
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSpecific binding and internalization: an investigation of fluorescent aptamer-gold nanoclusters and cells with fluorescence lifetime imaging microscopy†
Marina
Mutas
ab,
Christian
Strelow
a,
Tobias
Kipp
 *a and
Alf
Mews
a
*a and
Alf
Mews
a
aInstitute of Physical Chemistry, University of Hamburg, Grindelallee 117, Hamburg, Germany. E-mail: kipp@chemie.uni-hamburg.de; Tel: +49 40 42838 8277
bThe Hamburg Centre for Ultrafast Imaging, Luruper Chaussee 149, 22761 Hamburg, Germany
First published on 8th October 2018
Abstract
Fluorescent gold nanoclusters show promising properties for biological applications. We biofunctionalized fluorescent 11-mercaptoundecanoic-acid stabilized gold nanoclusters (AuNCs) with an aptamer to target the interleukin-6-receptor expressed on BaF3 cells specifically. Although the fluorescence emission of the AuNCs (535 nm) is in the same wavelength region as the autofluorescence of the cell, we are able to distinguish between nanoclusters and cells using the fluorescence decay time, which is much longer for the AuNCs (100 ns) than for the autofluorescence. After a first short incubation period we detected AuNCs specifically bound to the cell membrane by using two fluorescence lifetime imaging microscopy (FLIM) methods: gated and direct FLIM. After a second incubation period the previously bound AuNCs are internalized by the cells, as could be resolved solely by the direct FLIM. This proves the superior sensitivity of this method compared to gated FLIM. We find that the optical properties of AuNCs do not change upon binding to the cells, but exhibit a change when internalized into the cells, induced by an interaction between the AuNCs and cells.
1. Introduction
Fluorescent nanoclusters are promising novel fluorescent materials for biological applications. In particular, gold nanoclusters (AuNCs) combine promising chemical and physical properties like simple preparation, high photostability, good biocompatibility, and size-dependent emission wavelength making them useful in a variety of biomedical applications including sensing, diagnostics, and cell imaging.1–6 Size tunability is achieved only for very small Au5 to Au55 clusters.7,8 The specific choice of stabilizing ligands for AuNCs offers a further degree of freedom to tune the emission wavelength, which is also available for larger AuNCs.9,10 Furthermore, the ligands allow for a high degree of flexibility in terms of functional groups for biofunctionalization and targeting. Due to their strong affinity to noble metals, thiol molecules are commonly used as ligands for synthesizing, stabilizing, and functionalizing AuNCs.11,12Thiol-containing peptides, organosulfur compounds, polymers, and amino acids have already been used as stabilizing ligands for fluorescent AuNCs.13 These ligands additionally offer flexible functional groups for biofunctionalization and targeting for specific biomedical purposes.1,5,14–16
Glutathione stabilized fluorescent AuNCs, for example, show a high specificity to MCF7 breast cancer cells. Loaded with doxorubicin, an anti-cancer drug, the glutathione-AuNCs functioned as a therapeutic nanomaterial and only selectively killed the cancer cells.17 It has also been reported that small (1–2 nm in diameter) zwitterionic AuNCs show a high uptake rate with a combined low cytotoxicity in dendritic cells.18 Several other publications also demonstrate the possible application of fluorescent thiol-stabilized AuNCs for cancer-cell targeting and imaging.1,5,14,15 However, in all cases a high AuNC concentration (in a μM range) and a long incubation period (≥1 hour) were applied, making use of the general properties of cells to take up various types of nano-objects unspecifically.
For biomedical applications, high specificities are necessary to allow for an efficient targeted drug delivery. Specificity can be achieved by biofunctionalization of AuNCs with molecular targeting ligands that can bind to their target sites on the cancer cell membrane. Kong et al., for example, were able to image near-infrared emitting vitamin B12-R-AuNCs that were specifically internalized into Caco-2 cells by receptor-mediated endocytosis.19 Li and coworkers used a computer tomography and fluorescence imaging platform based on biofunctionalized and diatrizoic acid stabilized AuNCs to locate CL1–5 tumors in vivo. The orange-red fluorescence from AuNCs assisted in locating the tumor in mice under ultraviolet (UV) light.20
AuNCs stabilized with 11-mercaptoundecanoic acid (11-MUA), which are widely used as photochemical sensors for detecting H2O2, Cr3+, Cr6+, Hg2+, and pyrophosphatase,21–25 also show promising physiochemical characteristics for biomedical applications and cell targeting. Zhang et al., for example, synthesized AuNCs stabilized with 11-MUA and D-penicillamine (DPA) in ratios from 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :0 to 1:3, which exhibited emission wavelengths varying from 530 to 620 nm. They were able to locate 11-MUA/DPA-AuNCs attached to and internalized into HeLa cells after incubation for three hours.26 Tao et al. also applied 11-MUA-AuNCs with seven different dual-ligands for detecting targeted cancer cells with a fluorescence-based sensing platform.27 Lai and coworkers showed that 11-MUA-AuNCs with an emission wavelength of 600 nm strongly accumulate in EMT-6 and CT-26 cells without influencing the cancer cell proliferation. The localization of cancer cells loaded with 11-MUA-AuNCs in the lung and spleen tissue was possible by using just a UV lamp.28
:0 to 1:3, which exhibited emission wavelengths varying from 530 to 620 nm. They were able to locate 11-MUA/DPA-AuNCs attached to and internalized into HeLa cells after incubation for three hours.26 Tao et al. also applied 11-MUA-AuNCs with seven different dual-ligands for detecting targeted cancer cells with a fluorescence-based sensing platform.27 Lai and coworkers showed that 11-MUA-AuNCs with an emission wavelength of 600 nm strongly accumulate in EMT-6 and CT-26 cells without influencing the cancer cell proliferation. The localization of cancer cells loaded with 11-MUA-AuNCs in the lung and spleen tissue was possible by using just a UV lamp.28
Besides the outstanding and well-known biocompatibility of 11-MUA-AuNCs, no specific targeting using these clusters has been reported yet. In this work, specific interaction is achieved by biofunctionalizing 11-MUA-AuNCs with the aptamer AIR-3A that binds with a high affinity to the interleukin-6-receptor (IL-6R) expressed on the BaF3 cell membrane. We exploit the extraordinary long fluorescence lifetime of the aptamer-MUA-AuNCs (≈100 ns) compared to the short lifetime of the cellular autofluorescence (≈3 ns) to distinguish between attachment to and internalization into BaF3 cells by fluorescence lifetime imaging microscopy (FLIM). By using confocal FLIM we demonstrate binding of the biofunctionalized 11-MUA-AuNCs to cells after a short incubation time (<10 min). Internalization into the cells is detected after the cells, with aptamer-MUA-AuNCs bound to them, have been incubated for a second time without an additional supply of further AuNCs. Binding and internalization of the AuNCs are proven by evaluating 3D fluorescence stack scans throughout the cell with two different FLIM methods, gated FLIM and so-called direct FLIM, which differ in the representation of the fluorescence decay data. While the specific binding of AuNCs can be resolved by both methods, gated and direct FLIM, the specific internalization of the aptamer-MUA-AuNCs can only be visualized with the direct FLIM method. By analyzing the lifetime curves in detail, we find that the AuNCs exhibit an interaction with the cells after the second incubation step which is not present after the first incubation step. Our results show that 11-MUA-stabilized AuNCs functionalized with the AIR-3A aptamer have great potential for targeting of and selective internalization into cells.
2. Materials and methods
Chemical products were purchased from Sigma-Aldrich (Germany). Tetrakis-(hydroxy-methyl)-phosphonium chloride (THPC) was purchased from Acros (Renningen). DMEM and RPMI 1640 media were purchased from Pan Biotech (Germany). The NH2-linked and ATTO633-labeled aptamer AIR-3A was purchased from IBA (Göttingen). Ultrapure Milli-Q water was used for all syntheses and buffers.2.1. Synthesis, biofunctionalization and characterization of AuNCs
THPC-AuNCs were prepared first, followed by a ligand exchange with 11-MUA.25,29 For the THPC-AuNCs 45 mL H2O was mixed with 300 μL NaOH and a THPC solution (1 mL H2O, 18 μL 80% THPC) for 5 min. While stirring, 2 mL of a 25 mM chloroauric-acid solution was rapidly added and stirred for further 15 min. For the ligand exchange 5 mL freshly synthesized THPC-AuNCs were mixed with 1 mL borax buffer, 2 mL H2O, and 105-fold excess 11-MUA and left for three days without light. The resulting 11-MUA-AuNCs were washed several times with phosphate buffer (pH 7.8) in a centrifuge filter (7800 rpm, 10 min). The biofunctionalization of the 11-MUA-AuNCs with the aptamer AIR-3A (primary amine group at the 3′-terminus) was achieved via EDC- and NHS- mediated conjugation. The 11-MUA-AuNCs were incubated with 400 mM 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide and 100 mM N-hydroxysulfosuccinimide for 20 min. After several washing steps in a centrifuge filter (8000 rpm, 10 min), the 3′NH2-AIR-3A was cross-linked to the activated 11-MUA-AuNCs. The aptamer-AuNCs were washed again in a centrifuge filter (8000 rpm, 10 min) and stored at 4 °C.30 In both washing procedures, after each washing step, the filtrate was investigated by UV-Vis absorption spectroscopy and the washing procedure was stopped after the signal of the free educts disappeared. The aptamer-AuNC concentration was determined via inductively coupled plasma mass spectrometry (ICP-MS). Ensemble UV-Vis absorption and emission measurements of aptamer-AuNCs and 11-MUA-AuNCs were carried out with a Cary 5000 (Varian) and FluoroMax-4 (Horiba Jobin Yvon) spectrometer, respectively. Transmission electron microscopy (TEM) images were taken with the Jeol Philips CM 300. The average size of the AuNCs was determined manually by measuring 100 individual AuNCs using ImageJ.312.2. Cell culture and incubation with AuNCs
BaF3/IL6R/gp130/TNF suspension cells were grown in DMEM supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. PC3 cells were grown in RPMI 1640 medium supplemented with 10% fetal bovine serum, 100 U mL−1 penicillin and 100 μg mL−1 streptomycin. Cell cultures were kept at 37 °C in a 5% CO2 humidified incubator.For the incubation with aptamer-AuNCs/11-MUA-AuNCs each sample consisted of 5 × 106 cells which were washed three times (700 rcf, 4 °C, 4 min) with sterile PBS buffer and resuspended in 350 μL sterile PBS buffer. The aptamer-AuNCs/11-MUA-AuNCs (4.7 nM) were added to the cells, incubated for 10 min at 37 °C and again washed three times (700 rcf, 4 min). For the double incubated samples the cells were incubated with the aptamer-AuNCs/11-MUA-AuNCs for 10 min at 37 °C, washed three times, and incubated again for 10 min at 37 °C without adding AuNCs. For the confocal measurements the samples were fixed with paraformaldehyde (3.7%) for 20 min at room temperature, washed once, resuspended in H2O and dried on a substrate. For ICP-MS measurements the incubated cells were resuspended in lysis buffer (50 mM Tris-HCl, 1% Triton X-100, pH 10), sonicated and diluted with 12% HNO3 to 10 mL. The ICP-MS measurements were carried out with an Agilent 7700 ICP-MS. For the flow cytometry assay each sample consisted of 5 × 106 cells which were washed three times (700 rcf, 4 °C, 4 min) with sterile PBS buffer and resuspended in 350 μL sterile PBS buffer. ATTO633-labeled aptamer AIR-3A was added (0 and 50 pmol) and incubated for 10 min at 37 °C. After washing (three times, 700 rcf, 4 min), fluorescence intensities were determined with a FACScalibur (BD Bioscience, Germany). For the cell viability assay the BaF3 cells were diluted to 1 × 105 cells per mL with serum-free DMEM and incubated with 100 and 200 nM for 48 h. After 10 min, 24, and 48 h, living and dead cells were calculated using trypan blue and a Neubauer counting chamber.
