
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceExactly matched pore size for the intercalation of electrolyte ions determined using the tunable swelling of graphite oxide in supercapacitor electrodes†
Jinhua
Sun
a,
Artem
Iakunkov
a,
Anastasiia T.
Rebrikova
b and
Alexandr V.
Talyzin
 *a
*a
aDepartment of Physics, Umeå University, SE-90187 Umeå, Sweden. E-mail: alexandr.talyzin@umu.se
bDepartment of Chemistry, Moscow State University, Leninskie Gory 1-3, Moscow, Russia
First published on 25th October 2018
Abstract
The intercalation of solvent molecules and ions into sub-nanometer-sized pores is one of the most disputed subjects in the electrochemical energy storage applications of porous materials. Here, we demonstrate that the temperature- and concentration-dependent swelling of graphite oxide (GO) can be used to determine the smallest pore size required for the intercalation of electrolyte ions into hydrophilic pores. The structure of Brodie graphite oxide (BGO) in acetonitrile can be temperature-switched between the ambient one-layer solvate with an interlayer distance of ∼8.9 Å and the two-layer solvate (∼12.5 Å) at low temperature, thus providing slit pores of approximately 2.5 and 6 Å. Using in situ synchrotron radiation X-ray diffraction (XRD) and the temperature dependence of capacitance in supercapacitor devices, we found that solvated tetraethylammonium tetrafluoroborate (TEA-BF4) ions do not penetrate into both the 2.5 and 6 Å slit pores formed by BGO interlayers. However, increasing the electrolyte concentration results in the formation of a new phase at low temperature. This phase shows a distinct interlayer distance of ∼15–16.6 Å, which corresponds to the insertion of partly desolvated TEA-BF4 ions. Therefore, the remarkable ability of the GO structure to adopt variable interlayer distances allows for the determination of pore sizes that are optimal for solvated TEA-BF4 ions (about 9–10 Å). The intercalation of TEA-BF4 ions into the BGO structure is also detected as an anomaly in the temperature dependence of supercapacitor performance. The BGO structure remains to be expanded, even after the removal of acetonitrile, adopting an interlayer distance of ∼10 Å.
1. Introduction
Carbon materials are considered to be promising for applications in almost every important energy-related field. The ultra-high surface areas of graphene-related materials (GRMs) and composites (e.g., activated graphene) are efficient for gas storage (e.g., hydrogen)1–3 and can accommodate large amounts of charges for electrochemical energy storage.4–8 However, the theoretically predicted high performance of graphene for energy storage has not yet been achieved experimentally. For example, the application of GRMs as electrode materials for electric double-layer capacitors (supercapacitors) resulted in lower performance compared to the maximum theoretical prediction.9,10 This may be related to many factors, including the inaccessibility of small (sub-nanometer-sized) pores. The specific capacitance of a supercapacitor is correlated to the accessible surface area of the porous carbon material.11 In this regard, creating pores and tuning the pore size are efficient strategies to increase the intrinsic surface area and improve energy storage capacity.12 Known high-surface-area carbon materials have a wide variety of structures, surface chemistries, and pore sizes (from angstrom scale to more than several micrometers). To be useful for charge storage, the pore size should be sufficiently large to enable ion penetration. However, the most favorable pore size remains unclear and is highly debated at the moment.13–20 In this respect, graphite oxide (GO) provides a rare opportunity to study the insertion of ions into slit pores that can be precisely tuned in the sub-nanometer scale using swelling in solvents commonly used for the preparation of electrolytes in energy storage devices (e.g. water and acetonitrile).21–23 Moreover, the sizes of slit pores provided by the interlayers of GO are “flexible” as they depend on the nature of the solvent along with the sizes of intercalated ions/molecules.2,24–28Graphite/graphene oxides are commonly produced via the strong oxidation of graphite (e.g., using the Brodie or Hummers methods29,30) and inherit the layered structure of pristine graphite. The attached oxygen functional groups on both sides of the GO sheets result in the larger separation of graphene layers with the formation of a well-ordered structure with interlayer distances of ∼6.5 Å for Brodie GO (BGO) and ∼7.5 Å for Hummers GO (HGO). Numerous studies have demonstrated that the interlayer distance of BGO/HGO can be tuned to angstrom precision via swelling in different solvents under varying temperature and high pressure22–26,31–33 and via the intercalation of differently sized organic molecular pillars.2,34 The swelling properties of GO strongly depend on the oxidation method. HGO typically shows a poorly ordered structure and gradual changes in d(001) upon the variation of temperature and pressure.24,35 In this case, the gradual shift in d(001) should not be interpreted as a true change in the interlayer distance in the GO structure; it should only be seen as a change in the average value due to the effects of interstratification and intrastratification.25 BGO is more homogeneously oxidized and exhibits a set of solvates with crystalline swelling, which provides distinct structures for the intercalation of one, two or more layers of intercalated solvents.24,25 The interlayer space of BGO solvates can be then determined with angstrom precision, while the temperature and pressure changes can be used as stimuli to switch between the structures with solvent-filled pores of different sizes.
The swelling of BGO in liquid polar solvents under ambient conditions results in increase of the interlayer distance from 6.7 to 8.84 Å in methanol (one layer),36 8.99 Å in acetonitrile (one layer),24 9.24 Å in ethanol (one layer),36 9.5 Å in propanol (one layer), and 23.4 Å in 1-octanol (four layers).25 Cooling these BGO/solvent materials was found to result in phase transitions with stepwise increase of d(001) to 12.14 Å in methanol, 12.45 Å in acetonitrile, 13.34 Å in ethanol, and 27.9 Å in 1-octanol. Acetonitrile is a solvent of specific interest since it is commonly used to prepare electrolytes for application in electrochemical devices (supercapacitors).37 Based on the swelling phase transition, the slit pore size of BGO can be switched between 2.3 Å (one acetonitrile layer) and ∼6.5 Å (two layers of acetonitrile) by simple cooling/heating.24 The stepwise change in the GO interlayer distance provides us with a model system to study a broad range of scientific issues related to the intercalation of electrolytes into sub-nanometer sized pores. It is clear that the solvent confined within the sub-nanometer space might have properties significantly different compared to bulk solvent. Furthermore, ions surrounded by solvation will undergo partial desolvation due to the effects of sorption and interaction with GO layers. In fact, the smallest size of pores available for the sorption of ions is highly debated at the moment.13,16,19,20,38,39 Several issues have been studied with sometimes contradictory experimental results, including: the penetration of ions into the smallest pores in the completely or partly de-solvated state; the relationship between the size of desolvated ions and the smallest size of pores that the ions can penetrate; and the sorption properties of hydrophilic and hydrophobic pores with respect to polar solvents.13
Extensive research efforts have been dedicated to understanding the arrangement and distribution of ions in electrode nanopores.40–42 For example, a recently proposed charging mechanism in supercapacitors indicates that the maximum capacitance of porous carbon can be achieved when the average pore size matches the size of a desolvated ion.41,42 Therefore, many recent studies have focused on synthesizing complex carbon materials with monodisperse pore sizes (e.g., carbide-derived carbon) that are equal to the sizes of bare ions.19,40 However, the lack of long-range order in the pores of activated carbon makes structural characterization rather challenging. In this respect, BGO provides the advantage of precisely tuning and easily switching pore sizes in a relatively well-ordered structure that can be easily characterized by XRD methods.
