
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Tuning coiled coil stability with histidine-metal coordination†
Isabell
Tunn
 a,
Alberto S.
de Léon
a,
Kerstin G.
Blank
*a and
Matthew J.
Harrington
*ab
a,
Alberto S.
de Léon
a,
Kerstin G.
Blank
*a and
Matthew J.
Harrington
*ab
aMax Planck Institute of Colloids and Interfaces, Science Park Potsdam-Golm, 14424 Potsdam, Germany. E-mail: Kerstin.Blank@mpikg.mpg.de
bMcGill University, 801 Sherbrooke St West, Montreal, Quebec H3A 0B8, Canada. E-mail: Matt.Harrington@mcgill.ca
First published on 26th November 2018
Abstract
Coiled coils (CCs) have emerged as versatile building blocks for the synthesis of nanostructures, drug delivery systems and biomimetic hydrogels. Bioengineering metal coordination sites into the terminal ends of a synthetic coiled coil (CC), we generate a nanoscale biological building block with tunable stability. The reversible coordination of Ni2+ thermodynamically stabilizes the CC, as shown with circular dichroism spectroscopy. Using atomic force microscopy-based single-molecule force spectroscopy, it is further shown that Ni2+-binding reinforces the CC mechanically, increasing the barrier height for dissociation. When used as a dynamic crosslink in polyethyleneglycol-based hydrogels, the single-molecule stability of the CC is directly transferred to the bulk material and determines its viscoelastic properties. This reversibly tunable CC, thus, highlights an effective strategy for rationally engineering the single-molecule properties of biomolecular building blocks, which can be translated to the emergent properties of biomimetic materials, as well as other CC containing molecular assemblies.
Introduction
The supramolecular assembly of proteinaceous building blocks via non-covalent interactions is emerging as an effective strategy for producing biocompatible hydrogels1 as well as nanoarchitectures with increased structural and functional complexity.2,3 However, molecular control of their thermodynamic and mechanical stability remains a significant hurdle. Here, we employ a rational bottom-up approach towards bioengineering a tunable peptide building block. We combine two prominent material design paradigms found in natural supramolecular materials – namely, the coiled coil (CC) protein-folding motif and histidine (His)–metal crosslinking. The CC protein-folding motif is a common conformational feature of proteins with essential mechanical function in mammalian tissues (e.g. in molecular motors and extracellular matrix components)1 and further appears as a frequent building block for many proteinaceous biological materials (e.g. in wool, hagfish slime and gastropod egg capsules).7 CCs consist of two to seven α-helices wrapped around each other to form a superhelix.1 The structure and conformation of CCs is determined by a specific repeat of seven amino acids (abcdefg), called a heptad. The overall stability of the CC superhelix originates from hydrophobic interactions at positions a and d, ionic interactions at positions e and g, as well as the stability of the individual α-helices, which can be tuned by including amino acids with different helix propensity8 especially in the solvent-exposed positions b, c and f.His–metal coordination bonds are strong, non-covalent interactions with self-healing properties, observed, for example, in mussel byssal threads and spider fangs.9,10 Rationally engineering His–metal coordination sites into proteins has already been demonstrated to increase thermodynamic and mechanical stability.11–14 Here, we introduce His–metal coordination sites into the sequence of a