
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comment on “Causation or only correlation? Application of causal inference graphs for evaluating causality in nano-QSAR models” by N. Sizochenko, A. Gajewicz, J. Leszczynski and T. Puzyn, Nanoscale, 2016, 8, 7203
Domonkos Attila
Tasi
 *a,
József
Csontos
b,
Balázs
Nagy
c,
Zoltán
Kónya
c and
Gyula
Tasi
c
*a,
József
Csontos
b,
Balázs
Nagy
c,
Zoltán
Kónya
c and
Gyula
Tasi
c
aDepartment of Physical Chemistry and Materials Science, University of Szeged, Rerrich Béla tér 1, H-6720 Szeged, Hungary. E-mail: dtasi@chem.u-szeged.hu
bDepartment of Physical Chemistry and Materials Science, Budapest University of Technology and Economics, P.O. Box 91, H-1521 Budapest, Hungary
cDepartment of Applied and Environmental Chemistry, University of Szeged, Rerrich Béla tér 1, H-6720 Szeged, Hungary
First published on 16th October 2018
Abstract
In this comment we show that the accuracy of a recent nano-QSAR model for toxicity predictions of metal oxide nanoparticles towards bacteria E. coli can be greatly improved. On one hand, the experimental ionization energies of metal atoms could be substituted for the erroneous semi-empirically derived heat of formation values of metal ions as descriptors to construct a more reliable nano-QSAR model based on weighted linear least-squares fittings. On the other hand, if no experimental data is available, a model relying on ionization energy descriptors from quantum chemical calculations could also be used producing exactly the same toxicity values as the experimental model.
In a recent paper,1 Leszczynski et al. have studied causality relations among toxicity data of metal oxide nanoparticles (NPs) towards bacteria E. coli and several descriptors characterizing the whole chemical structures of metal oxide NPs and their atomic constituents (e.g., standard enthalpies of formation of gaseous metal ions, in the same oxidation states as in the oxides, charges, radii and polarization powers of the metal ions in question, etc.). For the computation, the PM6 semi-empirical quantum chemical method2 was used. Of the twelve quantum chemical descriptors studied, however, the standard enthalpy of formation of gaseous metal ions (ΔHMe+) turned out to be the most reliable one.3 Causality analysis revealed that there was a strong causality relation between these descriptors and the toxicity data of the metal oxide NPs.1 Finally, they concluded that the enthalpy of formation of metal ions was the most relevant descriptor determining the toxicity of the metal oxide NPs.
Let us consider the ionization process of a gaseous metal atom:
| Me → Meq+ + qe− | (1) |
According to this process, the standard enthalpy of formation of the metal ion is as follows:
| ΔHMe+ = ΔHMe + IE | (2) |
| Metal oxide | Meq+ | pMIC50a | ΔHMe (exp)b | ΔHMe+ (exp)b | ΔHMe+ (PM6)a | ΔHMe+ (PM6)c | ΔHMe+ (RQC)c | IE (exp)d | IE (RQC)c |
|---|---|---|---|---|---|---|---|---|---|
| a Ref. 1. b Ref. 4. c This work. d By eqn (2) using ΔHMe (exp) and ΔHMe+ (exp) from this table. | |||||||||
| Al2O3 | Al(III) | 2.49 | 78.87 | 1307.12 | 1187.83 | 1187.83 | 1304.22 | 1228.25 | 1227.10 |
| Bi2O3 | Bi(III) | 2.82 | 49.50 | 1192.12 | 1137.40 | 1137.40 | 1163.22 | 1142.62 | 1113.72 |
| CoO | Co(II) | 3.51 | 101.51 | 677.22 | 601.80 | 589.30 | 671.23 | 575.71 | 579.83 |
| Cr2O3 | Cr(III) | 2.51 | 94.79 | 1344.95 | 1268.70 | 1250.18 | 1350.52 | 1250.16 | 1250.05 |
| CuO | Cu(II) | 3.20 | 80.64 | 726.77 | 706.25 | 706.25 | 729.90 | 646.13 | 649.94 |
| Fe2O3 | Fe(III) | 2.29 | 99.50 | 1361.88 | 1408.29 | 1277.12 | 1354.70 | 1262.38 | 1257.98 |
