Intramolecular nucleophilic addition of carbanions generated from N-benzylamides to cyclopropenes†
Received
18th August 2017
, Accepted 8th December 2017
First published on 8th December 2017
Abstract
An unusual reaction is described, involving a formal intramolecular nucleophilic substitution of bromocyclopropanes with nitrogen ylides generated in situ from N-benzyl carboxamides. It is shown that this reaction involves cyclopropene intermediates and allows for the facile and expeditious preparation of 3-azabicyclo[3.1.0]hexan-2-one scaffolds.
Introduction
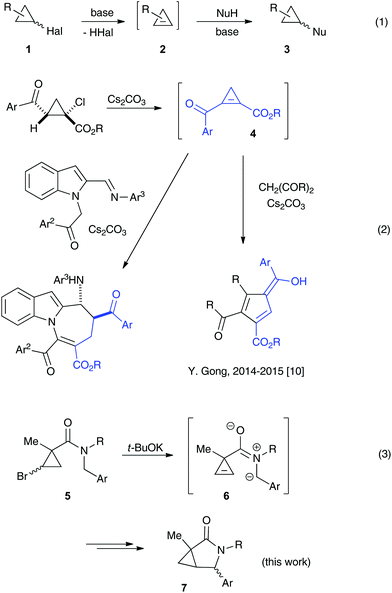
The base-assisted additions of heteroatom nucleophiles to cyclopropenes 2 generated in situ from stable halocyclopropanes 1 have emerged as a convenient route towards complex cyclopropyl scaffolds,1 complementary to existing transition metal-catalyzed methodologies2,3 (Scheme 1, eqn (1)). Oxygen-,4 nitrogen-,5 sulfur-,6 or halogen7-based entities have been successfully added, in either an inter- or an intramolecular fashion.8 The employment of carbon-based nucleophilic species in non-catalyzed transformations of these types has thus far been less abundant. The apparent challenges associated with a strong basicity of organometallic reagents on the one hand, and a lower reactivity of stabilized carbon nucleophiles, such as enolates, toward non-conjugate cyclopropenes on the other hand, have limited the application of this chemistry. Within this scope, the addition of strongly nucleophilic organometallic reagents to cyclopropenes has been known since the 1970s.9 A regioselective variant of these chemistry exploiting bis-metallated cyclopropyl intermediates was later shown by Marek.10 More recently, Gong demonstrated the Michael addition of enolates to highly activated conjugate cyclopropenylketone 4 generated in situ, which was accompanied by the cleavage of the three-membered ring (Scheme 1, eqn (2)).11 Herein, we wish to report an intramolecular, ring-retentive 5-exo-trig cyclization of non-conjugated cyclopropenes 6 with nitrogen ylides, generated from N-benzylcarboxamides 5 in the presence of relatively mild alkoxide bases (Scheme 1, eqn (3)). This process allowed for the straightforward and highly expeditious assembly of biologically relevant 3-azabicyclo[3.1.0]hexan-2-one scaffolds, although in only moderate yields and selectivity.
 |
| | Scheme 1 | |
Results and discussion
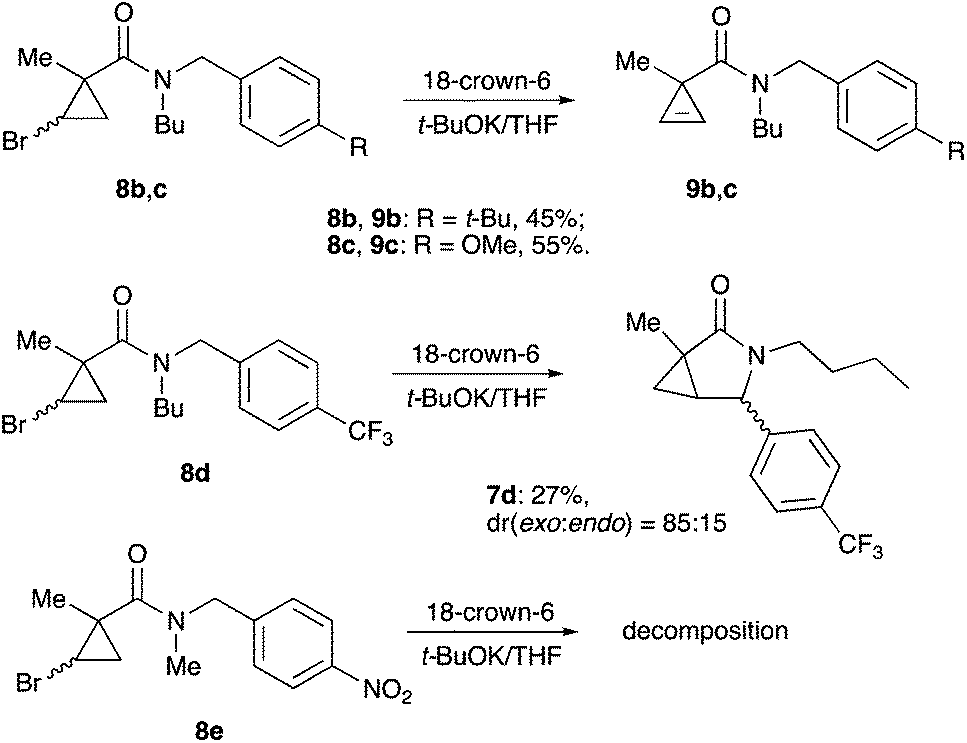
One of the challenges we encountered while developing the formal nucleophilic substitution of bromocyclopropanes was limitations in the existing synthetic approaches to cyclopropenes. Accordingly, a large part of our efforts was focused on expanding the scope of well-established methods to broaden the range of available strained olefins.12 Thus, we demonstrated that a very efficient 1,2-dehydrohalogenation of bromocyclopropanes could be carried out in THF in the presence of catalytic amounts of 18-crown-6 ether.13 This modification allowed for more convenient isolation and improved overall yields of cyclopropenes compared to the classical protocol in dry DMSO. This proved to be particularly beneficial for the synthesis of functionalized cyclopropenes, such as tertiary carboxamides 9 bearing an alkyl group and an electron rich aryl group.13 Recently, we probed this procedure for the preparation of N,N-benzyl substituted cyclopropene 9a from the corresponding bromocyclopropane 8 (R1 = R2 = Bn) (Scheme 2). Surprisingly, instead of olefin 9a, a mixture of diastereomeric lactams 7a was produced in ca. 60% yield.14 This was a pleasant surprise, since this scaffold occurs in nature15 and has significant importance for medicinal chemistry16 and synthetic methodology.17 Employing excess freshly sublimed tert-butoxide and carrying out the reaction under strictly anhydrous conditions allowed for the improvement of the yield up to 80%, but did not affect the diastereomeric composition of the product (Scheme 2). This unexpected 5-exo-trig cyclization was apparently triggered by the base-assisted deprotonation at the benzylic position of cyclopropene intermediate 9a (Scheme 3). The formation of anionic species in an α-position to nitrogen in carboxamides is well precedented.18 Stoichiometric deprotonation for intermolecular alkylation normally requires strong organometallic bases such as t-BuLi or n-BuLi.18 For intramolecular reactions, however, the use of LDA and even t-BuOK has also been reported, particularly successful in the Hurtly arylation.19
