
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceQuantitative determination of a model organic/insulator/metal interface structure†
Martin
Schwarz
a,
David A.
Duncan
 b,
Manuela
Garnica‡
a,
Jacob
Ducke
a,
Peter S.
Deimel
a,
Pardeep K.
Thakur
b,
Tien-Lin
Lee
b,
Francesco
Allegretti
*a and
Willi
Auwärter
*a
b,
Manuela
Garnica‡
a,
Jacob
Ducke
a,
Peter S.
Deimel
a,
Pardeep K.
Thakur
b,
Tien-Lin
Lee
b,
Francesco
Allegretti
*a and
Willi
Auwärter
*a
aPhysics Department, Technical University of Munich, 85748 Garching, Germany. E-mail: wau@tum.de; francesco.allegretti@tum.de
bDiamond Light Source, Harwell Science and Innovation Campus, Didcot OX11 0DE, UK
First published on 16th November 2018
Abstract
By combining X-ray photoelectron spectroscopy, X-ray standing waves and scanning tunneling microscopy, we investigate the geometric and electronic structure of a prototypical organic/insulator/metal interface, namely cobalt porphine on monolayer hexagonal boron nitride (h-BN) on Cu(111). Specifically, we determine the adsorption height of the organic molecule and show that the original planar molecular conformation is preserved in contrast to the adsorption on Cu(111). In addition, we highlight the electronic decoupling provided by the h-BN spacer layer and find that the h-BN–metal separation is not significantly modified by the molecular adsorption. Finally, we find indication of a temperature dependence of the adsorption height, which might be a signature of strongly-anisotropic thermal vibrations of the weakly bonded molecules.
Two-dimensional (2D) epitaxial materials such as graphene and hexagonal boron nitride (h-BN) have increasingly been employed as decoupling layers in recent years, as they can template the adsorption and steer the self-assembly of atoms, molecules and clusters.1–3 Research in this field is driven by both the ambition to tune the electronic properties of the 2D materials, e.g. via a periodic potential modulation by molecular superstructures,4–8 or charge transfer doping,9–13 as well as an interest in the characterization of fundamental intrinsic properties of adsorbates without the often appreciable perturbation induced by the underlying metal substrate.1,14–18 In particular, the strong hybridization of molecular orbitals with metal states can be prevented on h-BN,19–22 similar to other insulating spacer layers,23–26 which effectively decouple the adsorbate from the substrate.
The adsorption geometry of organic molecules on metal substrates was subject to various experimental and theoretical studies in the last decade.27–30 In particular, atomic force microscopy and the X-ray standing wave (XSW) technique have proven their potential to yield a detailed and quantitative structure determination, including molecular adsorbates.31–35 However, on metal-supported h-BN only few experimental studies address the geometric interface structure of adsorbates,36,37 and an approach to obtain quantitative information on the molecular conformation of large organic molecules on h-BN is still lacking. This is surprising in view of the manifold studies addressing the electronic properties of organic/h-BN/metal systems.18,20,21,37–39 A precise knowledge of the geometric structure of the organic/h-BN/metal interface is crucial to elucidate the physical properties of the hybrid system. Moreover, this information is highly relevant for benchmarking the various density functional theory (DFT) calculations and molecular dynamics simulations investigating adsorbates on h-BN sheets.10,40–49 Additionally, the effect that adsorbed metal–organic molecules exert on the 2D layer has yet to be addressed experimentally on the atomic scale.8
Herein, we report the first quantitative determination of the adsorption height and the conformation of an organic molecular adsorbate on an h-BN monolayer. We choose the tetrapyrrole compound cobalt porphine (Co–P, see inset of Fig. 1c) on h-BN/Cu(111) as a model system to study the adsorption geometry and the interaction of the molecule with the substrate in a combined experimental approach based on X-ray photoelectron spectroscopy (XPS), XSW, and scanning tunneling microscopy (STM). The geometric structure of both pristine h-BN and Co–P on Cu(111) have been separately determined in a quantitative fashion in prior works.50,51 The h-BN/Cu interface is characterized by coexisting moiré-like superstructures with periodicities ranging from 1 to 15 nm, providing hexagonal arrays of so-called pores separated by wire regions.50,52 The h-BN layer was found to display an average adsorption height of 3.38 ± 0.04 Å and exhibits a moderate geometric corrugation of 0.3–0.7 Å.50 Co–P molecules on Cu(111) were determined to adsorb in a single site, with the Co atom bridging surface Cu atoms at a height of 2.25 ± 0.04 Å.51 The molecular backbone of the Co–P molecule on Cu(111) presents an asymmetric saddle-shape conformation as well as a strong interaction between the Co atom and the copper substrate. The new results presented here indicate that photoemission-based techniques are capable of characterizing adsorbates on insulating epitaxial monolayers quantitatively and shine a light on the degree of interfacial interaction still present in those systems. Furthermore, we highlight a temperature-dependence of the adsorption height of the molecule on h-BN/Cu(111). Knowledge of the interfacial structure of semiconductor/insulator/metal junction is essential for applications in future nanoscale hybrid devices. For example, the self-assembly of magnetic molecules into ordered arrays and their decoupling from the metallic substrate are potentially relevant in the fields of data storage and spintronics.53,54
 | ||
| Fig. 1 Co–P on h-BN/Cu(111). STM images at 6 K display the preferred adsorption of Co–P molecules in the pores of the h-BN layer on Cu(111) for coverages of (a) ∼0.15 ML, and (b) ∼0.3 ML. (c) The XP spectrum of the Co 2p3/2 core level (recorded at 50 K, coverage ∼0.15 ML) exhibits a complex line shape due to the occurrence of multiplet splitting and a satellite structure. Scan parameters: (a) Ub = 1.0 V, It = 41 pA, (b) Ub = 1.23 V, It = 120 pA. The inset shows a structural model of the Co–P molecule (gray: carbon, blue: nitrogen, orange: cobalt, and white: hydrogen). | ||
STM images, taken at 6 K, for Co–P molecules adsorbed on monolayer h-BN on Cu(111) show an adsorption behavior in regular quasi-hexagonal arrays, which follows the moiré-like superstructure. Specifically, we identify preferential adsorption starting in the pores of the h-BN layer (Fig. 1a and b), which were determined in our prior study to be closer to the metal substrate and present a lower local work function than the wire regions.50,52 Site-selective adsorption was observed previously and assigned to the electrostatic potential landscape of the h-BN/metal interface.19,55 Depending on the moiré size and coverage, mainly single molecules (Fig. 1a) or aggregates (Fig. 1b) can be trapped inside the pores. Increasing deposition of molecules finally leads to a densely packed, uniform coverage across the whole h-BN layer (Fig. S1†). The latter defines one monolayer (ML) of molecules (∼0.75 molecules per nm2) according to the convention adopted here.
