
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceElucidation of interfacial pH behaviour at the cell/substrate nanogap for in situ monitoring of cellular respiration†
Hiroto
Satake
,
Akiko
Saito
and
Toshiya
Sakata
 *
*
Department of Materials Engineering, School of Engineering, The University of Tokyo, 7-3-1 Hongo, Bunkyo-ku, Tokyo, Japan 113-8656. E-mail: sakata@biofet.t.u-tokyo.ac.jp
First published on 21st May 2018
Abstract
In situ monitoring of cellular metabolism is useful for elucidating dynamic functions of living cells. In our previous studies, cellular respiration was continuously monitored as a change in pH at the cell/electrode nanoscale interface (i.e., interfacial pH) using an ion-sensitive field-effect transistor (ISFET). However, such interfacial pH behaviour on the nanoscale has not been confirmed using other methods such as fluorescence imaging. In this study, we have clarified the interfacial pH behaviour at a cell/substrate nanogap using a laser scanning confocal fluorescence microscope. The phospholipid fluorescein used as a pH indicator was fixed to the plasma membrane on the external side of a cell by inserting its lipophilic alkyl chain into the membrane, and used to observe the change in interfacial pH. As a result, hydrogen ions generated by cellular respiration were gradually accumulated at the cell/substrate nanogap, resulting in a decrease in pH. Moreover, the interfacial pH between the plasma membrane and the substrate became lower than the pH near the surface of cells not in contact with the substrate. The data obtained in this study support the idea that potentiometric ion sensors such as ISFETs can detect a cellular-metabolism-induced change in pH at a cell/electrode nanogap in real time.
1. Introduction
Cellular respiration involves a series of metabolic reactions to produce adenosine triphosphate (ATP) by the uptake of nutrients such as glucose and oxygen, which is followed by the release of waste products such as carbon dioxide and lactic acid. In particular, mitochondria play an important role in cellular respiration to produce ATP via the citric acid cycle, electron transfer system, and oxidative phosphorylation in aerobic respiration, the degradation of which has recently been focused on in relation to various conditions such as diabetes, Alzheimer's disease, and aging.1–3 For living cells, in situ monitoring of momentarily varying cellular behaviour is required to elucidate the mechanisms of disease development and investigate the effects of drugs.As one of the technologies to continuously monitor cellular respiration, an ion-sensitive field-effect transistor (ISFET) with a cell-coupled gate can monitor it as a change in pH at the cell/gate electrode nanoscale interface in real time. In our previous works, the cellular respiration activities of rat pancreatic β cells, a single mouse embryo, and bovine chondrocytes or the allergic responses of mast cells on a gate were monitored noninvasively, quantitatively, and continuously as the change in pH using cell-coupled gate ISFET sensors.4–7 Additionally, similar electrical responses were also obtained for other living cells, as shown in section S1 (ESI†). Since the gate insulator used as an electrode usually consists of an oxide with hydroxyl groups at the surface in a solution, the ISFET sensors are sensitive to changes in the concentration of positively charged hydrogen ions based on the equilibrium reaction (–OH2+ ↔ –OH ↔ –O−); consequently, they can be utilized as pH sensors (Fig. S3 in ESI†).8,9 Thus, the pH variation due to cellular respiration can be monitored at the cell/gate interface on the basis of the amount of carbon dioxide or lactic acid generated by cellular respiration and dissolved in a medium. In aerobic respiration, carbon dioxide dissolves in a solution, resulting in the generation of hydrogen ions, and in anaerobic respiration, lactic acid, which exhibits acidity, is released through glycolysis. Additionally, other previous papers reported that the electrical communication between semiconductors and nerve cells was based on the interaction of ion channels with a chip,10–13 that is, the ionic behaviour at a cell/gate interface.
Here, we need to focus on the charge behaviour of ions at the nanoscale interface between the plasma membrane and the gate surface, which is different from that in a bulk solution, to directly detect cell functions. This is because cells are alive and their functions should be directly monitored in real time. That is, the pH at the cell/gate nanoscale interface, the interfacial pH, reflects cellular functions such as metabolism in situ. Indeed, previous works showed a gap of approximately 50–150 nm at the cell/substrate interface (right of Fig. S2†), where focal or nonfocal regions of contact between membrane proteins and substrates were observed by total-internal-reflection fluorescence microscopy (TIRFM).14–16 The TIRFM technique utilizes an evanescent electromagnetic field, which penetrates into the cell and decays exponentially with increasing perpendicular distance z from the cell/substrate interface, for the excitation of fluorophores. Therefore, fluorophores located within or close to the plasma membrane are selectively excited. The interfacial pH is considered to be the pH localized at the cell/substrate nanogap and depends on the state of cells (i.e., living). Therefore, we assume that cellular respiration can be monitored as the change in interfacial pH at the nanogap using the ISFET sensor, which should be capable of continuously monitoring the change in pH around a cell (Fig. S2†). However, the change in interfacial pH at the nanogap between the cell and the substrate has not been investigated using other methods without an ISFET sensor; therefore, analysis of the interfacial pH behaviour at the nanogap is performed using fluorescence imaging in this study.
