalactofuranosyl chains.
22 was chosen as a key galactofuranosyl building block. Its main feature was the presence of Fmoc-protection at O(6) which could be selectively removed by deblocking either a 6-OH or 5-OH group depending on the reaction conditions.22 This made building block 16 suitable for assembling both types of linkages in the oligogalactofuranosyl chains.
The synthesis of glycosyl donor 16 was performed according to the procedure using pyranoside-into-furanoside (PIF) rearrangement,23,24 first described in 2014. Starting pyranoside 14 was initially transformed into the corresponding furanoside 15 and then subjected to subsequent per-O-benzoylation, deallylation and imidation (15 → 16). The glycosylation of 3-trifluoroacetamidopropanol with donor 16 in the presence of TMSOTf gave 17, a precursor to disaccharides 1 and 3 with β-(1→6)- and β-(1→5)-bonds, respectively (Scheme 1).
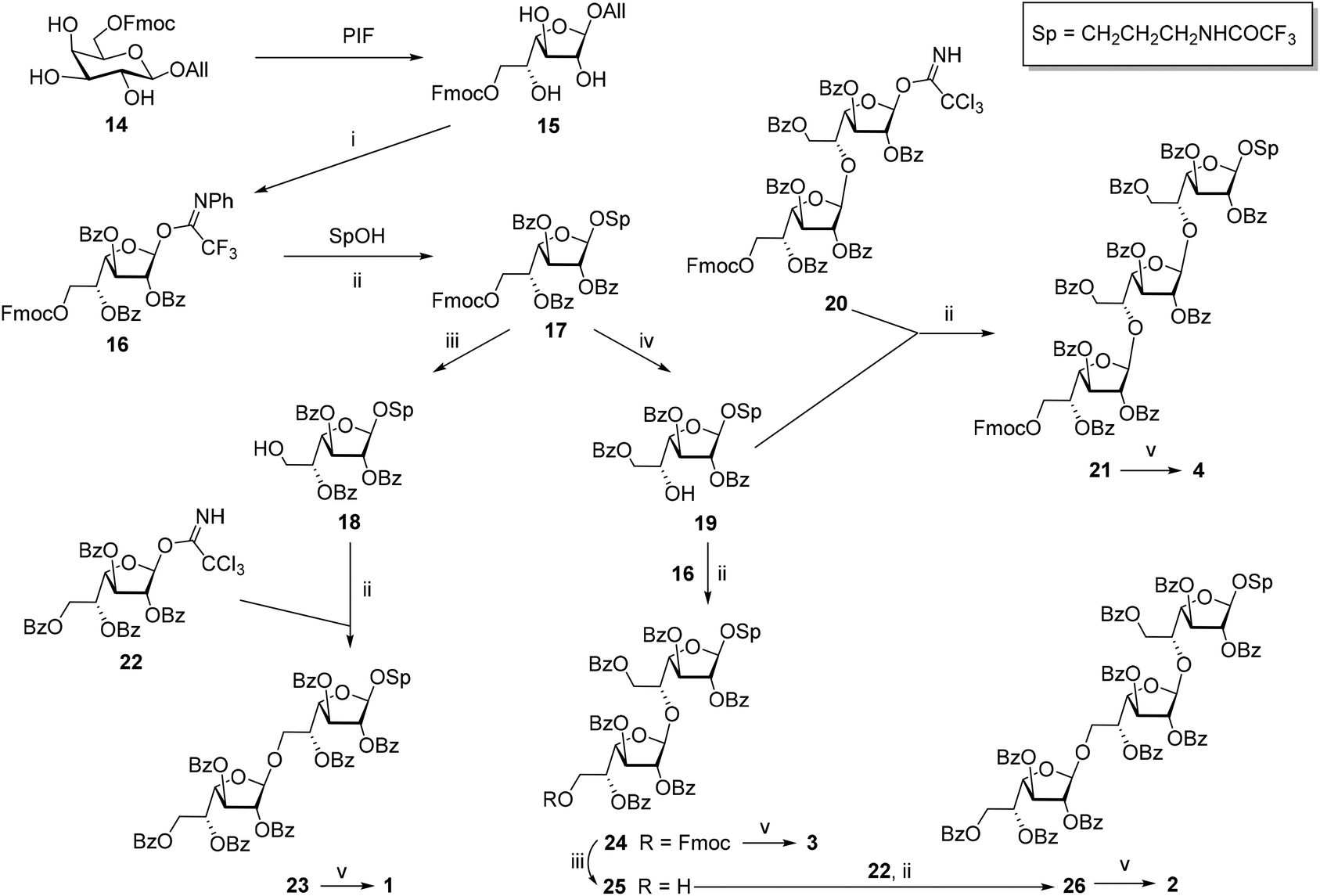 | ||
| Scheme 1 Synthesis of the model oligosaccharides 1–4. Reagents and conditions: PIF: (1) Py·SO3, HSO3Cl, DMF, 20 °C, 16 h, then an excess of NH4HCO3 aq.; (2) IR-120(H+), dioxane–DMF, 60 °C, 45 min, 66% on 2 steps; (i) (1) BzCl, Py, CH2Cl2, 20 °C, 3 h; (2) PdCl2, MeOH:CH2Cl2, 20 °C, 2.5 h; (3) CF3C(NPh)Cl, Cs2CO3, CH2Cl2, 20 °C, 1 h, 60% on 3 steps; (ii) TMSOTf, MS300 AW, CH2Cl2, −80 → −10 °C, 50 min, 87% for 17, 96% for 21, 71% for 23, 94% for 24, 76% for 26; (iii) morpholine, DMF, rt, 35 min, 86% for 18, 78% for 25; (iv) pyrrolidine, CH2Cl2, rt, 40 min, 92%; (v): MeONa, MeOH–H2O, rt, overnight, 82% for 1, 89% for 2, 83% for 3, 89% for 4. | ||
The removal of Fmoc-protection in monosaccharide 17 by treatment with morpholine in DMF gave product 18 with the free OH-group at C-6, which was further glycosylated with imidate 2225 yielding β-linked disaccharide 23. The configuration of the newly formed bond was confirmed by the singlet shape of the H(1)′-signal (JH1′,H2′ < 1.0 Hz) and the characteristic low-field chemical shift of C(1)′ (105.86 ppm) in the NMR spectra. One-step deblocking of the obtained disaccharide 23 gave target compound 1.
Alternatively, the removal of Fmoc-protection in 17 under conditions activating O(5) → O(6) benzoyl migration22 (pyrrolidine in CH2Cl2) afforded 5-OH derivative 19 in 92% yield. The structure of product 19 was confirmed by 1H NMR chemical shifts (4.43 ppm for H(5), and 4.51 and 4.62 ppm for H(6) in 19vs. 5.61 ppm for H(5) and 4.05 ppm for both H(6) in 18). The glycosylation of acceptor 19 with donors 16 and 2022 in the presence of TMSOTf gave β-linked di- 24 and trisaccharide 21, respectively. Following the deblocking of the obtained oligosaccharides resulted in the formation of target compounds 3 and 4.
Disaccharide 24 was also used for the preparation of trisaccharide 2 with alternating (1→6)- and (1→5)-linkages. For this purpose, Fmoc-protection in 24 was removed under conditions preventing benzoate migration (morpholine in DMF) to give acceptor 25 with the free hydroxyl group at O′(6) which was further glycosylated with imidate 22. The deblocking of the obtained protected derivative 26 afforded target trisaccharide 2.
Hetero-oligosaccharides 5, 6, 8 and 13 were prepared using mannoside acceptors 27, 30 and 32 (Scheme 2). For the preparation of the oligosaccharides with the (1→3)-linkage between galactofuranose and mannose, the glycosylations of acceptor 2721 with either monosaccharide 2225 or disaccharide 2022 donors were performed. The disaccharide with the (1→6)-linkage was prepared by the glycosylation of acceptor 3021 with donor 22.22 The trisaccharide with the (1→2)-linkage was prepared by the glycosylation of acceptor 3226 with donor 16. All the couplings proceeded with good yields and excellent β-stereoselectivity. The deblocking of the resulting protected precursors 28, 29, 31 and 33 gave target model oligosaccharides 5, 6, 8 and 13, respectively.
