
Hollow SnO2 nanospheres with oxygen vacancies entrapped by a N-doped graphene network as robust anode materials for lithium-ion batteries†
Naiteng
Wu
 a,
Wuzhou
Du
a,
Xu
Gao
b,
Liang
Zhao
c,
Guilong
Liu
a,
Xianming
Liu
*a,
Hao
Wu
*b and
Yan-Bing
He
*c
a,
Wuzhou
Du
a,
Xu
Gao
b,
Liang
Zhao
c,
Guilong
Liu
a,
Xianming
Liu
*a,
Hao
Wu
*b and
Yan-Bing
He
*c
aKey Laboratory of Function-oriented Porous Materials, College of Chemistry and Chemical Engineering, Luoyang Normal University, Luoyang, 471934, P. R. China. E-mail: myclxm@163.com
bCollege of Materials Science and Engineering, Sichuan University, Chengdu 610064, P. R. China. E-mail: hao.wu@scu.edu.cn
cEngineering Laboratory for the Next Generation Power and Energy Storage Batteries, Graduate School at Shenzhen, Tsinghua University, Shenzhen 518055, PR China. E-mail: he.yanbing@sz.tsinghua.edu.cn
First published on 19th May 2018
Abstract
The practical application of tin dioxide (SnO2) in lithium-ion batteries has been greatly hindered by its large volumetric expansion and low conductivity. Thus, a rational design of the size, geometry and the pore structure of SnO2-based nanomaterials is still a dire demand. To this end, herein we report an effective approach for engineering hollow-structured SnO2 nanospheres with adequate surface oxygen vacancies simultaneously wrapped by a nitrogen-doped graphene network (SnO2−x/N-rGO) through an electrostatic adsorption-induced self-assembly together with a thermal reduction process. The close electrostatic attraction achieved a tight and uniform combination of positively charged SnO2 nanospheres with negatively charged graphene oxide (GO), which can alleviate the aggregation and volume expansion of the entrapped SnO2 nanospheres. Subsequent thermal treatment not only ensures a significant reduction of the GO sheets accompanying nitrogen-doping, but also induces the generation of oxygen vacancies on the surface of the SnO2 hollow nanospheres, together building up a long-range and bicontinuous transfer channel for rapid electron and ion transport. Because of these structural merits, the as-built SnO2−x/N-rGO composite used as the anode material exhibits excellent robust cycling stability (∼912 mA h g−1 after 500 cycles at 0.5 A g−1 and 652 mA h g−1 after 200 cycles at 1 A g−1) and superior rate capability (309 mA h g−1 at 10 A g−1). This facile fabrication strategy may pave the way for the construction of high performance SnO2-based anode materials for potential application in advanced lithium-ion batteries.
Introduction
SnO2 has been extensively considered as a potential anode material for next-generation lithium-ion batteries (LIBs), which is attributed to its high theoretical specific capacity and low cost.1–4 However, as a typical semiconductor, low electrical conductivity and large volume changes during the alloying–dealloying process are the two intrinsic drawbacks which hinder the practical application of SnO2-based anode materials.5,6 The low electrical conductivity usually means an unsatisfactory rate performance.7 In addition, more than 300% volume expansion not only hinders the formation of a stable solid electrolyte interphase (SEI) film, but also peels the electrode materials off the copper foil, resulting in rapid capacity fading during the cyclic process.8,9The design and development of nanostructure materials has been considered as an effective approach to address the above issues.8,10 Numerous nanostructures, such as quantum dots,11,12 nanowires,13–15 nanotubes,16 nanosheets,17 nanospheres,7,18 nanocage19 and nanoparticles5 have been synthesized to improve the electrochemical performances of an SnO2 anode. Particularly, a hollow structure with a large void space can buffer the volume expansion and alleviate the volumetric strain significantly during repeated cycles.20–23 The integration of the anode material and the carbon-based material is another approach for buffering the volume changes, meanwhile increasing the electrical conductivity of the composite materials.3,24–27 Notably, nitrogen-doped carbon coating can also effectively enhance the electronic conductivity and the SEI film's stability, and decrease the charge transfer resistance at the electrode–electrolyte interface, synchronously.14,28,29 Furthermore, a graphene nanosheet, as a good coating carbon material to modify the nanomaterials, has been targeted for the improvement of the electrochemical performance of SnO2-based materials owing to its excellent conductivity and high flexibility.18,30–34 However, due to the easy aggregation of nanoparticles and the re-stacking of the graphene nanosheets, the composites with a tight and uniform combination between SnO2 and graphene are often hindered by the tedious and unscalable preparation.
Recently, introduction of oxygen vacancies into the electrode materials has been devoted to improving the electrochemical performance.35–37 A black tin oxide nanostructure reported by Dong et al. has demonstrated that the presence of numerous oxygen vacancies greatly enhanced the electrical conductivity.6 The SnO2-Mn-graphite composite-induced oxygen vacancies by the Mn reduction has greatly improved the initial coulombic efficiency and cycling stability.38 In this respect, the elaborate design and scalable preparation of a tight and uniform combined hollow SnO2/graphene composite with numerous oxygen vacancies would enhance the lithium storage properties for its further application in LIBs.
In this work, an electrostatic adsorption-induced self-assembly together with a thermal reduction process was developed to encapsulate SnO2 hollow nanospheres with oxygen vacancies in the N-doped graphene hierarchical network (Fig. 1). The surface charge of the SnO2 hollow nanospheres was modified with polydimethyl diallyl ammonium chloride (PDDA) to make the SnO2 hollow nanospheres carry a positive charge (Fig. S1†). Graphene oxide (GO, the aqueous solution of GO usually presents negative electricity39) sheets were driven in close combination on the surface of the PDDA-modified SnO2 due to electrostatic attraction. In the subsequent N-doped and thermal reduction process, the SnO2 hollow nanospheres were tightly encapsulated in the 3D N-doped graphene hierarchical network (denoted as SnO2−x/N-rGO). The tight coating layer, stretchy graphene network and the hollow structure endow the SnO2−x/N-rGO with the enhanced capability to buffer the volume expansion. Furthermore, the graphene network would improve its electron conductivity intrinsically. Because of the structural superiority, the as-prepared SnO2−x exhibits a significantly improved cycling stability and rate capability compared to the other SnO2 counterparts.