Fluorescence imaging has become a widely used tool to monitor biological processes inside or outside living cells. A number of intracellular fluorescent probes have recently been developed for nucleic acid staining in genomics research,17 to meet the requirements of the rapidly expanding field of proteomics,18 and for the monitoring of cell viability and cytotoxicity.19–21 In particular, endosomal fluorescent pH probes were designed to monitor the acidification of vesicles during endocytosis.22 On the other hand, using pH-sensitive dyes, extracellular pH dynamics was examined to clarify the H+ hypothesis of lateral feedback inhibition in the outer retina23 and to measure lateral H+ conduction along lipid monolayers.24–27 In these probes, fluorescein-labeled phospholipids were utilized as extracellular pH indicators to measure the pH adjacent to membrane surfaces, because the phospholipid unit was easily inserted and stably maintained in the cell membrane.24–27 Therefore, the interfacial pH behaviour at the nanogap between the cell and the substrate should be analyzed using the lipophilic pH indicator (Fig. 1), to support the data obtained using the ISFET sensors.
 | ||
| Fig. 1 Schematic illustration of hydrogen ion behaviour around cell cultured on substrate. The phospholipid fluorescein was used as an extracellular pH indicator (Probe) and fixed to the plasma membrane on the external side of a cell. Two regions of interfacial pH are considered around a cell on a substrate: one is the interface between the cell and the substrate, and the other is the interface between the cell and the bulk solution. A nanogap is found between the cell and the substrate, where hydrogen ions generated from cellular respiration are accumulated and prevented from diffusing to the bulk solution. On the other hand, hydrogen ions generated near the plasma membrane not in contact with the substrate easily diffuse to the bulk solution. | ||
In this study, we examined the interfacial pH behaviour at a cell/substrate nanogap using laser scanning confocal fluorescence microscopy. Here, phospholipid fluorescein [N-(fluorescein-5-thio-carbamyl)-1,2-dihexadecanoyl-sn-glycero-3-phosphoethanolamine, DHPE] used as an extracellular pH indicator was fixed to the plasma membrane on the external side of a cell by inserting its lipophilic alkyl chain into the membrane, and used to observe the change in interfacial pH.
2. Materials and methods
Cell culture and transfer to glass-bottomed dish
For respiratory monitoring by fluorescence imaging, bovine chondrocytes were utilized as a model of normal cells, while human cervical carcinoma (HeLa) cells were used as a model of cancer cells.After the pre-culture of chondrocytes (section S2 in ESI†), for fluorescence imaging, they were collected by trypsin treatment and transferred to a glass-bottomed dish (Iwaki) at a density of 2 × 104 cells per cm2 and cultivated with Leibovitz's L-15 medium, which is designed to support cell growth in environments without CO2 equilibration, without phenol red (Gibco) including 10% fetal bovine serum (FBS, Sigma-Aldrich), 50 U mL−1 penicillin, 50 μg mL−1 streptomycin, 5 μg mL−1 l-ascorbic acid phosphate magnesium salt (APM, Wako), and 1% (v/v) insulin transferrin selenium ethanolamine solution (ITS, Gibco) in an incubator (37 °C) for 24 h. APM and ITS were used as biochemical stimulants of metabolic reactions such as the cellular respiration of chondrocytes.28–33
HeLa cells were cultured in Dulbecco's Modified Eagle's Medium (DMEM, Gibco) with 10% FBS including 50 U mL−1 penicillin and 50 μg mL−1 streptomycin in an incubator (37 °C, 5% CO2) for 4 days as the pre-culture, and then transferred to a glass-bottomed dish for fluorescence imaging. Here L-15 medium was used in the same way as in the culture of chondrocytes, except for the addition of APM and ITS, and was kept at 37 °C for 24 h. The number of cells seeded on the dish was controlled to 2.5 × 104 cells per mL × 2 mL for HeLa cells.
Cell staining
Before cell staining, all of the living cells used in this study were washed with Ringer solution, which consisted of 126 mM NaCl, 4 mM KCl, 3 mM CaCl2, 1 mM MgCl2, 10 mM HEPES, and 15 mM glucose, the pH of which was adjusted to 7.4 with 1 M NaOH. A stock solution of fluorescein DHPE (Thermofisher) was prepared by dissolving 1 mg of fluorescein DHPE in 1 mL of ethanol and stored in a freezer (−30 °C). The stock fluorescein DHPE solution was diluted with Ringer solution to 10 μM for living chondrocytes and 30 μM for HeLa cells and used for incubation in the dark for 30 min at room temperature. After the incubation, the cells were washed with Ringer solution three times, and then a suitable culture medium for each cell were added into the glass-bottomed dish. The culture medium was covered with a mineral oil to prevent it from evaporating.For cell fixation, chondrocytes were utilized after pre-culture. The DHPE solution was diluted with Ringer solution to 30 μM and used for incubation in the dark for 30 min at room temperature. After washing the incubated cells with Ringer solution and Dulbecco's phosphate-buffered saline (DPBS, Gibco), 4% paraformaldehyde phosphate buffer solution (Wako) was added for fixation into the glass-bottomed dish with chondrocytes. The fixed cells were utilized as control cells to analyze the interfacial pH at the cell/substrate nanogap.