The intrinsic properties of electrode materials (e.g., surface area, pores size, conductivity, and geometry) play key roles in the energy and power density of supercapacitors.13 However, the practical performances of electrochemical energy storage devices are strongly affected by external factors such as cell configuration, temperature, and pressure.43–45 Supercapacitor performance is known to decay sharply with decreasing temperature due to the severe reduction in electrolyte conductivity and sluggish diffusion of ions within the electrolytes, although the rearrangement of ions at the electrode/electrolyte interface is very fast during the charging/discharging process. However, the penetration of ions into electrode pores and supercapacitor performance are most often investigated under ambient conditions; only a few temperature-dependent studies have been reported.46,47
Here, we present a systematic study of the temperature- and concentration-dependent intercalation of an acetonitrile-based electrolyte [tetraethylammonium tetrafluoroborate (TEA-BF4)] into the interlayers of BGO. In situ synchrotron radiation XRD and temperature-dependent electrochemical characterization of the BGO electrode reveal the intercalation of electrolyte ions into the BGO structure at low temperature and high TEA-BF4 concentration. BGO intercalated with electrolyte ions adapts an interlayer distance of ∼15–16 Å (depending on temperature), which is distinctly different from the interlayer distances of the common one-layered (∼8.9 Å) and two-layered (∼12.5 Å) BGO/acetonitrile solvate structures. Once the ions are inserted into pores at low temperature, the intercalated BGO structure cannot be recovered, even after the complete evaporation of acetonitrile solvent. The BGO structure remains to be intercalated with ions even in the solvent-free state, providing a structure with an interlayer distance of ∼10 Å. The insertion of electrolyte ions into the BGO structure is also detected as an anomaly in the temperature dependence of electrode capacitance in supercapacitor cells. The temperature- and concentration-dependent intercalation of ions into sub-nanometer-sized pores revealed in our study provides a possible way to tune the pore size and activate impermeable sub-nanometer-sized pores in layered electrochemical energy storage materials.
2. Experimental
2.1. Synthesis of GO using the Brodie method
BGO was synthesized using a slightly modified Brodie procedure, resulting in the complete oxidation of graphite powder in a one-step procedure. One-step oxidation results in BGO as a dark-brown powder with C/O = 2.8 according to X-ray photoelectron spectroscopy (XPS; Fig. S8 in ESI†) and d(001) = 6.5 Å according to XRD. As shown in our earlier studies, BGO synthesized in one step shows swelling properties and phase transitions similar to those of BGO synthesized using two oxidation steps. The detailed characterization of BGO powder is reported in our previous studies.322.2. In situ XRD characterization
In situ XRD data of BGO powder immersed in 2, 1, and 0.5 M TEA-BF4/acetonitrile upon cooling and heating were recorded on the ID 22 beamline (λ = 0.46794 Å) of the European Synchrotron Radiation Facility (ESRF) in Grenoble, France. BGO powders immersed in different electrolytes were sealed in glass capillaries with diameters of 0.7 mm. It is impossible to control the exact weight proportion between the material and electrolyte inside of small-diameter capillaries. Typically, the volume of solution was several times larger than the volume of BGO powder to ensure maximal swelling and excess electrolyte relative to the sorption capacity of the material.Sample heating and cooling were performed using an Oxford CryoStream system. Heating to 84 °C made one side of the sealed glass capillary leak due to the high inner pressure caused by the evaporation of acetonitrile. XRD scans were recorded using a short exposure time (30–60 s), and a new spot was selected for every scan to avoid the degradation of samples under the X-ray beam. The temperature at each step was allowed to stabilize for 5–10 min. Note that the swelling of BGO in liquid acetonitrile occurs rapidly and achieves saturation within 1–2 min.24 Part of the XRD data was recorded using an in-house Panalytical X'Pert Diffractometer with CuKα radiation. Cooling and heating were performed using a TTK450 stage with thermal equilibration being achieved for a few minutes at each step.
2.3. Electrochemical measurements
BGO powders were used as the active materials in the working electrode without adding any binder or conductive additives. A Pt coil and Ag/Ag+ electrode were used as the counter and reference electrodes, respectively, in a three-electrode system. Electrochemical measurements were performed in a sealed cell with the same volume of electrolyte for 2, 1, and 0.5 M TEA-BF4/acetonitrile. The sealing of the cell prevents the evaporation of acetonitrile and ensures that the concentration of TEA-BF4 in acetonitrile is constant. The cooling and heating of the electrochemical cell was controlled by a cooling system (JULABO F25, Germany) with the precision of ±1 °C. The in situ method allows us to study the working electrode at low temperatures without disturbing the setup of the electrochemical cell. Electrochemical data were recorded using an Autolab PGSTAT204/FRA32M module. Electrochemical impedance spectra (EIS) data were collected with an AC perturbation of 5 mV over the frequency range of 100 kHz to 10 mHz. Capacitance was calculated according to cyclic voltammetry (CV) results based on the slope of charge (Q) versus voltage (V).45 Charge was calculated by integrating the area of the CV curve. It should be noted that cooling 2 M TEA-BF4/acetonitrile below −40 °C resulted in the precipitation of TEA-BF4 from the solution, as detected visually in electrochemical cells during preliminary tests. For saturated solutions, crystallization was observed at −10 °C. Therefore, temperature-dependent capacitance measurements were stopped at −5 °C to prevent crystallization. The crystallization of electrolyte at low temperatures is an undesirable effect as it adversely affects the performances of supercapacitor devices. Electrolyte crystallization was not observed in XRD experiments, even at low temperatures, most likely due to strong overcooling in the small-diameter capillaries.3. Results
3.1. Temperature- and concentration-dependent intercalation of ions into flexible slit pores of GO
The interlayer distance of air-dried BGO powder is about 6.5 Å according to XRD data (Fig. S4†). In agreement with our previous results,24 immersing BGO into acetonitrile at ambient temperature increases d(001) to 8.9 Å due to swelling (Fig. S1†). BGO immersed in excess acetonitrile shows a phase transition upon cooling at around −10 °C along with an increase in interlayer distance up to 12.5 Å (Fig. S1†). This phase transition is attributed to the intercalation of an additional layer of acetonitrile molecules. Therefore, a one-layer solvate phase is found for BGO in acetonitrile at ambient temperature, while a two-layered phase is present at low temperature.24Thanks to the phase transition, BGO provides the possibility to study the insertion of electrolyte ions in two phases with distinctly different interlayer distances. The interlayer space of the low-temperature phase (∼12.5 Å) should be able to accommodate ions with larger size compared to the ambient-temperature phase. Therefore, we anticipated that some ions that do not penetrate into the BGO structure at ambient temperature will be inserted into the expanded interlayers of BGO at low temperature. Moreover, the insertion of ions into the BGO structure can be expected at modify some parameters of the phase transition (e.g., the temperature point of the transition and interlayer distance). Therefore, we conducted temperature-dependent in situ XRD characterization of BGO swelling in an acetonitrile-based electrolyte commonly used in supercapacitors (TEA-BF4). Indeed, we found that the swelling transition of BGO is strongly affected by the concentration of the electrolyte solution.
The BGO powder immersed in relatively diluted TEA-BF4 electrolyte (0.5 M) showed swelling properties similar to those of BGO in pure acetonitrile at both ambient temperature and upon cooling, as evidenced by the in situ XRD data (Fig. 1a and Fig. S1†). The phase transition caused by the intercalation of a second layer of acetonitrile molecules was observed for BGO in 0.5 M electrolyte solution almost at the same temperature as in pure acetonitrile (Fig. 1d). However, when the concentration of TEA-BF4 in acetonitrile was increased to 1 M, the temperature dependence of BGO swelling changed dramatically, with a new phase detected below −40 °C (Fig. 1b). The interlayer distance of BGO in 1 M electrolyte solution at room temperature remained similar to that in pure acetonitrile (8.96 Å), and the phase transition at −10 °C was also observed as in pure acetonitrile. However, the pattern recorded at −40 °C showed two (001) reflections with distinctly different d-spacings of 12.78 Å (as expected for the two-layered solvate) and 13.36 Å (new phase). The new broad peak with higher d(001) showed a further shift upon decreasing the temperature, reaching 16.5 Å at −60 °C (Fig. 1e).
