
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe role of catalytic residue pKa on the hydrolysis/transglycosylation partition in family 3 β-glucosidases†
Inacrist
Geronimo
 a,
Christina M.
Payne‡
b and
Mats
Sandgren
*a
a,
Christina M.
Payne‡
b and
Mats
Sandgren
*a
aDepartment of Molecular Sciences, Swedish University of Agricultural Sciences, Box 7015, 750 07 Uppsala, Sweden. E-mail: mats.sandgren@slu.se
bDepartment of Chemical and Materials Engineering, University of Kentucky, Lexington, Kentucky 40506-0046, USA
First published on 18th December 2017
Abstract
β-Glucosidases (βgls) primarily catalyze the hydrolysis of the terminal glycosidic bond at the non-reducing end of β-glucosides, although glycosidic bond synthesis (called transglycosylation) can also occur in the presence of another acceptor. In the final reaction step, the glucose product or another substrate competes with water for transfer to the glycosyl-enzyme intermediate. The factors governing the balance between the two pathways are not fully known; however, the involvement of ionizable residues in binding and catalysis suggests that their pKa may play a role. Through constant pH molecular dynamics simulations of a glycoside hydrolase Family 3 (GH3) βgl, we showed that the pKa of the catalytic acid/base residue, E441, is low (∼2) during either reaction due to E441–R125–E128 and E441–R125–E166 hydrogen bond networks. The low basicity of E441 would reduce its ability to deprotonate the acceptor. This may be less critical for transglycosylation because sugars have a lower deprotonation enthalpy than water. Moreover, their acidity would be increased by hydrogen bonding with R169 at the acceptor binding site. In contrast, no such interaction was observed for catalytic water. The results are likely applicable to other GH3 βgls because R125, E128, E166, and R169 are conserved. As these enzymes are commonly used in biomass degradation, there is interest in developing variants with enhanced hydrolytic activity. This may be accomplished by elevating the acid/base residue pKa by disrupting its hydrogen bond networks and reducing the affinity and reactivity of a sugar acceptor by mutating R169.
Introduction
β-Glucosidases (βgls) hydrolyze terminal, non-reducing β-D-glucosyl residues to release β-D-glucose.1–3 βgls, except those from glycoside hydrolase (GH) Family 9, employ the retaining hydrolytic/transglycosidic mechanism, wherein the generated product adopts the same stereochemistry as the substrate (Scheme 1).3 The acid/base residue of βgls protonates the glycosidic oxygen, while the nucleophile attacks the anomeric carbon to form a glycosyl-enzyme intermediate (GEI), which then undergoes hydrolysis.4–7 A parallel reaction, transglycosylation, may occur to a lesser extent in the presence of another suitable acceptor.8 The competition between the two pathways is usually expressed in terms of the ratio of their respective rate constants called the hydrolysis/transglycosylation (H/T) partition. βgls are an important component of biomass-degrading enzyme cocktails because they relieve product inhibition of endoglucanases and cellobiohydrolases through hydrolysis of cellobiose to glucose.9,10 In the context of biomass degradation, a high H/T partition is a preferable characteristic of βgls. However, there is also growing interest in exploiting the transglycosidic activity of βgls, particularly those from hyperthermophilic bacteria, in the synthesis of valuable oligosaccharides and glucoconjugates that are used as surfactants and food ingredients.8,11,12 | ||
| Scheme 1 Retaining mechanism of β-glucosidases. In the final reaction step, hydrolysis occurs if ROH is water, and transglycosylation if ROH is another acceptor, usually a sugar. | ||
Protein engineering of βgls for a specific industrial application, therefore, requires detailed knowledge of the molecular factors governing the H/T partition. Site-directed mutagenesis studies have been performed to investigate the role of (1) affinity at the acceptor binding sites (i.e., +1 site in Scheme 1), (2) stabilizing interactions of the GEI at the −1 site, and (3) access of the GEI to catalytic water.8 In addition, experimental studies suggest a correlation between H/T partition and the pKa of active site residues. Kinetic data for a GH Family 3 (GH3) βgl from Aspergillus niger13 and mutants of a GH Family 1 (GH1) βgl from Thermatoga neapolitana14 indicated that hydrolytic activity decreases with increasing pH, while transglycosidic activity is pH independent. Among the hypotheses suggested to account for pH dependence are conformational change in the active site region and ionization of a residue, usually tyrosine, involved in hydrolysis.13,15 As for the latter, a quantum mechanics/molecular mechanics study on a rice GH1 βgl showed that a tyrosine near the nucleophile forms a hydrogen bond with the O5 atom of the glycosyl moiety at the hydrolysis transition state, affecting the character of the C1–O5 bond.16 The dependence of the H/T partition on the pKa of the acid/base residue, which serves as an indication of its ability to activate water, was inferred from a site-directed mutagenesis study of another GH, an α-amylase from Bacillus stearothermophilus.17 In that study, the A289F and A289Y mutants exhibited increased transglycosidic activity, which could not be solely attributed to hydrophobicity since the water accessibility of the active site appeared to be unaffected by substitution. The shift in the alkaline limb of the pH-activity profile to lower pH indicated reduced pKa of the acid/base residue caused by changes in its electrostatic environment upon mutation.17
The pKa of active site residues has not been explored as extensively as other molecular factors, possibly owing to the challenges of experimentally determining this parameter for βgls. NMR titration, which can be used to obtain the pKa of individual residues, is not feasible for βgls because of their large size.18 For example, xylanases, for which this method has been successfully used, have a molecular weight of 20 kDa,19,20 while the βgls secreted by Trichoderma reesei have estimated molecular weights ranging from 55 to 130 kDa.21 Derivation of the apparent pKa values of the catalytic residues from the pH-activity profile22 is also not trivial because interaction with other ionizable residues in the active site causes deviation from ideal titration behavior.23 An alternative approach to these experimental methods is constant pH molecular dynamics (CpHMD), which captures fluctuations in the protonation state of ionizable residues caused by changes in solution pH and local conformation. In CpHMD, the ionizable residues are continuously switched between the protonated (0) and deprotonated (1) states through the introduction of a titration coordinate, λ. λ is propagated in implicit solvent using the generalized Born (GB) model,24 while spatial coordinates are propagated in explicit solvent. Conformational dynamics and sampling of protonation states are coupled through a hybrid effective energy term, comprised of van der Waals, Coulombic, and GB electrostatic solvation free energies, that depends on λ. The convergence of pKa values is accelerated by enhancing the sampling of protonation states with pH-based replica exchange (pH-REX), wherein independent simulations at different pH conditions are run in parallel. A pair of replicas adjacent in pH periodically swaps atomic coordinates via Hamiltonian exchange.25–27 This method has been previously demonstrated to be capable of pKa determination in GH6 and GH7 cellobiohydrolases.28,29
This study investigated the role of active site residue pKas on the H/T partition of the Hypocrea jecorina (teleomorph of T. reesei) GH3 βgl, Cel3A (HjCel3A), using CpHMD/pH-REX. HjCel3A, along with HjCel3B, exhibits the highest activity against cellobiose among the H. jecorina GH3 βgls21 and was found to improve the conversion of various cellulosic substrates by nearly 10%.30 However, its hydrolytic activity decreases at high cellobiose or glucose concentration because of increased transglycosidic activity,21 which appears to be prevalent among GH3 βgls.31 Simulations at the optimum pH of HjCel3A (pH 5.030) were performed to determine the differences in the active site conformation, acceptor-residue interactions, and solvent availability between the two reactions. The pKa values of ionizable active site residues (Fig. 1) at the GEI state (in the absence and presence of a glucose acceptor) were calculated, and their impact on the mechanism was examined. Residues that may modulate the pKa of the acid/base residue, E441, were identified through analysis of hydrogen bond interactions and charge coupling and verified by simulations of mutants.
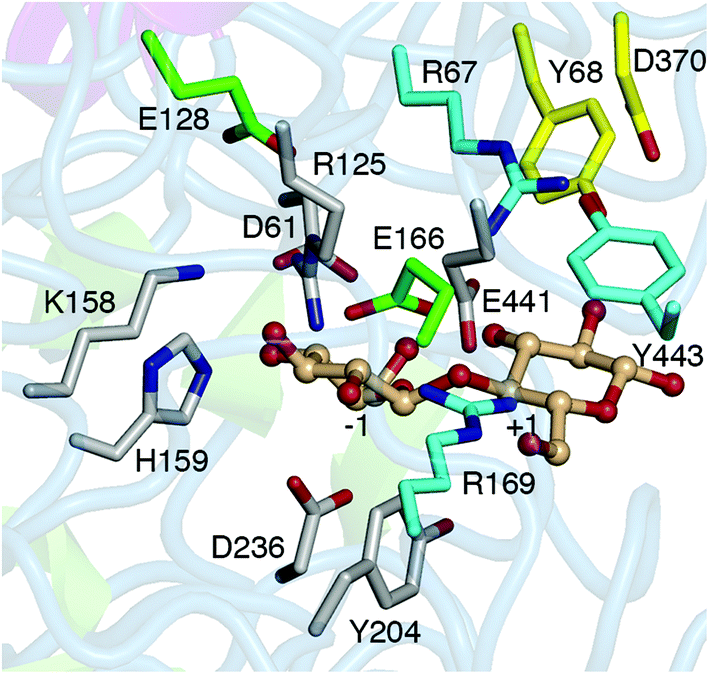 | ||
| Fig. 1 Ionizable residues in the HjCel3A active site. The catalytic residues are D236 (nucleophile) and E441 (acid/base residue). Residues in the −1 site (D61, R125, K158, H159, and Y204) are shown in gray, those in the +1 site (R67, R169, and Y443) in cyan, and those in the putative +2 site (Y68 and D370) in yellow. Ionizable residues near the catalytic residues (E128 and E166) are shown in green. | ||
Computational methods
All calculations were performed using CHARMM version c37b1.32 System preparation and equilibration are described in ESI.† The CHARMM22 force field with CMAP correction32–36 was used to describe the protein since this force field has been tested with the GB model with simple switching (GBSW).37 The C36 carbohydrate force field38–41 was used to describe glucose. Partial charges and force field parameters for the glycosyl–aspartate bond in GEI was obtained by analogy to similar, existing patches in CHARMM (provided as ESI†). Water was modeled using the TIP3P force field.42,43Constant pH molecular dynamics
All aspartate (38 excluding D236, which is covalently bound to the glycosyl in the GEI), glutamate (16), and histidine (9) residues of HjCel3A were titrated in the simulations. λ was propagated using Langevin dynamics with a collision frequency of 5.0 ps−1 and was updated every 10 steps. Solvation forces on λ were calculated using the GBSW model, with the input radii taken from ref. 44 and 45. Three independent production simulations, each with a new set of velocities, were run for 10 ns using the same parameters as equilibration. Structural changes in the active site, as will be discussed in the Results and discussion section, precluded longer simulations times. Temperature, energy, density, root-mean-square deviation (RMSD), and root-mean-square fluctuation (RMSF) plots are shown in Fig. S1–S4.† Hydrogen bond occupancy and lifetime were calculated at 4 ps intervals using a distance cutoff of 2.4 Å and angle cutoff of 160°.46 A distance cutoff of 3.4 Å was used in counting the number of water molecules within the first solvation shell of E441. The correlation between two protonation events was determined by calculating the Pearson correlation coefficient of a pair of λ values.pKa calculation from pH replica exchange
The equilibrated structure was used as the starting point for the pH-REX calculations. Eighteen replicas (1.0, 1.3, 1.6, 1.9, 2.2, 2.5, 2.8, 3.1, 3.4, 3.7, 4.0, 4.3, 4.6, 5.1, 5.6, 6.1, 6.6, and 7.1) were simulated using the same parameters as described for CpHMD. The average exchange success rate was only about 30% due to the large system (∼98![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 atoms, 63 residues titrated) (Fig. S5†).47 At pH < 3, glucose tends to diffuse from the shallow substrate pocket owing to significant structural changes caused by protonation of carboxylate residues in the active site. The center-of-mass distance between the GEI moiety and glucose was, therefore, restrained in all replica simulations using a harmonic potential with an amplitude of 10.0 kcal mol−1 Å−1 and offset distance of 6.0 Å. pH exchange was attempted every 500 steps or 1 ps, and a total of 10 ns was run to ensure convergence (Fig. S6†). The fraction of deprotonated states, S, at each pH was fit to the generalized Henderson–Hasselbalch equation using a non-linear, least-squares fitting algorithm to determine the pKa (eqn (1)). The Hill coefficient, n, is included in the fit to account for the coupling between ionizable sites but has minimal effect on the calculated pKa.26