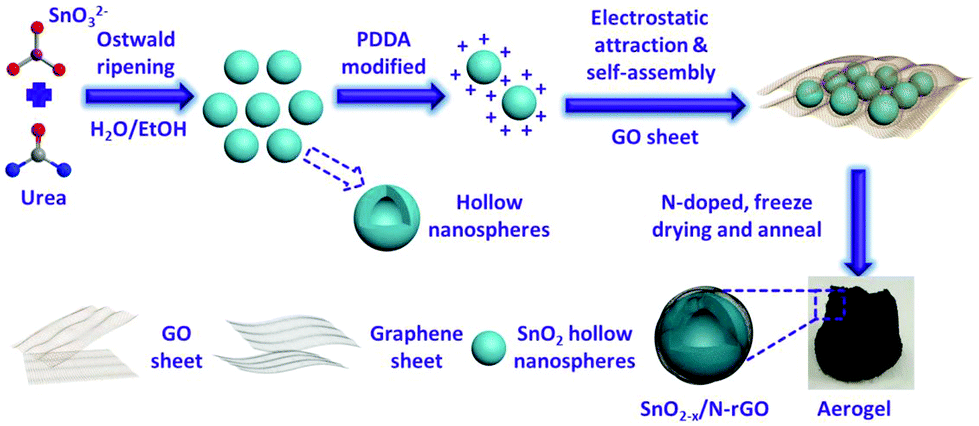 | ||
| Fig. 1 Schematic illustration of the preparation process of the 3D hierarchical SnO2−x/N-rGO network. | ||
Experimental
Material synthesis
Characterization
X-ray diffraction (XRD, Bruker D8 with Cu Kα radiation) analysis was carried out to determine the crystal structure of the samples. The morphology and the structure of the samples were investigated by SEM (SEM, Hitachi S4800) and transmission electron microscopy (TEM, JEOL JEM2100, equipped with an EDX spectrometer). X-ray photoelectron spectroscopy (XPS, EscaLab 250Xi) was performed to determine the valence state of the products. N2 adsorption/desorption measurements were conducted with a Tristar II 3020 instrument. To investigate the content of the coating carbon, thermal gravimetric analysis was conducted on a thermal analyzer (SII TG/DTA6300) in air at a heating rate of 5 °C min−1. NanoZS90 (Malvern Instruments) was used to measure the zeta potential.Electrochemical measurements
Half-coin cells were taken to test the lithium storage capability of the as-prepared SnO2. The assembly process of the electrode and the cells is the same as our previous studies.40,41 Particularly, the mass ratio of the active materials, acetylene black and polyvinylidene fluoride (PVDF, as a binder) is 7![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2:1. The average mass loading of the active material is 1.3 mg cm−2. The Neware CT-3008 W was used to record the data of charge–discharge. The potential range of the test process is from 0.005 to 3.0 V (vs. Li/Li+). The electrochemical impedance spectroscopy (EIS) of the cells after 500 cycles were performed in the frequency range from 100 kHz to 10 mHz with an AC amplitude of 5 mV using a Parstat 4000+ (Princeton Applied Research, USA) electrochemical workstation. Cyclic voltammetry (CV) measurements were also conducted on the Parstat 4000+ workstation at a scanning rate of 0.1 mV s−1 in the potential range from 0.005 to 3.0 V (vs. Li+/Li).
:2:1. The average mass loading of the active material is 1.3 mg cm−2. The Neware CT-3008 W was used to record the data of charge–discharge. The potential range of the test process is from 0.005 to 3.0 V (vs. Li/Li+). The electrochemical impedance spectroscopy (EIS) of the cells after 500 cycles were performed in the frequency range from 100 kHz to 10 mHz with an AC amplitude of 5 mV using a Parstat 4000+ (Princeton Applied Research, USA) electrochemical workstation. Cyclic voltammetry (CV) measurements were also conducted on the Parstat 4000+ workstation at a scanning rate of 0.1 mV s−1 in the potential range from 0.005 to 3.0 V (vs. Li+/Li).
Results and discussion
SEM and TEM were carried out to characterize the morphology and the detailed structure of the as-prepared SnO2-based samples. As shown in Fig. 2a and b, SnO2 exhibits the regular spherical morphology with the diameter in the range of 100–300 nm. The broken sphere pointed by the green arrow suggests that the as-prepared spheres process the hollow structure. The hollow structures of the SnO2 nanospheres are clearly demonstrated by the TEM image (as shown in the inset of Fig. 2a and Fig. S2a†) showing the distinct contrast between the hollow and the shell with about 50 nm thickness. After the carbon coating process, the SnO2/C exhibits a similar hollow structure as bare SnO2. However, the surface of SnO2/C is rough, because of the crystallization of the primary particles (Fig. 2b and Fig. S2b†). The SEM images of SnO2−x/N-rGO are shown in Fig. 2c and d. In the panoramic SEM image the SnO2−x hollow spheres are well encapsulated by the graphene network. The magnified SEM image (Fig. 2d) confirms that the SnO2−x was completely wrapped by the graphene sheets. The TEM images of SnO2−x/N-rGO (Fig. 2e and Fig. S2c†) exhibit the obvious hollow structure with about 200 nm diameter and 30 nm thickness. Besides, the hollow spheres were well encapsulated by the monolayer rGO sheet. This observation is consistent with the SEM results. The void space and the graphene coating layer can be clearly seen from the magnified TEM image (Fig. 2f), which would enhance the