
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Inducing nematic ordering of cellulose nanofibers using osmotic dehydration†
Valentina
Guccini‡
ab,
Shun
Yu‡¶
*ab,
Michael
Agthe§
a,
Korneliya
Gordeyeva
a,
Yulia
Trushkina
a,
Andreas
Fall¶
a,
Christina
Schütz||
 ab and
Germán
Salazar-Alvarez
*ab
ab and
Germán
Salazar-Alvarez
*ab
aDepartment of Materials and Environmental Chemistry, Arrhenius Laboratory, Stockholm University, SE-10691 Stockholm, Sweden. E-mail: shun.yu@ri.se; gersal@protonmail.com
bWallenberg Wood Science Center (WWSC), Teknikringen 58, SE-100 44, Stockholm, Sweden
First published on 29th November 2018
Abstract
The formation of nematically-ordered cellulose nanofiber (CNF) suspensions with an order parameter fmax ≈ 0.8 is studied by polarized optical microscopy, small-angle X-ray scattering (SAXS), and rheological measurements as a function of CNF concentration. The wide range of CNF concentrations, from 0.5 wt% to 4.9 wt%, is obtained using osmotic dehydration. The rheological measurements show a strong entangled network over all the concentration range whereas SAXS measurements indicate that at concentrations >1.05 wt% the CNF suspension crosses an isotropic-anisotropic transition that is accompanied by a dramatic increase of the optical birefringence. The resulting nanostructures are modelled as mass fractal structures that converge into co-existing nematically-ordered regions and network-like regions where the correlation distances decrease with concentration. The use of rapid, upscalable osmotic dehydration is an effective method to increase the concentration of CNF suspensions while partly circumventing the gel/glass formation. The facile formation of highly ordered fibers can result in materials with interesting macroscopic properties.
Introduction
The will to end the dependence on fossil fuel-based chemicals and components has driven the re-exploitation of ancient materials from renewable resources, such as cellulose, silk, collagen, etc.1 Particularly, cellulose nanomaterials extracted from plants are currently very attractive due to their excellent intrinsic optical and mechanical properties and the potential novel applications that uses these characteristics.2,3 However, despite the extraordinary intrinsic properties of nanocelluloses at the nanoscopic level, the translation of the properties to the macroscopic level is not straightforward and, e.g., their mechanical properties remain more than an order of magnitude below the expected values.2There are a number of cellulose nanomaterials (also referred to as nanocelluloses)3 whence cellulose nanocrystals (CNC, highly crystalline and mechanically rigid) and cellulose nanofibers (CNF, medium crystallinity and more flexible) stand out (for a more detailed description of such cellulose nanomaterials see ref. 2 and refs therein). In the case of CNC, it has long been known that the assembly of the crystals in a chiral nematic phase, observed in both synthetic4 and natural5 materials, results in structural colors that can be controlled to tailor the visual appearance.6 Regarding CNF, their assembly into ordered phases has remained elusive7 as its flexibility leads to more entanglements by constraining their freedom of motion which together with the influence of surface charges leads to the formation of arrested states (glasses or gels) at very low concentrations.8
Some novel routes to circumvent the agglomeration have been recently put forward to induce the nematic ordering of CNF into 1D filaments, for instance, Håkansson et al.9 and Mittal et al.10 showed that flow fields can be used to align CNF achieving a maximum order parameter fmax ≈ 0.4 and fmax ≈ 0.5, respectively, whereas Torres-Rendon et al. used wet-stretching of wet-spun (fmax ≈ 0.6)11 and Mohammadi et al. used extrusion followed by coagulation (fmax ≈ 0.6).12 Sehaqui et al. used cold drawing of wet films to achieve fmax ≈ 0.7.13 Zhao et al. used the slow evaporation of water to form 2D films of nematically-aligned CNF14 whereas Kobayashi et al. used the slow neutralization of the surface charges in CNF to bring about a liquid crystalline order in 3D.15 However, the order parameter was not determined in either case. In all the above-mentioned cases there was a dramatic improvement of the mechanical/optical properties of the obtained materials. This calls for the development of controllable methods that can bring about ordering of the fibers. In the previous work, it has been shown that a key element in achieving the assembly of the nanofibers is to reduce the network formation in the gel or glass form of the arrested states by promoting the orientation of CNF while controlling the concentration.
Osmotic dehydration (OD) shares some features with the flow field orientation of CNF as there is also a unidirectional flow of water. OD has been widely used to dehydrate food at industrial level,16,17 desalination, purification and biofuel separation.18 In this work, we used OD as facile approach to prepare nematically-ordered CNFs suspensions in a time efficient manner. In the following we describe how we fabricate ordered CNF suspensions with nominal concentrations (C) up to 4.9 wt% (fmax ≈ 0.8) without the addition of any substance or coagulant. We show their concentration-dependent structural evolution using polarized optical microscopy, rheological measurements and small-angle X-ray scattering.
Results and discussion, experimental
The carboxylated CNF suspensions with initial concentration 0.5 wt% (surface charge 0.6 mmol g−1, mean nominal aspect ratio ā0 = 125, see ESI Fig. S1†) were sealed in dialysis bags and immersed in a 50 wt% poly(ethylene glycol) solution during different lengths of time to obtain CNF suspensions with concentrations up to 4.9 wt% (ESI Fig. S2†).Fig. 1 summarizes the macroscopic characteristic of CNF suspensions under polarized optical microscopy (POM) and their rheological properties. Fig. 1A, shows the CNF suspensions in the concentration range 0.5 ≤ C ≤ 4.9 wt% between cross-polarizers. At lower concentrations, i.e., C ≤ 0.5 wt%, only small birefringent regions typical of nematic ordering19 are visible. As the concentration increases so does the intensity of the birefringence, displaying even iridescent colors at high concentrations (>1.7 wt%). The images show that there are a number of domains in the suspension of up to several mm in size. Fig. 1B shows the order parameter obtained from the birefringence of the suspensions fPOM. At 0.5 wt% there is already some reduced birefringence that indicates the early nematic ordering that increases readily towards 1 wt%. A plateau on the ordering is reached above 1.7 wt% but, as the POM images show, the size of the domains increases suggesting that nematic domains start coalescing. The values for the early onset of birefringence agree reasonably well with earlier reports. For instance, Saito et al. reported birefringence attributed to the nematic structuring of nanocellulose network in CNF suspensions already at 0.1 wt%, likely due to the larger nominal aspect ratio of the fibers (a0 ≈ 330).14,20
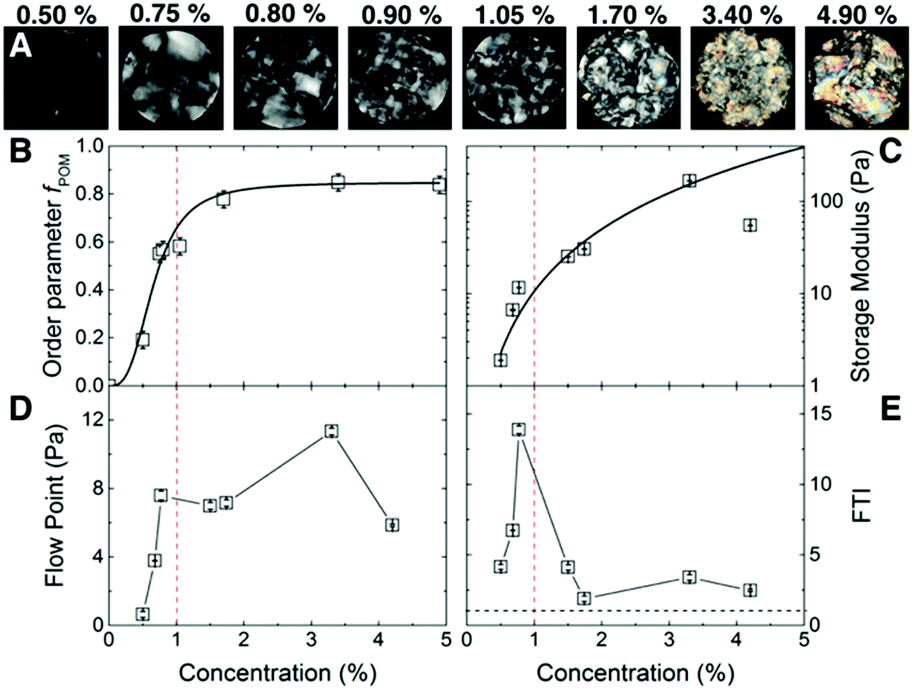 | ||
| Fig. 1 Macroscopic optical and rheological properties of ordered CNF suspensions. (A) Images of the CNF suspensions between cross-polars. The number indicates the concentration (wt%). The field of view is 85 mm. (B) Order parameter, fPOM determined from the birefringent images shown in (A), the line is a logistic function used as a guide to the eye. (C) Storage modulus (G′) as a function of CNF solid content. The line shows the curve G′ = ACα with α = 2.2 ± 0.2 as discussed in the text. (D) Flow point, i.e., G′ = G′′. (E) Variation of flow transition index, FTI, e.g., ratio between the flow point (τf) and the yield point (τy), as a function of concentration. The black dashed line in (E) corresponds to τf/τy = 1. The red dashed line in (B–E) shows the concentration value of 1 wt%. | ||
Fig. 1C shows the storage modulus (G′) of CNF suspensions as a function of the C. We correlated the strength of the suspension with the value of the G′ in the linear-viscoelastic range (≈0.1 to 10% strain, see ESI Fig. S3†). The amplitude sweep reveals that in the linear-viscoelastic range the value of G′ of all the samples is higher than the loss modulus (G′′) indicating a network behavior of a viscoelastic solid (see ESI Fig. S3†). The networks have a large storage modulus of up to 100 Pa at relatively low concentrations. Indeed, the data in the range up to 3.4 wt% follows a power law behavior G′ ∝ Cα with an exponent α = 2.2 ± 0.2, consistent with an entangled network of semiflexible polymers21,22 and earlier experimental studies on cellulose fibers.7,23 The combination of surface charge and aspect ratio of suspensions (i.e., the effective aspect ratio, aeff) studied in the present work (excluding the highest concentration) lies within the limits of the critical concentrations given by the excluded volume of the fibers24 and the threshold for the formation of arrested states (see ESI Fig. S4†),8 which may also result in the formation of a strong network.21,22
Fig. 1D shows the flow point (τf) of the CNF suspension, at which the G′ is equal to G′′ in the shear stress amplitude sweep. After this point G′ < G′′ and the suspensions are characterized by liquid-like character. The τf steeply increases from 0.5 to 0.8 wt% after which reaches a plateau, followed by a further increase at 3.4 wt% and a decrease at 4.9 wt%. Before the flow point (G′ > G′′) the CNF suspensions are characterized by interconnected network structure that is able to dissipate the shear stress by elastic deformation. The fact that the G′′ shows a peak in the amplitude sweep (see ESI S3†) suggests that this structure does not collapse suddenly but gradually until the final collapse at the flow point.25 Passed this point the CNF samples flow with viscous liquid behavior and the CNF are entangled. The decrease of the flow point at 4.9 wt% is related to the decrease of suspension strength, probably due to (random or ordered) agglomeration phenomena between the nanofibres.