Fluorescence imaging using laser scanning confocal microscopy
An LSM 510 laser scanning confocal microscope (Carl Zeiss Co., Ltd) was used for fluorescence imaging of stained chondrocytes. An Ar laser (458 and 488 nm) was used as the excitation laser. A long-pass filter (505 nm) was used as the emission filter for each excitation wavelength. An oil-immersion objective (×63, numerical aperture = 1.4) was used and the pinhole size was set at one Airy disk unit. z-Stack images of stained chondrocytes were obtained by time-lapse imaging every 30 min for a total of 270 min at 37 °C in air. An optical slice was set with an interval of 0.38 μm in the normal direction from a glass substrate.Ratiometric analysis of fluorescence intensity
Fluorescence images were analyzed using ZEN lite software (Carl Zeiss Co., Ltd). Ratiometric analysis of fluorescence intensity was carried out using the ratio of the emission intensity at 488 nm to that at 458 nm. As shown in Fig. 1, the fluorescence images were analyzed focusing on the following two regions: one was the interface between the cell and the glass substrate around the center of a cell, and the other was the interface between the cell and the bulk solution around the surface of the plasma membrane that was not in contact with the substrate. The interfacial pH in the former region was estimated from the peak fluorescence intensity obtained in an optical slice, which was set as z = 0–0.38 μm, and that in the latter region was estimated from the fluorescence intensities within four optical slices (z = 0.76–2.28 μm), which were between the middle and bottom of a cell placed on the substrate. The fluorescence intensities estimated as described above were calculated by subtracting the background intensity in the region without cells from each fluorescence intensity on cells.Calibration of fluorescence intensity for interfacial pH
The pH of L-15 (10% FBS, 50 U mL−1 penicillin, and 50 μg mL−1 streptomycin) was adjusted from 6.40 to 7.68 by adding 1 M HCl or 1 M NaOH; the pH was measured using a conventional pH meter (HORIBA). The z-stack images of chondrocytes stained with fluorescein DHPE in L-15 were taken by the same protocol as in the above-mentioned time-lapse measurement at different pH values. In this case, an optical slice was moved around the substrate surface (under the cell/substrate interface) to the top of spread cells to find the peak fluorescence intensity, where the cell/substrate interface with a nanogap was found. From the peak intensity, the fluorescence ratio was also calculated from the ratio of emission intensity at 488 nm to that at 458 nm at the cell/substrate interface around the center of a cell. All experiments for pH calibration were performed at 37 °C.3. Results and discussion
Calibration of fluorescence intensity based on fluorescein DHPE for interfacial pH measurement
The phospholipid fluorescein DHPE is fixed as the pH indicator at the plasma membrane of cells by inserting its lipophilic alkyl chain into the plasma membrane, the fluorescein moiety of which is located on the external side of a cell. The z-stack image in Fig. 2(a) indicates fluorescein DHPE molecules observed around the plasma membrane of chondrocytes near the glass substrate as well as the bulk solution. Basically, most of the fluorescein DHPE molecules were stably tethered to the plasma membrane for approximately 5 h, but some of the molecules were moved to the cytoplasm by endocytosis; therefore, the number of fluorescent molecules at the plasma membrane was assumed to decrease with time. This effect could be eliminated by calculating the relative change in the fluorescence.34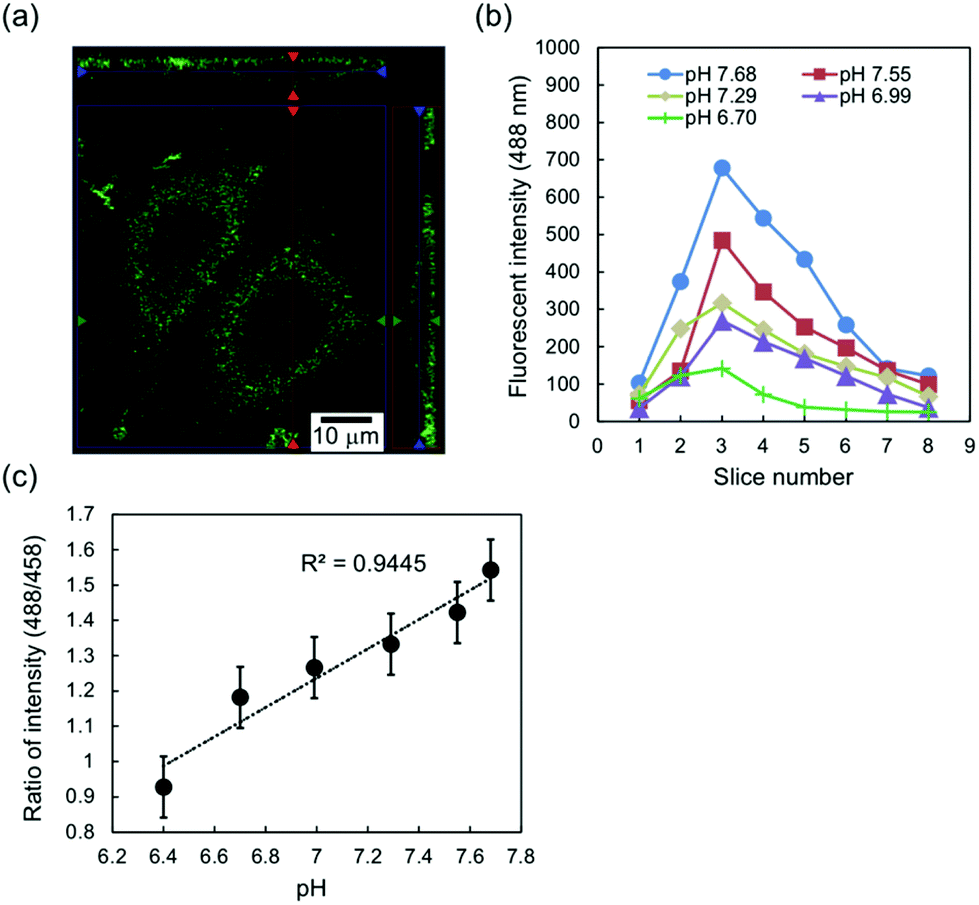 | ||
| Fig. 2 Calibration of fluorescence intensity for interfacial pH. (a) z-Stack image of chondrocytes stained by fluorescein DHPE in the L-15 medium. The fluorescence image was observed along the plasma membrane. (b) Fluorescence intensities at 488 nm excitation along the z-axis around the center of a cell. The z-axis indicates the normal direction of the cell/substrate interface. The width of an optical slice was 0.38 μm. In this case, the sliced fluorescence images were observed near the substrate surface to find the peak fluorescence intensity; therefore, the optical slice with the peak intensity, which corresponded to optical slice 3 here, was defined as z = 0–0.38 μm in Fig. 3, 4, and 5 and includes the cell/substrate interface. (c) Calibration curve of fluorescence peak intensity for interfacial pH measured by ratiometric analysis (488 nm/458 nm). The data are shown as the average of five cells in the L-15 medium at different pH values. | ||
Fig. 2(b) shows the typical fluorescence intensities per pixel (0.28 μm × 0.28 μm) at 488 nm excitation for optical slices around the center of a one stained chondrocyte in Leibovitz's L-15 medium at different pH values. The obtained fluorescence intensities were plotted for each optical slice along the normal direction of the substrate surface. As shown in Fig. 2(b), the peak intensities were found at slice 3 even with the change in pH, because the fluorescein DHPE molecules were localized at the plasma membrane around the cell/substrate interface within optical slice 3. Here, the gap between the cell and the glass substrate was approximately 50–150 nm, which was estimated from previous studies.14–16 That is, the fluorescein DHPE molecules at the plasma membrane in the nanogap were included in an optical slice. Therefore, the interfacial pH was defined as the pH in the optical slice with the peak fluorescence intensity in this study.
Additionally, the effect of photobleaching of fluorescein DHPE, which is the attenuation of fluorescence intensity, was considered to determine the actual pH by a ratiometric measurement.34Fig. 2(c) shows the calibration curve of the ratio of fluorescence intensities (488 nm/458 nm) for the interfacial pH on the basis of the peak fluorescence intensity shown in Fig. 2(b). The average fluorescence intensity ratio at the cell/glass interface for five cells decreased with decreasing pH, which was actually expected from the change in interfacial pH based on cellular respiration, and showed a linear relationship with interfacial pH in the range of about 6 to 8, which was assumed under the cell culture condition (pH 7.4). Thus, the calibration curve shown in Fig. 2(c) can be utilized to estimate changes in interfacial pH in cultured cells.
Change in interfacial pH between cell and substrate caused by cellular respiration
Fig. 3 shows the change in interfacial pH (ΔpH) between cells and a glass substrate over the incubation time. In this case, ΔpH was regarded as the change in pH in a closed nanospace, that is, the nanogap between the cells and the substrate, which was in the range of 50–150 nm, as described in Introduction. Corresponding to the change in the 488 nm/458 nm peak intensity ratio around the boundary between the chondrocytes and the glass substrate (Fig. S4 in ESI†), the interfacial pH was calculated on the basis of the calibration curve shown in Fig. 2(c). Indeed, ΔpH for the chondrocyte/substrate nanogap decreased from the initial pH of the medium (about 7.4) to −0.3 approximately 2 h after the cultivation in L-15 medium including ITS and APM growth factors, and subsequently saturated at 5 h, as shown in Fig. 3(b), although ΔpH for the fixed cell/substrate nanogap hardly changed during incubation because the fixed cells were not alive (Fig. 3(a)). The decrease in the pH was because carbon dioxide or lactic acid was released from chondrocytes as products of the respiration of cells stimulated following the addition of ITS and APM, resulting in the generation of hydrogen ions. It is well known that the respiration activity of chondrocytes is dominantly conducted through the glycolysis pathway even in an aerobic environment.35,36 However, the metabolic phenotype of chondrocytes is likely to shift from the glycolysis pathway to oxidative phosphorylation during monolayer cultivation on a substrate;36 therefore, hydrogen ions are considered to have been mainly generated by the dissolving of carbon dioxide released from cells in aerobic respiration. This result was in good agreement with the previous result and the supporting data shown in Fig. S3† obtained by pH measurement using the chondrocyte-based ISFET,6 which can specifically detect a change in pH at the oxidized gate surface, confirming that the ISFET detected the decrease in pH at the cell/gate nanoscale interface caused by cellular respiration.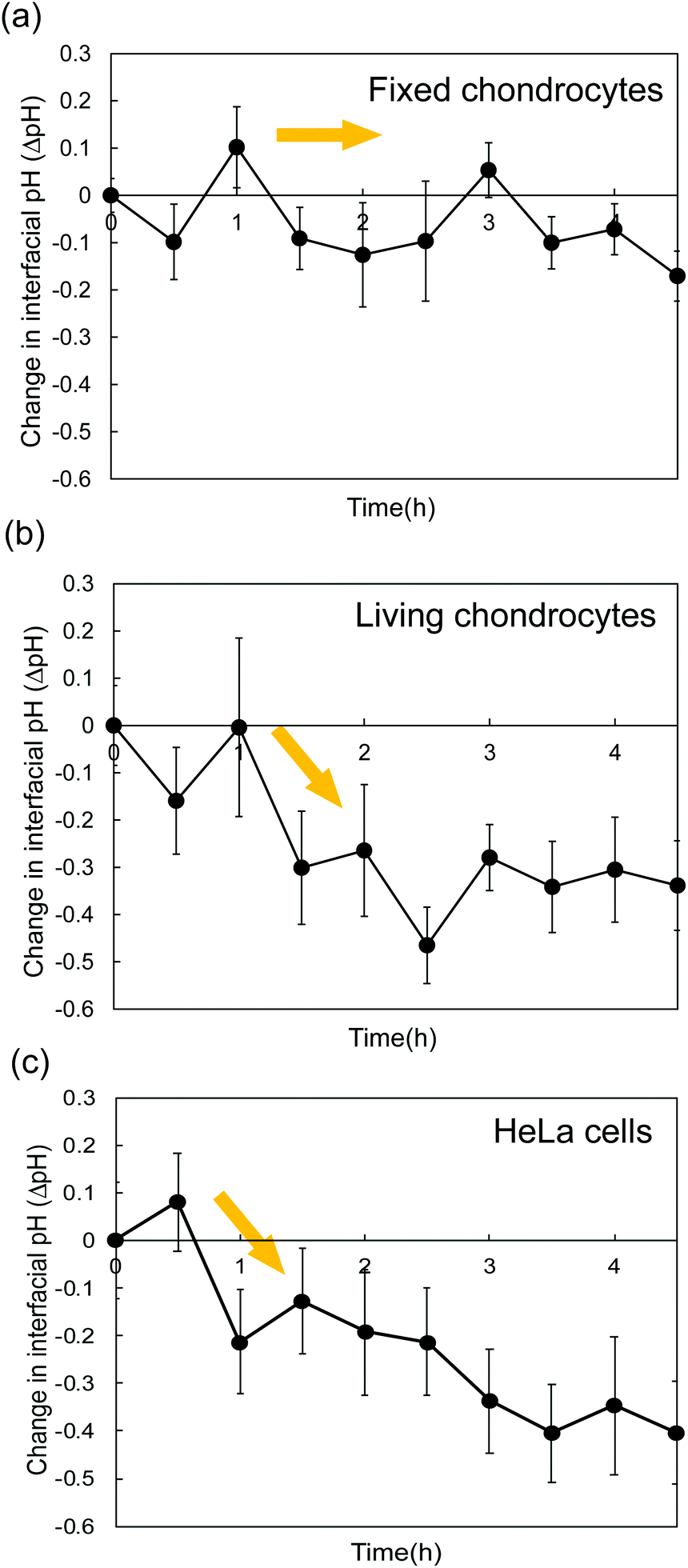 | ||
| Fig. 3 Change in interfacial pH at the interface between cell and substrate as a function of incubation time. Interfacial pH was calculated on the basis of the ratio of fluorescence intensities (Fig. S4 in ESI†) and the calibration curve (Fig. 2(c) for living chondrocytes, Fig. S5(a)† for fixed chondrocytes, and Fig. S5(b)† for HeLa cells). The data were the average of seven cells. (a) Fixed chondrocytes. (b) Living chondrocytes. (c) HeLa cells. | ||
Moreover, a similar trend of ΔpH was found for the HeLa cell/substrate nanogap, as shown in Fig. 3(c). Cancer cells such as HeLa cells are vigorously activated by suppressing oxidative phosphorylation in mitochondria so as not to induce apoptosis, resulting in proliferation and metastasis. Therefore, ΔpH based on the metabolic disorder of cancer cells should be larger than that for normal cells (Fig. S3†). This is why ΔpH for the HeLa cell/substrate nanogap shown in Fig. 3(c) appeared to gradually decrease even after several hours of cultivation (ΔpH < −0.3), which can also be concluded from the change in the interfacial potential (ΔVout) shown in Fig. S3,† different from normal cells such as chondrocytes.
Change in pH around a cell on substrate
In this study, chondrocytes were cultured on a glass substrate, where two regions for the measurement of interfacial pH can be found in the culture medium around a cell: at the nanogap between the cell and the substrate, and near the surface of a cell not in contact with the substrate (Fig. 1). Here, the interfacial pH in the former region, shown in Fig. 3(b), was compared with that in the latter region, as shown in Fig. 4. In this case, the pH in the former region was defined as the pH calculated within the optical slice of z = 0–0.38 μm, whereas the latter was defined as that within the optical slices of z = 0.76–2.28 μm. Indeed, the pH values in the former region were lower than those in the latter region (ratios < 1) regardless of the incubation time of the cell cultures (Fig. 4). This result was supported by Fig. S6 in ESI,† where the interfacial pH near the substrate was lower than that away from the substrate at 1.5 h. This is because hydrogen ions generated by cellular respiration were accumulated and concentrated at the cell/substrate interface, that is, in the closed nanospace between the cell and the substrate. Moreover, the ratio of interfacial pH increased with increasing incubation time from 0.938 (7.00 ± 0.093/7.47 ± 0.045) at 1.5 h to 0.964 (7.05 ± 0.198/7.32 ± 0.087) at 4.5 h (Fig. 4). In fact, the interfacial pH between the cell and the substrate initially decreased but subsequently remained relatively constant to 4.5 h (Fig. 3(b)) while the interfacial pH between the cell and bulk solution gradually decreased owing to the change in the rate of release of hydrogen ions from cells. When the ratio approaches 1, the difference between the interfacial pH values is reduced, that is, the interfacial pH between the plasma membrane and the substrate becomes equal to the pH near the surface of cells not in contact with the substrate. That is, the pH in the closed nanospace would have been maintained according to the balance between the concentrations of generated and diffused hydrogen ions. | ||
| Fig. 4 Comparison of interfacial pH at cell/substrate nanogap with that near surface of spread cell not in contact with substrate. The optical slice with the peak intensity was set as z = 0 to 0.38 μm for cells spread on a glass substrate to evaluate the interfacial pH at the cell/substrate interface, while the optical slice was moved from z = 0.76 to 2.28 μm to evaluate the interfacial pH near the plasma membrane not in contact with the substrate. A ratio of less than 1 indicates that more hydrogen ions were accumulated at the cell/substrate interface with a nanogap. The data were the average for three cells. | ||
On the other hand, the interfacial pH between the cell and the substrate was affected by the change in the morphology of the cells. Basically, chondrocytes tightly adhered and flatly spread on the glass substrate. However, for part of the cells, cell division induced a change in cell morphology from a flat morphology at 0 h to a spherical shape at 4.5 h, as shown in Fig. 5(a). In this case, there was no significant difference in interfacial pH values between the two regions at 4.5 h (ratio ≅ 1) in Fig. 5(b). This is also because hydrogen ions produced from cellular respiration were concentrated in the closed nanospace between a flat cell and the substrate, but they easily diffused from the interface between a spherical cell and the substrate during cell division. Nevertheless, the cells that spread were mostly found on the substrate regardless of cultivation time.6 Thus, the pH change around the cell/gate interface induced by cellular respiration, which is maintained in a closed nanospace, should be easily monitored in real time using the ISFET sensor. This means that a platform based on the cell-coupled gate ISFET sensor is suitable for systems for in situ monitoring of cellular respiration, which is related to various cellular functions in the fields of cell biology, medicine, and pharmaceutical discovery.
 | ||
| Fig. 5 Effect of cell morphology on interfacial pH. (a) Cross-sectional fluorescence image of a cell. Spread cells morphologically changed from flat (at 0 h) to spherical (at 4.5 h) owing to cell division, following the pre-culture for 24 h. (b) Comparison of interfacial pH at cell/substrate nanogap with that near surface of cell not in contact with substrate. The interfacial pH values became almost equal at 4.5 h as a result of the change in the cell morphology of from flat to spherical. | ||
The pKa of fluorescein DHPE is affected by the surface potential at the plasma membrane, which depends on the adsorption of ionic species in media onto the plasma membrane.37,38 In this study, we eliminated the effect of adsorbed proteins on the surface potential at the plasma membrane by pre-cultivating cells for 24 h before staining. However, a change in the surface potential at the plasma membrane may be induced in long-term measurements when the extracellular matrix (ECM), which is synthesized by chondrocytes during cultivation for a few days,6 interacts with the plasma membrane, resulting in a shift in the pKa of the pH indicator. This is why we set the observation time of fluorescein DHPE to be as short as possible (∼5 h) after staining the cells.
However, a change in the surface potential is not a concern when a cell-based ISFET is utilized to monitor cellular respiration for a long time. A cell culture medium includes various proteins, ions, and growth factors, which nonspecifically adhere to the gate surface of an ISFET sensor. Therefore, the ISFET sensor is assumed to be insensitive to most chemicals in a cell culture medium, although it actually showed good performance only in terms of pH responsivity (Fig. S7 in ESI†). Even in a cell culture medium, the change in the interfacial pH between the cell and the gate must be specifically detected using the ISFET sensor with an oxidized gate, regardless of the further nonspecific adsorption of proteins and so forth. This is also because hydrogen ions are very small and easily penetrate through such chemicals adsorbed onto the oxidized gate surface with hydroxyl groups. In short, the ISFET sensor is very simple method of monitoring cellular functions in a real-time and label-free manner, compared with fluorescence imaging.
Thus, the fluorescence imaging in this study provides the first proof-of-concept demonstration of monitoring pH behaviour around a cell cultured on a substrate, whereas a platform based on electrical methods, such as one based on ISFETs, is suitable for more quantitative pH measurements of cellular functions such as metabolic reactions in situ for a long time. This means that it is very important for the elucidation of cellular metabolism to use in situ and label-free measurement systems such as ISFETs, although such detected data of cellular functions have to be complementarily verified on the nanoscale by other methods such as fluorescence imaging.
4. Conclusions
In this study, the change in the extracellular pH around chondrocytes cultured on a glass substrate was measured using a laser scanning confocal fluorescence microscope. The change in pH at the interface between the cell and the substrate (i.e., interfacial pH) was focused on because it was assumed to affect the signals of the pH-responsive ISFET sensor. As a result, the acidification caused by cellular respiration was monitored in the cell/substrate nanogap by analyzing the fluorescence images obtained, the results of which confirmed that the ISFET sensor detected the change in interfacial pH between the cell and the substrate. Moreover, when flat chondrocytes were observed, hydrogen ions generated from cellular respiration were accumulated and concentrated around the cell/substrate interface, which induced the earlier and amplified detection of the change in pH than that at the interface between the cell and the bulk solution away from the substrate. Thus, a platform based on electrical methods such as one based on ISFETs is suitable for more quantitative pH measurement of cellular functions such as metabolic reactions in situ based on interfacial pH.Conflicts of interest
There are no conflicts to declare.Acknowledgements
We would like to thank Prof. K. Kushiro of the University of Tokyo for the use of the laser scanning confocal fluorescence microscope.Notes and references
- J. A. Maassen, L. M. Hart, E. v. Essen, R. J. Heine, G. Nijpels, R. S. J. Tafrechi, A. K. Raap, G. M. C. Janssen and H. H. P. J. Lemkes, Diabetes, 2004, 53, 103 CrossRef
.
- P. I. Moreira, C. Carvalho, X. Zhu, M. A. Smith and G. Perry, Biochim. Biophys. Acta, 2010, 1802, 2 CrossRef PubMed
- E. J. Lesnefsky, Q. Chen and C. L. Hoppel, Circ. Res., 2016, 118, 1593 CrossRef PubMed
- T. Sakata and H. Sugimoto, Jpn. J. Appl. Phys., 2011, 50, 020216 CrossRef
- T. Sakata, A. Saito, J. Mizuno, H. Sugimoto, K. Noguchi, E. Kikuchi and H. Inui, Anal. Chem., 2013, 85, 6633 CrossRef PubMed
- H. Satake, A. Saito, S. Mizuno, T. Kajisa and T. Sakata, Jpn. J. Appl. Phys., 2017, 56, 04CM03 CrossRef
- H. Yang, M. Honda, A. Akiko, T. Kajisa, Y. Yanase and T. Sakata, Anal. Chem., 2017, 89, 12918 CrossRef PubMed
- P. Bergveld, IEEE Trans. Biomed. Eng., 1972, BME-19, 342 CrossRef PubMed
- T. Matsuo and M. Esashi, Oyo Buturi, 1980, 49, 586 Search PubMed
- P. Fromherz, A. Offenhausser, T. Vetter and J. Weis, Science, 1991, 252, 1290 Search PubMed
- F. Patolsky, B. P. Timko, G. Yu, Y. Fang, A. B. Greytak, G. Zheng and C. M. Lieber, Science, 2006, 313, 1100–1104 CrossRef PubMed
- P. Fromherz, Ann. N. Y. Acad. Sci., 2006, 1093, 143 CrossRef PubMed
- B. Tian, J. Liu, T. Dvir, L. Jin, J. H. Tsui, Q. Qing, Z. Suo, R. Langer, D. S. Kohane and C. M. Lieber, Nat. Mater., 2012, 11, 986 CrossRef PubMed
- J. S. Burmeister, L. A. Olivier, W. M. Reichert and G. A. Truskey, Biomaterials, 1998, 19, 307 CrossRef PubMed
- K. Stock, R. Sailer, W. S. L. Strauss, M. Lyttek, R. Steiner and H. Schneckenburger, J. Microsc., 2003, 211, 19 CrossRef PubMed
- M. C. D. Santos, R. Deturche, C. Vezy and R. Jaffiol, Biophys. J., 2016, 111, 1316 CrossRef PubMed
-
The Molecular probes Handbook: A Guide to Fluorescent Probes and Labelling Technologies, ed. I. Johnson and M. T. Z. Spence, Life Technologies Corporation, 11th edn, 2010, ch. 8 Search PubMed
- I. Miller, J. Crawford and E. Gianazza, Proteomics, 2006, 6, 5385 CrossRef PubMed
- K. H. Jones and J. A. Senft, J. Histochem. Cytochem., 1985, 33, 77 CrossRef PubMed
- X. M. Wang, P. I. Terasaki, G. W. Rankin Jr., D. Chia, H. P. Zhong and S. Hardy, Hum. Immunol., 1993, 37, 264 CrossRef PubMed
- K. Sakamaki,
et al.
, Biochim. Biophys. Acta, Mol. Cell Res., 2016, 1863, 2766 CrossRef PubMed
- D. S. Richardson, C. Gregor, F. R. Winter, N. T. Urban, S. J. Sahl, K. I. Willig and S. W. Hell, Nat. Commun., 2017, 8, 577 CrossRef PubMed
- J. Jacoby, M. A. Kreitzer, S. Alford, H. Qian, B. K. Tchernookova, E. R. Naylor and R. P. Malchow, J. Neurophysiol., 2012, 107, 868 CrossRef PubMed
- J. Teissié, M. Prats, A. LeMassu, L. C. Stewart and M. Kates, Biochemistry, 1990, 29, 59 CrossRef
- B. Gabriel and J. Teissie, J. Am. Chem. Soc., 1991, 113, 8818 CrossRef
- M. Prats, J. F. Tocanne and J. Teissie, Eur. J. Biochem., 1987, 162, 379 CrossRef PubMed
- M. Prats, J. Teissié and J.-F. Tocanne, Nature, 1986, 322, 756 CrossRef
- K. H. Chua, B. S. Aminuddin, N. H. Fuizina and B. H. I. Ruszymah, Eur. Cells Mater., 2005, 9, 58 CrossRef PubMed
- P. C. Yaeger, T. L. Masi, J. L. de Oritz, F. Binette, R. Tubo and J. M. McPherson, Exp. Cell Res., 1997, 237, 318 CrossRef PubMed
- S. D. Waldman, C. G. Spiteri, M. D. Grynpas, R. M. Pillar and R. A. Kandel, J. Orthop. Res., 2003, 21, 590 CrossRef PubMed
- B. D. Elder and K. A. Athanasiou, Tissue Eng., 2009, 15, 42 Search PubMed
- T. Ikenoue, M. C. Trindade, M. S. Lee, E. Y. Lin, D. J. Schurman, S. B. Goodman and R. L. Smith, J. Orthop. Res., 2003, 21, 110 CrossRef PubMed
- S. Mizuno, T. Tateisi, T. Ushida and J. Glowacki, J. Cell. Physiol., 2002, 193, 319 CrossRef PubMed
- I. Kurtz, J. Clin. Invest., 1987, 80, 928 CrossRef PubMed
- J. P. G. Urban, A. C. Hall and K. A. Gehl, J. Cell. Physiol., 1993, 154, 262 CrossRef PubMed
- H. K. Heywood and D. A. Lee, Biochem. Biophys. Res. Commun., 2008, 373, 224 CrossRef PubMed
- J. Wall, F. Ayoub and P. O'shea, J. Cell Sci., 1995, 108, 2673 Search PubMed
- P. M. Matos, S. Goncalves and N. C. Santos, J. Pept. Sci., 2008, 14, 407 CrossRef PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr02950d |
| This journal is © The Royal Society of Chemistry 2018 |