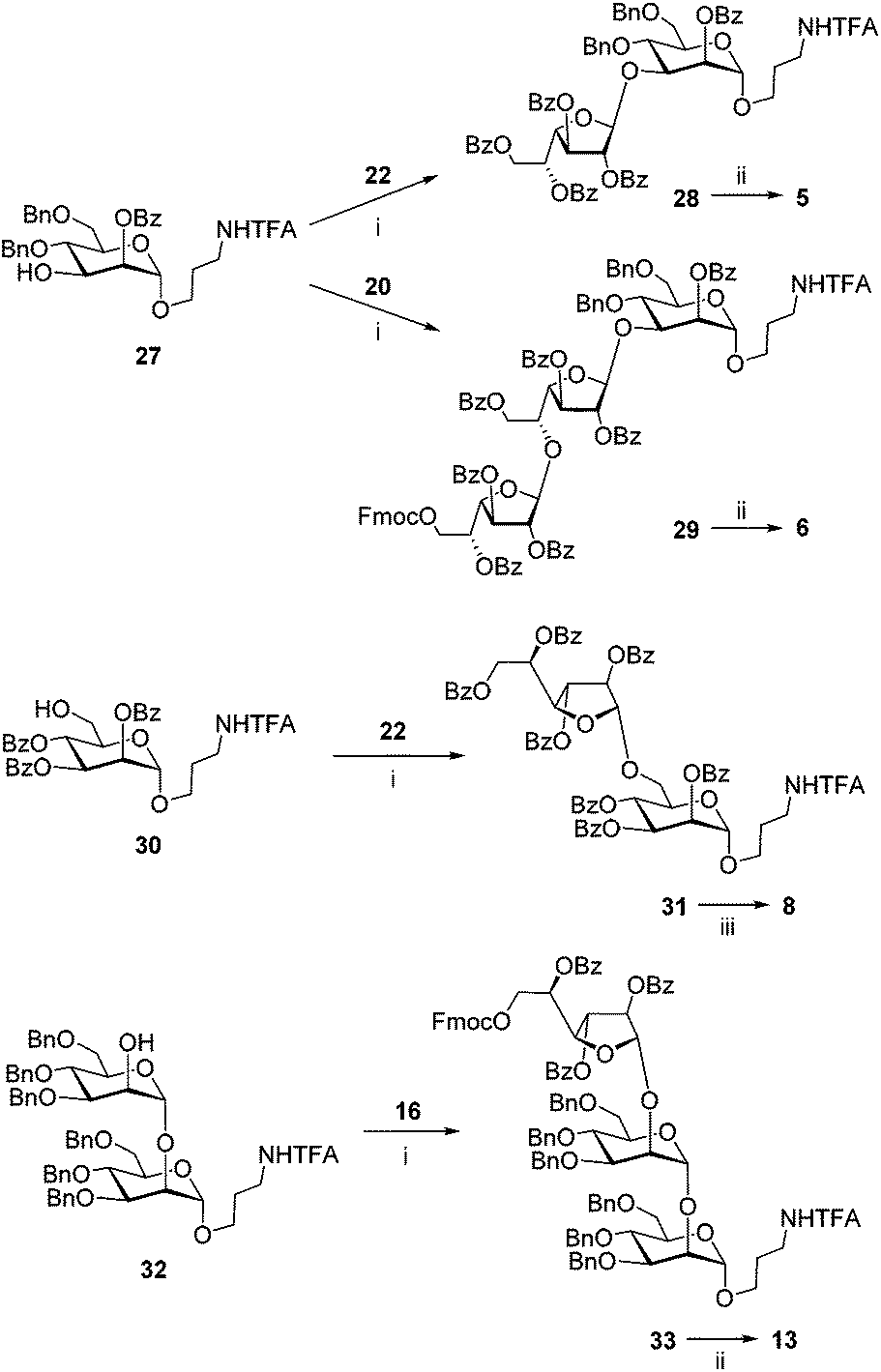 | ||
| Scheme 2 Synthesis of the model oligosaccharides 5, 6, 8 and 13. Reagents and conditions: (i) TMSOTf, MS300 AW, CH2Cl2, −80 → −10 °C, 50 min, 95% for 28, 67% for 29, 90% for 31, 77% for 33; (ii) (1) H2, Pd/C, EtOAc, MeOH, rt, 1 h; (2) NaOH, MeOH–H2O, rt, 1 h, 62% for 5 80% for 6, 80% for 13; (iii) NaOMe, MeOH–H2O, rt, overnight, 77%. | ||
NMR analysis of oligosaccharides
A complete signal assignment in the 1H and 13C NMR spectra of oligosaccharides 1–13 was successfully performed employing 2D NMR experiments: COSY, TOCSY, ROESY, HSQC, and HMBC. The chemical shifts were referenced to CH3CN (δ1H – 2.06; 13C – 1.47 ppm) used as an internal standard. The 13C chemical shifts of the studied compounds are summarized in Table 2 (for 1H chemical shifts see Table S1 in the ESI†). The 13C chemical shifts were measured using 1H-decoupled 1D 13C NMR spectra, and the 1H chemical shifts of overlapping signals were established from 2D 1H–13C HSQC spectra using the centres of the corresponding correlation signals.| # | Unit | C(1) | C(2) | C(3) | C(4) | C(5) | C(6) |
|---|---|---|---|---|---|---|---|
a Aglycon signals δ (ppm): Manp-O![[C with combining low line]](https://www.rsc.org/images/entities/i_char_0043_0332.gif) H2-CH2-CH2-NH2 – 65.68, Manp-OCH2-H2-CH2-NH2 – 27.21, Manp-OCH2-CH2-H2-NH2 – 38.13; Galf-OH2-CH2-CH2-NH2 – 66.31, Galf-OCH2-H2-CH2-NH2 – 27.19, Galf-OCH2-CH2-H2-NH2 – 38.44, (internal standard H3CN – 1.47);
b Two internal residues.
c Four internal residues.
d,e The assignment in this pair of chemical shifts may be reversed. H2-CH2-CH2-NH2 – 65.68, Manp-OCH2-H2-CH2-NH2 – 27.21, Manp-OCH2-CH2-H2-NH2 – 38.13; Galf-OH2-CH2-CH2-NH2 – 66.31, Galf-OCH2-H2-CH2-NH2 – 27.19, Galf-OCH2-CH2-H2-NH2 – 38.44, (internal standard H3CN – 1.47);
b Two internal residues.
c Four internal residues.
d,e The assignment in this pair of chemical shifts may be reversed. |
|||||||
| 1 | β-D-Galf-OH | 101.57 | 81.92 | 76.35 | 82.58 | 71.27 | 63.33 |
| β-D-Galf-(1→ | 108.40 | 81.58 | 77.40 | 83.62 | 71.45 | 63.30 | |
| →6)-β-D-Galf-O(CH2)3NH2 | 107.68 | 81.30 | 77.17 | 84.04 | 70.23 | 69.58 | |
| 2 | β-D-Galf-(1→ | 108.49 | 81.59 | 77.37 | 83.56 | 71.45 | 63.34 |
| →6)-β-D-Galf-(1→ | 107.51 | 81.83 | 77.28 | 83.67 | 70.16 | 69.89 | |
| →5)-β-D-Galf-O(CH2)3NH2 | 107.69 | 81.44 | 77.08 | 82.83 | 76.48 | 61.35 | |
| 3 | β-D-Galf-(1→ | 107.58 | 81.84 | 77.17 | 83.38 | 71.20 | 63.38 |
| →5)-β-D-Galf-O(CH2)3NH2 | 107.65 | 81.43 | 76.99 | 82.69 | 76.53 | 61.41 | |
| 4 | β-D-Galf-(1→ | 107.69 | 81.96d | 77.15 | 83.33 | 71.19 | 63.43 |
| →5)-β-D-Galf-(1→ | 107.50 | 81.89e | 77.08d | 82.29 | 76.23 | 61.70 | |
| →5)-β-D-Galf-O(CH2)3NH2 | 107.69 | 81.47 | 77.06e | 82.75 | 76.66 | 61.39 | |
| 5 | β-D-Galf-(1→ | 105.02 | 81.97 | 77.70 | 83.66 | 71.45 | 63.44 |
| →3)-α-D-Manp-O(CH2)3NH2 | 100.20 | 67.28 | 76.10 | 65.66 | 73.43 | 61.45 | |
| 6 | β-D-Galf-(1→ | 107.81 | 82.00 | 77.20 | 83.36 | 71.25 | 63.48 |
| →5)-β-D-Galf-(1→ | 105.07 | 81.94 | 77.62 | 82.77 | 76.60 | 61.86 | |
| →3)-α-D-Manp-O(CH2)3NH2 | 100.25 | 67.34 | 76.26 | 65.68 | 73.45 | 61.69 | |
| 7 | β-D-Galf-(1→ | 107.74 | 82.02d | 77.06d | 83.37 | 71.24 | 63.48 |
| →5)-β-D-Galf-(1→b | 107.74 | 82.05e | 77.12e | 82.19 | 76.22 | 61.69 | |
| 77.19 | 82.26 | 76.36 | |||||
| →5)-β-D-Galf-(1→ | 105.12 | 81.94 | 77.65 | 82.73 | 76.63 | 61.79 | |
| →3)-α-D-Manp-O(CH2)3NH2 | 100.24 | 67.36 | 76.29 | 65.70 | 73.45 | 61.84 | |
| 8 | β-D-Galf-(1→ | 108.41 | 81.50 | 77.31 | 83.56 | 71.44 | 63.28 |
| →6)-α-D-Manp-O(CH2)3NH2 | 100.39 | 70.45 | 71.03 | 67.25 | 72.30 | 67.41 | |
| 9 | β-D-Galf-(1→ | 107.63 | 81.87 | 77.17 | 83.33 | 71.18 | 63.43d |
| →5)-β-D-Galf-(1→ | 108.37 | 81.65 | 77.23 | 82.49 | 76.50 | 61.55e | |
| →6)-α-D-Manp-O(CH2)3NH2 | 100.47 | 70.52 | 71.13 | 67.27 | 72.39 | 67.45 | |
| 10 | β-D-Galf-(1→ | 107.67 | 81.86d | 77.10 | 83.26 | 71.11 | 63.40 |
| →5)-β-D-Galf-(1→b | 107.53 | 81.95e | 76.98 | 82.14 | 76.15 | 61.73 | |
| 107.57 | 81.97 | 77.10 | 76.27 | ||||
| →5)-β-D-Galf-(1→ | 108.37 | 81.66 | 77.29 | 82.46 | 76.60 | 61.51 | |
| →6)-α-D-Manp-O(CH2)3NH2 | 100.46 | 70.51 | 71.15 | 67.26 | 72.37 | 67.43 | |
| 11 | β-D-Galf-(1→ | 107.68 | 81.87d | 77.10 | 83.27 | 71.16 | 63.41 |
| →5)-β-D-Galf-(1→c | 107.54 | 81.96e | 76.98 | 82.15 | 76.25 | 61.70 | |
| 107.58 | 81.98 | 77.04 | 76.22 | 61.73 | |||
| 77.11 | |||||||
| →5)-β-D-Galf-(1→ | 108.38 | 81.67 | 77.29 | 82.46 | 76.59 | 61.50 | |
| →6)-α-D-Manp-O(CH2)3NH2 | 100.47 | 70.51 | 71.13 | 67.25 | 72.38 | 67.44 | |
| 12 | β-D-Galf-(1→ | 107.68 | 81.85 | 77.13 | 83.26 | 71.15 | 63.42 |
| →5)-β-D-Galf-(1→ | 108.43 | 81.69d | 77.32 | 82.54d | 76.47 | 61.63 | |
| →6)-β-D-Galf-(1→ | 107.51 | 81.85 | 77.23 | 83.67 | 70.20 | 69.91 | |
| →5)-β-D-Galf-(1→ | 108.43 | 81.63e | 77.32 | 82.49e | 76.36 | 61.50 | |
| →6)-α-D-Manp-O(CH2)3NH2 | 100.47 | 70.51 | 71.13 | 67.26 | 72.37 | 67.44 | |
| 13 | β-D-Galf-(1→ | 106.40 | 81.64 | 77.17 | 83.49 | 71.29 | 63.36 |
| →2)-α-D-Manp-(1→ | 100.25 | 75.42 | 70.13 | 67.80 | 74.03 | 61.64 | |
| →2)-α-D-Manp-O(CH2)3NH2 | 98.81 | 79.43 | 70.79 | 67.59 | 73.51 | 61.60 | |
It can be noted that the assignment of signals summarized in Table 2 has certain, often quite substantial deviations from the previously reported chemical shifts (see Introduction), in particular for the signals of C(1), C(6) and some other carbons. Typically, in the previous papers dedicated to the synthesis of Galf-containing oligosaccharides, NMR signal assignment procedures were described only briefly and the resulting attribution was probably tentative or based on possibly erroneous data from older studies.
The studied series of oligosaccharides (1–13) contained 10 types of galactofuranosyl residues which differed in (1) the type of the glycoside linkage in them and (2) the type of the carbohydrate unit as the aglycon (Fig. 3). The variation in chemical shifts among different compounds for the same type of residue did not exceed ±0.1 ppm. The analysis of chemical shifts did not reveal any chain length influence or influence from distant residues which allowed the use of these data for the spectral investigation of different types of oligo- and polysaccharides.
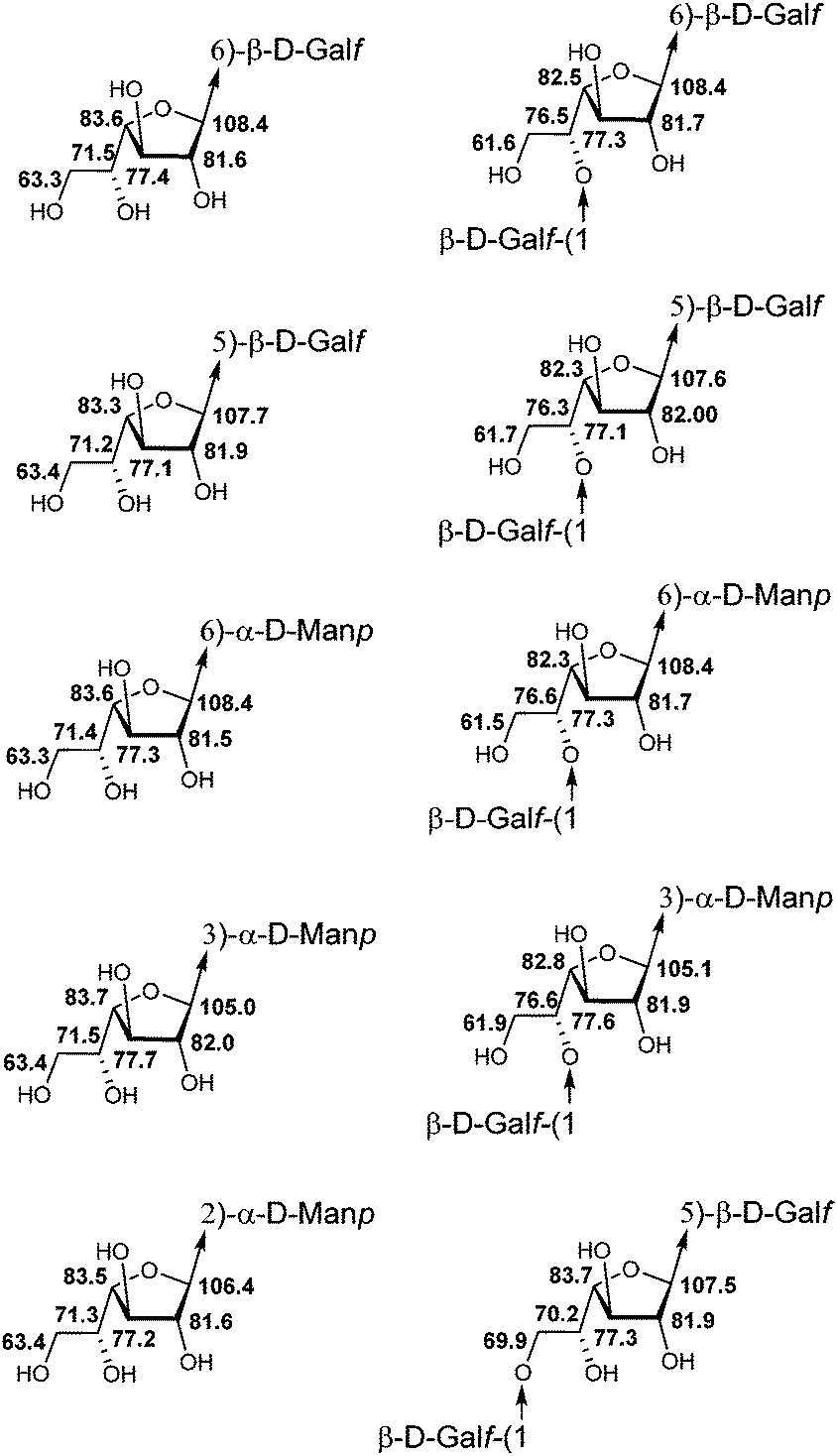 | ||
| Fig. 3 Averaged 13C NMR chemical shifts (δ, ppm; D2O) of galactofuranosyl residues of different fragments of galactomannan chains. | ||
An important feature for the analysis of the NMR spectra of large carbohydrate chains is the determination of 13C NMR glycosylation effects.27 They are the differences in chemical shifts for the corresponding carbon atoms within a residue taken as a part of an oligo- or polysaccharide and for the same residue in the parent mono- or oligosaccharide, non-glycosylated at the position under consideration. The differences in chemical shifts for atoms involved directly in the glycosidic linkage are called the α-effects of glycosylation.
The differences in chemical shifts for atoms bonded to the substituted carbon are the β-effects of glycosylation. To calculate the glycosylation effects, the 13C NMR chemical shifts of β-galactofuranose (detected as minor signals in the spectrum of D-galactose solution in D2O), α-D-Man-O(CH2)3NH228 and α-D-Man-(1→2)-α-D-Man-O(CH2)3NH226 were used. For example, Fig. 4 shows two monosaccharides used for the calculation of the glycosylation effects in the spectrum of disaccharide 5.
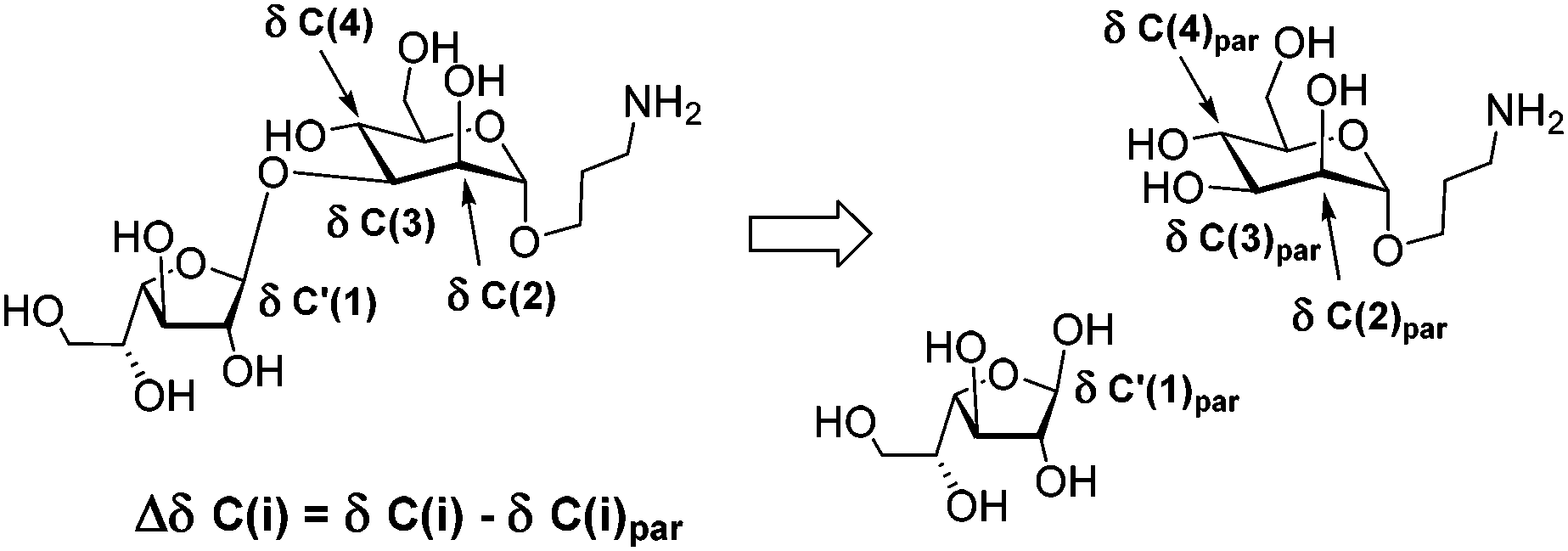 | ||
| Fig. 4 Illustration of the calculation of 13C NMR glycosylation effects (Δδ) in disaccharide 5. | ||
The α-effects for anomeric carbon atoms are most informative for the elucidation of monosaccharide sequences in oligo- or polysaccharide chains. They are dependent on the direction of the linkage and the stereochemical configuration of the anomeric center involved, and are not influenced by the type of substitution in the glycosylating residue under consideration. The exceptions to this rule are 2-O-substituted residues due to the interference of the β-effects of the substitution with the α-effects of glycosylation.