| In2O3 | In(III) | 2.81 | 58.15 | 1273.13 | 1271.13 | 1271.13 | 1266.01 | 1214.98 | 1207.86 |
| La2O3 | La(III) | 2.87 | 103.01 | 928.88 | 1017.22 | 1017.22 | 924.19 | 825.87 | 821.18 |
| NiO | Ni(II) | 3.45 | 102.70 | 697.86 | 596.70 | 562.81 | 690.70 | 595.16 | 599.48 |
| Sb2O3 | Sb(III) | 2.64 | 62.69 | 1228.13 | 1233.06 | 1233.06 | 1225.75 | 1165.44 | 1162.99 |
| SiO2 | Si(IV) | 2.20 | 107.55 | 2485.84 | 1686.38 | 1686.38 | 2480.21 | 2378.29 | 2375.95 |
| SnO2 | Sn(IV) | 2.01 | 71.99 | 2221.55 | 1717.32 | 1717.32 | 2210.28 | 2149.56 | 2138.29 |
| TiO2 | Ti(IV) | 1.74 | 113.05 | 2215.31 | 1575.73 | 1575.73 | 2211.74 | 2102.26 | 2091.88 |
| V2O3 | V(III) | 3.14 | 122.90 | 1291.50 | 1097.73 | 1054.30 | 1288.89 | 1168.60 | 1161.14 |
| Y2O3 | Y(III) | 2.87 | 100.69 | 999.16 | 837.15 | 837.15 | 994.55 | 898.47 | 893.86 |
| ZnO | Zn(II) | 3.45 | 31.17 | 662.07 | 662.44 | 662.44 | 654.48 | 630.90 | 638.24 |
| ZrO2 | Zr(IV) | 2.15 | 145.51 | 1922.65 | 1357.66 | 1357.66 | 1914.32 | 1777.14 | 1768.81 |
A careful analysis of their published data1 reveals that 9 of the 17 PM6 standard enthalpies of formation are in error. In five cases – Co(II), Cr(III), Fe(III), Ni(II), V(III) – the spin multiplicities assigned for the ground states of atomic ions differ from the real ones, and the standard enthalpies of formation obtained are too high. The largest deviation is more than 130 kcal mol−1 for Fe(III). For ions with the highest positive charges, Si(IV), Sn(IV), Ti(IV), Zr(IV), the computed standard enthalpies of formation are too low: the differences are more than 500 kcal mol−1 with respect to the experimental values. It is especially high for Si(IV): ∼800 kcal mol−1. These errors warrant us to replace the PM6 standard enthalpies of formation in question with experimental or more sophisticated computed ones.
The quality of the experimental standard enthalpies of formation and ionization energies can be guessed with the help of our computed results. Table 1 also contains our computed standard enthalpies of formation and ionization energies for ions and atoms, respectively, by relativistic quantum chemistry (RQC). The def2-TZVPP and def2-QZVPP basis sets of Ahlrichs and associates,5,6 and for bismuth the segmented all-electron relativistically contracted (SARC) basis set of Neese and coworkers7 were utilized in the quantum chemical calculations. The Hartree–Fock energies were assumed to be converged with the def2-QZVPP basis set. However, the MP2, CCSD, CCSD(T), CCSDT, and CCSDT(Q) correlation energies were extrapolated to the complete basis set limit using the inverse cubic formula of Helgaker and coworkers8 along with the def2-TZVPP and def2-QZVPP basis sets.
For third- and fourth-row elements scalar relativistic effects were described with the help of the second-order Douglas–Kroll–Hess (DKH) Hamiltonian.9,10 In these calculations the so-called point charge nucleus model was utilized. For elements below the fourth row, scalar relativistic contributions were included by means of the Stuttgart-Dresden effective core potentials (ECP).11,12 Nonrelativistic and ECP results were obtained with the MRCC suite of quantum chemical programs,13 while DKH computations were performed using the ORCA package.14 In the calculations all electrons were correlated, except, of course, the core electrons described by ECPs. We also repeated the PM6 calculations for all heat of formation values of metal ions with the proper settings. For these calculations the MOPAC2016 package15 was used.
Fig. 1 shows the correlation between the experimental and our corrected PM6- and RQC-computed ionization energies. The exceptionally good performance of the RQC over PM6 can clearly be seen. (For RQC the regression coefficient is 0.995 ± 0.002; Pearson's correlation coefficient (r) is 0.9999; and the standard deviation (sr) is 7.90 kcal mol−1. For PM6 the regression coefficient is 0.804 ± 0.067; Pearson's correlation coefficient (r) is 0.9373; and the standard deviation (sr) is 177.07 kcal mol−1.) The same is true for the computed standard enthalpies of formation. It is worth noting that due to fortuitous error cancellations, energy differences, e.g., ionization energies, can be more precisely obtained by quantum chemistry than discrete energy values, e.g., standard enthalpies of formation. That is why ionization energies are more favorable than enthalpies of formation as descriptors.
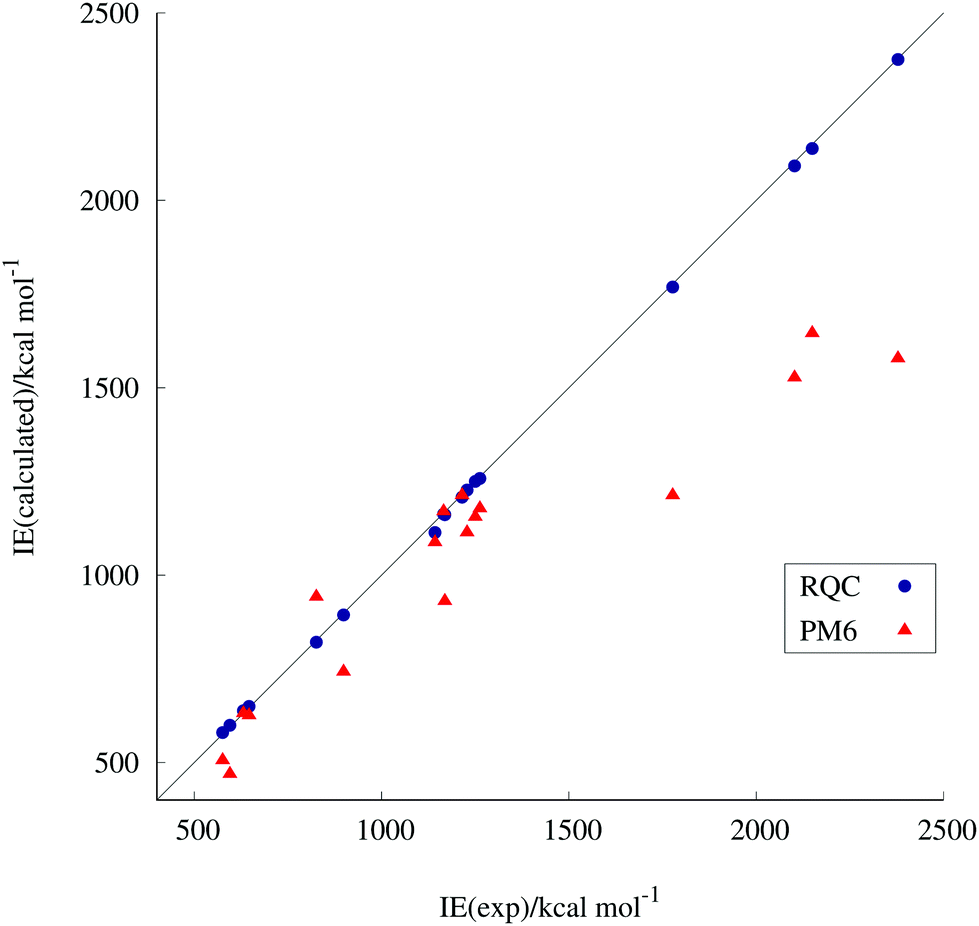 | ||
| Fig. 1 Correlation between the experimental and calculated ionization energies. | ||
PRESS and R statistics16 performed on the set of toxicity data in relation to the ionization energies revealed that two toxicity values were outliers (SiO2 and V2O3). After removing them, our training set consists of data for 15 metal oxide NPs.
