
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Partial magnetic ordering in one-dimensional arrays of endofullerene single-molecule magnet peapods†
Stanislav M.
Avdoshenko
 *a,
Fabian
Fritz
bc,
Christin
Schlesier
a,
Aram
Kostanyan
de,
Jan
Dreiser
e,
Martina
Luysberg
f,
Alexey A.
Popov
a,
Carola
Meyer
bc and
Rasmus
Westerström
*g
*a,
Fabian
Fritz
bc,
Christin
Schlesier
a,
Aram
Kostanyan
de,
Jan
Dreiser
e,
Martina
Luysberg
f,
Alexey A.
Popov
a,
Carola
Meyer
bc and
Rasmus
Westerström
*g
aLeibniz Institute for Solid State and Materials Research (IFW), 01069 Dresden, Germany
bDepartment of Physics, University Osnabrück, 49076 Osnabrück, Germany
cPeter Grünberg Institute (PGI-6), Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
dPhysik-Institut, Universität Zürich, Winterthurerstrasse 190, 8057 Zürich, Switzerland
eSwiss Light Source, Paul Scherrer Institute, 5232 Villigen PSI, Switzerland
fErnst Ruska-Centre for Microscopy and Spectroscopy with Electrons, Forschungszentrum Jülich GmbH, 52425 Jülich, Germany
gSynchrotron Radiation Research, Lund University, 22100 Lund, Sweden. E-mail: rasmus.westerstrom@sljus.lu.se
First published on 14th August 2018
Abstract
The magnetic ordering and bistability of one-dimensional chains of endofullerene Dy2ScN@C80 single-molecule magnets (SMMs) packed inside single-walled carbon nanotubes (SWCNTs) have been studied using high-resolution transmission electron microscopy (HRTEM), X-ray magnetic circular dichroism (XMCD), and ab initio calculations. X-ray absorption measurements reveal that the orientation of the encapsulated endofullerenes differs from the isotropic distribution in the bulk sample, indicating a partial ordering of the endofullerenes inside the SWCNTs. The effect of the one-dimensional packing was further investigated by ab initio calculations, demonstrating that for specific tube diameters, the encapsulation is leading to energetically preferential orientations of the endohedral clusters. Additionally, element-specific magnetization curves reveal a decreased magnetic bistability of the encapsulated Dy2ScN@C80 SMMs compared to the bulk analog.
The discovery of the giant magnetoresistance effect (GMR)1,2 in the late 1980s marks the start of the field of spintronics. A decade later came the first report on spin-dependent transport via a carbon nanotube (CNT)3 which opened the door for molecular spintronics. Studies of nanotube spintronics have demonstrated that spin valves with a large magnetoresistance ratio can be achieved by using magnetic La(1−x)SrxMnO3 (LSMO) contacts.4 More recently, a study demonstrated that the need for magnetic contacts could be eliminated by magnetically coupling single-molecule magnets (SMMs) to the conducting channel of a carbon nanotube.5 In that study, a supramolecular spin valve was created with a spin-dependent transport solely determined by the magnetic properties of TbPc2 SMMs6 grafted to the outside wall of the single-walled CNT (SWCNT). The SMM-SWCNT device demonstrated the feasibility of an electronic readout of a single Tb3+ electronic and nuclear spin,7 a first step towards the application of SMMs in spintronic devices8 and quantum computing.9 An alternative route towards functionalizing CNTs is by filling the hollow interior with SMMs. The only published study of encapsulated SMMs has reported on the formation of amorphous structures of Mn12 clusters inside large diameter multi-walled CNTs.10 Endofullerenes encapsulating rare-earth based clusters constitute a new class of SMMs.11–14 Some SWCNTs have a diameter similar to that of the endofullerenes. The cylindrical interior of the SWCNT is thus ideally suited for encapsulating one-dimensional arrays of endofullerenes15 in so-called peapod structures.16
Here we report on the first study of one-dimensional arrays of SMMs packed into SWCNTs. The study was performed using high-resolution transmission electron microscopy (HRTEM), X-ray magnetic circular dichroism (XMCD), and ab initio calculations. The presented X-ray absorption spectroscopy (XAS) data demonstrates that the average orientation of the magnetic Dy2ScN-units of the encapsulated endofullerenes differs from the isotropic distribution in a bulk sample, indicating a partial ordering inside the nanotubes. A partial ordering is further supported by ab initio calculations which demonstrate that, depending on the specific tube diameter, the encapsulation results in energetically preferential orientations of the endohedral clusters. Nitride clusterfullerene SMMs have anisotropic magnetic clusters, and a preferred orientation inside the nanotubes implies a one-dimensional magnetic ordering with aligned molecular moments. The experimental and computational results thus demonstrate the feasibility of producing fullerene SMM-SWCNT hybrid devices consisting of well-protected and aligned molecular spin chains with possible applications in spintronics8 and quantum computation.17,18
Additionally, element-specific magnetization curves recorded using X-ray magnetic circular dichroism demonstrate a decreased average magnetic bistability of the encapsulated Dy2ScN@C80 endofullerenes.
1. Experimental and computational section
For the fabrication of the peapods, single-walled carbon-nanotubes (Carbon Solutions, Inc.) were chosen with a diameter distribution of 1.55 nm ± 0.1 nm to achieve one-dimensional filling. Raman spectra taken with a HeNe-Laser (λ = 632 nm), however, showed a radial-breathing mode (RBM) signal at 185 cm−1. This is a clear indication that metallic SWCNTs with diameters as small as 1.3 nm are present in the sample.19 The SWCNTs were oxidized in air for 20 min at 550 °C to create defects through which the fullerenes can enter the SWCNTs.20 Then the SWCNTs were sonicated in methanol for 10 min and deposited onto Cu TEM grids. These grids were used for high-resolution transmission electron microscopy as well as for the X-ray absorption measurements. For the filling, the grids were positioned in a glass container above the endofullerenes. Supercritical CO2 (50 °C, 150 bar) was used as a solvent, following the method of Khlobystov et al.21 To increase the filling yield the CO2 pressure was varied 40 times between 100 and 150 bar. After the process, the grids with the filled SWCNTs were washed with carbon disulfide to remove excess fullerenes. The HRTEM measurements were done using the spherical and chromatic aberration corrected FEI Titan 60–300 Ultimate (“PICO”)22 at an acceleration voltage of 80 kV. A slight underfocus was used to achieve high resolution. The Dy2ScN@C80 bulk sample was prepared by dissolving the endofullerenes in toluene and spray coated onto an aluminum sample holder. The X-ray absorption measurements were carried out at the X-Treme beamline23 of the Swiss Light Source. Absorption spectra were acquired by measuring the total electron yield (TEY) in the on-the-fly mode24 while applying a magnetic field parallel to the X-ray beam. The SQUID measurements were performed using a Quantum Design MPMS3 Vibrating Sample Magnetometer (VSM).Static density functional theory (DFT) calculations were performed with the Vienna ab initio simulation package (VASP),25,26 which was used as optimization driver at the PBE level of theory.27 All optimizations were performed in Γ-point. During the optimizations, the energy-cut-off was set to 400 Ry and it correlates with pseudopotential choice,26 and all optimizations were run until residual forces became smaller than 0.005 Å eV−1. We used in-house Python scripts to analyze and visualize the results. Born–Oppenheimer molecular dynamics simulations were performed at the PBE-D/DZVP level using CP2K code.28–30 The velocity Verlet algorithm was employed with the time step of 0.5 fs and Nosé–Hoover thermostat (three chains, thermostat coupling constant 100 fs) set at 300 K. Overall integration time for each system was ∼0.5 ns. Molecular structures, and BOMD trajectories were visualized using VMD package.31 Molecular theoretical XAS and XMCD spectra for the Dy M4,5-edge were simulated based on the point charge model implemented in MULTIX code.32 In these calculations, the Dy atoms assumed to be trivalent (Dy3+) and point charges of surrounding atoms are derived from molecular charge densities using Bader charge analysis for each molecule in the set of 144 isomers of Y2ScN@C80@CNT(12,8) (small).33 A set of semiempirical parameters was used to match experimental M4–M5 split (spin-orbital couplings of 0.95 for core, and of 0.85 for valence states) and energy spread of the M5 edge (on-site Coulomb of 0.75, see ref. 32 for details). To account for the experimental broadening, the theoretical spectra were convoluted with a Gaussian of σ = 0.5. The magnetic field strength and modeling temperature were set to the experimental values of 6.5 T and 2.0 K respectively. In all simulations, the X-ray beam is collinear to the magnetic field applied perpendicular to the tube-axis, see Fig. 5(a).
2. Results and discussion
The HRTEM image presented in Fig. 1 shows the one-dimensional alignment of Dy2ScN@C80 endofullerenes inside a SWCNT of the sample recorded after the X-ray measurements. The C80 fullerene cages are visible as circles inside the SWCNT, demonstrating that there is no significant damage from the X-ray exposure. The image was taken using an exposure time of 0.5 s. Fullerenes and endohedral clusters appear a bit blurry due to their comparably fast movement (rotation) caused by the electron irradiation during the exposure.34 | ||
| Fig. 1 (a) Ball-and-stick-model of Dy2ScN@C80. (b) HRTEM image of a one-dimensional array of Dy2ScN@C80 endofullerenes inside a SWCNT. For clarification, the dashed lines give the position of the walls of the SWCNT, and the arrows point to the circular endofullerenes. | ||
Fig. 2 displays X-ray absorption spectra from a bulk and a peapod sample measured at a temperature of ∼2 K and with an external magnetic field of 6.5 T applied along the X-ray beam. The spectra were recorded over the Dy M4,5-edge by exciting 3d core electrons to the 4f valence shell using right (I+) and left (I−) circular polarized photons. The difference in absorption between the two helicities and the resulting XMCD spectra (I+ − I−) contains information about the magnetic properties of the 4f valence shell of the absorbing Dy3+ ions. Applying a sum rule analysis35,36 to the displayed data provides absolute values for the spin 〈Sz〉 and orbital 〈Lz〉 moments and Table 1 shows the extracted values. The number of holes in the 4f shell was taken to be nh = 5 and 〈Tz〉 was calculated according to ref. 36. The ratio of the extracted orbital and spin angular moments is lower than, but close to the value of 5/2.5 = 2 expected from a 6H15/2 Hund's ground state. The determined magnetic moments of 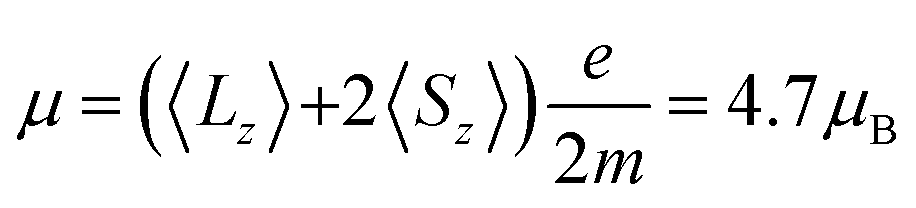 for the bulk sample and μ = 5.2μB for the peapods, correspond roughly to half of the saturated magnetization of 10μB of an isotropic paramagnet with J = 15/2 and gJ = 20/15. The reduced magnetic moments are a consequence of the anisotropic mJ = 15/2 ground state with a magnetic easy-axis directed along the Dy–N bonds.37 Observing magnetic moments reduced by a factor of one-half is therefore expected for systems with an isotropic distribution of endohedral clusters.11 As was observed for Er3N@C80@SWCNTs,15 the comparable results from the bulk and peapod sample indicate that the anisotropic properties of the individual Dy3+ ions are preserved, further demonstrating that the endofullerenes remain intact after the encapsulation into SWCNTs and the subsequent X-ray exposure.
for the bulk sample and μ = 5.2μB for the peapods, correspond roughly to half of the saturated magnetization of 10μB of an isotropic paramagnet with J = 15/2 and gJ = 20/15. The reduced magnetic moments are a consequence of the anisotropic mJ = 15/2 ground state with a magnetic easy-axis directed along the Dy–N bonds.37 Observing magnetic moments reduced by a factor of one-half is therefore expected for systems with an isotropic distribution of endohedral clusters.11 As was observed for Er3N@C80@SWCNTs,15 the comparable results from the bulk and peapod sample indicate that the anisotropic properties of the individual Dy3+ ions are preserved, further demonstrating that the endofullerenes remain intact after the encapsulation into SWCNTs and the subsequent X-ray exposure.