The C(1) glycosylation effects for differently linked β-Galf residues are shown in Table 3. It can be seen that their values are different for 2- (4.8 ppm, entry 1), 3- (3.4 ppm, entry 2), 5- (6.1 ppm, entry 4) and 6-linked (6.8 ppm, entries 3 and 5) glycosylating furanoses, but the two latter disaccharides are not distinguishable on the basis of the glycosylation effects for C(1). Unfortunately, H(1) chemical shifts for the glycosylating Galf residues in these two disaccharide fragments are also very close (5.02–5.04 ppm, Table S1†). In this case, more information can be given by the α-effects of substitution at C(6) of the glycosylated sugar residues, α-D-Manp (5.8 ppm) and β-D-Galf (6.5 ppm). Additionally, the positions and integral intensities of H(6a,b)/C(6) correlation peaks in the HSQC spectra can also be used for the quantitative assessment of the relative content of the two disaccharide fragments in the polymer. However, this two-dimensional approach requires full analysis and certain assignment of NMR spectra.
| # | Linkage | α′-Effect, C(1) | α-Effect | β-Effects |
|---|---|---|---|---|
| 1 | β-D-Galf-(1→2)-α-D-Man | 4.8 | (C2): 4.9 | (C1): −2.7 |
| (C3): −0.8 | ||||
| 2 | β-D-Galf-(1→3)-α-D-Man | 3.4 | (C3): 4.9 | (C2): −3.3 |
| (C4): −1.7 | ||||
| 3 | β-D-Galf-(1→6)-α-D-Man | 6.8 | (C6): 5.8 | (C5): −1.1 |
| 4 | β-D-Galf-(1→5)-β-D-Galf | 6.1 | (C5): 5.0 | (C4): −1.0 |
| (C6): −1.7 | ||||
| 5 | β-D-Galf-(1→6)-β-D-Galf | 6.8 | (C6): 6.5 | (C5): −1.0 |
NMR analysis of natural polysaccharides
All types of differently substituted Galf residues were characterized by specific 13C chemical shift patterns and 13C NMR glycosylation effects which permitted their unambiguous identification in the 13C-NMR spectra of natural polysaccharides. These signals could be regarded as “structure reporting” signals29,30 suitable for the identification of the characteristic fragments of galactomannans and related polysaccharides which are usually available in very limited amounts and thus produce spectra with low signal intensities.For example, the C(1) resonances of β-galactofuranoside residues represented the highest interest as structure reporting signals because they are located in a specific range (105–109 ppm) different from the anomeric signals of mannopyranoside units. The chemical shifts of β-galactofuranoside residues were strongly influenced by the nature of monosaccharide substituents at the anomeric site connected via β-(1→2)-, β-(1→3)-, β-(1→5)-, or β-(1→6)-linkages as illustrated by 13C NMR data for model oligosaccharides (Table 4). They coincided well with the corresponding chemical shifts in the spectrum of galactomannan.7,11 A small deviation (of 0.9 ppm) was observed only in the case of anomeric carbon in the β-galactofuranoside unit connected via the (1→3)-linkage to the (1→2)-mannosylated D-Man residue of the mannan backbone (see entry 2 in Table 4) and could be attributed to 2,3-vicinal branching.
| # | Terminal β-D-Galf-units with different aglycon counterparts | C(1) in model oligo-saccharide | C(1) in poly-saccharide (lit. data) |
|---|---|---|---|
| a Each chemical shift taken from ref. 7 has been decremented by 0.20 ppm for consistency with our chemical shift reference. b (1→6)-Mannosylated D-Man unit. c (1→2)-Mannosylated D-Man unit. | |||
| 1 | β-D-Galf-(1→2)-D-Man | 106.4 | 106.57a |
| 2 | β-D-Galf-(1→3)-D-Man | 105.0 | 105.411b |
| 105.911c |
|||
| 3 | β-D-Galf-(1→5)-D-Galf | 107.6 | 107.77a |
| 4 | β-D-Galf-(1→6)-D-Galf/Man | 108.4 | 108.57a |
To illustrate the applicability of the above mentioned structure reporting signals for the investigation of fungal galactomannans, we analysed the spectrum of galactomannan reported by Latgé et al.4,6 Despite the fact that the presence of the β-D-Galf-(1→3)-D-Man fragment was claimed by the authors, the signal of the anomeric carbon of the corresponding β-D-Galf-unit was not observed in the spectrum (Fig. 1 in ref. 4). Hence, in the present study, the 13C NMR spectrum was recorded for the galactomannan obtained under the reported conditions4 and it contained the discussed signal. Due to its low intensity in the 1D 13C NMR spectrum, the presence of the β-(1→3)-linked galactofuranoside unit was detected by a 2D HSQC spectral protocol (Fig. 5A).
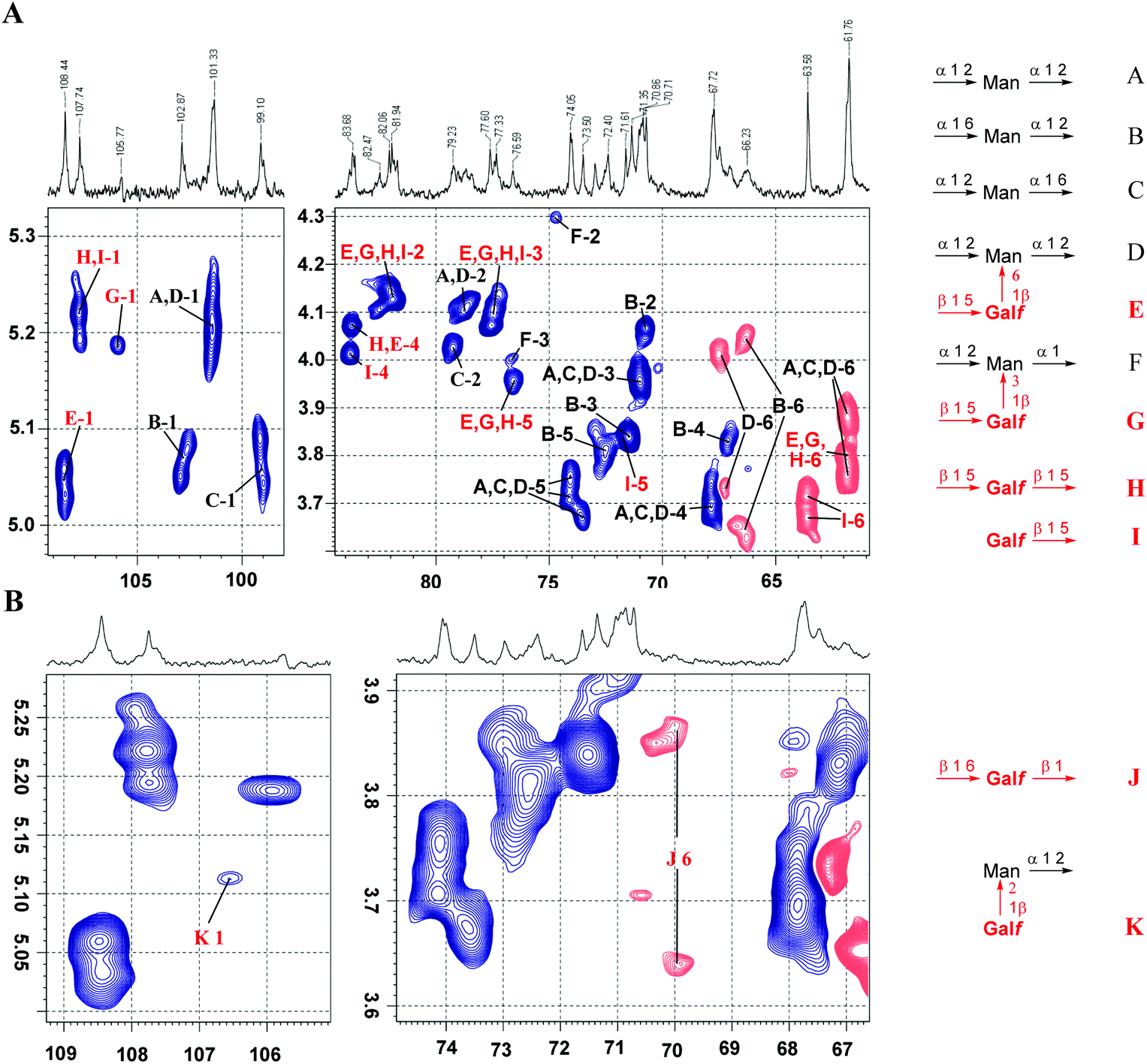 | ||
| Fig. 5 Application of structure reporting signals for the analysis of natural galactomannan from A. fumigatus. (A) Overview of the HSQC spectrum with the assignment of major signals: anomeric (left) and polyol (right) region. (B) Analysis of low intensity signals reporting the structural elements as shown in Fig. 1B. The blue color indicates the positive phase signals referred to as CH or CH3, whereas the red color indicates the negative phase signals referred to as CH2 groups. | ||
The full assignment of the signals in the HSQC spectrum (Fig. 5A) revealed the presence of all the types of carbohydrate residues reported by Latgé et al.4,6 However, the careful analysis of low intensity signals (Fig. 5B) suggested the presence of a very minor portion of structural elements reported by Shibata et al.:7 the (1→2)-linkage between β-D-Galf and α-D-Man residues and O(6)-glycosylated β-D-Galf residues (see Fig. 1B). Their presence was confirmed by characteristic 1H and 13C chemical shifts that perfectly matched the data obtained for the corresponding model compounds 13 and 12.