alleviated ability of the volumetric strain and the conductivity of the SnO2−x hollow spheres, simultaneously. The HRTEM image of SnO2−x/N-rGO displays clear lattice fringes with an interplanar distance of about 0.33 nm, assigned to the d-spacing of the (110) facet of the tetragonal SnO2.7,42 Furthermore, the EDX mapping results of SnO2−x/N-rGO exhibit a homogeneous distribution of the elements Sn, O, C and N, revealing the long-range and bi-continuous transfer channel for a rapid electron and ion transport. The tight and uniform combination of SnO2−x/N-rGO can be constructed on a large scale through the electrostatic attraction and self-assembly route according to the SEM and TEM analyses. The N2 absorption/desorption isotherms and the BET results have been shown in Fig. S3.† As measured, the specific surface area of the bare SnO2 is about 34.43 m2 g−1, while the specific surface area of SnO2/C and SnO2−x/N-rGO has increased to 38.86 and 103.4 m2 g−1, respectively. The increased specific surface area endows SnO2−x/N-rGO with more contact area with the electrolyte to improve the electrochemical performance.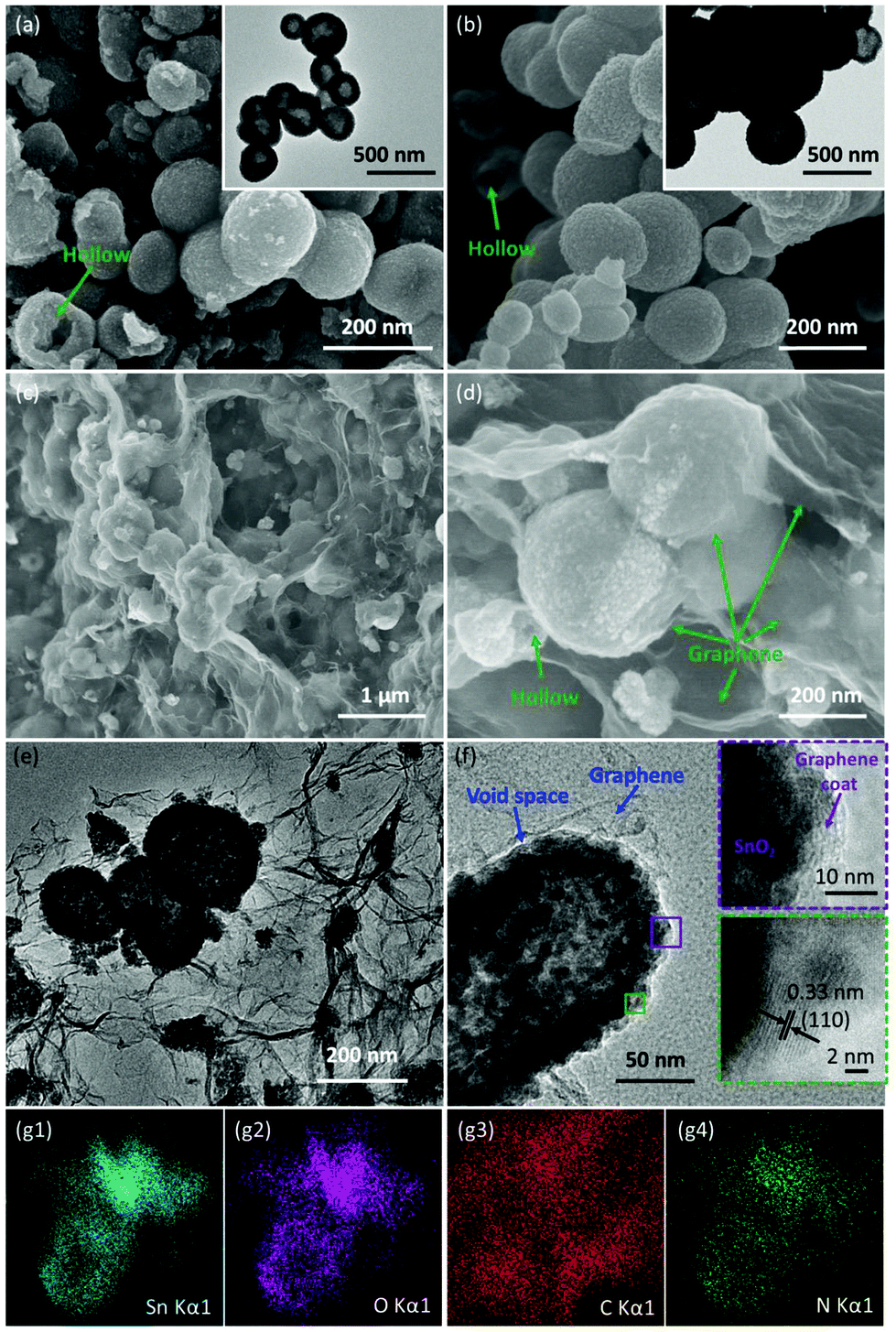 | ||
| Fig. 2 (a) and (b) SEM images of SnO2 hollow nanospheres and SnO2/C, and the corresponding TEM images in the inset; (c) and (d) panoramic and magnified SEM images of the as-prepared SnO2−x/N-rGO; (e) and (f) TEM images of SnO2−x/N-rGO, and the insets in (f) show the HRTEM images; (g1–4) EDX elemental mapping images of SnO2−x/N-rGO. | ||
XRD was carried out to identify the phase composition and crystal structure of the as-prepared SnO2-based samples. As shown in Fig. 3a, all the diffraction peaks of the samples can be clearly assigned to tetragonal SnO2 (JCPDS: 41-1445). The diffraction peaks of (110), (101) and (211) became sharper after the carbon coating and graphene wrapping, indicating the enhanced crystallinity of the modified SnO2-based materials. The composition and the chemical valence state of the as-prepared samples were obtained by XPS. As shown in Fig. 3b, the survey scan XPS spectra of SnO2/C and SnO2−x/N-rGO present Sn 3s, Sn 3p, O 1s, Sn 3d, C 1s, Sn 4s, Sn 4p and Sn 4d, with no evidence of impurities. In particular, there is an obvious peak of N 1s in the SnO2−x/N-rGO sample, suggesting that the nitrogen element was successfully doped in the lattice of the rGO sheets.39,43,44 The following high-resolution N 1s spectrum (Fig. 3c) reveals three components located at 398.5, 400.3 and 402.7 eV corresponding to the pyridinic, pyrrolic and graphitic nitrogen atoms, respectively.28,29,39 Furthermore, the N-doped level is about 5.9 at% revealed by the XPS analysis. The high-resolution C 1s XPS spectrum of SnO2−x/N-rGO (Fig. 3d) shows three peaks located at 284.7, 285.5 and 286.9 eV for C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C, C–N and CO, respectively.45 The low intensity of the CO peak indicates the high reduction degree of graphene oxides. Besides, the presence of C–N peaks further proves the nitrogen doping in the rGO lattice, which would improve the conductivity of the SnO2−x/N-rGO electrodes.46,47 The high-resolution Sn 3d spectra of bare SnO2 and SnO2−x/N-rGO are depicted in the inset of Fig. 3e. The locations of Sn 3d for SnO2−x/N-rGO exhibit the obvious right shift compared with the bare SnO2, which suggested the formation of a low valence state tin on the surface.6,8 The high-resolution Sn 3d XPS spectrum for SnO2−x /N-rGO (Fig. 3e) indicates that most of the tin element on the near-surface is Sn2+, due to the partial surface reduction of SnO2 by the rGO sheets. The existence of Sn2+ could induce the formation of numerous oxygen vacancies which enhanced the conductivity of SnO2-based materials.35,38 The TG evaluation was carried out to determine the carbon content in SnO2/C and SnO2−x/N-rGO in air at a heating rate of 5 °C min−1. As shown in Fig. S4,† the weight loss before 100 °C is due to the removal of absorbed water, and the subsequent weight loss of 11.3% and 19.4% may correspond to the combustion of the amorphous carbon coating layer and the rGO wrapping of SnO2/C and SnO2−x/N-rGO, respectively.