Scanning tunneling spectroscopy (STS) measurements on Co–P/h-BN/Cu(111) (Fig. S2b† and ref. 56) only show Co-related states above the Fermi level on any region of the h-BN moiré, clearly refuting the idea of a charge transfer to the Co–P molecules, in agreement with a report on Co-metalated functionalized porphyrins.57 Notably, this observation is in contrast to Co-Phthalocyanine on h-BN/Ir(111),18,20 where a shift of the Co resonance below the Fermi level (i.e. a charging of the molecule) has been observed for molecules adsorbed in the pores. However, the position of the Co-related unoccupied resonance does vary as a function of the lateral position of the Co–P molecule on the h-BN moiré (Fig. S2c†).
In both the low (∼0.15 ML) and the high (∼0.95 ML) Co–P coverage regimes, the Co 2p3/2 core-level line in XPS exhibits a composite structure comprising three features, which are labelled Co1, Co2, and Co3. An example of these spectra, measured at low coverage and 50 K is shown in Fig. 1c (a comparable spectrum for the high coverage regime and at 300 K is shown in Fig. S3†). The observed line shape is characteristic of a Co2+ ion in metal complexes.58–60 Specifically, the two features at the lowest binding energy (780.3 eV and 781.1 eV) are assigned to multiplet splitting due to the coupling of the core hole, left by the photoemission process, to unpaired electrons in the Co 3d shell. The highest binding energy feature (Co3, Eb = 782.7 eV), is assigned to a shake-up satellite in agreement with previous reports.58–61 A similar deconvolution into three components has also been proposed for the Fe 2p3/2 core level of Fe2+ ions in porphyrins and phthalocyanines.59,62,63
The XPS binding energy of the Co1 component (780.3 eV) is significantly greater than that found for Co–P/Cu(111) (778.2 eV),51 Co-TPP/Ag(111) (778.2 eV)61,64 and metallic Co (778.0 eV).65 The binding energy of Co–P/Cu(111) was ascribed to adsorption-related charge transfer from the copper substrate into the Co ion.51 Therefore, the significant increase in binding energy here and the observed line shape are both indicative of a +2 oxidation state and signal a dramatic decrease in any charge transfer from the substrate. Furthermore, the Co 2p binding energy of Co1 is very similar to that reported for a multilayer of Co-TPP (780.5 eV), where the thickness of the organic layer attenuates the substrate-induced core-hole screening effects and the charge transfer. Note also that the C 1s and N 1s XP spectra (Fig. S4†) both present a smaller upshift in binding energy of 0.4 eV, compared to Co-P/Cu(111),51 suggesting that the charge transfer into the molecule directly on the metal surface primarily affects the Co center (as discussed in our prior study).51 The XPS data are summarized in the Table S1 in the ESI.†
Roughly the same line shape is observed in both the low and high coverage regimes (Fig. 1c compared to Fig S3†), albeit with a different intensity ratio of Co2 to Co1. Specifically, at a coverage of ∼0.15 ML the Co2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :Co1 ratio was ∼2:5, while at a coverage of ∼0.95 ML it increased to ∼2:3. Although this change in the relative peak intensity might suggest coverage-related adsorption sites, this interpretation is refuted by the XSW analysis presented below. Moreover, despite the origin of the effect being unclear, coverage-dependent variations of the intensity ratio in the multiplet structure have been previously reported.58,59,62 No differences were observed, instead, in the Co2:Co1 ratio as a function of temperature (50 K to 300 K).
:Co1 ratio was ∼2:5, while at a coverage of ∼0.95 ML it increased to ∼2:3. Although this change in the relative peak intensity might suggest coverage-related adsorption sites, this interpretation is refuted by the XSW analysis presented below. Moreover, despite the origin of the effect being unclear, coverage-dependent variations of the intensity ratio in the multiplet structure have been previously reported.58,59,62 No differences were observed, instead, in the Co2:Co1 ratio as a function of temperature (50 K to 300 K).
The XSW data obtained from the photoelectron yield of the integrated area of the Co 2p3/2 core level (comprising both multiplet features Co1 and Co2) for the low coverage regime at 300 K are shown in Fig. 2a, and the corresponding profiles from the N 1s, C 1s and B 1s core levels are shown in Figs. S5.† The coherent fractions and positions, obtained from fitting these data, are summarized in Table 1. Notably, the Co 2p line shape in XPS was not observed to vary through the standing wave condition (Fig. S6 and Table S2†), demonstrating that the two components Co1 and Co2 most likely originate from multiplet splitting and thus are related to the same, single Co species exhibiting a well-defined adsorption height. Therefore, in the following, the integrated intensity from Co1 and Co2 is considered.
