
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA dual-signal amplification platform for sensitive fluorescence biosensing of leukemia-derived exosomes†
Lin
Huang
 a,
Dian-Bing
Wang
b,
Netrapal
Singh
bc,
Fang
Yang
a,
Ning
Gu
*a and
Xian-En
Zhang
*b
a,
Dian-Bing
Wang
b,
Netrapal
Singh
bc,
Fang
Yang
a,
Ning
Gu
*a and
Xian-En
Zhang
*b
aSchool of Biological Science and Medical Engineering, Southeast University, Nanjing 210096, PR China. E-mail: guning@seu.edu.cn
bNational Laboratory of Biomacromolecules, CAS center for Excellence in Biomacromolecules, Institute of Biophysics, Chinese Academy of Sciences, Beijing 100101, China. E-mail: zhangxe@ibp.ac.cn
cInstitute for Synthetic Biology Research, Shenzhen Institutes of Advanced Technology, Chinese Academy of Sciences, Shenzhen 518055, P.R. China
First published on 23rd October 2018
Abstract
Exosomes as nanosized biomarkers hold great potential for the diagnosis of cancer. However, the low concentration of cancer-derived exosomes present in biofluids makes early diagnosis strenuous. Here, we developed a fluorescent biosensing platform, namely a dual signal amplification, for the ultrasensitive detection of leukemia cell-derived exosomes. The protocol consists of three steps: first, leukemia-derived exosomes containing CD63 and nucleolin were captured by anti-CD63 antibody modified magnetic bead conjugates (MB-CD63); then, a DNA primer comprising a nucleolin-recognition aptamer (AS1411) was applied to bind the exosomes which further initiated a rolling circle amplification (RCA) reaction to generate many repeat sequences for hybridization with gold nanoparticle (GNP)–DNA–fluorescent dye (FAM) conjugates (GNP–DNA–FAM); finally, nicking endonuclease (Nb·BbvCI) assisted target recycling was introduced. As a result, FAM was released from GNP–DNA–FAM conjugates, transformed from the quenching state to the emission state and thus fluorescence signals continuously accumulated. With this dual signal amplification platform, as low as 1 × 102 particles per μL exosomes could be detected. Furthermore, we have successfully applied this method for the detection of exosomes in spiked serum samples, indicating a promising tool for clinical application.
Introduction
Leukemia, a heterogeneous group of hematopoietic malignancies is one of the most common lethal forms of cancer, where the bone marrow produces abnormal white blood cells. An early diagnosis of leukemia is crucial to avoid unnecessary morbidity and mortality. Additionally, it is necessary to monitor the minimal residual disease (MRD) which is a critical factor in determining the prognosis of leukemia patients after treatment.1 Diagnosis of leukemia is primarily performed on peripheral blood and bone marrow, which relies upon a multiparametric approach involving a number of different pathology disciplines. Various conventional and modern molecular technologies are being increasingly employed to help refine diagnosis.2,3 The clinical diagnosis of leukemia mainly includes two methods: bone marrow aspiration for biopsy and peripheral blood analysis by complete blood count (CBC) and blood smear.4–7 The former method is a rigorous invasive procedure involving complications and the latter is non-invasive but usually insensitive for early diagnosis. Therefore, there is a need to develop a less invasive, simple and sensitive diagnostic platform for the early diagnosis of leukemia.In recent years, exosomes, nanosized vesicles secreted by mammalian cells, have received close attention. Exosomes carry large amounts of proteins, lipids, DNA, RNA, and other biological molecules, which could provide information about the originating tumor.8,9 Since extraction of exosomes from body fluids is a relatively simple procedure, they are attractive for the early diagnosis of cancers, instead of the traditional tissue biopsy.10
So far, a large number of techniques, such as nanoparticle tracking analysis,11 flow cytometry,12 enzyme-linked immuno sorbent assay (ELISA),13 hybridization chain reaction (HCR)-based colorimetric sensor,14 electrochemical detection,15 microfluidic-based technology,16etc. have been used for the detection of exosomes. These methods show prospects for clinical application, but there are still some problems. For example, the operation is complex and skillful or the detection sensitivity is not high enough. Different types of sensors based on the direct interaction of the signal probe and biomarker on the exosomes were recently developed.17–19 However, these methods require a sufficient number of markers on the exosome surface to produce signals. Unfortunately, the quantity of disease related markers on exosomes is varied and lower.20 So, detection of exosomes with high efficiency remains challenging.
Recently, we have identified nucleolin as a potential target on the surface of leukemia cell-derived exosomes by mass spectrum analysis (data not shown). This protein is also mentioned to be overexpressed on leukemia cells, having a high affinity for the AS1411 aptamer.21 Upon these findings, we try to realize the specific and ultrasensitive detection of leukemia-derived exosomes by the combined use of aptamer recognition, magnetic enrichment and rolling circle amplification (RCA).22,23 The proposed method is a dual signal amplification platform, which consists of three main steps (Fig. 1). First, leukemia cell-derived exosomes containing CD63 and nucleolin were captured by MB-CD63. Then, a DNA primer comprising the nucleolin-recognition aptamer (AS1411) was applied to bind the exosomes and initiated the RCA reaction to generate many repeat sequences for hybridization with GNP–DNA–FAM. Finally, nicking endonuclease (Nb·BbvCI) assisted target recycling was introduced leading to the continuous release of FAM from GNP–DNA–FAM conjugates. Thus, the fluorescence signal can be dramatically enhanced by transforming FAM from the quenching state to the emission state.