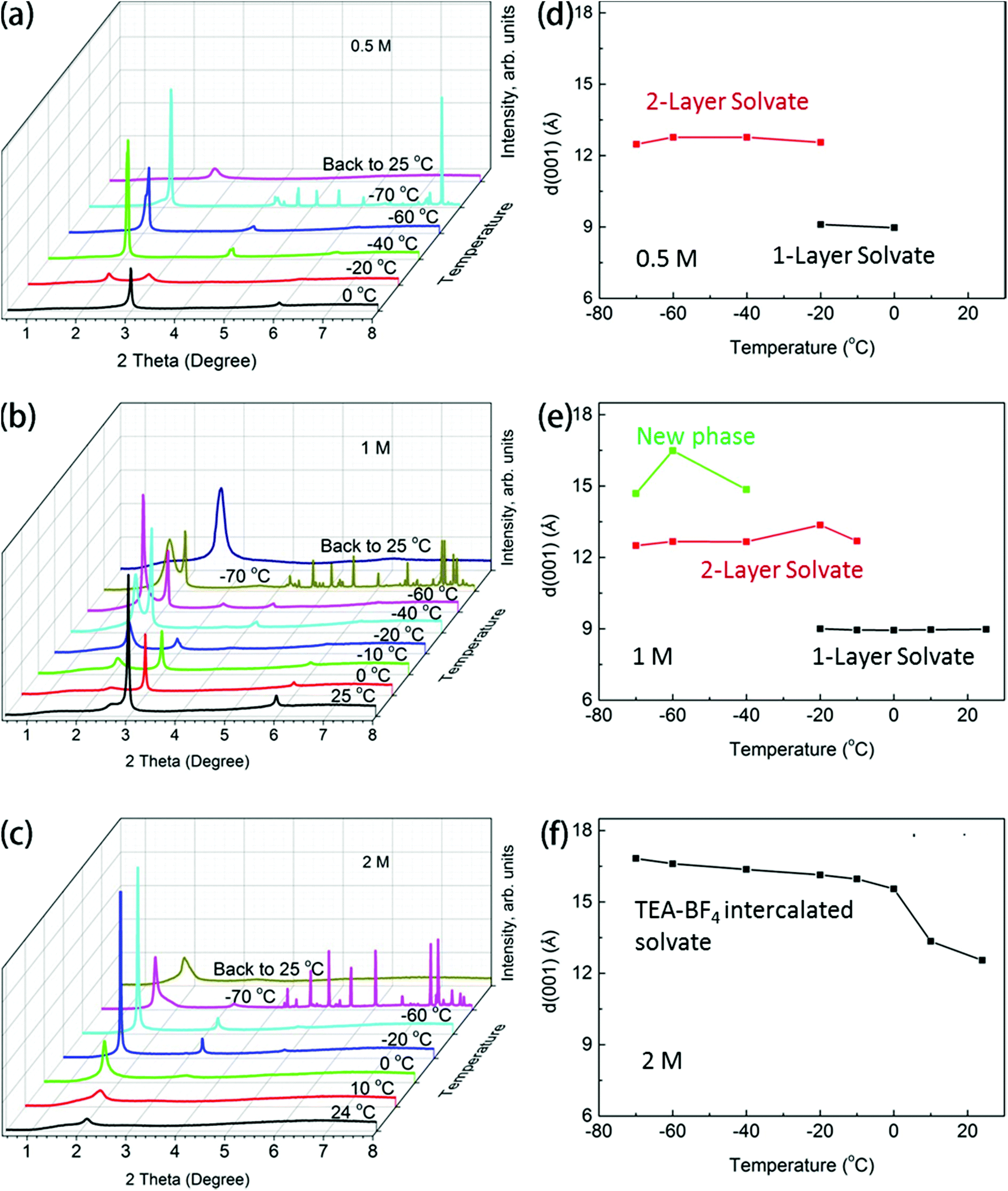 | ||
| Fig. 1 Temperature-dependent phase transition of BGO in different concentrations of electrolyte. Synchrotron XRD (λ = 0.46794 Å) patterns recorded for BGO in (a) 0.5 M TEA-BF4 electrolyte, (b) 1 M TEA-BF4 electrolyte, and (c) 2 M TEA-BF4 electrolyte upon cooling until the freezing of acetonitrile. Temperature dependence of the (001) d-spacing for BGO in (d) 0.5 M TEA-BF4 electrolyte, (e) 1 M TEA-BF4 electrolyte, and (f) 2 M TEA-BF4 electrolyte. The electrolyte freezes at −70 °C, as evidenced by the appearance of new XRD reflections. | ||
As shown in Fig. 1c and f, the further increase in electrolyte concentration to 2 M results in the formation of a two-layered BGO–acetonitrile structure at ambient temperature. The transition from this phase into the new expanded phase with d(001) = 16.6 Å is observed below 0 °C. The structure of BGO at low temperature is better ordered, as evidenced by a sharp (001) diffraction peak and the appearance of higher-order reflections from the (00l) set. The last experiment was performed by cooling BGO immersed in saturated TEA-BF4 acetonitrile solution (Fig. S2†). The transition into the expanded BGO phase with d(001) = 16.6 Å was observed at even higher temperature (15 °C). The crystallization of electrolyte was not observed in these XRD experiments (no additional reflections from TEA-BF4 are found in Fig. 1) performed in thin capillaries, likely due to the overcooling of concentrated solution.
We performed an additional experiment to check for possible kinetic effects. The new phase with d(001) = 16.6 Å was not observed at ambient temperature, even after the prolonged soaking of BGO in concentrated electrolyte solution (Fig. S3†).
The results shown in Fig. 1 provide evidence for the concentration-dependence phase transition of BGO into a phase with an interlayer distance of ∼16.6 Å, which exceeds the d(001) of two-layered BGO/acetonitrile solvate by ∼3.2 Å (Fig. 2 and 3).
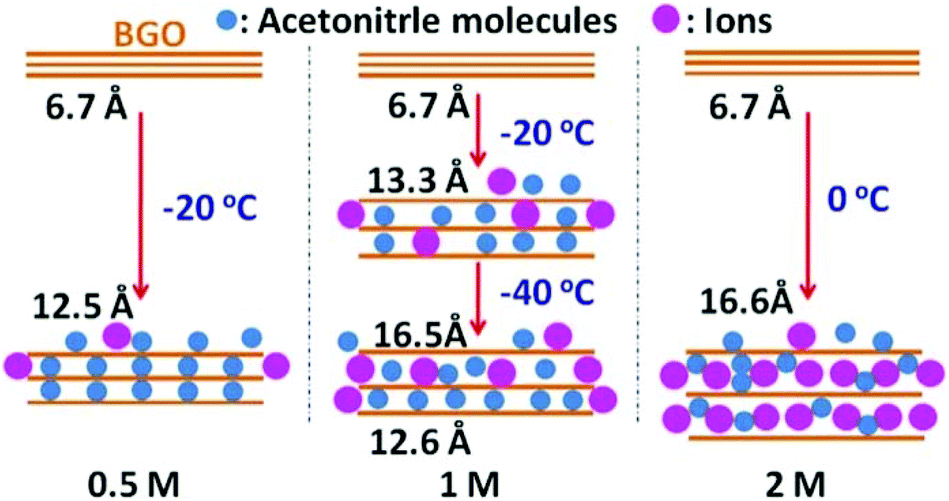 | ||
| Fig. 2 Schematic illustration of the temperature- and concentration-dependent intercalation of solvated TEA-BF4 ions into BGO. The BGO interlayer distance in 0.5, 1, and 2 M TEA-BF4/acetonitrile expands upon cooling due to the temperature-dependent intercalation of acetonitrile molecules and ions. | ||
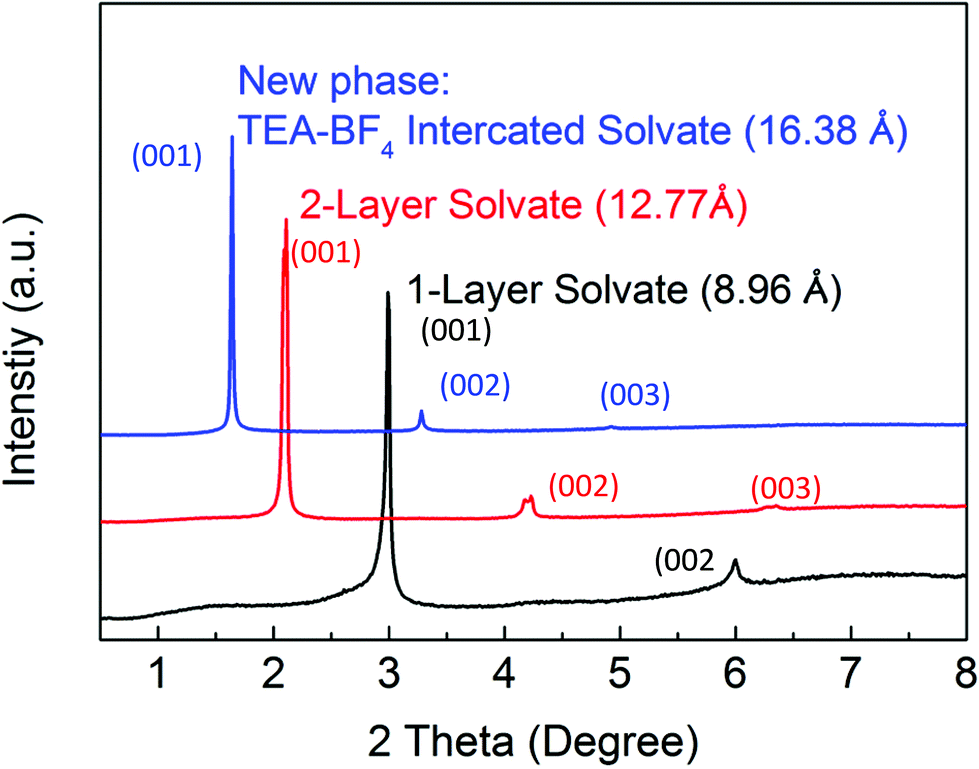 | ||
| Fig. 3 XRD patterns representing three different phases of BGO immersed in electrolytes with different concentrations and characterized by different interlayer distances d(001). Synchrotron XRD (λ = 0.46794 Å) patterns recorded for BGO in 0.5 M TEA-BF4 electrolyte at 0 °C (black curve), 0.5 M TEA-BF4 electrolyte at −40 °C (red curve), and 2 M TEA-BF4 electrolyte at −40 °C (blue curve). | ||
Here, we suggest that the expanded phase observed in concentrated electrolyte solution at low temperature is explained by the intercalation of electrolyte ions into the interlayer space of the BGO structure (Fig. 2). The TEA-BF4 dissociates in acetonitrile, providing TEA+ and BF4− ions. Assuming that the ions are surrounded with solvation shells, their sizes should be about 13 and 11.6 Å, respectively.38 The sizes of solvated TEA+ and BF4− ions are larger than the interlayer distance of acetonitrile-swelled BGO at both ambient temperature (∼9 Å) and temperatures above the swelling phase transition [d(001) ∼ 12.8 Å]. However, we suggest that higher concentrations of solution (>1 M) result in the insertion of solvated ions into the BGO structure, resulting in the formation of a new expanded phase with d(001) = 16.6 Å. The phase transformations observed upon cooling in solution with insufficiently high concentration (1 M) are then explained by the co-existence of two BGO phases: one intercalated only with acetonitrile molecules (∼12.8 Å), and a second intercalated with solvated ions (∼16.6 Å; Fig. 1e and 2). The sorption of electrolyte ions from liquid leads to a decrease in the local concentration of electrolyte solution, and intercalation does not occur at low concentration. Analysis of the full width at half maximum (FWHM) and integral intensity of the (001) reflections as function of temperature are in agreement with earlier observations (Fig. S9†). The general trends observed in these data are an increase in integral intensity and decrease in the FWHM values for the low-temperature phases of BGO solvates compared to higher temperature phases. The same trend was reported for the swelling transitions of BGO in acetonitrile and some other solvents in previous publications.24,48
Alternatively, the data shown in Fig. 1e and f [phase with d(001) ∼ 16.6 Å] could be explained by the insertion of a third acetonitrile layer in highly concentrated electrolyte solutions and the formation of a BGO solvate with three layers of solvent. However, this alternative explanation can be ruled out based on the results of additional experiments, as presented below.
Our previous experiments with GO immersed in a series of pure polar solvents revealed that the lattice expansion of GO observed upon cooling is reversible upon heating. The phase transition occurs due to the insertion and removal of solvent layers into and out of confined GO interlayer spaces.22,24,25,36 Therefore, if the formation of a new phase with d(001) = 16.6 Å at low temperature is connected to a phase transition from a two-layered to a three-layered solvate, the phase transition should be reversible; thus, heating back to ambient temperature would be expected to result in the de-insertion of acetonitrile solvent layers and the recovery of the pristine one-layered solvate at ambient temperature. However, our experiments reveal that the transition into the new phase with an interlayer distance of 16.6 Å at low temperature is only partly reversible upon heating back to ambient temperature and depends on the concentration of ions in the electrolyte (Fig. 4a).
 | ||
| Fig. 4 Irreversible intercalation of ions into BGO. (a) Interlayer distances of pure BGO powder compared to those of BGO immersed in 0.5, 1, and 2 M TEA-BF4/acetonitrile at 25 °C, after cooling to −70 °C followed by heating back to 25 °C (under excess solution), after air drying at 25 °C, and after cooling to −70 °C and vacuum drying. (b) Schematic illustration of the intercalation of TEA-BF4 electrolyte ions into the BGO interlayer space at low temperature and after vacuum drying or air heating, which removes all solvent from the BGO structure. | ||
The interlayer distance of BGO in TEA-BF4/acetonitrile measured after cooling followed by heating back to 25 °C (14.5 Å for a solvent concentration of 2 M) is larger than those of the one-layered and two-layered BGO–acetonitrile solvates (8.9 and 12.5 Å, respectively; Fig. 4). This suggests that the electrolyte ions absorbed by the low-temperature phase of BGO remain trapped upon heating back to room temperature, even when part of the solvent escapes from the interlayer space. We then attempted to record XRD data for BGO immersed in electrolytes at temperatures from ambient temperature up to the boiling point of acetonitrile (84 °C). However, the sealed capillary did not withstand the heating, and all the solvent evaporated. Remarkably, the BGO structure remained expanded relative to precursor material even after air drying of the sample which resulted in the decrease of the interlayer distance from 14.5 Å to ∼10 Å. Note that the swelling of BGO in pure acetonitrile is completely reversible, and the pristine structure with d(001) = 6.5 Å is recovered once acetonitrile is evaporated. Also note that the actual concentration of 0.5 M TEA-BF4/acetonitrile increases due to the evaporation of acetonitrile, resulting in the intercalation of ions with the same behavior as the samples in 1 M and 2 M TEA-BF4/acetonitrile.