C, C–N and CO, respectively.45 The low intensity of the CO peak indicates the high reduction degree of graphene oxides. Besides, the presence of C–N peaks further proves the nitrogen doping in the rGO lattice, which would improve the conductivity of the SnO2−x/N-rGO electrodes.46,47 The high-resolution Sn 3d spectra of bare SnO2 and SnO2−x/N-rGO are depicted in the inset of Fig. 3e. The locations of Sn 3d for SnO2−x/N-rGO exhibit the obvious right shift compared with the bare SnO2, which suggested the formation of a low valence state tin on the surface.6,8 The high-resolution Sn 3d XPS spectrum for SnO2−x /N-rGO (Fig. 3e) indicates that most of the tin element on the near-surface is Sn2+, due to the partial surface reduction of SnO2 by the rGO sheets. The existence of Sn2+ could induce the formation of numerous oxygen vacancies which enhanced the conductivity of SnO2-based materials.35,38 The TG evaluation was carried out to determine the carbon content in SnO2/C and SnO2−x/N-rGO in air at a heating rate of 5 °C min−1. As shown in Fig. S4,† the weight loss before 100 °C is due to the removal of absorbed water, and the subsequent weight loss of 11.3% and 19.4% may correspond to the combustion of the amorphous carbon coating layer and the rGO wrapping of SnO2/C and SnO2−x/N-rGO, respectively.
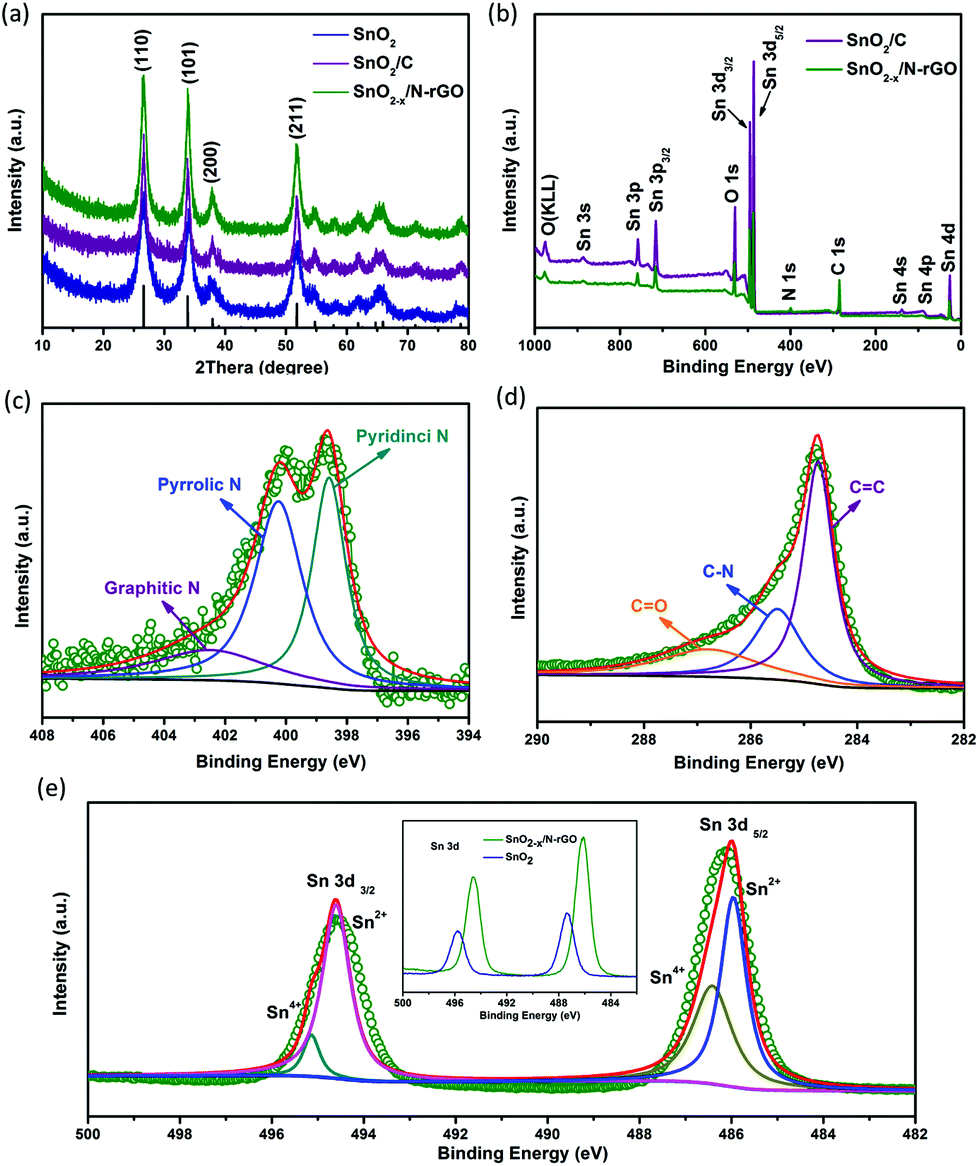 | ||
| Fig. 3 (a) XRD patterns of SnO2, SnO2/C and SnO2−x/N-rGO; (b) XPS spectrum of SnO2/C and SnO2−x/N-rGO; and the high-resolution spectrum of (c) C 1s, (d) N 1s and (e) Sn 3d for the as-prepared SnO2−x/N-rGO, and the inset of (e) shows the Sn 3d spectrum of SnO2/C and SnO2−x/N-rGO. | ||
The as-prepared 3D hierarchical SnO2−x/N-rGO network was assembled as the anode of the CR2032 cells to evaluate the lithium storage properties. The specific capacity is calculated according to the total weight of the SnO2-based electrode materials. The typical discharge–charge curves of SnO2−x/N-rGO in the voltage range of 0.005–3.0 V at a current density of 0.1 A g−1 are shown in Fig. 4a. The initial discharge and charge capacity of SnO2−x/N-rGO are 1271 and 773 mA h g−1, respectively, corresponding to a coulombic efficiency of 60.8%. The low initial coulombic efficiency is ascribed to the formation of the irreversible amorphous lithium oxides and the SEI film and the decomposition of the electrolyte.38 After the first cycle, the discharge–charge curves of SnO2−x/N-rGO are well overlapped, suggesting the formation of a stable SEI film on the wrapping graphene sheet surface. The coulombic efficiency increases to 93.9%, 95.4% and 98.1% in the 2nd, 3rd and 4th cycles, respectively. The discharge–charge curves of the counterparts are shown in Fig. S5.† The initial coulombic efficiency of SnO2 and SnO2/C is 51.7 and 60%, respectively. The poorer initial coulombic efficiency of the bare SnO2 may be attributed to the inferior reversibility of the conversion reaction in the lithiated SnO2. Besides, the nonoverlapping discharge–charge curves of SnO2 and SnO2/C at the subsequent cycles imply that a stable SEI film was not formed.13 The tight graphene coating layers together promote the reaction reversibility and prevent the particles coarsening successfully during the charge–discharge process.