Fig. 1E shows the flow transition index, FTI = τf/τy, at which the suspensions still show network behavior but irreversible deformations are present together with the reversible-elastic behavior. The FTI shows a maximum towards 1 wt%, followed by a decrease in which it approaches 1 as the solid content increases. The τf/τy characterizes the breaking behavior of the CNF network structure. Values closer to 1 shows that the CNF suspensions embrittle,25 typical of ordered (crystalline) systems. Indeed, the POM images show an increasing fraction of the nematic phase at C ≥ 1 wt%, and together with the rheological measurements, they both point towards the formation of arrested states in competition with the formation of the nematic phase. At 4.9 wt% the ordered fraction may be surpassing the arrested fraction, resulting in the weakening of the suspension through phase separation.
The rheological measurements show that the CNF behaves as a viscoelastic network. We carried small-angle X-ray scattering studies to study the structural characteristics of the suspensions. 2D SAXS pattern of CNF suspensions are displayed in Fig. 2A. In general, all the 2D patterns appears rather isotropic below 1.05 wt% where there is a slight increase of the anisotropy at higher concentration. Such isotropic-to-anisotropic transition could be highlighted via the azimuthal plot in Fig. 2B. At each azimuthal angle, ϕ, the intensity is integrated from the radial region of q = 0.30 ± 0.05 nm−1. When the concentration is below 1.05%, the scattering intensity is uniformly distributed along the azimuthal angles, while at higher concentrations such intensities show modulation as a function of azimuthal angle ϕ, indicating preferred orientation. The observed order parameter, f, gradually increases with the concentration. This observation corresponds to a small portion of the nematic domains aligned along the same direction where the majority of the nematic domains are randomly oriented. This per se agrees with the appearance of nematic phases observed with POM where the images show a collection of randomly oriented domains.
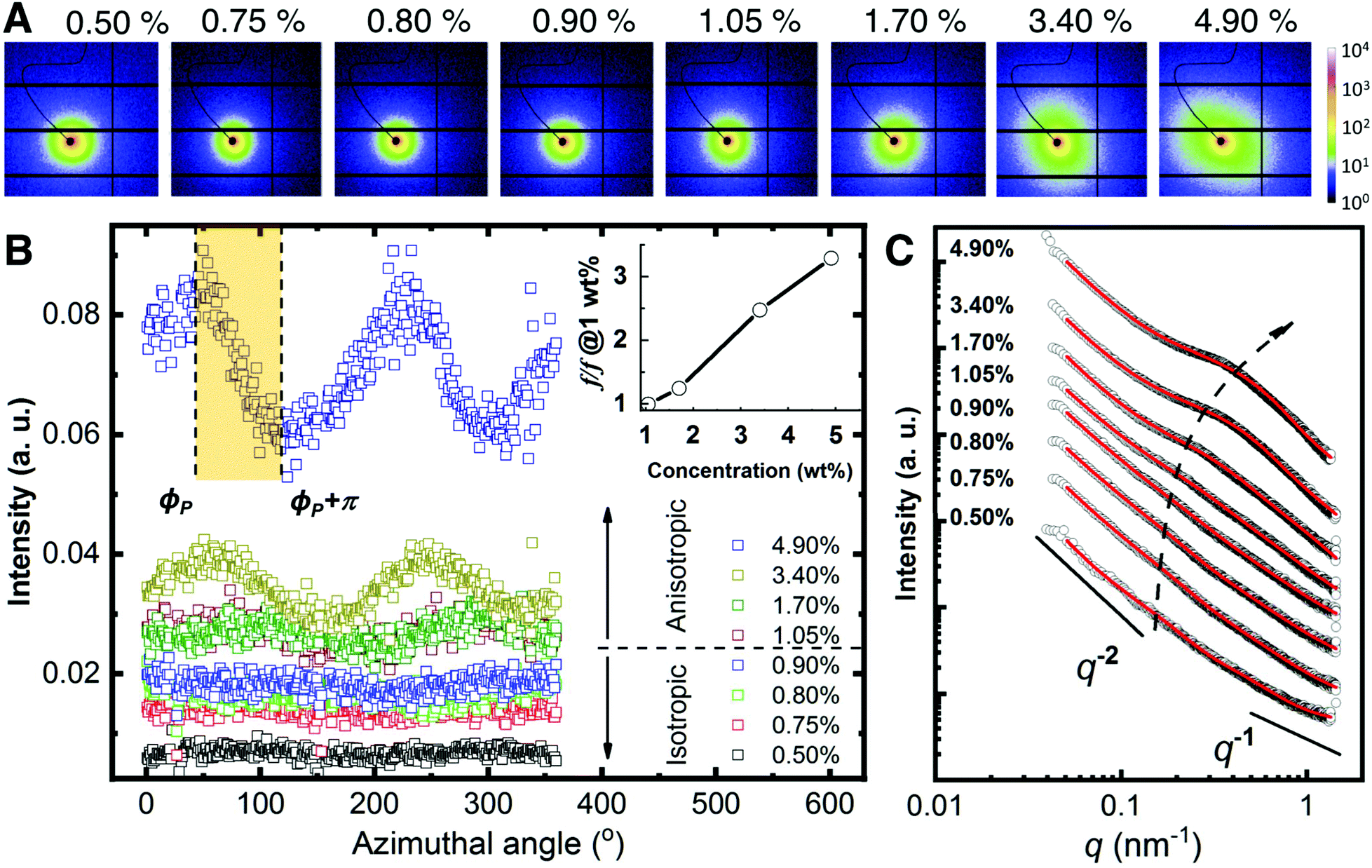 | ||
| Fig. 2 Small-angle X-ray scattering analysis. (A) 2D SAXS patterns upon increasing CNF concentration. (B) Azimuthal plots from the data in A. The scattering intensity of the region q = 0.30 ± 0.05 nm−1 is plotted as a function of azimuthal angles for the different concentrations. The shadowed region shows an example where the Hermans orientation factor (f) is calculated. Insert: The relative increase of f w.r.t. that at 1.05 wt%. (C) Radial integration line profiles of the images shown in A. Black circles are experimental data and red solid lines are fitting results using eqn (1). The curves are vertically shifted to highlight the structure transition. The dashed arrow is a guide to the eye. | ||
The information on the hierarchical structures of CNF suspension and their dependence on the concentration can be extracted from 1D SAXS profile in Fig. 2C. Given the small values of anisotropy in the 2D SAXS patterns, the SAXS line profiles were obtained by integrating the 2D SAXS radially 0–360°.