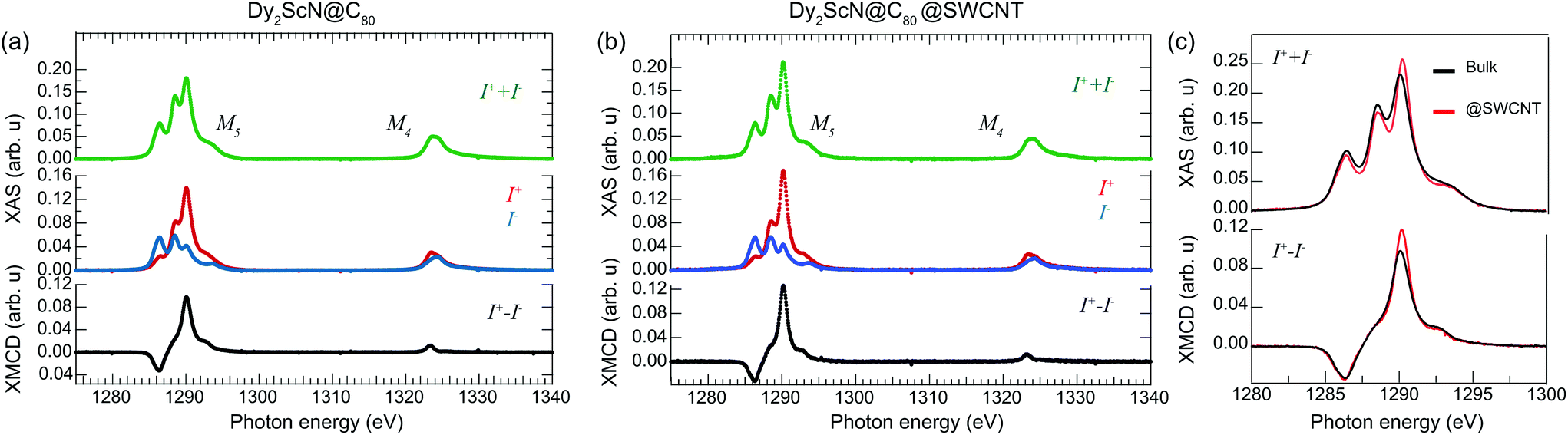 | ||
| Fig. 2 X-ray absorption spectra recorded using right (I+) and left (I−) circular polarized X-rays from samples consisting of (a) Dy2ScN@C80 endofullerenes in the bulk phase, and (b) encapsulated in SWCNTs. (c) A comparison of the normalized total absorption and XMCD spectra from the two systems. The temperature was set to 2 K, and an external magnetic field of 6.5 T was applied parallel to the X-ray beam and the surface normal of the samples. The data was normalized to the integrated total absorption (I+ + I−) over the displayed energy range. | ||
| Sample | 〈Sz〉 (ℏ) | 〈Lz〉 (ℏ) | 〈Lz〉/〈Sz〉 | 〈Lz〉 + 2〈Sz〉 (ℏ) |
|---|---|---|---|---|
| Dy2ScN@C80-bulk | 1.3 ± 0.1 | 2.1 ± 0.1 | 1.6 ± 0.1 | 4.7 ± 0.1 |
| Dy2ScN@C80@SWCNT | 1.4 ± 0.1 | 2.4 ± 0.2 | 1.7 ± 0.2 | 5.2 ± 0.2 |
We return to the total absorption (I+ + I−) displayed in Fig. 2. The Dy M5 multiplet structure reflects the average orientation of the endohedral clusters with respect to the polarization vector of the X-rays.38 Comparing the total absorption at the Dy M5 edge from the bulk and the peapod sample in Fig. 2(c) reveals a difference in the multiplet structure. A deviation of ∼9% is determined by integrating the absolute value of the difference between the two background subtracted and normalized spectra. The significant deviation from the isotropic bulk sample indicates that the endofullerenes are not completely randomly oriented inside the nanotubes. The magnitude of the XMCD signal is proportional to the projection of the magnetic moment of the absorbing Dy3+ ion onto the direction of the impinging X-rays. Because of the uniaxial anisotropy along the Dy–N bond, a deviation from the isotropic bulk sample should therefore also be observed in the XMCD signal. Comparing XMCD spectra in Fig. 2(c) and the extracted moments in Table 1 reveals a 22% larger maximum XMCD signal at the M5-edge and a 10% larger magnetic moment from the peapod sample, consistent with a partial ordering inside the nanotubes. It has been demonstrated that the torque exerted on the anisotropic Ho3+ moment in HoLu2N@C80 from an external magnetic field can lead to a thermally activated endohedral hopping motion that tends to align the cluster parallel to the field above a freezing temperature TF ∼ 55 K.39 In the present case, the system was cooled from room temperature to 2 K in zero field, and it is not expected that the applied field influences the partial ordering along the tubes. To understand the origin of the partial ordering we turn to ab initio calculations.
The fullerene cage C80-Ih is a quasi-spherical molecule, which gives rise to an almost isotropic distribution of cluster orientations inside the fullerene in the M3N@C80 molecule. As a result, at room temperature in solution, the M3N cluster is known to rotate quasi-freely inside the carbon cage. At the same time, chemical functionalization of the fullerene cage or supramolecular assembly (such as in co-crystals with Ni-octaethylporphyrin molecules40) will cause a partial freeze in the cluster rotation. That is, the cluster gains preferential orientation inside the cage governed by the external interactions. It is not known which effects are to be expected from a carbon nanotube on the M3N@C80 molecule inside. To address this problem, we performed extended computational studies of the Y2ScN@C80 molecule placed inside a carbon nanotube. It is well known that due to the “buried” nature of the f-shell, f-electrons rarely participate in chemical bonding. Therefore, the structural properties of lanthanide metallofullerenes can be well predicted by an analogous trivalent atom with close ionic radius. As ionic radii of Dy3+ and Y3+ are very close (0.91 Å and 0.90 Å, respectively41), the Y2ScN cluster closely matches the structural and dynamic properties of the Dy2ScN cluster inside the fullerene cage.
