
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The potential of magnetic hyperthermia for triggering the differentiation of cancer cells
Sandhya
Moise
 *ab,
James M.
Byrne
c,
Alicia J.
El Haj
bd and
Neil D.
Telling
b
*ab,
James M.
Byrne
c,
Alicia J.
El Haj
bd and
Neil D.
Telling
b
aDepartment of Chemical Engineering, University of Bath, Bath BA2 7AY, UK. E-mail: s.moise@bath.ac.uk
bInstitute for Science and Technology in Medicine (ISTM), Keele University, Stoke-on-Trent ST4 7QB, UK
cCentre for Applied Geoscience (ZAG), University of Tuebingen, Tuebingen 72076, Germany
dHealthcare Technologies Institute, University of Birmingham, Heritage building Edgbaston, West Midlands B15 2TT, UK
First published on 29th October 2018
Abstract
Magnetic hyperthermia is a potential technique for cancer therapy that exploits heat generated by magnetic nanoparticles to kill cancerous cells. Many studies have shown that magnetic hyperthermia is effective at killing cancer cells both in vitro and in vivo, however little attention has been paid to the cellular functioning of the surviving cells. We report here new evidence demonstrating the onset of thermally triggered differentiation in osteosarcoma cancer cells that survive magnetic hyperthermia treatment. This raises the possibility that in addition to causing cell death, magnetic hyperthermia could induce surviving cancer cells to form more mature cell types and thereby inhibit their capacity to self-renew. Such processes could prove to be as important as cell death when considering magnetic hyperthermia for treating cancer.
Magnetic nanoparticles (MNPs) can remotely transduce external field energy into thermal energy. This property of the MNPs to induce local heating under the influence of an alternating magnetic field, known as magnetic hyperthermia, has been exploited in various biomedical applications.1–3 In particular magnetic hyperthermia is being explored as a method for treating tumours in cancer therapy. By functionalizing the MNP surface with cancer-cell specific antigens and by applying a suitable high frequency alternating magnetic field (typically 50–1000 kHz) to the region of interest, it is possible to precisely target and eliminate cancer cells without affecting healthy tissue.4,5
At temperatures higher than 41 °C, the survival rate of mammalian cells decreases with increasing exposure time.6,7 Magnetic hyperthermia adopts such an approach and triggers heat-related cell death to eliminate cells within a cancerous tumour. While much current research is focussing on developing advanced particles with excellent heating properties and studying their effect on cell viability following hyperthermia,8,9 there is very little understanding of other responses of the cancer cells to the treatment.
Chemical, spatial and physical cues, including temperature have been shown to affect cellular behaviour. For instance, such cues have been shown to trigger the maturation of stem cells towards more specialized cell types, a phenomenon known as differentiation.10–15 Both in embryonic and adult stem cells, studies have found evidence of mild heat-shock affecting cellular proliferation and differentiation.11,16–18 Both single and repetitive heat-shock treatments were found to trigger differentiation down various lineages such as chondro- (cartilage) and osteogenic (bone).10,11 In osteogenic differentiation (bone-forming), heat-shock was found to enhance the production of the enzyme alkaline phosphatase (ALP), an early marker for osteogenesis.11,17
Differentiation therapy is an emerging research field for treating cancer where the aim is to cause differentiation of tumour cells so as to decrease their proliferative capacity. This therapy is targeted towards a subset of cancer cells that can self-renew, referred to as cancer stem cells (CSC). Their unregulated self-renewal property is considered to drive the growth and spread of the tumour.19,20 CSCs could arise from malignant transformation of normal stem cells or due to progenitor cells acquiring the ability to continuously self-renew.21 Channelling these cells to differentiate into more mature phenotypes results in a reduction of their self-renewal capacity, similar to their non-transformed stem cell-counterparts.22
In this study we assessed the effect of heat-shock from magnetic hyperthermia treatment on cancer cells, focusing on the fate of the surviving cells. For this, we used a bone-cancer cell-line (MG-63)23,24 as our model system and determined both cell viability and differentiation state following the treatment. MG-63 cells have been used as a model cell line for assessing various cancer drugs and cancer therapies including differentiation therapy using retinoic acid.25 Unlike their healthy osteoblastic counterparts, these cells are not terminally differentiated and only show basal levels of expression of osteogenic markers such as RUNX2, Osterix or ALP.26–28 Following hyperthermia treatment we assessed changes in their differentiation state by measuring the degree of up-regulation of ALP. For bone-related cells such as the MG-63 cells, an increased expression of ALP indicates commitment to differentiate down the osteogenic (bone) lineage.29,30
For this study we used citric acid coated zinc doped magnetite nanoparticles (Zn0.4Fe2.6O4) to produce heat via magnetic hyperthermia, as they were found previously to show the strongest magnetic response in cellular environments (as measured via AC susceptometry) compared to similar particles,31 and with citric acid coating show minimal cytotoxicity at the concentrations of interest.32 The citric acid coating also ensures that the MNPs remain stably suspended, thus allowing homogenous heating of the entire sample volume.