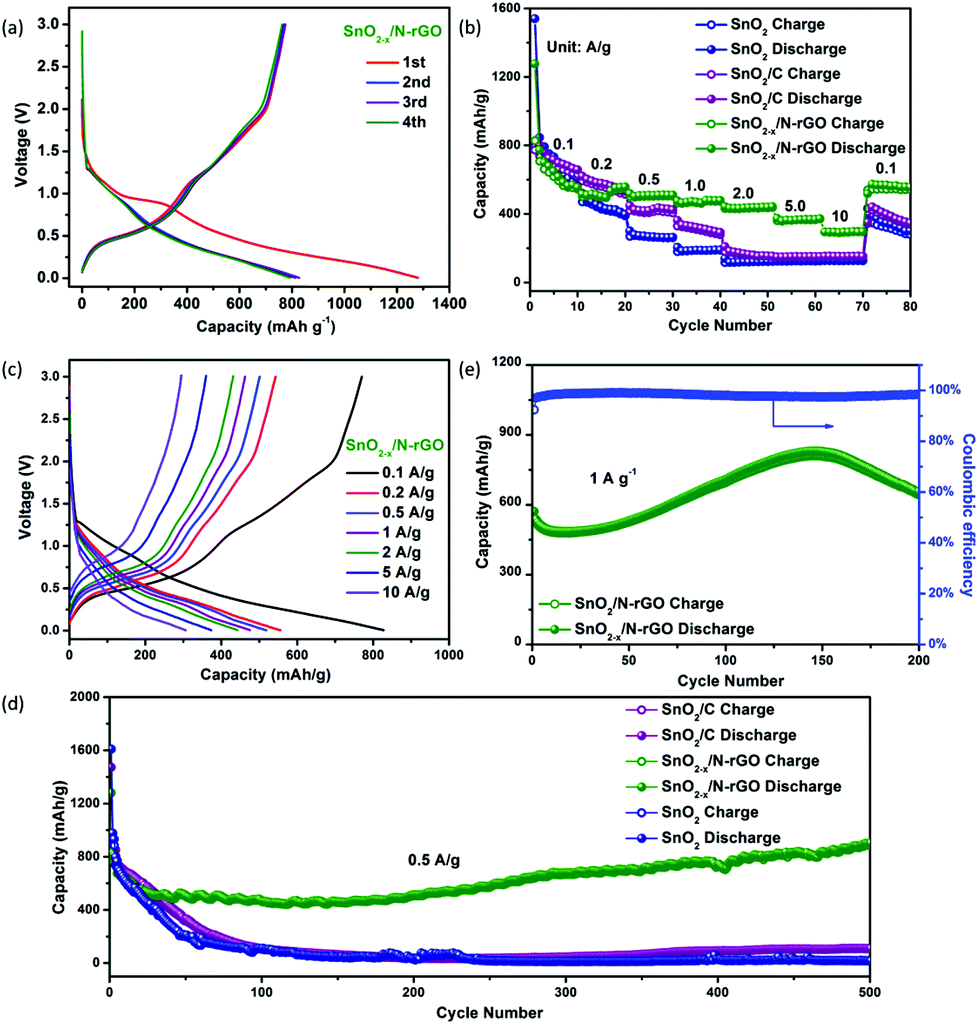 | ||
| Fig. 4 (a) The first four discharge–charge curves of the SnO2−x/N-rGO electrode at 0.1 A g−1; (b) rate performances and (c) charge–discharge curves of the as-prepared SnO2, SnO2/C and SnO2−x/N-rGO samples at different current densities; (d) cycling properties of the as-prepared cells at a current rate of 0.5 A g−1 (e) cycling performance of SnO2−x/N-rGO at 1 A g−1. | ||
The rate performances of the as-prepared SnO2-based electrodes at different current densities are depicted in Fig. 4b. The SnO2−x/N-rGO delivers a stable reversible discharge capacity of 564, 507, 483, 434, 371 and 309 mA h g−1 at 0.2, 0.5, 1, 2, 5, and 10 A g−1, respectively, which is much higher than its counterparts, especially at high current densities. Fig. 4c depicts the charge–discharge curves of the SnO2−x/N-rGO cells at different current densities. These curves show similar shapes at different rates, which indicate the superior rate capability of the SnO2−x/N-rGO electrodes. The electronic conductivity of the anode materials which is a critical issue for its rate capability had also been enhanced by the graphene network. As shown in Fig. 4d, the initial discharge capacity of SnO2−x/N-rGO is about 827 mA h g−1. After the activation process, the capacity of SnO2−x/N-rGO gradually reached as high as 912 mA h g−1 at the end of 500 cycles at a current density of 0.5 A g−1. This reversible capacity is the highest among the three types of SnO2 electrodes, which only delivers about 17 (SnO2) and 110 mA h g−1 (SnO2/C), respectively. Besides, the stable and close to 100% coulombic efficiency of SnO2−x/N-rGO further demonstrates that the 3D hierarchical graphene network enhances the reversibility of the SnO2 electrode during the tedious cycles (Fig. S6†). The increasing capacity of SnO2−x/N-rGO would be attributed to the increasing contract area between the porous electrode and the electrolyte, which could be developed and exposed gradually during the cycling process, further leading to the enhancement of the interfacial Li+ storage ability and reversible capacity.48–51 Furthermore, the long-term cyclic evaluation at 1 A g−1 is depicted in Fig. 4e. Impressively, SnO2−x/N-rGO maintains a 652 mA h g−1 at the end of 200 cycles. The electrochemical performances of the as-prepared SnO2−x hollow spheres wrapped in the 3D N-doped graphene network are more competitive compared to those in the previous studies on the composites of the SnO2 nanostructures and graphene (detailed data are listed in Table S1†). The excellent robust cycling performance of SnO2−x/N-rGO would be attributed to the tight and uniform construction of the SnO2 hollow structure and the graphene sheets which suppressed the side reaction and volume expansion during the cycles.
The first three CV curves of SnO2−x/N-rGO are depicted in Fig. 5a to investigate their kinetic behavior. The cathodic peak located at 0.8 V corresponds to the initial reduction of SnO2 and the generation of the SEI film.3 The peak observed at 0.1 V represents the alloying reaction between Li and Sn.30,52 At the subsequent anodic process, the peaks located at 0.56 and 1.25 V indicate the dealloying process of LixSn and the reversible reaction between SnO2 and Li, respectively.31,53,54 Compared with the CV curves of SnO2 and SnO2/C, SnO2−x/N-rGO exhibits a better reversibility and lower polarization when observed from the symmetrical and overlapping CV curves after the first cycle (Fig. S7†). In addition, EIS measurements were used to investigate the origin of the SnO2−x/N-rGO excellent cycling performance. The Nyquist plots of SnO2, SnO2/C and SnO2−x/N-rGO cells after 500 cycles at 0.5 A g−1 are shown in Fig. 5b. All the Nyquist plots could be divided into three components, including the intercept at the Z′-axis in the high frequency region, the semicircle in the high-middle frequency region and the oblique line in the low frequency region, which represent the resistance of the electrolyte (Rs), the resistance of the charge transfer (Rct) and the Warburg impedance related to the lithium-ion diffusion (DLi), respectively.55–57 The smallest Rct of SnO2−x/N-rGO (55.7 Ω, Table S2†) compared to that of SnO2 (534.4 Ω) and SnO2/C (124.6 Ω) indicates the reduction of the unsatisfactory side reactions on the electrode/electrolyte interface. The tight and stretchy graphene coating layer may decrease the contact area of the anode material and the electrolyte, and avoid the repeated formation of the SEI films. Furthermore, the Warburg factors (σ) of the electrodes, as a crucial parameter for DLi according to the previous literature studies,6,58 are fitted by the relationship between Z′ and ω−0.5 (Fig. S8†) and listed in Table S2.† The smallest σ of SnO2−x/N-rGO means the largest DLi and the fast lithium intercalation/deintercalation during the long-term cycles, which is attributed to the high conductivity of the SnO2-based electrode by the hierarchical graphene network wrapping. The CV and EIS analyses demonstrate that the elaborate construction of SnO2−x/N-rGO endows the SnO2 electrode with the improved reversibility of the kinetic behavior and the electronic conductivity, simultaneously.
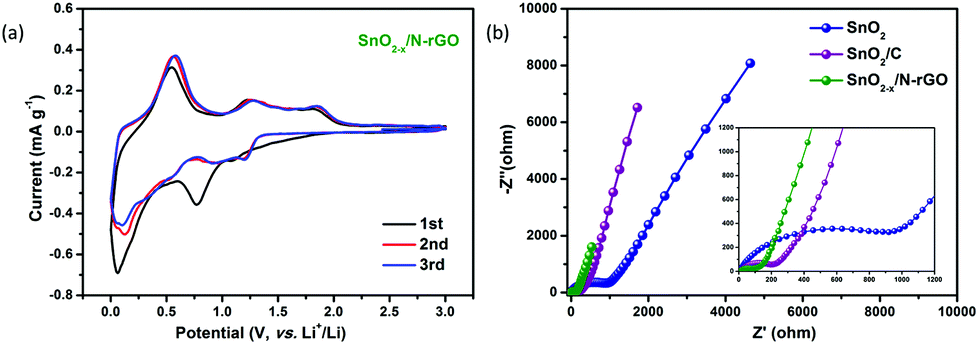 | ||
| Fig. 5 (a) CV curves of the SnO2−x/N-rGO electrode in the voltage range from 0.005 to 3.0 V at a scanning rate of 0.1 mV s−1; (b) Nyquist plots SnO2, SnO2/C and SnO2−x/N-rGO after 500 cycles. | ||
The correlation between the cycling performance and the structural features of the as-prepared SnO2 and SnO2−x/N-rGO cathode materials has been confirmed by the SEM characterization after the 500th cycle. As shown in Fig. 6a and b, the initial spherical morphology of the SnO2−x/N-rGO has been well maintained at the end of the 500 cycles. The volumetric expansion during the repeated cycles of the SnO2−x/N-rGO electrode has been restricted to the flexible graphene sheets. The structural integrity of the electrode materials is the precondition for the cycling stability.39,59 In contrast, the bare SnO2 electrode without the constraint of the graphene sheets exhibits an obvious amorphization and volumetric expansion after the long-term cycles (Fig. 6c and d). The initial hollow spherical structure of the bare SnO2 has collapsed, and the cycled SnO2 electrode displays the morphology of the solid particle packaged in an amorphous shell, which is commonly considered as the unstable SEI film and the degraded structure. The schematic diagram of the lithiation process of SnO2−x/N-rGO and SnO2 is shown in Fig. 6e and f, respectively. The amorphization of the primary particles during the alloying–dealloying process of the SnO2−x/N-rGO was inhibited by the synergistic effect of the tight protective layer. Furthermore, the comparison of the electrode disk thickness at the initial and cycled status had also been carried out. As shown in Fig. S9,† the thickness swelling of the SnO2 electrode was about 139.5%, which is much bigger than that of SnO2−x/N-rGO (106.4%). The post-mortem analyses further indicate that the hierarchical graphene network may tolerate the stress from a large volumetric expansion.