Conclusions
A series of model oligosaccharides representing key structural fragments of Aspergillus galactomannans was synthesised and studied by NMR spectroscopy. The obtained NMR database on 13C NMR chemical shifts for different galactomannan fragments is shown to be useful for the verification of the previously established structures and further development of a non-destructive approach towards the structural analysis of galactomannan related polysaccharides based on the calculation of their 13C NMR spectra by additive schemes.31–35Experimental
General methods
All solvents were distilled and dried if necessary according to standard procedures (CH2Cl2, MeOH, toluene, and EtOAc) or purchased as dry (DMF and CH3CN, Sigma-Aldrich). Commercial chemicals were used without purification unless noted. All glycosylation reactions were carried out using dry solvents under an Ar atmosphere. Molecular sieves for glycosylation reactions were activated prior to application at 180 °C under vacuum from an oil pump for 2 h. Analytical thin-layer chromatography (TLC) was performed on Silica Gel 60 F254 aluminium sheets (Merck), and visualization was accomplished using UV light or by charring at ∼150 °C with 10% (v/v) H3PO4 in ethanol. Column chromatography was performed on Silica Gel 60, 40–63 μm (Merck). Gel-filtration was performed on a TSK-40 HW(S) column (400 × 17 mm) by elution with 0.1 M AcOH in water at a flow rate of 0.5 mL min−1. 1H and 13C NMR spectra were recorded on Bruker AMX-400, Bruker DRX-500, and Bruker AV-600 spectrometers. The spectra of compounds 1–13 were recorded in a Shigemi NMR microtube in D2O (0.25 mL). Chemical shifts were referenced to residual solvent signals. For samples in D2O, acetonitrile was used as an internal standard. The average sample concentration was 30 μmol mL−1. The signal assignment in the 1H and 13C NMR spectra was made using COSY, TOCSY, and 1H–13C HSQC and HMBC techniques. High-resolution mass spectra (HR MS) were recorded on a Bruker micrOTOF II instrument using electrospray ionization (ESI). The measurements were performed in a positive ion mode (interface capillary voltage −4500 V); mass range from m/z 50 to m/z 3000 Da; external or internal calibration was made with Electrospray Calibrant Solution (Fluka). A syringe injection was used for solutions in a mixture of acetonitrile and water (50:50 v/v, flow rate 3 μL min−1). Nitrogen was applied as a dry gas; the interface temperature was set at 180 °C.
:1 → 3:1) to give the glycosylation product.
:EtOAc 10:1). 1H NMR (400 MHz, CDCl3) δ 8.10–7.13 (m, 23H, Fmoc, Bz), 5.98–5.92 (m, 1H, H-5), 5.65 (d, J = 5.5 Hz, 1H, H-3), 5.39 (s, 1H, H-2), 5.29 (s, 1H, H-1), 4.71–4.62 (m, 3H, H-4, 2 × H-6), 4.39 (dd, J = 10.3, 7.6 Hz, 1H, Fmoc-CH2), 4.31 (dd, J = 10.3, 7.5 Hz, 1H, Fmoc-CH2), 4.21 (t, J = 7.5 Hz, 1H, Fmoc-CH), 3.98–3.94 (m, 1H, OCHH′CH2), 3.67–3.57 (m, 2H, OCHH′CH2, CH2CHH′NH), 3.54–3.48 (m, 1H, CH2CHH′NH), 2.01–1.90 (m, 2H, CH2CH2CH2). 13C NMR (101 MHz, CDCl3) δ 133.82 (Ar), 133.75 (Ar), 133.54 (Ar), 130.18 (Ar), 130.01 (Ar), 128.64 (Ar), 128.02 (Ar), 127.31 (Ar), 125.35 (Ar), 125.30 (Ar), 120.15 (Ar), 106.53 (C-1), 82.90 (C-2), 81.22 (C-4), 77.29 (C-3), 70.42 (Fmoc-CH2), 70.10 (C-5), 66.61 (OCH2CH2), 66.23 (C-6), 46.75 (Fmoc-CH), 38.21 (CH2CH2NH), 28.29 (CH2CH2CH2). HRMS (ESI): M = C47H40F3NO12. Calcd m/z for [M + Na]+ 890.2395, found 890.2386.
:1 → 3:1) to afford the 6-OH product.
:EtOAc 5:1). 1H NMR (400 MHz, CDCl3) δ 8.07 (d, J = 7.3 Hz, 2H, o-Ph), 8.03 (d, J = 7.3 Hz, 2H, o-Ph), 7.95 (d, J = 7.2 Hz, 2H, o-Ph), 7.61–7.28 (m, 9H, Ph), 5.66–5.58 (m, 2H, H-3, H-5), 5.41 (d, J = 1.5 Hz, 1H, H-2), 5.30 (s, 1H, H-1), 4.72 (dd, J = 5.5, 3.7 Hz, 1H, H-4), 4.10–3.99 (m, 2H, H-6a, H-6b), 3.95 (dt, J = 10.5, 5.2 Hz, 1H, OCH2), 3.70–3.47 (m, 3H, OCH2, CH2N), 2.69 (br. s, 1H, OH), 2.02–1.86 (m, 2H, OCH2CH2CH2N). 13C NMR (100 MHz, CDCl3) δ = 166.23 (PhC(O)), 165.88 (PhC(O)), 133.69, 133.64, 133.34, 129.91, 129.88, 129.85, 129.48, 128.82, 128.74, 128.53, 128.43 (Ar), 106.24 (C-1), 82.78 (C-2), 81.41 (C-4), 77.26 (C-3), 73.06 (C-5), 66.19 (OCH2), 61.87 (C-6), 37.90 (CH2N), 28.30 (OCH2CH2CH2N). HRMS (ESI): M = C32H30F3NO10. Calcd m/z for [M + Na]+ 668.1714, found 668.1707.
:EtOAc 5:1) to afford the compound 19 (33 mg, 92%). Rf = 0.17 (toluene:EtOAc 5:1). 1H NMR (600 MHz, CDCl3) δ 8.12–8.00 (m, 6H, arom.), 7.63–7.39 (m, 9H, arom.), 7.30–7.20 (m, 2H, arom.), 5.72 (dd, J = 5.6, 2.2 Hz, 1H, H-3), 5.43 (d, J = 2.3 Hz, 1H, H-2), 5.22 (s, 1H, H-1), 4.62 (dd, J = 11.6, 6.3 Hz, 1H, H-6), 4.51 (dd, J = 11.6, 4.7 Hz, 1H, H-6′), 4.44–4.42 (m, 1H, H-5), 4.40 (dd, J = 5.6, 2.4 Hz, 1H, H-4), 3.89 (m, 1H, OCHH′CH2), 3.63–3.53 (m, 2H, OCHH′CH2, CH2CHH′NH), 3.47–3.45 (m, 1H, CH2CHH′NH), 2.82 (d, J = 8.4 Hz, 1H, 5-OH), 1.95–1.84 (m, 2H, CH2CH2CH2). 13C NMR (150 MHz, CDCl3) δ 165.79 (arom.), 133.74 (arom.), 133.23 (arom.), 129.90 (arom.), 129.83 (arom.), 129.64 (arom.), 128.61 (arom.), 128.56 (arom.), 128.42 (arom.), 106.27 (C-1), 82.71 (C-4), 82.25 (C-2), 77.49 (C-3), 68.81 (C-5), 66.33 (OCH2CH2), 65.97 (C-6), 38.04 (CH2CH2NH), 28.19 (CH2CH2CH2). HRMS (ESI): M = C32H30F3NO10. Calcd m/z for [M + Na]+ 668.1714, found 668.1714.
:EtOAc 5:1). 1H NMR (400 MHz, CDCl3) δ 8.08–7.09 (m, 53H, Ph), 6.00–5.95 (m, 1H, H-5III), 5.87 (dd, J = 5.5, 1.8 Hz, 1H, H-3I), 5.84 (d, J = 5.0 Hz, 1H, H-3II), 5.79 (s, 1H, H-1III), 5.70 (s, 1H, H-1II), 5.67–5.64 (m, 2H, H-2II, H-2III), 5.56 (d, J = 4.1 Hz, 1H, H-3III), 5.35 (d, J = 1.7 Hz, 1H, H-2I), 5.17 (s, 1H, H-1I), 4.95 (dd, J = 5.1, 3.4 Hz, 1H, H-4III), 4.82 (dd, J = 4.9, 3.2 Hz, 1H, H-4II), 4.80–4.58 (m, 7H, H-6aII, H-6aI, H-6aIII, H-6bII, H-6bI, H-5II, H-5I), 4.55–4.47 (m, 2H, H-4I, H-6bIII), 4.23 (dd, J = 10.0, 7.3 Hz, 1H, Fmoc-CH2), 4.17–4.05 (m, 2H, Fmoc-CH2, Fmoc-CH), 3.86 (dt, J = 10.3, 5.3 Hz, 1H, OCH2), 3.61–3.49 (m, 2H, CH2N, OCH2), 3.47–3.38 (m, 1H, CH2N), 1.90–1.79 (m, 2H, OCH2CH2CH2N). 13C NMR (100 MHz, CDCl3) δ = 166.19, 165.84, 165.68, 165.54, 165.29, 165.20, 154.76, 143.41, 143.25, 141.15 (quat. Ph), 133.63, 133.52, 133.44, 133.30, 133.25, 133.17, 133.11, 133.05, 132.99, 130.03, 129.81, 129.76, 129.70, 129.66, 129.61, 129.38, 128.85, 128.63, 128.54, 128.44, 128.30, 128.10, 127.74, 127.10, 126.35, 125.26, 125.20, 120.45, 119.88 (Ph), 106.00 (C-1I), 105.53 (C-1II), 105.47 (C-1III), 83.42 (C-4II), 82.48 (C-2I), 82.26 (C-4I), 82.02 (C-2III), 81.90 (C-2II), 81.74 (C-4III), 77.66 (C-3III), 77.22 (C-3II), 76.57 (C-3I), 73.69 (C-5I), 73.17 (C-5II), 70.17 (C-5III), 70.10 (Fmoc-CH2), 66.71 (C-6III), 66.30 (OCH2), 65.35 (C-6II), 63.98 (C-6I), 46.54 (Fmoc-CH), 38.15 (CH2N), 28.14 (OCH2CH2CH2N). HRMS (ESI): M = C101H88F3NO26. Calcd m/z for [M + Na]+ 1810.5439, found 1810.5402.