In order to expose cells to a uniform temperature we developed an approach based on MNP extracellular heating. In this approach, cells grown as monolayers on sterile PLA films were placed vertically into 5 ml sample tubes (Fig. 1a) and completely immersed in cell-culture media that was pre-loaded with stably suspended MNPs. A sterile fiber optic temperature probe was also immersed into the solution in order to measure the media temperature in situ. The sample tubes were placed at the centre of a coil in a custom built magnetic hyperthermia instrument. In the case of the 37 °C control, the sample tube containing cells and nanoparticles was placed in the magnetic hyperthermia instrument for 30 min without turning on the magnetic field. In this case, the cooling water in the coil was set such that the ambient temperature in the sample volume was 37 °C. For the heat treated samples, cells were exposed to a 30 min magnetic hyperthermia treatment at time point zero, with a repeated treatment performed at 48 hours. After each treatment the cells were removed and placed in fresh MNP-free media to avoid interactions between the cells and MNPs that could influence cell behaviour.
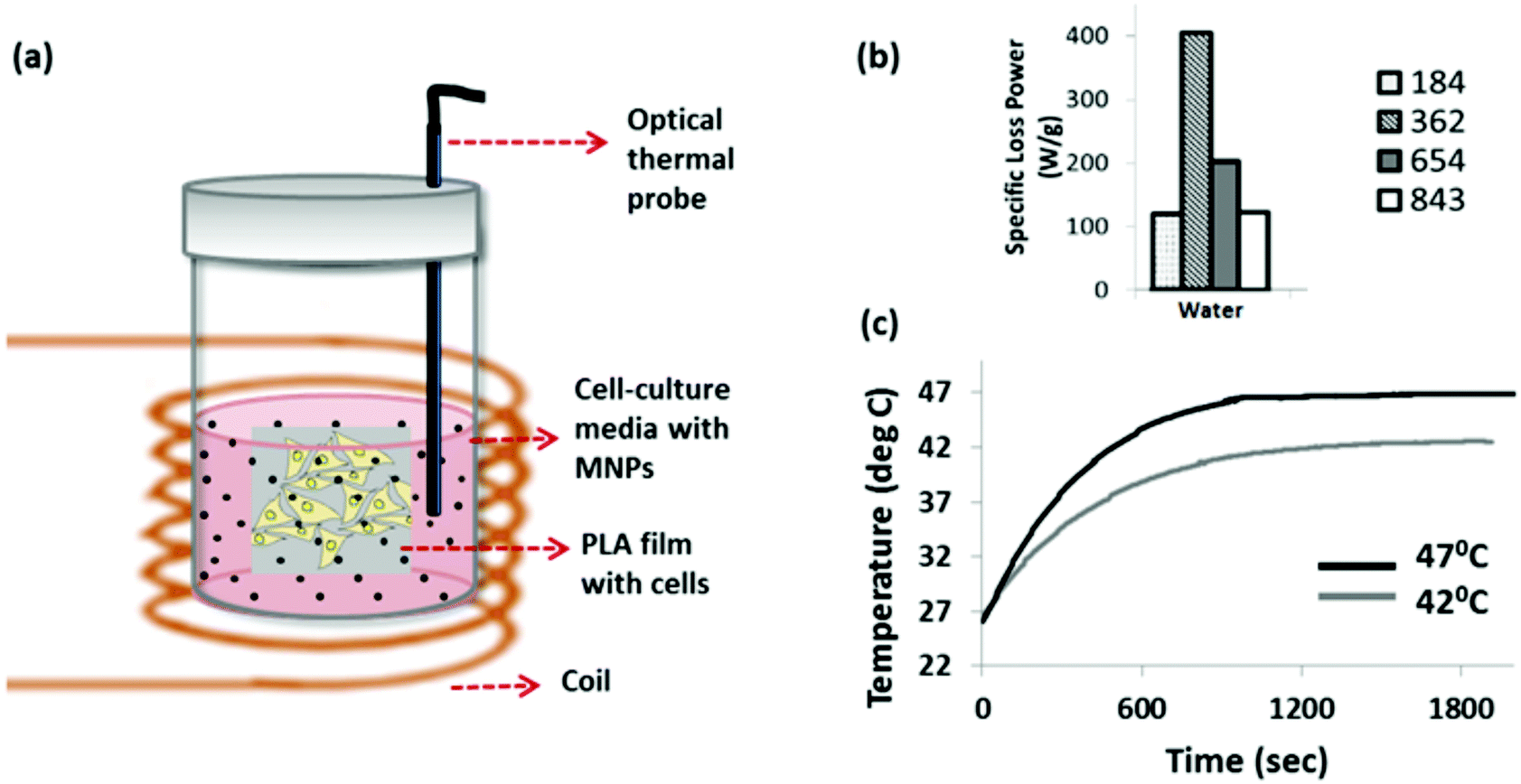 | ||
| Fig. 1 (a) Schematic showing the set-up for cellular magnetic hyperthermia experiments. Monolayer of cells grown on PLA films were placed vertically into a solution of 10% v/v MNP containing cell-culture media. An optical probe was immersed into this solution to measure temperature in situ. (b) Measured Specific Loss Power (SLP) values of aqueous suspensions of Zn0.4Fe2.6O4 nanoparticles at different field frequencies (184–843 kHz). (c) In situ temperature measurements during cellular magnetic hyperthermia using the optical thermal probe, using a magnetic field of 29.4 mT at 362 kHz. | ||
Results
Prior to the cellular experiments the optimum field conditions required to maximise the heating power of the MNPs were determined. This was done by measuring the heating of the aqueous suspensions of the MNPs. The heating power (i.e. the specific loss power, also known as the specific absorption rate) was measured at different field strengths and frequencies as shown in Fig. 1b. The combination of field strength and frequency parameters were limited due to the limitations of the coil-capacitor combination compatible with our hyperthermia instrument. At 29.4 mT and 362 kHz, the citric acid-coated Zn0.4Fe2.6O4 nanoparticle suspension showed a maximum heating response, as reflected by its specific loss power value (SLP) of 182 W g−1 (Fig. 1b). We chose this magnetic field frequency for all the cellular experiments, but controlled the magnetic field strength by adjusting the coil voltage in order to reach target temperatures of 42 °C for mild heat-shock, and 47 °C for severe heat-shock in the solutions which the cells were immersed in. An example measurement for this is shown in Fig. 1c.The changes in morphology of the cells following the hyperthermia treatment at 0 and after the repeated treatment at 48 hours, were observed by bright-field imaging, as shown in Fig. 2. Cells were imaged at 24, 72 and 120 hours after the start of the experiment. The MG-63 cells are adherent cells with a well-defined triangular morphology. This can be observed clearly