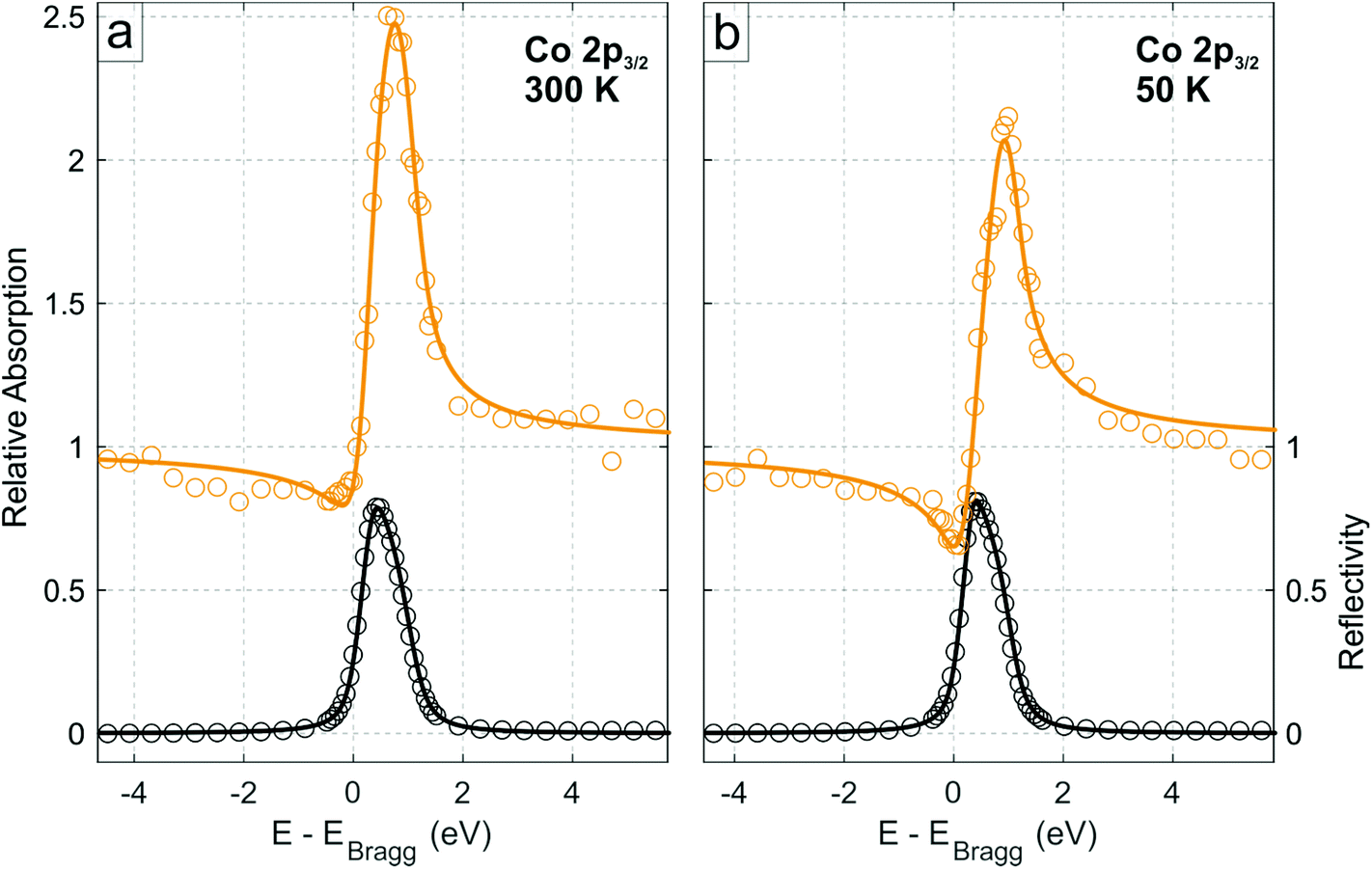 | ||
| Fig. 2 Normal incidence XSW Co 2p3/2 absorption profiles of Co–P on h-BN/Cu(111) in the low coverage regime (∼0.15 ML) at the (111) Bragg reflection, acquired at (a) 300 K and (b) 50 K, respectively, integrated over both the Co1 and Co2 species (see Fig. 1c). Solid lines are fits to the data. Black data points are the reflectivity curve. | ||
111, the coherent position p111 (in units of the (111) interplanar distance of Cu(111)) and the mean adsorption height ![[h with combining macron]](https://www.rsc.org/images/entities/i_char_0068_0304.gif) for all atomic species in the Co–P molecule and the h-BN layer
for all atomic species in the Co–P molecule and the h-BN layer
| Core-level | Low coverage, 300 K | Low coverage, 50 K | |||||
|---|---|---|---|---|---|---|---|
|
f111 |
p 111 | Adsorption height (Å) |
f111 |
p 111 | Adsorption height (Å) |
||
| Co–P | C 1s (C–C) | 0.32 ± 0.04 | 0.13 ± 0.02 | 6.53 ± 0.04 | 0.49 ± 0.04 | 0.06 ± 0.02 | 6.37 ± 0.04 |
| C 1s (C–N) | 0.37 ± 0.04 | 0.10 ± 0.02 | 6.47 ± 0.04 | 0.53 ± 0.04 | 0.05 ± 0.02 | 6.35 ± 0.04 | |
| N 1s (NCo–P) | 0.37 ± 0.08 | 0.12 ± 0.03 | 6.51 ± 0.06 | 0.47 ± 0.05 | 0.00 ± 0.04 | 6.24 ± 0.08 | |
| Co 2p (Co1 + Co2) | 0.58 ± 0.04 | 0.09 ± 0.02 | 6.45 ± 0.04 | 0.67 ± 0.04 | 0.97 ± 0.02 | 6.18 ± 0.04 | |
| h-BN | B 1s (B0) | 0.69 ± 0.04 | 0.63 ± 0.02 | 3.40 ± 0.04 | 0.58 ± 0.04 | 0.58 ± 0.02 | 3.29 ± 0.04 |
| N 1s (N0) | 0.69 ± 0.02 | 0.61 ± 0.01 | 3.36 ± 0.02 | 0.58 ± 0.02 | 0.58 ± 0.01 | 3.29 ± 0.02 | |
The boron nitride layer, when compared to our prior results,50 is found to be at the same height above the Cu(111) surface as without adsorbed Co–P (with/without Co–P: 3.39 ± 0.04/3.40 ± 0.04 Å for B and 3.37 ± 0.04/3.36 ± 0.02 Å for N), and only a minor decrease in the coherent fraction of the two species is observed. The Co center is found to be 6.45 ± 0.04 Å above the Cu(111) surface, i.e. 3.07 ± 0.05 Å above the average h-BN position, with a coherent fraction that is slightly lower than that of the h-BN layer underneath (Fig. 3). The molecular backbone of Co–P (Fig. S2a†) appears to be significantly more planar than on the bare Cu(111) surface.51 Specifically, no difference is observed in the coherent fractions between the N atoms and the C–N carbon atoms in Co–P, whereas on the bare Cu(111) surface a difference of 0.19 ± 0.05 was found.51 For Co–P on bare Cu(111), the Co center was found to lie below the organic backbone of the molecule.51 Similarly, such a displacement may also be present when adsorbed on the h-BN layer, but it is less than the precision