In all cases, I(q) displays a power-law dependence on the scattering vector at q ≤ 0.1 nm−1 with an exponent close to −2. This exponent indicates an entangled network22 that can be approximated with a fractal system, consistent with the rheological measurements (see Fig. 1C). At higher concentrations, two additional features become increasingly pronounced in the region q ≥ 0.1 nm−1 that shift towards higher q values as the concentration increases. Given the dominant contribution from the interference between CNF in the network, a shape-independent model (eqn (1)) by combining power-law function and two-stage gel model26,27 has been employed to fit the experimental data, as an analog to the traditional scattering model of polymer gels.28–30 In eqn (1), the power-law function with a Porod exponent n mainly contributes to the intensity variation in the low q region, e.g. large features. The two-stage network model in the high q region are modeled with a Gaussian function derived from the Guinier function and attributed to entangled region with a characteristic size of Ξ.26,31
The third term is in a form of Lorentzian function or so-called Ornstein–Zernicke function with a correlation distance ξ, which depicts the electron density correlation γ(r) in the system via a modulated exponent function  . For polymer gels, ξ typically describes the correlation length of polymer–polymer chains interaction due to thermal fluctuation.26 The detailed fitting parameters can be found in the ESI (Table S1†).
. For polymer gels, ξ typically describes the correlation length of polymer–polymer chains interaction due to thermal fluctuation.26 The detailed fitting parameters can be found in the ESI (Table S1†).
 | (1) |
The dependence of Ξ and ξ correlation distances on the concentration can be revealed by a log–log plot in Fig. 3A. As the concentration increases, both Ξ and ξ decrease, indicating that CNF become more closely packed. This trend can be fitted via power-law decay functions with the exponents −0.62 (Ξ) and −0.35 (ξ), respectively. For ideal semi-diluted polymer solution, the exponent of such power-law decay is predicted as −0.75.32 In this case, neither of the exponents is close to this value. Generally, when a system is compressed isotropically in 3D, the separation distance will decrease as C−1/3.33 According to the fitting results, the exponent of ξ (−0.35) is very close to −1/3. Considering the nematic phases are randomly oriented in 3D to a large extent in our system, the decrease of ξ reflects a reduction of fiber to fiber distance in the nematic phase, which is equivalent to the isotropic shrinkage of a general 3D CNF network. The size of the agglomerate, Ξ, is scaled with ξ by a power 1.77 (0.62/0.35), indicating a fractal nature. Such power-law change leads to an interpretation that the agglomerates experience approximately affine contraction.32 The change of Ξ and ξ is also illustrated in Fig. 3B, where the length of Ξ and ξ reduces as the concentration increases regardless the way of the entire network connection.
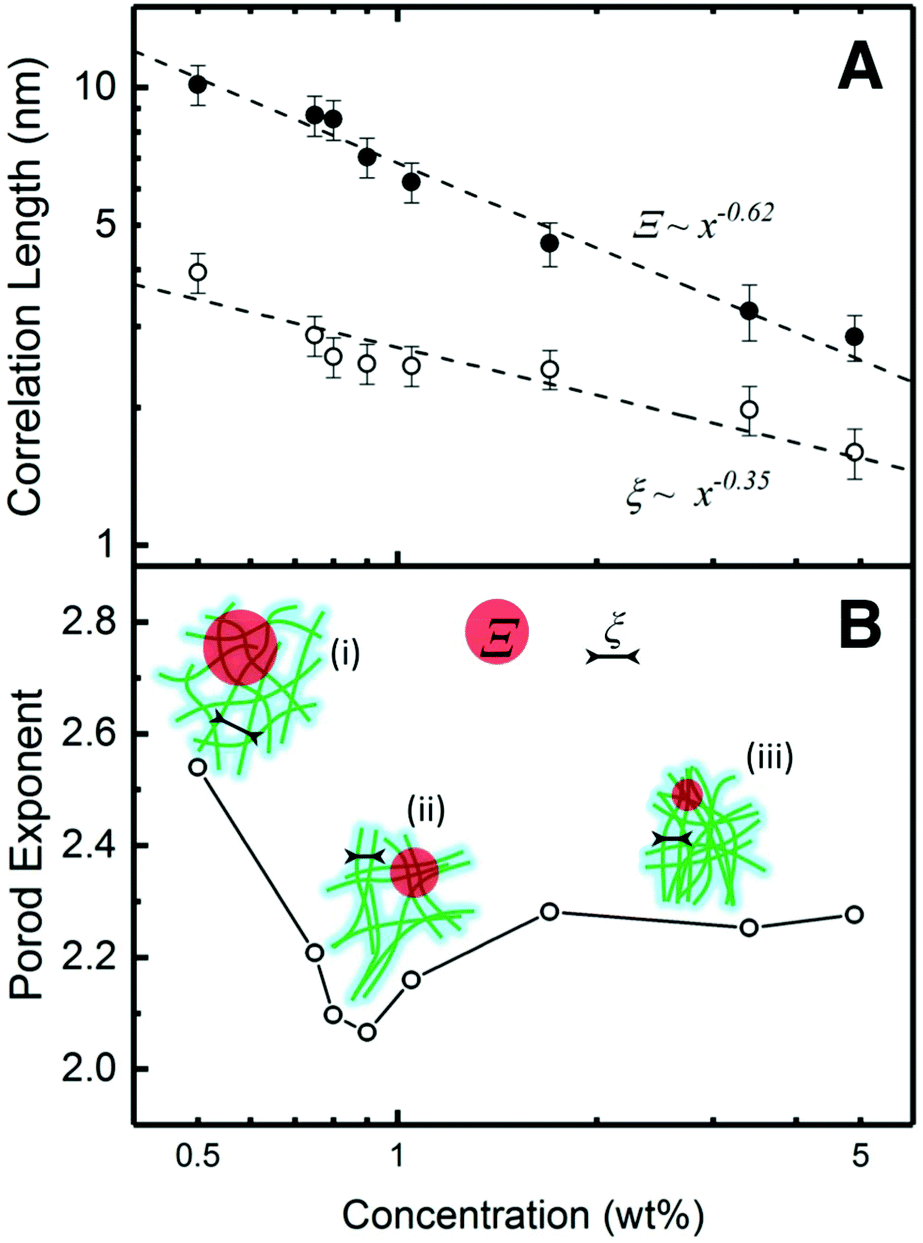 | ||
| Fig. 3 SAXS-derived correlation lengths. (A) the development of Ξ and ξ as a function of gel concentration. The dashed lines are power-law fitting. (B) The Porod exponents or mass fractal dimension change as a function of concentration. The schematics illustrate the structural characteristics where green lines represent CNF, red region the entangled areas with size Ξ, and black lines the correlation length ξ. Three types of CNFs connections (i–iii) illustrates the structures with different fractal number. | ||
The power-law term in eqn (1) describe the complexity of the connections of the CNF suspensions, which is typically assigned to the fractal structure in finite scale. Shown in Fig. 3B, the Porod exponents are plotted as function of CNF concentrations. The Porod exponent, n, is assigned to a mass fractal dimension dm when 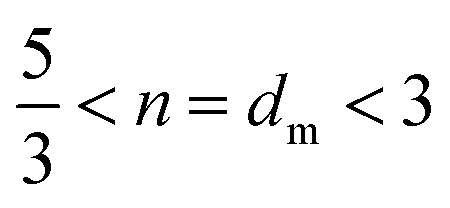 and related to surface fractal dimension, ds, by n = 6 − ds, when 3 < n < 4.34 The values of n in all our cases are between 2.0 and 2.6, indicating the existence of mass fractal CNF networks with the fractal dimension equal to the Porod exponent. For a mass fractal nanofibers structure at the given concentration, the larger fractal dimension it has, the more complex connection it may have. The fractal dimension follows an interesting trend as the concentration increases, that it first decreases from 2.5 till a minimum ≈2.1 at C ≈ 1.0 wt% and then increases till a plateau around 2.3, which is independent to the further increased concentration. The transition point around 1.0 wt% is reminiscent of the transition from isotropic to anisotropic scattering patterns in Fig. 2B. Similar fractal dimension change has been reported for the structures of nanoparticles agglomerates35,36 and whey protein networks37 at fixed concentration, showing a decrease followed by an increasing as a function of systematically increased ionic strengths. Such phenomenon was interpreted as a consequence of the change of the self-assembly kinetics between the fast diffusion-limited cluster aggregates (DLCA) and the slow reaction-limited cluster aggregation (RLCA). When increasing the amount of salts into the initial systems, the additives first screen the intrinsic surface charges of nanoparticles and proteins until they are neutralized and then flip the sign of their surface charge by an excess of amount. As a result, the net repulsion first decreases when surface charges are neutralized and then is overcome by the attraction, suggesting a transition from a glass to a gel state.
and related to surface fractal dimension, ds, by n = 6 − ds, when 3 < n < 4.34 The values of n in all our cases are between 2.0 and 2.6, indicating the existence of mass fractal CNF networks with the fractal dimension equal to the Porod exponent. For a mass fractal nanofibers structure at the given concentration, the larger fractal dimension it has, the more complex connection it may have. The fractal dimension follows an interesting trend as the concentration increases, that it first decreases from 2.5 till a minimum ≈2.1 at C ≈ 1.0 wt% and then increases till a plateau around 2.3, which is independent to the further increased concentration. The transition point around 1.0 wt% is reminiscent of the transition from isotropic to anisotropic scattering patterns in Fig. 2B. Similar fractal dimension change has been reported for the structures of nanoparticles agglomerates35,36 and whey protein networks37 at fixed concentration, showing a decrease followed by an increasing as a function of systematically increased ionic strengths. Such phenomenon was interpreted as a consequence of the change of the self-assembly kinetics between the fast diffusion-limited cluster aggregates (DLCA) and the slow reaction-limited cluster aggregation (RLCA). When increasing the amount of salts into the initial systems, the additives first screen the intrinsic surface charges of nanoparticles and proteins until they are neutralized and then flip the sign of their surface charge by an excess of amount. As a result, the net repulsion first decreases when surface charges are neutralized and then is overcome by the attraction, suggesting a transition from a glass to a gel state.