To understand the influence of the CNT encapsulation on the Dy2ScN cluster ordering and consequently the magnetic ordering, we have built the three model peapod systems Y2ScN@C80@CNT(14,7), Y2ScN@C80@CNT(12,8), and Y2ScN@C80@CNT(11,1). The two semiconducting chiral nanotubes were chosen to represent two different situations which the fullerene molecule may experience in the peapod. The inner diameter of the CNT(14,7) is 14.7 Å. This nanotube provides a tight fit of the Y2ScN@C80 molecule with comfortable π–π contact distance between fullerene and tube carbons of 3.2–3.4 Å. The system is therefore dubbed as B (“big”) in the following discussion. The inner diameter of CNT(12,8) is 13.6 Å, and the fullerene diameter has to be squeezed by 5% to fit the space inside the nanotube. The Y2ScN@C80@CNT(12,8) system is therefore dubbed as S (“small”). Importantly, the next smaller chiral CNT would be too small to fit the Y2ScN@C80 molecule in, and the next bigger one would be too roomy to have any effect on the Y2ScN cluster ordering. The only considered metallic tube M (“metal”) has a diameter of 14.9 Å which is similar to the CNT(14,7) and it provides a perfect fitting condition for the Y2ScN@C80 molecule inside. Having this nanotube included must help to understand the effect of near-zero-gap on the inner cluster ordering. Thus, this theoretical portfolio of CNTs covers experimentally expectable sizes (1.4–1.6 Å). In each system (S, B, and M), 144 conformational isomers were generated by 3D rotation of Y2ScN with respect to the cluster origin. The Euler's angles increments were of δϕ = 60°, δθ = 60°, and δψ = 40° which provides a representative structural cast, if not a sufficient sampling. All these systems were optimized at the PBE level of theory using the plane wave program VASP.
Fig. 3 shows the statistic of the optimized structures. Reportedly, in the free M3N@C80 molecule, there are three local minima for the cluster inside the fullerene cage (see ESI†). However, as one can see, the CNT confinement has produced multiple local minima. Moreover, the confinement has spread the energy spectra to higher energy end in case of B and M and even higher for the S-structures. Apparently, this is due to the fact that significant structural changes are prohibited by higher barriers in these structures and local nature of optimization procedure. Thus, only the closest minimum to the original structural guess has been located. The inner cluster/cage orientations in all these optimized structures are similar to those in the isolated molecule (see ESI†), but chemical compression by CNTs results in the spread of the relative energies (i.e. different orientations of the inner cluster with respect to the nanotube walls gain different energies). Thus, the encapsulation of the fullerene molecule inside the peapod results in the appearance of the energetically preferential orientations, and hence can lead to a partial ordering. Fig. 3 shows that the effect is stronger for the S-system than for the M-system or B-system. Moreover, as charge rearrangements (the charge transfer from CNT onto Y2ScNC80) in all systems are very similar we have concluded that the ordering is mostly driven by compression due to the confinement.
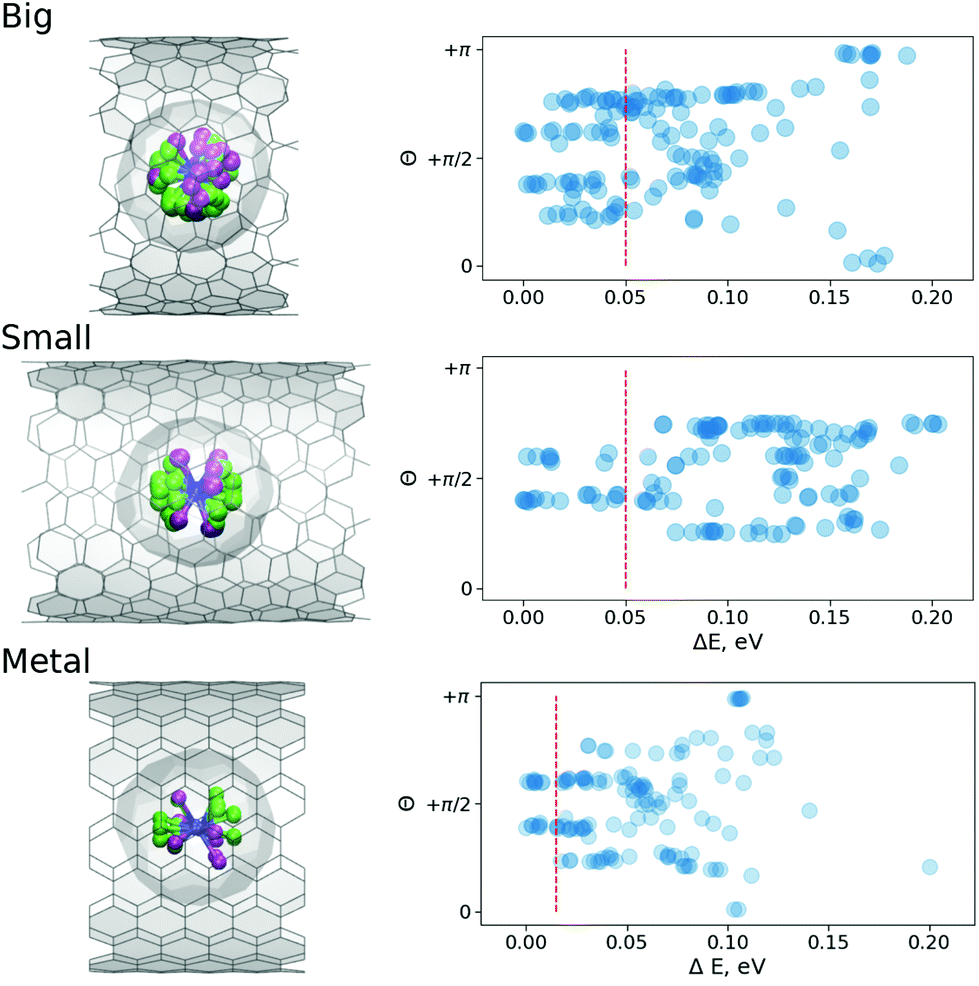 | ||
| Fig. 3 (Left) Superposed subset of optimized structures with the relative energy below the 0.05 eV thresholds (0.01 eV for the M-system), where Y-site is captured in light green and Sc-site in purple. (Right) Relative energy of different conformers of Y2ScNC80@CNT(14,7) (Big), Y2ScNC80@CNT(12,8) (Small) and Y2ScNC80@CNT(11,11) (Metal) systems as function of angle θ between the normal to the Y2ScN cluster plane and z-axis (aligned along the tube); the red dashed lines denote the energy threshold used to plot superposed structures in the left panel. | ||
As Fig. 3 shows, the energy distributions in the peapods cover the range of 0.2 eV, which is twice as big as for the isolated fullerene molecule. The value corresponds to 8kBT for T = 300 K, the angle distribution at room temperature should be more or less uniform. However, as the sample in the XMCD experiment is cooled down to He temperature, a less uniform angular distribution can be expected. From Fig. 3 one can see that if the accessible energy window is limited to 0.05 eV, in the S-system the preferential orientation of the normal to the cluster plane is orthogonal to the CNT axis. Therefore, the S-system structures will likely produce XMCD response with the ordering effects. For B-system structures, the similar energy window covers a broader range of angles, and so, it is safe to assume that the XMCD response will be a powder-like. Very similar conclusions can be made based on the molecular dynamic (MD) studies discussed below.
Unfortunately, at experimentally relevant temperatures of XMCD measurements (∼2 K) it would be hard to achieve any statistical significant integration of MD trajectories. At the same time, room temperature trajectories propagated for 5–10 ps would be sufficient to sample the local phase space. Moreover, if at room temperature the system would show any preferential geometric arrangements, it is reasonable to assume that after the cooling the preferential conformations would dominate the low-temperature phase. To study the dynamics of the endohedral cluster in the peapods we used the same 144 structures as initial guesses for 144 independent MD propagations with NVT ensemble at T = 300 K. Each geometry (in S- and B-system) was integrated for about 5–6 ps with 1 fs time step. This makes an overall integration time of 0.5 ns. Fig. 4 shows a superposition of all accessed geometries of the Y2ScN cluster in the S- and B-systems. Visually, the B-system has a more homogeneous spatial distribution of the positions of metal atoms (where Y-site is captured in light green and Sc-site in purple), and in the S-system the Y position is dominated in the front (which again points to a restricted rotation of the cluster in the S-systems). The histogram of the angle distribution between the normal to the cluster plane and the z-axis (θ) shows the same result in a numerical form (Fig. 4 (right)). According to Fig. 4, at room temperature, the B-system has a broader distribution of the θ-angles, while in the S-system the range of accessible angles is narrower and is biased towards θ = π/2.
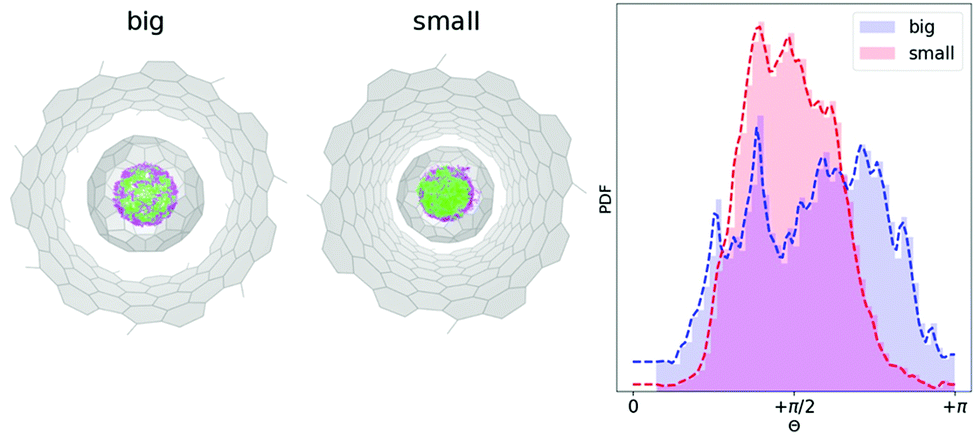 | ||
| Fig. 4 (Left) Superposed 144 Born–Oppenheimer MD trajectories (each propagated 5 ps) for Y2ScNC80@CNT(14,7) (big) and Y2ScNC80@CNT(12,8) (small). The Y and Sc atoms are captured in light green and purple respectively. (Right) Angle distribution θ (see Fig. 3) of the normal to the Y2ScN cluster-plane and z-axis (the CNT-axis) along joint 0.5 ns MD trajectory at T = 300 K. | ||
At this point, one can justifiably conclude that the confinement of Dy2ScN@C80 inside a CNT is likely to orient the clusters so that one Dy-I within some accuracy is placed on the tube-axis while the second Dy-II is placed within the geometrical constraints (Dy–N–Dy angle 120°) with a freedom to rotate around the tube-axis (Θrot in Fig. 5(a) and 3). In our experimental setup, the incoming X-ray beam (as well as magnetic field Bz) is always perpendicular to the tube-axis and due to cylindrical symmetry of this simplified problem, there are only two distinct XAS/XMCD spectra to be expected, with ground state magnetic moment perpendicular (90°) to the incoming X-ray beam and with 60° angle. These two spectra are shown in Fig. 5(b). Naturally, an average of these spectra would be different from an isotropic one due to a limited sampling. However, as our structural modelling indicates, positions of Dy-I and Dy-II would deviate from the proposed idealistic positions within a certain energy window. Fig. 5(c) shows a comparison between CNT-confined spectra and an isotropic spectrum (includes all 144 considered structures) for different sizes of the selected window. In fact, the average XAS spectrum obtained based on seven most stable structures (Fig. 5(c), energy ΔE ≤ 0.01 eV) reassembles closely experimental observations [Fig. 2(c)]. Having this window just 0.1 eV broad, theoretically, it would be impossible to tell the difference between @SWCNT and isotropic cases. This further suggests that the peapod system is likely to be under thermodynamic control and thus only most stable structures are allowed to form.
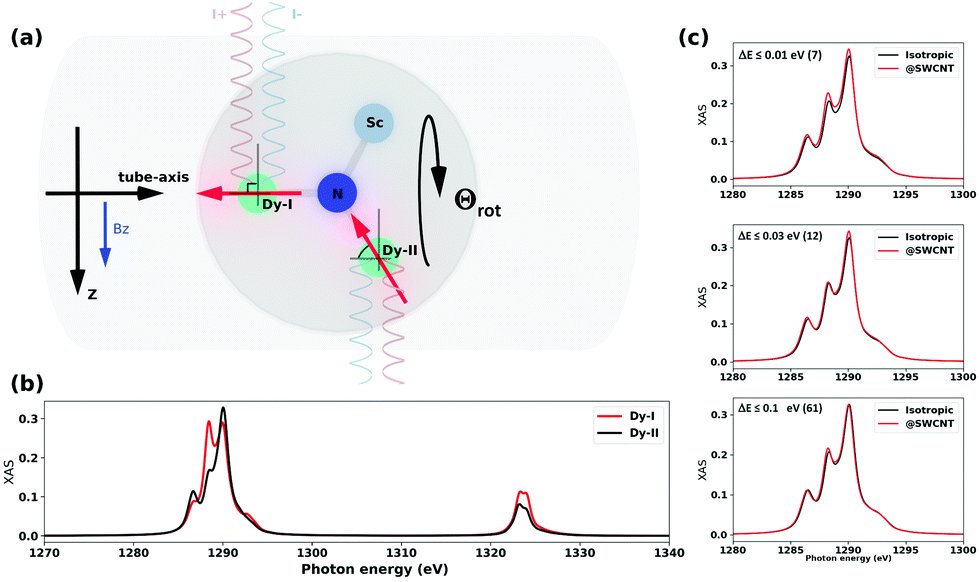 | ||
| Fig. 5 (a) Schematic representation of the peapods along the tube axis. The direction of magnetic field (Bz) and direction of X-ray beams (I+ and I−) with respect to most probable positions of Dy-ions (Dy-I, Dy-II with the magnetic moments’ orientation in ground states i.e. Jz = 15/2). (b) Theoretical XAS spectra for two Dy-sites as shown in (a), the model point charges qN = −2 and qSc,Dy = +2 (these charges provide a correct structure of 4I15/2 multiplets). Note that rotation of the Dy2ScN cluster around (Dy-I)–N bond (Θrot) would not change Dy-II spectrum. (c) Theoretical XAS for average overall 144 confirmation (isotropic) and well-optimized structures within relative energy window (≤0.01 eV (7), ≤0.03 eV (12), ≤0.1 eV (61)). The number in the round brackets indicates how many structures fall into the selected window (see Fig. 3). | ||
Recording the maximum XMCD signal at the Dy M5-edge while sweeping the magnetic field probes the response of the Dy3+ moments to external magnetic field changes. Fig. 6(a) displays an element-specific magnetization curve recorded from the bulk sample. The magnetization curve exhibits the characteristic low-temperature hysteresis of Dy2ScN@C80 with a significant remanent magnetization and coercive field.37 In strong contrast, the same measurement performed on the peapod sample yields a magnetization curve that, within the error bars, does not show a clear hysteresis. Hysteresis is observed in the system when the relaxation of the magnetization is slow compared to the measurement time. If instead, the system has time to relax to an equilibrium, hysteresis will not be observed and the magnetization curve will be reminiscent of a Brillouin function with a shape determined by the temperature.37 To verify that the absence of hysteresis in the peapod sample is not caused by a higher local temperature due to poor thermal conductance across the TEM grid, the data is compared to an equilibrium magnetization curve recorded with an average field sweep rate of 0.4 mT s−1 at 4 K from a bulk sample using a SQUID, see Fig. 6(b). This comparison is not taking into account the small anisotropy in the peapod sample which would translate into a slightly larger average moment and thereby modify the slope of the magnetization curve. However, a 10% increase in the effective magnetic moment would only correspond to a change in the temperature of a few percent. Therefore, the steeper slope observed in the magnetization curve recorded from the peapod sample in Fig. 6(b) demonstrates that there is no significant difference in the temperature between the two systems when performing the XMCD measurements, and there must be additional effects that are influencing the magnetic bistability of the encapsulated endofullerenes. Reduced magnetic bistability was observed in a sub-monolayer of Dy2ScN@C80 on a metal surface.38 Depending on the chirality, nanotubes can have zero bandgaps, and it is possible that encapsulating endofullerenes in metallic SWCNTs yield a similar effect. Substantial alteration of the magnetic properties has also been observed when depositing Fe4 SMMs on graphene.42 However, the nanotubes used in this study consist of a mixture of semiconducting and metallic tubes of different chiralities, and interaction with conducting electrons cannot be the only mechanism contributing to the observed decreased magnetic bistability. In the peapod sample, a fraction of the encapsulated endofullerenes could also be affected by dipole–dipole interaction with residue metallic catalyst nanoparticles. Though, such interactions should only have a minor influence on the total magnetic response of the system. The exposure to high photon flux can result in X-ray induced demagnetization.43 However, in the present case, the photon flux was the same for the two systems and this effect should not contribute to the observed differences between the two systems.