 | ||
| Fig. 1 Schematic illustration of the dual-signal amplification-based platform for the ultrasensitive detection of exosomes. The primer contains the RCA primer (labeled in black) and AS1411 aptamer (labeled in gray). | ||
Experimental section
Reagents
All the oligonucleotides used in this study were synthesized, modified and purified by Sangon Biotech Co., Ltd (Shanghai, China). The base sequences and relevant modification details of oligonucleotides are given as follows.Padlock: 5′phosphate-TGTCTTCGCCTGTCCGATGCTCTTCCTTGAAACTTCTTCCTTGCTGAGGGACTAAGCACC-3′.
DNA primer (bold characters in the sequence are the aptamer for nucleolin): 5′-GGTGGTGGTGGTTGTGGTGGTGGTGGGTGGCGTAAAGGGAGCATCGGACAGGCGAAGACAGGTGCTTAGT-3′.
DNA–FAM: 5′SH-(CH2)6 AAATTGCTGAGGGAC-3′FAM.
Streptavidin-coated magnetic beads (1 μm, cat. no. 65001) were purchased from Thermo Fisher Scientific Co Ltd (MA, USA). Biotinylated mouse anti-human CD63 monoclonal antibody (ab134331) was obtained from Abcam (Cambridge, United Kingdom). Different enzymes, i.e. Phi29 DNA polymerase, T4 DNA ligase and Nb·BbvCI were received from New England Biolabs Co Ltd (Beijing, China). Human cancerous cell line HL-60 was purchased from The Cell Bank, Chinese Academy of Sciences (Shanghai, China). The other reagents such as tris(2-carboxyethyl) phosphine hydrochloride (TCEP) and chloroauric acid (HAuCl4) were purchased from Sigma-Aldrich (St Louis, MO, USA). The buffers used in this work are as follows: (1) phosphate-buffered saline (PBS) buffer containing 8 mM Na2HPO4, 2 mM NaH2PO4, 2.6 mM KCl, and 136 mM NaCl, pH 7.4; (2) blocking buffer (PBSB) containing 1% (w/v) BSA in PBS buffer; (3) RCA buffer (50 mM Tris-HCl, 4 mM DTT, 10 mM MgCl2, and 10 mM (NH4)2SO4, pH 7.5); (4) TBST buffer (1% (w/v) BSA, 10 mM Tris-HCl and 50 mM NaCl, pH 7.5).
Cell culture and isolation of exosomes
The HL-60 cells were maintained in RPMI supplemented with 10% fetal bovine serum (FBS) containing 1% penicillin and streptomycin at 37 °C with 5% CO2 under a humidified atmosphere. To avoid the cross-contamination by serum exosomes, the cells were then cultured in RPMI containing 10% exosome-depleted FBS for 48 h and then harvested from a 225 cm2 flask at 80–90% confluency. Subsequently, the exosomes were isolated from the culture medium of HL-60 cells by a standard ultracentrifugation method as described in the previous report.24 Briefly, the removal of intact cells from the cell culture medium was done by centrifugation at 300g for 10 min, and then repeated centrifugation was carried out at 2000g for 20 min and 11![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g for 45 min to remove all the cell debris and large extracellular vesicles (EVs). In the next step, the supernatant was centrifuged at 100000g for 2 h to acquire intact exosomes. Finally, the sediment exosomes were washed with PBS once and re-suspended in PBS. The number of exosomes was quantified by nanoparticle tracking analysis (NTA) (NS500 instrument, Malvern Instruments, USA).
000g for 45 min to remove all the cell debris and large extracellular vesicles (EVs). In the next step, the supernatant was centrifuged at 100000g for 2 h to acquire intact exosomes. Finally, the sediment exosomes were washed with PBS once and re-suspended in PBS. The number of exosomes was quantified by nanoparticle tracking analysis (NTA) (NS500 instrument, Malvern Instruments, USA).
Transmission electron microscopy (TEM)
For TEM analysis, the optimal concentration of exosomes was directly adsorbed on 300-mesh carbon grids and subsequently dried for 5 min at room temperature (RT), and followed by staining with 2.5% uranyl acetate for 50 s. After the removal of the excess stain, TEM was used to analyze the morphology of HL-60-cell derived exosomes under high vacuum with a working voltage of 100 kV (Tecnai Spirit, FEI, USA).Western blotting analysis
The western blotting experiment was carried out to confirm the presence of the CD63 protein in exosomes. Here, the exosomes and cells were lysed in a SDS sample buffer and boiled for 10 min for protein denaturation, and then the lysates were separated by 12% SDS-polyacrylamide gel electrophoresis. After electrophoresis, the proteins in the gel were transferred onto an activated polyvinylidene fluoride (PVDF) membrane (Millipore, USA). After blocking the protein blot with 5% non-fat dry milk in TBST, the PVDF membranes were incubated with the mouse anti-β actin and anti-CD63 primary antibodies (1:1000) for 2 h at RT. Subsequently, the PVDF membranes were washed three times with TBST and incubated with goat anti-mouse horseradish peroxidase (HRP)-conjugated secondary antibodies for 1 h at RT. Finally, the immunoblots were imaged with enhanced chemiluminescent reagents using an Amersham imager 600 system (GE Healthcare, USA).
Preparation of gold nanoparticle (GNP)–DNA–fluorescence dye (FAM) conjugates (GNP–DNA–FAM)
To prepare the gold nanoparticle (GNP), a previously reported method was followed.25 Briefly, 0.01% HAuCl4 (50 mL) was heated to boiling under magnetic stirring. Then, 1 mL of sodium citrate (1%) was rapidly added into the above-mentioned boiling aqueous solution. After the appearance of a red colour, the mixture was further stirred and boiled for an additional 5 min to ensure the uniform size of grown nanoparticles. Finally, the solution was cooled to RT and stored at 4 °C for further use. The immobilization of DNA–FAM on GNPs was done according to a previously reported procedure.26 In brief, the thiolated DNA–FAM was firstly activated by TCEP for 30 min at 37 °C. Then, the different concentrations of DNA–FAM were incubated with 1 mL GNP (1 nM) solution overnight at RT. Subsequently, 50 μL of 10% Tween 20 was added to the above-mentioned solution to reduce the aggregation of GNPs, and then the salt-aging process with 20 μL of 3 M NaCl was performed at repeated intervals of 20 min under a cycle of 20 s sonication each time. After incubation for another 48 h, the excess reagents in the reaction mixture were removed by centrifugation at 12000g for 10 min. The pellet was resuspended in 100 μL of PBS (10 mM, pH 7.4) containing 0.01% Tween 20 and stored at 4 °C.
The average number of DNA–FAM per GNP was quantified according to the previous protocol.26 The DNA–FAM complex was firstly released from the GNP by treating with mercaptoethanol (20 mM). After overnight incubation at RT, the solution was centrifuged at 13000g for 10 min to remove the GNP, and the fluorescence of the released DNA–FAM strands was determined with a Cytation 3 Imaging Reader (Biotek, USA). The fluorescence intensity was compared to a calibration curve that was prepared with known concentrations of DNA–FAM and converted to molar concentrations of DNA–FAM. The average number of DNA–FAM per GNP was then calculated according to the concentration of GNP–DNA–FAM. UV-vis spectrometry was used to determine the concentration of the GNP–DNA–FAM.
Exosome detection by dual signal amplification
A dual signal amplification-based platform for exosome detection was carried out in 200 μL Eppendorf tubes and 5% BSA and PBS were used for blocking and washing steps throughout the detection protocol. Anti-CD63 antibody-magnetic bead conjugates (MB-CD63) were obtained by attaching the biotinylated CD63 antibody to streptavidin-modified magnetic beads. Briefly, 5 μL of the streptavidin-MB suspension (10 mg mL−1) was washed three times with 100 μL of PBS buffer to remove the preservative. After washing, 50 μL of the biotinylated CD63 antibody (40 μg mL−1) in PBS was added and incubated for 30 min at RT with gentle rotation. The unreacted biotinylated CD63 antibody was washed away with PBST. To start the RCA-based integrated platform for the detection of HL-60 cell-derived exosomes, 50 μL of ten-fold serially diluted cell-derived exosomes (10 particles per μL to 1 × 107 particles per μL) in PBS were mixed with 50 μL of MB-CD63 conjugates for 30 min at RT with gentle rotation. An external magnetic field was applied to remove the unbound entities and was followed by a washing step. After washing, the DNA primer-padlock was added into the above-mentioned solution to bind exosomes and incubated at RT for 30 min. After another washing step, the ligation reaction was performed by adding 10 U T4 DNA ligase to the above-mentioned mixture. After completion of the ligation, RCA was performed by the Phi29 DNA polymerase (20 U) in the amplification mixture containing 1 × RCA buffer and 0.5 mM dNTPs at RT for 60 min. The first round of amplification by RCA was followed by magnetic separation. Then, 100 μL of 0.5 nM GNP–DNA–FAM and nicking endonuclease (20 U) were added to the mixture (60 min at RT) to generate the second round of amplification. Finally, the fluorescence was measured at 488 nm for excitation and 519 nm for emission using a Cytation 3 Imaging Reader (Biotek, USA).Results and discussion
The principle of a dual signal amplification-based platform for exosome detection
Aptamers have emerged as an excellent alternative to antibodies as recognition elements in sensors27 which exhibit many advantages, including fast, reproducible synthesis, easy modifications, long-term stability28 and moreover, these may be easily combined with nucleic acid-based techniques. Taking these into account, we exploited AS1411 as the aptamer for recognizing a novel protein marker i.e. ‘nucleolin’ present on the surface of HL-60 cell-derived exosomes. The AS1411 aptamer has high affinity to nucleolin which was identified to be expressed on the surface of leukemia-derived exosomes from the results of the mass spectrum and flow cytometry (Fig. S1†). The schematic diagram of a dual signal amplification-based platform for the detection of exosomes is shown in Fig. 1. Firstly, the exosomes were captured by MB-CD63, targeting CD63, a surface marker of exosomes. After magnetic separation, a DNA primer (RCA primer + AS1411 aptamer) was used to recognize the exosomes. Subsequently, the padlock was added into the above-mentioned complex and ligation was performed by adding the 10 U T4 DNA ligase. Under the conditions of RCA, the DNA primer could be elongated with many repeat sequences. To produce the detection signal, GNP–DNA–FAM conjugates were hybridized with RCA products to form the recognition sites of Nb·BbvCI. Before releasing, the fluorescence of FAM could be quenched by GNPs due to its specific optical effect. Then, Nb·BbvCI would nick the recognition site and repeatedly release the FAM from the surface of GNPs, resulting in the enhanced fluorescence signal. Consequently, the fluorescence signal demonstrated a good linear relationship with the concentration of exosomes, and the sensitivity of this method was improved 180 fold as compared to the sensitivity observed without amplification (Fig. S2†). The result of Fig. S2† was obtained by detecting the exosomes without the second round of the signal amplification step. Briefly, the RCA reaction was induced on the surface of exosomes which were captured by MB-CD63. Then, the DNA–FAM was hybridized with the RCA product to produce the fluorescence signal.Characterization and quantification of exosomes
Exosomes derived from HL-60 cells were firstly isolated from the cell culture supernatant by ultracentrifugation. TEM analysis revealed a cup-shaped morphology of the exosomes (Fig. 2A) as has been reported previously.15 An average diameter (∼150 nm) of purified exosomes also corresponds to the reported distribution range.29 Here, in order to further confirm the successful isolation of exosomes, the expression of CD63 was also analyzed by western blotting. The presence of specific bands on the western blot confirmed that CD63 protein is present both in the cells and exosome lysates (Fig. 2B). In addition, the average size and number of purified exosomes were measured by NTA. The result showed that the concentration was 2.8 × 108 particles per μL with the size distribution at approximately 164 nm (Fig. 2C). We found that nucleolin was present in several leukemia cell line-derived exosomes by mass spectrum analysis (data not shown), indicating that nucleolin could be used as a potential target for exosome detection. In order to further confirm the presence of nucleolin on the surface of leukemia cell-derived exosomes, fluorescence activated cell sorting (FACS) was used to characterize the exosomes. 293 T cell-derived exosomes and the isotype of the antibody were used as controls. The result indicated that leukemia cell-derived exosomes contain this protein (Fig. S1†).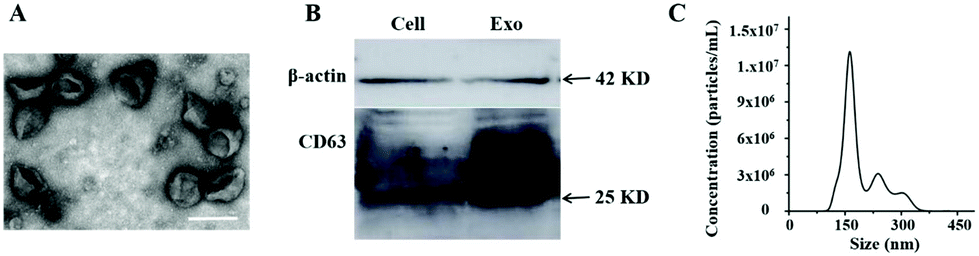 | ||
| Fig. 2 The characterization of HL-60 cell-derived exosomes isolated from the culture supernatant by ultracentrifugation. (A) TEM image of exosomes. Scale bar: 200 nm. (B) Western blotting image of β-actin and CD63 proteins from HL-60 cells and exosome lysates. (C) Nanoparticle tracking analysis (NTA) of exosomes. | ||
Characterization of dual-signal amplification
In this system, the RCA and Nb·BbvCI based reaction is a key step for signal amplification. Therefore, the products of the RCA and Nb·BbvCI-based reaction in the reaction mixture were analyzed by agarose gel electrophoresis. As shown in Fig. 3A, the presence of the DNA band in lane 4 with some retardation indicates that the designed DNA primer (lane 2, primer only) had hybridized with the padlock (lane 3, padlock only), and the DNA band in lane 5 represents the product of RCA with high molecular weight. In lane 8, the band of DNA–FAM shifted more quickly than lane 6, indicating that DNA–FAM have hybridized with the RCA product and then released by Nb·BbvCI. In addition, we accomplished the analysis of exosomes with a dual signal amplification platform. The result showed that the signal of exosome detection was dramatically increased due to the enormous repeat sequences produced by RCA for the nicking endonuclease-based signal amplification (Fig. 3B). The signal of the biosensor without RCA and nicking endonuclease-based signal amplification only showed lower fluorescence. These results indicated that this dual signal amplification platform could be successfully used to detect exosomes.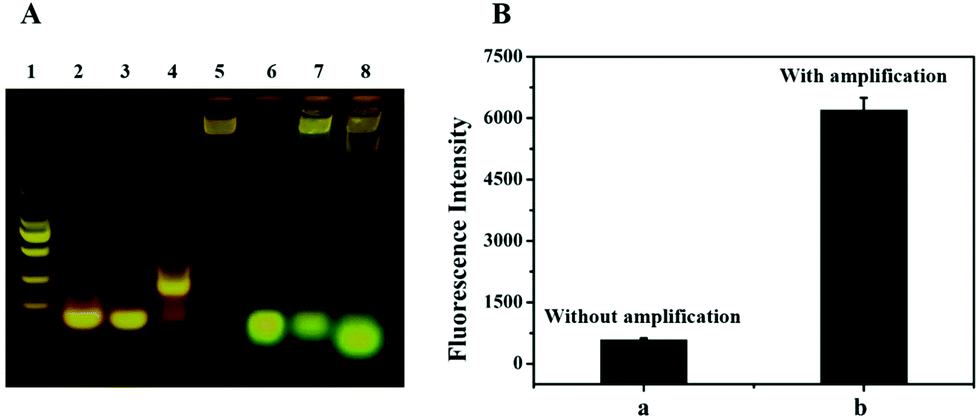 | ||
| Fig. 3 The confirmation of RCA products and signal amplification. (A) Agarose gel electrophoresis analysis of RCA products. Lane 1: marker; Lane 2: primer; Lane 3: padlock; Lane 4: primer + padlock; Lane 5: RCA products; Lane 6: DNA–FAM; Lane 7: RCA products + DNA–FAM; Lane 8: RCA products + DNA–FAM + Nb·BbvCI. (B) Fluorescence intensity of biosensing with and without signal amplification. | ||
Optimization of experimental conditions
To achieve the optimal performance of a biosensor for exosome detection, RCA time, Nb·BbvCI nicking time and the concentrations of DNA–FAM conjugated on the GNPs were optimized. The RCA time was important for the amplification of the detection signal. Theoretically, along with the extension of the RCA time, the primer could be prolonged with more repeat sequences resulting in the production of more recognition sites for Nb·BbvCI to amplify the signal. However, the maximum fluorescence intensity was achieved at 60 min, after that there was no increase in fluorescence (Fig. 4A). This may be caused by the steric hindrance for the hybridization of GNP–DNA–FAM with the longer RCA products, or the inactivation of the Phi29 DNA polymerase.30 Here, another important factor is the nicking time of Nb·BbvCI for the release of FAM from GNP–DNA–FAM conjugates. As shown in Fig. 4B, the fluorescence intensity had been increasing with the increasing nicking time of Nb·BbvCI and reached a plateau after 60 min. This result indicated that the optimum sensing response was obtained at 60 min and a further increase in the reaction time did not improve the sensing response. Therefore, the optimal nicking time of Nb·BbvCI for signal generation was chosen as 60 min. The concentration of DNA–FAM attached on the GNP was optimized to achieve a stronger sensing signal. Fig. 4C shows the agarose gel electrophoresis analysis of GNPs with different ratios of DNA–FAM, and the different migration of the GNPs indicates that the DNA–FAM has been successfully conjugated on the GNP with different numbers. TEM analysis of GNP–DNA–FAM conjugates showed a halo around GNP which also indicates that DNA–FAM had been modified on the GNP (Fig. 4C, inset image). Furthermore, the maximum signal was achieved at a ratio of 1:500 (GNP:DNA–FAM) (Fig. 4D), and thus this concentration (∼55 molecules per particle) was adopted for further research. In addition, we also image the exosomes after the RCA reaction and GNP–DNA–FAM hybridization by TEM. As shown in Fig. 4D (inset image), we can see that many GNPs (black sphere) have been successfully assembled on the exosomes by the RCA products.
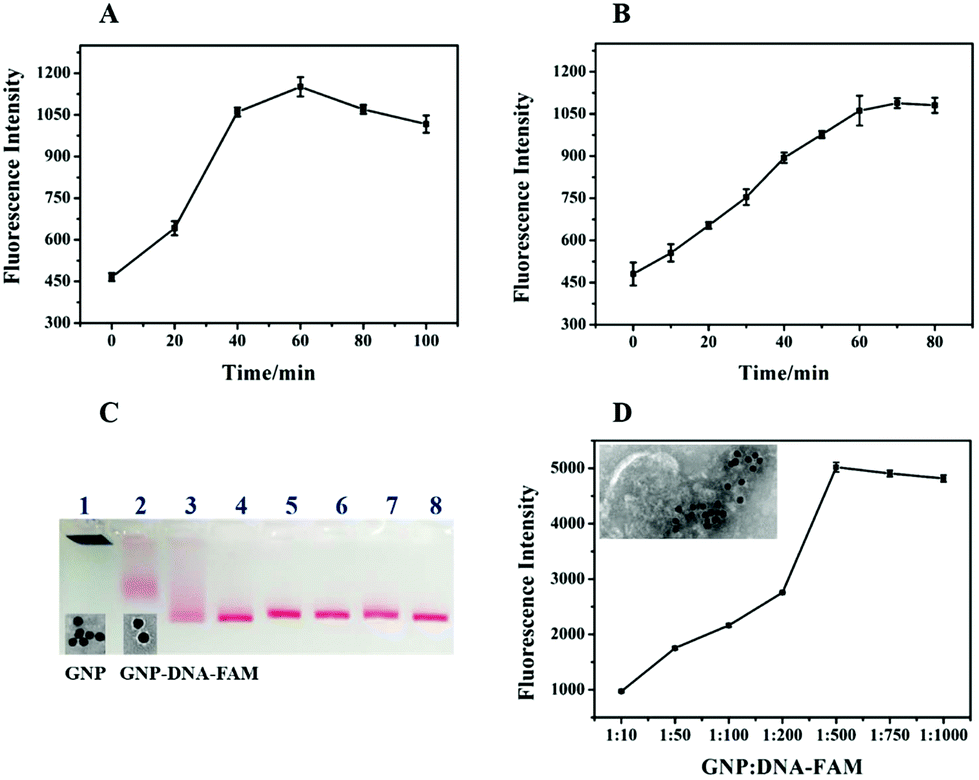 | ||
| Fig. 4 Optimization of (A) RCA reaction time and (B) the time for nicking by Nb·BbvCI. (C) Agarose gel electrophoresis analysis of GNP with different ratios of DNA–FAM. GNP:DNA–FAM = 1:0, 1:10, 1:50, 1:100, 1:200, 1:500, 1:750 and 1:1000 (Line 1–8). The inserted image is the transmission electron microscopy (TEM) results of the GNP without or with DNA–FAM modification. (D) The fluorescence biosensing with different ratios of GNP and DNA–FAM. The inserted image is the TEM image of the GNP assembly on the exosome by RCA products. | ||
Analytical performance of fluorescent biosensor
To evaluate the analytical performance of a RCA-based biosensor, HL-60 cell-derived exosomes with different concentrations were added to the reaction system to determine the detection range and sensitivity under the optimized conditions. As shown in Fig. 5A, the fluorescence intensity of the sensor gradually increased with the increasing concentration of exosomes. Based on the response of the fluorescence signal, a calibration curve with a good linear relationship in the range from 103 to 105 particles per μL was obtained with a correlation coefficient (R2) of 0.998 (Fig. 5B). In addition, the limit of detection as low as 1 × 102 particles per μL was determined based on the 3σ method (where σ is the standard deviation of a blank solution, n = 10), which was 10000 times lower than the commercial kit (Immunoassays from Exosome Antibodies, Array & ELISA Kits, System Biosciences). This improved the sensing performance that may have resulted from RCA and nicking endonuclease-assisted signal amplification.
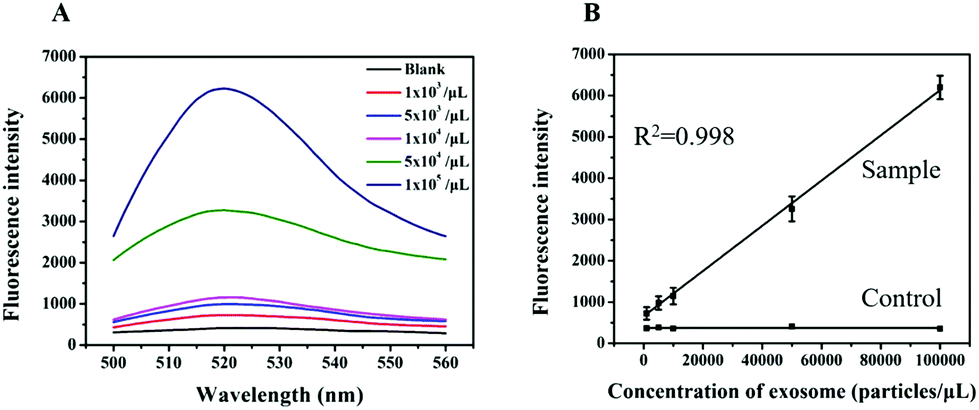 | ||
| Fig. 5 (A) The fluorescence response of the biosensor for exosomes at different concentrations. (B) Standard curve of the fluorescence intensity versus the concentration of exosomes (particles per μL). | ||
To further investigate the feasibility of our method, we assessed the specificity of the method using exosomes derived from 293 T cells and HL-60 cells. Flow cytometric analysis showed that there were fewer number of nucleolin proteins on the exosome derived from 293 T cells as compared to that from the HL-60 cells (Fig. S1†). According to the principle of this method, RCA could only be produced on the surface of nucleolin-positive exosomes with the induction of nicking endonuclease-based signal amplification. Thus, we analyzed the fluorescence intensity of our biosensor with appropriate controls such as the nucleolin positive HL-60 cell-derived exosomes (positive control) and nucleolin negative 293 T cell-derived exosomes (negative control). As shown in Fig. 6A, the signal was dramatically increased in the presence of HL-60 cell-derived exosomes, whereas the 293 T cell-derived exosomes could not induce a high signal response as compared to the blank group. This result also suggested that more nucleolin proteins were present on cancerous exosomes as compared to those on noncancerous exosomes. We also evaluated the selectivity of the primer probe with the response of a sensor for exosomes lacking the AS1411 aptamer sequence in the primer probe. The results showed that the fluorescence intensity was almost the same as that in the blank control which conferred no false signal amplification (Fig. 6A).
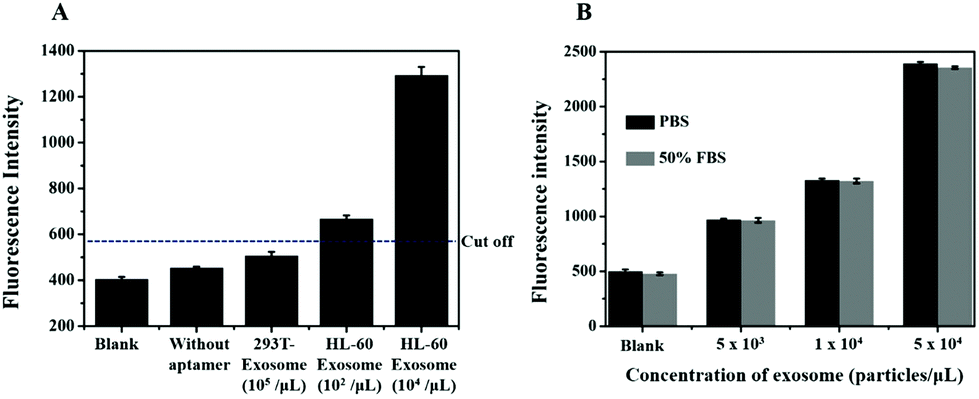 | ||
| Fig. 6 (A) The specificity of the biosensor. (B) Detection of the exosomes in spiked FBS by the proposed method. The cut-off value is shown as a horizontal dotted line. | ||
Furthermore, exosomes spiked in the serum sample at different concentrations were also analyzed to determine the sample matrix effect. Firstly, exosomes with a known concentration were spiked into 50% EV-depleted FBS, and then the signal response of the exosomes in serum was analyzed. The result showed that the signal of the exosomes spiked in FBS was almost the same as that in PBS (Fig. 6B). The results indicated that there was no sample matrix effect and therefore, the proposed method can work well in complex biological samples and holds great potential for future use in clinical samples with high sensitivity and specificity.
Conclusions
In this work, we proposed a dual-signal amplification method to detect leukemia derived nanosized exosomes with the combined use of aptamer recognition, magnetic enrichment and rolling cycle amplification. The method features high selectivity and sensitivity with a detection limit as low as 1 × 102 exosomes per μL and avoids the sampling of bone marrow. The protocol could provide an extraordinary option for the non-invasive liquid biopsy and be promising for the early diagnosis of leukemia.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the Strategic Priority Research Program of the Chinese Academy of Sciences (XDB29050000) and the National Key Research and Development Program of China (No. 2017YFA0205500). We gratefully acknowledge the help of Prof. Hai-Yan Xu for providing the research material.Notes and references
- M. E. Percival, C. Lai, E. Estey and C. S. Hourigan, Blood Rev., 2017, 31, 185–192 CrossRef PubMed.
- L. Belov, O. de la Vega, C. G. dos Remedios, S. P. Mulligan and R. I. Christopherson, Cancer Res., 2001, 61, 4483–4489 CAS.
- T. J. Ley, E. R. Mardis, L. Ding, B. Fulton, M. D. McLellan, K. Chen, D. Dooling, B. H. Dunford-Shore, S. McGrath, M. Hickenbotham, L. Cook, R. Abbott, D. E. Larson, D. C. Koboldt, C. Pohl, S. Smith, A. Hawkins, S. Abbott, D. Locke, L. W. Hillier, T. Miner, L. Fulton, V. Magrini, T. Wylie, J. Glasscock, J. Conyers, N. Sander, X. Q. Shi, J. R. Osborne, P. Minx, D. Gordon, A. Chinwalla, Y. Zhao, R. E. Ries, J. E. Payton, P. Westervelt, M. H. Tomasson, M. Watson, J. Baty, J. Ivanovich, S. Heath, W. D. Shannon, R. Nagarajan, M. J. Walter, D. C. Link, T. A. Graubert, J. F. DiPersio and R. K. Wilson, Nature, 2008, 456, 66–72 CrossRef CAS PubMed.
- A. V. Moorman, C. J. Harrison, G. A. N. Buck, S. M. Richards, L. M. Secker-Walker, M. Martineau, G. H. Vance, A. M. Cherry, R. R. Higgins, A. K. Fielding, L. Foroni, E. Paietta, M. S. Tallman, M. R. Litzow, P. H. Wiernik, J. M. Rowe, A. H. Goldstone, G. W. Dewald and MRC, NCRI, A. L. W. P. U and E. C. O. Grp, Blood, 2007, 109, 3189–3197 CrossRef CAS PubMed.
- J. Gabert, E. Beillard, V. H. J. van der Velden, W. Bi, D. Grimwade, N. Pallisgaard, G. Barbany, G. Cazzaniga, J. M. Cayuela, H. Cave, F. Pane, J. L. E. Aerts, D. De Micheli, X. Thirion, V. Pradel, M. Gonzalez, S. Viehmann, M. Malec, G. Saglio and J. J. M. van Dongen, Leukemia, 2003, 17, 2318–2357 CrossRef CAS PubMed.
- R. Paredes-Aguilera, L. Romero-Guzman, N. Lopez-Santiago, L. Burbano-Ceron, O. Camacho-Del Monte and S. Nieto-Martinez, Am. J. Hematol., 2001, 68, 69–74 CrossRef CAS PubMed.
- J. Mason and M. Griffiths, Expert Rev. Mol. Diagn., 2012, 12, 511–526 CrossRef CAS PubMed.
- G. van Niel, G. D'Angelo and G. Raposo, Nat. Rev. Mol. Cell Biol., 2018, 19, 213–228 CrossRef CAS PubMed.
- M. Boyiadzis and T. L. Whiteside, Leukemia, 2017, 31, 1259–1268 CrossRef CAS PubMed.
- Y. H. Soung, S. Ford, V. Zhang and J. Chung, Cancers, 2017, 9, 8 CrossRef PubMed.
- R. A. Dragovic, C. Gardiner, A. S. Brooks, D. S. Tannetta, D. J. P. Ferguson, P. Hole, B. Carr, C. W. G. Redman, A. L. Harris, P. J. Dobson, P. Harrison and I. L. Sargent, Nanomedicine, 2011, 7, 780–788 CrossRef CAS PubMed.
- E. N. M. Nolte-'t Hoen, E. J. van der Vlist, M. Aalberts, H. C. H. Mertens, B. J. Bosch, W. Bartelink, E. Mastrobattista, E. V. B. van Gaal, W. Stoorvogel, G. J. A. Arkesteijn and M. H. M. Wauben, Nanomedicine, 2012, 8, 712–720 CrossRef PubMed.
- M. Logozzi, A. De Milito, L. Lugini, M. Borghi, L. Calabro, M. Spada, M. Perdicchio, M. L. Marino, C. Federici, E. Iessi, D. Brambilla, G. Venturi, F. Lozupone, M. Santinami, V. Huber, M. Maio, L. Rivoltini and S. Fais, PLoS One, 2009, 4, e5219 CrossRef PubMed.
- F. He, H. Liu, X. G. Guo, B. C. Yin and B. C. Ye, Anal. Chem., 2017, 89, 12968–12975 CrossRef CAS PubMed.
- H. L. Dong, H. F. Chen, J. Q. Jiang, H. Zhang, C. X. Cai and Q. M. Shen, Anal. Chem., 2018, 90, 4507–4513 CrossRef CAS PubMed.
- P. Zhang, M. He and Y. Zeng, Lab Chip, 2016, 16, 3033–3042 RSC.
- Y. K. Xia, M. M. Liu, L. L. Wang, A. Yan, W. H. He, M. Chen, J. M. Lan, J. X. Xu, L. H. Guan and J. H. Chen, Biosens. Bioelectron., 2017, 92, 8–15 CrossRef CAS PubMed.
- X. Doldan, P. Fagundez, A. Cayota, J. Laiz and J. P. Tosar, Anal. Chem., 2016, 88, 10466–10473 CrossRef CAS PubMed.
- X. S. Chen, J. M. Lan, Y. X. Liu, L. Li, L. Yan, Y. K. Xia, F. Wu, C. Y. Li, S. R. Li and J. H. Chen, Biosens. Bioelectron., 2018, 102, 582–588 CrossRef CAS PubMed.
- S. A. Melo, L. B. Luecke, C. Kahlert, A. F. Fernandez, S. T. Gammon, J. Kaye, V. S. LeBleu, E. A. Mittendorf, J. Weitz, N. Rahbari, C. Reissfelder, C. Pilarsky, M. F. Fraga, D. Piwnica-Worms and R. Kalluri, Nature, 2015, 523, 177–U182 CrossRef CAS PubMed.
- C. R. Ireson and L. R. Kelland, Mol. Cancer Ther., 2006, 5, 2957–2962 CrossRef CAS PubMed.
- Y. M. Qiao, Y. C. Guo, X. E. Zhang, Y. F. Zhou, Z. P. Zhang, H. P. Wei, R. F. Yang and D. B. Wang, Biotechnol. Lett., 2007, 29, 1939–1946 CrossRef CAS PubMed.
- L. Huang, J. J. Wu, L. Zheng, H. S. Qian, F. Xue, Y. C. Wu, D. D. Pan, S. B. Adeloju and W. Chen, Anal. Chem., 2013, 85, 10842–10849 CrossRef CAS PubMed.
- C. Thery, S. Amigorena, G. Raposo and A. Clayton, Current protocols in cell biology, 2006, ch. 3, Unit 3 22 Search PubMed.
- L. Huang, L. Zheng, Y. J. Chen, F. Xue, L. Cheng, S. B. Adeloju and W. Chen, Biosens. Bioelectron., 2015, 66, 431–437 CrossRef CAS PubMed.
- H. Q. Zhang, M. D. Lai, A. Zuehlke, H. Y. Peng, X. F. Li and X. C. Le, Angew. Chem., Int. Ed., 2015, 54, 14326–14330 CrossRef CAS PubMed.
- S. Tombelli, M. Minunni and A. Mascini, Biosens. Bioelectron., 2005, 20, 2424–2434 CrossRef CAS PubMed.
- X. H. Fang and W. H. Tan, Acc. Chem. Res., 2010, 43, 48–57 CrossRef CAS PubMed.
- M. Tkach and C. Thery, Cell, 2016, 164, 1226–1232 CrossRef CAS PubMed.
- Y. Zhu, H. J. Wang, L. Wang, J. Zhu and W. Jiang, ACS Appl. Mater. Interfaces, 2016, 8, 2573–2581 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional figures. See DOI: 10.1039/c8nr07720g |
| This journal is © The Royal Society of Chemistry 2018 |