of the results presented here (0.06 ± 0.07 Å). The adsorption height of the molecule relative to the average h-BN layer position (3.07 ± 0.06 Å [Co] – 3.15 ± 0.06 Å [C–C]) is significantly less than the summation of the van der Waals radii of Co–B (4.3 Å) and C–B (3.7 Å), indicating that the Co–P is not completely free-standing on the h-BN. However, the increased planarity of the molecule and the still significantly larger adsorption heights do suggest that any interaction between the h-BN and the Co–P is weak. Interestingly, a selective decoupling of non-planar Co-TPP molecules adsorbed on a Cu3N spacer layer on Cu(110) has been reported, featuring strong interaction of the Co center with the support highlighted by a short Co–N distance (2.72 Å).66
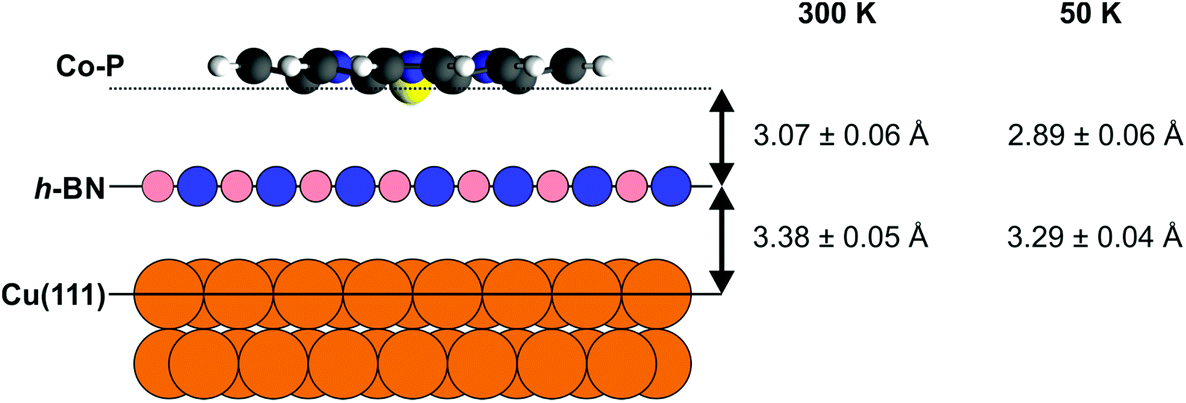 | ||
| Fig. 3 Schematic model of the Co–P molecule on h-BN/Cu(111) with the indicated average adsorption heights of the h-BN layer and the Co center for low coverage (∼0.15 ML) at 300 K and at 50 K, respectively. Note that the vertical corrugation of the h-BN layer was not considered in the sketch and only the average position is taken. | ||
The XSW results for the high coverage regime (shown in Fig. S7,† coherent fractions and heights in Table S3†) again indicate no effect on the h-BN layer from Co–P adsorption, and suggest a highly planar Co–P molecule. However, a subtle increase in adsorption height of the whole molecule is observed (an average increase of 0.11 ± 0.03 Å across all four species) and a dramatic decrease in the coherent fraction of the Co ion (0.27 ± 0.06).
Finally, XSW measurements were performed on the low coverage phase at 50 K (Fig. 2b and Fig. S8,† the deduced coherent fractions and positions are summarized in Table 1), in an attempt to measure a regime that is more similar to that under which the STM measurements were performed. As can be seen in the comparison between the 300 K and 50 K Co 2p XSW measurements in Fig. 2, a significant difference in the maximum photoemission yield of the Co 2p3/2 during XSW measurements is observed. Specifically the adsorption height of the Co ions, with respect to the Cu(111) surface, has decreased by 0.27 ± 0.06 Å (∼30 times greater than the thermal expansion of Cu over the same temperature range), with a similar reduction in adsorption height for all species in the molecule. The h-BN layer itself is also reduced in adsorption height by 0.09 ± 0.04 Å (thus the height of the Co above the h-BN layer is reduced by 0.18 ± 0.07 Å). A similar difference is observed by cooling the high coverage phase to 200 K (Table S3†).
One potential explanation for the difference in adsorption height as a function of temperature and coverage could be found in the presented STM measurements (Fig. 1a). At 6 K, the STM measurements indicate that the Co–P island growth starts on the pore areas of the h-BN, which have been previously determined to lie closer to the substrate than the rest of the h-BN layer.50 Obviously, at higher coverage the molecule cannot only occupy the pores, thus a slight increase in adsorption height is not surprising. Equally, the rather large adsorption height between the Co–P and the h-BN (≥2.89 Å) is suggestive of a weak bonding of the molecule (although the preferential adsorption in the pores still indicates an effect of the corrugated surface potential) and thus it is likely that, at 300 K, the molecule is highly mobile on the surface. In other words, at least at low coverage, by cooling the sample to 50 K (and to a lesser extent, 200 K) we are potentially quenching the mobility of the Co–P on the surface and “freezing” out the molecule into the pores, which results in a lower adsorption height (Fig. 3).
Another, more likely argument that could explain, at least in part, the observed difference as a function of the temperature are anisotropic thermal vibrations. As the Co–P molecule is clearly weakly bound, but not free-standing, it is feasible that an increase in adsorption height only results in a small attractive force pulling it back towards the equilibrium height, but that a similar decrease in adsorption height could result in a strong repulsive force pushing it back, i.e. a strongly asymmetrical Lennard-Jones potential. Therefore, although the modal adsorption height at both temperatures may be the same, the mean adsorption height, which is what is measured by XSW, may vary. Certainly, the coherent fraction of the molecule increases significantly (≥0.09 ± 0.06 for all species) when cooling to 50 K, which could indicate a notable decrease in the amplitude of the thermal vibrations. Notably, no conclusive effect on the adsorption height was detected in more strongly bonded porphyrins on Cu(111).67 These finite temperature effects, on the mean adsorption height, are very intriguing, as quantitative structural parameters are a popular benchmark for DFT calculations,29,31,68 but DFT calculations are, by their nature, performed for the system at 0 K. Addressing this so-called “finite temperature effect” has become of increasing interest over the last decade,33,69 in order to provide DFT formalizations that yield more realistic results.
Previous DFT calculations of organic molecules adsorbed on a free-standing monolayer of h-BN have predicted significantly larger adsorption heights compared to those measured experimentally here, e.g.: benzene/h-BN (3.29 Å),44 borazine/h-BN (3.24 Å),44 TCNQ/h-BN (3.49 Å),10 functionalized porphyrins/h-BN (3.37 Å).41 Even DFT calculations including the metal support (Phthalocyanines on h-BN/Rh(111), ∼3.25 Å)55 yielded adsorption heights exceeding our experimental values. Thus, all predicted adsorption heights are greater than the experimentally measured adsorption height above the h-BN layer. However, it is important to note that in our prior work50 we observed a corrugation in the h-BN layer (∼0.4 Å) and that our STM images (Fig. 1) indicate a preferred Co–P adsorption in the pore regions of the h-BN layer. Yet, even were the Co–P molecules solely adsorbed in these low-lying sites, only the room temperature measurements would show a good agreement with theory – the adsorption height at 50 K would still be far closer to the h-BN layer than expected from these 0 K calculations. Although we cannot address whether the difference is due to the specificity of the adsorbed molecular species,70 a general underestimation of the molecule/h-BN interaction strength, or the influence of the underlying support,71 this discrepancy calls for further systematic experimental and theoretical studies on the effect of both support and temperature.
In summary, based on the STM observations we propose a Co–P island formation starting on the pore areas of the h-BN layer, in line with reports of free-base porphine (2H-P) molecules on h-BN/Cu(111).19 We suggest that this growth mode is due to the previously reported electronic surface potential modulation of the moderately corrugated h-BN layer.50,52 The binding energy of the Co 2p XP spectra signals little, if any, direct electronic influence of the h-BN or the underlying Cu(111) surface on the Co–P molecule. The XSW measurements indicate a more planar adsorption of the Co–P molecule, than is observed on the bare Cu(111) surface,51 and an adsorption height that exhibits a pronounced temperature dependence and suggests weak physisorption. The observed adsorption height (3.07 ± 0.06 Å) is significantly shorter than the associated van der Waals radii and thus, although the interaction between the molecule and the h-BN is generally weak, the Co–P does not form a fully free-standing molecular layer, which is further evidenced by the aforementioned preference for adsorbing in the pores of the h-BN. Nonetheless, the suppression of the charge transfer from the Cu(111) surface into the Co–P molecule and the increased molecular adsorption height suggest that a monolayer of h-BN act as a suitable buffer layer between organic adsorbates and metal substrates. This is important as in numerous studies the metal substrates have been observed to quench the many interesting electronic and chemical properties of, e.g., organic metal complexes.72–74 Furthermore, the minimization of the influence of the metal substrate suggests that the h-BN monolayer could well be used as a powerful model system for insulating substrates – which are typically challenging to measure with electron-based techniques.
Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work is supported by the European Research Council Consolidator Grant NanoSurfs (No. 615233) and the Munich-Centre for Advanced Photonics (MAP). We thank Johannes V. Barth for fruitful discussions. M. G. would like to acknowledge funding by the H2020-MSCA-IF-2014 program under GA no. 658070 (2DNano). W. A. acknowledges funding by the Deutsche Forschungsgemeinschaft via a Heisenberg professorship. We thank Diamond Light Source for the award of beam time and funding (SI14624-2).References
- A. Kumar, K. Banerjee and P. Liljeroth, Nanotechnology, 2017, 28, 82001 CrossRef PubMed.