To ensure the complete evaporation of acetonitrile, we also vacuum dried a BGO sample intercalated with 2 M electrolyte solution at −70 °C and then heated back to ambient temperature. However, pristine BGO could not be recovered, even under prolonged vacuum treatment. The d(001) value of the vacuum-dried samples (10.5 Å) is about 4 Å larger than that of the precursor BGO, which can only be explained by the intercalation of electrolyte ions in the desolvated state (Fig. 4a and b and Fig. S4†). Once the intercalation of ions into the BGO structure occurs at low temperature, the ions cannot be removed via the evaporation of solvent, as evidenced by the XRD data. However, the intercalated TEA-BF4 can be dissolved away almost completely by repeated washing with acetonitrile (see Fig. S4 in the ESI† file).
3.2. Effect of ion intercalation on the electrochemical performance of a supercapacitor
Evidence supporting the intercalation of electrolyte ions into the swelled BGO structure was found by the temperature-dependent analysis of supercapacitor electrochemical performance. If ions do not penetrate into the interlayers of BGO, the total accessible surface area of this material should be rather low, with only external surface area available for the sorption of ions. If the ions begin to penetrate between BGO sheets at low temperatures, the accessible surface area will increase significantly, resulting in an increase in the measured capacitance. However, it should be noted that the capacitance of a supercapacitor is adversely affected by cooling.The in situ, low-temperature electrochemical characterization of the BGO electrode in 0.5, 1, or 2 M TEA-BF4/acetonitrile was performed using a three-electrode cell. The temperature inside the cooling cell was controlled with a precision of ±1 °C without disturbing the setup of the electrochemical cell. The CV curves of BGO electrodes recorded at ambient temperature at scan rates of 5 and 10 mV s−1 showed quasi-rectangular shapes, indicating typical capacitive behavior (Fig. 5 and Fig. S6†). The capacitance observed in these experiments is low due to the poor conductivity of BGO and agrees with earlier reported values for GO electrodes in supercapacitors.21,49 As expected, the capacitance gradually decreases with decreasing temperature due to the severely reduced ionic conductivity.
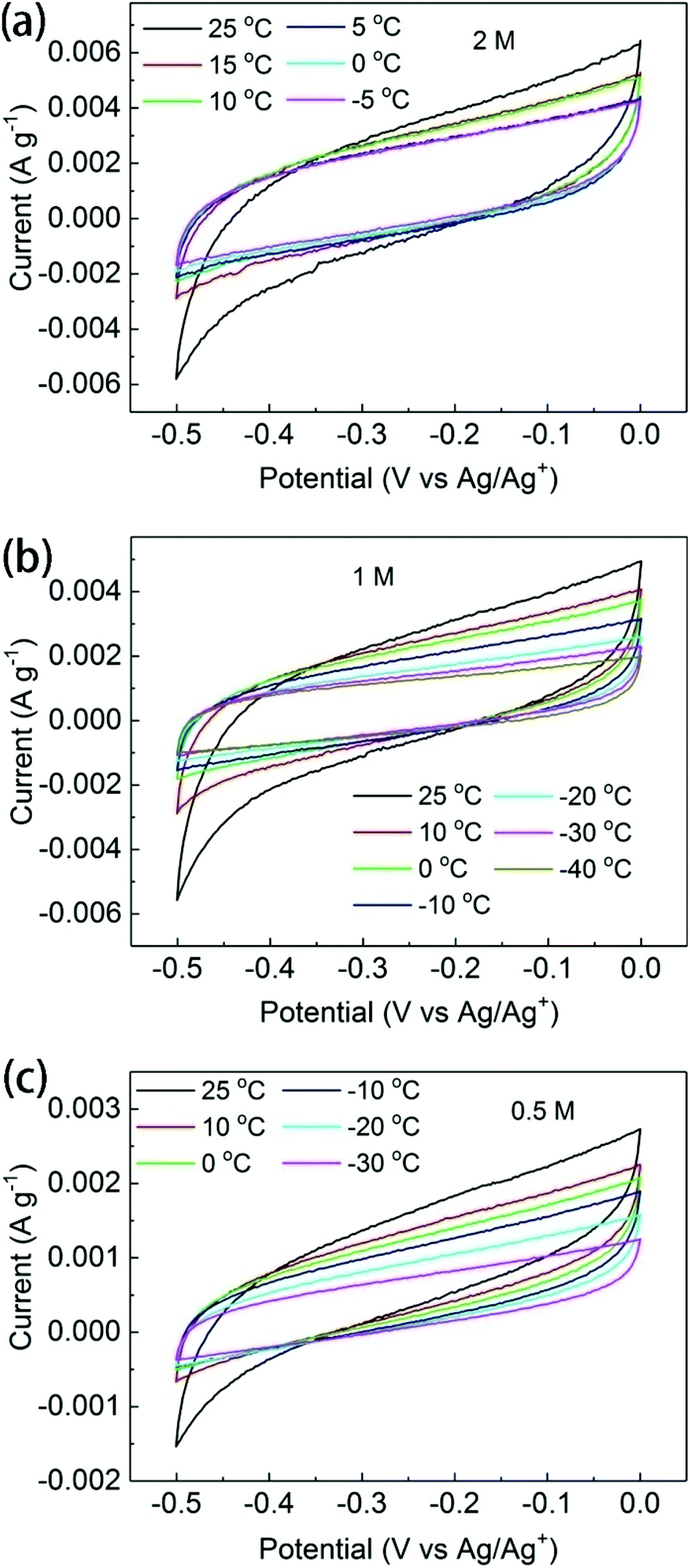 | ||
| Fig. 5 Electrochemical characterization under low temperature. CV curves of BGO electrodes in (a) 2 M, (b) 1 M, and (c) 0.5 M TEA-BF4/acetonitrile at scanning rate of 5 mV s−1. | ||
The temperature dependence of the electrochemical performance of BGO was found to be significantly different in concentrated and diluted electrolyte solutions, as shown in Fig. 6. The temperature dependence of specific capacitance recorded using BGO electrodes in 0.5 M electrolyte solution is almost linear. The phase transition between the one-layered and two-layered solvates of BGO, which occurs at around −20 °C (see Fig. 1a), has no effect on the performance of the supercapacitor. However, the capacitance of the BGO electrode in 1 M and 2 M TEA-BF4/acetonitrile solutions showed sharp changes in the slope of temperature dependence at −20 °C and 5 °C, respectively. Moreover, the change in slope is stronger for solutions with higher concentration (Fig. 6).