000 atoms, 63 residues titrated) (Fig. S5†).47 At pH < 3, glucose tends to diffuse from the shallow substrate pocket owing to significant structural changes caused by protonation of carboxylate residues in the active site. The center-of-mass distance between the GEI moiety and glucose was, therefore, restrained in all replica simulations using a harmonic potential with an amplitude of 10.0 kcal mol−1 Å−1 and offset distance of 6.0 Å. pH exchange was attempted every 500 steps or 1 ps, and a total of 10 ns was run to ensure convergence (Fig. S6†). The fraction of deprotonated states, S, at each pH was fit to the generalized Henderson–Hasselbalch equation using a non-linear, least-squares fitting algorithm to determine the pKa (eqn (1)). The Hill coefficient, n, is included in the fit to account for the coupling between ionizable sites but has minimal effect on the calculated pKa.26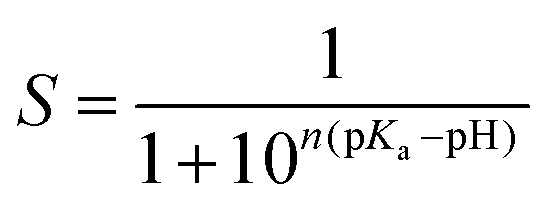 | (1) |
Results and discussion
CpHMD/pH-REX simulations were performed to determine whether the balance between the hydrolytic and transglycosidic activities of HjCel3A could be linked to pKa differences between the two reactions. Does E441 have a higher pKa, and therefore, a stronger base, in one reaction? Are the protonation states of ionizable active site residues at the −1 site such that the glycosyl moiety is more stabilized in one reaction? Hydrolysis and transglycosylation were modeled using HjCel3A-GEI in the absence and presence of a glucose or cellobiose acceptor in the +1 site, respectively. The active site residues titrated included E441, D61, and H159 in the −1 site and D370 in the putative +2 site. R125, K158, and Y204 in the −1 site, R169, R67, and Y443 in the +1 site, and Y68 in the putative +2 site were not expected to titrate within the pH range of study (pH 1.0–7.1) because of their high intrinsic pKa. The titrations of E128 and E166 were also monitored because of their proximity to the catalytic residues (Fig. 1).Hydrolysis—HjCel3A-GEI
The titration curves of ionizable active site residues in HjCel3A-GEI, with no acceptor other than water, are shown in Fig. S7.† The pKa and n values of the residues are summarized in Table 1, with the exception of H159, which did not titrate at all pH values simulated. A very low pKa (2.6 ± 0.2) was obtained for E441. Of the two glutamate residues near E441, E166 has a similarly low pKa (2.3 ± 0.1), while E128, located deeper in the active site, has a higher pKa (3.9 ± 0.4). The Hill coefficient provides an indication of the cooperativity between protonation sites (i.e., the proton affinity of one site is enhanced by protonation of another site); protonation is anti-cooperative if n < 1 and cooperative if n > 1.48 The moderate deviation of the n values from 1 indicates that the titration of the active site residues is not strongly coupled. The exception is E128, although the poor fit of the data in the titration curve indicates that the low n may be due to poor protonation state sampling (Fig. S7†) rather than deviation from ideal Henderson–Hasselbalch behavior.47| Residue | Without sugar | With glucose | ||
|---|---|---|---|---|
| pKa | n | pKa | n | |
| a Mean and standard deviation from the last 5 ns of simulation were calculated by averaging over 1 ns blocks. b Values not reported because of poor fit to the Henderson–Hasselbalch equation (Fig. S15). | ||||
| D61 | 1.6 ± 0.1 | 0.9 ± 0.5 | —b | —b |
| E128 | 3.9 ± 0.4 | 0.5 ± 0.1 | 4.8 ± 0.2 | 3.3 ± 0.8 |
| E166 | 2.3 ± 0.1 | 0.8 ± 0.4 | 3.5 ± 0.1 | 0.6 ± 0.3 |
| D370 | 4.1 ± 0.4 | 1.1 ± 0.6 | 4.2 ± 0.3 | 3.0 ± 2.1 |
| E441 | 2.6 ± 0.2 | 0.7 ± 0.1 | 2.0 ± 0.2 | 0.9 ± 0.4 |
Three independent simulations were performed at the optimum pH of HjCel3A (pH 5) to determine the impact of active site conformation, hydrogen bonding, charge coupling, and water availability on hydrolytic activity. A detailed description of each simulation is provided in ESI† and only general observations are discussed herein. The average structures from each simulation are similar (backbone RMSD of ∼0.5 Å), all of which showed R169 breaking its interaction with E441. Y204 also moved away from the −1 site to form a cation–π interaction with R169 (Fig. 2, Movie S1†). The active site conformation changed once the hydrogen bond network around E441 (involving R125, E128, E166, R169, and S384) was broken and the E441 side chain had moved away from the glycosyl moiety (Fig. S4 and S8†).