- J. M. MacLeod and F. Rosei, Small, 2014, 10, 1038–1049 CrossRef CAS PubMed.
- J. Yang, K. Kim, Y. Lee, K. Kim, W. C. Lee and J. Park, FlatChem, 2017, 5, 50–68 CrossRef CAS.
- C.-H. Park, L. Yang, Y.-W. Son, M. L. Cohen and S. G. Louie, Nat. Phys., 2008, 4, 213–217 Search PubMed.
- S. Wickenburg, J. Lu, J. Lischner, H.-Z. Tsai, A. A. Omrani, A. Riss, C. Karrasch, A. Bradley, H. S. Jung, R. Khajeh, D. Wong, K. Watanabe, T. Taniguchi, A. Zettl, A. H. C. Neto, S. G. Louie and M. F. Crommie, Nat. Commun., 2016, 7, 13553 Search PubMed.
- M. Gobbi, S. Bonacchi, J. X. Lian, Y. Liu, X.-Y. Wang, M.-A. Stoeckel, M. A. Squillaci, G. D'Avino, A. Narita, K. Müllen, X. Feng, Y. Olivier, D. Beljonne, P. Samorì and E. Orgiu, Nat. Commun., 2017, 8, 14767 Search PubMed.
- J. Cervenka, A. Budi, N. Dontschuk, A. Stacey, A. Tadich, K. J. Rietwyk, A. Schenk, M. T. Edmonds, Y. Yin, N. Medhekar, M. Kalbac and C. I. Pakes, Nanoscale, 2015, 7, 1471–1478 RSC.
- Q. Cai, A. Du, G. Gao, S. Mateti, B. C. C. Cowie, D. Qian, S. Zhang, Y. Lu, L. Fu, T. Taniguchi, S. Huang, Y. Chen, R. S. Ruoff and L. H. Li, Adv. Funct. Mater., 2016, 26, 8202–8210 CrossRef CAS.
- B. Cai, S. Zhang, Z. Yan and H. Zeng, ChemNanoMat, 2015, 1, 542–557 CrossRef CAS.
- Q. Tang, Z. Zhou and Z. Chen, J. Phys. Chem. C, 2011, 115, 18531–18537 CrossRef CAS.
- W. Chen, S. Chen, D. C. Qi, X. Y. Gao and A. T. S. Wee, J. Am. Chem. Soc., 2007, 129, 10418–10422 CrossRef CAS PubMed.
- C. Coletti, C. Riedl, D. S. Lee, B. Krauss, L. Patthey, K. von Klitzing, J. H. Smet and U. Starke, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 235401 CrossRef.
- R. Voggu, B. Das, C. S. Rout and C. N. R. Rao, J. Phys.: Condens. Matter, 2008, 20, 472204 CrossRef.
- J. Uihlein, M. Polek, M. Glaser, H. Adler, R. Ovsyannikov, M. Bauer, M. Ivanovic, A. B. Preobrajenski, A. V. Generalov, T. Chassé and H. Peisert, J. Phys. Chem. C, 2015, 119, 15240–15247 CrossRef CAS.
- S. Kahle, Z. Deng, N. Malinowski, C. Tonnoir, A. Forment-Aliaga, N. Thontasen, G. Rinke, D. Le, V. Turkowski, T. S. Rahman, S. Rauschenbach, M. Ternes and K. Kern, Nano Lett., 2012, 12, 518–521 CrossRef CAS PubMed.
- P. Erler, P. Schmitt, N. Barth, A. Irmler, S. Bouvron, T. Huhn, U. Groth, F. Pauly, L. Gragnaniello and M. Fonin, Nano Lett., 2015, 15, 4546–4552 CrossRef CAS PubMed.
- S. Bose, A. M. García-García, M. M. Ugeda, J. D. Urbina, C. H. Michaelis, I. Brihuega and K. Kern, Nat. Mater., 2010, 9, 550–554 CrossRef CAS PubMed.
- F. Schulz, M. Ijäs, R. Drost, S. K. Hämäläinen, A. Harju, A. P. Seitsonen and P. Liljeroth, Nat. Phys., 2015, 11, 229–234 Search PubMed.
- S. Joshi, F. Bischoff, R. Koitz, D. Écija, K. Seufert, A. P. Seitsonen, J. Hutter, K. Diller, J. I. Urgel, H. Sachdev, J. V. Barth and W. Auwärter, ACS Nano, 2014, 8, 430–442 CrossRef CAS PubMed.
- F. Schulz, R. Drost, S. K. Hämäläinen and P. Liljeroth, ACS Nano, 2013, 7, 11121–11128 CrossRef CAS PubMed.
- L. Liu, T. Dienel, R. Widmer and O. Gröning, ACS Nano, 2015, 9, 10125–10132 CrossRef CAS PubMed.
- R. Forker, T. Dienel, A. Krause, M. Gruenewald, M. Meissner, T. Kirchhuebel, O. Gröning and T. Fritz, Phys. Rev. B, 2016, 93, 165426 CrossRef.
- C. F. Hirjibehedin, C. P. Lutz and A. J. Heinrich, Science, 2006, 312, 1021–1024 CrossRef CAS PubMed.
- J. Repp, G. Meyer, S. M. Stojkovic, A. Gourdon and C. Joachim, Phys. Rev. Lett., 2005, 94, 026803 CrossRef PubMed.
- J. Cho, J. Smerdon, L. Gao, O. Süzer, J. R. Guest and N. P. Guisinger, Nano Lett., 2012, 12, 3018–3024 CrossRef CAS PubMed.
- J. Repp, G. Meyer, F. E. Olsson and M. Persson, Science, 2004, 305, 493–495 CrossRef CAS PubMed.
- C. Bürker, N. Ferri, A. Tkatchenko, A. Gerlach, J. Niederhausen, T. Hosokai, S. Duhm, J. Zegenhagen, N. Koch and F. Schreiber, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 165443 CrossRef.
- W. Auwärter, F. Klappenberger, A. Weber-Bargioni, A. Schiffrin, T. Strunskus, C. Wöll, Y. Pennec, A. Riemann and J. V. Barth, J. Am. Chem. Soc., 2007, 129, 11279–11285 CrossRef PubMed.
- F. Albrecht, F. Bischoff, W. Auwärter, J. V. Barth and J. Repp, Nano Lett., 2016, 16, 7703–7709 CrossRef CAS PubMed.
- F. S. Tautz, Prog. Surf. Sci., 2007, 82, 479–520 CrossRef CAS.
- B. Schuler, W. Liu, A. Tkatchenko, N. Moll, G. Meyer, A. Mistry, D. Fox and L. Gross, Phys. Rev. Lett., 2013, 111, 106103 CrossRef PubMed.
- C. Stadler, S. Hansen, F. Pollinger, C. Kumpf, E. Umbach, T.-L. Lee and J. Zegenhagen, Phys. Rev. B: Condens. Matter Mater. Phys., 2006, 74, 35404 CrossRef.
- G. Mercurio, R. J. Maurer, W. Liu, S. Hagen, F. Leyssner, P. Tegeder, J. Meyer, A. Tkatchenko, S. Soubatch, K. Reuter and F. S. Tautz, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 88, 35421 CrossRef.
- C. Stadler, S. Hansen, I. Kröger, C. Kumpf and E. Umbach, Nat. Phys., 2009, 5, 153–158 Search PubMed.
- B. Stadtmüller, D. Lüftner, M. Willenbockel, E. M. Reinisch, T. Sueyoshi, G. Koller, S. Soubatch, M. G. Ramsey, P. Puschnig, F. S. Tautz and C. Kumpf, Nat. Commun., 2014, 5, 3685 Search PubMed.
- M. Ohtomo, Y. Yamauchi, X. Sun, A. A. Kuzubov, N. S. Mikhaleva, P. V. Avramov, S. Entani, Y. Matsumoto, H. Naramoto and S. Sakai, Nanoscale, 2017, 9, 2369–2375 RSC.
- M. L. Ng, A. B. Preobrajenski, A. A. Zakharov, A. S. Vinogradov, S. A. Krasnikov, A. A. Cafolla and N. Mårtensson, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 115449 CrossRef.
- M. Muntwiler, W. Auwärter, A. P. Seitsonen, J. Osterwalder and T. Greber, Phys. Rev. B: Condens. Matter Mater. Phys., 2005, 71, 121402 CrossRef.
- C.-A. Palma, S. Joshi, T. Hoh, D. Écija, J. V. Barth and W. Auwärter, Nano Lett., 2015, 15, 2242–2248 CrossRef CAS PubMed.
- T. Dienel, J. Gomez Diaz, A. P. Seitsonen, R. Widmer, M. Iannuzzi, K. Radican, H. Sachdev, K. Müllen, J. Hutter and O. Gröning, ACS Nano, 2014, 8, 6571–6579 CrossRef CAS PubMed.
- V. V. Korolkov, S. A. Svatek, A. Summerfield, J. Kerfoot, L. Yang, T. Taniguchi, K. Watanabe, N. R. Champness, N. A. Besley and P. H. Beton, ACS Nano, 2015, 9, 10347–10355 CrossRef CAS PubMed.
- J.-H. Lee, Y.-K. Choi, H.-J. Kim, R. H. Scheicher and J.-H. Cho, J. Phys. Chem. C, 2013, 117, 13435–13441 CrossRef CAS.
- Y. S. Al-Hamdani, M. Ma, D. Alfè, O. A. von Lilienfeld and A. Michaelides, J. Chem. Phys., 2015, 142, 181101 CrossRef PubMed.
- V. Caciuc, N. Atodiresei, M. Callsen, P. Lazić and S. Blügel, J. Phys.: Condens. Matter, 2012, 24, 424214 CrossRef PubMed.
- Y.-X. Yu, J. Mater. Chem. A, 2014, 2, 8910–8917 RSC.
- X. Chen, S. Jia, N. Ding, J. Shi and Z. Wang, Environ. Sci.: Nano, 2016, 3, 1493–1503 RSC.
- Y. Zhang, J. Qiao, S. Gao, F. Hu, D. He, B. Wu, Z. Yang, B. Xu, Y. Li, Y. Shi, W. Ji, P. Wang, X. Wang, M. Xiao, H. Xu, J.-B. Xu and X. Wang, Phys. Rev. Lett., 2016, 116, 16602 CrossRef PubMed.
- A. Matković, J. Genser, D. Lüftner, M. Kratzer, R. Gajić, P. Puschnig and C. Teichert, Sci. Rep., 2016, 6, 38519 CrossRef PubMed.
- Y. Zhao, Q. Wu, Q. Chen and J. Wang, J. Phys. Chem. Lett., 2015, 6, 4518–4524 CrossRef CAS PubMed.