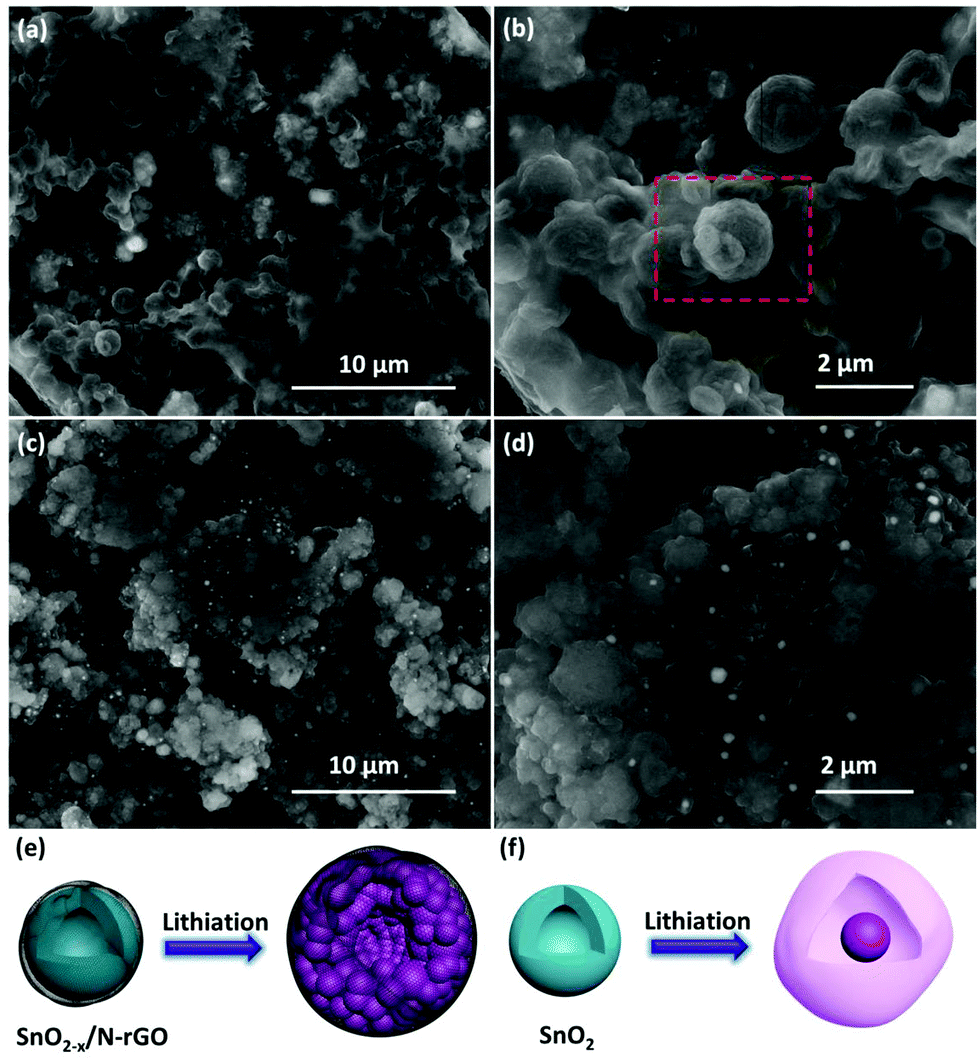 | ||
| Fig. 6 SEM images of the 500 cycles of (a, b) SnO2−x/N-rGO and(c, d) the SnO2 electrode; schematic diagrams of the lithiation process of (e) SnO2−x/N-rGO and (f) SnO2. | ||
Conclusions
In summary, SnO2 hollow nanospheres with oxygen vacancies encapsulated by a N-doped graphene hierarchical network have been designed and synthesized by using a facile and scalable approach. The SnO2−x hollow nanospheres which were in a tight and uniform combination with the rGO through the electrostatic attraction had efficiently alleviated the aggregation and volume expansion during the cycles. Besides, the oxygen vacancies which were induced by the reduction of N-doped rGO, together with the stretchy graphene network, constructed the rapid transfer channel for the electrons and lithium ions. Owing to the above structural features, the as-prepared SnO2−x/N-rGO exhibits excellent lithium storage properties, including an enhanced reversible capacity, more robust cycling stability (912 mA h g−1 at the end of 500 cycles at 0.5 A g−1 and 652 mA h g−1 at the end of 200 cycles at 1 A g−1) and quick charge/discharge capability (309 mA h g−1 at 10 A g−1) than its bare and carbon coated counterparts. This study shows this elaborate fabrication as an efficient strategy to construct SnO2-based anode materials with excellent properties for use in advanced lithium ion batteries.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (No. 21373107), Colleges and Universities in Henan Province Key Science and Research Project (No. 18A430002), the Guangdong special support program (2015TQ01N401), and the Production-Study-Research Cooperation Project of Guangdong Province (No. 2014B090901021).Notes and references
- Y. Zhao, L. P. Wang, M. T. Sougrati, Z. Feng, Y. Leconte, A. Fisher, M. Srinivasan and Z. Xu, Adv. Energy Mater., 2017, 7, 1601424 CrossRef.
- B. Ahmed, D. H. Anjum, Y. Gogotsi and H. N. Alshareef, Nano Energy, 2017, 34, 249–256 CrossRef.
- B. Huang, X. Li, Y. Pei, S. Li, X. Cao, R. C. Massé and G. Cao, Small, 2016, 12, 1945–1955 CrossRef PubMed.
- Z. Xu, X. Liu, Y. Luo, L. Zhou and J. Kim, Prog. Mater. Sci., 2017, 90, 1–44 CrossRef.
- G. Xia, N. Li, D. Li, R. Liu, C. Wang, Q. Li, X. Lü, J. S. Spendelow, J. Zhang and G. Wu, ACS Appl. Mater. Interfaces, 2013, 5, 8607–8614 Search PubMed.
- W. Dong, J. Xu, C. Wang, Y. Lu, X. Liu, X. Wang, X. Yuan, Z. Wang, T. Lin, M. Sui, I. Chen and F. Huang, Adv. Mater., 2017, 29, 1700136 CrossRef PubMed.
- Y. J. Hong, M. Y. Son and Y. C. Kang, Adv. Mater., 2013, 25, 2279–2283 CrossRef PubMed.
- R. Hu, Y. Ouyang, T. Liang, H. Wang, J. Liu, J. Chen, C. Yang, L. Yang and M. Zhu, Adv. Mater., 2017, 29, 1605006 CrossRef PubMed.
- J. Liu, W. K. Pang, T. Zhou, L. Chen, Y. Wang, V. K. Peterson, Z. Yang, Z. Guo and Y. Xia, Energy Environ. Sci., 2017, 10, 1456–1464 Search PubMed.
- L. Zhang, W. Fan and T. Liu, Nanoscale, 2016, 8, 16387–16394 RSC.
- C. Zhu, S. Zhu, K. Zhang, Z. Hui, H. Pan, Z. Chen, Y. Li, D. Zhang and D. Wang, Sci. Rep., 2016, 6, 25829–25836 CrossRef PubMed.
- K. Zhao, L. Zhang, R. Xia, Y. Dong, W. Xu, C. Niu, L. He, M. Yan, L. Qu and L. Mai, Small, 2016, 12, 588–594 CrossRef PubMed.
- D. Zhou, W. Song and L. Fan, ACS Appl. Mater. Interfaces, 2015, 7, 21472–21478 Search PubMed.
- S. Wang, Y. Yang, C. Jiang, Y. Hong, W. Quan, Z. Zhang and Z. Tang, J. Mater. Chem. A, 2016, 4, 12714–12719 Search PubMed.
- C. Guan, X. Wang, Q. Zhang, Z. Fan, H. Zhang and H. J. Fan, Nano Lett., 2014, 14, 4852–4858 CrossRef PubMed.
- J. Wang, N. Du, H. Zhang, J. Yu and D. Yang, J. Phys. Chem. C, 2011, 115, 11302–11305 Search PubMed.
- G. Sun, F. Qi, Y. Li, N. Wu, J. Cao, S. Zhang, X. Wang, G. Yi, H. Bala and Z. Zhang, Mater. Lett., 2014, 118, 69–71 CrossRef.
- C. Miao, M. Liu, Y. He, X. Qin, L. Tang, B. Huang, R. Li, B. Li and F. Kang, Energy Storage Mater., 2016, 3, 98–105 CrossRef.
- Z. Wang, D. Luan, F. Y. C. Boey and X. W. Lou, J. Am. Chem. Soc., 2011, 133, 4738–4741 CrossRef PubMed.
- X. W. Lou, Y. Wang, C. Yuan, J. Y. Lee and L. A. Archer, Adv. Mater., 2006, 18, 2325–2329 CrossRef.
- X. Hu, G. Zeng, J. Chen, C. Lu and Z. Wen, J. Mater. Chem. A, 2017, 5, 4535–4542 Search PubMed.