well-characterized synthetic heterodimeric CC (A4B4).5,6 Considering that the terminal helical turns of CCs are more dynamic,15 we therefore hypothesize that bridging these helical turns with metal coordination bonds (Fig. 1A) will have a stabilizing effect on the entire CC. To achieve this, we introduce His at the solvent exposed b and f positions of the terminal heptads of the CC (HA4HB4), spacing the His residues in a distance of i, i + 4 (Fig. 1B). This ensures a proper geometric configuration for forming chelate complexes with transition metals.11,16 In addition to the expected thermodynamic stabilization, we further hypothesize that metal-coordinated helical turns will also affect the mechanical stability of CCs. Using molecular dynamics (MD) simulations, we have recently investigated the mechanistic response of a heterodimeric 4-heptad CC to an externally applied shear force. These simulations suggest that the applied force causes the consecutive unfolding of helical turns, most likely initiated at the termini where the force is applied.6 Introducing His–metal bridges into the mechanically loaded heptads (Fig. 1A and B) is thus expected to stabilize the helices against mechanically induced unfolding, allowing us to experimentally test and verify the predicted mechanism. To investigate the possible effect of metal coordination, we focus on Ni2+ as the coordinating metal, since it has been shown to exhibit slow bond dynamics combined with rupture forces ranging from 40 pN to 300 pN.17,18
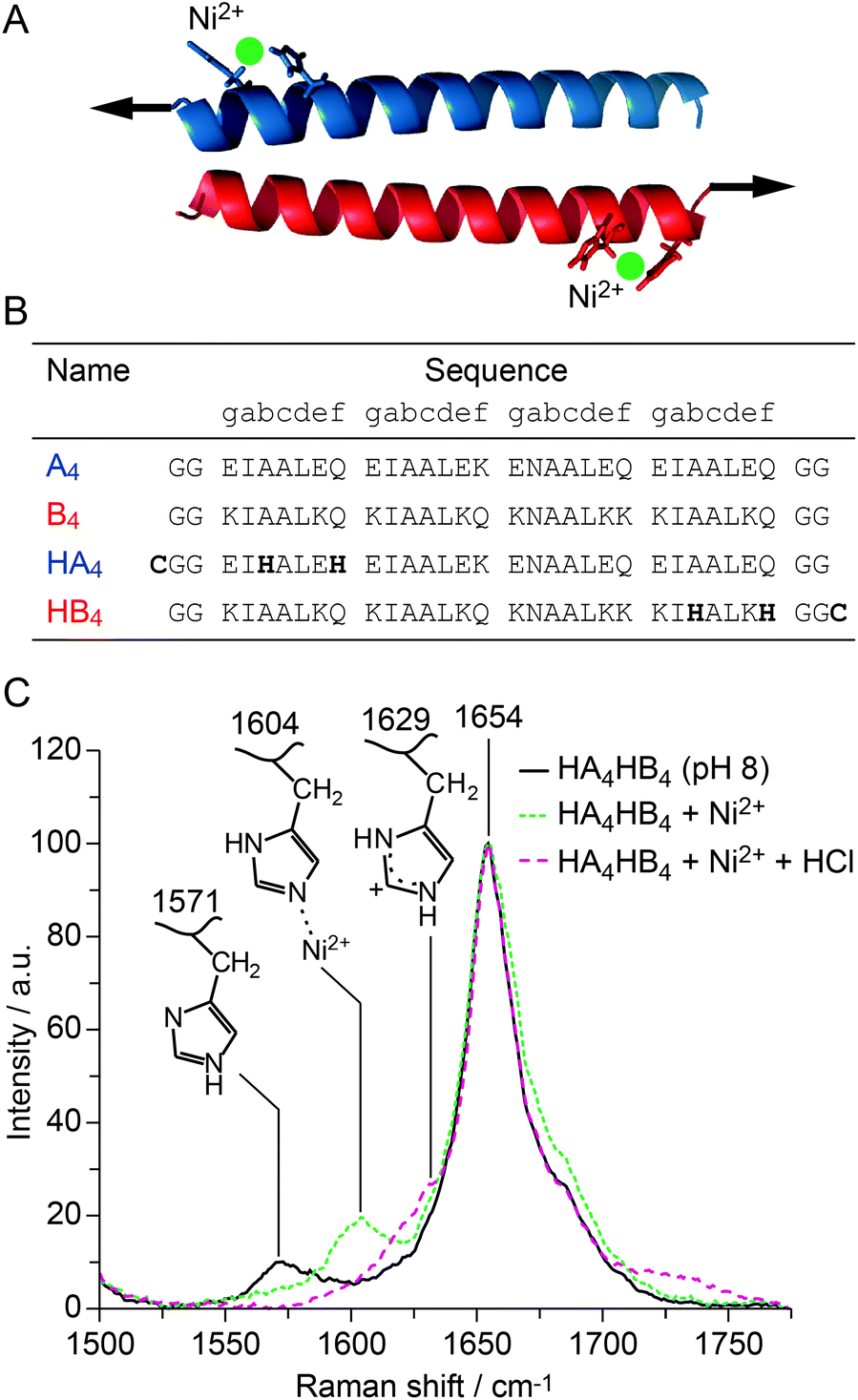 | ||
Fig. 1 Design and structural characterization of the His-modified CC. (A) Structural model of the CC generated with CC-builder,4 showing the His–metal coordination bridges. Arrows indicate the direction and point of origin of the applied shear force. (B) Amino acid sequence of the reference peptides A4 and B4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 5,6 as well as the His-modified peptides HA4 and HB4. The terminal cysteines serve for coupling the peptides to maleimide-functionalized PEG. They were absent in the peptides used for structural and thermodynamic characterization. (C) Raman spectra of thin films of HA4HB4 formed at pH 8 without metals, with 2:1 His:Ni2+, and with Ni2+ at acidic pH. 5,6 as well as the His-modified peptides HA4 and HB4. The terminal cysteines serve for coupling the peptides to maleimide-functionalized PEG. They were absent in the peptides used for structural and thermodynamic characterization. (C) Raman spectra of thin films of HA4HB4 formed at pH 8 without metals, with 2:1 His:Ni2+, and with Ni2+ at acidic pH. | ||
Results and discussion
Raman spectroscopy was used to investigate both the secondary structure of the CC (characteristic amide vibrational bands19) and the ability of His to coordinate Ni2+. Histidine shows distinct vibrational modes that are extremely sensitive to the protonation and coordination state of its functional group.20 The resulting Raman peaks, originating from C4![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C5 vibrations of the imidazole ring, are expected in the range from 1550–1640 cm−1. The Raman spectrum of HA4HB4 shows an amide I peak at 1654 cm−1, confirming the α-helical secondary structure of the CC (Fig. 1C). The peak at 1571 cm−1 is assigned to deprotonated His at pH 8.0.19,20 Upon addition of NiCl2 in a ratio of 2:1 His:Ni2+, the His peak shifts to 1604 cm−1 due to His–Ni2+ coordination. After acidifying the solution, the dominant peak at 1629 cm−1 indicates that His becomes protonated and no longer coordinates Ni2+. This clearly reveals that the interaction between His and Ni2+ is reversible. HA4HB4 was further investigated using circular dichroism (CD) spectroscopy, which shows the expected spectrum with two minima at 208 nm and 222 nm, characteristic for an α-helical structure (Fig. S1A†). CD spectroscopy was also used to investigate the thermodynamic stability of the CC in the presence and the absence of NiCl2, performing thermal unfolding experiments (Fig. S1B†). His–Ni2+ coordination (1:3 His:Ni2+ ratio) increased the melting temperature (Tm) of HA4HB4 from 76.4 ± 0.9 °C to 80.2 ± 0.6 °C. In contrast, no Ni2+-induced stabilization was observed in the reference CC A4B4, which lacks His (Fig. 1B and Fig. S2†). A comparable thermodynamic stabilization induced by His–metal coordination was described previously for several other proteins.11,21
C5 vibrations of the imidazole ring, are expected in the range from 1550–1640 cm−1. The Raman spectrum of HA4HB4 shows an amide I peak at 1654 cm−1, confirming the α-helical secondary structure of the CC (Fig. 1C). The peak at 1571 cm−1 is assigned to deprotonated His at pH 8.0.19,20 Upon addition of NiCl2 in a ratio of 2:1 His:Ni2+, the His peak shifts to 1604 cm−1 due to His–Ni2+ coordination. After acidifying the solution, the dominant peak at 1629 cm−1 indicates that His becomes protonated and no longer coordinates Ni2+. This clearly reveals that the interaction between His and Ni2+ is reversible. HA4HB4 was further investigated using circular dichroism (CD) spectroscopy, which shows the expected spectrum with two minima at 208 nm and 222 nm, characteristic for an α-helical structure (Fig. S1A†). CD spectroscopy was also used to investigate the thermodynamic stability of the CC in the presence and the absence of NiCl2, performing thermal unfolding experiments (Fig. S1B†). His–Ni2+ coordination (1:3 His:Ni2+ ratio) increased the melting temperature (Tm) of HA4HB4 from 76.4 ± 0.9 °C to 80.2 ± 0.6 °C. In contrast, no Ni2+-induced stabilization was observed in the reference CC A4B4, which lacks His (Fig. 1B and Fig. S2†). A comparable thermodynamic stabilization induced by His–metal coordination was described previously for several other proteins.11,21
To obtain more detailed insight into the stabilizing effect of His–Ni2+ coordination, single-molecule force spectroscopy (SMFS) was performed. SMFS directly probes whether His–Ni2+ coordination mechanically stabilizes the CC and, more importantly, provides information about the effect of bridging the terminal helical turns on the energy landscape of the CC interaction. The two peptides HA4 and HB4 were immobilized separately, either to the AFM cantilever or to a glass surface (Fig. 2A). For this purpose, cysteine was introduced at the respective termini and the peptides were coupled to surfaces functionalized with maleimide-terminated polyethyleneglycol (PEG).22Fig. 2B shows typical force–distance curves recorded at a retract speed of 400 nm s−1 in the presence and absence of 1 mM NiCl2. The resulting rupture force histograms show that His–Ni2+ coordination increases the rupture force of HA4HB4 by ∼10 pN (Fig. S3†). Adding an excess of chelating agent, such as ethylenediaminetetraacetic acid (EDTA), reduces the rupture force by approximately the same value, supporting the observation that the stabilizing effect of His–Ni2+ coordination is fully reversible (Fig. 2B). This result is a first indication that the computationally proposed mechanism of mechanical CC unfolding is reproduced in our SMFS experiments and that the CC can be stabilized against shear forces when bridging the terminal helical turns at the points of force application.
 | ||
| Fig. 2 Single-molecule force spectroscopy of HA4HB4. (A) Experimental setup. The individual CC-forming peptides were coupled to either the cantilever or the glass surface via a PEG linker. A maleimide-functionalized PEG was used for coupling the Cys-terminated peptides. (B) Typical force distance curves of the His-modified CC in the absence and presence of 1 mM Ni2+ and after washing with 10 mM EDTA. (C) Results of the dynamic SMFS measurements of the His-modified CC in the presence and absence of 1 mM Ni2+. The most probable rupture forces were plotted versus the logarithm of the most probable loading rates. Three data sets (triangles, squares, circles) were obtained per condition, using three different cantilevers and surfaces. The lines are fits to the Bell–Evans model. The parameters koff and Δx obtained from the fits to the single data sets are shown in Table S2.† The mean values ± SEM are reported in this figure. | ||
To investigate the dynamic mechanical response of HA4HB4 and to understand how His–Ni2+ coordination affects the energy landscape of the CC, the measurements were repeated at different retract speeds and the most probable rupture forces (FR) and the corresponding loading rates (Ḟ) were determined (see Fig. S4, S5 and Table S1† for representative data sets obtained in the absence and the presence of 1 mM NiCl2). Fig. 2C reveals a linear dependency of FR on the logarithm of Ḟ, which can be described with the Bell–Evans model (see ESI†).23
Fitting the data sets with the Bell–Evans equation yields the extrapolated thermal off-rate at zero force (koff) and the potential width (Δx). Notably, the mean koff in the presence of 1 mM Ni2+ (2.5 × 10−3 s−1) is almost one order of magnitude lower than in the absence of 1 mM Ni2+ (11.0 × 10−3 s−1), indicating that the barrier height to dissociation is increased when Ni2+ is bound (Fig. 2B). In contrast, the potential width Δx is not significantly different in the absence and presence of Ni2+ (Table S2†), suggesting that the mechanical dissociation pathway of the CC is not changed. Considering the proposed mechanism, this again confirms the stabilizing effect of the His–Ni2+ bridge. We propose that stabilization primarily affects the individual helices.11,13,14 As a secondary effect, the reduced conformational freedom of one helix is transferred to the second helix via the hydrophobic core, thereby synergistically increasing the stability of the entire CC. This interpretation is supported by the observation that the presence of a third helix (CC trimer) greatly stabilizes the CC against shear forces.24
It remains an open question what exactly causes the increased energy barrier. It may originate from the simultaneous rupture of the His–Ni2+ bond and the helix stabilizing hydrogen bonds. Alternatively, the His–Ni2+ stabilized heptads may remain intact, forcing the CC to start uncoiling at internal heptads, which are stabilized by a larger number of hydrogen bonds. No information is available about the unbinding and rebinding kinetics of the His–Ni2+ bridge and the rupture forces reported for His:Ni2+ bonds lie in the range between 40 and 300 pN.9 This force range overlaps with the rupture forces of the CC so that no direct conclusion is possible about which scenario is more likely. Additional experiments using an irreversible bridge, such as a covalent staple,25,26 would be required to unambiguously answer this question. Despite this remaining question, the results show that His–Ni2+ coordination stabilizes HA4HB4 thermodynamically and mechanically and that our rational bottom-up approach is valid.
Based on the evidence above, the engineered CC has an excellent potential to be used as a building block for the synthesis of biomimetic materials and supramolecular assemblies with tunable stimuli-responsive behaviors. As a proof-of-concept demonstration that molecular level tunability can be translated into bulk-scale dynamic properties, we used HA4HB4 as a supramolecular crosslink for the production of polymer–peptide hybrid hydrogels. Cysteine-containing HA4 and HB4 were each separately coupled to maleimide-terminated star-shaped polyethyleneglycol (sPEG, 4 arms) and gels were formed by mixing the two in a stoichiometric ratio (Fig. 3 inset). Using oscillatory rheology, the viscoelastic properties of the CC-crosslinked hydrogel were investigated in the absence and presence of Ni2+ (ratio of 1:1 His:Ni2+) and after adding 10 mM EDTA to the Ni2+-containing hydrogel. Amplitude sweeps show a linear viscoelastic behavior up to at least 100% strain (Fig. S6†). At strains above 100% the storage modulus G′ starts to decrease while the loss modulus G′′ increases, indicating the onset of CC rupture. Due to the reversible nature of the CC crosslinks, the mechanical properties fully recover when the strain is lowered to 1% (Fig. S6†), indicating dynamic self-healing. Frequency sweeps performed at 1% strain (Fig. 3) give insights into this dynamic behavior. As described by the Maxwell model27 for reversibly crosslinked networks, the relaxation time τ was obtained from the crossover frequency (ωCR) of the G′ and G′′ curves of all three hydrogels. A comparison of τ shows that the addition of Ni2+ increases the mean relaxation time by ∼3-fold from 92 ± 12 s to 258 ± 36 s – an effect that is fully reversed upon treatment with EDTA (τ = 44 ± 4 s). Notably, the relaxation time, which provides information about the average supramolecular lifetime of the CC crosslinks,27 shows a similar trend as koff determined with SMFS. Thus, it stands to reason that reducing koff for single CC crosslinks via metal chelation, effectively increases the bond lifetime of the crosslink and shifts ωCR to lower values. This direct correlation is additional proof for the functionality of the engineered CC building block and highlights that engineering the molecular parameters of hydrogel building blocks directly determines bulk material properties.
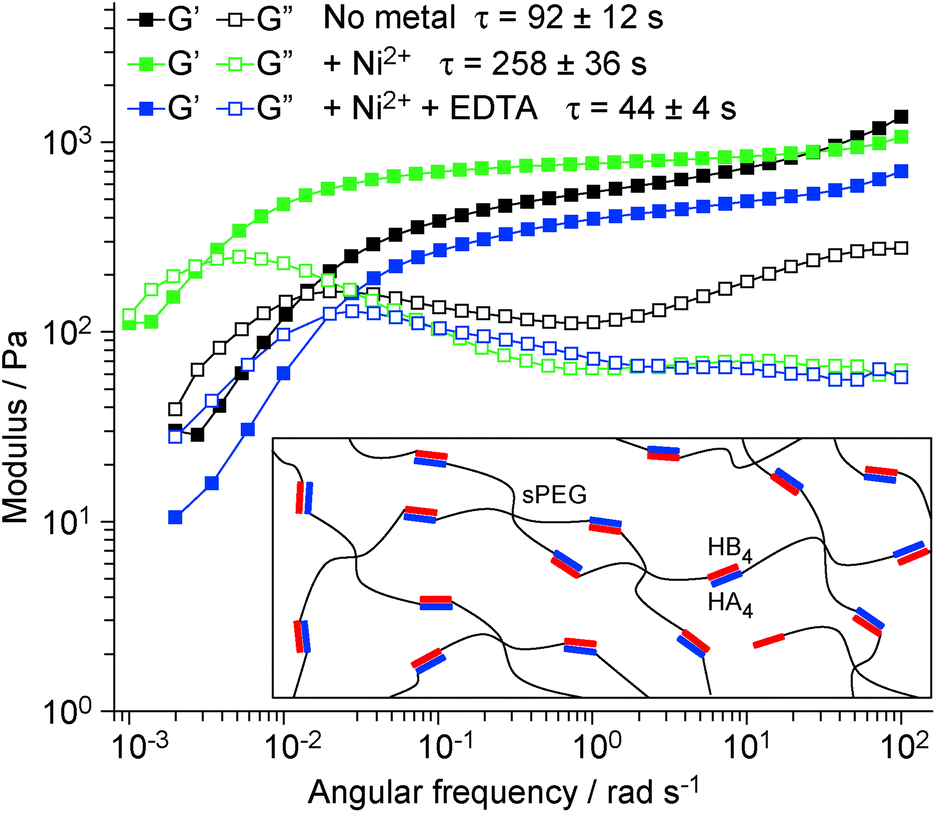 | ||
| Fig. 3 Oscillatory shear rheology of HA4HB4-crosslinked sPEG hydrogels. Frequency sweep of the CC-crosslinked hydrogel at a constant strain amplitude of 1%, without metals, with 1:1 His:Ni2+ and 1:1 His:Ni2+ + EDTA (10 mM). G′ is the storage modulus and G′′ is the loss modulus. The crossover of G′ and G′′ (ωCR) yields the relaxation time (ωCR = 1/τ). Inset: Schematic representation of the hydrogel structure. | ||
Conclusions
In summary, the presented rational design approach sheds new insights into the factors controlling CC stability. It allowed us to test and validate the prediction that bridging individual helical turns stabilizes the entire CC, thermodynamically and mechanically. These important mechanistic insights enabled us to reversibly tune the stability of the CC and provide a proof-of-concept demonstration of the potential of this versatile nanomechanical building block for biomimetic materials. Specifically, when used as a hydrogel crosslink, the CC molecular properties directly determine the performance of the bulk material in a metal-dependent manner. This provides a solid starting point for the development of a library of tunable CC building blocks with the potential of synthesizing well-controlled and dynamic hydrogels. Future studies will focus on expanding the tunable range, repositioning the stabilizing bridge to other heptads or using other transition metal ions, such as Cu2+, Co2+ and Zn2+ as well as covalent staples.27Conflicts of interest
There are no conflicts to declare.Acknowledgements
The project was financially supported by the Max Planck Society and the International Max Planck Research School (IMPRS) on Multiscale Bio-Systems. ASL acknowledges support from a postdoctoral fellowship of the Alexander von Humboldt Foundation. Open Access funding provided by the Max Planck Society.Notes and references
- B. Apostolovic, M. Danial and H.-A. Klok, Chem. Soc. Rev., 2010, 39, 3541 RSC
.
- G. C. Pugh, J. R. Burns and S. Howorka, Nat. Rev. Chem., 2018, 2, 113 CrossRef CAS
- A. Ljubetič, I. Drobnak, H. Gradišar and R. Jerala, Chem. Commun., 2016, 52, 5220 RSC
- C. W. Wood, M. Bruning, A. Á. Ibarra, G. J. Bartlett, A. R. Thomson, R. B. Sessions, R. L. Brady and D. N. Woolfson, Bioinformatics, 2014, 30, 3029 CrossRef CAS PubMed
- F. Thomas, A. L. Boyle, A. J. Burton and D. N. Woolfson, J. Am. Chem. Soc., 2013, 135, 5161 CrossRef CAS PubMed
- M. Goktas, C. Luo, R. M. A. Sullan, A. E. Bergues-Pupo, R. Lipowsky, A. Vila Verde and K. G. Blank, Chem. Sci., 2018, 9, 4610 RSC
- A. Miserez and P. A. Guerette, Chem. Soc. Rev., 2013, 42, 1973 RSC
- J. R. Litowski and R. S. Hodges, J. Biol. Chem., 2002, 277, 37272 CrossRef CAS PubMed
- E. Degtyar, M. J. Harrington, Y. Politi and P. Fratzl, Angew. Chem., Int. Ed., 2014, 53, 12026 CrossRef CAS PubMed
- C. N. Z. Schmitt, Y. Politi, A. Reinecke and M. J. Harrington, Biomacromolecules, 2015, 16, 2852 CrossRef CAS PubMed
- F. H. Arnold and B. L. Haymore, Science, 1991, 252, 1796 CrossRef CAS PubMed
- Y. Cao, T. Yoo and H. Li, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 11152 CrossRef CAS PubMed
- W. D. Kohn, C. M. Kay, B. D. Sykes and R. S. Hodges, J. Am. Chem. Soc., 1998, 120, 1124 CrossRef CAS
- T. Miura, T. Satoh and H. Takeuchi, Biochim. Biophys. Acta, 1998, 1384, 171 CrossRef CAS
- G. De Crescenzo, J. R. Litowski, R. S. Hodges and M. D. O'Connor-McCourt, Biochemistry, 2003, 42, 1754 CrossRef CAS PubMed
- S. Knecht, D. Ricklin, A. N. Eberle and B. Ernst, J. Mol. Recognit., 2009, 22, 270 CrossRef CAS PubMed
- M. Conti, G. Falini and B. Samorì, Angew. Chem., Int. Ed., 2000, 39, 215 CrossRef CAS PubMed
- L. Schmitt, M. Ludwig, H. E. Gaub and R. Tampé, Biophys. J., 2000, 78, 3275 CrossRef CAS PubMed
- Z. Movasaghi, S. Rehman and D. I. U. Rehman, Appl. Spectrosc. Rev., 2007, 42, 493 CrossRef CAS
- H. Takeuchi, Biopolymers, 2003, 72, 305 CrossRef CAS PubMed
- M. R. Ghadiri and C. Choi, J. Am. Chem. Soc., 1990, 112, 1630 CrossRef CAS
- J. L. Zimmermann, T. Nicolaus, G. Neuert and K. Blank, Nat. Protoc., 2010, 5, 975 CrossRef CAS PubMed
- E. Evans, Annu. Rev. Biophys. Biomol. Struct., 2001, 30, 105 CrossRef CAS PubMed
- A. E. Bergues-Pupo, K. G. Blank, R. Lipowsky and A. Vila Verde, Phys. Chem. Chem. Phys., 2018 10.1039/c8cp04896g
- Y.-W. Kim and G. L. Verdine, Bioorg. Med. Chem. Lett., 2009, 19, 2533 CrossRef CAS PubMed
- G. L. Verdine and G. J. Hilinski, Methods Enzymol., 2012, 503, 3 CAS
- S. C. Grindy, R. Learsch, D. Mozhdehi, J. Cheng, D. G. Barrett, Z. Guan, P. B. Messersmith and N. Holten-Andersen, Nat. Mater., 2015, 14, 1210 CrossRef CAS PubMed
Footnote |
| † Electronic supplementary information (ESI) available: Methods and data of the Raman, CD, SMFS and rheology experiments. See DOI: 10.1039/c8nr07259k |
| This journal is © The Royal Society of Chemistry 2018 |