
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Exploiting the biomolecular corona: pre-coating of nanoparticles enables controlled cellular interactions†
Johanna
Simon
 ab,
Laura K.
Müller
b,
Maria
Kokkinopoulou
b,
Ingo
Lieberwirth
b,
Svenja
Morsbach
b,
Katharina
Landfester
b and
Volker
Mailänder
*ab
ab,
Laura K.
Müller
b,
Maria
Kokkinopoulou
b,
Ingo
Lieberwirth
b,
Svenja
Morsbach
b,
Katharina
Landfester
b and
Volker
Mailänder
*ab
aDermatology Clinic, University Medical Center of the Johannes Gutenberg-University Mainz, Langenbeckstr. 1, 55131 Mainz, Germany. E-mail: volker.mailaender@unimedizin-mainz.de
bMax Planck Institute for Polymer Research, Ackermannweg 10, 55128 Mainz, Germany
First published on 24th May 2018
Abstract
Formation of the biomolecular corona ultimately determines the successful application of nanoparticles in vivo. Adsorption of biomolecules such as proteins is an inevitable process that takes place instantaneously upon contact with physiological fluid (e.g. blood). Therefore, strategies are needed to control this process in order to improve the properties of the nanoparticles and to allow targeted drug delivery. Here, we show that the design of the protein corona by a pre-formed protein corona with tailored properties enables targeted cellular interactions. Nanoparticles were pre-coated with immunoglobulin depleted plasma to create and design a protein corona that reduces cellular uptake by immune cells. It was proven that a pre-formed protein corona remains stable even after nanoparticles were re-introduced to plasma. This opens up the great potential to exploit protein corona formation, which will significantly influence the development of novel nanomaterials.
Introduction
Nanoparticle (NP) based drug delivery systems hold great promise as they offer the possibility for targeted cell interactions.1 This eventually improves the drug bioavailability,2 minimizes toxic side effects3 and reduces the drug dose.4 Despite the recent progress in the development of novel nanoparticles, the majority of those systems show a low targeting efficiency in vivo.5Rapid recognition of nanoparticles by immune cells causes their fast clearance from the blood stream and therefore prevents interactions with targeted cells.6,7 This process is mainly governed by the adsorption of blood proteins towards the nanoparticles’ surfaces. The adsorption happens immediately when nanoparticles enter the blood stream and has been termed ‘biomolecular or protein corona formation’.8–10 Specific proteins such as immunoglobulins11 and complement proteins12 are known to mediate interactions with phagocytic cells and hereby influence the clearance process (‘opsonization’).13,14 There are different reports, which could prove a direct recognition of corona proteins by specific cell-receptors. For example, Lara et al. developed an immuno-mapping technique and was herby able to show that corona proteins (e.g. immunoglobuline G) present functional motifs, which allow an interaction with FcγI receptors.15 Other studies indicated that corona proteins are denatured upon binding towards the nanoparticles’ surface and therefore, the unfolded protein can be recognized via scavenger receptors.16,17
To overcome the issue of opsonization, several approaches have been developed in order to mask the nanoparticles from phagocytic cells.18 Coating the nanoparticles’ surface with hydrophilic polymers e.g. poly(ethylene glycol) (PEG) is the common strategy to prolong the blood circulation (‘stealth effect’) and hereby enabling the nanoparticle to reach the desired target.19 This effect has been widely described to be a result of reduced protein interactions with PEGylated surfaces.20 However, several reports indicated that PEGylation cannot completely abolish protein adsorption.21,22 On top of this, Hamad and colleagues23 could show that PEG can even trigger the activation of the complement system.
Therefore, alternative strategies are needed to improve the properties of targeted nanoparticles in vivo.24 Various studies have investigated the effect of corona formation on nanoparticle targeting.25 It was found that nanoparticles can recruit specific proteins from plasma, which eventually promote interactions with targeted cells.26 Adsorption of apolipoproteins to polysorbate 80-coated nanoparticles was shown to enhance the transport of nanoparticles across the blood brain barrier.27 Additionally, Caracciolo et al. saw that the adsorption of vitronectin can enhance cellular interactions with cancer cells.28 Further, we already showed that due to a pre-formed protein corona nanoparticle aggregation in blood plasma is prevented.29 Next to this, other studies demonstrated that the cytotoxicity of nanoparticles was strongly reduced if nanoparticles were pre-coated with cell culture medium containing FBS.30,31 This highlights the great potential to exploit corona formation.
To take this one-step forward, we aimed to create a pre-formed protein corona, which enables controlled cellular interactions. Here, it was intended to engineer nanoparticles with stealth properties mediated only through corona proteins. Immunoglobulins (IgG) are one major class of proteins (‘opsonins’), which are known to enhance cellular uptake by macrophages via interactions with the Fc receptor.32 Therefore, we removed IgG from human plasma via affinity chromatography. This protein fraction (‘IgG depleted plasma’) was chosen as a protein coating in order to create a pre-formed protein corona, which should prevent interactions with macrophages. To explore the stealth properties of the pre-formed protein, we analyzed the cellular interactions of uncoated and pre-coated nanoparticles with a macrophage cell line (RAW264.7). Next, the major aspect of our work focused on the question, whether the pre-adsorbed corona proteins are exchanged or covered by other proteins over time, when pre-coated nanoparticles are re-exposed to human plasma. This question needs to be addressed in order to reveal if nanoparticle pre-coating is an effective strategy to enable controlled cellular interactions in vivo. To answer this question, we carried out a detailed proteomic analysis (LC-MS). Additionally, dynamic light scattering (DLS) and isothermal titration (ITC) experiments were used to study the direct interaction of uncoated and pre-coated nanoparticles with plasma proteins.
Taking this together, we could prove that a pre-formed protein corona remains stable, which further implies that pre-coating of nanoparticles is a feasible method to engineer the protein corona and to obtain tailored protein corona properties for targeted cell interactions.
Results and discussion
As a model system, polystyrene nanoparticles (PS-NPs) stabilized with the PEG-based surfactant Lutensol AT50 were used in this study. Additionally, human plasma was depleted of immunoglobulins (IgG) as previously described.29 ‘IgG depleted plasma’ was isolated from human plasma via affinity chromatography using a Protein A column. The protein composition was determined by LC-MS and we were able to reduce the total amount of IgG from 28% down to 4% (Fig. S1†). Then, carboxy- and amino functionalized PS-NPs were incubated with IgG depleted plasma and characterized before and after incubation regarding size and ζ-potential (Table 1 and Fig. S2†).| PS-COOH | PS-NH2 | |||
|---|---|---|---|---|
| Protein coating | Uncoated | Coated | Uncoated | Coated |
| Diameter | 130 ± 13 nm | 164 ± 16 nm | 150 ± 15 nm | 162 ± 16 nm |
| ζ-Potential | −27 ± 8 mV | −17 ± 9 mV | +4 ± 1 mV | −29 ± 6 mV |
Dynamic light scattering experiments (DLS) indicate a minor size increase (∼20 nm) after protein coating, which is attributed to protein corona formation.29,33 Additional transmission electron microscopy images (TEM) confirmed the protein layer surrounding the nanoparticles as found in previous reports33 (Fig. 1A). To characterize the biological identity of nanoparticles after protein coating, the exact protein corona composition was identified by LC-MS (Fig. 1B and C).
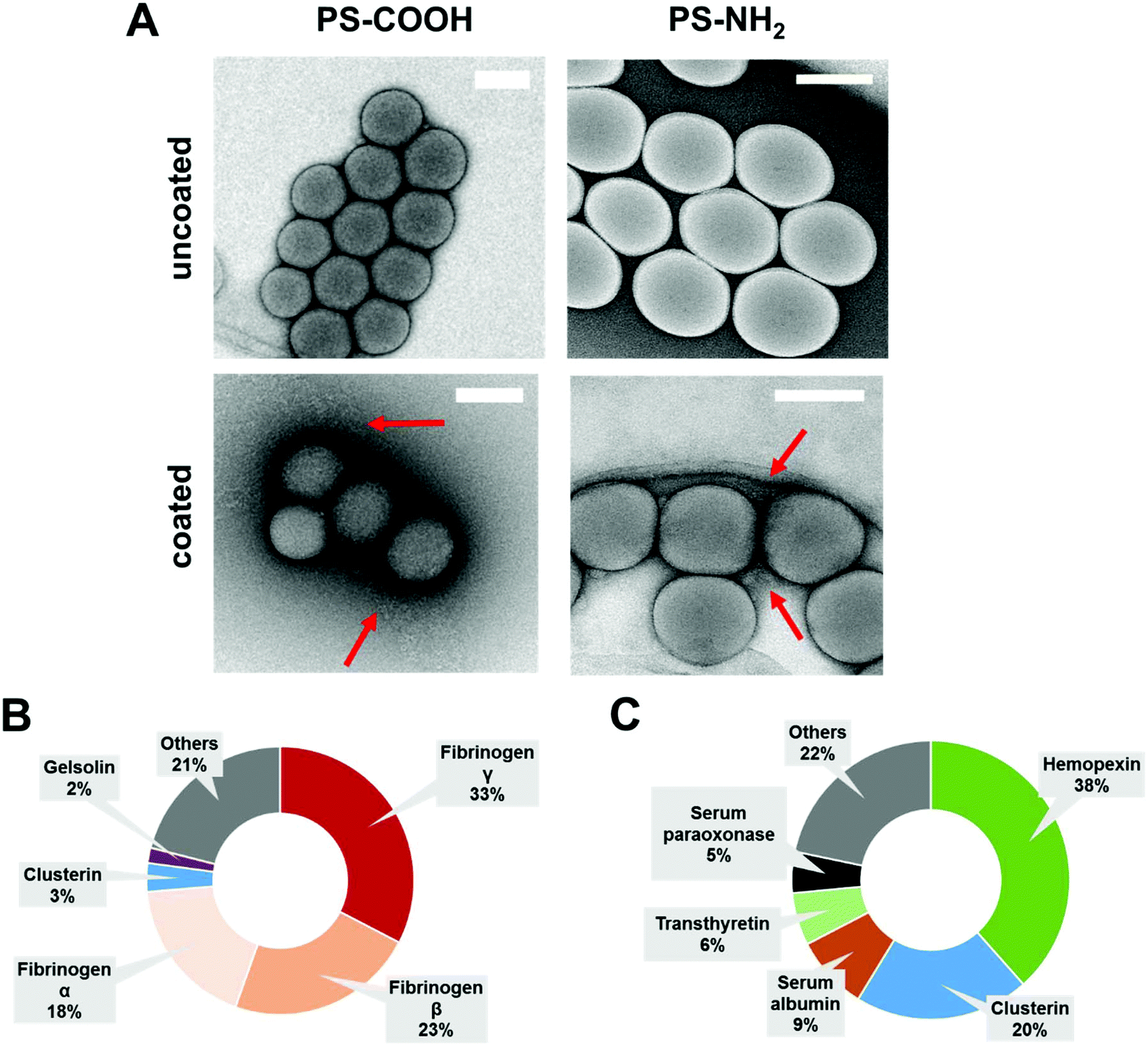 | ||
| Fig. 1 Biological identity of PS-COOH and PS-NH2 NPs. (A) Representative TEM images of PS-NPs (PS-COOH and PS-NH2) before (uncoated) and after (coated) pre-incubation with IgG depleted plasma. Red arrows highlight the lose protein corona network surrounding nanoparticles. Scale bar: 100 nm. LC-MS analysis of the protein composition after incubation with IgG depleted plasma. TOP 5 most abundant corona proteins are shown (B = PS-COOH and C = PS-NH2). | ||
Therefore, nanoparticles were incubated with IgG depleted plasma to allow corona formation, and afterwards centrifuged and washed (see material/methods).9 As shown before, the surface functionalization of the nanoparticles strongly influences the protein adsorption pattern.34,35 Coated carboxy-functionalized nanoparticles (PS-COOH) were surrounded by a protein layer, which was strongly enriched by fibrinogen (∼74%). In contrast, hemopexin (∼38%) and clusterin (∼20%) were the major hard corona proteins for coated amino-functionalized nanoparticles (PS-NH2). A full list of all identified proteins giving the relative values in % and absolute amounts in fmol is supplemented in a separate Excel File Table S1.†
IgG depleted plasma was chosen as protein coating as it was intended to reduce the interaction of pre-coated nanoparticle with phagocytic cells in order to obtain stealth properties (Fig. 2A). Therefore, cellular uptake towards macrophages (RAW264.7) of uncoated and pre-coated nanoparticles was analyzed via flow cytometry (Fig. 2B) and confocal laser scanning microscopy (Fig. 2C).
 | ||
| Fig. 2 (A) Schematic overview: Exploiting protein corona formation. NPs are pre-coated with IgG depleted plasma to create an artificial protein corona, which reduces interactions with phagocytic cells. Macrophages (RAW264.7) were incubated with coated or pre-coated PS-COOH NPs (75 μg mL−1) for 2 h at 37 °C. Cell uptake was analyzed by flow cytometry (B) and confocal laser scanning microscopy (C). Cells were kept in cell culture medium with (+) or without plasma proteins (−). Scale bar: 5 μm. | ||
To present the principal mechanism, we first summarized a detailed analysis for PS-COOH nanoparticles (Fig. 2 and 3). In the following section, we additionally focused on the investigations for PS-NH2 nanoparticles (Fig. 4).
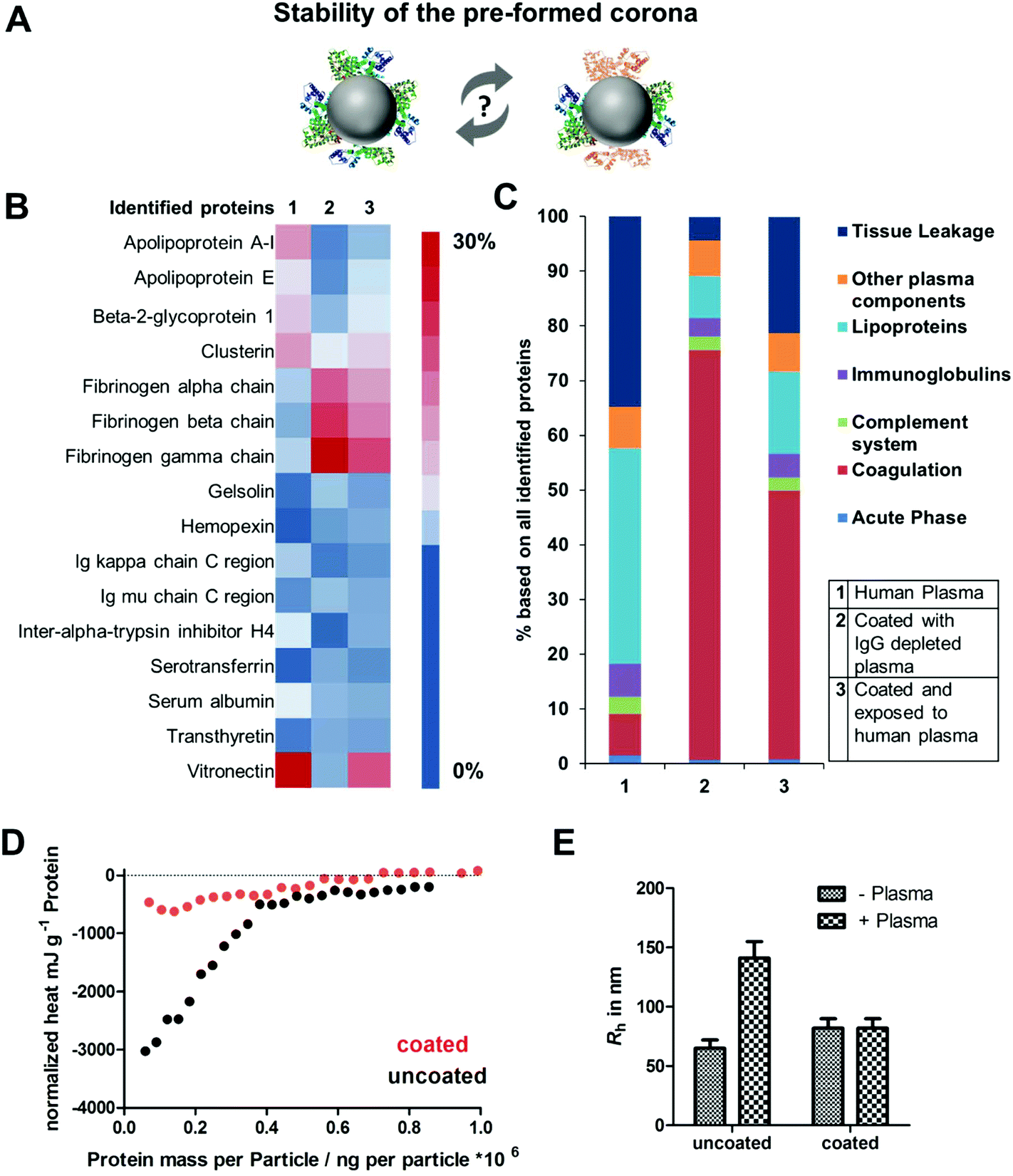 | ||
| Fig. 3 (A) Investigating the stability of a pre-formed protein corona. (B) LC-MS analysis of the most abundant (amount >1%) corona proteins after plasma incubation (1), pre-coating (2) or if pre-coated NPs were re-introduced to plasma (3). (C) Protein classification of all identified proteins. (D) ITC measurements. Titration of plasma to uncoated and pre-coated PS-COOH NPs. One representative measurement out of five replicates each is shown exemplarily. (E) Multi-angle dynamic light scattering (DLS) analysis of uncoated and pre-coated PS-COOH NPs in human plasma. Hydrodynamic radii (Rh) is given in nm. | ||
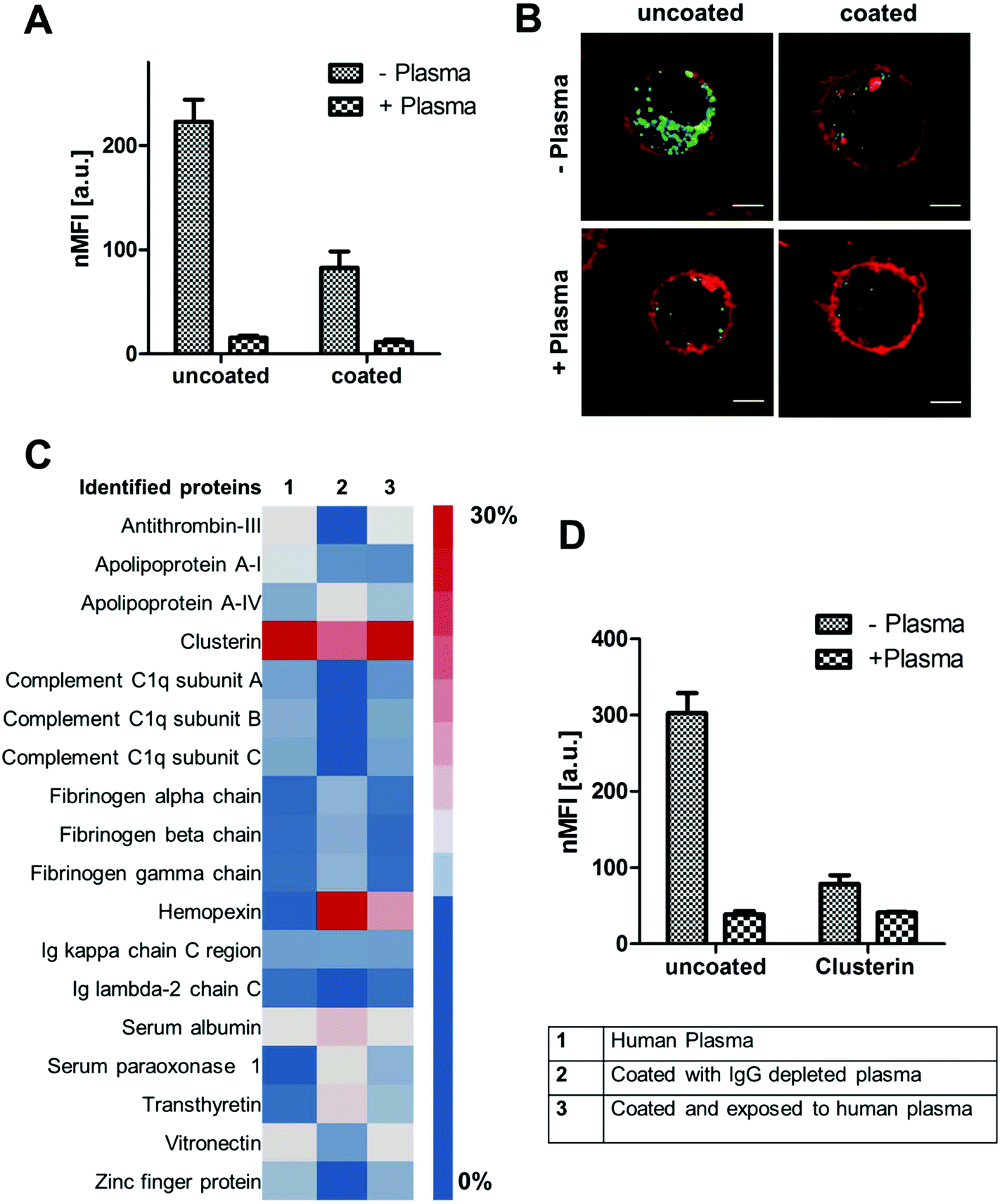 | ||
| Fig. 4 (A) + (B) Cellular uptake of uncoated and pre-coated PS-NH2 NPs towards macrophages (RAW 264.7) analyzed by flow cytometry and confocal laser scanning microscopy. Cells were kept in cell culture medium with (+) or without (−) plasma proteins. (C) LC-MS analysis of the corona composition after plasma incubation (1), pre-coating (2) or if pre-coated NPs were re-introduced to plasma (3). (D) Flow cytometry analysis of uncoated or clusterin coated PS-NH2 NPs in the presence (+) or absence of human plasma (−). | ||
Carboxy-functionalized nanoparticles were coated with IgG depleted plasma and applied to cell culture medium without additional proteins (marked as −plasma) or supplemented with plasma proteins (marked +plasma). This offered the possibility to study the influence of the pre-coating on the cellular uptake of nanoparticles and to investigate if the preformed protein corona (coating with IgG depleted plasma) is exchanged or remains stable after re-incubation with whole plasma (Fig. 2).
Flow cytometry analysis (Fig. 2B) and confocal laser scanning microscopy images clearly indicated the rapid uptake of uncoated PS-COOH nanoparticles in all cases (with or without plasma). Hence, cellular interactions of PS-COOH NPs pre-coated with IgG depleted plasma were strongly decreased (Fig. 2C). Most importantly, even if pre-coated NPs were re-introduced to plasma, cellular uptake was still reduced. This highlights that the obtained stealth properties due to the pre-formed protein corona were preserved meaning that pre-coating allowed us to control cellular uptake.
Based on this, we now wanted to focus on the interaction between plasma proteins and uncoated or pre-coated nanoparticles once they were introduced to whole plasma. Here, it was questioned how pre-coating influences protein corona formation (Fig. 3A). The hard protein corona of uncoated as well as pre-coated nanoparticles formed after incubation in full plasma was isolated via repetitive centrifugation and analyzed by Pierce Assay (Tables S2†), SDS PAGE (Fig. S3†) and LC-MS (Fig. 3B). We found that uncoated PS-COOH, which were incubated with full plasma, were surrounded by a protein corona, which was strongly enriched with vitronectin (∼33%). In contrast to this, we did not identify vitronectin as being highly abundant in the corona formed with IgG depleted plasma. Here, the hard corona of pre-coated nanoparticles was enriched with fibrinogen (Fig. 3B).
Next, we saw that there was no significant change in the protein adsorption pattern if pre-coated PS-COOH nanoparticles were re-introduced into whole plasma (Fig. 3C). This underlines the stability of the pre-formed protein corona (Fig. 3C). Hence, we noted minor differences in the corona composition. Re-adsorption of other plasma proteins (e.g. vitronectin) had occurred. Nevertheless, this did not affect cellular uptake decisively (Fig. 2).
To further investigate the properties of the pre-formed corona, isothermal titration calorimetry experiments (ITC) were carried out (Fig. 3D and raw data heat rates see Fig. S5†). Plasma was titrated to uncoated and coated PS-COOH NPs. With ITC it is possible to study the binding or adsorption behavior of proteins towards nanoparticles in situ.9,36 We observed a strong binding of plasma proteins towards uncoated PS-COOH NPs (black circles). This interaction was diminished when NPs were pre-coated (red circles), which further highlights the stealth properties of the pre-formed protein corona. This is in accordance with the cellular uptake that was shown to be strongly reduced if nanoparticles were pre-coated (Fig. 2).
As described in previous work, interactions of NPs with plasma proteins can cause aggregation of NPs hereby highly affecting their in vivo biodistribution.37 With multi angle dynamic light scattering (DLS) it is possible to monitor the size of nanoparticles incubated with plasma.29,38 This method allows studying direct interactions of proteins and nanoparticles without applying any washing step. When uncoated PS-COOH nanoparticles were applied to plasma, an overall size increase of ∼100 nm was measured indicating aggregation formation (Fig. 3E and raw data Fig. S6 and S7†). In strong contrast to this, we did not observe any size increase for pre-coated PS-COOH nanoparticles after plasma incubation. This underlines that the pre-formed protein corona remains stable after re-exposure to plasma.
Overall, we were able to present here a set of different analytical methods that can be generally applied to investigate if a defined pre-coating can enable targeted cell interaction. For the here chosen model system using IgG depleted plasma we were able to prove that pre-coating offers the possibility to tailor the biological properties of the nanoparticles.
As highlighted above (Fig. 1) and intensively studied in literature the nanoparticle charge can significantly influence protein corona formation34 and cellular uptake behavior.17 Therefore, we additionally explored the influence of pre-coating with IgG depleted plasma for amino-functionalized nanoparticles (PS-NH2) (Fig. 4). First, cellular internalization of uncoated and pre-coated PS-NH2 towards macrophages was studied followed by a detailed proteomic investigation of the hard protein corona.
For PS-NH2 nanoparticles we found that only in the absence of proteins (−plasma) nanoparticles are highly internalized (Fig. 4A and B). If plasma proteins were present (+plasma), cellular uptake was strongly reduced. Coated PS-NH2 nanoparticles (with IgG depleted plasma) displayed a significantly lower internalization rate compared to uncoated PS-NH2 in both cases (+with or −without plasma).
Analyzing the protein corona of uncoated PS-NH2 indicates a strong enrichment of clusterin (∼60%). Lower amounts of clusterin (∼20%) were observed for nanoparticles incubated with IgG depleted plasma (Fig. 4C). Clusterin has been identified as major corona protein of PEGylated nanoparticles19 and it was found that clusterin reduces interactions with macrophages.22 The here presented PS nanoparticles are stabilized with the non-ionic surfactant Lutensol AT50, which has a PEG analog structure and therefore the interaction with clusterin is favored.
We were able to confirm that via pre-coating with clusterin cellular uptake of the here investigated PS-NH2 was strongly decreased (Fig. 4D). However, it has to be noted that the uptake levels of PS-NH2 after plasma incubation were even lower meaning that also other corona proteins, their orientation or even other biomolecules (e.g. sugar, lipids) may contribute to this effect. Overall, this demonstrates that PS-NH2 nanoparticles are surrounded by a protein corona with natural stealth properties as due to plasma coating interactions with phagocytic cells are reduced.
Conclusion
Here, we demonstrate that pre-coating of nanoparticles allows the defined design of the protein corona and offers the possibility to regulate cellular interaction. IgG depleted plasma was chosen as a coating system as it was intended to reduce the interaction of pre-coated nanoparticles with phagocytic cells. Overall, we were able to show that due to the pre-formed protein corona the interactions with macrophages were strongly reduced. On top of that, it was confirmed that the obtained stealth properties are preserved even if pre-coated protein-corona engineered nanoparticles are re-introduced to plasma. This proves that directing corona formation is a feasible strategy to control cellular interactions.Experimental
Nanoparticle synthesis
Polystyrene nanoparticle stabilized with the non-ionic surfactant Lutensol AT50 (BASF, Ludwigshafen) were synthesized as previously reported.33,39 The fluorescent dye Bodipy40 was in-cooperated for cellular uptake studies.Briefly, nanoparticles were synthesized via free-radical copolymerization miniemulsion technique. For amino-functionalized nanoparticles 2-aminoethyl methacrylate hydrochloride (AEMH, 90%, Sigma-Aldrich) was used whereas for carboxy-functionalized nanoparticles acrylic acid AA (99%, Sigma-Aldrich) was chosen.
Dynamic light scattering (DLS)
Multi-angle dynamic light scattering experiments were performed with an ALV-CGS 8F SLS/DLS 5022F goniometer equipped with eight simultaneously working correlators, eight photodiode detectors and a HeNe laser (632.8 nm, 25 mW output power). Experiments were performed at 37 °C. Nanoparticle dispersions (1 μL, 10 mg mL−1) were measured in 1 mL of filtered cell culture medium. Data was analyzed according to the method from Rausch et al.37,38ζ potential
The zeta (ζ) potential of the different polystyrene nanoparticle (10 μL, 10 mg mL−1) was measured in a 1 mM potassium chloride solution (1 mL) with a Zeta Sizer Nano Series (Malvern Instrument). Nanoparticle coated with proteins were prepared as described in the section protein corona preparation (below) before ζ potential measurements.Transmission electron microscopy (TEM)
To visualize the protein corona, nanoparticles were diluted with 1 mL water and further placed onto a lacey grid. Samples were stained with 4% uranyl acetate according to the method from Kokkinopoulou et al.33,41 Images were taken with a Ultrascan 1000 (Gatan) charge-coupled device (CCD) camera.Human blood plasma
Human plasma was collected by the Department of Transfusion Medicine Mainz from healthy donors after obtaining informed consent as requested by the local ethics committee. All experiments followed institutional guidelines and were performed in accordance with relevant laws and guidelines, local and global like the Declaration of Helsinki. Human plasma was stored at −80 °C. Before usage, human plasma pooled (5–10 donors) was centrifuged at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g (30 min) to remove protein aggregates.
000g (30 min) to remove protein aggregates.
Plasma fractionation
Immunoglobulins (IgG) were removed from human plasma according to our previous established method.29 A modified HPLC system was operated with a ToyoScreen AF-rProtein A HC-650F column (Tosoh Bioscience) using 0.01 M Tris*HCl as running buffer. Human plasma was diluted 1:3 with running buffer, filtered and applied to the HPLC system. Bound IgG was recovered from the column using 0.2 M citric acid.
Pre-coating with IgG depleted plasma
Nanoparticles were incubated with IgG depleted plasma in a defined ratio between surface area and protein concentration. In previous studies, varying protein concentration were used for pre-coating. We could demonstrate a direct correlation between the amount of protein used for pre-coating and the cellular uptake behavior.29 Therefore, nanoparticles (0.05 m2) were incubated with 5 mg of IgG depleted plasma for 1 h, 37 °C to allow corona formation. This amount was sufficient to significantly reduce cellular uptake by macrophages. After pre-coating, nanoparticles were centrifuged (20000g, 30 min, 4 °C) to remove unbound proteins.
SDS-PAGE
Hard corona proteins were separated using 10% Bis–Tris-Protein Gels and NuPAGE MES and SDS Running Buffer. Proteins (8 μg in 26 μL) were mixed with NuPage Reducing Agent (4 μL) and NuPage LDS Samples Buffer (10 μL). The gel was run for 1.5 h at 100 V with SeeBlue Plus2 Pre-Stained (Invitrogen) as molecular marker. Gels were stained with SimplyBlue SafeStain overnight and destained with water (all reagents Thermo Fisher Scientific).Protein quantification
The protein concentration was determined via Pierce 660 nm protein Assay (Thermo Fisher Scientific) according to manufacturer's instructions. Bovine serum albumin was used at standard and the absorption was measured at 660 nm with a Tecan infinite M1000 plate reader.LC-MS analysis
Protein samples were prepared for proteomic analysis as described.22,29,42 Briefly, proteins were precipitated using ProteoExtract protein precipitation kit (CalBioChem) according to the manufacturer's instructions. The resulting protein pellet was resuspended with RapiGest SF (Waters) in ammonium bicarbonate (50 mM). Samples were reduced (dithiothreitol, Sigma 5 mM, 45 min, 56 °C) and alkylated (idoacetoamide, Sigma 15 mM, 1 h, dark). A protein:trypsin ratio of 50:1 was chosen for tryptic digestion (18 h, 37 °C). The reaction was quenched with 2 μL hydrochloric acid (Sigma).
Samples were spiked with 50 fmol μL−1 Hi3 E. coli (Waters) for absolute protein quantification and diluted with 0.1% formic acid. Tryptic peptides were applied towards a nanoACQUITY UPLC system which was coupled with a Synapt G2- Si mass spectrometer. Electrospray ionization (ESI) was conducted in positive ion mode with a NanoLockSpray source. Measurements were performed in resolution mode and data-independent acquisition (MSE) experiments were carried. Data was analyzed with MassLynx 4.1
For protein identification Progenesis GI (2.0) was used. Identified peptides were search against a reviewed human database downloaded from Uniprot. The analysis was carried out using the following criteria: one missed cleavage, max protein mass 600 kDa, fixed modifications for cysteine and carbamidomethyl, variable oxidation for methionine and a false discovery rate of 4%. For peptide identification, three fragments need to be identified whereas protein identification requires five fragments and two peptides. The TOP3/Hi343 quantification approach was chosen to determine the amount of fmol for each protein.
Cell culture
RAW 264.7 cells were culture in Dulbecco's modified eagle medium (DMEM) supplemented with 10% FBS, 100 U ml−1 penicillin, 100 mg ml−1 streptomycin and 2 mM glutamine (all reagents Thermo Fisher Scientific).Flow cytometry and confocal laser scanning microscopy
RAW 264.7 cells (150000 cells per well) were seeded out in 24 well plates (1 mL) for flow cytometry analysis. For confocal images, cells were seeded out in μ-Dish 35 mm (ibidi) at a density of (100000 cells per mL−1, 800 μL). After overnight incubation at 37 °C, cells were washed and cell culture medium without (−) or with human plasma (+) was added.
For nanoparticle uptake analysis, uncoated or pre-coated nanoparticles were applied to cells in medium without (−) or with plasma proteins (+) at a concentration of 75 μg mL−1 for 2 h. Afterwards, cells were washed with PBS and detached with 2.5% trypsin from cell culture wells. Flow cytometry measurements were performed with a CyFlow ML cytometer (Partec, Germany). The fluorescent dye Bodipy was excited with a 488 nm laser. Data was analyzed with FCS Express V4 software (DeNovo Software, USA). Values are expressed as median fluorescence intensity (MFI) as mean of at least three independent experiments.
Confocal laser scanning microscopy (cLSM) images were taken o LSM SP5 STED Leica Laser Scanning Confocal Microscope (Leica, Germany). Nanoparticles were excited with an argon laser (488 nm) and are pseudocolored in green. The cell membrane was stained with CellMaskOrange (2.5 μg mL−1, Invitrogen). The dye was excited with a laser DPSS 561 nm laser and the membrane was pseudocolored in red.
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
The authors thank Katja Klein for the synthesis of the nanoparticles and gratefully acknowledge the financial support from Deutsche Forschungsgemeinschaft (SFB1066 TP Q1, Q2 and B11).References
- E. Blanco, H. Shen and M. Ferrari, Nat. Biotechnol., 2015, 33, 941 CrossRef PubMed.
- J.-R. Peng and Z.-Y. Qian, Nanomedicine, 2014, 9, 747–750 CrossRef PubMed.
- L. Liu, Q. Ye, M. Lu, Y.-C. Lo, Y.-H. Hsu, M.-C. Wei, Y.-H. Chen, S.-C. Lo, S.-J. Wang and D. J. Bain, Sci. Rep., 2015, 5, 10881 CrossRef PubMed.
- J. Della Rocca, D. Liu and W. Lin, Nanomedicine, 2012, 7, 303–305 CrossRef PubMed.
- K. Park, ACS Nano, 2013, 7, 7442–7447 CrossRef PubMed.
- H. H. Gustafson, D. Holt-Casper, D. W. Grainger and H. Ghandehari, Nano Today, 2015, 10, 487–510 CrossRef PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Aberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef PubMed.
- P. C. Ke, S. Lin, W. J. Parak, T. P. Davis and F. Caruso, ACS Nano, 2017, 11, 11773–11776 CrossRef PubMed.
- S. Winzen, S. Schoettler, G. Baier, C. Rosenauer, V. Mailaender, K. Landfester and K. Mohr, Nanoscale, 2015, 7, 2992–3001 RSC.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779 CrossRef PubMed.
- M. Daeron, Curr. Top. Microbiol., 2014, 382, 131–164 Search PubMed.
- L. Shen, S. Tenzer, W. Storck, D. Hobernik, V. K. Raker, K. Fischer, S. Decker, A. Dzionek, S. Krauthauser, M. Diken, A. Nikolaev, J. Maxeiner, P. Schuster, C. Kappel, A. Verschoor, H. Schild, S. Grabbe and M. Bros, J. Allergy Clin. Immunol., 2018 DOI:10.1016/j.jaci.2017.08.049.
- D. E. Owens III and N. A. Peppas, Int. J. Pharm., 2006, 307, 93–102 CrossRef PubMed.
- V. Mirshafiee, R. Kim, S. Park, M. Mahmoudi and M. L. Kraft, Biomaterials, 2016, 75, 295–304 CrossRef PubMed.
- S. Lara, F. Alnasser, E. Polo, D. Garry, M. C. Lo Giudice, D. R. Hristov, L. Rocks, A. Salvati, Y. Yan and K. A. Dawson, ACS Nano, 2017, 11, 1884–1893 CrossRef PubMed.
- Y. Yan, K. T. Gause, M. M. Kamphuis, C.-S. Ang, N. M. O'Brien-Simpson, J. C. Lenzo, E. C. Reynolds, E. C. Nice and F. Caruso, ACS Nano, 2013, 7, 10960–10970 CrossRef PubMed.
- C. C. Fleischer and C. K. Payne, J. Phys. Chem. B, 2014, 118, 14017–14026 CrossRef PubMed.
- Z. Amoozgar and Y. Yeo, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2012, 4, 219–233 CrossRef PubMed.
- B. Kang, P. Okwieka, S. Schöttler, S. Winzen, J. Langhanki, K. Mohr, T. Opatz, V. Mailänder, K. Landfester and F. R. Wurm, Angew. Chem., Int. Ed., 2015, 54(25), 7436–7440 CrossRef PubMed.
- A. Wörz, B. Berchtold, K. Moosmann, O. Prucker and J. Rühe, J. Mater. Chem., 2012, 22, 19547–19561 RSC.
- H. R. Kim, K. Andrieux, C. Delomenie, H. Chacun, M. Appel, D. Desmaële, F. Taran, D. Georgin, P. Couvreur and M. Taverna, Electrophoresis, 2007, 28, 2252–2261 CrossRef PubMed.
- S. Schöttler, G. Becker, S. Winzen, T. Steinbach, K. Mohr, K. Landfester, V. Mailander and F. R. Wurm, Nat. Nanotechnol., 2016, 11, 372–377 CrossRef PubMed.
- I. Hamad, A. Hunter, J. Szebeni and S. M. Moghimi, Mol. Immunol., 2008, 46, 225–232 CrossRef PubMed.
- N. Bertrand, P. Grenier, M. Mahmoudi, E. M. Lima, E. A. Appel, F. Dormont, J.-M. Lim, R. Karnik, R. Langer and O. C. Farokhzad, Nat. Commun., 2017, 8, 777 CrossRef PubMed.
- Q. Dai, N. Bertleff-Zieschang, J. A. Braunger, M. Björnmalm, C. Cortez-Jugo and F. Caruso, Adv. Healthcare Mater., 2017, 7(1), 1700575 CrossRef PubMed.
- T. Takeuchi, Y. Kitayama, R. Sasao, T. Yamada, K. Toh, Y. Matsumoto and K. Kataoka, Angew. Chem., Int. Ed., 2017, 56(25), 7088–7092 CrossRef PubMed.
- J. Kreuter, D. Shamenkov, V. Petrov, P. Ramge, K. Cychutek, C. Koch-Brandt and R. Alyautdin, J. Drug Targeting, 2002, 10, 317–325 CrossRef PubMed.
- G. Caracciolo, F. Cardarelli, D. Pozzi, F. Salomone, G. Maccari, G. Bardi, A. L. Capriotti, C. Cavaliere, M. Papi and A. Laganà, ACS Appl. Mater. Interfaces, 2013, 5, 13171–13179 Search PubMed.
- L. K. Müller, J. Simon, S. Schöttler, K. Landfester, V. Mailänder and K. Mohr, RSC Adv., 2016, 6, 96495–96509 RSC.
- H. Yin, R. Chen, P. S. Casey, P. C. Ke, T. P. Davis and C. Chen, RSC Adv., 2015, 5, 73963–73973 RSC.
- W. Hu, C. Peng, M. Lv, X. Li, Y. Zhang, N. Chen, C. Fan and Q. Huang, ACS Nano, 2011, 5, 3693–3700 CrossRef PubMed.
- S. Akula, S. Mohammadamin and L. Hellman, PLoS One, 2014, 9, e96903 Search PubMed.
- M. Kokkinopoulou, J. Simon, K. Landfester, V. Mailänder and I. Lieberwirth, Nanoscale, 2017, 9(25), 8858–8870 RSC.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef PubMed.
- S. Tenzer, D. Docter, J. Kuharev, A. Musyanovych, V. Fetz, R. Hecht, F. Schlenk, D. Fischer, K. Kiouptsi and C. Reinhardt, Nat. Nanotechnol., 2013, 8, 772–781 CrossRef PubMed.
- J. Müller, K. N. Bauer, D. Prozeller, J. Simon, V. Mailänder, F. R. Wurm, S. Winzen and K. Landfester, Biomaterials, 2017, 115, 1–8 CrossRef PubMed.
- K. Mohr, M. Sommer, G. Baier, S. Schöttler, P. Okwieka, S. Tenzer, K. Landfester, V. Mailänder, M. Schmidt and R. G. Meyer, J. Nanomed. Nanotechnol., 2014, 5(2), 1000193 Search PubMed.
- K. Rausch, A. Reuter, K. Fischer and M. Schmidt, Biomacromolecules, 2010, 11, 2836–2839 CrossRef PubMed.
- A. Musyanovych, R. Rossmanith, C. Tontsch and K. Landfester, Langmuir, 2007, 23, 5367–5376 CrossRef PubMed.
- I. García-Moreno, A. Costela, L. Campo, R. Sastre, F. Amat-Guerri, M. Liras, F. López Arbeloa, J. Bañuelos Prieto and I. López Arbeloa, J. Phys. Chem. A, 2004, 108, 3315–3323 CrossRef.
- P. Renz, M. Kokkinopoulou, K. Landfester and I. Lieberwirth, Macromol. Chem. Phys., 2016, 217, 1879–1885 CrossRef.
- D. Hofmann, S. Tenzer, M. B. Bannwarth, C. Messerschmidt, S.-F. Glaser, H. Schild, K. Landfester and V. Mailaender, ACS Nano, 2014, 8, 10077–10088 CrossRef PubMed.
- J. C. Silva, M. V. Gorenstein, G. Z. Li, J. P. Vissers and S. J. Geromanos, Mol. Cell. Proteomics, 2006, 5, 144–156 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr03331e |
| This journal is © The Royal Society of Chemistry 2018 |