The higher fractal dimension associated with C = 0.5 wt% is due to a dominant net repulsion associated with the deprotonated carboxylate groups on CNF surface, leading to high degree of random connection in the CNF network (state (i) of the schematics in Fig. 3B). Whereas the low concentration limited the absolute number of connections in a unit volume, and thus lead to the low strength (low value of G′). As the concentration of CNF (and the counterions) increases, the decay of the Debye length leads to a net attraction increase. Then, the CNF network collapses from a structure with higher fractal dimension at low concentrations to a structure with lower fractal dimension via adjacent CNF attracting to each other as state (ii) in Fig. 3B. Such CNF attraction certainly aids the formation of nematic domains. By assuming that some of the CNF in the network are more mobile than others, they would move and stay aligned with other less mobile CNF when the concentration is increased. The fractal dimension will continue decreasing until all the mobile ones are attracted to immobile one and reaches minimum, in this case, ca. 1.05 wt%. During this process, the density of the contact points of the network keep increasing, rendering a stronger suspension with larger G′. Further increasing the concentration will force the networks to condense into higher fractal dimensions because the CNF attraction is frustrated by the depleted number of mobile CNF. However, the rigidity of the network consisting of CNF rigid bundles will generate mechanical resistance to densification. When a mechanical balance is reached, the fractal dimension of network will remain unchanged.
Conclusions
In summary, we characterized the concentration-dependent structural evolution of carboxylated CNF suspension by macroscopic techniques, such as polarized optical imaging and rheological measurements; as well as SAXS. The macroscopic optical and rheological properties show that the CNF suspensions consist of randomly oriented nematic domains coexisting with a random CNF network. Towards C ≥ 1 wt% the nematic ordering readily increases reaching a maximum order parameter ≈ 0.8. Using SAXS we show that, at the microscopic level, CNF appears inside (anisotropic) nematic domains and in an (isotropic) entangled network that can be approximated with a fractal system. As the concentration increases, the correlation distance shows that CNF become more closely packed, which is equivalent to a reduction of fiber to fiber distance in the nematic phase and the isotropic shrinkage of the 3D CNF network. Our work shows that the formation of nematic domains in the CNF suspension is promoted by the increase of solid content by osmotic dehydration and the dynamics involving the charge screening between nanofibers. The technique has a potential to prepare ordered materials in a facile fashion while circumventing the formation of arrested states.Experimental
Carboxylated CNFs with surface charge of 0.6 mmol g−1 were prepared by TEMPO-mediated oxidation, according to the protocol of Saito et al.20 The dimensions of the obtained CNFs are 2.4 ± 0.7 nm in height and 299 ± 168 nm in length (see ESI Fig. S1†).The concentration of CNFs in aqueous media was increased from the initial 0.5 wt% to a maximum of 4.9 wt% by controlling the time of osmotic dehydration using an initial osmotic pressure π ≈ 2.8 MPa. Typically, a dialysis bag containing 30 g of the initial CNF suspension was sealed from one extremity and then immersed in a 50 wt% poly(ethylene glycol) solution under magnetic stirring. The CNF suspension was manually mixed every 20 min so as to prevent a gradient build-up (see ESI Fig. S2† for details). The concentration of the CNF suspension was determined by thermogravimetric analysis using a PerkinElmer TGA 7 under nitrogen atmosphere with a heating rate of 1 °C min−1. The average error over four measurements was about 0.01 wt%.
The birefringence of the CNF suspensions was measured using a homemade setup with cross-polars and a digital camera. The CNF samples were deposited on a clean glass slide and constrained within a 8.3 mm-diameter Viton o-ring and mounted vertically. The intensity of the birefringent images under cross-polars, I, was extracted using the program ImageJ.38 CNF suspensions up-concentrated using air-drying did not show the same optical features and were not carefully studied. The order parameter fPOM was calculated according to Håkansson39 as:
 | (2) |
The rheological characterization of the CNF samples was carried out with and Anton Paar Physica MCR 301 rheometer in oscillatory mode by using a plate-plate geometry at 23 °C. Amplitude sweep measurements were done at a frequency of 5 rad s−1 with amplitude scanning from 0.01% to 1000% using a gap size of 1 mm.
Small-angle X-ray scattering (SAXS) measurements were carried out at P03 “MiNaXS” beamline40 at PETRA III storage ring, Deutsche Elektronen-Synchrotron (DESY), Hamburg. The X-ray energy of the beam with a cross section of 42 × 20 μm2 was set to 13.0 keV and the sample-to-detector distance was calibrated as 5386.2 ± 0.1 mm using dry rat-tail collagen. The CNF samples were loaded into borosilicate capillaries (Hilgenberg, GmbH) with a diameter of 2 mm and a wall thickness of 10 μm. Beam damage to the samples was carefully scrutinized the changes to the intensity and scattering profile on consecutive images exposed 0.1 s. 2D SAXS patterns were collected via a Dectris Pilatus 1 M detector using exposure times of 0.4 s. Data reductions and analyses were carried out using the program DPDAK.41 The SAXS modelling and fitting were done using the program SasView 4.1.42 To quantify the degree of orientation from the SAXS curves, we use the Hermans orientation factor, f, (see (eqn (3))) as previously described.9 The reference direction (i.e. φ = 0) is set at one of the symmetric peak positions ϕP of the azimuthal curve and integrating in the region [ϕP, ϕP + π].
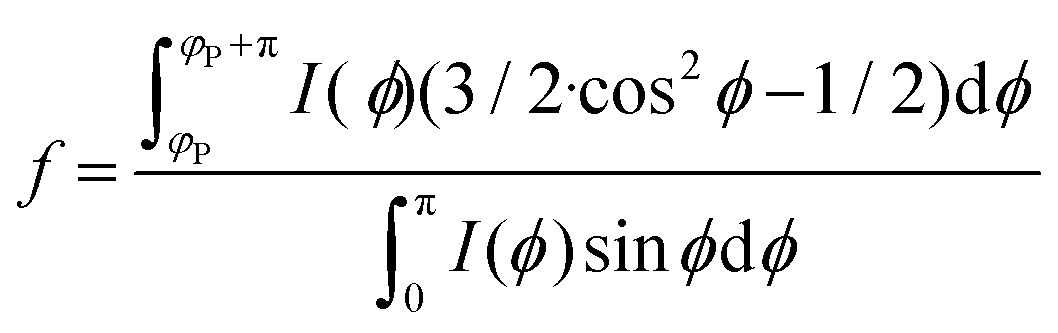 | (3) |
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank Yingxin Liu and María del Mar González del Campo Barbera Rodríguez for their help preparing CNF. The Knut and Alice Wallenberg foundation is acknowledged for the financial support. We acknowledge DESY (Hamburg, Germany), a member of the Helmholtz Association HGF, for the provision of experimental facilities. Parts of this research were carried out at PETRA III and we would like to thank Matthias Schwartzkopf for assistance in using the beamline P03. The research leading to this publication has been supported by the project CALIPSOplus (Grant Agreement No. 730872) from the EU Framework Programme for Research and Innovation HORIZON 2020. This work benefited from the use of the SasView application, originally developed under NSF award DMR-0520547. SasView contains code developed with funding from the European Union's Horizon 2020 research and innovation programme under the SINE2020 project, grant agreement no. 654000.Notes and references
- B. Wicklein and G. Salazar-Alvarez, J. Mater. Chem. A, 2013, 1, 5469 RSC.
- R. J. Moon, A. Martini, J. Nairn, J. Simonsen and J. Youngblood, Chem. Soc. Rev., 2011, 40, 3941–3994 RSC.
- D. Klemm, F. Kramer, S. Moritz, T. Lindström, M. Ankerfors, D. Gray and A. Dorris, Angew. Chem., Int. Ed., 2011, 50, 5438–5466 CrossRef CAS PubMed.
- J.-F. Revol, H. Bradford, J. Giasson, R. H. Marchessault and D. G. Gray, Int. J. Biol. Macromol., 1992, 14, 170–172 CrossRef CAS PubMed.
- S. Vignolini, P. J. Rudall, A. V. Rowland, A. Reed, E. Moyroud, R. B. Faden, J. J. Baumberg, B. J. Glover and U. Steiner, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15712–15715 CrossRef CAS PubMed.
- R. M. Parker, G. Guidetti, C. A. Williams, T. Zhao, A. Narkevicius, S. Vignolini and B. Frka-Petesic, Adv. Mater., 2018, 30, 1704477 CrossRef PubMed.
- T. Saito, T. Uematsu, S. Kimura, T. Enomae and A. Isogai, Soft Matter, 2011, 7, 8804 RSC.
- M. Nordenström, A. Fall, G. Nyström and L. Wågberg, Langmuir, 2017, 33, 9772–9780 CrossRef PubMed.
- K. M. O. Håkansson, A. B. Fall, F. Lundell, S. Yu, C. Krywka, S. V. Roth, G. Santoro, M. Kvick, L. Prahl Wittberg, L. Wågberg and L. D. Söderberg, Nat. Commun., 2014, 5, 4018 CrossRef PubMed.
- N. Mittal, F. Ansari, V. K. Gowda, C. Brouzet, P. Chen, P. T. Larsson, S. V. Roth, F. Lundell, L. Wågberg, N. A. Kotov and L. D. Söderberg, ACS Nano, 2018, 12, 6378–6388 CrossRef CAS PubMed.
- J. G. Torres-Rendon, F. H. Schacher, S. Ifuku and A. Walther, Biomacromolecules, 2014, 15, 2709–2717 CrossRef CAS PubMed.
- P. Mohammadi, M. S. Toivonen, O. Ikkala, W. Wagermaier and M. B. Linder, Sci. Rep., 2017, 7, 11860 CrossRef PubMed.
- H. Sehaqui, N. Ezekiel Mushi, S. Morimune, M. Salajkova, T. Nishino and L. A. Berglund, ACS Appl. Mater. Interfaces, 2012, 4, 1043–1049 CrossRef CAS PubMed.
- M. Zhao, F. Ansari, M. Takeuchi, M. Shimizu, T. Saito, L. A. Berglund and A. Isogai, Nanoscale Horiz., 2018, 3, 28–34 RSC.
- Y. Kobayashi, T. Saito and A. Isogai, Angew. Chem., Int. Ed., 2014, 53, 10394–10397 CrossRef CAS PubMed.
- K. A. Taiwo, A. Angersbach, B. I. O. Ade-Omowaye and D. Knorr, J. Agric. Food Chem., 2001, 49, 2804–2811 CrossRef CAS PubMed.
- A. Rózek, I. Achaerandio, M. P. Almajano, C. Güell, F. López and M. Ferrando, J. Agric. Food Chem., 2007, 55, 5147–5155 CrossRef PubMed.
- L. A. Hoover, W. A. Phillip, A. Tiraferri, N. Y. Yip and M. Elimelech, Environ. Sci. Technol., 2011, 45, 9824–9830 CrossRef CAS PubMed.
- J. P. F. Lagerwall, C. Schütz, M. Salajkova, J. Noh, J. Hyun Park, G. Scalia and L. Bergström, NPG Asia Mater., 2014, 6, e80 CrossRef CAS.
- T. Saito, S. Kimura, Y. Nishiyama and A. Isogai, Biomacromolecules, 2007, 8, 2485–2491 CrossRef CAS PubMed.
- F. C. MacKintosh, J. Käs and P. A. Janmey, Phys. Rev. Lett., 1995, 75, 4425–4428 CrossRef CAS PubMed.
- O. Nechyporchuk, M. N. Belgacem and F. Pignon, Biomacromolecules, 2016, 17, 2311–2320 CrossRef CAS PubMed.
- D. Tatsumi, S. Ishioka and T. Matsumoto, Nihon Reoroji Gakkaishi, 2002, 30, 27–32 CrossRef CAS.
- S. G. Mason, Pulp Pap. Mag. Can., 1950, 51, 94–98 CAS.
- T. G. Mezger, The Rheology Handbook: For Users of Rotational and Oscillatory Rheometers, Vincentz Network, 2014 Search PubMed.
- G. Evmenenko, E. Theunissen, K. Mortensen and H. Reynaers, Polymer, 2001, 42, 2907–2913 CrossRef CAS.
- C. A. Maestri, M. Abrami, S. Hazan, E. Chistè, Y. Golan, J. Rohrer, A. Bernkop-Schnürch, M. Grassi, M. Scarpa and P. Bettotti, Sci. Rep., 2017, 7, 11129 Search PubMed.
- M. Shibayama, Soft Matter, 2012, 8, 8030 RSC.
- M. Shibayama, Polym. J., 2010, 43, 18–34 CrossRef.
- F. Yeh, E. L. Sokolov, T. Walter and B. Chu, Langmuir, 1998, 14, 4350–4358 CrossRef CAS.
- M. Shibayama, T. Tanaka and C. C. Han, J. Chem. Phys., 1992, 97, 6829–6841 CrossRef CAS.
- D. a. Head, A. J. Levine and F. C. MacKintosh, Phys. Rev. Lett., 2003, 91, 108102 CrossRef PubMed.
- Y. Liu, D. Stoeckel, K. Gordeyeva, M. Agthe, C. Schütz, A. B. Fall and L. Bergström, ACS Macro Lett., 2018, 172–177 CrossRef.
- J. Teixeira, J. Appl. Crystallogr., 1988, 21, 781–785 CrossRef.
- W. Y. Shih, J. Liu, W. H. Shih and I. A. Aksay, J. Stat. Phys., 1991, 62, 961–984 CrossRef.
- J. Liu, W. Y. Shih, M. Sarikaya and I. A. Aksay, Phys. Rev. A, 1990, 41, 3206–3213 CrossRef CAS.
- S. Ikeda, E. A. Foegeding and T. Hagiwara, Langmuir, 1999, 15, 8584–8589 CrossRef CAS.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- K. M. O. Håkansson, RSC Adv., 2015, 5, 18601–18608 RSC.
- A. Buffet, A. Rothkirch, R. Döhrmann, V. Körstgens, M. M. Abul Kashem, J. Perlich, G. Herzog, M. Schwartzkopf, R. Gehrke, P. Müller-Buschbaum and S. V. Roth, J. Synchrotron Radiat., 2012, 19, 647–653 CrossRef CAS PubMed.
- G. Benecke, W. Wagermaier, C. Li, M. Schwartzkopf, G. Flucke, R. Hoerth, I. Zizak, M. Burghammer, E. Metwalli, P. Müller-Buschbaum, M. Trebbin, S. Förster, O. Paris, S. V. Roth and P. Fratzl, J. Appl. Crystallogr., 2014, 47, 1797–1803 CrossRef CAS PubMed.
- M. Doucet, J. H. Cho, G. Alina, J. Bakker, W. Bouwman, P. Butler, K. Campbell, M. Gonzales, R. Heenan, A. Jackson, P. Juhas, S. King, P. Kienzle, J. Krzywon, A. Markvardsen, T. Nielsen, L. O'Driscoll, W. Potrzebowski, R. F. Leal, T. Richter, P. Rozycko, T. Snow and A. Washington, Zenodo, 2017 Search PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on morphological characterization, the osmotic dehydration setup, additional data on the rheological characterization of the suspensions and the fittings parameters of the SAXS. See DOI: 10.1039/c8nr08194h |
| ‡ These authors contributed equally to the work. |
| § Current address: Center for Free-Electron Laser Science, University of Hamburg, 22761 Hamburg, Germany. |
| ¶ Current address: RISE Bioeconomy, Box 5604, 114 86 Stockholm, Sweden. |
| || Current address: Physics and Materials Science Research Unit, University of Luxembourg, 1511 Luxembourg, Luxembourg. |
| This journal is © The Royal Society of Chemistry 2018 |