- R. Liu, D. Li, C. Wang, N. Li, Q. Li, X. Lü, J. S. Spendelow and G. Wu, Nano Energy, 2014, 6, 73–81 CrossRef.
- Y. Dong, M. Yu, Z. Wang, Y. Liu, X. Wang, Z. Zhao and J. Qiu, Adv. Funct. Mater., 2016, 26, 7590–7598 CrossRef.
- Y. Huang, Q. Pan, H. Wang, C. Ji, X. Wu, Z. He and Q. Li, J. Mater. Chem. A, 2016, 4, 7185–7189 Search PubMed.
- X. Wang, J. Feng, Y. Bai, Q. Zhang and Y. Yin, Chem. Rev., 2016, 116, 10983–11060 CrossRef PubMed.
- M. Xu, F. Wang, Y. Zhang, S. Yang, M. Zhao and X. Song, Nanoscale, 2013, 5, 8067 RSC.
- X. Feng, J. Yang, Y. Bie, J. Wang, Y. Nuli and W. Lu, Nanoscale, 2014, 6, 12532–12539 RSC.
- J. Gu, Z. Du, C. Zhang and S. Yang, Adv. Energy Mater., 2016, 6, 1600917–1600923 CrossRef.
- X. Cui, S. Yang, X. Yan, J. Leng, S. Shuang, P. M. Ajayan and Z. Zhang, Adv. Funct. Mater., 2016, 26, 5708–5717 CrossRef.
- S. Shi, T. Deng, M. Zhang and G. Yang, Electrochim. Acta, 2017, 246, 1104–1111 CrossRef.
- S. Li, W. Xie, S. Wang, X. Jiang, S. Peng and D. He, J. Mater. Chem. A, 2014, 2, 17139–17145 Search PubMed.
- X. Li, X. Zhang, R. Wang, Z. Su, J. Sha and P. Liu, J. Power Sources, 2016, 336, 298–306 CrossRef.
- J. Zhu, G. Zhang, X. Yu, Q. Li, B. Lu and Z. Xu, Nano Energy, 2014, 3, 80–87 CrossRef.
- H. He, W. Fua, H. Wang, H. Wang, C. Jin, H. J. Fan and Z. Liu, Nano Energy, 2017, 34, 449–455 CrossRef.
- T. Zhai, S. Xie, M. Yu, P. Fang, C. Liang, X. Lu and Y. Tong, Nano Energy, 2014, 8, 255–263 CrossRef.
- L. Tao, C. Lin, S. Dou, S. Feng, D. Chen, D. Liu, J. Huo, Z. Xia and S. Wang, Nano Energy, 2017, 41, 417–425 CrossRef.
- N. Wu, W. Du, G. Liu, Z. Zhou, H. Fu, Q. Tang, X. Liu and Y. He, ACS Appl. Mater. Interfaces, 2017, 9, 43681–43687 Search PubMed.
- R. Hu, Y. Ouyang, T. Liang, X. Tang, B. Yuan, J. Liu, L. Zhang, L. Yang and M. Zhu, Energy Environ. Sci., 2017, 10, 2017–2029 Search PubMed.
- Y. Zhang, P. Chen, X. Gao, B. Wang, H. Liu, H. Wu, H. Liu and S. Dou, Adv. Funct. Mater., 2016, 26, 7754–7765 CrossRef.
- N. Wu, H. Wu, W. Yuan, S. Liu, J. Liao and Y. Zhang, J. Mater. Chem. A, 2015, 3, 13648–13652 Search PubMed.
- N. Wu, Y. Zhang, Y. Wei, H. Liu and H. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 25361–25368 Search PubMed.
- X. Huang, X. Zhou, L. Zhou, K. Qian, Y. Wang, Z. Liu and C. Yu, ChemPhysChem, 2011, 12, 278–281 CrossRef PubMed.
- J. Yu, Y. Zhong, W. Zhou and Z. Shao, J. Power Sources, 2017, 338, 26–33 CrossRef.
- L. Chen, S. Ma, S. Lu, Y. Feng, J. Zhang, S. Xin and S. Yu, Nano Res., 2017, 10, 1–11 CrossRef.
- C. Liu, C. Zhang, H. Fu, X. Nan and G. Cao, Adv. Energy Mater., 2017, 7, 1601127 CrossRef.
- Z. Zhang, L. Wang, Y. Li, Y. Wang, J. Zhang, G. Guan, Z. Pan, G. Zheng and H. Peng, Adv. Energy Mater., 2017, 7, 1601814 CrossRef.
- W. J. Lee, J. Lim and S. O. Kim, Small Methods, 2017, 1, 1600014 CrossRef.
- X. Zhou, L. J. Wan and Y. G. Guo, Adv. Mater., 2013, 25, 2152–2157 CrossRef PubMed.
- Y. Sun, X. Hu, W. Luo, F. Xia and Y. Huang, Adv. Funct. Mater., 2013, 23, 2436–2444 CrossRef.
- S. Kang, X. Chen and J. Niu, Nano Lett., 2018, 18, 467–474 CrossRef PubMed.
- W. Wen, J. M. Wu, Y. Z. Jiang, L. L. Lai and J. Song, Chem, 2017, 2, 404–416 Search PubMed.
- J. S. Chen and X. W. D. Lou, Mater. Today, 2012, 15, 246–254 CrossRef.
- M. Gao, X. Chen, H. Pan, L. Xiang, F. Wu and Y. Liu, Electrochim. Acta, 2010, 55, 9067–9074 CrossRef.
- R. Demircakan, Y. Hu, M. Antonietti, J. Maier and M. Titirici, Chem. Mater., 2008, 20, 1227–1229 CrossRef.
- N. Wu, H. Wu, J. Kim, X. Liu and Y. Zhang, ChemElectroChem, 2018, 5, 78–83 CrossRef.
- L. Huang, J. Cheng, X. Li, D. Yuan, W. Ni, G. Qu, Q. Guan, Y. Zhang and B. Wang, J. Mater. Chem. A, 2015, 3, 4049–4057 Search PubMed.
- H. Wang, Q. Pan, Q. Wu, X. Zhang, Y. Huang, A. Lushington, Q. Li and X. Sun, J. Mater. Chem. A, 2017, 9, 4576–4582 Search PubMed.
- N. Wu, Y. Zhang, Y. Guo, S. Liu, H. Liu and H. Wu, ACS Appl. Mater. Interfaces, 2016, 8, 2723–2731 Search PubMed.
- L. Tang, Y. He, C. Wang, S. Wang, M. Wagemaker, B. Li, Q. Yang and F. Kang, Adv. Sci., 2017, 4, 1600311 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Fig. S1–S9 and Tables S1 and S2. See DOI: 10.1039/c8nr02290a |
| This journal is © The Royal Society of Chemistry 2018 |