- M. Schwarz, A. Riss, M. Garnica, J. Ducke, P. S. Deimel, D. A. Duncan, P. K. Thakur, T.-L. Lee, A. P. Seitsonen, J. V. Barth, F. Allegretti and W. Auwärter, ACS Nano, 2017, 11, 9151–9161 CrossRef CAS PubMed.
- M. Schwarz, M. Garnica, D. A. Duncan, A. Pérez Paz, J. Ducke, P. S. Deimel, P. K. Thakur, T.-L. Lee, A. Rubio, J. V. Barth, F. Allegretti and W. Auwärter, J. Phys. Chem. C, 2018, 122, 5452–5461 CrossRef CAS.
- S. Joshi, D. Écija, R. Koitz, M. Iannuzzi, A. P. Seitsonen, J. Hutter, H. Sachdev, S. Vijayaraghavan, F. Bischoff, K. Seufert, J. V. Barth and W. Auwärter, Nano Lett., 2012, 12, 5821–5828 CrossRef CAS PubMed.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef CAS PubMed.
- B. D. Terris and T. Thomson, J. Phys. D: Appl. Phys., 2005, 38, R199–R222 CrossRef CAS.
- M. Iannuzzi, F. Tran, R. Widmer, T. Dienel, K. Radican, Y. Ding, J. Hutter and O. Gröning, Phys. Chem. Chem. Phys., 2014, 16, 12374–12384 RSC.
- J. Ducke, A. Riss, A. Pérez Paz, K. Seufert, M. Schwarz, M. Garnica, A. Rubio and W. Auwärter, ACS Nano, 2018, 12, 2677–2684 CrossRef CAS PubMed.
- J. I. Urgel, M. Schwarz, M. Garnica, D. Stassen, D. Bonifazi, D. Écija, J. V. Barth and W. Auwärter, J. Am. Chem. Soc., 2015, 137, 2420–2423 CrossRef CAS PubMed.
- Y. Bai, M. Sekita, M. Schmid, T. Bischof, H.-P. Steinrück and J. M. Gottfried, Phys. Chem. Chem. Phys., 2010, 12, 4336–4344 RSC.
- L. Massimi, M. Angelucci, P. Gargiani, M. G. Betti, S. Montoro and C. Mariani, J. Chem. Phys., 2014, 140, 244704 CrossRef PubMed.
- G. Mette, D. Sutter, Y. Gurdal, S. Schnidrig, B. Probst, M. Iannuzzi, J. Hutter, R. Alberto and J. Osterwalder, Nanoscale, 2016, 8, 7958–7968 RSC.
- T. Lukasczyk, K. Flechtner, L. R. Merte, N. Jux, F. Maier, J. M. Gottfried and H.-P. Steinrück, J. Phys. Chem. C, 2007, 111, 3090–3098 CrossRef CAS.
- M. Schmid, J. Zirzlmeier, H.-P. Steinrück and J. M. Gottfried, J. Phys. Chem. C, 2011, 115, 17028–17035 CrossRef CAS.
- C. Isvoranu, B. Wang, K. Schulte, E. Ataman, J. Knudsen, J. N. Andersen, M. L. Bocquet and J. Schnadt, J. Phys.: Condens. Matter, 2010, 22, 472002 CrossRef CAS PubMed.
- W. Auwärter, K. Seufert, F. Klappenberger, J. Reichert, A. Weber-Bargioni, A. Verdini, D. Cvetko, M. Dell'Angela, L. Floreano, A. Cossaro, G. Bavdek, A. Morgante, A. P. Seitsonen and J. V. Barth, Phys. Rev. B: Condens. Matter Mater. Phys., 2010, 81, 245403 CrossRef.
- N. S. McIntyre and M. G. Cook, Anal. Chem., 2002, 47, 2208–2213 CrossRef.
- V. C. Zoldan, R. Faccio, C. Gao and A. A. Pasa, J. Phys. Chem. C, 2013, 117, 15984–15990 CrossRef CAS.
- C. Bürker, A. Franco-Cañellas, K. Broch, T.-L. Lee, A. Gerlach and F. Schreiber, J. Phys. Chem. C, 2014, 118, 13659–13666 CrossRef.
- R. J. Maurer, V. G. Ruiz, J. Camarillo-Cisneros, W. Liu, N. Ferri, K. Reuter and A. Tkatchenko, Prog. Surf. Sci., 2016, 91, 72–100 CrossRef CAS.
- R. J. Maurer, W. Liu, I. Poltavsky, T. Stecher, H. Oberhofer, K. Reuter and A. Tkatchenko, Phys. Rev. Lett., 2016, 116, 146101 CrossRef PubMed.
- G. Kim, S. C. Jung and Y.-K. Han, Curr. Appl. Phys., 2013, 13, 2059–2063 CrossRef.
- Y. Ding, M. Iannuzzi and J. Hutter, J. Phys. Chem. C, 2011, 115, 13685–13692 CrossRef CAS.
- W. Auwärter, D. Écija, F. Klappenberger and J. V. Barth, Nat. Chem., 2015, 7, 105–120 CrossRef PubMed.
- J. M. Gottfried, Surf. Sci. Rep., 2015, 70, 259–379 CrossRef CAS.
- D. A. Duncan, P. S. Deimel, A. Wiengarten, R. Han, R. G. Acres, W. Auwärter, P. Feulner, A. C. Papageorgiou, F. Allegretti and J. V. Barth, Chem. Commun., 2015, 51, 9483–9486 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, additional STM, XPS and XSW data. See DOI: 10.1039/c8nr06387g |
| ‡ Current address: Instituto Madrileño de Estudios Avanzados en Nanociencia (IMDEA-Nanociencia), Cantoblanco, 28049 Madrid, Spain |
| This journal is © The Royal Society of Chemistry 2018 |