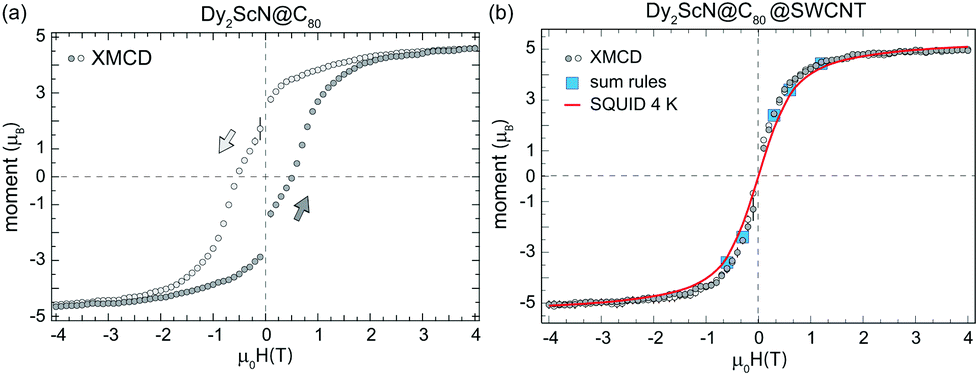 | ||
| Fig. 6 Element specific magnetization curves recorded at the Dy M5-edge with the temperature set to 2 K, a field sweep rate of 33 mT s−1, the X-ray beam parallel to the sample normal, and a photon flux of 5 × 1010 ph per s per mm2 from Dy2ScN@C80 endofullerens in the bulk phase (a) and encapsulated in SWCNTs (b). The magnetization curves correspond to the average of several independent measurements and the error bars shown as black bars are the standard deviation at each external magnetic field. The magnetization curves are scaled to the magnetic moments extracted by applying a sum-rule analysis to the XMCD data recorded at ±6.5 T (Table 1). The drop in magnetization at zero field in (a) is a consequence of the time of 30 s which it takes for the magnet to switch polarity. The 4 K magnetization curve in (b) was recorded using a SQUID with an effective field sweep rate of 0.4 mT s−1. | ||
3. Conclusions
We have performed an HRTEM, XAS, and a computational study of one-dimensional arrays of Dy2ScN@C80 SMMs packed inside SWCNTs. X-ray absorption spectra recorded at the Dy M5-edge from the encapsulated Dy2ScN@C80 endofullerenes display deviation from that of an isotropic bulk sample which indicates a partial ordering of the endohedral clusters inside the nanotubes. Calculations performed on structurally analogous Y2ScN@C80 molecules packed into SWCNTs demonstrate that the encapsulation results in energetically preferential orientations of the endohedral clusters and that the ordering effect depends on the relative size of the fullerene cage and the diameter of the nanotubes. When compared to a bulk sample, element-specific magnetization curves recorded at ∼2 K from SWCNTs filled with Dy2ScN@C80 SMMs reveal a decreased magnetic bistability and vanishing hysteresis for the encapsulated endofullerenes. In the case of endofullerene SMMs, a preferential orientation of the endohedral units implies alignment of the molecular anisotropy axis and magnetic ordering. Thus, the presented results demonstrate the feasibility of forming ordered and well protected one-dimensional spin chains that could possibly be addressed through the creation of SMM/SWCNT hybrid devices.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financed by the Swedish Research Council (Grant No. 2015-00455), Sklodowska Curie Actions co-founding project INCA 600398, and the Volkswagen Stiftung program “Integration of Molecular Components in Functional Macroscopic Systems” (Grant No. 86393 and 9200). C. M. acknowledges funding by the DFG project ME 32375/6-1 and the “Niedersächsisches Vorab” program of the Niedersächsisches Ministerium für Wissenschaft und Kultur. S. M. A. acknowledges funding by the Marie Sklodowska-Curie actions, EU project SamSD (grant agreement number 748635). A. A. P. acknowledges the European Research Council under the European Unions Horizon 2020 research and innovation programme (grant no 648295 “GraM3”). We thank G. Manoharan for Raman measurements on the pristine SWCNTs and Thomas Greber for access to the MPMS3.References
- G. Binasch, P. Grünberg, F. Saurenbach and W. Zinn, Phys. Rev. B: Condens. Matter Mater. Phys., 1989, 39, 4828 CrossRef.
- M. N. Baibich, J. Broto, A. Fert, N. F. van Dau, F. Petroff, P. Etienne, G. Creuzet, A. Friederich and J. Chazelas, Phys. Rev. Lett., 1988, 61, 2472 CrossRef PubMed.
- K. Tsukagoshi, B. W. Alphenaar and H. Ago, Nature, 1999, 401, 572–574 CrossRef.
- L. E. Hueso, J. M. Pruneda, V. Ferrari, G. Burnell D, J. P. Valdés-Herrera, B. D. Simons, P. B. Littlewood, E. Artacho, A. Fert and N. D. Mathur, Nature, 2007, 445, 410–413 CrossRef PubMed.
- M. Urdampilleta, S. Klyatskaya, J. Cleuziou, M. Ruben and W. Wernsdorfer, Nat. Mater., 2011, 10, 502–506 CrossRef PubMed.
- N. Ishikawa, M. Sugita, T. Ishikawa, S.-y. Koshihara and Y. Kaizu, J. Am. Chem. Soc., 2003, 125, 8694–8695 CrossRef PubMed.
- M. Urdampilleta, S. Klyatskaya, M. Ruben and W. Wernsdorfer, Phys. Rev. B: Condens. Matter Mater. Phys., 2013, 87, 195412 CrossRef.
- L. Bogani and W. Wernsdorfer, Nat. Mater., 2008, 7, 179–186 CrossRef PubMed.
- M. N. Leuenberger and D. Loss, Nature, 2001, 410, 789–793 CrossRef PubMed.
- M. d. C. Giménez-López, F. Moro, A. La Torre, C. J. Gómez-García, P. D. Brown, J. van Slageren and A. N. Khlobystov, Nat. Commun., 2011, 2, 407 CrossRef PubMed.
- R. Westerström, J. Dreiser, C. Piamonteze, M. Muntwiler, S. Weyeneth, H. Brune, S. Rusponi, F. Nolting, A. Popov, S. Yang, L. Dunsch and T. Greber, J. Am. Chem. Soc., 2012, 134, 9840–9843 CrossRef PubMed.
- K. Junghans, C. Schlesier, A. Kostanyan, N. A. Samoylova, Q. Deng, M. Rosenkranz, S. Schiemenz, R. Westerström, T. Greber, B. Büchner and A. A. Popov, Angew. Chem., Int. Ed., 2015, 54, 13411–13415 CrossRef PubMed.
- F. Liu, C.-L. Gau, Q. Deng, X. Zhu, A. Kostanyan, R. Westerström, S. Wang, Y.-Z. Tan, J. Tao, S.-Y. Xie, A. Popov, T. Greber and S. Yang, J. Am. Chem. Soc., 2016, 138, 14764–14771 CrossRef PubMed.
- F. Liu, D. Krylov, L. Spree, S. Avdoshenko, N. A. Samoylova, M. Rosenkranz, A. Kostanyan, T. Greber, A. Wolter, B. Büchner and A. A. Popov, Nat. Commun., 2017, 8, 16098 CrossRef PubMed.
- F. Fritz, R. Westerström, A. Kostanyan, C. Schlesier, J. Dreiser, B. Watts, L. Houben, M. Luysberg, S. M. Avdoshenko, A. A. Popov, C. M. Schneider and C. Meyer, Nanotechnology, 2017, 28, 435703 CrossRef PubMed.
- B. W. Smith, M. Monthioux and D. E. Luzzi, Nature, 1998, 396, 323–324 CrossRef.
- W. Harneit, Phys. Rev. A, 2002, 65, 032322 CrossRef.
- W. Harneit, C. Meyer, A. Weidinger, D. Suter and J. Twamley, Phys. Status Solidi B, 2002, 233, 453–461 CrossRef.
- C. Fantini, A. Jorio, M. Souza, M. S. Strano, M. S. Dresselhaus and M. A. Pimenta, Phys. Rev. Lett., 2004, 93, 147406 CrossRef PubMed.
- C. Meyer, C. Spudat, L. Houben and C. M. Schneider, Nanotechnology, 2009, 065603 CrossRef PubMed.
- A. N. Khlobystov, D. A. Britz, J. Wang, S. A. O'Neil, M. Poliakoff and G. A. D. Briggs, J. Mater. Chem., 2004, 14, 2852–2857 RSC.
- Ernst Ruska-Centre for Microscopy and Spectroscopy with Electrons (ER-C), et al. , J. Large-Scale Res. Facil., 2015, 1, A34 CrossRef.
- C. Piamonteze, U. Flechsig, S. Rusponi, J. Dreiser, J. Heidler, M. Schmidt, R. Wetter, M. Calvi, T. Schmidt, H. Pruchova, J. Krempasky, C. Quitmann, H. Brune and F. Nolting, J. Synchrotron Radiat., 2012, 19, 661–674 CrossRef PubMed.
- J. Krempaský, U. Flechsig, T. Korhonen, D. Zimoch, C. Quitmann and F. Nolting, AIP Conf. Proc., 2010, 1234, 705–708 CrossRef.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 47, 558–561 CrossRef.
- G. Kresse and D. Joubert, Phys. Rev. B: Condens. Matter Mater. Phys., 1999, 59, 1758–1775 CrossRef.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef.
- J. Hutter, M. Iannuzzi, F. Schiffmann and J. VandeVondele, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2014, 4, 15–25 Search PubMed.
- C. Hartwigsen, S. Goedecker and J. Hutter, Phys. Rev. B: Condens. Matter Mater. Phys., 1998, 58, 3641 CrossRef.
- J. VandeVondele and J. Hutter, J. Chem. Phys., 2007, 127, 114105 CrossRef PubMed.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef PubMed.
- A. Uldry, F. Vernay and B. Delley, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 125133 CrossRef.
- G. Henkelman, A. Arnaldsson and H. Jónsson, Comput. Mater. Sci., 2006, 36, 354–360 CrossRef.
- M. d. C. Giménez-López, A. Chuvilin, U. Kaiser and A. N. Khlobystov, Chem. Commun., 2011, 47, 2116–2118 RSC.
- B. T. Thole, P. Carra, F. Sette and G. van der Laan, Phys. Rev. Lett., 1992, 68, 1943–1946 CrossRef PubMed.
- P. Carra, B. T. Thole, M. Altarelli and X. Wang, Phys. Rev. Lett., 1993, 70, 694–697 CrossRef PubMed.
- R. Westerström, J. Dreiser, C. Piamonteze, M. Muntwiler, S. Weyeneth, K. Krämer, S.-X. Liu, S. Decurtins, A. Popov, S. Yang, L. Dunsch and T. Greber, Phys. Rev. B: Condens. Matter Mater. Phys., 2014, 89, 060406 CrossRef.
- R. Westerström, A.-C. Uldry, R. Stania, J. Dreiser, C. Piamonteze, M. Muntwiler, F. Matsui, S. Rusponi, H. Brune, S. Yang, A. Popov, B. Büchner, B. Delley and T. Greber, Phys. Rev. Lett., 2015, 114, 087201 CrossRef PubMed.
- A. Kostanyan, R. Westerström, Y. Zhang, D. Kunhardt, R. Stania, B. Büchner, A. A. Popov and T. Greber, Phys. Rev. Lett., 2017, 119, 237202 CrossRef PubMed.
- M. M. Olmstead, T. Zuo, H. C. Dorn, T. Li and A. L. Balch, Inorg. Chim. Acta, 2017, 468, 321–326 CrossRef.
- R. D. Shannon, Acta Crystallogr., Sect. A: Cryst. Phys., Diffr., Theor. Gen. Crystallogr., 1976, 32, 751–767 CrossRef.
- C. Cervetti, A. Rettori, M. G. Pini, A. Cornia, A. Repollés, F. Luis, M. Dressel, S. Rauschenbach, K. Kern, M. Burghard and L. Bogani, Nat. Mater, 2016, 15, 164–168 CrossRef PubMed.
- J. Dreiser, R. Westerström, C. Piamonteze, F. Nolting, S. Rusponi, H. Brune, S. Yang, A. Popov, L. Dunsch and T. Greber, Appl. Phys. Lett., 2014, 105, 032411 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/C8NR05386C |
| This journal is © The Royal Society of Chemistry 2018 |