for the cells maintained at 37 °C (Fig. 2a) which are seen to proliferate and increase in confluence at the later time-points. For the cells heated to 42 °C by hyperthermia, although a majority of the cells are viable at 24 hours following the first treatment (Fig. 2d), a significant population of detached cells can be seen after the second treatment at the 72 hours time point (Fig. 2e). By 120 hours this sample shows stress-related changes in the cellular morphology such as shrinkage as well as rounding up with minimal or no cells left attached (Fig. 2e and f). For the strongest heat-shock treated sample (47 °C), loss in a healthy morphology is observed even after a single treatment (Fig. 2g). Very few cells remain at further time points following the second treatment (Fig. 2h and i).
 | ||
| Fig. 2 Bright field images of MG-63 cells maintained at 37 °C (a–c) or exposed to two 30 min magnetic hyperthermia treatments in expansion media: one at time point 0 hours and the second at 48 hours, reaching controlled temperatures of 42 °C (d–f), or 47 °C (g–i) during each treatment. Samples a, d and g were imaged at 24 hours; b, e and h were imaged at 72 hours and c, f and i at 120 hours, from the start of the experiment. Scale bar is 100 μm. | ||
To assess osteogenic differentiation, the expression of the enzyme alkaline phosphatase was measured on these samples at various time points (Fig. 3). Fig. 3a shows the total DNA content (quantified using the PicoGreen™ assay) as a proportional measure of cell numbers33 for each of the sample at the different time points. While no significant difference is observed between the 37 °C sample and 42 °C until 72 hours, at 120 hours significant decrease in cell numbers is observed as also perceived in the micrographs (Fig. 2). At the highest heat-shock condition of 47 °C a qualitative reduction in cell number is seen by 24 hours, but this only becomes significant by 72 hours and beyond.
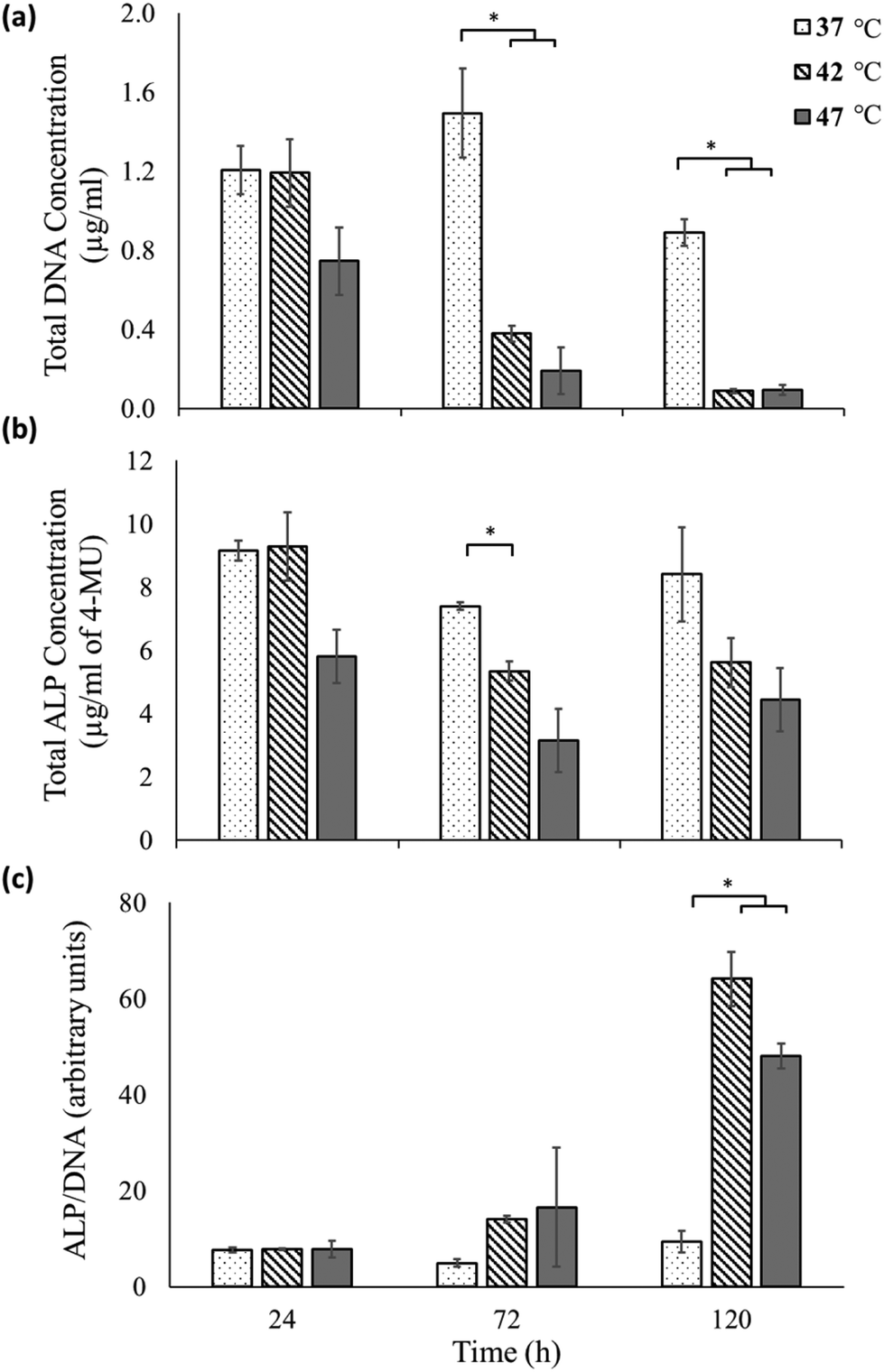 | ||
| Fig. 3 Effect of heat-shock treatment on the cell number and alkaline phosphatase (ALP) expression: MG-63 cells exposed to temperatures of 37, 42 and 47 °C in expansion media for two 30 min treatments, one at time point 0 hours and the second at time point 48 hours. Total DNA content (a), total ALP content (b), and the normalised ALP/DNA (proportional to ALP/cell) (c), were measured at 24, 72 and 120 hours. Error bars represent standard error for n = 3 and line over columns indicates groups that were significantly different from each other (Tukey test, p < 0.05) with the star indicating significance. | ||
Fig. 3b shows the total ALP expression of the samples as measured using the 4-MU assay. At each time point of measurement, the ALP measurements reflect the trend observed for total DNA measurements of each sample. However a significant difference in ALP expression between untreated and heat treated samples is only observed at 72 hours between the 37 °C and the 42 °C sample.