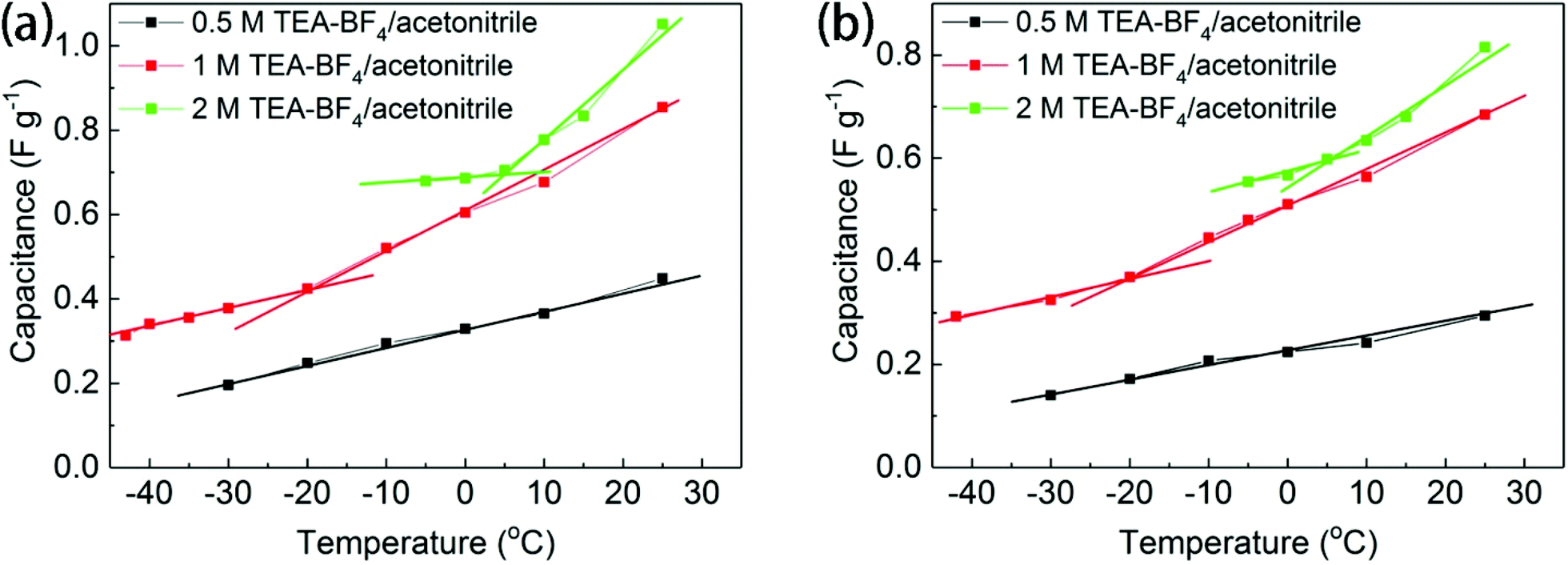 | ||
| Fig. 6 Temperature- and concentration-dependent electrochemical performances of BGO electrodes. Comparison of the capacitances of BGO electrodes in 2, 1, and 0.5 M TEA-BF4/acetonitrile recorded under different temperatures at scan rates of (a) 5 mV s−1 and (b) 10 mV s−1. | ||
The change in the slope of the temperature dependence of capacitance occurs in the same temperature region where the new expanded BGO phase is detected by XRD [d(001) = 14.9–15.5 Å]. As suggested above, this expanded phase is formed when solvated electrolyte ions are intercalated into the BGO structure. Once the slit pores of the BGO structure become accessible to the electrolyte ions, a change occurs in the electrode capacitance. Similar results were observed when the scanning rate was increased to 10 mV s−1 (Fig. 6b), although the change in the slope of temperature dependence was smaller than at the scanning rate of 5 mV s−1. This result is related to the retarded diffusion of ions under a fast scanning rate. The Nyquist plots of BGO electrodes immersed in 0.5 M, 1 M and 2 M electrolyte solutions were recorded at different temperatures from ambient temperature down to −40 °C; however, no anomalies specific to the phase transitions were observed between differently expanded BGO phases (Fig. S7†).
4. Discussion
Following interpretation of the data presented above is suggested:• The hydrophilic slit pores formed by BGO sheets are too small for the penetration of electrolyte ions in both the one-layered and two-layered BGO/acetonitrile structures [d(001) = 8.9 Å and d(001) = 12.5 Å, respectively].
• The intercalation of ions into the BGO structure occurs at low temperatures but only in highly concentrated TEA-BF4 solutions (1 M and 2 M). The distinct BGO structure with an interlayer distance of ∼15–16 Å is formed to accommodate solvated electrolyte ions.
• The ions remain to be intercalated into the BGO structure, even after the complete evaporation of acetonitrile by air heating or vacuum treatment.
The results presented above illustrate the advantages of GO for studying the relationship between pore size and ions with respect to the possibility of pore filling. Most other porous materials provide rigid pores with fixed diameters.4,13,20 The effect of ion size on pore filling can be estimated only by using many ions with different sizes or testing many materials with different pore sizes to verify the smallest pore size sufficient for the insertion of one single type of ion. For example, this was done by Centeno et al., who tested 28 types of porous carbons to establish the critical size of 0.7 nm as the penetration “cut-off”.20 Also note that these carbon materials are typically amorphous for XRD, and the pore size is determined based on gas sorption isotherms. The analysis of pore size distribution using gas sorption isotherms is not trivial and depends strongly on the selected model, which is a subject of controversy.20,37,47 It is also questionable whether the results obtained using gas sorption isotherms are valid for the analysis of pore filling with liquids and ions.
BGO provides a unique structure with “flexible” pore size that can be easily determined using XRD and tuned to match the sizes of specific ions (e.g., TEA+ and BF4− in this study). It is not pore size that defines the size of ions that can penetrate inside the pores, but the size of solvated ion that helps to define the size of the “pores” provided by GO sheets. Our results obtained using BGO are in good agreement with a recently reported study performed using multilayered HGO papers prepared via the vacuum filtration of Hummers GO dispersions.21 The slow swelling of HGO papers in propylene carbonate was demonstrated to result in peak capacitance corresponding to an average interlayer distance of ∼15 Å, as indicated by XRD. The advantage of BGO used in our study is the existence of well-defined phases with distinct structures and interlayer distances. HGO papers (often referred to as membranes) tested by Galhena et al.21 are different materials, and the gradual changes in d(001) are a result of averaging over many layers along with the effects of interstratification and intrastratification.26 Inhomogeneous hydration/solvation is typical in HGO powders, and swelling with slow kinetics is typical in GO membranes/papers due to the slow diffusion of solvent and ions through the “labyrinth path” of densely packed and overlapped GO flakes.50
Considering that we used different types of GO and different solvents, the agreement between our experiments and results of Galhena et al.21 is remarkably good. Both studies provide identical interlayer distances optimal for the insertion of solvated (∼15 Å) and desolvated (10–11 Å) TEA+/BF4− ions. This corresponds to a “pore” size of ∼7.5 Å being sufficient to accommodate solvated ions and ∼3.5–4.5 Å sufficient for desolvated ions.
Based on a recently proposed charge storage mechanism, the highest capacitance of a given porous carbon is achieved by using the electrolyte with ion size matched to the average pore size of the electrode material.13,19 Determining the favorable pore size is desirable since it makes it possible to synthesize electrode materials with specific monodisperse pore sizes. The anomaly in capacitance found in our experiments near the temperature point of ion intercalation into pores confirms that even sub-nanometer-sized pores contribute to the overall electrode capacitance.
Further experiments are required to understand the detailed mechanism of ion insertion into BGO interlayers (e.g., why intercalation occurs only at specific and relatively high electrolyte concentrations). It is typically assumed that increasing the electrolyte concentration has a simple effect on supercapacitor capacitance resulting from the increased number of ions. However, our results indicate that smaller pores could possibly be filled with ions at higher electrolyte concentrations.
It should be noted that developing a detailed understanding of GO solvate structures remains challenging due to the uncertain compositions, structures and relative proportions of various functional groups in precursor GO. As a result, the structures of GO solvates are also strongly disordered. For example, it is not clear why only BGO shows sharp phase transitions between different solvate phases, whereas HGO shows only gradual changes over broad temperature intervals.51,52 As proposed in our previous publication, swollen GO–acetonitrile can be considered as a solid solution of solvent in GO.52 In this case, swollen BGO has to be considered as a “solid solvate with temperature-independent composition” rather than a “solid solution with variable composition”, as in the case of HGO. The phase transition between two-layered and one-layered acetonitrile BGO solvates can be then described as incongruent melting. The amount of acetonitrile inserted into these two BGO solvates was quantitatively estimated using the isopiestic method and differential scanning calorimetry (DSC) to be 0.58 and 0.25 g g−1 (grams of solvent per gram of GO) for the two-layered and one-layered solvates, respectively.52 When electrolyte ions are introduced into the system, the phase that forms in solution can possibly be considered as a mixed solid solution of solvated ions and solvent in BGO. The size of solvated ions is somewhat larger than thickness of two layers of acetonitrile, which allows a distinct phase with d(001) ∼ 16 Å to be observed by XRD. The solvent-free BGO phase formed after the intercalation of TEA-BF4 and evaporation of acetonitrile can be then considered as a solid solution of TEA-BF4 in GO. In principle, the amount of TEA-BF4 intercalated into BGO in equilibrium with solution could be different compared to in solvent-free TEA-BF4/BGO if some of the ions escape the BGO structure together with acetonitrile during solvent evaporation and drying. The amount of TEA-BF4 intercalated into BGO after the evaporation of acetonitrile can be estimated based on XRD data. The increase in the interlayer distance from 6.5 Å in precursor BGO to 12.6 Å can be attributed to the intercalation of TEA-BF4 in volumetric proportion of 3(BGO)![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2(TEA-BF4). Using known densities of GO (2.0 g cm−3, measured using the gravimetric He buoyancy method) and TEA-BF4 (1.2 g cm−3),53 an approximate sorption of ∼0.4 g g−1 (TE-BF4/BGO) can be calculated. The TEA-BF4 intercalated into the BGO structure can be almost completely removed by repeated washing in acetonitrile (see Fig. S4 in the ESI† file). Thus, it can be concluded that the ions are relatively weakly bound to GO sheets, similar to the non-covalent intercalation of sugar alcohols observed in our earlier studies.54 The detailed characterization of solvent-free TEA-BF4/BGO materials is beyond the scope of this study, which focused primarily on ion penetration into slit pores in the GO structure.
:2(TEA-BF4). Using known densities of GO (2.0 g cm−3, measured using the gravimetric He buoyancy method) and TEA-BF4 (1.2 g cm−3),53 an approximate sorption of ∼0.4 g g−1 (TE-BF4/BGO) can be calculated. The TEA-BF4 intercalated into the BGO structure can be almost completely removed by repeated washing in acetonitrile (see Fig. S4 in the ESI† file). Thus, it can be concluded that the ions are relatively weakly bound to GO sheets, similar to the non-covalent intercalation of sugar alcohols observed in our earlier studies.54 The detailed characterization of solvent-free TEA-BF4/BGO materials is beyond the scope of this study, which focused primarily on ion penetration into slit pores in the GO structure.
5. Conclusions
The interlayer distance of BGO can be tuned using swelling in polar solvents, thus providing a rare example of flexible pores that adapt to the sizes of electrolyte ions under certain temperatures and electrolyte concentrations. The BGO structure in TEA-BF4/acetonitrile electrolyte can be switched between three distinctly different phases by changing the temperature and solution concentration. Ion intercalation into the BGO structure is not observed for 0.5 M solutions in the entire temperature range of our experiments (down to −60 °C). However, a distinctly different BGO structure is observed at low temperatures when the material is immersed in highly concentrated TEA-BF4/acetonitrile solutions. We attribute this new phase, which has an interlayer distance of ∼15–16 Å, to the intercalation of solvated ions into the “flexible pores” of the BGO structure. The interlayer structure corresponds to a pore size of about 9–10 Å. This pore size can be considered as the smallest pore size sufficient for the accommodation of solvated TEA-BF4 ions.The ions intercalated into BGO at low temperatures are not removed when the solvent is evaporated by air heating or vacuum drying. The desolvated BGO structure with intercalated TEA+/BF4− ions has an interlayer distance of ∼10 Å. It can be concluded that the smallest size of hydrophilic pore sufficient for the insertion (intercalation) of solvated ions is about 7.5 Å, while pores with sizes of about 3.5 Å are sufficient to accommodate desolvated TEA+/BF4− ions. Our results also indicate that the smallest sub-nanometer-sized pores might show concentration-dependent barriers to filling.
An anomaly in the temperature dependence of electrode capacitance was observed near the temperature point where BGO slit pores become filled with electrolyte ions. The change in the slope of temperature dependence is attributed to the intercalation of ions into the expanded interlayer space of the BGO electrode acting against the negative effects of reduced ionic conductivity on the charge storage performance at low temperatures. Our findings regarding the temperature-dependent intercalation of ions provide a possible way to tune the pore size and activate impermeable sub-nanopores in layered electrochemical energy storage materials.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the European Union Horizon 2020 research and innovation program under grant agreement no. 696656 and 785219, Kempestiftelserna and the Swedish Research Council (no. 2017-04173), the Åforsk Foundation (nr. 17-501) and RFBR grant no. 18-33-00439. We thank the support from ESRF for experiments performed at ID22 and technical assistance from Wilson Mogodi.References
- J. H. Sun, A. Klechikov, C. Moise, M. Prodana, M. Enachescu and A. V. Talyzin, Angew. Chem., Int. Ed., 2018, 57, 1034–1038 CrossRef CAS PubMed
.
- J. H. Sun, F. Morales-Lara, A. Klechikov, A. V. Talyzin, I. A. Baburin, G. Seifert, F. Cardano, M. Baldrighi, M. Frasconi and S. Giordani, Carbon, 2017, 120, 145–156 CrossRef CAS
- A. Klechikov, G. Mercier, T. Sharifi, I. A. Baburin, G. Seifert and A. V. Talyzin, Chem. Commun., 2015, 51, 15280–15283 RSC
- Y. W. Zhu, S. Murali, M. D. Stoller, K. J. Ganesh, W. W. Cai, P. J. Ferreira, A. Pirkle, R. M. Wallace, K. A. Cychosz, M. Thommes, D. Su, E. A. Stach and R. S. Ruoff, Science, 2011, 332, 1537–1541 CrossRef CAS PubMed
- M. Sevilla and R. Mokaya, Energy Environ. Sci., 2014, 7, 1250–1280 RSC
- M. D. Stoller, S. J. Park, Y. W. Zhu, J. H. An and R. S. Ruoff, Nano Lett., 2008, 8, 3498–3502 CrossRef CAS PubMed
- W. Z. Ouyang, J. H. Sun, J. Memon, C. Wang, J. X. Geng and Y. Huang, Carbon, 2013, 62, 501–509 CrossRef CAS
- Y. B. Ding, W. Bai, J. H. Sun, Y. Wu, M. A. Memon, C. Wang, C. B. Liu, Y. Huang and J. X. Geng, ACS Appl. Mater. Interfaces, 2016, 8, 12165–12175 CrossRef CAS PubMed
- W. Lv, Z. J. Li, Y. Q. Deng, Q. H. Yang and F. Y. Kang, Energy Storage Mater., 2016, 2, 107–138 CrossRef
- M. F. El-Kady, Y. L. Shao and R. B. Kaner, Nat. Rev. Mater., 2016, 1, 16033 CrossRef CAS
- L. L. Zhang and X. S. Zhao, Chem. Soc. Rev., 2009, 38, 2520–2531 RSC
- J. M. Luo, W. K. Zhang, H. D. Yuan, C. B. Jin, L. Y. Zhang, H. Huang, C. Liang, Y. Xia, J. Zhang, Y. P. Gan and X. Y. Tao, ACS Nano, 2017, 11, 2459–2469 CrossRef CAS PubMed
- A. C. Forse, C. Merlet, J. M. Griffin and C. P. Grey, J. Am. Chem. Soc., 2016, 138, 5731–5744 CrossRef CAS PubMed
- C. Largeot, C. Portet, J. Chmiola, P. L. Taberna, Y. Gogotsi and P. Simon, J. Am. Chem. Soc., 2008, 130, 2730–2731 CrossRef CAS PubMed
- E. Raymundo-Pinero, K. Kierzek, J. Machnikowski and F. Beguin, Carbon, 2006, 44, 2498–2507 CrossRef CAS
- J. Chmiola, G. Yushin, R. Dash and Y. Gogotsi, J. Power Sources, 2006, 158, 765–772 CrossRef CAS
- E. Frackowiak, Phys. Chem. Chem. Phys., 2007, 9, 1774–1785 RSC
- P. Simon and Y. Gogotsi, Nat. Mater., 2008, 7, 845–854 CrossRef CAS PubMed
- J. Chmiola, G. Yushin, Y. Gogotsi, C. Portet, P. Simon and P. L. Taberna, Science, 2006, 313, 1760–1763 CrossRef CAS PubMed
- T. A. Centeno, O. Sereda and F. Stoeckli, Phys. Chem. Chem. Phys., 2011, 13, 12403–12406 RSC
- D. T. L. Galhena, B. C. Bayer, S. Hofmann and G. A. J. Amaratunga, ACS Nano, 2016, 10, 747–754 CrossRef CAS PubMed
- A. V. Talyzin, S. M. Luzan, T. Szabo, D. Chernyshev and V. Dmitriev, Carbon, 2011, 49, 1894–1899 CrossRef CAS
- A. V. Talyzin, B. Sundqvist, T. Szabo and V. Dmitriev, J. Phys. Chem. Lett., 2011, 2, 309–313 CrossRef CAS
- A. V. Talyzin, A. Klechikov, M. Korobov, A. T. Rebrikova, N. V. Avramenko, M. F. Gholami, N. Severin and J. P. Rabe, Nanoscale, 2015, 7, 12625–12630 RSC
- A. Klechikov, J. H. Sun, I. A. Baburin, G. Seifert, A. T. Rebrikova, N. V. Avramenko, M. V. Korobov and A. V. Talyzin, Nanoscale, 2017, 9, 6929–6936 RSC
- B. Rezania, N. Severin, A. V. Talyzin and J. P. Rabe, Nano Lett., 2014, 14, 3993–3998 CrossRef CAS PubMed
- A. V. Talyzin, B. Sundqvist, T. Szabo, I. Dekany and V. Dmitriev, J. Am. Chem. Soc., 2009, 131, 18445–18449 CrossRef CAS PubMed
- L. Chen, G. S. Shi, J. Shen, B. Q. Peng, B. W. Zhang, Y. Z. Wang, F. G. Bian, J. J. Wang, D. Y. Li, Z. Qian, G. Xu, G. P. Liu, J. R. Zeng, L. J. Zhang, Y. Z. Yang, G. Q. Zhou, M. H. Wu, W. Q. Jin, J. Y. Li and H. P. Fang, Nature, 2017, 550, 380–383 CrossRef CAS PubMed
- B. Brodie, Ann. Chim. Phys., 1860, 59, 466–472 Search PubMed
- U. Hofmann, A. Frenzel and E. Csalán, Justus Liebigs Ann. Chem., 1934, 510, 1–41 CrossRef CAS
- A. V. Talyzin, V. L. Solozhenko, O. O. Kurakevych, T. Szabo, I. Dekany, A. Kurnosov and V. Dmitriev, Angew. Chem., Int. Ed., 2008, 47, 8268–8271 CrossRef CAS PubMed
- S. J. You, S. M. Luzan, T. Szabo and A. V. Talyzin, Carbon, 2013, 52, 171–180 CrossRef CAS
- J. Zhu, C. M. Andres, J. D. Xu, A. Ramamoorthy, T. Tsotsis and N. A. Kotov, ACS Nano, 2012, 6, 8357–8365 CrossRef CAS PubMed
- G. Mercier, A. Klechikov, M. Hedenstrom, D. Johnels, I. A. Baburin, G. Seifert, R. Mysyk and A. V. Talyzin, J. Phys. Chem. C, 2015, 119, 27179–27191 CrossRef CAS
- S. J. You, B. Sundqvist and A. V. Talyzin, ACS Nano, 2013, 7, 1395–1399 CrossRef CAS PubMed
- S. J. You, S. Luzan, J. C. Yu, B. Sundqvist and A. V. Talyzin, J. Phys. Chem. Lett., 2012, 3, 812–817 CrossRef CAS PubMed
- M. Deschamps, E. Gilbert, P. Azais, E. Raymundo-Pinero, M. R. Ammar, P. Simon, D. Massiot and F. Beguin, Nat. Mater., 2013, 12, 351–358 CrossRef CAS PubMed
- M. M. Hantel, V. Presser, R. Kotz and Y. Gogotsi, Electrochem. Commun., 2011, 13, 1221–1224 CrossRef CAS
- P. Kleszyk, P. Ratajczak, P. Skowron, J. Jagiello, Q. Abbas, E. Frackowiak and F. Beguin, Carbon, 2015, 81, 148–157 CrossRef CAS
- S. Kondrat, C. R. Perez, V. Presser, Y. Gogotsi and A. A. Kornyshev, Energy Environ. Sci., 2012, 5, 6474–6479 RSC
- C. Prehal, D. Weingarth, E. Perre, R. T. Lechner, H. Amenitsch, O. Paris and V. Presser, Energy Environ. Sci., 2015, 8, 1725–1735 RSC
- C. Prehal, C. Koczwara, N. Jackel, A. Schreiber, M. Burian, H. Amenitsch, M. A. Hartmann, V. Presser and O. Paris, Nat. Energy, 2017, 2, 16215 CrossRef CAS
- L. L. Liu, Z. Q. Niu and J. Chen, Chem. Soc. Rev., 2016, 45, 4340–4363 RSC
- Y. Korenblit, A. Kajdos, W. C. West, M. C. Smart, E. J. Brandon, A. Kvit, J. Jagiello and G. Yushin, Adv. Funct. Mater., 2012, 22, 1655–1662 CrossRef CAS
- W. Y. Tsai, R. Y. Lin, S. Murali, L. L. Zhang, J. K. McDonough, R. S. Ruoff, P. L. Taberna, Y. Gogotsi and P. Simon, Nano Energy, 2013, 2, 403–411 CrossRef CAS
- M. R. Lukatskaya, O. Mashtalir, C. E. Ren, Y. Dall'Agnese, P. Rozier, P. L. Taberna, M. Naguib, P. Simon, M. W. Barsoum and Y. Gogotsi, Science, 2013, 341, 1502–1505 CrossRef CAS PubMed
- C. Decaux, C. M. Ghimbeu, M. Dahbi, M. Anouti, D. Lemordant, F. Beguin, C. Vix-Guterl and E. Raymundo-Pinero, J. Power Sources, 2014, 263, 130–140 CrossRef CAS
- C. Cabrillo, F. Barroso-Bujans, R. Fernandez-Perea, F. Fernandez-Alonso, D. Bowron and F. J. Bermejo, Carbon, 2016, 100, 546–555 CrossRef CAS
- D. R. Dreyer, S. Park, C. W. Bielawski and R. S. Ruoff, Chem. Soc. Rev., 2010, 39, 228–240 RSC
- A. Klechikov, J. C. Yu, D. Thomas, T. Sharifi and A. V. Talyzin, Nanoscale, 2015, 7, 15374–15384 RSC
- A. V. Talyzin, G. Mercier, A. Klechikov, M. Hedenstrom, D. Johnels, D. Wei, D. Cotton, A. Opitz and E. Moons, Carbon, 2017, 115, 430–440 CrossRef CAS
- M. V. Korobov, A. V. Talyzin, A. T. Rebrikova, E. A. Shilayeva, N. V. Avramenko, A. N. Gagarin and N. B. Ferapontov, Carbon, 2016, 102, 297–303 CrossRef CAS
- G. Giuseppetti, C. Tadini, P. Ferloni, G. Zabinska and S. Torre, Z. Kristallogr., 1994, 209, 509–511 CAS
- A. Klechikov, S. You, L. Lackner, J. Sun, A. Iakunkov, A. T. Rebrikova, M. Korobov, I. Baburin, G. Seifert and A. V. Talyzin, Carbon, 2018, 140, 157–163 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr07469k |
| This journal is © The Royal Society of Chemistry 2018 |