 | ||
| Fig. 2 Representative structures of HjCel3A-GEI in the absence (gray) and presence (green) of a glucose acceptor from the pH 5 simulations. A. View facing the substrate pocket entrance. B. View from side of the pocket. The positions of the active site residues are similar in the two structures except for R169 and Y204. In the absence of glucose, R169 and Y204, which form a cation–π interaction, are positioned further away from E441 and the glycosyl moiety. In the presence of glucose, Y204 is hydrogen bonded to W237, while R169 forms a hydrogen bond network with E441 and glucose. | ||
Consistent with the calculated pKa values, E441 was predominantly deprotonated (while oriented toward the glycosyl moiety) in the pH 5 simulations (Fig. S9–S11†). Its protonation state did not appear to be correlated to that of the neighboring glutamate residues, with only one of the three simulations indicating a weak positive correlation (r ∼ 0.2) with E128 (Table S1†). Nevertheless, both E128 and E166 formed a hydrogen bond network with E441 through R125 in all three simulations (Table S2†). The Glu–Arg–Glu motif is also observed in the acidic xylanase from A. kawachii (pH optima of 2), albeit with one Glu being the nucleophile. The second Glu is postulated to further strengthen the Glu–Arg interaction.49 This hypothesis would be consistent with the weak positive correlation observed in this study; protonation of E128 would weaken the E441–R125 hydrogen bond and increase the proton affinity of E441. The E128–R125–E441 and E166–R125–E441 hydrogen bond networks account for the low pKa of E441,50 which would presumably reduce its ability to deprotonate the catalytic water.17 For comparison, the experimental pKa of the acid/base residue (E172) in the trapped GEI of a GH Family 11 xylanase from B. circulans, which also follows the retaining mechanism (Scheme 1), is higher at 4.2 units.19 Unlike HjCel3A, the only residues within hydrogen bonding distance of E172 in the GEI crystal structure (PDB ID 1BVV51) of the xylanase are N35 and Y80. The predicted pKa of the GEI of the GH1 βgls BGLA and BGLB from B. polymyxa, based on the crystal structures (PDB IDs 1E4I52 and 2JIE,53 respectively), are 6.1 and 5.4 units, respectively.54 The acid/base residue has a hydrophobic environment as well, comprising tryptophan, methionine, and asparagine residues.
Experimental studies on retaining βgl and β-galactosidases suggest that stability of the glycosyl or galactosyl moiety at the −1 site is more critical to hydrolysis than transglycosylation.55–57 Talens-Perales, et al. also hypothesized in their β-galactosidase study that the greater mobility of the galactosyl moiety would facilitate its transfer to a sugar acceptor.57 In HjCel3A, the glycosyl moiety was stabilized by hydrogen bonding to D61, doubly protonated H159, and S384 (Table S2†). The hydrogen bonds to R125 and K158 found in the HjCel3A crystal structure with glucose at the −1 site (PDB ID 3ZYZ30) were not observed in any of the simulations. Two to four water molecules were found within the first hydration shell of E441 (Fig. S12A†), occupying the area where the R169 and Y204 side chains were originally positioned in the crystal structure. The population of trajectories having a reactive configuration was determined using distance criteria based on QM/MM calculations of retaining GHs – dOH2-C1 < 3.8 Å for the attack of water on the anomeric carbon (C1 atom) of the glycosyl moiety and dH*-OE* < 2 Å for the proton transfer from water to E441.16,58,59 Fig. S13A–S13C† show that the population of reactive configurations was ∼5% at most. As discussed earlier, the E441 side chain moves farther from the glycosyl moiety once the hydrogen bond network around it is broken (Fig. S4 and S8†).
Water-bridged hydrogen bond interactions were also analyzed to locate possible binding sites for the catalytic water. R169 could position a water molecule for proton transfer to E441 (Movie S1†), as well as place it close enough to attack the anomeric carbon. However, the occupancy of this water-bridged interaction was rather low (<10%). In some GHs, such as α-amylases and endoxylanases, a tyrosine residue in the catalytic center acts as the anchoring point for water.60,61 In HjCel3A, this residue could be Y204, located next to the nucleophile D236. However, two of the three simulations showed only a very short-lived (<1% occupancy) water-bridged hydrogen interaction of the Y204 hydroxyl group with the E441 carboxylate oxygen atoms, which occurred as the C–Cα–Cβ–Cγ dihedral of Y204 rotated from ∼160° to ∼−(90–100)° (Fig. S14A, Movie S1†). Unlike the tyrosine residue found in GH1 βgl, Y204 is not positioned in front of the O5 atom of the glycosyl moiety (Fig. 1); thus, it is also unlikely to play a role in the stabilization of the oxocarbenium-like transition state.16
Transglycosylation—HjCel3A-GEI with glucose acceptor
The titration curves of ionizable active site residues in the presence of a glucose acceptor in the +1 site are shown in Fig. S15,† while the pKa and n values are summarized in Table 1. Reliable values for D61 could not be obtained because of the very poor fit of data at pH < 4, while H159 also did not titrate (Fig. S15†). The pKa of E441 is lower at 2.0 ± 0.2, while that of E128 and E166 are higher at 4.8 ± 0.2 and 3.5 ± 0.1, respectively, compared to HjCel3A-GEI without glucose. The n values of E128 and D370 are much greater than 1, indicating that these residues are protonated cooperatively.The glucose acceptor was placed at the +1 site with its O4 atom positioned for transfer to the anomeric carbon of the glycosyl moiety at the −1 site (Fig. S16A†). This initial structure represents the case where the glucose product is retained in the active site after cleavage of the cellobiose glycosidic bond. However, the structure equilibrated to one wherein the O6 atom, previously hydrogen bonded to Y204, moved closer to the anomeric carbon, while the HO4 hydrogen formed a hydrogen bond with the O6 atom of the glycosyl moiety (Fig. S16B†). Rather than reformation of cellobiose through transglycosylation, this equilibrated conformation would lead to formation of gentiobiose, which happens to be one of the major transglycosylation products obtained for HjCel3A.21 Unlike in the pH-REX simulations, glucose was not restrained during the pH 5 simulations to avoid bias in the analysis of interactions. The binding of glucose proved to be unstable, as it remained bound in the +1 site for 10 ns in only one of the three simulations. This may be attributed to disruption of hydrogen bond interactions, discussed in more detail in ESI.† Analyses of hydrogen bonds and λ correlation were, therefore, performed using only trajectories where glucose was in the +1 site.
The average structures from each simulation did not differ significantly, even in the positions of the side chains of the active site residues (backbone RMSD of 0.4–0.5 Å). The major difference from the active site conformation of HjCel3A-GEI without glucose is that R169 and Y204 remained close to their initial positions in the +1 and −1 sites. There was no cation–π interaction between the two residues in two of the simulations because the hydrogen bonding between Y204 and W237 (35% occupancy) prevented the former from moving closer and orienting parallel to R169 (Fig. 2, Movie S2†). The retention of the R169 and Y204 positions at the +1/−1 sites, together with the presence of glucose, limited the solvent access of E441 and the glycosyl moiety (Fig. S12B†). As for the other ionizable residues in the +1 site, the positions of R67 and Y443 were similar in the presence and absence of glucose. Both residues were hydrogen bonded to D370 located in loop B close to the active site loop (loop C) (Fig. S2A†). However, D370 can also adopt the protonated state because its pKa is close to the optimum pH, which would break its interaction with either residue.