The total DNA measurements indicate that the heat-shock treatment had a negative effect on the cells, with their corresponding total ALP production lower than that of the untreated sample. However, the total ALP measurement does not take into account the reduction in cell viability following hyperthermia treatments, as indicated in the data shown in Fig. 2 and 3a. In fact the ALP production normalized to the total DNA content (proportional to the ALP produced by each live cell34), reveals a very different picture (Fig. 3c). At 24 hours, there is minimal ALP production per live cell in the 37, 42 and 47 °C conditions with no significant difference between them. At 72 hours, an increase in the ALP/cell of the 42 °C sample is found compared to the untreated 37 °C sample although this doesn't become significant until 120 hours. At 120 hours, the expression of ALP/cell is significantly elevated for both the heat-shock treated samples compared to the untreated sample.
These results can also be appreciated qualitatively by observing that the total ALP expressed by heat-shock treated cells (Fig. 3b) shows a less dramatic reduction for increasing time points than the corresponding live cell count (Fig. 3a). Thus the surviving cells must have up-regulated their ALP expression, as is shown quantitatively in Fig. 3c.
Discussion
The results shown here suggest that magnetic hyperthermia can induce differentiation in cancer cells, in addition to reducing cell viability. It could therefore be considered as an alternative to current chemical based methods for inducing differentiation in cancer therapy. To date, the most extensively studied differentiation factor for CSC is all-trans-retinoic acid which acts by triggering expression of differentiation-related genes. However, the in vitro efficiency of retinoic acid to trigger differentiation has not been replicated in vivo.35 In addition, although it is generally well tolerated, it could lead to complications collectively referred to as the ‘retinoic acid syndrome’.36Bulk heating has previously been shown to trigger differentiation in MG-63 cells.17 In the work by Shui & Scutt (2001),17 the cells were exposed to 1 hour heat treatment at different temperatures from 39 to 42.5 °C by placing them in a temperature-controlled water bath. The expression of ALP per 104 cells, 96 hours following the treatment, was found to linearly increase with the treatment temperature. These results indicate that temperature-triggered differentiation is not restricted to magnetic hyperthermia. However, the advantage of magnetic hyperthermia in comparison to bulk heating would be the ability to remotely target specific regions within a cell and to apply localized heating at the cellular level without affecting neighbouring tissue.37
During hyperthermia treatment, at temperatures above a threshold of 45 °C, cells predominantly undergo necrosis, whilst they are more susceptible to undergo apoptosis below this threshold.38,39 Triggering cell-death via apoptosis is preferred to necrosis as the latter can result in inflammation and metastasis.40–42 In our study we found that both mild (42 °C) and more extreme (47 °C) magnetic hyperthermia mediated heat-shock had a significant impact on the viability of the MG-63 cells, as seen by the DNA measurements. On the other hand, the surviving cells express significantly higher levels of ALP in both cases. The upregulation of ALP expression by the surviving cells in the 42 °C sample indicates that it is possible to trigger the differentiation of cancerous cells via magnetic hyperthermia even at modest temperatures.
MG-63 cells, apart from being a good model cell line for assessing various cancer therapies, are of additional interest as they have been found to harbour a sub-population of cancer stem cells (CD133+/Nestin).43–46 Although in this work the effect of hyperthermia has been studied on the population as a whole, future studies can look at identifying the CSC and non-CSC subpopulations of the MG-63 cells and investigating the differential effects of magnetic hyperthermia on each subtype. This approach would be able to clearly define the responses of these cells to heat and the potential of magnetic hyperthermia as an alternative cancer differentiation therapy targeting CSCs.
In this initial study we have explored an early marker for differentiation of osteosarcoma cells down the osteogenic lineage. Further work is now needed to understand how MNP-mediated heat-shock affects long-term lineage commitment of the surviving cells, as well the effects of the combination of duration and frequency of the heat treatment in different cell types and down different lineages. However these results are the first demonstration showing the potential of MNP-mediated hyperthermia as a differentiation therapy. As heat is only generated in the proximity of the MNPs, magnetic hyperthermia is a local effect and so should not adversely influence healthy tissue surrounding the tumour. By functionalizing the MNPs with CSC-specific markers which distinguish the CSCs from non-tumorigenic cells, an even more targeted approach could be adopted. In addition, by combining traditional radio- or chemotherapy to target the non-CSC population, together with MNP-mediated hyperthermia using MNPs with higher heating potential47 to target the CSC population, it may be possible to develop more efficient and curative therapies for cancer treatment.
Conclusion
We investigated the effect of magnetic hyperthermia on the proliferative and differentiation state of the bone-cancer cell line MG-63. Our results show that, apart from triggering heat-related cell death, magnetic hyperthermia also provides an additional stimulus to the surviving cells to further differentiate to form a more mature phenotype. In addition, our study shows that mild hyperthermia of 42 °C is sufficient to trigger differentiation commitment, with such mild heat-shock likely to trigger apoptosis rather than necrotic cell death. Further work is needed to understand how MNP-mediated heat-shock affects long-term lineage commitment of the surviving cells in different cell types down different lineages. However, the high degree of localised temperature control possible with the magnetic hyperthermia technique, means that this method has huge potential to be used as either an alternative or complementary approach to existing differentiation therapies.Experimental section
Preparation and characterisation of MNP suspensions
The Zn0.4Fe2.6O4 MNPs were produced via microbial iron reduction of zinc-doped ferrihydrite according to the methods described in detail previously.48 Following this, the MNPs were coated with citric acid to prepare stable aqueous suspensions using procedures described in detail elsewhere.9,49 The suspensions were sterilized using 0.2 μm filters prior to being resuspended in cell-culture media for the hyperthermia experiments. MNP suspension concentrations were measured using the Ferrozine method for iron. Briefly, the nanoparticle suspensions were digested in concentrated nitric acid (70%; Sigma, UK) at high temperatures (>60 °C) overnight and were treated with equal amounts of 6.6 M of the reducing agent hydroxylamine hydrochloride (Sigma, UK) for 150 min at room temperature. This reduces Fe(III) to Fe(II) as the Ferrozine assay is specific for the latter.To quantify the heating properties of the Zn0.4Fe2.6O4 MNPs, 1 ml of the aqueous suspensions were transferred to 1.5 ml centrifuge tubes. The tubes were placed in the centre of a custom built magnetic hyperthermia instrument comprising of a temperature-regulated coil capable of generating high-frequency (50–1000 kHz) AC magnetic fields of up to 30 mT. By using different coil-capacitor combinations, different field strengths were attained. Water at a pre-set temperature was circulated through the coil to prevent the coil from over-heating and to regulate the non-magnetic heating of the sample. The temperature was recorded in the sample space at the centre of the coil by inserting an optical probe into the sample.
The specific loss power (SLP) was calculated using the expression
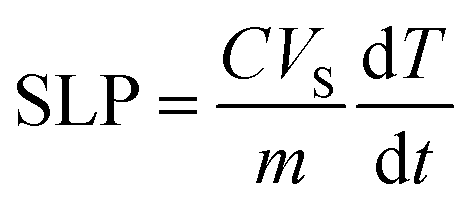
Cell culture
MG-63 cells, an osteosarcoma cell line (Lonza, UK) were expanded in T-flasks in expansion media consisting of 4.5 g L−1 glucose Dulbecco's Modified Eagle's medium (Lonza, UK) supplemented with 10% foetal bovine serum (FBS), 1% penicillin/streptomycin (antibiotics and antimycotics) and 1% L-glutamine to obtain required cell numbers.PLA films were cut to required size (1 × 1 cm2) and sterilized by soaking in 70% ethanol overnight followed by UV sterilization. They were rinsed well with phosphate buffered saline to remove any remaining ethanol and soaked in expansion media overnight in well plates. This step was done to promote formation of a protein layer by the FBS and enhance cellular attachment.