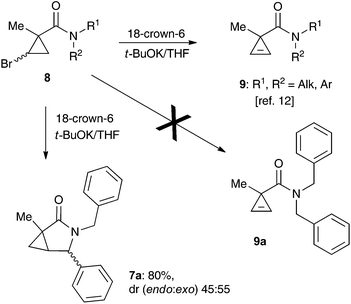 |
| | Scheme 2 | |
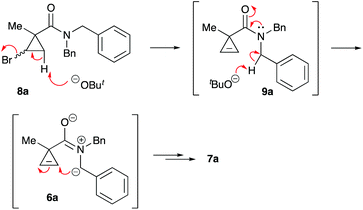 |
| | Scheme 3 | |
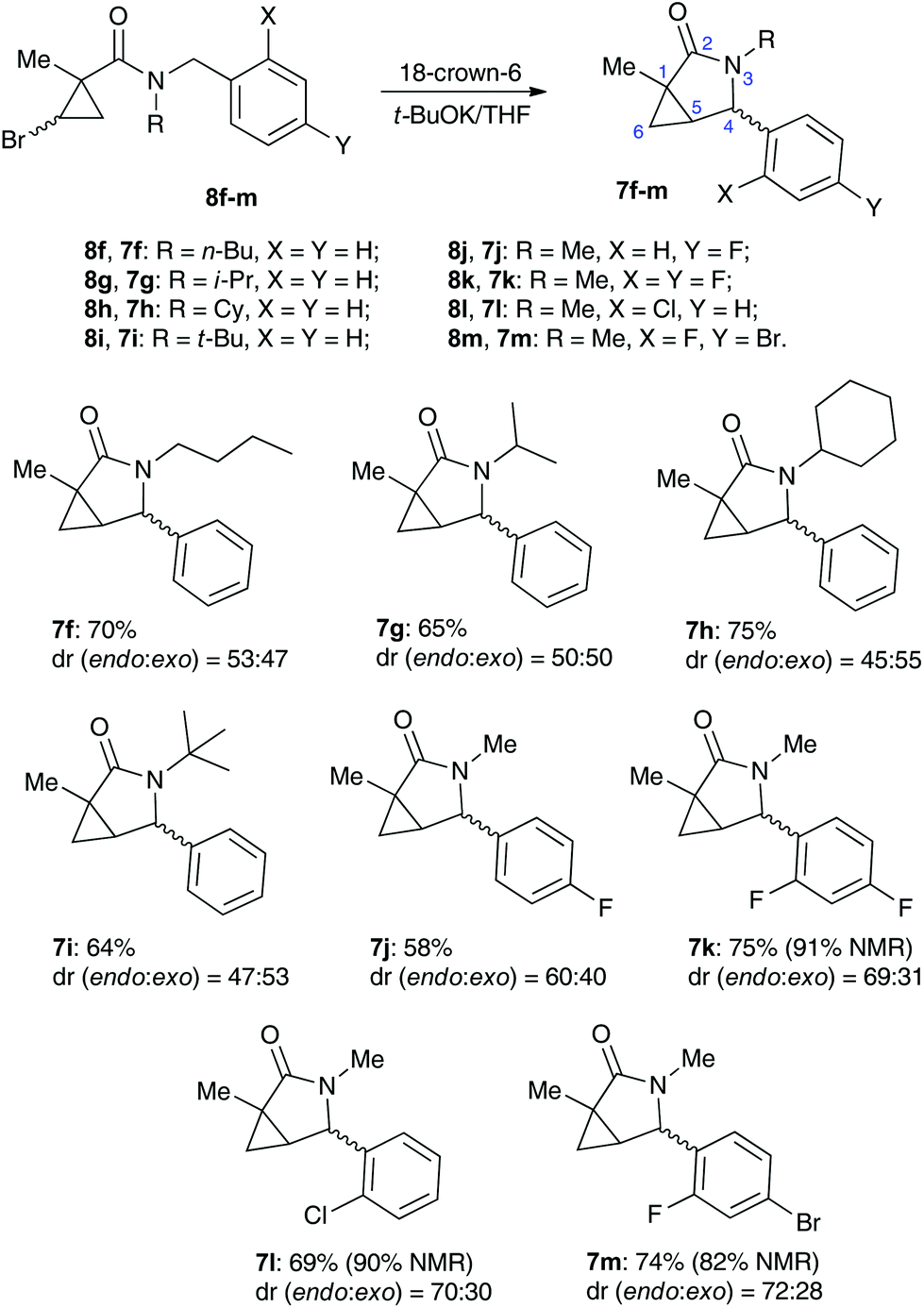
We reasoned that lowering the C–H acidity of the benzylic group could help shut down the 5-exo-trig cyclization pathway and divert it to the desired dehydrohalogenation. Indeed, bromocyclopropanes 8b,c possessing electron-donating substituents in an aromatic ring produced the corresponding cyclopropenes 9b,c as sole isolable products in moderate yields (Scheme 4). Conversely, the stabilization of the benzylic anion by a strong electron-withdrawing substituent resulted in reduced nucleophilicity, which made the cyclization inefficient. Thus, the reaction of 8d, bearing a CF3 group in the para-position, afforded a very poor yield of bicyclic product 7d,20 while p-NO2 analog 8e simply decomposed under the reaction conditions (Scheme 4). All of the substrates possessing neutral or moderately electron-withdrawing substituents reacted smoothly affording the corresponding 3-azabicyclo[3.1.0]hexan-2-ones in moderate to high yields (Scheme 5). Remarkably, this reaction demonstrated high tolerance to steric hindrance at the nitrogen atom, as we were able to efficiently cyclize the substrates bearing (a) primary, Me (8j–8m) and n-Bu (8f), (b) secondary, i-Pr and Cy (8h,g, respectively), and (c) tertiary, t-Bu (8i) groups. Interestingly, steric hindrance on the nitrogen atom influences diastereoselectivity; however, this effect is quite weak.
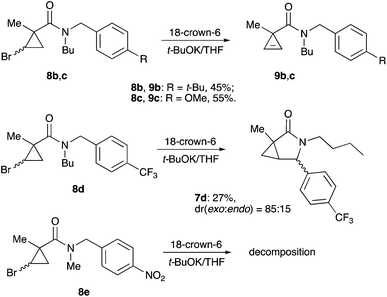 |
| | Scheme 4 | |
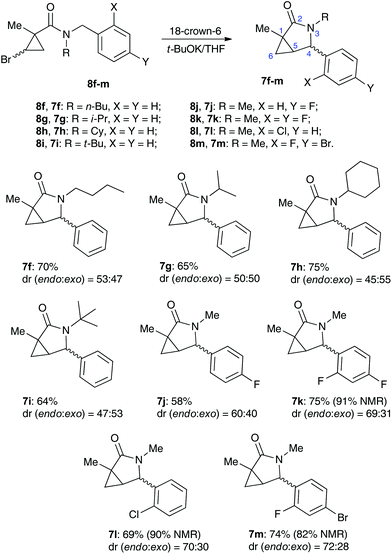 |
| | Scheme 5 | |
It should be emphasized that all of the bicyclic products 7 were obtained as mixtures of endo- and exo-diastereomers. This was not surprising, taking into account the relatively high acidity of the tertiary C–H group at C-4, and the possibility of a facile base-assisted epimerization under the reaction conditions. The cyclization of ylide 6, generated by the deprotonation of cyclopropenylamide 9, should provide cyclopropyl anion 10 (Scheme 6). Subsequent protonation affords a mixture of exo-7 and endo-7 products, and their initial ratio depends on stereo-electronic factors at the cyclization step. However, the final product ratio is determined by a thermodynamic equilibrium that occurs via stabilized cyclic ylide 11. The exo-/endo-ratio change can be monitored in time by GC (shown for compound 7m, Fig. 1). In some cases, such as with ortho-chlorinated derivative 7l, deprotonation at C-4 cannot be achieved efficiently due to steric hindrance, so the final diastereomeric ratio matches that for the initial kinetic distribution (Fig. 2).21
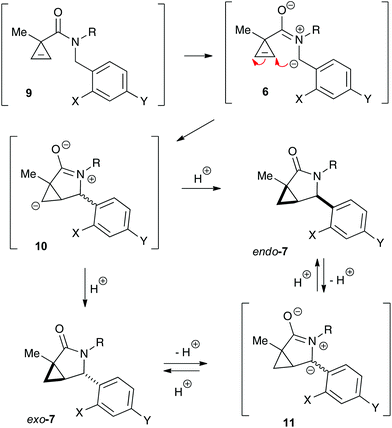 |
| | Scheme 6 | |
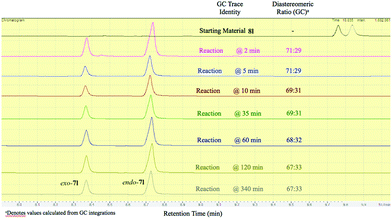 |
| | Fig. 1 Real-time GC monitoring of endo-7m![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :exo-7m equilibrium in the reaction mixture containing bromocyclopropane 8m and t-BuOK in THF at 30 °C. :exo-7m equilibrium in the reaction mixture containing bromocyclopropane 8m and t-BuOK in THF at 30 °C. | |
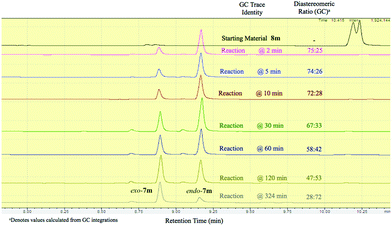 |
| | Fig. 2 Real-time GC monitoring of endo-7l:exo-7l equilibrium in the reaction mixture containing bromocyclopropane 8l and t-BuOK in THF at 30 °C. | |
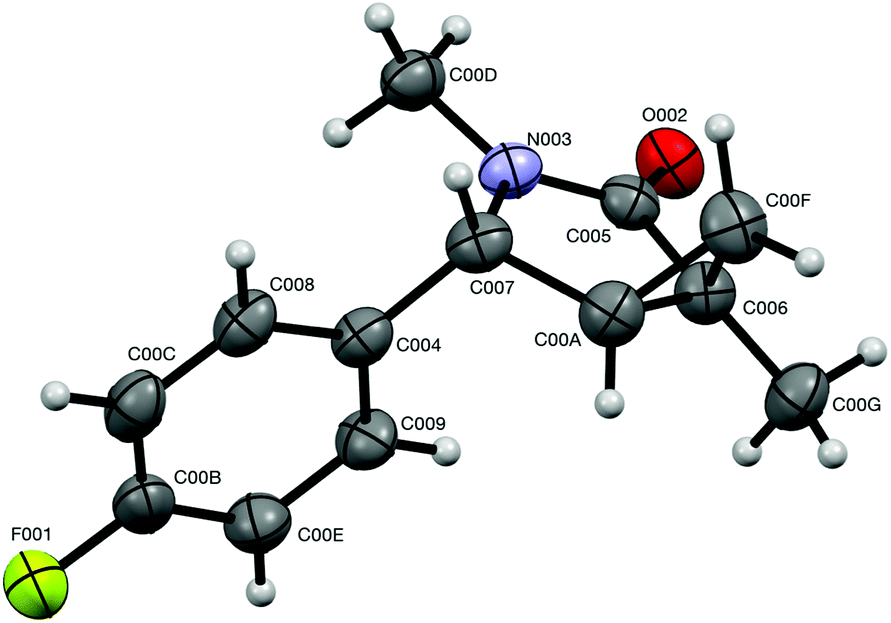
It should also be pointed out that the difference in the thermodynamic stabilities of endo- and exo-diastereomers in five-membered scaffold 7 is not large enough to warrant high degrees of diastereoselectivity. Our DFT modeling showed that exo-7j is more stable than endo-7j by only 2.386 × 10−3 amu (1.50 kcal mol−1), which corresponds to the best achievable dr of ca. 70:30.22 Also, this modeling helped to assign relative configurations of the diastereomeric bicyclic products. Indeed, calculation showed that dihedral angles between (C-4)–H and (C-5)–H bonds in endo- and exo-isomers are 36.8° and 94.4°, respectively.22 This suggests that the value of the corresponding vicinal spin–spin coupling constants for endo-isomers should be larger. Indeed, the benzylic proton signals in the 1H NMR spectra appeared as doublets for the isomers (endo) and as singlets for another one (exo). Also, the relative configuration of exo-7j was independently and unambiguously assigned by single-crystal X-ray diffraction (Fig. 3).