E441 was predominantly deprotonated in only two of the three simulations (Fig. S17–S19†). The protonation of E441 was not strongly correlated (r < |0.5|) to that of E128 and E166 (Table S3†). The E128–R125–E441 and E166–R125–E441 hydrogen bond networks were also observed, and R169 was in close proximity to E441 because of its hydrogen bonds to E166 and glucose. Taken together, this would account for the lower pKa of E441 in the presence of glucose. However, hydrogen bond analysis could not explain the higher pKa of E128 and E166; in fact, E166 has an additional hydrogen bond with R169 (22% occupancy). The shift in the pKa values was possibly caused by increased hydrophobicity at the active site due to glucose. E166 was predominantly deprotonated during the simulations (Fig. S17–S19†). Although the pKa of E128 is similar to the simulation pH, the protonated state was sampled often in only one of the simulations (Fig. S18†). Contrary to the calculated Hill coefficient, a negative correlation with the neighboring residue, H159, was obtained from all simulations (Table S3†). The trajectories showed that when H159 is deprotonated at the Nε atom, R125 replaces it as the hydrogen bond partner of the O2/O3 atom of the glycosyl moiety. Consequently, the interaction of E128 with R125 is broken, leading the former to adopt the protonated state and directly interact with E441 (Movie S2†). In the absence of glucose, the correlation between E128 and H159 was not observed (Table S1†), as the latter was not responsible for breaking the E128–R125 hydrogen bond.
The glycosyl intermediate is stabilized by hydrogen bonds with D61, doubly protonated H159, and S384, as in the absence of glucose. Glucose was held at the +1 site by hydrogen bonding of the O6 atom with R169 and E441 (Table S4†). Because of this hydrogen bond network, reactive configurations leading to gentiobiose formation (attack by the O6 atom) have a much higher population than those leading to cellobiose formation (attack by the O4 atom) or hydrolysis (Fig. S13†). Glucose has a lower pKa (12.28 at 25 °C) than water.62 Using the experimental homolytic bond enthalpy, DH298(RH), electron affinity, EA0(R), and ionization energy, IE0(H),63,64 the estimated deprotonation enthalpy (eqn (2))63 is 379/377 kcal mol−1 for a 1°/2° alcohol compared to 390 kcal mol−1 for water.
| ΔacidH298(RH) ≈ DH298(RH) − EA0(R) + IE0(H) | (2) |
Moreover, the acidity of HO6 is presumably enhanced by hydrogen bonding with R169 because the excess charge that would develop on oxygen during proton transfer could be transferred to this residue.65 The relative ease of glucose deprotonation would account for the transglycosidic activity of HjCel3A, despite the unstable binding of glucose in the +1 site observed during the simulations.
Transglycosylation—HjCel3A-GEI with cellobiose acceptor
Simulations of HjCel3A-GEI with a cellobiose acceptor were also performed to investigate the effect of another glucose unit at the +2 site on the position, interactions, and protonation state of residues at the −1 and +1 sites. The non-reducing end of cellobiose was positioned similarly at the +1 site as the glucose acceptor. Doing so placed the reducing end in a stacking interaction with F260, though too far from D370 to form a hydrogen bond. The equilibrated structure is similar to that of HjCel3A-GEI with glucose, in which the O6 atom of the non-reducing end has moved closer to the anomeric carbon of the glycosyl moiety. The reducing end, on the other hand, has bent to form a T-shaped interaction with F260 (Fig. S16C†).Details of the three simulations performed are described in ESI.† The resulting average structures have a backbone RMSD of 0.4 Å and a representative structure is shown in Fig. 3. The positions and hydrogen bond interactions of R169, Y204, R67, Y443, and D370 are similar to the HjCel3A-GEI-glucose model. The E166–R125–E441 hydrogen bond network was formed in all simulations. On the other hand, the E128–R125–E441 hydrogen bond network was observed in only one simulation because in the other two, E128 frequently interacted directly with E441 and adopted the protonated state. As the pH-REX simulations of HjCel3A-GEI in the presence of a glucose acceptor showed, E128 has a pKa close to the solution pH. Nevertheless, E441 remained predominantly deprotonated (Fig. S20–S22†). Thus, the E441 pKa would likely be similarly low in the presence of a cellobiose acceptor. No strong correlation between the protonation states of any of the residues was observed (Table S5†).
 | ||
| Fig. 3 Representative structures of HjCel3A-GEI with glucose (gray) and cellobiose (green) acceptor from the pH 5 simulations. The positions of residues around the +1 acceptor site (R169, Y204, R67, Y443, and D370) are similar in the 2 models. However, the non-reducing end of cellobiose is not hydrogen bonded to E441 unlike glucose. The reducing end of cellobiose interacts mainly with F260. | ||
The glycosyl moiety also had hydrogen bonds with D61, H159, and S384. On the other hand, the O6 atom of the non-reducing end of cellobiose only had a hydrogen bond with R169 and not with E441 (Table S6†). This could explain the slight difference in the first hydration shell of E441 between the two transglycosylation models (Fig. S12†). Additionally, the HO4 atom of the non-reducing end was hydrogen bonded to the O6 atom of the glycosyl moiety (Table S6†). This interaction was not observed in the HjCel3A-GEI-glucose model. Trajectories fulfilling both distance criteria for a reactive configuration, that leading to formation of 6-O-β-D-glucopyranosyl-4-O-β-D-glucopyranosyl-D-glucopyranose, were found only in one of the simulations. While trajectories having an O4 atom-anomeric carbon distance of 3.5–4 Å (leading to the observed major product, cellotriose21) have relatively high population, the HO4 atom–E441 carboxylate oxygen distances were long (∼6 Å) (Fig. S13J–S13O†). As the hydrogen bond analysis showed, the HO4 atom is already hydrogen bonded to the glycosyl moiety. Alternative initial structures may need to be used to obtain a higher population of configurations leading to cellotriose formation. However, since the protonation states of ionizable residues at the −1 and +1 sites do not appear to be highly sensitive to the presence of an acceptor at the positive sites, additional simulations using new structures were not performed.
Mutational effects on E441 pKa
Structural comparisons with available crystal structures of other GH3 βgls showed that the ionizable residues at the −1 and +1 sites, as well as E128 and E166, are conserved. The exception is the barley β-D-glucan glucohydrolase isoenzyme Exo1, which has N219 for E166 and E220 for R169 (Fig. S23†).66 Thus, the pKa of the acid/base residue of GH3 βgls may be expected to be low in general. As this may be a contributing factor to reduced hydrolytic activity, mutations that can elevate the pKa were investigated. E128 and E166 were selected not only because they may modulate the E441 pKa through the Glu–Arg–Glu hydrogen bond network discussed above, but also because substitutions at these positions are unlikely to affect binding of the cellobiose substrate. CpHMD simulations (pH 5) of the E128 mutants without a sugar acceptor (Fig. S24 and S25†) show the breaking of the E441–R125 hydrogen bond upon mutation of E128 to a neutral residue (Fig. 4, Tables S7 and S8†). Sampling of the E441 protonated state is enhanced in the E128Q mutant (Fig. S26†), which has a relatively low occupancy for the E441–S384 hydrogen bond (Table S8†). Comparison of representative structures of the wild-type and E128Q HjCel3A-GEI indicates that this is due to the shift in the positions of the loops containing E441 and S384 upon mutation (Fig. 4). The E166A and E166Q mutants were also investigated, but simulations indicate that substitution at this position would not elevate the E441 pKa (Fig. S27†). | ||
| Fig. 4 Representative structures of the GEI of wild-type HjCel3A (gray) and E128Q mutant (green) from the pH 5 simulations. The substitution breaks the E128–R125–E441 and E166–R125–E441 hydrogen bond networks and shifts the positions of the loops containing the acid/base residue E441 and its hydrogen bond partner, S384. These conditions led to enhanced sampling of the E441 protonation state. | ||
Conclusions
CpHMD/pH-REX simulations showed that the pKa of the catalytic acid/base residue of HjCel3A, E441, is very low regardless of whether the acceptor molecule is water (hydrolysis) or a sugar (transglycosylation). The protonation states of ionizable residues found at the substrate −1 binding site, where the glycosyl moiety is located, are also similar in both cases. Thus, the pKa factor alone could not account for the reduced hydrolytic activity/increased transglycosidic activity of HjCel3A at high cellobiose or glucose concentration. Active site conformation and acceptor-residue interactions must also be considered. In the absence of a sugar acceptor, the residues R169 and Y204, which form a cation–π interaction, moved away from the +1/−1 sites, and there appeared to be no residues other than E441 that can anchor and/or activate catalytic water for proton transfer. In contrast, the glucose or cellobiose acceptor was anchored at the +1 site by R169 through hydrogen bonding. Such interaction would, moreover, increase the acidity of the sugar and make proton transfer favorable despite the low basicity of E441. Therefore, two possible approaches can be adopted to enhance the hydrolytic activity of HjCel3A. First, elevate the pKa of E441 to enhance its ability to activate water by mutating neighboring residues. For example, substitution of E128 with neutral residues breaks both the E128–R125–E441 and E166–R125–E441 hydrogen bond networks and was shown by simulations to improve the proton affinity of E441. Second, reduce the +1 site affinity and reactivity of a sugar acceptor by substituting R169. The effect of these predicted mutations on the pH-activity profile and H/T partition of HjCel3A will be experimentally verified in the future as part of our ongoing comprehensive study exploring various governing factors of H/T partition. The findings of this study are also expected to be applicable to other GH3 βgls because the ionizable active site residues are generally conserved in this family of βgls. Furthermore, the consistency of our findings with the experimental study of α-amylase from B. stearothermophilus suggests that pure transglycosylases can be potentially engineered from βgls by altering the electrostatic environment of the catalytic acid/base residue.Conflicts of interest
There are no conflicts to declare.Acknowledgements
Acknowledgement is made to the Swedish Governmental Agency for Innovation Systems (VINNOVA, Grant No. 2015-04835) for support of this research. Simulations were performed on resources provided by the Swedish National Infrastructure for Computing (SNIC) at High Performance Computing Center North (HPC2N) and National Science Foundation (NSF) Extreme Science and Engineering Discovery Environment (XSEDE). XSEDE is supported by NSF Grant No. ACI-1548562 (Gordon cluster under allocation MCB090159).67 This material is also based upon work supported by (while CMP is serving at) the NSF. Any opinion, findings, and conclusions or recommendations expressed in this material are those of the authors and do not necessarily reflect the views of the NSF.Notes and references
- Carbohydrate Active Enzymes Database, http://www.cazy.org/ (accessed April 2017).
- V. Lombard, H. Golaconda Ramulu, E. Drula, P. M. Coutinho and B. Henrissat, Nucleic Acids Res., 2014, 42, D490–D495 CrossRef CAS PubMed.
- J. R. Ketudat Cairns and A. Esen, Cell. Mol. Life Sci., 2010, 67, 3389–3405 CrossRef CAS PubMed.
- D. E. Koshland, Biol. Rev., 1953, 28, 416–436 CrossRef CAS.
- G. J. Davies, L. Mackenzie, A. Varrot, M. Dauter, A. M. Brzozowski, M. Schülein and S. G. Withers, Biochemistry, 1998, 37, 11707–11713 CrossRef CAS PubMed.
- C. M. Payne, B. C. Knott, H. B. Mayes, H. Hansson, M. E. Himmel, M. Sandgren, J. Ståhlberg and G. T. Beckham, Chem. Rev., 2015, 115, 1308–1448 CrossRef CAS PubMed.
- A. Ardèvol and C. Rovira, J. Am. Chem. Soc., 2015, 137, 7528–7547 CrossRef PubMed.
- B. Bissaro, P. Monsan, R. Faure and M. J. O'Donohue, Biochem. J., 2015, 467, 17–35 CrossRef CAS PubMed.
- A. Sørensen, M. Lübeck, P. Lübeck and B. Ahring, Biomolecules, 2013, 3, 612–631 CrossRef PubMed.