The cell suspensions were prepared via trypsinization, and immediately seeded onto the PLA films in expansion media at ∼80% confluency and allowed to attach overnight. For the heat-shock treatments, the 3 cell-seeded PLA films were removed from the well plates and placed vertically into 5 ml bijou tubes. A fourth empty film was placed in the tube for stability. The MNP suspension was resuspended in HEPES buffered DMEM medium (pH stable without gas regulation; Gibco, UK) containing 20% FBS, 2% antibiotics/antimycotics, 1% L-glutamine and 1% non-essential amino acids at a concentration of 0.3 mg ml−1 (10% v/v). 1.5 ml of this solution was added slowly into the tubes so as to not disturb the films or cause shear stress to the cells and tubes were closed tight with a screw cap.
In vitro magnetic hyperthermia
The closed tubes containing the PLA films were placed within the coil of the magnetic hyperthermia instrument. The water cooling in the magnetic hyperthermia system coil was set at a temperature that allowed the solution placed within the coil to be maintained at 37 °C in the absence of MNP induced heating. For the heat-shock treated samples, the cells were exposed to 362 kHz and varying field strength (by adjusting the applied voltage) to achieve the target temperatures. To reach 42 °C, the voltage was set to 40 ± 5 V and current 10.2 ± 1.5 A. For 47 °C, the voltage was set to 51.1 V and the current 13 ± 0.2 A. Cells were maintained at the target temperatures for 30 min at time 0 and for a second 30 min exposure at 48 hours. Temperature was measured using an optical temperature probe. Following each treatment, the PLA films with cells were immediately transferred to fresh well plates and incubated in expansion media for further assays. Bright-field images of the cells were taken at different time points after hyperthermia treatment, using an Olympus IX83 microscope fitted with a camera using the Fluorview 10 software.DNA and ALP quantification
At each time point, wells were washed with PBS to remove cell debris and any secreted ALP to ensure that only the intracellular ALP was measured. This was followed by cell lysis using 0.1% Triton-X in water. Cell lysates were used for both ALP and DNA assays. The total DNA concentration was quantified using the Quanti-iT™ PicoGreen® dsDNA assay kit (ThermoFisher Scientific, UK). In brief, 20 μl of cell lysate was mixed with 50 μl of 1× PicoGreen (PG) diluted in 1× TE buffer. Samples were incubated with PG for 5 min at room temperature in the dark. The fluorescence of PG is enhanced when bound to double stranded DNA. The fluorescence intensities of the samples following incubation were measured at excitation/emission wavelengths of 485/530 nm on a BioTek synergy2 plate reader using the GEN 1.05 software.Alkaline phosphatase expression
Total intracellular ALP content was measured via the 4-methylumbelliferyl phosphate (4-MUP) Liquid Substrate System (Sigma-Aldrich, UK). ALP cleaves the 4-MUP in alkaline conditions to release the fluorescent product 4-MU. In brief, cell lysates were incubated with equal volumes of 4-MUP reagent for 30 min in the dark at 37 °C. Following this, the fluorescence was measured in a plate reader (BioTek Instrument Inc.) at excitation/emission wavelengths of 360/440 nm. For normalizing to the number of cells, the total ALP content was divided by the total DNA content for each sample.Statistics
Each column in Fig. 3 represent the mean (n = 3) and the error bars are the standard error of the mean. A one-way ANOVA was performed in conjunction with Tukey post-hoc test between samples at each time point as the data was shown to be normally distributed and having equal variance using the Shapiro–Wilk and Levene's test respectively.Conflicts of interest
There are no conflicts to declare.Acknowledgements
S. Moise was supported by the EPSRC Centre for Innovative Manufacturing in Regenerative Medicine (EP/H028277/1).References
- M. Johannsen, U. Gneveckow, L. Eckelt, A. Feussner, N. Waldofner, R. Scholz, S. Deger, P. Wust, S. A. Loening and A. Jordan, Int. J. Hyperthermia, 2005, 21, 637–647 CrossRef CAS PubMed.
- A. Levy, A. Dayan, M. Ben-David and I. Gannot, Nanomedicine, 2010, 6, 786–796 CrossRef CAS PubMed.
- H. Oliveira, E. Perez-Andres, J. Thevenot, O. Sandre, E. Berra and S. Lecommandoux, J. Controlled Release, 2013, 169, 165–170 CrossRef CAS PubMed.
- V. F. Cardoso, A. Francesko, C. Ribeiro, M. Bañobre-López, P. Martins and S. Lanceros-Mendez, Adv. Healthcare Mater., 2018, 7, 1700845 CrossRef PubMed.