:EtOAc 5:1). 1H NMR (600 MHz, CDCl3) δ 8.07–7.75 (m, 14H, o-Ph), 7.56–7.21 (m, 21H, Ph), 6.09 (dt, J = 7.4, 3.8 Hz, 1H, H-5), 5.87 (td, J = 6.3, 3.7 Hz, 1H, H-5), 5.66 (dd, J = 5.4, 1.5 Hz, 1H, H-3), 5.60 (d, J = 5.0 Hz, 1H, H-3), 5.44 (d, J = 0.9 Hz, 1H, H-2), 5.40 (s, 1H, H-1), 5.34 (d, J = 1.6 Hz, 1H, H-2), 5.24 (s, 1H, H-1), 4.81–4.70 (m, 4H, 2 × H-4, H-6aII, H-6bII), 4.18 (dd, J = 10.3, 6.7 Hz, 1H, H-6aI), 4.01 (dd, J = 10.3, 6.1 Hz, 1H, H-6bI), 3.90 (dt, J = 10.4, 5.2 Hz, 1H, OCH2), 3.62–3.49 (m, 2H, OCH2, CH2N), 3.45–3.38 (m, 1H, CH2N), 1.86–1.73 (m, 2H, OCH2CH2CH2N). 13C NMR (150 MHz, CDCl3) δ = 166.22, 165.97, 165.72, 165.70, 165.67, 165.63, 165.34 (PhC(O)), 133.58, 133.54, 133.48, 133.34, 133.24, 133.07, 129.96, 129.92, 129.85, 129.80, 129.73, 129.56, 129.51, 129.42, 128.87, 128.75, 128.70, 128.51, 128.46, 128.44, 128.41, 128.36, 128.32 (Ph), 106.56 (C-1), 105.86 (C-1), 83.06 (C-4), 81.96 (C-2), 81.78 (C-2), 81.34 (C-4), 77.51 (C-3), 77.12 (C-3), 70.78 (C-5), 70.18 (C-5), 66.64 (C-6), 65.28 (OCH2), 63.67 (C-6×), 37.97 (CH2N), 27.99 (OCH2CH2CH2N). HRMS (ESI): M = C66H56F3NO19. Calcd m/z for [M + Na]+ 1246.3291, found 1246.3265.
:EtOAc 5:1). 1H NMR (500 MHz, CDCl3) δ 8.07–7.68 (m, 12H, arom.), 7.56–7.14 (m, 26H, arom.), 6.07–6.00 (m, 1H, H-5II), 5.88 (d, J = 5.7 Hz, 1H, H-3), 5.76 (s, 1H, H-1), 5.64 (s, 1H, H-2), 5.58 (d, J = 4.7 Hz, 1H, H-3), 5.37 (s, 1H, H-2), 5.20 (s, 1H. H-1), 4.98 (t, J = 3.9 Hz, 1H, H-4), 4.76 (dd, J = 11.7, 4.1 Hz, 1H, H-6a), 4.71–4.52 (m, 5H, 3 × H-6, H-5I, H-4), 4.28 (dd, J = 10.1, 7.9 Hz, 1H, Fmoc-CH2), 4.23–4.17 (m, 1H, Fmoc-CH2), 4.11 (t, J = 7.5 Hz, 1H, Fmoc-CH), 3.88 (dt, J = 10.0, 5.1 Hz, 1H, OCH2), 3.60–3.42 (m, 3H, OCH2, CH2N), 1.91–1.77 (m, 2H, OCH2CH2CH2N). 13C NMR (125 MHz, CDCl3) δ = 166.18, 165.96, 165.72, 165.59, 165.52, 165.25 (PhC(O)), 154.83, 143.34, 143.17, 141.17, 133.63, 133.34, 133.22, 133.13, 130.02, 129.82, 129.78, 129.66, 129.57, 129.39, 128.74, 128.61, 128.52, 128.39, 128.35, 128.22, 127.79, 127.77, 127.12, 127.06, 125.22, 125.16, 119.92 (Ph), 106.14 (C-1), 105.54 (C-1), 82.81 (C-4), 82.19 (C-2), 81.97 (C-2), 81.77 (C-4), 77.72 (C-3), 77.23 (C-3), 76.53 (C-5), 73.39 (C-5), 70.17 (Fmoc-CH2), 66.58 (C-6), 66.47 (OCH2), 64.23 (C-6), 46.53 (Fmoc-CH), 38.08 (CH2N), 28.04 (OCH2CH2CH2N). HRMS (ESI): M = C74H62F3NO20. Calcd m/z for [M + Na]+ 1364.3709, found 1364.3705.
:EtOAc 3:1). 1H NMR (600 MHz, CDCl3) δ 8.06–7.81 (m, 12H, o-Ph), 7.60–7.19 (m, 18H, Ph), 5.89 (dd, J = 5.7, 2.3 Hz, 1H, H-3), 5.76 (s, 1H, H-1), 5.66 (d, J = 1.4 Hz, 1H, H-2), 5.64–5.60 (m, 1H, H-5II), 5.60–5.57 (m, 1H, H-3), 5.37 (d, J = 2.3 Hz, 1H, H-2), 5.21 (s, 1H, H-1), 5.05 (dd, J = 5.3, 2.8 Hz, 1H, H-4), 4.78 (dd, J = 11.8, 4.3 Hz, 1H, H-6aI), 4.71 (dd, J = 11.8, 6.8 Hz, 1H, H-6bI), 4.62–4.57 (m, 2H, H-4, H-5I), 4.05–3.88 (m, 3H, H-6aII, H-6bII, OCH2), 3.62–3.45 (m, 3H, OCH2, CH2N), 3.23 (br. s, 1H, OH), 1.97–1.81 (m, 2H, OCH2CH2CH2N). 13C NMR (150 MHz, CDCl3) δ = 166.29, 166.23, 166.03, 165.63, 165.31 (PhC(O)), 133.88, 133.67, 133.35, 133.17, 129.91, 129.89, 129.84, 129.79, 129.65, 128.85, 128.76, 128.61, 128.41, 128.38, 128.35, 128.22 (Ph), 106.24 (C-1), 105.86 (C-1), 82.74 (C-4), 82.35 (C-2), 82.02 (C-2), 81.63 (C-4), 77.69 (C-3), 73.66 (C-3), 72.97 (C-5), 66.42 (OCH2), 64.43 (C-6), 61.34 (C-6), 38.08 (CH2N), 28.11 (OCH2CH2CH2N). HRMS (ESI): M = C59H52F3NO18. Calcd m/z for [M + Na]+ 1142.3029, found 1142.3030.
:EtOAc 5:1). 1H NMR (400 MHz, CDCl3) δ 8.04–7.72 (m, 20H, o-Ph), 7.53–7.13 (m, 30H, Ph), 6.10–6.02 (m, 1H, H-5), 5.99 (dt, J = 7.7, 4.0 Hz, 1H, H-5), 5.87 (dd, J = 5.8, 2.3 Hz, 1H, H-3), 5.75 (s, 1H, H-2), 5.64–5.61 (m, 2H, H-2, H-3), 5.54 (d, J = 5.0 Hz, 1H, H-3), 5.35–5.34 (m, 2H, 2 × H-2), 5.24 (s, 1H, H-1), 5.18 (s, 1H, H-1), 4.94 (t, J = 4.1 Hz, 1H, H-4), 4.79–4.56 (m, 6H, H-4, 4 × H-6, H-5I), 4.53 (dd, J = 5.7, 3.9 Hz, 1H, H-4), 4.19–4.10 (m, 1H, H-6aII), 4.01 (dd, J = 11.2, 7.4 Hz, 1H, H-6bII), 3.87 (dt, J = 10.1, 5.0 Hz, 1H, OCH2), 3.61–3.36 (m, 4H, OCH2, CH2N), 1.89–1.66 (m, 2H, OCH2CH2CH2N). 13C NMR (100 MHz, CDCl3) δ = 166.17, 166.09, 165.88, 165.75, 165.62, 165.54, 165.25, 165.13 (PhC(O)), 133.61, 133.54, 133.27, 133.22, 133.11, 132.96, 129.89, 129.78, 129.74, 129.69, 129.66, 129.60, 129.50, 128.88, 128.86, 128.84, 128.79, 128.77, 128.66, 128.57, 128.48, 128.37, 128.32, 128.28, 128.16 (Ph), 106.61 (C-1), 106.09 (C-1), 105.46 (C-1), 82.84 (C-4), 82.74 (C-2), 81.95 (2 × C-4), 81.88 (2 × C-2), 77.77 (C-3), 77.48 (C-3), 76.48 (C-3), 73.17 (C-4), 71.75 (C-5), 70.42 (C-5), 67.43 (C-6II), 66.41 (OCH2), 64.25 (C-6), 63.82 (C-6), 38.08 (CH2N), 28.05 (OCH2CH2CH2N). HRMS (ESI): M = C93H78F3NO27. Calcd m/z for [M + Na]+ 1720.4606, found 1720.4585.
:EtOAc 10:1). 1H NMR (600 MHz, CDCl3) δ 8.11–7.07 (m, 35H), 6.05 (ddd, 1H, H-5II), 5.72 (s, 1H, H-1II), 5.67 (dd, J1,2 < 2 Hz, J2,3 = 3.0 Hz, 1H, H-2I), 5.58 (dd, J2,3 = 1.4 Hz, J3,4 = 5.8 Hz, 1H, H-3II), 5.55 (d, 1H, H-2II), 5.01 (d, 1H, H-1I), 4.93 (d, J = 11.3 Hz, 1H, PhCHH′), 4.73 (dd, J4,5 = 2.8 Hz, 1H, H-4II), 4.72(dd, J5,6 = 7.65 Hz, J6,6′ = 11.5 Hz, 1H, H-6aII), 4.69 (d, J = 11.9 Hz, 1H, PhCHH′), 4.66 (dd, J5,6′ = 5.15 Hz, 1H, H-6bII), 4.57 (d, 1H, PhCHH′), 4.53 (d, 1H, PhCHH′), 4.50 (dd, J3,4 = 9.5 Hz, 1H, H-3I), 4.11 (t, J = 9.3 Hz, 1H, H-4I), 3.85 (m, 1H, OCHH′CH2), 3.78–3.77 (m, 2H, H-5I, H-6aI), 3.72 (d, J = 8.5 Hz, 1H, H-6bI), 3.56 (m, 2H, OCHH′CH2, CHH′N), 3.44 (m, 1H, CHH′N), 1.90 (m, 2H, OCH2CH2CH2N). 13C NMR (151 MHz, CDCl3) δ 133.37, 133.28, 133.23, 133.16, 130.03, 129.93, 129.86, 129.80, 128.44, 128.37, 128.33, 128.27, 127.61, 127.57, 127.51, 127.47 (Ph), 102.28 (C-1II), 97.76 (C-1I), 82.36 (C-2II), 81.52 (C-4II), 78.04 (C-3II), 75.22 (PhCH2), 73.60 (C-3I), 73.45 (PhCH2), 73.36 (C-4I), 72.08 (C-5I), 69.69 (C-5II), 68.96 (C-6I), 67.99 (C-2I), 65.51 (OCH2), 63.55 (C-6II), 37.85 (CH2N), 28.45 (CH2CH2CH2). HRMS (ESI): M = C66H60F3NO17. Calcd m/z for [M + Na]+ 1218.3706, found 1218.370313.
:1 (1 mL), Pd(OH)2 was added and the mixture was vigorously stirred overnight under a H2 atmosphere. Then it was filtered through a Celite layer and concentrated in vacuo. To the residue, a solution of MeONa (0.1 M) in MeOH (0.6 mL) was added and the mixture was stirred for 1 h. Then, 1 drop of water was added and the reaction mixture was left overnight. Base was neutralized with AcOH (6 μL), and the reaction mixture was diluted with water and concentrated in vacuo. Gel chromatography on TSK-40 HW(S) followed by lyophilization afforded 5 (7.2 mg, 62%) as a white foam. HRMS (ESI): M = C15H29NO11. Calcd m/z for [M + H]+ 400.1813, found 400.1816.
:EtOAc 5:1). 1H NMR (600 MHz, CDCl3) δ 8.07–7.12 (m, 53H, arom.), 5.99–5.95 (m, 1H, H-5III), 5.74 (s, 1H, H-1III), 5.72 (dd, J3,4 = 6.3 Hz, 1.9 Hz, 1H, H-3II), 5.65 (d, J = 1.5 Hz, 1H, H-2III), 5.64–5.61 (m, 2H, H-2I, H-1II), 5.57 (dd, J = 5.5, 1.2 Hz, 1H, H-3III), 5.53 (d, J = 2.0 Hz, 1H, H-2II), 4.97 (dd, J = 5.4, 3.4 Hz, 1H, H-4III), 4.91–4.87 (m, 2H, H-1I, PhCH2), 4.67 (dd, J = 12.4, 4.1 Hz, 2H, H-6aIII), 4.65–4.58 (m, 3H, PhCH2, H-5II, H-6aII), 4.56 (dd, J = 6.2, 3.9 Hz, 1H, H-4II), 4.55–4.49 (m, 3H, H-6bII, H-6bIII, PhCH2), 4.45 (d, J = 11.9 Hz, 1H, PhCH2), 4.37 (dd, J = 9.5, 3.1 Hz, 1H, H-3I), 4.29 (dd, J = 10.5, 7.4 Hz, 1H, Fmoc-CH2), 4.24 (dd, J = 10.5, 7.6 Hz, 1H, Fmoc-CH2), 4.11 (t, J = 7.4 Hz, 1H, Fmoc-CH), 4.04 (t, J = 9.5 Hz, 1H, H-4I), 3.73–3.68 (m, 1H, OCH2), 3.59 (dd, J = 10.7, 3.9 Hz, 1H, H-6aI), 3.56–3.50 (m, 2H, H-6bI, H-5I), 3.48–3.41 (m, 2H, OCH2, NCH2), 3.37–3.30 (m, 1H, CH2N), 1.92–1.81 (m, 2H, OCH2CH2CH2N). 13C NMR (150 MHz, CDCl3) δ = 166.40, 165.85, 165.67, 165.54, 165.53, 165.20, 165.04 (quat. Ph), 133.30, 133.14, 133.05, 130.03, 129.86, 129.82, 129.65, 128.43, 128.35, 128.33, 128.29, 128.25, 127.72, 127.52, 127.44, 127.08, 125.18, 119.89 (Ph), 105.36 (C-1III), 102.11 (C-1II), 97.78 (C-1I), 82.43 (C-2II), 82.27 (C-2III), 82.10 (C-4II), 81.75 (C-4III), 77.73 (C-3II), 77.56 (C-3III), 75.11 (PhCH2), 73.74 (C-3I), 73.58 (C-5II), 73.44 (PhCH2), 73.18 (C-4I), 71.84 (C-5I), 70.25 (C-5III), 70.05 (Fmoc-CH2), 68.83 (C-6I), 67.79 (C-2I), 66.87 (C-6III), 65.45 (OCH2), 64.83 (C-6II), 46.61 (Fmoc-CH), 37.75 (CH2N), 28.45 (OCH2CH2CH2N). HRMS (ESI): M = C101H88NF3NO26. Calcd m/z for [M + Na]+ 1810.5439, found 1810.5402.
:EtOAc 10:1). 1H NMR (600 MHz, CDCl3) δ 8.14–7.15 (m, 35H), 6.06 (ddd, J4,5 = 3.4 Hz, J5,6 = 4.5 Hz, J5,6′ = 7.8 Hz, 1H, H-5II), 5.88 (dd, J2,3 = 3.1 Hz, J3,4 = 9.9 Hz, 1H, H-3I), 5.84 (m, 1H, H-4I), 5.68 (dd, J1,2 = 1.7 Hz, 1H, H-2I), 5.63 (dd, J2,3 = 1.0 Hz, 1H, H-3II), 5.56 (s, 1H, H-1II), 5.53 (d, 1H, H-2II), 5.08 (d, 1H, H-1I), 4.77–4.74 (m, 2H, H-6II, H-6′II), 4.69 (dd, 1H, H-4II), 4.35 (m, 1H, H-5I), 4.12–3.91 (m, 3H, H-6aI, H-6b′I, OCHH′), 3.74–3.49 (m, 3H, OCHH′, CH2N), 2.04 (m, 2H, CH2CH2CH2). 13C NMR (151 MHz, CDCl3) δ 133.65, 133.58, 133.54, 133.52, 133.31, 133.24, 133.13, 130.00, 129.91, 129.89, 129.79, 129.74, 128.65, 128.49, 128.44, 128.38, 128.32 (PhC(O)), 106.49 (C-1II), 97.52 (C-1I), 82.06 (C-2II), 81.26 (C-4II), 77.63 (C-3II), 71.34 (C-5I), 70.39 (C-2I), 70.19 (C-5II), 69.83 (C-3I), 67.15 (C-4I), 66.27 (OCH2), 66.07 (C-6I), 63.66 (C-6II), 37.61 (CH2N), 28.52 (CH2CH2CH2). HRMS (ESI): M = C66H56F3NO19. Calcd m/z for [M + Na]+ 1246.3291, found 1246.3255.
:EtOAc 5:1). 1H NMR (600 MHz, CDCl3) δ 8.12 (d, J = 7.2 Hz, 2H, o-Ph), 8.02 (d, J = 7.3 Hz, 2H, o-Ph), 7.81 (d, J = 7.3 Hz, 2H, o-Ph), 7.71 (dd, J = 7.6, 3.0 Hz, 2H, Ph), 7.52–7.06 (m, 44H, arom.), 6.97 (t, J = 7.4 Hz, 1H, Ph), 6.89 (br s, 1H, NH), 5.97–5.93 (m, 1H, H-5III), 5.50 (d, J = 4.1 Hz, 1H, H-3III), 5.48 (s, 1H, H-2III), 5.20 (s, 1H, H-1III), 5.10 (d, J = 1.5 Hz, 1H, H-1II), 4.88 (d, J = 1.6 Hz, 1H, H-1I), 4.82 (d, J = 10.8 Hz, 1H, PhCHH′), 4.74–4.44 (m, 11H, 10 × PhCHH′, H-6aIII), 4.34 (d, J = 10.9 Hz, 1H, PhCHH′), 4.29–4.15 (m, 4H, H-2II, Fmoc-CH2, H-6bIII), 4.09 (t, J = 7.6 Hz, 1H, Fmoc-CH), 4.03–4.00 (m, 1H, H-2I), 3.95–3.61 (m, 11H, H-3I, H-4I, H-5I, H-6aI, H-6bI, H-3II, H-4II, H-5II, H-6aII, H-6bII, OCHH′CH2), 3.43–3.17 (m, 3H, OCHH′CH2CH2NH), 1.77–1.66 (m, 2H, CH2CH2CH2). 13C NMR (151 MHz, CDCl3) δ 165.69, 165.29, 154.93, 143.43, 141.33, 138.44, 138.38, 133.54, 133.42, 133.26, 130.15, 129.98, 128.61–128.36, 128.09, 127.90, 127.71, 127.39, 127.27, 125.40, 125.36, 120.05 (arom.), 103.75 (C-1III), 99.40 (C-1II), 99.24 (C-1I), 82.50 (C-4III), 81.43 (C-2III), 79.90 (C-3I), 78.18 (C-3II), 77.91 (C-3III), 75.74, 75.34, 75.14, 75.06, 74.89, 73.70, 73.55, 72.69, 72.37 (6 × PhCH2, C-2I, C-4I, C-5I, C-2II, C-4II, C-5II), 70.57 (C-5III), 70.25 (Fmoc-CH2), 69.99 (C-6II), 69.59 (C-6I), 66.88 (C-6III), 66.02 (OCH2CH2), 46.72 (Fmoc-CH), 38.15 (CH2NH), 28.26 (OCH2CH2CH2NH). HRMS (ESI): M = C101H96F3NO22. Calcd m/z for [M + Na]+ 1754.6268, found 1754.6258.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by RSF grant 14-23-00199 (NEN). We thank Dr Thierry Fontaine for providing the galactomannan sample. We thank Dr A. O. Chizhov for recording high resolution mass spectra in the Department of Structural Studies of the Zelinsky Institute of Organic Chemistry, Moscow.References
- J. R. Köhler, B. Hube, R. Puccia, A. Casadevall and J. R. Perfect, Microbiol. Spectrum, 2017, 5 DOI:10.1128/microbiolspec.FUNK-0014-2016.
- R. R. Herbrecht, D. W. Denning, T. F. Patterson, J. E. Bennett, R. E. Greene, J.-W. Oestmann, W. V. Kern, K. A. Marr, P. Ribaud, O. Lortholary, R. Sylvester, R. H. Rubin, J. R. Wingard, P. Stark, C. Durand, D. Caillot, E. Thiel, P. H. Chandrasekar, M. R. Hodges, H. T. Schlamm, P. F. Troke and B. de Pauw, N. Engl. J. Med., 2002, 347, 408–415 CrossRef CAS PubMed.
- H. Kume, T. Yamazaki, T. Togano, M. Abe, H. Tanuma, S. Kawana and M. Okudaira, Med. Mycol. J., 2011, 52, 117–127 CrossRef PubMed.
- J.-P. Latgé, H. Kobayashi, J.-P. Debeaupuis, M. Diaquin, J. Sarfati, J.-M. Wieruszeski, E. Parra, J.-P. Bouchara and B. Fournet, Infect. Immun., 1994, 62, 5424–5433 Search PubMed.
- D. Stynen, A. Goris, J. Sarfati and J. P. Latgé, J. Clin. Microbiol., 1995, 33, 497–500 CAS.
- C. Costachel, B. Coddeville, J.-P. Latgé and T. Fontaine, J. Biol. Chem., 2005, 280, 39835–39842 CrossRef CAS PubMed.
- A. Kudoh, Y. Okawa and N. Shibata, Glycobiology, 2015, 25, 74–87 CrossRef CAS PubMed.
- A. W. Van Bruggen-Van Der Lugt, H. J. Kamphuis, G. A. De Ruiter, P. Mischnick, J. H. Van Boom and F. M. Rombouts, J. Bacteriol., 1992, 174, 6096–6102 CrossRef CAS PubMed.
- S. Guo, W. Mao, M. Yan, C. Zhao, N. Li, J. Shan, C. Lin, X. Liu, T. Guo, T. Guo and S. Wang, Carbohydr. Polym., 2014, 105, 325–333 CrossRef CAS PubMed.
- N. Shibata, T. Saitoh, Y. Tadokoro and Y. Okawa, Microbiology, 2009, 155, 3420–3429 CrossRef CAS PubMed.
- K. Ikuta, N. Shibata, J. S. Blake, M. V. Dahl, R. D. Nelson, K. Hisamichi, H. Kobayashi, S. Suzuki and Y. Okawa, Biochem. J., 1997, 323, 297–305 CrossRef CAS PubMed.
- R. P. Da Silva, C. Heiss, I. Black, P. Azadi, J. Q. Gerlach, L. R. Travassos, L. Joshi, M. Kilcoyne and R. Puccia, Sci. Rep., 2015, 5, 14213 CrossRef PubMed.
- E. A. Leitão, V. C. Bittencourt, R. M. Haido, A. P. Valente, J. Peter-Katalinic, M. Letzel, L. M. de Souza and E. Barreto-Bergter, Glycobiology, 2003, 13, 681–692 CrossRef PubMed.
- G. C. Completo and T. L. Lowary, J. Org. Chem., 2008, 73, 4513–4525 CrossRef CAS PubMed.
- A. K. Pathak, V. Pathak, L. Seitz, J. A. Maddry, S. S. Gurcha, G. S. Besra, W. J. Suling and R. C. Reynolds, Bioorg. Med. Chem., 2001, 9, 3129–3143 CrossRef CAS PubMed.
- L. Gandolfi-Donadío, C. Gallo-Rodriguez and R. M. de Lederkremer, J. Org. Chem., 2003, 68, 6928–6934 CrossRef PubMed.
- M. J. Tilve, C. R. Cori and C. Gallo-Rodriguez, J. Org. Chem., 2016, 81, 9585–9594 CrossRef CAS PubMed.
- L. Cattiaux, B. Sendid, M. Collot, E. Machez, D. Poulain and J.-M. Mallet, Bioorg. Med. Chem., 2011, 19, 547–555 CrossRef CAS PubMed.
- M. Fu, G. Zhang and J. Ning, Carbohydr. Res., 2005, 340, 25–30 CrossRef CAS PubMed.
- R. Euzen, V. Ferrières and D. Plusquellec, Carbohydr. Res., 2006, 341, 2759–2768 CrossRef CAS PubMed.
- D. A. Argunov, V. B. Krylov and N. E. Nifantiev, Org. Biomol. Chem., 2015, 13, 3255–3267 CAS.
- D. A. Argunov, V. B. Krylov and N. E. Nifantiev, Org. Lett., 2016, 18, 5504–5507 CrossRef CAS PubMed.
- V. B. Krylov, D. A. Argunov, D. Z. Vinnitskiy, S. A. Verkhnyatskaya, A. G. Gerbst, N. E. Ustyuzhanina, A. S. Dmitrenok, J. Huebner, O. Holst, H.-C. Siebert and N. E. Nifantiev, Chem. – Eur. J., 2014, 20, 16516–16522 CrossRef CAS PubMed.
- V. B. Krylov, D. A. Argunov, D. Z. Vinnitskiy, A. G. Gerbst, N. E. Ustyuzhanina, A. S. Dmitrenok and N. E. Nifantiev, Synlett, 2016, 1659–1664 CAS.
- C. Gallo-Rodriguez, L. Gandolfi and R. M. de Lederkremer, Org. Lett., 1999, 1, 245–248 CrossRef CAS PubMed.
- A. A. Karelin, Y. E. Tsvetkov, G. Kogan, S. Bystricky and N. E. Nifantiev, Russ. J. Bioorg. Chem., 2007, 33, 110–121 CrossRef CAS.
- A. S. Shashkov, G. M. Lipkind, Y. A. Knirel and N. K. Kochetkov, Magn. Reson. Chem., 1988, 26, 735–747 CrossRef CAS.
- B. Schumann, S. G. Parameswarappa, M. P. Lisboa, N. Kottari, F. Guidetti, C. L. Pereira and P. H. Seeberger, Angew. Chem., Int. Ed., 2016, 55, 14431–14434 CrossRef CAS PubMed.
- H. Van Halbeek, Biochem. Soc. Trans., 1984, 12, 601–605 CrossRef CAS PubMed.
- J. F. Vliegenthart, L. Dorland and H. van Halbeek, Adv. Carbohydr. Chem. Biochem., 1983, 41, 209–374 CrossRef CAS.
- G. M. Lipkind, A. S. Shashkov, Y. A. Knirel, E. V. Vinogradov and N. K. Kochetkov, Carbohydr. Res., 1988, 175, 59–75 CrossRef CAS PubMed.
- G. M. Lipkind, A. S. Shashkov, N. E. Nifant'ev and N. K. Kochetkov, Carbohydr. Res., 1992, 237, 11–22 CrossRef CAS PubMed.
- F. Lisacek, J. Mariethoz, D. Alocci, P. M. Rudd, J. L. Abrahams, M. P. Campbell, N. H. Packer, J. Ståhle, G. Widmalm, E. Mullen, B. Adamczyk, M. A. Rojas-Macias, C. Jin and N. G. Karlsson, Methods Mol. Biol., 2017, 1503, 235–264 Search PubMed.
- M. Lundborg and G. Widmalm, Anal. Chem., 2011, 83, 1514–1517 CrossRef CAS PubMed.
- A. G. Gerbst, V. B. Krylov, D. Z. Vinnitskiy, A. S. Dmitrenok, A. S. Shashkov and N. E. Nifantiev, Carbohydr. Res., 2015, 417, 1–10 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: NMR data and copies of 1H and 13C spectra. See DOI: 10.1039/c7ob02734f |
| This journal is © The Royal Society of Chemistry 2018 |