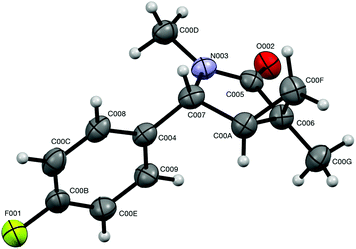 |
| | Fig. 3 ORTEP drawing of the crystal structure of compound exo-7j (CCDC 1575277†) showing atom numbering labels and 50% probability amplitude displacement ellipsoids. | |
Conclusion
A cascade, base-assisted dehydrohalogenation/5-exo-trig nucleophilic cyclization of stabilized benzylic anions to cyclopropenes was discovered. This reaction represents the first example of the non-catalytic addition of carbon nucleophiles to unactivated cyclopropenes. The obtained results are valuable as a proof of concept and are being applied in design of the diastereoselective cyclization of carbon-based nucleophiles to obtain six- and seven-membered ring systems. The latter models are expected to allow for better stereo-electronic control, due to a more substantial difference in the thermodynamic stabilities of the corresponding diastereomers. Synthetic and computational studies towards this goal are currently underway in our laboratories.
Experimental part
NMR spectra were recorded on a Bruker Avance DRX-500 spectrometer (500 MHz) equipped with a dual carbon/proton cryoprobe (CPDUL) or with a BBO probe or on a Bruker Avance DPX-400 spectrometer (400 MHz) equipped with a quadruple-band gradient probe (H/C/P/F QNP). 13C NMR spectra were recorded with broadband decoupling. IR spectra were recorded on a ThermoFisher Nicolet™ iS™ 5 FT-IR spectrometer. HRMS was carried out on a LCT Premier (Micromass Technologies) instrument employing ESI TOF detection techniques. Glassware used in moisture-free syntheses was flame-dried under vacuum prior to use. Column chromatography was carried out on silica gel (Sorbent Technologies, 40–63 mm). Pre-coated silica gel plates (Sorbent Technologies Silica XG 200 mm) were used for TLC analyses. Anhydrous THF and dichloromethane (DCM) were obtained by the distillation of a degassed commercially available HPLC-grade inhibitor-free solvent over calcium hydride and stored over 4 Å molecular sieves under nitrogen. Commercial potassium tert-butoxide was sublimed under vacuum prior to use. The syntheses of bromocyclopropanes 8a–m are described in the ESI.†22 All other reagents and solvents were purchased from commercial vendors and used as received.
(1R*,5S*)-3-Benzyl-1-methyl-4-phenyl-3-azabicyclo[3.1.0]hexan-2-one (7a)
Typical procedure I. An oven-dried Wheaton vial equipped with a Teflon septum cap was charged with freshly sublimed t-BuOK (315 mg, 2.80 mmol) and 18-crown-6 ether (18.5 mg, 0.07 mmol) in a nitrogen-filled glovebox. Anhydrous THF (2.22 mL) was then added to this vial and the solution was stirred to premix for 30 minutes. A solution of N,N-dibenzyl-2-bromo-1-methylcyclopropane-1-carboxamide (8a) (251 mg, 0.70 mmol) in anhydrous THF (1.48 mL) was added dropwise to the stirred reaction mixture, which was then stirred at 30 °C until starting materials were consumed (10 min for the reaction of 7a). The reaction was then quenched by pouring the mixture into brine (35 mL). The aqueous layer was extracted with EtOAc (2 × 20 mL). The combined organic phases were washed with brine (20 mL), dried with MgSO4, gravity filtered, and concentrated in vacuo. The crude material contains a mixture of diastereomers 45:55 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (2:1) afforded the titled product as a pale yellow oil (Rf 0.38). Yield 156 mg (0.56 mmol, 80%). endo-7a: 1H NMR (400 MHz, CDCl3) δ 7.42–7.32 (m, 3H), 7.31–7.21 (m, 3H), 7.20–7.14 (m, 2H), 7.09–7.01 (m, 2H), 5.06 (d, J = 15.0 Hz, 1H), 4.57 (d, J = 6.0 Hz, 1H), 3.50 (d, J = 14.5 Hz, 1H), 1.93–1.86 (m, 1H), 1.42 (s, 3H), 1.05 (t, J = 4.5 Hz, 1H), 0.64 (dd, J = 7.8, 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 177.6, 138.5, 136.6, 128.9 (2C), 128.9 (2C), 128.7 (2C), 128.0, 127.6, 127.0 (2C), 59.6, 44.2, 27.1, 26.6, 16.4, 15.2. exo-7a: 1H NMR (400 MHz, CDCl3) δ 7.42–7.32 (m, 3H), 7.31–7.21 (m, 3H), 7.20–7.14 (m, 2H), 7.09–7.01 (m, 2H), 5.00 (d, J = 14.9 Hz, 1H), 4.12 (s, 1H), 3.36 (d, J = 14.7 Hz, 1H), 1.58 (dd, J = 7.5, 3.9 Hz, 1H), 1.48 (s, 3H), 0.72 (t, J = 4.3 Hz, 1H), 0.92 (dd, J = 7.5, 4.6 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.5, 140.8, 137.2, 129.2 (2C), 128.8 (2C), 128.5, 128.5 (2C), 127.6, 127.0 (2C), 61.8, 43.9, 26.4, 25.6, 19.3, 15.1. FTIR (NaCl, cm−1): 3063, 3030, 2961, 2928, 2869, 1694, 1495, 1454, 1414, 1357, 1301, 1200, 1151, 1078, 1029, 941, 747, 761, 701, 622; HRMS (TOF ES): found 300.1378, calculated for C19H19NONa (M + Na) 300.1364 (4.7 ppm); EA found C 82.12, 82.00, H 6.76, 6.74, N 5.19, 5.12, calculated for C19H19NO: C 82.28, H 6.90, N 5.05.
(1R*,5S*)-3-Butyl-1-methyl-4-(4-(trifluoromethyl)phenyl)-3-azabicyclo[3.1.0]hexan-2-one (7d)
This compound was synthesized according to the Typical procedure I employing 2-bromo-N-butyl-1-methyl-N-(4-(trifluoromethyl)benzyl)cyclopropane-1-carboxamide (8d) (67 mg, 0.171 mmol), 18-crown-6 ether (4.5 mg, 0.017 mmol), and t-BuOK (77 mg, 0.68 mmol). The reaction mixture was stirred at rt for 5 min and then quenched with a saturated solution of ammonium chloride. The crude material contains an inseparable mixture of diastereomers 15:85 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (2:1) afforded the titled product as a colorless oil (Rf 0.33). Yield: 12.4 mg (0.046 mmol, 27%). endo-7d: 1H NMR (500 MHz, CDCl3) δ 7.64 (d, J = 8.2 Hz, 2H), 7.24 (d, J = 8.1 Hz, 2H), 4.90 (d, J = 6.2 Hz, 1H), 3.66 (ddd, J = 13.9, 8.9, 7.1 Hz, 1H), 2.50–2.43 (m, 1H), 1.96 (ddd, J = 7.7, 6.1, 4.0 Hz, 1H), 1.41 (s, 3H), 1.36–1.25 (m, 2H), 1.25–1.15 (m, 2H), 0.89 (t, J = 4.5 Hz, 1H), 0.85 (t, J = 7.3 Hz, 3H), 0.62 (dd, J = 7.8, 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 177.2, 143.0, 130.0 (q, 2JCF = 40.0 Hz), 126.9 (2C), 125.8 (q, 3JCF = 3.7 Hz, 2C), 124.0 (q, 1JCF = 271.9 Hz), 59.7, 40.2, 28.8, 28.2, 26.6, 20.1, 16.1, 15.0, 13.8. exo-7d: 1H NMR (500 MHz, CDCl3) δ 7.64 (d, J = 8.2 Hz, 2H), 7.33 (d, J = 8.0 Hz, 2H), 4.43 (s, 1H), 3.59 (dt, J = 14.0, 7.8 Hz, 1H), 2.46 (ddd, J = 13.8, 7.8, 5.5 Hz, 1H), 1.57 (dd, J = 7.5, 4.0 Hz, 1H), 1.43 (s, 3H), 1.36–1.25 (m, 2H), 1.25–1.15 (m, 2H), 1.00 (dd, J = 7.5, 4.7 Hz, 1H), 0.85 (t, J = 7.3 Hz, 3H), 0.80 (t, J = 4.3 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.5, 145.4, 130.6 (q, 2JCF = 32.7 Hz), 126.9 (2C), 126.1 (q, 3JCF = 3.9 Hz, 2C), 124.0 (q, 1JCF = 271.9 Hz), 62.2, 40.0, 29.6, 26.4, 25.8, 19.9, 19.9, 14.9, 13.7. 19F NMR (376 MHz, chloroform-d) δ −62.5, −62.6; FTIR (NaCl, cm−1): 2961, 2933, 2873, 1676, 1645, 1459, 1414, 1326, 1294, 1246, 1166, 1125, 1067, 1018, 959, 846, 756, 608; HRMS (TOF ES): found 312.1578, calculated for C17H21NOF3 (M + H) 312.1575 (1.0 ppm); EA found C 65.70, 65.45, H 6.38, 6.65, N 4.39, 4.78, calculated for C17H20F3NO: C 65.58, H 6.48, N 4.50.
(1R*,5S*)-3-Butyl-1-methyl-4-phenyl-3-azabicyclo[3.1.0]hexan-2-one (7f)
This compound was synthesized according to the Typical procedure I employing N-benzyl-2-bromo-N-butyl-1-methylcyclopropane-1-carboxamide (8f) (229 mg, 0.70 mmol), 18-crown-6 ether (18.5 mg, 0.07 mmol), and t-BuOK (314 mg, 2.80 mmol). The reaction mixture was stirred overnight at 30 °C. The crude material contains an inseparable mixture of diastereomers 53:47 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (3:1) afforded the titled product as a yellow oil (Rf 0.30). Yield 119.2 mg (0.49 mmol, 70%). endo-7f: 1H NMR (400 MHz, CDCl3) δ 7.41–7.29 (m, 3H), 7.15–7.09 (m, 2H), 4.85 (d, J = 6.0 Hz, 1H), 3.63 (ddd, J = 13.8, 8.6, 7.3 Hz, 1H), 2.50 (m, 1H), 1.93 (ddd, J = 7.7, 6.0, 3.9 Hz, 1H), 1.40 (s, 3H), 1.38–1.12 (m, 4H), 0.99–0.91 (m, 1H), 0.84 (t, J = 7.3 Hz, 3H), 0.59 (dd, J = 7.7, 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 177.4, 138.8, 128.8 (2C), 127.9, 126.8 (2C), 60.2, 40.2, 29.0, 26.9, 26.7, 20.2, 16.2, 15.2, 13.9. exo-7f: 1H NMR (400 MHz, CDCl3) δ 7.41–7.29 (m, 3H), 7.22–7.17 (m, 2H), 4.35 (s, 1H), 3.55 (dt, J = 13.9, 7.7 Hz, 1H), 2.50 (m, 1H), 1.59 (dd, J = 7.5, 3.9 Hz, 1H), 1.42 (s, 3H), 1.38–1.12 (m, 4H), 0.99–0.91 (m, 1H), 0.84 (t, J = 7.3 Hz, 3H), 0.76 (t, J = 4.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.6, 141.3, 129.1 (2C), 128.3, 126.7 (2C), 62.8, 39.9, 29.8, 26.7, 25.9, 20.0 (2C), 15.1, 13.8. FTIR (NaCl, cm−1): 2960, 2931, 2872, 1692, 1457, 1417, 1372, 1219, 1051, 756, 701. HRMS (TOF ES): found 266.1514, calculated for C16H21NONa (M + Na) 266.1521 (2.6 ppm); EA found C 78.81, 79.23, H 8.42, 8.50, N 5.92, 5.55, calculated for C16H21NO: C 78.97, H 8.70, N 5.76.
(1R*,5S*)-3-Isopropyl-1-methyl-4-phenyl-3-azabicyclo[3.1.0]hexan-2-one (7g)
This compound was synthesized according to the Typical procedure I employing N-benzyl-2-bromo-N-isopropyl-1-methylcyclopropane-1-carboxamide (8g) (217 mg, 0.70 mmol), 18-crown-6 ether (18.5 mg, 0.07 mmol), and t-BuOK (314 mg, 2.80 mmol). The reaction mixture was stirred at 30 °C for 3 h. The crude material contains a mixture of diastereomers 50:50 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (3:1) afforded the titled product as a pale yellow glass (Rf 0.36, 0.30). Yield 104.3 mg (0.455 mmol, 65%). Analytical samples of individual diastereomers were obtained by column chromatography on silica gel eluting with a CH2Cl2/EtOAc mixture (10:1). endo-7g: 1H NMR (400 MHz, CDCl3) δ 7.42–7.32 (m, 3H), 7.26–7.21 (m, 2H), 4.86 (d, J = 5.8 Hz, 1H), 3.47 (p, J = 6.8 Hz, 1H), 1.91 (ddd, J = 7.7, 5.9, 3.9 Hz, 1H), 1.38 (s, 3H), 1.32 (d, J = 6.9 Hz, 3H), 1.15 (d, J = 6.8 Hz, 3H), 1.08 (t, J = 4.4 Hz, 1H), 0.62 (dd, J = 7.7, 4.8 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 178.2, 140.4, 128.7 (2C), 128.0, 126.9 (2C), 61.4, 46.1, 27.3, 26.8, 20.0, 19.6, 16.2, 15.1. exo-7g: 1H NMR (500 MHz, CDCl3), δ1H NMR (500 MHz, CDCl3) δ 7.38–7.33 (m, 2H), 7.33–7.28 (m, 1H), 7.27–7.21 (m, 2H), 4.36 (s, 1H), 4.02 (p, J = 6.9 Hz, 1H), 1.49 (dd, J = 7.3, 3.9 Hz, 1H), 1.44 (s, 3H), 1.08 (d, J = 6.8 Hz, 3H), 0.91 (dd, J = 7.3, 4.6 Hz, 1H), 0.77 (d, J = 6.9 Hz, 3H), 0.66 (t, J = 4.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.8, 143.6, 128.9 (2C), 128.2, 126.7 (2C), 61.5, 44.7, 26.6, 26.2, 21.3, 20.4, 19.5, 15.0. FTIR (NaCl, cm−1): 2970, 2931, 1685, 1456, 1412, 1380, 1345, 1223, 1028, 956, 763, 738, 702. HRMS (TOF ES): found 252.1353, calculated for C15H19NONa (M + Na) 252.1364 (4.4 ppm); EA found C 78.47, 78.68, H 8.55, 8.18, N 6.07, 6.40, calculated for C15H19NO: C 78.56, H 8.35, N 6.11.
(1R*,5S*)-3-Cyclohexyl-1-methyl-4-phenyl-3-azabicyclo[3.1.0]hexan-2-one (7h)
This compound was synthesized according to the Typical procedure I employing N-benzyl-2-bromo-N-cyclohexyl-1-methylcyclopropane-1-carboxamide (8h) (245 mg, 0.70 mmol), 18-crown-6 ether (18.5 mg, 0.07 mmol), and t-BuOK (314 mg, 2.80 mmol). The reaction mixture was stirred overnight at 30 °C. The crude material contains an inseparable mixture of diastereomers 45:55 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (3:1) afforded the titled product as a colorless glass (Rf 0.38). Yield 141.3 mg (0.525 mmol, 75%). endo-7h1H NMR (400 MHz, CDCl3) δ 7.41–7.27 (m, 3H), 7.25–7.18 (m, 2H), 4.85 (d, J = 6.0 Hz, 1H), 3.10 (tt, J = 12.2, 3.5 Hz, 1H), 2.04–1.90 (m, 1H), 1.87 (ddd, J = 7.8, 6.0, 3.9 Hz, 1H), 1.67 (dd, J = 22.8, 11.2 Hz, 4H), 1.57–1.44 (m, 2H), 1.35 (s, 3H), 1.17–0.92 (m, 4H), 0.57 (dd, J = 7.8, 4.8 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 178.2, 140.6, 128.6 (2C), 127.9, 126.8 (2C), 61.2, 54.5, 30.1, 29.6, 27.5, 26.8, 26.3, 26.0, 25.5, 16.2, 15.1. exo-7h: 1H NMR (400 MHz, CDCl3) δ 7.41–7.27 (m, 3H), 7.25–7.18 (m, 2H), 4.37 (s, 1H), 3.65 (tt, J = 12.1, 3.8 Hz, 1H), 1.73–1.59 (m, 1H), 1.55–1.42 (m, 5H), 1.42 (s, 3H), 1.43–1.33 (m, 1H), 1.30–1.21 (m, 2H), 1.18–1.00 (m, 1H), 1.00–0.70 (m, 2H), 0.64 (t, J = 4.2 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.9, 143.9, 128.9 (2C), 128.0, 126.6 (2C), 61.6, 52.7, 31.9, 30.9, 26.8, 26.3, 25.9, 25.9, 25.5, 19.6, 15.0. FTIR (NaCl, cm−1): 2931, 2855, 1684, 1453, 1414, 1360, 1205, 1028, 894, 751, 736, 702, 624. HRMS (TOF ES): found 292.1664, calculated for C18H23NONa (M + Na) 292.1677 (4.4 ppm); EA found C 80.31, 80.34, H 8.89, 8.86, N 4.95, 5.14, calculated for C18H23NO: C 80.26, H 8.61, N 5.20.