- Y. Hu, S. Mignani, J. P. Majoral, M. Shen and X. Shi, Chem. Soc. Rev., 2018, 47, 1874–1900 RSC.
- K. M. Krishnan, IEEE Trans. Magn., 2010, 46, 2523–2558 CAS.
- G. M. Hahn, IEEE Trans. Biomed. Eng., 1984, 31, 3–8 CAS.
- M. K. Lima-Tenorio, E. A. Pineda, N. M. Ahmad, H. Fessi and A. Elaissari, Int. J. Pharm., 2015, 493, 313–327 CrossRef CAS PubMed.
- A. Espinosa, J. Kolosnjaj-Tabi, A. Abou-Hassan, A. Plan Sangnier, A. Curcio, A. K. A. Silva, R. Di Corato, S. Neveu, T. Pellegrino, L. M. Liz-Marzán and C. Wilhelm, Adv. Funct. Mater., 2018, 28, 1803660 CrossRef.
- J. Chen, C. Li and S. Wang, PLoS One, 2014, 9, e91561 CrossRef PubMed.
- J. Chen, Z. D. Shi, X. Ji, J. Morales, J. Zhang, N. Kaur and S. Wang, Tissue Eng., Part A, 2013, 19, 716–728 CrossRef CAS PubMed.
- A. J. Engler, S. Sen, H. L. Sweeney and D. E. Discher, Cell, 2006, 126, 677–689 CrossRef CAS PubMed.
- V. Du, N. Luciani, S. Richard, G. Mary, C. Gay, F. Mazuel, M. Reffay, P. Menasche, O. Agbulut and C. Wilhelm, Nat. Commun., 2017, 8, 400 CrossRef PubMed.
- J. M. Kanczler, H. S. Sura, J. Magnay, D. Green, R. O. C. Oreffo, J. P. Dobson and A. J. E. Haj, Tissue Eng., Part A, 2010, 16, 3241–3250 CrossRef CAS PubMed.
- M. Rotherham, J. R. Henstock, O. Qutachi and A. J. El Haj, Nanomedicine, 2018, 14, 173–184 CrossRef CAS PubMed.
- M. S. Choudhery, M. Badowski, A. Muise and D. T. Harris, Cytotherapy, 2015, 17, 359–368 CrossRef CAS PubMed.
- C. Shui and A. Scutt, J. Bone Miner. Res., 2001, 16, 731–741 CrossRef CAS PubMed.
- K. Byun, T. K. Kim, J. Oh, E. Bayarsaikhan, D. Kim, M. Y. Lee, C. G. Pack, D. Hwang and B. Lee, Stem Cell Res., 2013, 11, 1323–1334 CrossRef CAS PubMed.
- M. Al-Hajj, M. S. Wicha, A. Benito-Hernandez, S. J. Morrison and M. F. Clarke, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3983–3988 CrossRef CAS PubMed.
- S. K. Singh, C. Hawkins, I. D. Clarke, J. A. Squire, J. Bayani, T. Hide, R. M. Henkelman, M. D. Cusimano and P. B. Dirks, Nature, 2004, 432, 396–401 CrossRef CAS PubMed.
- A. Cozzio, E. Passegue, P. M. Ayton, H. Karsunky, M. L. Cleary and I. L. Weissman, Genes Dev., 2003, 17, 3029–3035 CrossRef CAS PubMed.
- R. Pardal, M. F. Clarke and S. J. Morrison, Nat. Rev. Cancer, 2003, 3, 895–902 CrossRef CAS PubMed.
- S. U. Lauvrak, E. Munthe, S. H. Kresse, E. W. Stratford, H. M. Namlos, L. A. Meza-Zepeda and O. Myklebost, Br. J. Cancer, 2013, 109, 2228–2236 CrossRef CAS PubMed.
- C. Pautke, M. Schieker, T. Tischer, A. Kolk, P. Neth, W. Mutschler and S. Milz, Anticancer Res., 2004, 24, 3743–3748 CAS.
- R. C. Haydon, L. Zhou, T. Feng, B. Breyer, H. Cheng, W. Jiang, A. Ishikawa, T. Peabody, A. Montag, M. A. Simon and T. C. He, Clin. Cancer Res., 2002, 8, 1288–1294 CAS.
- X. Luo, J. Chen, W. X. Song, N. Tang, J. Luo, Z. L. Deng, K. A. Sharff, G. He, Y. Bi, B. C. He, E. Bennett, J. Huang, Q. Kang, W. Jiang, Y. Su, G. H. Zhu, H. Yin, Y. He, Y. Wang, J. S. Souris, L. Chen, G. W. Zuo, A. G. Montag, R. R. Reid, R. C. Haydon, H. H. Luu and T. C. He, Lab. Invest., 2008, 88, 1264–1277 CrossRef CAS PubMed.
- D. M. Thomas, S. A. Johnson, N. A. Sims, M. K. Trivett, J. L. Slavin, B. P. Rubin, P. Waring, G. A. McArthur, C. R. Walkley, A. J. Holloway, D. Diyagama, J. E. Grim, B. E. Clurman, D. D. Bowtell, J. S. Lee, G. M. Gutierrez, D. M. Piscopo, S. A. Carty and P. W. Hinds, J. Cell Biol., 2004, 167, 925–934 CrossRef CAS PubMed.
- R. C. Haydon, H. H. Luu and T. C. He, Clin. Orthop. Relat. Res., 2007, 454, 237–246 CrossRef PubMed.
- Y. H. Kim, D. S. Yoon, H. O. Kim and J. W. Lee, Stem Cells Dev., 2012, 21, 2958–2968 CrossRef CAS PubMed.
- L. Carpio, J. Gladu, D. Goltzman and S. A. Rabbani, Am. J. Physiol. Endocrinol. Metab., 2001, 281, E489–E499 CrossRef CAS PubMed.
- E. Cespedes, J. M. Byrne, N. Farrow, S. Moise, V. S. Coker, M. Bencsik, J. R. Lloyd and N. D. Telling, Nanoscale, 2014, 6, 12958–12970 RSC.
- S. Moise, E. Cespedes, D. Soukup, J. M. Byrne, A. J. El Haj and N. D. Telling, Sci. Rep., 2017, 7, 39922 CrossRef CAS PubMed.
- V. M. Quent, D. Loessner, T. Friis, J. C. Reichert and D. W. Hutmacher, J. Cell. Mol. Med., 2010, 14, 1003–1013 CrossRef CAS PubMed.
- L. P. Silva, P. L. Lorenzi, P. Purwaha, V. Yong, D. H. Hawke and J. N. Weinstein, Anal. Chem., 2013, 85, 9536–9542 CrossRef CAS PubMed.
- L. A. Hammond, G. Brown, R. G. Keedwell, J. Durham and R. A. Chandraratna, Anticancer Drugs, 2002, 13, 781–790 CrossRef CAS PubMed.
- R. S. Larson and M. S. Tallman, Best Pract. Res., Clin. Haematol., 2003, 16, 453–461 CrossRef CAS PubMed.
- H. Huang, S. Delikanli, H. Zeng, D. M. Ferkey and A. Pralle, Nat. Nanotechnol., 2010, 5, 602 CrossRef CAS PubMed.
- K. L. O'Neill, D. W. Fairbairn, M. J. Smith and B. S. Poe, Apoptosis, 1998, 3, 369–375 CrossRef.
- B. V. Harmon, A. M. Corder, R. J. Collins, G. C. Gobe, J. Allen, D. J. Allan and J. F. Kerr, Int. J. Radiat. Biol., 1990, 58, 845–858 CrossRef CAS PubMed.
- T. Kobayashi, K. Kakimi, E. Nakayama and K. Jimbow, Nanomedicine, 2014, 9, 1715–1726 CrossRef CAS PubMed.
- D. Kanduc, A. Mittelman, R. Serpico, E. Sinigaglia, A. A. Sinha, C. Natale, R. Santacroce, M. G. Di Corcia, A. Lucchese, L. Dini, P. Pani, S. Santacroce, S. Simone, R. Bucci and E. Farber, Int. J. Oncol., 2002, 21, 165–170 CAS.
- R. D. Bonfil, O. D. Bustuoabad, R. A. Ruggiero, R. P. Meiss and C. D. Pasqualini, Clin. Exp. Metastasis, 1988, 6, 121–129 CrossRef CAS PubMed.
- V. Tirino, V. Desiderio, R. d'Aquino, F. De Francesco, G. Pirozzi, A. Graziano, U. Galderisi, C. Cavaliere, A. De Rosa, G. Papaccio and A. Giordano, PLoS One, 2008, 3, e3469 CrossRef PubMed.
- R. Di Fiore, A. Guercio, R. Puleio, P. Di Marco, R. Drago-Ferrante, A. D'Anneo, A. De Blasio, D. Carlisi, S. Di Bella, F. Pentimalli, I. M. Forte, A. Giordano, G. Tesoriere and R. Vento, J. Cell. Biochem., 2012, 113, 3380–3392 CrossRef CAS PubMed.
- L. Wang, P. Park and C. Y. Lin, Cancer Biol. Ther., 2009, 8, 543–552 CrossRef CAS PubMed.
- R. Veselska, M. Hermanova, T. Loja, P. Chlapek, I. Zambo, K. Vesely, K. Zitterbart and J. Sterba, BMC Cancer, 2008, 8, 300 CrossRef PubMed.
- A. Espinosa, M. Bugnet, G. Radtke, S. Neveu, G. A. Botton, C. Wilhelm and A. Abou-Hassan, Nanoscale, 2015, 7, 18872–18877 RSC.
- J. M. Byrne, V. S. Coker, E. Cespedes, P. L. Wincott, D. J. Vaughan, R. A. Pattrick, G. van der Laan, E. Arenholz, F. Tuna and M. Bencsik, Adv. Funct. Mater., 2014, 24, 2518–2529 CrossRef CAS.
- J. M. Byrne, V. S. Coker, S. Moise, P. L. Wincott, D. J. Vaughan, F. Tuna, E. Arenholz, G. van der Laan, R. A. Pattrick, J. R. Lloyd and N. D. Telling, J. R. Soc., Interface, 2013, 10, 20130134 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |