
Fabrication and understanding of Cu3Si-Si@carbon@graphene nanocomposites as high-performance anodes for lithium-ion batteries†

Abstract
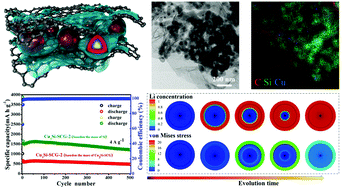
Besides silicon's low electronic conductivity, another critical issue for using silicon as the anode for lithium-ion batteries (LIBs) is the dramatic volume variation (>300%) during lithiation/delithiation processes, which can lead to rapid capacity fading and poor rate capability, thereby hampering silicon's practical applications in batteries. To mitigate these issues, herein, we report our findings on the design and understanding of a self-supported Cu3Si-Si@carbon@graphene (Cu3Si-SCG) nanocomposite anode. The nanocomposite is composed of Cu3Si-Si core and carbon shell with core/shell particles uniformly encapsulated by graphene nanosheets anchored directly on a Cu foil. In this design, the carbon shell, the highly elastic graphene nanosheet, and the formed conductive and inactive Cu3Si phase in Si serve as buffer media to suppress volume variation of Si during lithiation/delithiation processes and to facilitate the formation of a stable solid electrolyte interface (SEI) layer as well as to enable good transport kinetics. Chemomechanical simulation results quantitatively coincide with the in situ TEM observations of volume expansion and provide process details not seen in experiments. The optimized Cu3Si-SCG nanocomposite anode exhibits good rate performance and delivers reversible capacity of 483 mA h g−1 (based on the total weight of Cu3Si-SCG) after 500 cycles with capacity retention of about 80% at high current density of 4 A g−1, rendering the nanocomposite a desirable anode candidate for high-performance LIBs.
- This article is part of the themed collection: Nanoscale 10th Anniversary: Top Authors
 Please wait while we load your content...
Please wait while we load your content...