(1R*,5S*)-3-(tert-Butyl)-1-methyl-4-phenyl-3-azabicyclo[3.1.0]hexan-2-one (7i)
This compound was synthesized according to the Typical procedure I employing N-benzyl-2-bromo-N-(tert-butyl)-1-methylcyclopropane-1-carboxamide (8i) (227 mg, 0.70 mmol), 18-crown-6 ether (18.5 mg, 0.07 mmol), and t-BuOK (314 mg, 2.80 mmol). The reaction mixture was stirred overnight at 30 °C. The crude material contains an inseparable mixture of diastereomers 47:53 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (3:1) afforded the titled product as a colorless glass (Rf 0.44). Yield 109 mg (0.448 mmol, 64%). endo-7i: 1H NMR (500 MHz, CDCl3) δ 7.39–7.26 (m, 4H), 7.28–7.14 (m, 1H), 4.97 (d, J = 6.5 Hz, 1H), 1.88 (ddd, J = 7.9, 6.5, 3.7 Hz, 1H), 1.34 (s, 3H), 1.26 (s, 9H), 0.98 (t, J = 4.3 Hz, 1H), 0.49 (dd, J = 7.9, 4.8 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 178.8, 144.7, 128.5 (2C), 127.1, 125.5 (2C), 61.1, 54.4, 28.3 (3C), 28.3, 28.3, 16.1, 15.7. 1H NMR (500 MHz, CDCl3) δ 7.39–7.26 (m, 4H), 7.28–7.14 (m, 1H), 4.57 (s, 1H), 1.40 (dd, J = 7.2, 3.9 Hz, 1H), 1.38 (s, 3H), 1.24 (s, 9H), 0.84 (dd, J = 7.2, 4.3 Hz, 1H), 0.66 (t, J = 4.1 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 177.8, 143.7, 128.9 (2C), 127.7, 126.1 (2C), 62.7, 55.5, 28.1 (3C), 26.4, 26.0, 19.2, 14.9. FTIR (NaCl, cm−1): 2963, 2929, 2869, 1664, 1493, 1455, 1396, 1383, 1359, 1343, 1221, 1198, 1141, 950, 762, 740, 705; HRMS (TOF ES): found 266.1528, calculated for C16H21NONa (M + Na) 266.1521 (2.6 ppm); EA found C 79.05, 79.07, H 8.60, 8.95, N 5.92, 5.57, calculated for C16H21NO: C 78.97, H 8.70, N 5.76.
(1R*,5S*)-4-(4-Fluorophenyl)-1,3-dimethyl-3-azabicyclo[3.1.0]hexan-2-one (7j)
This compound was synthesized according to the Typical procedure I employing 2-bromo-N-(4-fluorobenzyl)-N,1-dimethylcyclopropane-1-carboxamide (8j) (51 mg, 0.171 mmol), 18-crown-6 ether (4.5 mg, 0.017 mmol), and t-BuOK (76 mg, 0.68 mmol). The reaction mixture was stirred at rt for 4 h. The crude material contains an inseparable mixture of diastereomers 60:40 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (3:1) afforded the titled product as a colorless glass (Rf 0.36). Yield 21.6 mg (0.099 mmol, 58%). endo-7j: 1H NMR (400 MHz, CDCl3) δ 7.11–7.02 (m, 4H), 4.68 (d, J = 5.9 Hz, 1H), 2.61 (s, 3H), 1.93 (ddd, J = 7.7, 5.9, 4.0 Hz, 1H), 1.40 (s, 3H), 0.90 (t, J = 4.6 Hz, 1H), 0.63 (dd, J = 7.8, 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.4, 162.4 (d, 1JCF = 246.1 Hz), 136.5 (d, 4JCF = 3.5 Hz), 128.0 (d, 3JCF = 8.1 Hz, 2C), 115.7 (d, 2JCF = 21.2 Hz, 2C), 62.2, 28.1, 26.9, 25.7, 16.3, 14.9. exo-7j: 1H NMR (400 MHz, CDCl3) δ 7.21–7.13 (m, 2H), 7.11–7.02 (m, 2H), 4.23 (s, 1H), 2.57 (s, 3H), 1.57 (dd, J = 7.6, 3.9 Hz, 1H), 1.42 (s, 3H), 0.96 (dd, J = 7.6, 4.7 Hz, 1H), 0.81 (t, J = 4.3 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.4, 162.6 (d, 1JCF = 247.0 Hz), 136.5 (d, 4JCF = 3.5 Hz), 128.1 (d, 3JCF = 8.2 Hz, 2C), 116.0 (d, 2JCF = 21.7 Hz, 2C), 64.5, 27.9, 26.6, 25.7, 20.0, 15.1. 19F NMR (376 MHz, CDCl3) δ −113.8, −114.6; FTIR (NaCl, cm−1): 2929, 1683, 1509, 1481, 1398, 1223, 1158, 1007, 845, 819, 752, 668, 647. HRMS (TOF ES): found 242.0960, calculated for C13H14NOFNa (M + Na) 242.0957 (1.2 ppm); EA found C 71.35, 71.27, H 6.18, 6.34, N 6.26, 6.33, calculated for C13H14FNO: C 71.21, H 6.44, N 6.39. Slow crystallization of the purified material from hexane afforded a crop of crystals of exo-7j suitable for X-ray analysis.
(1R*,5S*)-4-(2,4-Difluorophenyl)-1,3-dimethyl-3-azabicyclo[3.1.0]hexan-2-one (7k)
This compound was synthesized according to the Typical procedure I employing 2-bromo-N-(2,4-difluorobenzyl)-N,1-dimethylcyclopropane-1-carboxamide (8k) (54 mg, 0.171 mmol), 18-crown-6 ether (4.5 mg, 0.017 mmol), and t-BuOK (76 mg, 0.68 mmol). The reaction mixture was stirred at rt for 5 min and then quenched with a saturated solution of ammonium chloride. The crude material contains an inseparable mixture of diastereomers 69:31 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (2:3) afforded the titled product as a colorless oil (Rf 0.38). Yield 30.2 mg (0.127 mmol, 75%). endo-7k: 1H NMR (500 MHz, CDCl3) δ 6.93–6.79 (m, 3H), 4.95 (d, J = 5.9 Hz, 1H), 2.64 (s, 3H), 2.06 (ddd, J = 7.8, 5.9, 4.0 Hz, 1H), 1.38 (s, 3H), 0.77 (t, J = 4.5 Hz, 1H), 0.64 (dd, J = 7.8, 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 177.9, 162.3 (dd, 1JCF = 249.2 Hz, 3JCF = 13.2 Hz), 160.7 (dd, 1JCF = 248.8 Hz, 3JCF = 12.0 Hz), 127.8 (dd, 3JCF = 9.4 Hz, 3JCF = 5.8 Hz), 122.1 (dd, 2JCF = 13.2 Hz, 4JCF = 3.9 Hz), 111.3 (dd, 2JCF = 21.0 Hz, 4JCF = 3.6 Hz), 104.3 (t, 2JCF = 25.5 Hz), 56.0 (d, 3JCF = 4.5 Hz), 28.5, 26.5, 25.6, 16.5, 14.9. exo-7k: 1H NMR (500 MHz, CDCl3) δ 7.14–7.07 (m, 1H), 6.93–6.79 (m, 2H), 4.57 (s, 1H), 2.59 (s, 3H), 1.60 (dd, J = 7.6, 3.9 Hz, 1H), 1.40 (s, 3H), 0.98 (dd, J = 7.5, 4.7 Hz, 1H), 0.82 (t, J = 4.3 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.5, 162.7 (dd, 1JCF = 249.7 Hz, 3JCF = 12.2 Hz), 160.8 (dd, 1JCF = 249.7 Hz, 3JCF = 12.2 Hz), 128.8 (dd, 3JCF = 9.9 Hz, 3JCF = 5.5 Hz), 123.5 (dd, 2JCF = 13.2 Hz, 4JCF = 3.9 Hz), 112.0 (dd, 2JCF = 21.0 Hz, 4JCF = 3.7 Hz), 104.4 (t, 2JCF = 25.5 Hz), 58.2 (d, 3JCF = 3.5 Hz), 27.9, 25.7, 25.6, 20.0, 14.9. 19F NMR (376 MHz, CDCl3) δ −109.9 (d, 4JFF = 7.5 Hz), −111.1 (d, 4JFF = 7.3 Hz), −115.97 (d, 4JFF = 7.8 Hz), −116.03 (d, 4JFF = 7.3 Hz); FTIR (NaCl, cm−1): 2932, 1694, 1617, 1503, 1430, 1397, 1269, 1234, 1140, 1092, 974, 961, 850, 765, 610. HRMS (TOF ES): found 260.0862, calculated for C13H13NOF2Na (M + Na) 260.0863 (0.4 ppm); EA found C 65.92, 65.51, H 5.60, 5.67, N 6.14, 5.82, calculated for C13H13F2NO: C 65.81, H 5.52, N 5.90.
(1R*,5S*)-4-(2-Chlorophenyl)-1,3-dimethyl-3-azabicyclo[3.1.0]hexan-2-one (7l)
This compound was synthesized according to the Typical procedure I employing 2-bromo-N-(2-chlorobenzyl)-N,1-dimethylcyclopropane-1-carboxamide (8l) (54 mg, 0.171 mmol), 18-crown-6 ether (4.5 mg, 0.017 mmol), and t-BuOK (77 mg, 0.68 mmol). The reaction mixture was stirred at rt for 2 min and then quenched with a saturated solution of ammonium chloride. The crude material contains an inseparable mixture of diastereomers 70:30 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (2:3) afforded the titled product as a colorless oil (Rf 0.45). Yield 27.7 mg (0.118 mmol, 69%). endo-7l: 1H NMR (400 MHz, CDCl3) δ 7.47–7.39 (m, 1H), 7.33–7.22 (m, 2H), 6.86 (dd, J = 6.9, 2.4 Hz, 1H), 5.07 (d, J = 5.9 Hz, 1H), 2.67 (s, 3H), 2.24 (ddd, J = 7.8, 5.9, 4.0 Hz, 1H), 1.41 (s, 3H), 0.75 (t, J = 4.5 Hz, 1H), 0.60 (dd, J = 7.8, 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 178.3, 136.4, 133.1, 130.2, 128.8, 127.1, 127.0, 59.9, 28.8, 26.6, 24.9, 16.5, 15.2. exo-7l: 1H NMR (400 MHz, CDCl3) δ 7.47–7.39 (m, 1H), 7.33–7.22 (m, 2H), 7.11 (dd, J = 7.3, 2.1 Hz, 1H), 4.80 (s, 1H), 2.64 (s, 3H), 1.60 (dd, J = 7.5, 4.0 Hz, 1H), 1.38 (s, 3H), 0.99 (dd, J = 7.5, 4.7 Hz, 1H), 0.86 (t, J = 4.3 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 177.1, 138.0, 133.1, 130.2, 129.3, 127.7, 127.0, 61.3, 28.4, 25.2, 26.0, 20.1, 15.1. FTIR (NaCl, cm−1): 2929, 1698, 1471, 1445, 1395, 1384, 1340, 1232, 1035, 972, 757, 698; HRMS (TOF ES): found 258.0660, calculated for C13H14NOClNa (M + Na) 258.0662 (0.8 ppm); EA found C 66.21, 66.16, H 5.92, 6.10, N 5.64, 6.22, calculated for C13H14ClNO: C 66.24, H 5.99, N 5.94.
(1R*,5S*)-4-(4-Bromo-2-fluorophenyl)-1,3-dimethyl-3-azabicyclo[3.1.0]hexan-2-one (7m)
This compound was synthesized according to the Typical procedure I employing 2-bromo-N-(4-bromo-2-fluorobenzyl)-N,1-dimethylcyclopropane-1-carboxamide (8m) (65 mg, 0.171 mmol), 18-crown-6 ether (4.5 mg, 0.017 mmol), and t-BuOK (77 mg, 0.69 mmol). The reaction mixture was stirred at rt for 2 min and then quenched with a saturated solution of ammonium chloride. The crude material contains an inseparable mixture of diastereomers 72:28 (endo:exo). Purification by column chromatography eluting with a mixture of hexanes/EtOAc (1:1) afforded the titled product as a colorless oil (Rf 0.35). Yield 37.8 mg (0.127 mmol, 74%). endo-7m: 1H NMR (400 MHz, CDCl3) δ 7.33–7.19 (m, 2H), 6.74 (t, J = 8.2 Hz, 1H), 4.89 (d, J = 5.9 Hz, 1H), 2.60 (s, 3H), 2.03 (ddd, J = 8.0, 5.8, 3.9 Hz, 1H), 1.34 (s, 3H), 0.71 (t, J = 4.5 Hz, 1H), 0.60 (dd, J = 7.8, 5.0 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 178.0, 160.5 (d, J = 250.9 Hz), 128.2 (d, J = 4.7 Hz), 127.7 (d, J = 3.6 Hz), 125.6 (d, J = 12.8 Hz), 121.6 (d, J = 9.9 Hz), 119.6 (d, J = 24.5 Hz), 56.2 (d, J = 4.4 Hz), 28.6, 26.7, 25.5, 16.6, 15.0. exo-7m: 1H NMR (400 MHz, CDCl3) δ 7.33–7.19 (m, 2H), 6.96 (t, J = 8.0 Hz, 1H), 4.52 (s, 1H), 2.56 (s, 3H), 1.55 (dd, J = 7.5, 3.9 Hz, 1H), 1.35 (s, 3H), 0.95 (dd, J = 7.5, 4.8 Hz, 1H), 0.78 (t, J = 4.4 Hz, 1H); 13C NMR (126 MHz, CDCl3) δ 176.6, 160.6 (d, J = 253.3 Hz), 129.0 (d, J = 5.1 Hz), 128.3 (d, J = 4.7 Hz), 126.9 (d, J = 13.0 Hz), 122.3 (d, J = 9.8 Hz), 119.8 (d, J = 23.9 Hz), 58.3 (d, J = 3.6 Hz), 28.1, 25.8, 25.6, 20.1, 14.9. 19F NMR (376 MHz, chloroform-d) δ −116.4, −117.3; FTIR (NaCl, cm−1): 2961, 2930, 1695, 1605, 1574, 1483, 1396, 1220, 1077, 973, 883, 850, 757. HRMS (TOF ES): found 320.0056, calculated for C13H13NOFBrNa (M + Na) 320.0062 (1.9 ppm); EA found 52.21, 52.44, H 4.29, 4.57, N 4.90, 4.64, calculated for C13H13BrFNO: C 52.37, H 4.40, N 4.70.
N-Butyl-N-(4-(tert-butyl)benzyl)-1-methylcycloprop-2-ene-1-carboxamide (9b)
This compound was synthesized according to the Typical procedure I employing 2-bromo-N-butyl-N-(4-(tert-butyl)benzyl)-1-methylcyclopropane-1-carboxamide (8b) (266 mg, 0.70 mmol), 18-crown-6 ether (18.5 mg, 0.07 mmol), and t-BuOK (314 mg, 2.80 mmol). The reaction mixture was stirred at 25 °C for 47 h. The product was isolated by column chromatography eluting with a mixture of hexanes/EtOAc (3:2) as a yellow oil (Rf 0.39). Yield 72.2 mg (0.315 mmol, 45%). 1H NMR (500 MHz, CDCl3) δ 7.33 (br. s, 3H), 7.22 (br. s, 1H), 7.08 (br. s, 2H), 4.81–4.48 (m, 2H), 3.44–3.18 (m, 2H), [1.95 (br. s), 1.53–1.35 (m), ∑5H], 1.37 (br. s, 11H), 1.31 (br. s, 3H); 13C NMR (126 MHz, CDCl3) δ 176.3, (150.4, 149.9, 1C), (134.6, 134.2, 1C), (127.4, 126.5, 2C), 125.6 (2C), 115.8 (2C), (50.3, 44.0, 1C), 46.4, 34.5, 31.4 (3C), (30.6, 29.1, 1C), 24.1, 23.2, 20.1, 13.9; FTIR (NaCl, cm−1): 2960, 2869, 1625, 1514, 1463, 1410, 1365, 1269, 1104, 1005, 927, 819, 732, 617; HRMS (TOF ES): found 322.2147, calculated for C20H29NONa (M + Na) 322.2147 (0.0 ppm); EA found C 80.07, 80.50, H 9.75, 9.95, N 4.93, 4.97, calculated for C20H29NO: C 80.22, H 9.76, N 4.68.
N-Butyl-N-(4-methoxybenzyl)-1-methylcycloprop-2-ene-1-carboxamide (9c)
This compound was synthesized according to the Typical procedure I employing 2-bromo-N-butyl-N-(4-methoxybenzyl)-1-methylcyclopropane-1-carboxamide (8c) (248 mg, 0.70 mmol), 18-crown-6 ether (18.5 mg, 0.07 mmol), and t-BuOK (314 mg, 2.80 mmol). The reaction mixture was stirred at 25 °C for 27 h. The product was isolated by column chromatography eluting with a hexane/EtOAc mixture (1:1) as a yellow oil (Rf 0.39). Yield 105.3 mg (0.385 mmol, 55%). 1H NMR (500 MHz, CDCl3) δ [7.30 (br. s), 7.24 (br. s), 7.07 (br. s), ∑4H], 6.86 (br. s, 2H), 4.75–4.46 (m, 2H), 3.79 (s, 3H), 3.41–3.16 (m, 2H), [1.47 (br. s), 1.37 (s), 1.26 (br. s), ∑7H], 0.90 (br. s, 3H); 13C NMR (126 MHz, CDCl3) δ 176.2, 158.9, 129.1, 128.0, 115.8, 114.04 (2C), 55.3, 50.1, 46.1, 43.8, 30.5, 29.0, 24.0, 23.2, 20.1, 13.9; FTIR (NaCl, cm−1): 2958, 2933, 2872, 1615, 1513, 1464, 1417, 1302, 1247, 1175, 1104, 1033, 815, 621. HRMS (TOF ES): found 296.1637, calculated for C17H23NO2Na (M + Na) 296.1626 (3.7 ppm); EA found C 74.72, 74.89, H 8.75, 8.39, N 4.89, 5.19, calculated for C17H23NO2: C 74.69, H 8.48, N 5.12.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This work was financed by the Russian Foundation for Basic Research (grant #15-03-02661) and the Ministry of Education and Science of the Russian Federation (the project numbers 02.a03.21.0008 and #4.1196.2017/4.6). Support for NMR instruments used in this project was provided by the NIH Shared Instrumentation Grant #S10RR024664 and the NSF Major Research Instrumentation Grant #0329648.
References
- For reviews, see:
(a) A. Edwards, M. Rubina and M. Rubin, Curr. Org. Chem., 2016, 20, 1862–1877 CrossRef CAS;
(b) R. Vicente, Synthesis, 2016, 2343–2360 CrossRef CAS;
(c) Z.-B. Zhu, Y. Wei and M. Shi, Chem. Soc. Rev., 2011, 40, 5534–5563 RSC;
(d) M. Rubin, M. Rubina and V. Gevorgyan, Synthesis, 2006, 1221–1245 CAS;
(e) J. M. Fox and N. Yan, Curr. Org. Chem., 2005, 9, 719–732 CrossRef CAS.
- For hydrometallations, see:
(a) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2004, 126, 3688–3689 CrossRef CAS PubMed;
(b) Y. Lou, M. Horikawa, R. A. Kloster, N. A. Hawryluk and E. J. Corey, J. Am. Chem. Soc., 2004, 126, 8916–8918 CrossRef CAS PubMed;
(c) A. Parra, L. Amenos, M. Guisan-Ceinos, A. Lopez, J. L. Garcia Ruano and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833–15836 CrossRef CAS PubMed;
(d) M. Rubin and V. Gevorgyan, Synthesis, 2004, 796–800 CAS;
(e) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2003, 125, 7198–7199 CrossRef CAS PubMed. For carbometallations, see:
(f) D. S. Muller and I. Marek, J. Am. Chem. Soc., 2015, 137, 15414–15417 CrossRef CAS PubMed;
(g) M. Simaan, P. O. Delaye, M. Shi and I. Marek, Angew. Chem., Int. Ed., 2015, 54, 12345–12348 CrossRef CAS PubMed;
(h) D. Didier, P.-O. Delaye, M. Simaan, B. Island, G. Eppe, H. Eijsberg, A. Kleiner, P. Knochel and I. Marek, Chem. – Eur. J., 2014, 20, 1038–1048 CrossRef CAS PubMed;
(i) P.-O. Delaye, D. Didier and I. Marek, Angew. Chem., Int. Ed., 2013, 52, 5333–5337 CrossRef CAS PubMed;
(j) K. Kramer, P. Leong and M. Lautens, Org. Lett., 2011, 13, 819–821 CrossRef CAS PubMed;
(k) S. Simaan, A. Masarwa, E. Zohar, A. Stanger, P. Bertus and I. Marek, Chem. – Eur. J., 2009, 15, 8449–8464 CrossRef CAS PubMed;
(l) V. Tarwade, X. Liu, N. Yan and J. M. Fox, J. Am. Chem. Soc., 2009, 131, 5382–5383 CrossRef CAS PubMed;
(m) L. Dian, D. S. Müller and I. Marek, Angew. Chem., Int. Ed., 2017, 56, 6783–6787 CrossRef CAS PubMed;
(n) F.-G. Zhang, G. Eppe and I. Marek, Angew. Chem., Int. Ed., 2016, 55, 714–718 CrossRef CAS PubMed;
(o) D. S. Müller, V. Werner, S. Akyol, H.-G. Schmaltz and I. Marek, Org. Lett., 2017, 19, 3970–3973 CrossRef PubMed.
- For dimetallations, see:
(a) M. Rubina, M. Rubin and V. Gevorgyan, J. Am. Chem. Soc., 2002, 124, 11566–11567 CrossRef CAS PubMed;
(b) A. Trofimov, M. Rubina, M. Rubin and V. Gevorgyan, J. Org. Chem., 2007, 72, 8910–8920 CrossRef CAS PubMed;
(c) A. Parra, L. Amenos, M. Guisán-Ceinos, A. Lopez, R. Garcia, L. Jose and M. Tortosa, J. Am. Chem. Soc., 2014, 136, 15833–15836 CrossRef CAS PubMed;
(d) B. Tian, Q. Liu, X. Tong, P. Tian and G.-Q. Lin, Org. Chem. Front., 2014, 1, 1116–1122 RSC. For hydroformylation, see:
(e) W. M. Sherrill and M. Rubin, J. Am. Chem. Soc., 2008, 130, 13804–13809 CrossRef CAS PubMed. For hydroacylation, see:
(f) D. H. T. Phan, K. G. M. Kou and V. M. Dong, J. Am. Chem. Soc., 2010, 132, 16354–16355 CrossRef CAS PubMed. For hydrophosphorylation, see:
(g) B. K. Alnasleh, W. M. Sherrill and M. Rubin, Org. Lett., 2008, 10, 3231–3234 CrossRef CAS PubMed. For hydroamination, see:
(h) H. L. Teng, Y. Luo, B. Wang, L. Zhang, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2016, 55, 15406–15410 CrossRef CAS PubMed;
(i) Z. Li, J. Zhao, B. Sun, T. Zhou, M. Liu, S. Liu, M. Zhang and Q. Zhang, J. Am. Chem. Soc., 2017, 139, 11702–11705 CrossRef CAS PubMed. For hydroalkylation, see:
(j) Y. Luo, H.-L. Teng, M. Nishiura and Z. Hou, Angew. Chem., Int. Ed., 2017, 56, 9207–9210 CrossRef CAS PubMed. For carboamination, see:
(k) H. L. Teng, Y. Luo, M. Nishiura and Z. Hou, J. Am. Chem. Soc., 2017, 139, 16506–16509 CrossRef CAS PubMed.
-
(a) K. B. Wiberg, R. K. Barnes and J. Albin, J. Am. Chem. Soc., 1957, 79, 4994–4999 CrossRef CAS;
(b) J. E. Banning, A. R. Prosser, B. K. Alnasleh, J. Smarker, M. Rubina and M. Rubin, J. Org. Chem., 2011, 76, 3968–3986 CrossRef CAS PubMed;
(c) B. K. Alnasleh, W. M. Sherrill, M. Rubina, J. Banning and M. Rubin, J. Am. Chem. Soc., 2009, 131, 6906–6907 CrossRef CAS PubMed;
(d) J. E. Banning, A. R. Prosser and M. Rubin, Org. Lett., 2010, 12, 1488–1491 CrossRef CAS PubMed;
(e) P. Ryabchuk, A. Edwards, N. Gerasimchuk, M. Rubina and M. Rubin, Org. Lett., 2013, 15, 6010–6013 CrossRef CAS PubMed;
(f) K. N. Sedenkova, E. B. Averina, I. S. Borisov, Y. K. Grishin, V. B. Rybakov, T. S. Kuznetsova and N. S. Zefirov, Russ. J. Org. Chem., 2012, 48, 1265–1271 CrossRef CAS;
(g) M. Zhang, J. Guo and Y. Gong, Eur. J. Org. Chem., 2014, 1942–1950 CrossRef CAS;
(h) M. Zhang, F. Luo and Y. Gong, J. Org. Chem., 2014, 79, 1335–1343 CrossRef CAS PubMed;
(i) J. Hu, M. Zhang and Y. Gong, Eur. J. Org. Chem., 2015, 1970–1978 CrossRef CAS;
(j) P. Yamanushkin, M. Lu-Diaz, A. Edwards, N. A. Aksenov, M. Rubina and M. Rubin, Org. Biomol. Chem., 2017, 15, 8153–8156 RSC.
-
(a) J. E. Banning, J. Gentillon, P. G. Ryabchuk, A. R. Prosser, A. Rogers, A. Edwards, A. Holtzen, I. A. Babkov, M. Rubina and M. Rubin, J. Org. Chem., 2013, 78, 7601–7616 CrossRef CAS PubMed;
(b) E. C. Taylor and B. Hu, Synth. Commun., 1996, 26, 1041–1049 CrossRef CAS;
(c) K. N. Shavrin, V. D. Gvozdev, D. V. Budanov, S. V. Yurov and O. M. Nefedov, Mendeleev Commun., 2006, 16, 73–76 CrossRef;
(d) K. N. Shavrin, V. D. Gvozdev and O. M. Nefedov, Russ. Chem. Bull., 2010, 59, 396–404 CrossRef CAS;
(e) A. R. Prosser, J. E. Banning, M. Rubina and M. Rubin, Org. Lett., 2010, 12, 3968–3971 CrossRef CAS PubMed;
(f) P. Ryabchuk, M. Rubina, J. Xu and M. Rubin, Org. Lett., 2012, 14, 1752–1755 CrossRef CAS PubMed;
(g) Z. Huang, J. Hu and Y. Gong, Org. Biomol. Chem., 2015, 13, 8561–8566 RSC;
(h) Y. Zhu and Y. Gong, J. Org. Chem., 2015, 80, 490–498 CrossRef CAS PubMed;
(i) K. N. Shavrin, V. D. Gvozdev and O. M. Nefedov, Mendeleev Commun., 2008, 18, 300–301 CrossRef CAS;
(j) K. N. Shavrin, V. D. Gvozdev and O. M. Nefedov, Russ. Chem. Bull., 2010, 59, 1451–1458 CrossRef CAS.
- K. N. Shavrin, V. D. Gvozdev and O. M. Nefedov, Russ. Chem. Bull., 2009, 58, 2432–2436 CrossRef CAS.
- M. Zhang, Y. Gong and W. Wang, Eur. J. Org. Chem., 2013, 7372–7381 CrossRef CAS.
-
(a) P. Ryabchuk, J. P. Matheny, M. Rubina and M. Rubin, Org. Lett., 2016, 18, 6272–6275 CrossRef CAS PubMed;
(b) B. K. Alnasleh, M. Rubina and M. Rubin, Chem. Commun., 2016, 52, 7494–7496 RSC;
(c) A. Edwards, T. Bennin, M. Rubina and M. Rubin, RSC Adv., 2015, 5, 71849–71853 RSC.
-
(a) T. Y. Rudashevskaya and O. A. Nesmeyanova, Izv. Akad. Nauk SSSR, Ser. Khim., 1983, 1821–1824 CAS;
(b) T. Y. Rudashevskaya and O. A. Nesmeyanova, Izv. Akad. Nauk SSSR, Ser. Khim., 1979, 669–671 CAS;
(c) A. Padwa and M. W. Wannamaker, Tetrahedron Lett., 1986, 27, 5817–5820 CrossRef CAS.
- A. Levin and I. Marek, Chem. Commun., 2008, 44, 4300–4302 RSC.
-
(a) Y. Zhu, M. Zhang, H. Yuan and Y. Gong, Org. Biomol. Chem., 2014, 12, 8828–8831 RSC;
(b) J. Hu, Y. Liu and Y. Gong, Adv. Synth. Catal., 2015, 357, 2781–2787 CrossRef CAS.
-
(a) W. M. Sherrill, R. Kim and M. Rubin, Tetrahedron, 2008, 64, 8610–8617 CrossRef CAS;
(b) R. Kim, W. M. Sherrill and M. Rubin, Tetrahedron, 2010, 66, 4947–4953 CrossRef CAS.
- W. M. Sherrill, R. Kim and M. Rubin, Synthesis, 2009, 1477–1484 CAS.
- In an attempt to improve the selectivity, we also carried out these transformations at a lower temperature or in the presence of a smaller amount of base, but this proved impossible, as the first step, involving the generation of cyclopropenes via a base-assisted 1,2-elimination of the HBr reaction, did not proceed under these conditions. It should also be mentioned that unactivated benzylic groups do not demonstrate this type of reactivity, as their C–H acidity is not sufficient for such an anion to be generated in the presence of such a relatively weak base as t-BuOK. This was demonstrated, for example, in Fox's report, who used the derivatives of cyclopropene-3-carboxylic acid with Evans chiral auxiliary, derived from phenylalanine: L. Liao, E. Zhang, N. Yan, J. A. Golen and J. M. Fox, Tetrahedron, 2004, 60, 1803–1816 CrossRef CAS . In our case, such deprotonation is only possible because benzylic anions are generated next to carboxamide functionality, which is known to stabilize alpha-anions greatly due to the significant contribution of the nitrogen ylide resonance form.
- See, for example:
(a) S. Tanaka, Y. Honmura, S. Uesugi, E. Fukushi, K. Tanaka, H. Maeda, K. Kimura, T. Nehira and M. Hashimoto, J. Org. Chem., 2017, 82, 5574–5582 CrossRef CAS PubMed;
(b) P. Paya, M. Anastassiades, D. Mack, I. Sigalova, B. Tasdelen, J. Oliva and A. Barba, Anal. Bioanal. Chem., 2007, 389, 1697–1714 CrossRef CAS PubMed.
- See, for example:
(a) H. P. Cho, D. W. Engers, D. F. Venable, C. M. Niswender, C. W. Lindsley, P. J. Conn, K. A. Emmitte and A. L. Rodriguez, ACS Chem. Neurosci., 2014, 5, 597–610 CrossRef CAS PubMed;
(b) K. X. Chen, L. Nair, B. Vibulbhan, W. Yang, A. Arasappan, S. L. Bogen, S. Venkatraman, F. Bennett, W. Pan, M. L. Blackman, A. I. Padilla, A. Prongay, K.-C. Cheng, X. Tong, N.-Y. Shih and F. G. Njoroge, J. Med. Chem., 2009, 52, 1370–1379 CrossRef CAS PubMed.
- For recent examples on the synthesis of bicyclic lactam systems like 7via inter- or intramolecular cyclopropanations see:
(a) J. Tao, C. D. Estrada and G. K. Murphy, Chem. Commun., 2017, 53, 9004–9007 RSC. Concerted cycloadditions:
(b) S. McCabe and P. Wipf, Angew. Chem., Int. Ed., 2017, 56, 324–327 CrossRef CAS PubMed. Alkene or alkyne hydroaminations:
(c) D. C. Miller, G. J. Choi, H. S. Orbe and R. R. Knowles, J. Am. Chem. Soc., 2015, 137, 13492–13495 CrossRef CAS PubMed;
(d) B. de Carne-Carnavalet, C. Meyer, J. Cossy, B. Folleas, J.-L. Brayer and J.-P. Demoute, J. Org. Chem., 2013, 78, 5794–5799 CrossRef CAS PubMed. Radical cyclizations:
(e) M. K. Nielsen, B. J. Shields, J. Liu, M. J. Williams, M. J. Zacuto and A. G. Doyle, Angew. Chem., Int. Ed., 2017, 56, 7191–7194 CrossRef CAS PubMed. Cylizations, involving C–H activations:
(f) E. Hernando, J. Villalva, A. M. Martinez, I. Alonso, N. Rodriguez, R. Gomez Arrayas and J. C. Carretero, ACS Catal., 2016, 6, 6868–6882 CrossRef CAS See also ref. 3k for a novel approach involving the asymmetric carboamination of cyclopropenes.
-
(a) R. A. Bragg, J. Clayden and C. J. Menet, Tetrahedron Lett., 2002, 43, 1955–1959 CrossRef CAS;
(b) R. A. Bragg and J. Clayden, Tetrahedron Lett., 1999, 40, 8323–8326 CrossRef CAS;
(c) R. A. Bragg, J. Clayden, G. A. Morris and J. H. Pink, Chem. – Eur. J., 2002, 8, 1279–1289 CrossRef CAS PubMed;
(d) P. Beak and B. Lee, J. Org. Chem., 1989, 54, 458–464 CrossRef CAS;
(e) A. J. Burton, J. P. Graham and N. S. Simpkins, Synlett, 2000, 1640–1642 CAS;
(f) D. Seebach, J. J. Lohmann, M. A. Syfrig and M. Yoshifuji, Tetrahedron, 1983, 39, 1963–1974 CrossRef CAS;
(g) J. J. Lohmann, D. Seebach, M. A. Syfrig and M. Yoshifuji, Angew. Chem., 1981, 93, 125–126 CrossRef CAS;
(h) A. I. Meyers, K. B. Kunnen and W. C. Still, J. Am. Chem. Soc., 1987, 109, 4405–4407 CrossRef CAS.
-
(a) A. Couture, E. Deniau, D. Ionescu and P. Grandclaudon, Tetrahedron Lett., 1998, 39, 2319–2320 CrossRef CAS;
(b) B. S. Bhakuni, A. Yadav, S. Kumar, S. Patel, S. Sharma and S. Kumar, J. Org. Chem., 2014, 79, 2944–2954 CrossRef CAS PubMed;
(c) J. Clayden, S. D. Hamilton and R. T. Mohammed, Org. Lett., 2005, 7, 3673–3676 CrossRef CAS PubMed;
(d) J. Clayden and C. J. Menet, Tetrahedron Lett., 2003, 44, 3059–3062 CrossRef CAS.
- Since exo-products are somewhat more stable than endo-isomers, the selective formation of exo-7d might be a result of thermodynamically-driven epimerization proceeding via a base-assisted deprotonation at the benzylic position. Unfortunately, this reaction has to be quenched after 5 min to obtain at least some isolated yield. Longer exposure to the base causes quick decomposition of both exo- and endo-products, which does not allow us to exploit this isomerization for achieving even better selectivity.
- Results for the detailed GC-monitoring for all other cyclization reactions are included in the ESI.† These were used to choose the best reaction duration and the appropriate quenching time for each particular product.
- See the ESI† for details.
Footnote |
| † Electronic supplementary information (ESI) available: 1H and 13C spectral data, GC traces, optimized geometries, and X-ray spectral data. CCDC 1575277. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c7ob02068f |
|
| This journal is © The Royal Society of Chemistry 2018 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *ab
*ab