- I. S. Ng, S.-W. Tsai, Y.-M. Ju, S.-M. Yu and T.-H. D. Ho, Bioresour. Technol., 2011, 102, 6073–6081 CrossRef CAS PubMed.
- Y. Bhatia, S. Mishra and V. S. Bisaria, Crit. Rev. Biotechnol., 2002, 22, 375–407 CrossRef CAS PubMed.
- B. M. de Roode, M. C. R. Franssen, A. V. D. Padt and R. M. Boom, Biotechnol. Prog., 2003, 19, 1391–1402 CrossRef PubMed.
- H. F. Seidle and R. E. Huber, Arch. Biochem. Biophys., 2005, 436, 254–264 CrossRef CAS PubMed.
- P. Lundemo, E. N. Karlsson and P. Adlercreutz, Appl. Microbiol. Biotechnol., 2017, 101, 1121–1131 CrossRef CAS PubMed.
- R. E. Huber, M. T. Gaunt, R. L. Sept and M. J. Babiak, Can. J. Biochem. Cell Biol., 1983, 61, 198–206 CAS.
- S. Badieyan, D. R. Bevan and C. Zhang, Biochemistry, 2012, 51, 8907–8918 CrossRef CAS PubMed.
- G. Saab-Rincón, G. del-Río, R. I. Santamaría, A. López-Munguía and X. Soberón, FEBS Lett., 1999, 453, 100–106 CrossRef.
- H. L. Frericks Schmidt, G. J. Shah, L. J. Sperling and C. M. Rienstra, J. Phys. Chem. Lett., 2010, 1, 1623–1628 CrossRef CAS PubMed.
- L. P. McIntosh, G. Hand, P. E. Johnson, M. D. Joshi, M. Körner, L. A. Plesniak, L. Ziser, W. W. Wakarchuk and S. G. Withers, Biochemistry, 1996, 35, 9958–9966 CrossRef CAS PubMed.
- M. D. Joshi, A. Hedberg and L. P. McIntosh, Protein Sci., 1997, 6, 2667–2670 CrossRef CAS PubMed.
- B. Guo, N. Sato, P. Biely, Y. Amano and K. Nozaki, Appl. Microbiol. Biotechnol., 2016, 100, 4959–4968 CrossRef CAS PubMed.
- M. D. Joshi, G. Sidhu, J. E. Nielsen, G. D. Brayer, S. G. Withers and L. P. McIntosh, Biochemistry, 2001, 40, 10115–10139 CrossRef CAS PubMed.
- A. Olivera-Nappa, B. A. Andrews and J. A. Asenjo, Biotechnol. Bioeng., 2004, 86, 573–586 CrossRef CAS PubMed.
- W. Im, M. S. Lee and C. L. Brooks, J. Comput. Chem., 2003, 24, 1691–1702 CrossRef CAS PubMed.
- M. S. Lee, F. R. Salsbury and C. L. Brooks, Proteins: Struct., Funct., Bioinf., 2004, 56, 738–752 CrossRef CAS PubMed.
- J. Khandogin and C. L. Brooks, Biophys. J., 2005, 89, 141–157 CrossRef CAS PubMed.
- J. A. Wallace and J. K. Shen, J. Chem. Theory Comput., 2011, 7, 2617–2629 CrossRef CAS PubMed.
- L. Bu, M. F. Crowley, M. E. Himmel and G. T. Beckham, J. Biol. Chem., 2013, 288, 12175–12186 CrossRef CAS PubMed.
- D. M. Granum, S. Vyas, S. V. Sambasivarao and C. M. Maupin, J. Phys. Chem. B, 2014, 118, 434–448 CrossRef CAS PubMed.
- S. Karkehabadi, K. E. Helmich, T. Kaper, H. Hansson, N.-E. Mikkelsen, M. Gudmundsson, K. Piens, M. Fujdala, G. Banerjee, J. S. Scott-Craig, J. D. Walton, G. N. Phillips and M. Sandgren, J. Biol. Chem., 2014, 289, 31624–31637 CrossRef CAS PubMed.
- C. Bohlin, E. Praestgaard, M. Baumann, K. Borch, J. Praestgaard, R. Monrad and P. Westh, Appl. Microbiol. Biotechnol., 2013, 97, 159–169 CrossRef CAS PubMed.
- B. R. Brooks, C. L. Brooks III, A. D. Mackerell Jr., L. Nilsson, R. J. Petrella, B. Roux, Y. Won, G. Archontis, C. Bartels, S. Boresch, A. Caflisch, L. Caves, Q. Cui, A. R. Dinner, M. Feig, S. Fischer, J. Gao, M. Hodoscek, W. Im, K. Kuczera, T. Lazaridis, J. Ma, V. Ovchinnikov, E. Paci, R. W. Pastor, C. B. Post, J. Z. Pu, M. Schaefer, B. Tidor, R. M. Venable, H. L. Woodcock, X. Wu, W. Yang, D. M. York and M. Karplus, J. Comput. Chem., 2009, 30, 1545–1614 CrossRef CAS PubMed.
- A. D. MacKerell, D. Bashford, M. Bellott, R. L. Dunbrack, J. D. Evanseck, M. J. Field, S. Fischer, J. Gao, H. Guo, S. Ha, D. Joseph-McCarthy, L. Kuchnir, K. Kuczera, F. T. K. Lau, C. Mattos, S. Michnick, T. Ngo, D. T. Nguyen, B. Prodhom, W. E. Reiher, B. Roux, M. Schlenkrich, J. C. Smith, R. Stote, J. Straub, M. Watanabe, J. Wiórkiewicz-Kuczera, D. Yin and M. Karplus, J. Phys. Chem. B, 1998, 102, 3586–3616 CrossRef CAS PubMed.
- M. Feig, A. D. MacKerell and C. L. Brooks, J. Phys. Chem. B, 2003, 107, 2831–2836 CrossRef CAS.
- A. D. Mackerell, M. Feig and C. L. Brooks, J. Comput. Chem., 2004, 25, 1400–1415 CrossRef CAS PubMed.
- A. D. MacKerell, M. Feig and C. L. Brooks, J. Am. Chem. Soc., 2004, 126, 698–699 CrossRef CAS PubMed.
- W. Im, M. Feig and C. L. Brooks III, Biophys. J., 2003, 85, 2900–2918 CrossRef CAS PubMed.
- O. Guvench, E. Hatcher, R. M. Venable, R. W. Pastor and A. D. MacKerell, J. Chem. Theory Comput., 2009, 5, 2353–2370 CrossRef CAS PubMed.
- O. Guvench, S. S. Mallajosyula, E. P. Raman, E. Hatcher, K. Vanommeslaeghe, T. J. Foster, F. W. Jamison and A. D. MacKerell, J. Chem. Theory Comput., 2011, 7, 3162–3180 CrossRef CAS PubMed.
- S. S. Mallajosyula, O. Guvench, E. Hatcher and A. D. MacKerell, J. Chem. Theory Comput., 2012, 8, 759–776 CrossRef CAS PubMed.
- E. P. Raman, O. Guvench and A. D. MacKerell, J. Phys. Chem. B, 2010, 114, 12981–12994 CrossRef CAS PubMed.
- S. R. Durell, B. R. Brooks and A. Ben-Naim, J. Phys. Chem., 1994, 98, 2198–2202 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- M. Nina, D. Beglov and B. Roux, J. Phys. Chem. B, 1997, 101, 5239–5248 CrossRef CAS.
- J. Chen, W. Im and C. L. Brooks, J. Am. Chem. Soc., 2006, 128, 3728–3736 CrossRef CAS PubMed.
- I. K. McDonald and J. M. Thornton, J. Mol. Biol., 1994, 238, 777–793 CrossRef CAS PubMed.
- J. M. Swails and A. E. Roitberg, J. Chem. Theory Comput., 2012, 8, 4393–4404 CrossRef CAS PubMed.
- G. Kaslik, W. M. Westler, L. Gráf and J. L. Markley, Arch. Biochem. Biophys., 1999, 362, 254–264 CrossRef CAS PubMed.
- J. Kongsted, U. Ryde, J. Wydra and J. H. Jensen, Biochemistry, 2007, 46, 13581–13592 CrossRef CAS PubMed.
- T. K. Harris and G. J. Turner, IUBMB Life, 2002, 53, 85–98 CrossRef CAS PubMed.
- G. Sidhu, S. G. Withers, N. T. Nguyen, L. P. McIntosh, L. Ziser and G. D. Brayer, Biochemistry, 1999, 38, 5346–5354 CrossRef CAS PubMed.
- J. Sanz-Aparicio, J. A. Hermoso, M. Martinez-Ripoll, B. Gonzalez, C. Lopez-Camacho and J. Polaina, Proteins, 1998, 33, 567–576 CrossRef CAS.
- P. Isorna, J. Polaina, L. Latorre-García, F. J. Cañada, B. González and J. Sanz-Aparicio, J. Mol. Biol., 2007, 371, 1204–1218 CrossRef CAS PubMed.
- P. Ramachandran, S. S. Jagtap, S. K. S. Patel, J. Li, Y. Chan Kang and J.-K. Lee, RSC Adv., 2016, 6, 48137–48144 RSC.
- D. Teze, J. Hendrickx, M. Czjzek, D. Ropartz, Y. H. Sanejouand, V. Tran, C. Tellier and M. Dion, Protein Eng., Des. Sel., 2014, 27, 13–19 CrossRef CAS PubMed.
- G. Placier, H. Watzlawick, C. Rabiller and R. Mattes, Appl. Environ. Microbiol., 2009, 75, 6312–6321 CrossRef CAS PubMed.
- D. Talens-Perales, J. Polaina and J. Marín-Navarro, J. Agric. Food Chem., 2016, 64, 2917–2924 CrossRef CAS PubMed.
- N. F. Brás, P. A. Fernandes and M. J. Ramos, J. Chem. Theory Comput., 2010, 6, 421–433 CrossRef PubMed.
- J. Wang, Q. Hou, X. Sheng, J. Gao, Y. Liu and C. Liu, Int. J. Quantum Chem., 2013, 113, 1071–1075 CrossRef CAS.
- M. Mizuno, T. Tonozuka, A. Uechi, A. Ohtaki, K. Ichikawa, S. Kamitori, A. Nishikawa and Y. Sakano, Eur. J. Biochem., 2004, 271, 2530–2538 CrossRef CAS PubMed.
- T. Collins, D. De Vos, A. Hoyoux, S. N. Savvides, C. Gerday, J. Van Beeumen and G. Feller, J. Mol. Biol., 2005, 354, 425–435 CrossRef CAS PubMed.
- Applications of Ion Chromatography for Pharmaceutical and Biological Products, ed. L. Bhattacharyya and J. S. Rohrer, John Wiley & Sons, Inc., Hoboken, NJ, USA, 2012, pp. 455–456 Search PubMed.
- S. J. Blanksby and G. B. Ellison, Acc. Chem. Res., 2003, 36, 255–263 CrossRef CAS PubMed.
- J. C. Rienstra-Kiracofe, G. S. Tschumper, H. F. Schaefer, S. Nandi and G. B. Ellison, Chem. Rev., 2002, 102, 231–282 CrossRef CAS PubMed.
- A. Shokri, A. Abedin, A. Fattahi and S. R. Kass, J. Am. Chem. Soc., 2012, 134, 10646–10650 CrossRef CAS PubMed.
- M. Hrmova, J. N. Varghese, R. De Gori, B. J. Smith, H. Driguez and G. B. Fincher, Structure, 2001, 9, 1005–1016 CrossRef CAS PubMed.
- J. Towns, T. Cockerill, M. Dahan, I. Foster, K. Gaither, A. Grimshaw, V. Hazlewood, S. Lathrop, D. Lifka, G. D. Peterson, R. Roskies, J. R. Scott and N. Wilkins-Diehr, Comput. Sci. Eng., 2014, 16, 62–74 CrossRef.
Footnotes |
| † Electronic supplementary information (ESI) available: Details of computational method, glycosyl-enzyme intermediate force field parameters, simulation analyses, and movies of trajectories. See DOI: 10.1039/c7ob02558k |
| ‡ Present Address: Directorate of Engineering, Division of Chemical, Bioengineering, Environmental and Transport Systems, National Science Foundation, Alexandria, Virginia 22314, USA. |
| This journal is © The Royal Society of Chemistry 2018 |