2.3. Fluorescence lifetime imaging microscopy
FLIM measurements were performed using a home built confocal laser scanning microscope. Laser light of a pulsed laser diode (ALS, Germany) operating at 405 nm is focused on the sample using an air objective (Epiplan-Apochromat 150×, Zeiss, Germany, NA = 0.95), which is mounted on a x–y–z-piezo table (Princeton Instruments, USA) with the 200 μm × 200 μm × 20 μm scanning range. The emitted fluorescence light is collected using the same objective, filtered using a 430 nm long-pass filter and focused on a photon counting module (PerkinElmer, Canada). The movement of the piezo table is controlled by a real-time computer-controlled measurement data acquisition system (ADwin gold, Jäger Messtechnik, Germany) that is synchronized to a time-correlated single-photon counting setup (PicoHarp 300, PicoQuant, Germany) operating in the time-tagged time-resolved single photon counting mode for data acquisition. The system's time resolution was found to be 410 ps (full width at half maximum of the instrumental response function). Typical scans were carried out with 60 × 60 × 10 pixels in a sample volume of 15 μm × 15 μm × 4.5 μm, with a pulse repetition rate of 2.5 MHz and a time averaged laser power of around 12 nW. The bin time (recording time for every pixel) was set to 0.1 s. FLIM data were processed using a self-written script in MATLAB (The MathWorks Inc., USA).Confocal fluorescence emission spectra were recorded with an Andor Shamrock 303i spectrograph and an Andor iXon Ultra CCD.
3. Results and discussion
3.1. Biofunctionalized fluorescent AuNCs
We biofunctionalized 11-MUA-AuNCs, having a diameter of 1.9 nm, with the aptamer AIR-3A, which specifically binds to the IL-6R.32,33 The TEM image and size distribution of the 11-MUA-AuNCs as well as the IL-6R verification on the BaF3 cells by flow cytometry are given in the ESI in Fig. S1(a) and S2(d).† The aptamer AIR-3A was conjugated to the carboxylic group of the 11-MUA ligand by EDC and Sulfo-NHS cross-linking as described in the Materials and methods section.30Fig. 1(a) shows the UV-Vis absorption spectra recorded after each step during biofunctionalization: 11-MUA-AuNCs (blue curve), Sulfo-NHS-activated MUA-AuNCs (red curve), and aptamer-MUA-AuNCs (green curve). The chemical structures of the ligands attached to the AuNCs, corresponding to each biofunctionalization step, are shown in Fig. 1(b). All curves in Fig. 1(a) show an absorbance maximum at a wavelength of 370 nm, which is characteristic of 11-MUA-AuNCs.25 The terminal carboxylic group of the 11-MUA ligand is activated by EDC/Sulfo-NHS forming the NHS ester [see the second scheme in Fig. 1(b)]. The corresponding UV-Vis spectrum of the activated MUA-AuNCs (red curve) shows, in addition to the absorbance maximum at 370 nm, a shoulder at a wavelength of 270 nm originating from the NHS ring. Through a nucleophilic attack the NHS ester is then easily displaced and a stable amide bond is formed.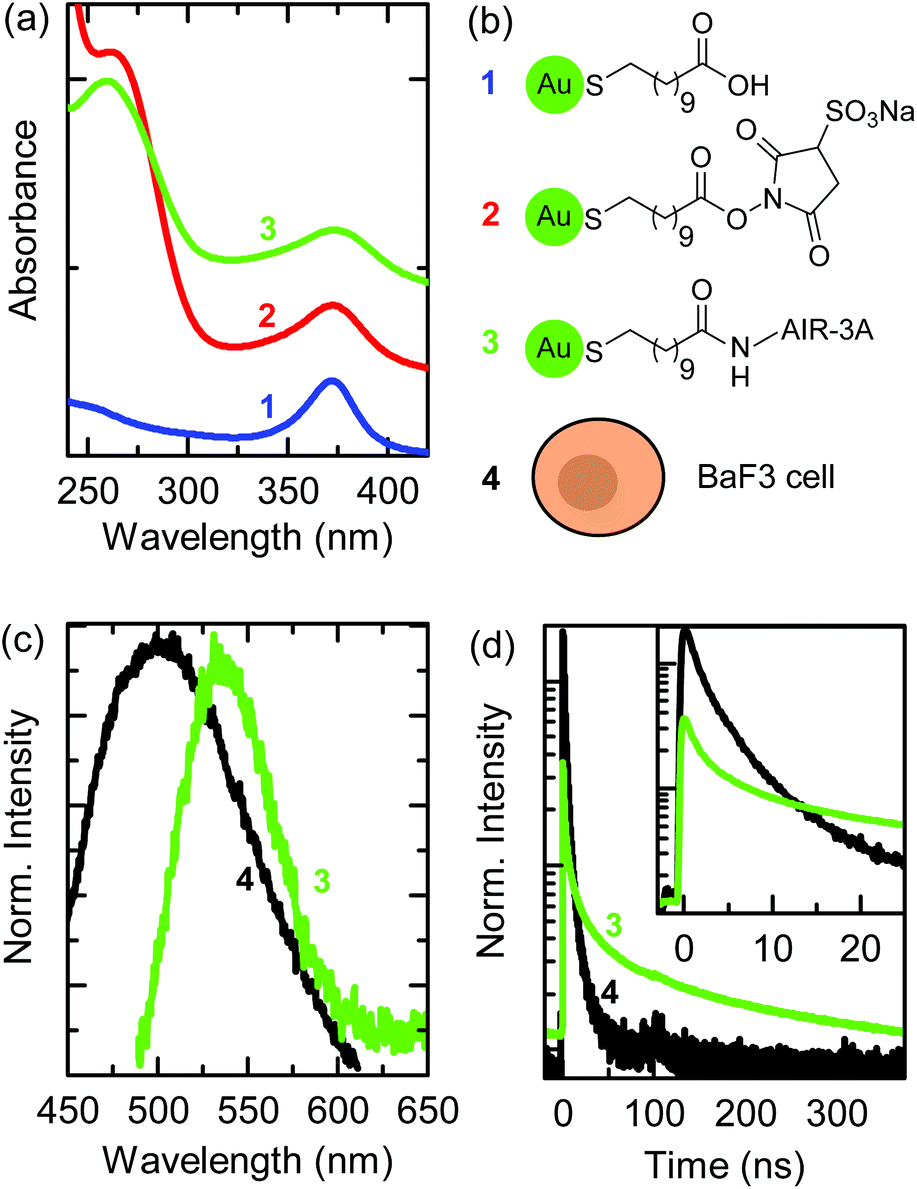 | ||
| Fig. 1 (a) UV-Vis absorption spectra of aqueous solutions during biofunctionalization of the AuNCs: 11-MUA-AuNCs (blue), EDC and Sulfo-NHS-activated MUA-AuNCs (red), and aptamer-AuNCs (green). (b) Chemical structures of AuNC ligands and illustration of a BaF3 cell. (c) Fluorescence spectra (λexc = 405 nm) of a BaF3 cell (autofluorescence, black) and of aptamer-AuNCs. (d) Fluorescence decay curves of the BaF3 cell's autofluorescence and of aptamer-AuNCs normalized to the area. The inset shows the first 20 ns of the fluorescence decay curves after excitation. Numbers that label the spectra in (a, c, and d) are linked to the labeled sketches in (b). | ||
The green curve in Fig. 1(a) shows the UV-Vis absorption spectrum after the cross-linking of the AIR-3A aptamer to the MUA ligand. The absorption shoulder at a wavelength of 270 nm of the Sulfo-NHS ester vanished and the characteristic absorption maximum for RNA sequences appeared at 260 nm.34 The difference in the wavelength of the absorption feature of the Sulfo-NHS ester (shoulder in spectrum 2) and of the aptamer (peak in spectrum 3) is small but sufficient to prove the successful biofunctionalization. The biofunctionalized AIR-3A-MUA-AuNCs are called aptamer-AuNCs in the following.
Fig. 1(c) shows the fluorescence spectra of aptamer-AuNCs (green curve) and the autofluorescence of a BaF3 cell (black curve). The aptamer-AuNCs display a fluorescence peaking at a wavelength of 535 nm, which is similar to unfunctionalized 11-MUA-AuNCs (see Fig. S1(b)†). The fluorescence emission strongly overlaps with the autofluorescence spectrum of BaF3 cells making it difficult to distinguish between emission from the aptamer-AuNCs and the cells.
Fig. 1(d) displays the fluorescence decay curves of the aptamer-AuNCs (green) and a BaF3 cell (black). The fluorescence decay of aptamer-AuNCs exhibits an average lifetime of around 110 ns while the autofluorescence only shows average lifetimes between 2 and 3 ns. This significant difference in lifetime enables us to distinguish between the autofluorescence of the cell and the emission of the AuNCs by FLIM. In contrast to common fluorescence microscopy, where different fluorophores are distinguished by their emission wavelengths, FLIM produces images encoding different arrival times of photons after pulsed laser excitation.35,36
3.2. Selective attachment during the first incubation
For a specific interaction we incubated BaF3 cells with aptamer-AuNCs at 37 °C for 10 min. Dark field and scanning electron microscopy images as well as the atomic force microscopy cross section of a BaF3 cell are given in ESI Fig. S2.† Also, we verified the biocompatibility of the 11-MUA-AuNCs by a simple cell viability assay (see ESI, Fig. S1(c)†). The preparation procedure is shown in Fig. 2(a), more details are given in the Materials and methods section. In a previous work, we found that an incubation time of 10 min showed the highest specificity for 7 nm-sized aptamer-biofunctionalized, non-fluorescent gold nanoparticles for the IL-6R expressed on the BaF3 cells.37 The investigation by ICP-MS did not allow for the localization of the nanoparticles. | ||
| Fig. 2 Sketches of (a) the incubation process of BaF3 cells with aptamer-AuNCs and (b) the three measured axial (z) levels within the cell. BaF3 cells were incubated for 10 min at 37 °C with aptamer-AuNCs, then washed three times to remove any excess of AuNCs, and fixed. (c) Gated and (d) direct FLIM images of a BaF3 cell with aptamer-AuNCs at axial levels z = 0.0, 1.0, and 3.0 μm. (e) Fluorescence decay curves corresponding to the areas encircled in (d) (1–3; orange, red, blue) fitted by decay curves (grey) calculated by superimposing the autofluorescence curve (black) and fluorescence curve of aptamer-AuNCs (green) with adjusted weighting ratios. Scale bars are 2 μm. | ||
Fig. 2(c) and (d) show the FLIM images of a BaF3 cell incubated with aptamer-AuNCs after washing and fixation. Exemplarily, the FLIM images for the three axial levels of 0.0, 1.0, and 3.0 μm above the substrate are shown. The heights are chosen to be on the substrate, within the cell, and within the upper part of the cell, respectively, as shown in Fig. 2(b). The images of all 10 stack scans are given in the ESI in Fig. S3.†
For FLIM we measure pixelwise single BaF3 cells confocally and record in each pixel the fluorescence decay curve. The exact setup is described in the Materials and methods section. To analyze a data set we utilize two different methods that lead to gated and direct FLIM images. Before discussing the resulting FLIM images in Fig. 2(c) and (d), we want to describe both methods briefly.
In the case of gated FLIM the signal of every pixel in our confocal scan is temporally filtered using a certain time gate. This means the confocal image is built up only by photon counts of a specific part of the fluorescence decay curve. As addressed in Fig. 1(d), it becomes obvious that in the first 30 ns after excitation the decay curve consists of mixed signals from the fluorescence of the cell and the AuNCs, whereas the last part of the decay curve contains almost exclusively photons of the fluorescence of the AuNCs. Based on the decay curves, two gates were chosen as follows: in the short timeframe all photon counts between 0 ns and 25 ns are considered. For the normalized decay curves in Fig. 1(d) already 95% of the photons of the cell's autofluorescence (black curve) are in the short timeframe, while for the fluorescence of the aptamer-AuNCs (green curve) only 27% of all photons are emitted in this period. Due to the presence of both fluorescence types shortly after excitation, the second timeframe is more important for the localization of AuNCs. Here, choosing a gate between 50 and 400 ns leads to a collection of around 70% of the photons of the fluorescence of AuNCs while less than 5% of the photons of the autofluorescence are detected. Photons passing through the corresponding gates are counted for each pixel resulting in two different intensity maps.
In contrast, the direct FLIM method leads to one image per scan, but each image point contains two pieces of information. The first information is the overall number of photons counted at a particular position. This overall intensity is encoded into the brightness of a direct FLIM image. The second information is the time after which 50% of the overall number of photons of a certain image point is emitted. This information is a measure of the fluorescence lifetime. It is encoded into the color of a direct FLIM image. The direct FLIM method shows to be very robust in the case of low photon counts in the decay curve, which is typical of either low emission intensities or short integration times, e.g., when performing fast image acquisitions.
In Fig. 2(c) the gated FLIM images with the short timeframe are dominated by signals of a blurred circular shape typical of the autofluorescence of the BaF3 cell (images of a cell without any AuNCs are given in the ESI in Fig. S4† for comparison). Additionally, sharp features are visible. These features become very prominent in the gated FLIM images with the long timeframe while the blurred circular-shaped intensity distribution of the auto-fluorescence is suppressed and only remains as a background signal. Obviously, with the long timeframe only a few photons of the autofluorescence are detected. The high-intensity spots in the gated FLIM images with the long timeframe clearly prove the signals arising from the long-lived fluorescence of the aptamer-AuNCs. Importantly, this already demonstrates the successful binding of the AuNCs to the cell. We also performed control measurements similar to the ones shown in Fig. 2(c): (i) BaF3 cells incubated with 11-MUA-AuNCs without aptamer functionalization and (ii) PC3 control cells incubated with aptamer-AuNCs. In both cases, the long-gated FLIM images do not show any sharp features, proving a strong specificity of the aptamer-AuNCs (see ESI, Fig. S5 and S6†). Regarding the spatial distribution of sharp features in the long-gated images, we find that they are typically localized in patches around those areas that exhibit strong signals in short-gated images (see Fig. 2(c) for z = 1.0 μm), basically representing autofluorescence emission from the cell. Sharp features in the center part of the cell only appear for larger z values, as can be observed in Fig. 2(c) for z = 3.0 μm.
Fig. 2(d) shows the direct FLIM images that were obtained from the same data set as the gated FLIM images. The total intensity in every pixel is encoded into the brightness whereas a red-to-green color gradient represents the lifetime, as can be seen from the corresponding color map depicted on the right side of Fig. 2(d).
The direct FLIM images clearly show the combination of the two main aspects observed in the gated FLIM images: the rounded shape of the signals from the autofluorescence of the BaF3 cell and the sharp detailed features from the fluorescence of the AuNCs. With the chosen colormap the cell itself is displayed in red, which corresponds to lifetimes of around 3 ns or less. The dominant sharp features appear as green-colored spots, representing lifetimes of 6 ns or longer, with a high intensity. In addition, yellow-colored parts in the image (e.g., the spot in the center part of the direct FLIM image for z = 1.0 μm) indicate lifetimes around 4.5 ns.
Note that in our depiction of direct FLIM images, the lower border of the lifetime is set to 3 ns, corresponding to the autofluorescence of the cell. The higher border has to be optimized in terms of the highest contrast between the signals from different parts of the image. In the direct FLIM images in Fig. 2(d) this results in a cut-off lifetime of 6 ns above which all lifetimes are encoded green.
Both the above-mentioned control measurements with non-functionalized 11-MUA-AuNCs and with the PC3 cells reveal no AuNC signal also in direct FLIM images (Fig. S5 and S6†). Thus, also the direct FLIM method confirms the high specificity of the aptamer-AuNCs to the IL-6R expressed on the BaF3 cells. Furthermore, the spatial distribution of the AuNC fluorescence signals in the direct FLIM images in Fig. 2(d) indicates that (i) aptamer-AuNCs are attached as islands of many AuNCs to the BaF3 cells and (ii) they seem to be mainly attached to the surface of the cells.
Concerning (i) we performed additional reference measurements. To clarify that the sharp features do not arise from single AuNCs we investigated individualized AuNCs. Combining confocal fluorescence measurements with atomic force microscopy and element specific scanning electron microscopy we could estimate that the detection limit lies above a few hundreds of AuNCs in one patch (see ESI Fig. S7†). Although we were not able to quantify the intensity of individual AuNCs, this proves that the sharp features arise from many AuNCs. This conclusion is also supported by ICP-MS measurements. We determined the amount of gold to be 1.04 × 106 AuNCs per cell, assuming a AuNC diameter of 1.9 nm. To illustrate the meaning of this number, we also calculated the fraction of the cell surface that would be covered with AuNCs if all these AuNCs would be arranged in a monolayer on top of the cell surface. We found this value to be 7.3% assuming a cell with a diameter of 10 μm, which is a reasonable result with respect to the FLIM images in Fig. 2(c) and (d).
Regarding (ii), our results strongly imply a specific binding of aptamer-AuNCs to the surface of BaF3 cells after the first incubation step: the sharp features that are dominant in gated FLIM images with a long timeframe and appear green in direct FLIM images are always located around the region of strong autofluorescence, the latter visible only in gated FLIM images with a short timeframe and colored red in the direct FLIM. If the patches of AuNCs were located inside the cell, they should become visible for intermediate z-positions and disappear for higher z-positions. At the same time autofluorescence of the cell should be present at lower and higher z-positions than the position of AuNCs. We would also expect that the color in the direct FLIM images of AuNCs inside a cell should be different from AuNCs on the cell membrane since the emission of the AuNCs inside the cell is mixed with a higher amount of autofluorescence.
With gated and direct FLIM we are able to visualize the specific binding of biofunctionalized AuNCs to the IL-6R expressed on BaF3 cells after only 10 min at 37 °C. Since no internalized AuNCs could be observed in gated and direct FLIM images, we introduced a second incubation step and investigated further.
3.3. Internalization after a second incubation
In order to address internalization of AuNCs in more detail we expanded the above-described experiment by a further incubation step. Fig. 3(a) schematically shows the preparation procedure. Principally, the first steps of the preparation were the same as for the single incubated samples: after incubation and washing, which removes unbound aptamer-AuNCs, the majority of the AuNCs is attached to the cell membrane and (nearly) no AuNCs are internalized into the cell. In the next step we incubated again for 10 min at 37 °C, but without adding aptamer-AuNCs. The idea behind this is that the previously attached aptamer-AuNCs might get internalized as shown in Fig. 3(a).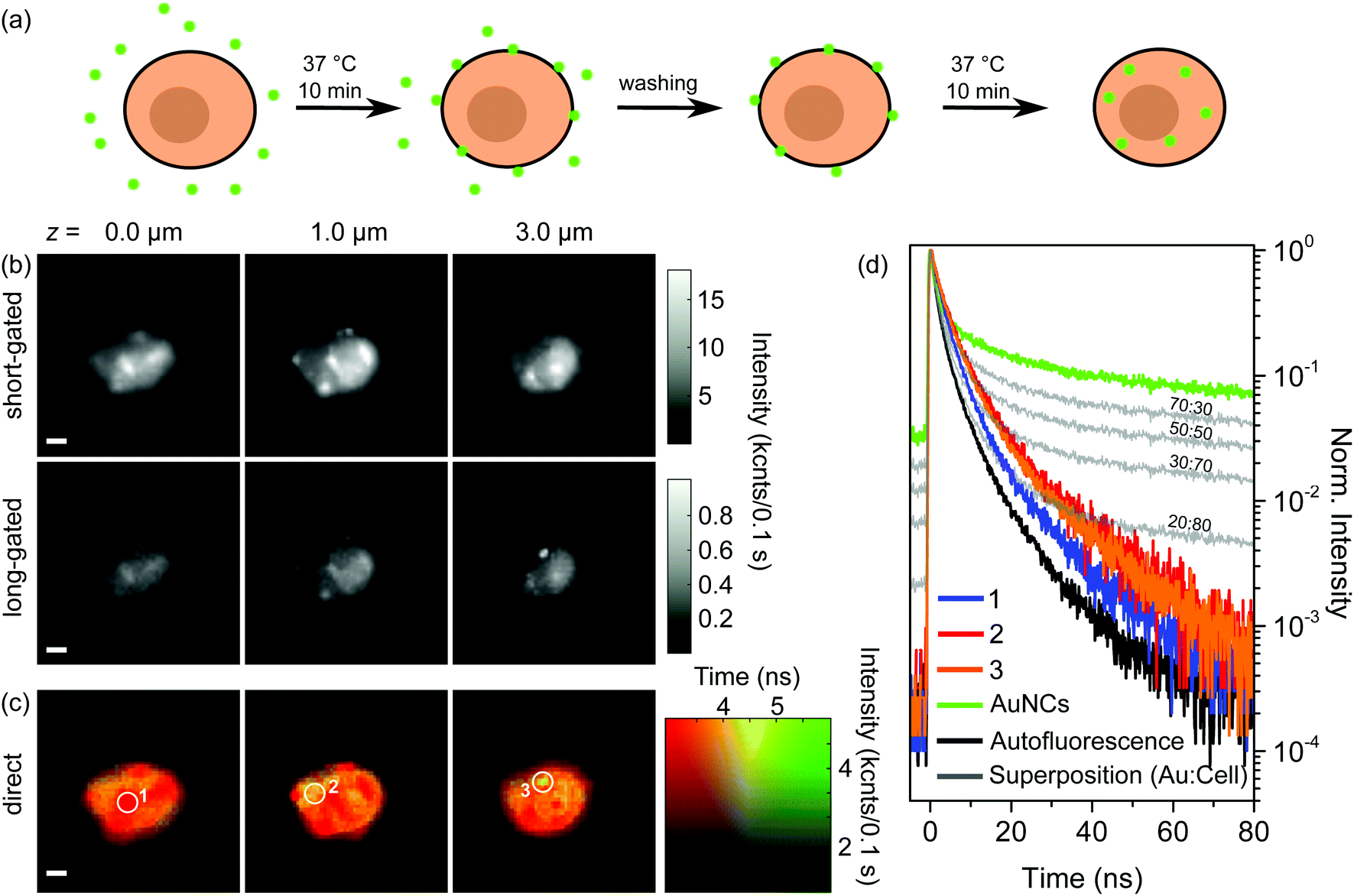 | ||
| Fig. 3 (a) Sketches of the incubation process of BaF3 cells with aptamer-AuNCs. BaF3 cells were incubated for 10 min at 37 °C in a 4.7 nM aptamer-AuNC solution, washed three times to remove any excess of AuNCs, and then again incubated for 10 min at 37 °C without any addition of aptamer-AuNCs. (b) Gated and (c) direct FLIM images of two times incubated BaF3 cell aptamer-AuNCs at axial z = 0.0, 1.0, and 3.0 μm. (d) Fluorescence decay curves corresponding to the encircled areas in (c) (1–3, orange, red, blue) compared to decay curves (grey) calculated by superimposing the autofluorescence curve (black) and fluorescence curve of aptamer-AuNCs (green) with adjusted weighting ratios. Scale bar 2 μm. | ||
In Fig. 3(b) and (c) we show the results of gated and direct FLIM measurements of a BaF3 cell after the second incubation step. The three heights above the substrate, the particular timeframes for gated FLIM images, and the time and intensity scaling for direct FLIM images were chosen to be the same as for Fig. 2(c) and (d). The FLIM images of all measured heights (10 levels between 0.0 and 4.5 μm) are shown in the ESI in Fig. S8.†
In the FLIM images with short timeframes in Fig. 3(b) we find again the blurred circular-shaped pattern typical of the autofluorescence signal of the cell. Additionally, a few sharp features of AuNCs are visible, similar to the situation in Fig. 2(c). In contrast, some of the features also appear in the center part of the cell (cf.Fig. 2(c) and 3 (b) at z = 1.0 μm). In gated FLIM images with a long gate the sharp features do not become as prominent as in the corresponding Fig. 2(c). Moreover, the overall intensity is strongly reduced. Note that the intensity range is scaled down by a factor of more than 15 compared to the image with a short timeframe. Only for a height of 3.0 μm, which corresponds to the top of the cell, a sharp feature becomes apparent. In the gated FLIM images, we observe no attached aptamer-AuNCs except for the one spot on top of the cell. Also, we cannot identify any internalized AuNCs. Consequently, from the gated FLIM images in Fig. 3(b) we are not able to make a statement concerning the localization of the AuNCs.
An explanation for not being able to localize the AuNCs could be the detachment of AuNCs from the IL-6R during the second incubation step. As a control experiment we performed ICP-MS measurements, similar to already described above. Instead of 1.04 × 106 AuNCs per cell that we received for the single incubation, we obtained a value of 0.73 × 106 AuNCs per cell. This means that instead of 16% of the total amount of gold, we still measure 11% after the second incubation step. The ICP-MS investigation shows that the vast majority of aptamer-AuNCs are still on or in the cells. An extensive detachment of AuNCs as being the reason for the absence of the sharp features in long-gated FLIM images can be excluded. The slightly decreased amount of the AuNCs after the second incubation step can easily be explained by the additional washing step and exocytosis. Hence, aptamer-AuNCs are still on and in the cells. Another scenario that could explain the absence of the sharp features could be that aptamer-AuNCs are still bound to the cell membrane while they lost their fluorescence properties due to chemical degradation during the second incubation step. We reviewed this possibility with control experiments: exposing aptamer-AuNCs for 20 min at 37 °C just in buffer or with cells in a single incubation step still showed fluorescence behaviour. Furthermore, since the treatment of aptamer-AuNCs and cells during the second incubation step is the same as during the first incubation step we can exclude a chemical degradation during the second incubation step.
Summarizing the results for the gated FLIM images, we can only indirectly conclude that a considerable part of the previously attached aptamer-AuNCs has been internalized during the second incubation step, but a direct proof is missing. Fig. 3(c) shows the direct FLIM images. The first and most important observation is that in the direct FLIM representation, instead of predominant sharp green features found after the first incubation step, most of the cells are now colored orange, indicating lifetimes of around 4.5 ns. Furthermore, the orange-colored regions are found for all heights. Only a few parts of the cell show a red color indicating dominant autofluorescence with a lifetime of about 3 ns or less. Interestingly, these regions coincide with sharp features visible in the gated FILM images, in clear contrast to the images after the first incubation step, where sharp features were colored green. Only a single small green spot is visible for a height of 3.0 μm (white circle), coinciding with the sharp feature prominent in the FLIM image with a long timeframe at z = 3.0 μm.
The first fundamental conclusion from observing an orange color in direct FLIM images is that aptamer-AuNCs are still emitting fluorescence. Furthermore, since we observe orange color for all heights, we conclude that aptamer-AuNCs have to be inside the cell. This is direct proof of successful internalization of aptamer-AuNCs into the BaF3 cells, which is not visible in the gated FLIM images. The orange color appears in rather large regions of the cell. This shows that the AuNCs are not strongly localized in only a few regions inside the cell. Based on our results we propose the following course of events: during the first incubation step, islands of aptamer-AuNCs are specifically bound to the surface of the cells (visible as green spots in direct FLIM images) while nearly no or only a few AuNCs are internalized into the cell. During the second incubation step, the previously bound patches are internalized into the cell. We expect the AuNCs to be internalized by receptor-mediated endocytosis.32,33,38 One might speculate that the AuNCs are located in many endosomes as well as lysosomes with their spatial distribution being connected to the orange-colored regions observed in the direct FLIM images. Instead of internalization, a few of the bound AuNCs could also have detached from the IL-6R and eventually bound again to another receptor (which could explain the small green spot on top of the cell in Fig. 3(c)).
Summarizing the different FLIM methods, we find that gated FLIM was not capable of giving direct proof of the internalization of AuNCs into the cell, while the direct FLIM method is. It is worth mentioning again that both FLIM methods rely on the very same data set that is differently expressed.
3.4. Cell–AuNC interaction
We now want to analyze the lifetime data for selected points of FLIM images in more detail to gain further insight into the optical properties of AuNCs either attached to or internalized into the cells. Fig. 2(e) shows the decay curves of the selected positions marked by white circles in the direct FLIM images in Fig. 2(d), measured after the first incubation step. They are shown together with decay curves of the fluorescence of aptamer-AuNCs alone (green) and BaF3 cells alone (black). By superimposing both latter decay curves with different weighting-factor ratios, we fitted the decay curves of the selected position.The fit curves are depicted in grey together with the weighting factor ratios. We observe excellent agreement between the measured and fitted curves, which indicates that the signals after the first incubation step are just different superpositions of signals from AuNC emission and autofluorescence. Fig. 3(d) shows the decay curves of the selected positions marked by white circles in the direct FLIM images in Fig. 3(c), measured after the second incubation step. Here, the curves are compared to selected decay curves that result from superpositions of the aptamer-AuNC and cell fluorescence decays with different weighting factors. These curves are again depicted in grey together with the weighting factor ratios. It can clearly be observed that the decay curves of the selected positions cannot be fitted by a superposition of pure aptamer-AuNC and autofluorescence emission. The decays are much faster than the aptamer-AuNC emission but also slower than the autofluorescence.
We conclude that, unlike in the case of AuNCs attached to the cell, the fluorescence-decay dynamics of AuNCs changes when they are internalized into the cell. This means that there is an interaction between AuNCs and the cell, which does not take place when AuNCs are just attached to the cell.
As a last point we want to discuss the nature of the observed interaction. One explanation could be that a change of the pH value inside the endosomes during endocytosis leads to the shortening of the fluorescence lifetime. This was already reported by Shang et al. for dihydrolipoic acid stabilized AuNCs.39 We performed test measurements on 11-MUA-AuNCs at different pH values that only showed a weak influence of the pH value on the fluorescence lifetime, much less than that observed here after the second incubation step (see ESI, Fig. S9†). Thus, we can exclude this explanation. Another possible explanation of the fluorescence lifetime shortening of aptamer-AuNCs could be the Förster resonance energy transfer (FRET) effect. Here, after internalization, biomolecules that cause autofluorescence, like flavin adenine dinucleotide (FAD),36 could come in close proximity (in the order of 10 nm) to the AuNCs and act as acceptors for an energy transfer from the aptamer-AuNCs. Thus, the long lifetime of the AuNC emission could be shortened by the extra decay channel opened by the FRET effect. Other possibilities for the change in the lifetime are photon reabsorption and quenching.36 Further experiments would be necessary to unambiguously clarify the exact mechanisms, which is not in the scope of this paper. Nevertheless, the onset of the interaction of aptamer-AuNCs with the cell after the second incubation step is an interesting topic for future investigations. It also strongly supports the observed differentiation of specific attachment during the first incubation step and the internalization during the second incubation step.
4. Conclusions
In conclusion, we fabricated fluorescing aptamer-AuNCs that specifically bind to the IL-6R on the membrane of BaF3 cells. By applying a two-step incubation procedure we could show that during the first incubation step AuNCs specifically bind to the surface of the cells, whereas already attached AuNCs get internalized into the cells during the second incubation step in the absence of further AuNCs. After the first incubation step aptamer-AuNCs specifically bound in patches of many AuNCs on the cell membrane. The patches are identified as sharp intense features in gated FLIM images with a long timeframe, whereas in direct FLIM images, sharp features with a longer fluorescence lifetime become visible. After the second incubation step the patches disappeared and instead the whole cell volume is filled with AuNCs. In direct FLIM images this is visualized as an intermediate lifetime in all heights of the confocal scan while the presence of the AuNCs cannot be verified by the gated FLIM method. This demonstrates the high sensitivity of the direct FLIM method to small and widely spread amounts of AuNCs. We also investigated the fluorescence decay curves in detail after the two incubation steps. Here, we found that after the first incubation step the decays are superpositions of pure AuNC emission and autofluorescence, whereas after the second incubation step the decay curves cannot be fitted as a superposition any more. We attribute this to an interaction of AuNCs with biomolecules inside the cell that is not possible when AuNCs are only attached to the surface.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work has been supported by the excellence cluster ‘The Hamburg Centre for Ultrafast Imaging – Structure, Dynamics and Control of Matter at the Atomic Scale’ of the Deutsche Forschungsgemeinschaft via EXC1074.References
- L. Polavarapu, M. Manna and Q.-H. Xu, Nanoscale, 2011, 3, 429–434 RSC.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, Am. Chem. Soc., 2013, 7, 2509–2521 CAS.
- I. Chakraborty and T. Pradeep, Chem. Rev., 2017, 117, 8208–8271 CrossRef CAS PubMed.
- P. Bian, J. Zhou, Y. Liu and Z. Ma, Nanoscale, 2013, 5, 6161 RSC.
- A. Cantelli, G. Battistelli, G. Guidetti, J. Manzi, M. Di Giosia and M. Montalti, Dyes Pigm., 2016, 135, 64–79 CrossRef CAS.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 5–8 Search PubMed.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed.
- J. Sun and Y. Jin, J. Mater. Chem. C, 2014, 2, 8000–8011 RSC.
- M. S. Devadas, J. Kim, E. Sinn, D. Lee, T. G. Iii and G. Ramakrishna, J. Phys. Chem. C, 2010, 114, 22417–22423 CrossRef CAS.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- H. Häkkinen, Nat. Chem., 2012, 4, 443–455 CrossRef PubMed.
- N. Goswami, K. Zheng and J. Xie, Nanoscale, 2014, 6, 13328–13347 RSC.
- E. Oh, F. K. Fatemi, M. Currie, J. B. Delehanty, T. Pons, A. Fragola, S. Lévêque-Fort, R. Goswami, K. Susumu, A. L. Huston and I. L. Medintz, Part. Part. Syst. Charact., 2013, 30, 453–466 CrossRef CAS.
- S. Roy, A. Baral, R. Bhattacharjee, B. Jana, A. Datta, S. Ghosh and A. Banerjee, Nanoscale, 2015, 7, 1912–1920 RSC.
- X. D. Zhang, Z. Luo, J. Chen, X. Shen, S. Song, Y. Sun, S. Fan, F. Fan, D. T. Leong and J. Xie, Adv. Mater., 2014, 26, 4565–4568 CrossRef CAS PubMed.
- S. Chattoraj, A. Amin, B. Jana, S. Mohapatra, S. Ghosh and K. Bhattacharyya, ChemPhysChem, 2016, 17, 253–259 CrossRef CAS PubMed.
- T. D. Fernández, J. R. Pearson, M. P. Leal, M. J. Torres, M. Blanca, C. Mayorga and X. Le Guével, Biomaterials, 2015, 43, 1–12 CrossRef PubMed.
- Y. Kong, J. Chen, F. Gao, R. Brydson, B. Johnson, G. Heath, Y. Zhang, L. Wu and D. Zhou, Nanoscale, 2013, 5, 1009–1017 RSC.
- C.-H. Li, T.-R. Kuo, H.-J. Su, W.-Y. Lai, P.-C. Yang, J.-S. Chen, D.-Y. Wang, Y.-C. Wu and C.-C. Chen, Sci. Rep., 2015, 5, 15675 CrossRef CAS PubMed.
- H. C. Chang, Y. F. Chang, N. C. Fan and J. A. A. Ho, ACS Appl. Mater. Interfaces, 2014, 6, 18824–18831 CrossRef CAS PubMed.
- J. Sun, F. Yang, D. Zhao and X. Yang, Anal. Chem., 2014, 86, 7883–7889 CrossRef CAS PubMed.
- J. Sun, J. Zhang and Y. Jin, J. Mater. Chem. C, 2013, 1, 138–143 RSC.
- H.-Y. Chang, H.-T. Chang, Y.-L. Hung, T.-M. Hsiung, Y.-W. Lin and C.-C. Huang, RSC Adv., 2013, 3, 4588 RSC.
- C. C. Huang, Z. Yang, K. H. Lee and H. T. Chang, Angew. Chem., Int. Ed., 2007, 46, 6824–6828 CrossRef CAS PubMed.
- Z. Zhang, L. Xu, H. Li and J. Kong, RSC Adv., 2013, 3, 59–63 RSC.
- Y. Tao, M. Li and D. T. Auguste, Biomaterials, 2017, 116, 21–33 CrossRef CAS PubMed.
- S.-F. Lai, C.-C. Chien, W.-C. Chen, H.-H. Chen, Y.-Y. Chen, C.-L. Wang, Y. Hwu, C. Yang, C. Chen, K. Liang, C. Petibois, H.-R. Tan, E.-S. Tok and G. Margaritondo, Biotechnol. Adv., 2013, 31, 362–368 CrossRef CAS PubMed.
- D. G. Duff, A. Baiker and P. P. Edwards, J. Chem. Soc., Chem. Commun., 1993, 272, 96 RSC.
- O. C. Farokhzad, J. Cheng, B. A. Teply, I. Sherifi, S. Jon, P. W. Kantoff, J. P. Richie and R. Langer, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6315–6320 CrossRef CAS PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- C. Meyer, K. Eydeler, E. Magbanua, T. Zivkovic, N. Piganeau, I. Lorenzen, J. Grötzinger, G. Mayer, S. Rose-John and U. Hahn, RNA Biol., 2012, 9, 67–80 CrossRef CAS PubMed.
- C. Meyer, K. Berg, K. Eydeler-Haeder, I. Lorenzen, J. Grötzinger, S. Rose-John and U. Hahn, RNA Biol., 2014, 11, 57–65 CrossRef CAS PubMed.
- A. Fleck and D. Begg, Biochim. Biophys. Acta, Nucleic Acids Protein Synth., 1965, 108, 333–339 CrossRef CAS.
- W. Becker, J. Microsc., 2012, 247, 119–136 CrossRef CAS PubMed.
- M. M. Y. Berezin and S. Achilefu, Chem. Rev., 2010, 110, 2641–2684 CrossRef CAS PubMed.
- L. Prisner, N. Bohn, U. Hahn and A. Mews, Nanoscale, 2017, 9, 14486–14498 RSC.
- S. Kruspe, C. Meyer and U. Hahn, Mol. Ther.–Nucleic Acids, 2014, 2, e143 CrossRef PubMed.
- L. Shang, L. Yang, H. Wang and G. U. Nienhaus, Small, 2016, 12, 868–873 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C8NR06639F |
| This journal is © The Royal Society of Chemistry 2018 |