Let us perform simple linear regressions16 based on ordinary linear least-squares method (OLLS), with model function pMIC50 = aIE + b, for the toxicity data using ionization energies (or standard enthalpies of formation) as descriptors. The first four lines of Table 2 show the statistics for these fittings. According to Table 2, all the models have almost equal goodness. It is known that the application of the OLLS method requires error-free independent variables and a dependent variable with uniform error. We can also perform, however, weighted linear least-squares (WLLS) fittings16 with the following weights for the toxicity data:
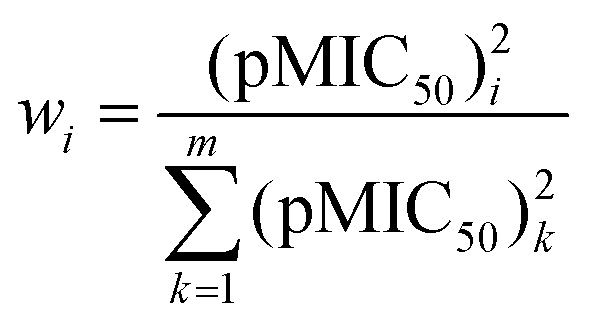 | (3) |
| Descriptor | Method | s r | r | â |
![[b with combining circumflex]](https://www.rsc.org/images/entities/i_char_0062_0302.gif)
|
F |
|---|---|---|---|---|---|---|
| a s r : standard deviation; r: Pearson's correlation coefficient; a and b: regression coefficients; F: F-test statistic. | ||||||
| IE (exp) | OLLS | 0.1798 | −0.9469 | −0.510 ± 0.048 | 2.721 ± 0.046 | 113 |
| ΔHMe+ (exp) | OLLS | 0.1728 | −0.9510 | −0.512 ± 0.046 | 2.721 ± 0.045 | 123 |
| IE (RQC) | OLLS | 0.1799 | −0.9468 | −0.510 ± 0.048 | 2.721 ± 0.046 | 112 |
| ΔHMe+ (RQC) | OLLS | 0.1732 | −0.9508 | −0.512 ± 0.046 | 2.721 ± 0.045 | 122 |
| IE (exp) | WLLS | 0.0442 | −0.9521 | −0.549 ± 0.053 | 2.731 ± 0.048 | 126 |
| IE (RQC) | WLLS | 0.0444 | −0.9517 | −0.550 ± 0.053 | 2.730 ± 0.048 | 125 |
It can be seen that 0 < wi ≤ 1, and higher toxicity involves higher precision. The last two rows of Table 2 have been obtained via WLLS fittings using eqn (3) as weights. It is to be seen that the two models are equivalent in their performance.
Because, in several cases, the PM6 results carry considerable errors neither the OLLS nor the WLLS method can be used for linear regression.
Recently, Leszczynski et al. have also considered two metal oxide NPs which do not belong to the training set:17 Mn2O3 and WO3. Unfortunately, no experimental toxicity data are available for them. For the Mn atom, both experimental4 (1308.50 kcal mol−1) and theoretical ionization energies (1302.76 kcal mol−1, this study) are available. The reported predicted value for its toxicity is 2.84 based on PM6 standard enthalpy of formation.17 The models corresponding to the last two rows of Table 2 supply a slightly lower value: 2.58. As for WO3, no experimental ionization energy is available for W(VI). Our computed value is 4639.17 kcal mol−1. Considering the range of the ionization energies in Table 1, this is out of the domain of the training set. Since every model is only valid in its own IE range used to construct the model, in other words, only interpolation is allowed in this domain, therefore no reliable prediction for WO3 can be made via the nano-QSAR models. The same holds for SiO2, where the IE of Si atom is slightly out of range considering that both the experimental and RQC-computed values are around 2380 kcal mol−1. For V2O3, which was also an outlier, the experimental pMIC50 is 3.14, while our computed value is 2.73 with both the RQC- and the experimental-IE-based models. Assuming that our nano-QSAR models yield accurate toxicity for every NP, where the IE of the constituent metal atom is in the range of the training set, the experimental pMIC50 for V2O3 is very likely to be in error, and repetition of this toxicity measurement is recommended.
In summary, we discussed that the recently published PM6-based heat of formation values used as descriptors for nano-QSAR models were in error for many transition metal ions, therefore toxicity predictions for metal oxide NPs based on them are not reliable. In this comment we revealed that by using the weighted linear least-squares method, it is possible to construct better nano-QSAR models based on either experimentally or quantum chemically derived atomic ionization energies instead of the ionic enthalpies of formation from semi-empirical calculations.
Conflicts of interest
There are no conflicts to declare.References
- N. Sizochenko, A. Gajewicz, J. Leszczynski and T. Puzyn, Nanoscale, 2016, 8, 7203–7208 RSC.
- J. J. P. Stewart, J. Mol. Model., 2007, 13, 1173–1213 CrossRef CAS PubMed.
- T. Puzyn, B. Rasulev, A. Gajewicz, X. Hu, T. P. Dasari, A. Michalkova, H.-M. Hwang, A. Toropov, D. Leszczynska and J. Leszczynski, Nat. Nanotechnol., 2011, 6, 175 CrossRef CAS PubMed.
- W. M. Haynes, CRC handbook of chemistry and physics, 96th Edition (Internet Version 2016), CRC Press/Taylor and Francis, Boca Raton, FL, 2016 Search PubMed.
- A. Schäfer, H. Horn and R. Ahlrichs, J. Chem. Phys., 1992, 97, 2571–2577 CrossRef.
- F. Weigend and R. Ahlrichs, Phys. Chem. Chem. Phys., 2005, 7, 3297–3305 RSC.
- D. A. Pantazis and F. Neese, Theor. Chem. Acc., 2012, 131, 1292 Search PubMed.
- T. Helgaker, W. Klopper, H. Koch and J. Noga, J. Chem. Phys., 1997, 106, 9639–9646 CrossRef CAS.
- M. Douglas and N. M. Kroll, Ann. Phys., 1974, 82, 89–155 CAS.
- B. A. Hess, Phys. Rev. A, 1986, 33, 3742–3748 CrossRef CAS.
- D. Andrae, U. Häußermann, M. Dolg, H. Stoll and H. Preuß, Theor. Chim. Acta, 1990, 77, 123–141 CrossRef CAS.
- B. Metz, H. Stoll and M. Dolg, J. Chem. Phys., 2000, 113, 2563–2569 CrossRef CAS.
- Mrcc, a quantum chemical program suite written by M. Kállay, Z. Rolik, J. Csontos, P. Nagy, G. Samu, D. Mester, J. Csóka, B. Szabó, I. Ladjánszki, L. Szegedy, B. Ladóczki, K. Petrov, M. Farkas, P. D. Mezei, and B. Hégely. See also. Z. Rolik, L. Szegedy, I. Ladjánszki, B. Ladóczki and M. Kállay, J. Chem. Phys., 2013, 139, 094105, as well as: http://www.mrcc.hu.
- F. Neese, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2012, 2, 73–78 CAS.
- J. J. P. Stewart, MOPAC2016, Stewart Computational Chemistry, Colorado Springs, CO, USA, 2016. http://OpenMOPAC.net Search PubMed.
- R. H. Mayers, Classical and Modern Regression with Applications, Higher Education Press, Beijing, 2nd edn., 2005 Search PubMed.
- S. Kar, A. Gajewicz, K. Roy, J. Leszczynski and T. Puzyn, Ecotoxicol. Environ. Saf., 2016, 126, 238–244 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |