
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Characterization techniques for nanoparticles: comparison and complementarity upon studying nanoparticle properties
Stefanos
Mourdikoudis
 ab,
Roger M.
Pallares
ab and
Nguyen T. K.
Thanh
*ab
ab,
Roger M.
Pallares
ab and
Nguyen T. K.
Thanh
*ab
aBiophysics Group, Department of Physics and Astronomy, University College London, London, WC1E 6BT, UK. E-mail: ntk.thanh@ucl.ac.uk
bUCL Healthcare Biomagnetic and Nanomaterials Laboratories, 21 Albemarle Street, London W1S 4BS, UK
First published on 4th June 2018
Abstract
Nanostructures have attracted huge interest as a rapidly growing class of materials for many applications. Several techniques have been used to characterize the size, crystal structure, elemental composition and a variety of other physical properties of nanoparticles. In several cases, there are physical properties that can be evaluated by more than one technique. Different strengths and limitations of each technique complicate the choice of the most suitable method, while often a combinatorial characterization approach is needed. In addition, given that the significance of nanoparticles in basic research and applications is constantly increasing, it is necessary that researchers from separate fields overcome the challenges in the reproducible and reliable characterization of nanomaterials, after their synthesis and further process (e.g. annealing) stages. The principal objective of this review is to summarize the present knowledge on the use, advances, advantages and weaknesses of a large number of experimental techniques that are available for the characterization of nanoparticles. Different characterization techniques are classified according to the concept/group of the technique used, the information they can provide, or the materials that they are destined for. We describe the main characteristics of the techniques and their operation principles and we give various examples of their use, presenting them in a comparative mode, when possible, in relation to the property studied in each case.
 Stefanos Mourdikoudis | Dr Stefanos Mourdikoudis is a chemical engineer who obtained his PhD degree from the Department of Physics, Aristotle University of Thessaloniki in Greece in 2009. Apart from his native country, he also worked in post-doctoral projects in France and Spain. Currently he is working at the University College London (UCL) as a research associate. His current research activity involves mostly work on the synthesis and characterization of magnetic nanoparticles. The variety of nanostructures he prepares are either directed for specific applications or simply inspired from curiosity to explore new protocols and characterize the resulting products. |
 Roger M. Pallares | Roger M. Pallares received his BSc and MSc degrees in chemistry from the Ramon Llull University (Barcelona, Spain) in 2009 and 2011, respectively. After a year working on the growth of 2D nanomaterials at NTT Basic Research Laboratories (Atsugi, Japan), he started a joint doctoral program between the University College London (UCL, London, UK) and the Agency for Science, Technology and Research (Singapore), obtaining a PhD in materials science from UCL in 2016. He is currently a postdoctoral fellow at Northwestern University (Evanston, IL). His research interests focus on the use of nanomaterials for biomedical applications. |
 Nguyen T. K. Thanh | Professor Nguyễn Thi Kim Thanh, FRSC, MInstP (http://www.ntk-thanh.co.uk), held a prestigious Royal Society University Research Fellowship (2005–2014). She was appointed a full professor in nanomaterials in 2013 at the Biophysics Group, Department of Physics and Astronomy, University College London, UK. She leads a very dynamic group conducting cutting edge interdisciplinary and innovative research on the design and synthesis of nanomaterials for biomedical applications from diagnostics to treatment of diseases such as cancer. She has published nearly 100 peer reviewed journal articles and book chapters with over 5000 citations so far. She has been a visiting professor at various universities in France, Japan, China and Singapore. She has been an invited speaker at over 200 institutes and scientific meetings. |
1. Introduction
Nanoscale materials often present properties different from their bulk counterparts, as their high surface-to-volume ratio results in an exponential increase of the reactivity at the molecular level. Such properties include electronic, optical and chemical properties, while the mechanical characteristics of the nanoparticles (NPs) may also differ extensively.1 This enables them to be an object of intensive studies due to their academic interest and the prospective technological applications in various fields. Such nanostructures may be synthesized by a wide number of methods, which involve mechanical, chemical and other pathways.2 Nowadays, many more types of nanomaterials are synthesized than only a decade ago, and in higher amounts than before, requiring the development of more precise and credible protocols for their characterization. However, such characterization is sometimes incomplete. This is because of the inherent difficulties of nanoscale materials to be properly analysed, compared to the bulk materials (e.g. too small size and low quantity in some cases following laboratory-scale production). In addition, the multidisciplinary aspects of nanoscience and nanotechnology do not permit every research team to have easy access to a broad range of characterization facilities. In fact, quite often a wider characterization of NPs is necessary, requiring a comprehensive approach, by combining techniques in a complementary way. In this context, it is desirable to know the limitations and strengths of the different techniques, in order to know if in some cases the use of only one or two of them is enough to provide reliable information when studying a specific parameter (e.g. particle size). Nanoscience and nanotechnology are still undergoing constant growth, and the scientific community is rather aware that there may be certain differences between the way analytical characterization methods operate for nanomaterials, in comparison with their more ‘traditional’ modes of use for more ‘conventional’ (macroscopic) materials.3Herein we describe extensively the use of different methods for the characterization of NPs. These techniques are sometimes exclusive for the study of a particular property, while in other cases they are combined.4 We discuss all these techniques in a comparative way, considering factors such as their availability, cost, selectivity, precision, non-destructive nature, simplicity and affinity to certain compositions or materials. The techniques are analysed in depth, despite their big number presented herein. There are microscopy-based techniques (e.g. TEM, HRTEM, and AFM – the full names of the techniques are provided later in the text, when presenting each one of them), which provide information on the size, morphology and crystal structure of the nanomaterials. Other techniques are specialized for certain groups of materials, such as the magnetic techniques. Examples of these techniques are SQUID, VSM, FMR, and XMCD. Many other techniques provide further information on the structure, elemental composition, optical properties and other common and more specific physical properties of the nanoparticle samples. Examples of these techniques include X-ray, spectroscopy and scattering techniques.
This review is organized in different sections, which will present numerous distinct characterization techniques for NPs in relation to the properties studied (see Tables 1 and 2). The sections are categorized according to the different technique groups, as described above.
| Technique | Main information derived | Section | Fig. |
|---|---|---|---|
| XRD (group: X-ray based techniques) | Crystal structure, composition, crystalline grain size | 2.1 | |
| XAS (EXAFS, XANES) | X-ray absorption coefficient (element-specific) – chemical state of species, interatomic distances, Debye–Waller factors, also for non-crystalline NPs | 2.1 | |
| SAXS | Particle size, size distribution, growth kinetics | 2.1 | 1 |
| XPS | Electronic structure, elemental composition, oxidation states, ligand binding (surface-sensitive) | 2.1 | |
| FTIR (group: further techniques for structure/composition/main properties) | Surface composition, ligand binding | 2.2 | |
| NMR (all types) | Ligand density and arrangement, electronic core structure, atomic composition, influence of ligands on NP shape, NP size | 2.2 | |
| BET | Surface area | 2.2 | |
| TGA | Mass and composition of stabilizers | 2.2 | |
| LEIS | Thickness and chemical composition of self-assembled monolayers of NPs | 2.2 | |
| UV-Vis | Optical properties, size, concentration, agglomeration state, hints on NP shape | 2.2 | |
| PL spectroscopy | Optical properties – relation to structure features such as defects, size, composition | 2.2 | |
| DLS | Hydrodynamic size, detection of agglomerates | 2.2 | 2 |
| NTA | NP size and size distribution | 2.2 | 3 |
| DCS | NP size and size distribution | 2.2 | |
| ICP-MS | Elemental composition, size, size distribution, NP concentration | 2.2 | 4 |
| SIMS, ToF-SIMS, MALDI | Chemical information (surface-sensitive) on functional group, molecular orientation and conformation, surface topography, MALDI for NP size | 2.2 | 5 |
| RMM-MEMS, ζ-potential, pH, EPM, GPC, DSC, etc. | Please check the relevant parts of the manuscript | 2.2 | |
| SQUID-nanoSQUID (group: magnetic nanomaterials) | Magnetization saturation, magnetization remanence, blocking temperature | 2.3 | 6 |
| VSM | Similar to SQUID through M–H plots and ZFC-FC curves | 2.3 | |
| Mössbauer | Oxidation state, symmetry, surface spins, magnetic ordering of Fe atoms, magnetic anisotropy energy, thermal unblocking, distinguish between iron oxides | 2.3 | 7 |
| FMR | NP size, size distribution, shape, crystallographic imperfection, surface composition, M values, magnetic anisotropic constant, demagnetization field | 2.3 | |
| XMCD | Site symmetry and magnetic moments of transition metal ions in ferro- and ferri-magnetic materials, element specific | 2.3 | |
| Magnetic susceptibility, magnetophoretic mobility | Please check the relevant parts of the manuscript | 2.3 | |
| Superparamagnetic relaxometry | Core properties, hydrodynamic size distribution, detect and localize superparamagnetic NPs | 2.3 | 8 |
| TEM (group: microscopy techniques) | NP size, size monodispersity, shape, aggregation state, detect and localize/quantify NPs in matrices, study growth kinetics | 2.4 | 9 and 10 |
| HRTEM | All information by conventional TEM but also on the crystal structure of single particles. Distinguish monocrystalline, polycrystalline and amorphous NPs. Study defects | 2.4 | 11 and 12 |
| Liquid TEM | Depict NP growth in real time, study growth mechanism, single particle motion, superlattice formation | 2.4 | 13 |
| Cryo-TEM | Study complex growth mechanisms, aggregation pathways, good for molecular biology and colloid chemistry to avoid the presence of artefacts or destroyed samples | 2.4 | 14 |
| Electron diffraction | Crystal structure, lattice parameters, study order–disorder transformation, long-range order parameters | 2.4 | |
| STEM | Combined with HAADF, EDX for morphology study, crystal structure, elemental composition. Study the atomic structure of hetero-interfaces | 2.4 | |
| Aberration-corrected (STEM, TEM) | Atomic structure of NP clusters, especially bimetallic ones, as a function of composition, alloy homogeneity, phase segregation | 2.4 | 15 and 16 |
| EELS (EELS-STEM) | Type and quantity of atoms present, chemical state of atoms, collective interactions of atoms with neighbors, bulk plasmon resonance | 2.4 | 17 |
| Electron tomography | Realistic 3D particle visualization, snapshots, video, quantitative information down to the atomic scale | 2.4 | 18 and 19 |
| SEM-HRSEM, T-SEM-EDX | Morphology, dispersion of NPs in cells and other matrices/supports, precision in lateral dimensions of NPs, quick examination–elemental composition | 2.4 | 20 |
| EBSD | Structure, crystal orientation and phase of materials in SEM. Examine microstructures, reveal texture, defects, grain morphology, deformation | 2.4 | |
| AFM | NP size and shape in 3D mode, evaluate degree of covering of a surface with NP morphology, dispersion of NPs in cells and other matrices/supports, precision in lateral dimensions of NPs, quick examination–elemental composition | 2.4 | 21, 22 and 23 |
| MFM | Standard AFM imaging together with the information of magnetic moments of single NPs. Study magnetic NPs in the interior of cells. Discriminate from non-magnetic NPs | 2.4 | 21 |
| Entity characterized | Characterization techniques suitable |
|---|---|
| Size (structural properties) | TEM, XRD, DLS, NTA, SAXS, HRTEM, SEM, AFM, EXAFS, FMR, DCS, ICP-MS, UV-Vis, MALDI, NMR, TRPS, EPLS, magnetic susceptibility |
| Shape | TEM, HRTEM, AFM, EPLS, FMR, 3D-tomography |
| Elemental-chemical composition | XRD, XPS, ICP-MS, ICP-OES, SEM-EDX, NMR, MFM, LEIS |
| Crystal structure | XRD, EXAFS, HRTEM, electron diffraction, STEM |
| Size distribution | DCS, DLS, SAXS, NTA, ICP-MS, FMR, superparamagnetic relaxometry, DTA, TRPS, SEM |
| Chemical state–oxidation state | XAS, EELS, XPS, Mössbauer |
| Growth kinetics | SAXS, NMR, TEM, cryo-TEM, liquid-TEM |
| Ligand binding/composition/density/arrangement/mass, surface composition | XPS, FTIR, NMR, SIMS, FMR, TGA, SANS |
| Surface area, specific surface area | BET, liquid NMR |
| Surface charge | Zeta potential, EPM |
| Concentration | ICP-MS, UV-Vis, RMM-MEMS, PTA, DCS, TRPS |
| Agglomeration state | Zeta potential, DLS, DCS, UV-Vis, SEM, Cryo-TEM, TEM |
| Density | DCS, RMM-MEMS |
| Single particle properties | Sp-ICP-MS, MFM, HRTEM, liquid TEM |
| 3D visualization | 3D-tomography, AFM, SEM |
| Dispersion of NP in matrices/supports | SEM, AFM, TEM |
| Structural defects | HRTEM, EBSD |
| Detection of NPs | TEM, SEM, STEM, EBSD, magnetic susceptibility |
| Optical properties | UV-Vis-NIR, PL, EELS-STEM |
| Magnetic properties | SQUID, VSM, Mössbauer, MFM, FMR, XMCD, magnetic susceptibility |
2. Characterization of nanoparticles
Two of the main parameters studied in the characterization of NPs are size and shape. We can also measure size distribution, degree of aggregation, surface charge and surface area, and to some extent evaluate the surface chemistry.5 Size, size distribution and organic ligands present on the surface of the particles may affect other properties and possible applications of the NPs. In addition, the crystal structure of the NPs and their chemical composition are thoroughly investigated as a first step after nanoparticle synthesis. Until now, there were no standardized protocols for this aim. Credible and robust measurement methods for NPs will greatly affect the uptake of these materials in commercial applications and allow the industry to comply with regulation. Nevertheless, there are important challenges in the analysis of nanomaterials because of the interdisciplinary nature of the field, the absence of suitable reference materials for the calibration of analytical tools, the difficulties linked to the sample preparation for analysis and the interpretation of the data. In addition, there are unmet challenges in the characterization of NPs such as the measurement of their concentration in situ and on-line, especially in a scaled-up production, as well as their analysis in complex matrices. Waste and effluent from mass production will also need to be monitored.6 With the scale-up of nanoparticle manufacture, more reliable quantification techniques will be required. For this reason, it is crucial to characterize the nanomaterials prepared in several ways to the maximum extent. We do not only focus on the characterization of the nanoparticle core, but also on the surface ligands that influence the physical properties. In addition, we do not present only techniques that one might classify as ‘common’, but we also show examples of modern in situ operando techniques that are used to monitor the kinetics of nanoparticle formation and study through some recent advances in the topic the controlled defects that affect nanoparticle properties in a crucial manner.2.1 X-ray-based techniques
X-ray diffraction (XRD) is one of the most extensively used techniques for the characterization of NPs. Typically, XRD provides information regarding the crystalline structure, nature of the phase, lattice parameters and crystalline grain size. The latter parameter is estimated by using the Scherrer equation using the broadening of the most intense peak of an XRD measurement for a specific sample. An advantage of the XRD techniques, commonly performed in samples of powder form, usually after drying their corresponding colloidal solutions, is that it results in statistically representative, volume-averaged values. The composition of the particles can be determined by comparing the position and intensity of the peaks with the reference patterns available from the International Centre for Diffraction Data (ICDD, previously known as Joint Committee on Powder Diffraction Standards, JCPDS) database. However, it is not suitable for amorphous materials and the XRD peaks are too broad for particles with a size below 3 nm.Upadhyay et al. determined the average crystallite size of magnetite NPs using X-ray line broadening, and it was found to be in the range of 9–53 nm. The broadening of XRD peaks was mainly caused by particle/crystallite size and lattice strains other than instrumental broadening.7 The XRD-derived size is usually bigger than the so-called magnetic size, due to the fact that smaller domains are present in a particle where all moments are aligned in the same direction, even if the particle is single domain. On the contrary, the TEM-deduced size was higher than that calculated using XRD, for samples with very large particles; in fact, when the particle size is bigger than 50 nm, there are more than one crystal boundary on their surface. XRD cannot distinguish between the two boundaries; therefore the actual (TEM) size of certain samples can be in reality bigger than the 50–55 nm calculated by the Scherrer formula. Dai and co-workers prepared ultra-small Au NPs which were very likely to be more developed along the 〈111〉 direction (rather than the 〈220〉 one) as the peak corresponding to the former direction was much more intense in their XRD measurement.8 Similarly, Li and colleagues noticed that after preparing copper telluride nanostructures with different shapes (i.e. cubes, plates, and rods), the relative intensities between the different XRD peaks varied in relation to the particle shape.9
X-ray absorption spectroscopy (XAS) includes both extended X-ray absorption fine structure (EXAFS) and X-ray absorption near edge structure (XANES, also known as NEXAFS). XAS measures the X-ray absorption coefficient of a material as a function of energy. Each element has a set of characteristic absorption edges corresponding to the different binding energies of its electrons, giving XAS element selectivity. As a highly sensitive technique, EXAFS is a convenient way to identify the chemical state of species which may occur even in very low concentrations. Synchrotrons are usually needed to acquire XAS spectra; therefore it is not a routine or readily available technique. XANES probes the density of states of empty/partially filled electronic states by considering the excitation of an inner shell electron to those states that are permitted by dipole selection rules. Pugsley et al. used in situ XAS to examine the kinetics and mechanism of formation of germanium NPs upon the reaction of Mg2Ge and GeCl4.10 Actually, the EXAFS experiments and TEM results indicated the formation of GeO2 NPs along with the Ge NPs. The analysis of EXAFS yielded a first-neighbour Ge–Ge distance of 2.45 Å in good agreement with XRD. Moreover, Chen et al. applied in situ EXAFS for the inspection of structural changes around germanium atoms in GeO2 NPs. Surprisingly, they noticed that at high temperature GeS2 was formed as a product of the complete transformation of germanium dioxide, in the presence of a sulfur source.11 Requejo and co-workers investigated the effects of sulfur–palladium interaction on the structural and electronic properties of alkyl thiol-capped Pd NPs. The XANES and EXAFS analyses of the atomic structure and electronic properties of these NPs showed that the sulfidation of Pd clusters caused by the capping thiol molecules took place not only on the surface but also in the bulk.12
Energy dispersive EXAFS helps to determine both structural and kinetic parameters in supported metal catalysts for reactions occurring on a timescale of a few seconds. Such a fast operation enables the aforementioned technique to be used at temperatures higher than 200 °C, which would hinder the use of surface enhanced Raman spectroscopy (SERS), as the latter technique is not that fast under such conditions. Even on a timescale of tens of milliseconds, energy dispersive EXAFS can be used as a quantitatively suitable in situ probe of the dynamics of quick phase change in supported nanoparticulate metal catalysts.13
Bugaev and colleagues determined with EXAFS parameters the atomic structure of PtCu NPs in PtCu/C catalysts. EXAFS is one of the most convenient techniques for the structural analysis of NPs with sizes lower than 10 nm. It possesses a high spatial resolution and provides information on the nearest environment of an atom in a compound in the absence of long-range order. The parameters derived in that study were partial coordination numbers, interatomic distances and Debye–Waller factors.14 Moreover, Klasovsky and co-workers performed a physicochemical characterization of a new electron-conducting polymer (PANI) supported PtO2 catalyst by electron paramagnetic resonance (EPR), diffuse reflectance FTIR spectroscopy (DRIFTS) and EXAFS. The importance of in situ/operando techniques was highlighted toward a better comprehension of the working oxidation catalyst.15
In another study, Zhang and colleagues coated γ-Fe2O3 NPs with sodium dodecylbenzene sulphonate (DBS), stearic acid and hexadecyltrimethylammonium bromide (CTAB) surfactants by the microemulsion method. The role of the surfactants was investigated through EXAFS analysis and it was found that all samples had a tendency to extend the Fe–O bond length. All these molecules possess large spatial resistance, with the CTAB molecule having the largest one. The lattice distortion and disorder at the interfaces could play a significant role in hindering the fast nanoparticle growth.16 CuFe2O4 and CuFe2O4–MO2 (M = Sn, Ge) NPs were investigated by Bertagnolli and colleagues by means of EXAFS and XANES. The authors state the importance of EXAFS for the acquisition of information concerning the coordination number, the nature of the scattering atoms surrounding the absorbing atom, the interatomic distance between absorbing and backscattering atoms, as well as the Debye–Waller factor, which is related to a disorder because of static displacements and thermal vibrations.17 The Fourier transform (FT) of the EXAFS signal as a function of wavenumber is related to the radial distribution of backscattering atoms in real space x(r). The possible phase shifts during the EXAFS process and interference effects from different scattering channels cause the modification of the position of the peaks in the FT, which become no longer identical to the geometric distance between the backscattering atoms and the absorbing atom. As an alternative method aiming to tackle the drawbacks of the FT approach, the wavelet transform (WT) has been proposed, as reported by C. Schmitz Antoniak.18 The principal concept behind the WT is to replace the infinitely expanded periodic oscillations in a FT with located wavelets as a kernel for the integral transformation. More details of that approach can be found in ref. 18. EXAFS can also be used to study copper cation inversion in CuFe2O4 as a function of saturation magnetization. XANES is more helpful to determine the oxidation states, vacant orbitals, electronic configuration and site symmetry of the absorbing atom. XANES measurements were in agreement with EXAFS, both suggesting that iron (Fe) ions occupied more tetrahedral sites than octahedral sites. Overall, these researchers showed that the aforementioned investigation on their copper ferrite NPs illustrated that these nanostructures had a structure analogous to that of the corresponding bulk material. The incorporation of the tetravalent metal ions in the spinel structures did not modify the local environment around Cu and Fe ions.17
Moroz reviewed the X-ray diffraction structure diagnostics of nanomaterials and stated that a remarkable advantage of EXAFS over REDD (radial electron density distribution) is its selectivity, whereas REDD is better in providing accurate values of the interatomic distances; in that case, EXAFS provides interatomic distances corrected for the phase shift. Ideally, REDD should be combined with EXAFS, FTIR and microscopy techniques to acquire knowledge on the relation between the structure and physicochemical properties of nanomaterials.19 In another work, Gomes et al. combined XRD and EXAFS to determine the cation distribution and other structural parameters, comparing the NP-based sample spectrum with the standard bulk material spectrum of the Cu ferrite. Differences were found among the cation redistribution at the nanoparticle samples with regard to the ideal copper ferrite.20 CeO2 NPs were characterized with EXAFS by Zhu and co-workers. The authors emphasized on the suitability of the technique under discussion for their materials due to its element selectivity and nondependence on the long-range order of materials. From the acquired Debye–Waller factors and the Ce–O bond lengths, it was deduced that the surface or interface of the NPs coated with sodium bis(2-ethylhexyl) sulfosuccinate (AOT) surfactants was quite ordered; however the bond lengths were elongated.21 Swatsitang and colleagues analysed with EXAFS the impact of cation distribution on the magnetic properties of Co1−xNixFe2O4 NPs prepared by a hydrothermal method. The results implied that Co and Ni ions could occupy both the tetrahedral and octahedral sites with the preference to occupy the octahedral site more than the tetrahedral site, which is different from the bulk sample where all cations occupy only the octahedral site in an inverse spinel ferrite model structure.22 Furthermore, Zhang et al. synthesized Co@SiO2 core–shell NPs with the sol–gel approach. In situ XRD was used along with EXAFS to monitor the oxidation process of the Co cores after thermal treatment at 800 °C either in air or under an inert atmosphere. Interestingly, it was noticed that Co was oxidized in three steps no matter if air or N2 gas was employed during the annealing.23 Ni–P was another material in nanoscale form studied by EXAFS. In particular, EXAFS proved to be very robust for the screening of the initial crystallization behaviour of such amorphous NPs by probing the atomic-level structural change. Its combination with XRD, HRTEM and VSM helped to investigate in detail the structural changes of Ni–P NPs in both short-range and long-range order during heating at high temperatures. More specifically, XRD illustrated the crystalline phases and phase changes. HRTEM provided information on size, size distribution and shape. EXAFS provided insights regarding the changes of a local atomic structure and the chemical valence, especially for XRD-amorphous samples. VSM enabled the study of magnetic properties corresponding to different crystallization stages.24
Metal chalcogenides have also been analysed by EXAFS, as in the case of CdS NPs prepared by Rockenberger et al.25 They found EXAFS to be suitable for their samples since it does not rely on any long-range order, in contrast to XRD. EXAFS can also be used for liquid samples or even in cluster beams in the gaseous phase, permitting the identification of intercluster interactions by comparison with solid state measurements. It revealed that the stabilization of CdS NPs with 1.3–12 nm diameter affected the mean Cd–S distance. Unlike XRD, EXAFS is only sensitive to the local geometrical arrangement of neighboring atoms that surround the absorbing atom. O'Brien and colleagues investigated the local environment of phosphorus in the capping agent on the surface of CdSe quantum dots. The binding mode of the capping agents onto the surface was analysed, depending on the use of two distinct synthetic routes followed for its preparation (ligands: trioctylphosphine oxide and/or trioctylphosphine selenide).26 Furthermore, Lloyd and co-workers used various techniques, including EXAFS, to monitor the reduction of Se(IV) to Se(II) by a microbial whole-cell catalyst (Veillonella atypica). The reduction was found to proceed via an insoluble red amorphous Se(0) phase and the formation of metal selenide was shown by EXAFS analysis from both ex situ and in situ ways.27
Noble metal nanostructures, either monometallic or bimetallic, have also been studied by EXAFS. For example, the structural features of silver NPs embedded in silicate glass were investigated combining HRTEM and EXAFS techniques.28 Bugaev's group employed HRTEM, XRD, optical absorption and EXAFS to identify correlations between the plasmonic properties and the atomic structure of Ag NPs and their aggregates. The processing of the Ag K-edge EXAFS spectra resulted in the acquisition of values for the parameters of the atomic structure in Ag–Ag and Ag–O bonds averaged over the ionic and neutral states of Ag.29 The determination of the atomic structure of metallic Ag NPs, such as the type of point symmetry in the interior (core) region of small NPs, the nearest-neighbor Ag–Ag distances and the structure of the near-surface region, is a challenging problem for this system. The same group analysed by EXAFS the changes in the atomic structure of Ag NPs in soda-lime glass after annealing at 550 °C for 8 h.30 Brunsch and co-workers found that EXAFS showed a higher accuracy than HRTEM in the determination of lattice parameters for Ag NPs embedded in silicate glasses. The EXAFS results were averaged parameters of atom–atom correlations summed up for all detected particles, different from the data derived by HRTEM for single particles. Combining both techniques would be beneficial for precise structural characterization.31 The same researchers achieved the determination of the thermal expansion coefficient of similar glass-embedded silver NPs, in a wide range of temperatures. EXAFS allowed the precise demonstration of the changes of bond lengths and the stress state of their materials on the basis of a thermal expansion mismatch. The evaluation of the EXAFS data of small NPs typically provides a decreased coordination number, a dilatation or a contraction of the lattice structure and an increased Debye–Waller factor, together with an increased static disorder.32 In addition, EXAFS does not require operation under vacuum, in contrast to XPS. It was used by the Parkin group to investigate the phase change in silver speciation, during the photo-assisted growth of Ag from AgNO3.33 The most plausible transition from metallic silver to Ag2O was illustrated.
EXAFS has also been applied for the characterization of alloys such as PdxPty NPs since common analytical techniques, such as XRD, have a limited capacity to distinguish between the various compositions of the aforementioned alloy, i.e. these metals are perfectly miscible in any relative proportion and they both possess an fcc structure with similar lattice constants. Furthermore, structural information can also be obtained from EXAFS measurements, even though sometimes with lower precision and difficulty in extraction in comparison with the use of XRD.34 Ingham has written a comprehensive review describing what X-ray scattering techniques such as EXAFS, in situ XRD and small-angle X-ray scattering (SAXS) can offer in nanoparticle characterization.35 Beale and Weckhuysen have reported how the ratio between coordination numbers varied as a function of shape, through the EXAFS data. Their study concerned a series of nanoscale structures with several shapes and fcc, hcp, or bcc structures, with a maximum isotropic diameter of 3 nm.36 In the case of Pt–Ru nanoclusters, size could also be obtained from EXAFS analysis, due to the fact that the coordination number of nearest neighbors in NPs is a non-linear function of the particle diameter if the latter parameter lies below the range of 3–5 nm.37 Sokolov and co-workers characterized their Pd NPs – synthesized by two separate routes – by X-ray reflectivity, EXAFS and electron microscopy. The EXAFS-deduced size was lower than the TEM one if a cuboctahedral fcc structural model of Pd NPs surrounded by thiol was assumed. Compared to bulk Pd, lattice expansion was noticed in all types of NPs, by both HRTEM and EXAFS.38 Regarding cobalt NPs, a report by Cheng et al. showed that EXAFS was able to differentiate ε-Co from the fcc and hcp crystal structures.39
The combination of XANES and EXAFS for the characterization of CuO, Cu2O/CuO and CuO/TiO2 NPs was published by Sharma et al.40 These techniques probed the local electronic/atomic structure of these samples and the existence of the different oxide phases was evidenced. Iron oxide NPs were also studied by XANES: this technique provided information on the oxidation state and local structure of iron atoms, while SAXS analysis was useful for the size determination of the particles. The combination of time-resolved in situ SAXS and XANES measurements enabled the study of the formation of maghemite NPs in water, on a structural and chemical level. On the basis of the acquired data, a complex four-stage formation mechanism was proposed, using ferrous and ferric chlorides as well as triethanolamine. The acquired knowledge would facilitate the control of the formation of NPs in solution and tailor the properties of the final product.41 In another publication, Leveneur et al. investigated the nucleation and growth of Fe NPs in SiO2 by TEM, XPS and XANES. It was demonstrated that ion implantation initially resulted in the formation of dilute cationic Fe2+ species, while at higher dissolved iron concentrations, the formation of small metallic nuclei was noticed, which seed the nanocluster growth during prolonged implantation or annealing. XANES is a technique far more sensitive to coordination and bonding environment than XPS since it probes the unoccupied electronic states of atoms and therefore can provide information about the crystal field (octahedral, square pyramidal or tetrahedral) that the iron cations occupy. The complementary use of both XPS and XANES was considered to be handy for such types of nanostructures with complex compositions and various possible states for the valence of iron.42
Similar to the XRD method presented above, the SAXS technique allows elastic scattering processes into a given solid angle to be run; however the detector in SAXS covers only small scattering angles (normally lower than 1°).34 A scheme that illustrates an in situ setup which manages to record real-time SAXS/WAXS/UV–Vis measurements during the formation of Au NPs is displayed in Fig. 1. The pictured device conducts SAXS and WAXS and records the UV–Vis spectra at the same time in a given sample volume.43 WAXS (wide-angle X-ray scattering) is similar to SAXS, but the distance between the sample and the detector is smaller and therefore diffraction maxima at larger angles are observed. The authors investigated the nucleation and growth kinetics of gold NPs as a function of parameters such as concentration, temperature, ligand ratio and solvent type. Stuhn and co-workers characterized extensively polystyrene-grafted SiO2 NPs, using techniques such as SAXS, SANS, DLS, TGA and TEM. Small angle neutron scattering (SANS) provided direct access to the static structure of the polymer layer. Both SAXS and SANS can be used to measure the particle size; in that report, SAXS gave a 25.6 nm value, while a 23.3 nm NP size was derived by SANS. Although SANS and SAXS are very similar in various aspects (e.g. SANS uses elastic neutron scattering), the advantages of SANS over SAXS include its sensitivity to light elements, the possibility of isotope labelling and the strong scattering by magnetic moments.44 The ligand shells on small ZnO NPs were characterized by a combined SANS/SAXS approach. Standard in situ methods such as UV-Vis and SAXS are sensitive only to the ZnO core; however SANS probes the organic stabilizer in dispersion thanks to the high sensitivity of neutrons to H2. In the work under discussion, both techniques allowed the determination of the size distribution of the cores of the NPs and the distribution of the stabilizer molecules (acetate shell) simultaneously in the native solution.45
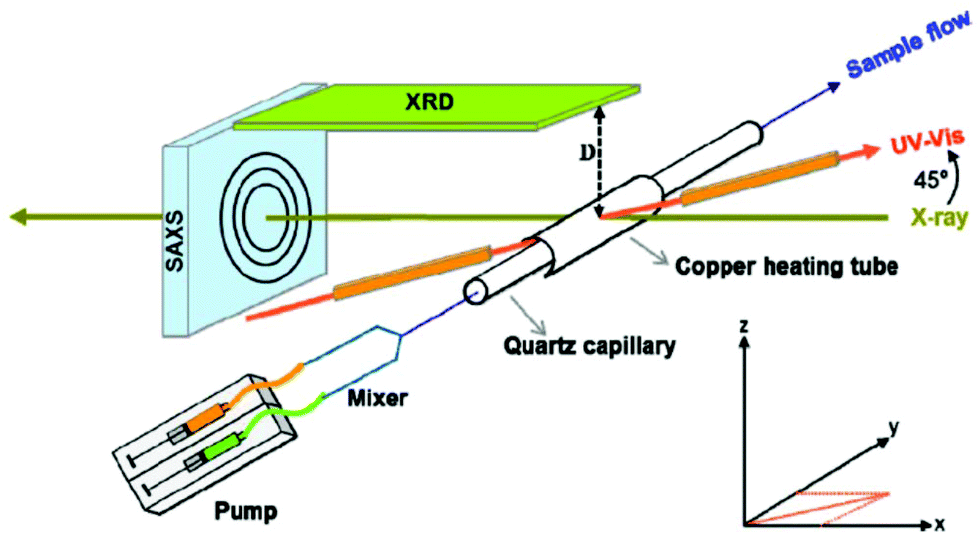 | ||
| Fig. 1 Schematic presentation of the in situ setup employed for real-time SAXS/WAXS/UV–Vis measurements during the formation of Au NPs. The setup measures SAXS, WAXS and the UV–Vis spectra simultaneously in the same sample volume. Reprinted with permission from ref. 43. Copyright 2015 American Chemical Society. | ||
Typically, SAXS is used to determine the particle size, size distribution, and shape. Regarding size values, SAXS results are more statistically average than TEM imaging. Wang et al. employed SAXS to investigate the structural change of Pt NPs with temperature.46 For certain temperatures, the size obtained by XRD was different from the corresponding SAXS value. This is because SAXS is sensitive to the size of the fluctuation region of electronic density, but XRD is sensitive to the size of the long-range order region. SAXS provides the actual particle size, while XRD yields the crystallite size. It is important to note that the different size values of SAXS and XRD are related to the growth mode of NPs during thermal treatment. The particle size acquired with SAXS was found to be a little bigger than that obtained from TEM. The reason is that Pt NPs were coated with PVP, and the scattering intensity due to the PVP coating cannot be easily removed.46 It has to be noted that SAXS is a low resolution technique and in certain cases further studies by XRD and/or electron diffraction techniques are indispensable for the characterization of NPs. In fact, Ti et al. have written a lengthy review article dedicated to the role of SAXS for nanoparticle research.47 In the case of PVA-stabilized Ag NPs, SAXS enables a more quantitative understanding of the correlations between the localised surface plasmon resonance (LSPR) behaviour and the aggregation phenomena. Structure–property correlations between the LSPR behaviour and the SAXS spectra are feasible as both are scattering phenomena, which happen in the sol state. Moreover, the size range of the NPs which show LSPR is similar to that measured by SAXS.48
Bulavin and colleagues reported a combined approach with SAXS, UV-Vis and QELS (quasi-elastic light scattering) to characterize silver sols in polymer matrices. SAXS analysis showed a monomodal scatterer size distribution, whereas QELS and UV-Vis yielded a multimodal particle size distribution. This discrepancy might come from the ability of the latter techniques to register large particles or aggregates within the range of 30–60 nm. These large particles and aggregates are not within the detection limits of SAXS. However, for relatively small particles, all the aforementioned methods were in good agreement for the evaluation of the particle size and polydispersity.49 In the case of Ag–Cu alloy NPs synthesized using Cu- and Ag-nitrates in water with hydrazine as the reductant and starch as the sol-stabilizing agent, SAXS demonstrated the formation of mass fractal aggregates. A bimodal size distribution was noticed, with the smaller aggregates having Ag-rich compositions, while larger aggregates with low mass fractal dimensions were Cu-rich. This bimodal composition mode was also evident in the LSPR spectra. It is important to note that in view of the length scale of the NP aggregates, which is related to LSPR changes, SAXS is the most suitable non-invasive technique for these studies. In typical invasive techniques, such as TEM and SEM, the substrate–particle interactions and the solvent drying kinetics may affect the nanostructures formed. These techniques cannot thus be helpful to analyse sol structures and explain adequately their LSPR behaviour.50 In another work, Hashimoto and co-workers focused on the kinetics of the reduction-reaction-induced self-assembling process of (Pd)n in the polystyrene-block-polyisoprene (PS-b-PI) matrix by time-resolved SAXS. Pd(acac)2 was used as precursor, and it was found that its reduction to Pd(0) obeyed the first-order kinetics in both the disordered and ordered PS-b-PI matrix.51
Takenaka and co-workers performed a ‘screening’ of the photoreduction synthesis of rhodium and palladium NPs in aqueous ethanol/PVP solution using in situ and time-resolved SAXS. The nucleation, growth and particle coalescence of metal atoms for the production of metal NPs were monitored successfully.52 In general, SAXS helps to quantify the mass or concentration of NPs as well as their size simultaneously, as a function of time. The evolution of size, size distribution, amount and total volume of these Rh and Pd NPs was quantitatively determined by SAXS. Moreover, the formation process of mesostructured PtRu NPs electrochemically reduced on a microemulsion lyotropic liquid-crystallic template was studied by in situ XRD, SAXS and XANES.53 These techniques, together with complementary measurements by SEM, FE-TEM and EDS, facilitated the understanding of the structural evolution, starting from the metallic precursors to the subsequent atom reduction, NP formation and aggregation, and finally mesostructure formation. Several insights were acquired, for instance, the degree of alloying between both metals was studied, and any composition distribution (e.g. Pt-rich core and Ru-rich shell) was attributed to the different reduction rates of the Pt and Ru precursors. In another work, LaGrow et al. used in situ synchrotron small-angle X-ray scattering to monitor the growth and interparticle interaction of Ni NPs in solution as a function of time, and for different trioctylphosphine/Ni precursor proportions in order to understand the influence of the TOP amount on the growth kinetics. It was found that the TOP/Ni ratio affected radically the final Ni NP size because of the action of TOP as a nucleating agent, together with the fact that TOP hindered the capacity of the nickel precursor to reach the NP surface.54
Metal oxide NPs have also been investigated extensively by SAXS. In particular, grazing-incident SAXS (GISAXS) and AFM were employed to study self-assembled iron oxide NPs prepared by a high-temperature solution phase reaction, as well as silicon dots produced by ion bombardment.55 GISAXS is a unique non-destructive technique, which detects the diffusely scattered X-ray intensity from nanoscale objects (through a large illumination area), providing information on properties such as NP size, shape and arrangement. AFM can fit well with GISAXS by delivering a localized morphological image of the surface of the material. Tobler and colleagues tried to shed light on the nucleation and growth steps of SiO2 NPs, through SAXS and DLS measurements. DLS is much more sensitive to the presence of aggregates in comparison with SAXS; thus it is more suitable to monitor the starting step of the aggregation process.56 The SAXS and DLS results confirmed that the rate of silica polymerization and nanoparticle formation was enhanced by increasing the ionic strength and silica concentration. SEM and TEM verified the results obtained by SAXS and DLS regarding the particle size and shape although under certain conditions (sample dehydration, exposure to high vacuum), the microscopy techniques yielded smaller NP size compared to SAXS and DLS. Titania NPs prepared by the reaction of TiCl4 with HCl were studied by a combined approach using SAXS, DLS and TEM. While DLS provides information only on the average hydrodynamic diameter of the particles and not on their internal features, SAXS and TEM were employed to unravel such details. The authors stated the importance of SAXS for the investigation of condensed and solid matter as well as for processes in colloidal systems.57 In another work, TiO2 NPs were studied by SAXS, DSC and WAXS. The capacity of SAXS to determine the structure of a nanocomposite polymer electrolyte was noted; in the work under discussion, (PEO)8ZnCl2 polymer electrolytes and nanocomposites were doped with 10% of TiO2 nanograins and γ-irradiated.58 The effect of the inserted nanoscale titania grains on the electrical, elastic and morphological properties of the nanocomposites, and the influence of γ-radiation from a Co-60 source were investigated with the aforementioned techniques. In contrast to WAXS, which showed lines and nanocrystallites only in low temperature crystalline phases, SAXS was able to demonstrate the existence of nanograins in both the low and high temperature phases. While WAXS evidences only nanocrystallites, SAXS records the presence of both crystalline and amorphous structures. All techniques together showed that this complex material showed a transition from a crystalline–amorphous phase to a highly conductive superionic one.
In another work, SiO2/TiO2 hollow NPs in the size range of 25–100 nm were studied by a combination of SAXS, GISAXS, SANS, TEM, DLS and other techniques. Experimental broadening of the scattering was negligible in the case of SAXS and GISAXS measurements, but it was not negligible in SANS.59 In general, the results for the NP size derived by all techniques were in reasonable agreement. SAXS/GISAXS provided accurate information on the inner diameter, outer diameter and size distribution. TEM sometimes overestimated the shell thickness, unless HRTEM was used. DLS was considered to be able to provide fast and cheap analysis, but SAXS was more reliable in determining polydispersity. ZnO NPs were also investigated by SAXS in various cases. More specifically, the structural evolution of zinc species toward ZnO NPs prepared with a sol–gel synthesis route using zinc oxy-acetate as the Zn source was studied by SAXS, UV-Vis and Quick-XAFS.60 The precursor led to the formation of NPs through a hydrolysis–condensation pathway in ethanol solution, induced by the addition of sodium hydroxide. ZnO/SiO2 nanocomposite thin films were investigated by GISAXS/WAXS/ellipsometry experiments. These measurements helped to evaluate the film structure (thickness, porosity, and density) as an evolution of temperature, since annealing was needed to acquire the final structure, starting with TEOS and ZnCl2 precursors.61 Post-synthetic thermal treatment improved the crystallinity of ZnO and helped the creation of oxygen-related defects from the grain boundaries, which could have radical influence on the photoluminescent behaviour. The above-mentioned measurements were also useful for the comprehension of the formation kinetics. Finally, zinc oxide NPs encapsulated into zeolite-Y were analysed through an in situ combined XRD, XAFS and SAXS approach. NaOH was employed to assist the encapsulation of zinc into the zeolite using aqueous zinc acetate as the Zn source. The particle sizes estimated by EXAFS and SAXS were in agreement with the cavity size of zeolite-Y.62
In another report, the clustering and dispersion behaviour of carboxylic acid-modified ZrO2 NPs in several solvents was evaluated by SAXS/SANS measurements. Such experiments helped to identify with precision the structural details of surface-modified NPs, including the sizes of the inorganic core and the organic shell, as well as their secondary clusters. Concerning the size measurement, the authors note that TEM provides solely the structural details in the dry form, which can differ from what occurs in the liquid state. On the other hand, DLS refers to the dispersed state, but a correct assignment of the refractive indexes for the NPs and the solvent is necessary to obtain reliable measurements. For these reasons, the authors highlighted the relatively better precision that SAXS/SANS can offer for the NP size determination.63 Tin oxide (SnO2) NPs were also investigated by SAXS: the effect of an acetylacetonate (acac) complexing ligand on the formation and growth of such particles, generated by the thermohydrolysis of SnCl4−n(acac)n at 70 °C, was analysed.64 SAXS and EXAFS were also employed to study the formation process of SnO2 NPs produced after dissolving tetrachloride pentahydrate in acid ethanol solution and subsequent heating at 70 °C.65 A five-step formation mechanism was suggested. Time-resolved SAXS experiments helped to monitor the evolution of the number and size of some intermediate species, known as nanoscopic polynuclear tin-oxo clusters.
X-ray photoelectron spectroscopy (XPS) is the most widely used analytical technique for surface chemical analysis, also employed for the characterization of nanoscale materials. Its underlying physical principle is the photoelectric effect.66 XPS is a powerful quantitative technique, useful to elucidate the electronic structure, elemental composition and oxidation states of elements in a material. It can also analyse the ligand exchange interactions and surface functionalization of NPs as well as core/shell structures, and it operates under ultra-high vacuum conditions. Nag and co-workers have published a review paper describing the role of XPS as an interesting means to study the internal heterostructures of NPs. For example, it has been used to investigate the environment-dependent crystal structure tuning of metal chalcogenide NPs of various sizes.67 It can also distinguish between core/shell and homogeneous alloy structures, and identify the bonding mode of ligands such as trioctylphosphine oxide (TOPO) onto the surface of metal chalcogenide NPs. For example, if TOPO bonds preferentially to the surface metal element, then the uncapped surface chalcogenide element may oxidize more easily upon exposure to air. In comparison with microscopy techniques, like TEM and TEM/EELS, which use lateral spatial resolution to identify elements in a direction vertical to the probing electron beam, XPS probes the composition of the material along the direction of the electron beam. Regarding core–shell NPs, Shard has published an article that reports a straightforward method to interpret the XPS data for such types of particles. It involves a direct and accurate empirical method to convert the XPS intensities into overlayer thicknesses, mostly suitable for spherical NPs.68 As further advantages of XPS the author mentions that it provides the depth information, similar to the size of NPs (up to 10 nm depth from the surface) and it does not significantly damage the samples. Two drawbacks of XPS analysis are the preparation of samples (i.e. dry solid form is required without contamination) and the interpretation of data.
In another study, the interaction of L-cysteine with naked Au NPs has been studied with XPS: that report aimed to provide experimental spectroscopic support to the kinetic models of catalyst deactivation, studying the role of low-coordinated Au atoms belonging to NP edges and corners.69 Furthermore, Minelli and co-workers wrote an article on the analysis of protein coatings on Au NPs by XPS and liquid-based particle sizing techniques. XPS is robust and useful to study proteins quantitatively, as well as peptides adsorbed at Au interfaces. It can also characterize the molecular interface of Au NPs. The chemical information from the NP surface analysed by XPS can be used to assess the thickness of NP coatings.70 Smirnov et al. used the so-called Davis’ method to determine the size of Au NPs in the planar model Au/C systems based on the data of XPS. The NP size values derived by XPS agreed well with those from the scanning tunneling miscroscope (STM) data, with the degree of similarity being related to the particle shape (e.g. sphere, hemisphere and truncated hemisphere).71 Tunc et al. presented a simple method by applying an external voltage stress during the XPS analysis of Au@SiO2 NPs; their method facilitated the detection, location and identification of charges developed on surface structures in a completely non-contact mode. Therefore, XPS provided information not only on the chemical identity but also on the dielectric properties of nanomaterials, by recording their charging/discharging behaviour.72 In another work, Polzonetti and colleagues used synchrotron XPS and NEXAFS to study the interaction at the molecule–metal interface and ligand arrangement in the molecular shell of Au NPs capped by aromatic thiols. The experimental results of both techniques were supported by density functional theory (DFT) calculations, illustrating the presence of a hybrid system in which the metallic Au core was surrounded by a shell of aromatic thiol molecules, whose thickness could be assessed by XPS.73 Castner and co-workers quantified the impact of nanoparticle coatings and non-uniformities on XPS analysis, for the case of Au@Ag core–shell NPs. They analysed the benefits of a complementary approach using XPS, STEM and simulated electron spectra for surface analysis (SESSA) simulations to characterize the structure and composition of NPs with nonideal geometries.74 For instance, STEM provided information concerning the metallic cores and shells, while XPS provided information regarding organic species and contaminants that were difficult to identify by STEM.
In another report, Ag NPs capped with taurine were investigated by SERS and XPS. The latter method together with DFT calculations showed that the gauche tautomer of taurine was the main component of the Ag NP surface.75 XPS confirmed the binding of taurine through the oxygen atoms of the sulfonate group, denoting the existence of 71% Ag–O in taurine-functionalized Ag NPs. This protocol provided a quantitative understanding of the interaction of the above molecule with Ag NPs. Furthermore, Ramstedt and Franklyn produced Ag NPs inside a poly(3-sulfopropyl methacrylate) brush and studied their formation process with TEM, UV-Vis and XPS. The apparent oxidation state of Ag in the different forms (NPs, films, and clusters) was investigated using XPS and a chemical state plot.76 In the case of bimetallic Ag/Pd colloids, prepared by galvanic replacement onto Ag colloids pre-synthesized by laser ablation, the final product was investigated by UV-Vis, XPS, SERS and ζ-potential. These measurements showed that the nanostructures were mainly coated with metallic Pd.77 Nevertheless, it was not easy to distinguish between Ag(0) and Ag(I) through XPS analysis. The galvanic replacement process using Pd(II) nitrate was monitored by the ζ-potential measurements on the basis of the fact that the charged species on the Ag surface had a progressively modified adsorption due to the oxidative action of air diluted in an aqueous medium.
CdS@Ag2S core/shell NPs prepared with the AOT/n-heptane/water microemulsion technique were characterized by XPS and SEM-EDX (EDX stands for energy-dispersive X-ray spectroscopy).78 The authors emphasize on the benefit of the high sensitivity of XPS, since every element has a particular set of peaks in the photoelectron spectrum at kinetic energies determined by the photon energy and the respective binding energies. The intensity of the peaks is a function of the concentration of the respective element. The XPS and SEM-EDX results supported the core–shell formation. In another work, Ag, Ni, and AgNi NPs synthesized by the derived seed-mediated growth method on a transparent conductive indium tin oxide substrate were studied by XPS, XRD and optical spectroscopy. XPS determined the oxidation states of Ag and Ni at the outer layers of the NPs. It was shown that the surface of Ag NPs was not oxidized, while Ni NPs were oxidized to nickel oxide and hydroxide.79 It is interesting to note that no peaks of NiO or Ni(OH)2 were detected in the XRD measurements, highlighting the utility of XPS to identify amorphous species. In the case of bimetallic AgNi NPs, fcc-Ag, silver in Ag(II) state and oxidized Ni atoms were detected. This indicated the presence of a Ag core@NiO–Ni(OH)2 shell structure.
Kalinkin et al. investigated the particle size influence in the oxidation of small Pt NPs on graphite with NiO2, using XPS and STM. The combination of these methods provided the most complete information regarding the composition, state and structure of the surface of the NPs and could facilitate the understanding of the origin of the size effect.80 In fact, only Pt NPs with a size smaller than 2.5 nm were found to oxidize to a mixture of PtO and PtO2 under the experimental conditions of that study. Chakroune et al. studied acetate- and thiol-coated Ru NPs with XPS, XAS and HRTEM. For NPs stored in polyol/acetate, surface oxidation limited to one monolayer and a surface coating with mainly acetate ions were evidenced by XPS measurements.81 For particles capped with thiol after being prepared in polyol, the formation of a Ru–S bond was shown for very small (2 nm) ruthenium particles. XANES and XPS were in agreement with charge transfer from Ru to S atoms. Rhodium NPs, prepared by reducing RhCl3·3H2O in a water/ethanol/PVP mixture, were characterized by several techniques, including XPS and NEXAFS. These techniques investigated the chemical states and indicated that the chlorine moiety derived from the precursor remained at the obtained NPs at both surface and bulk volume but heating may have caused its removal.82 In another work, Ir NPs were produced by decomposing [Ir(COD)Cl]2 in dichloromethane in the presence of an ionic liquid under a hydrogen atmosphere. XPS helped to identify the interaction of Ir(0) NP surface with ionic species of the imidazolium ionic liquid 1-ethyl-3-methylimidazolium ethylsulfate (EMI·EtSO4).83
Zerovalent Fe NPs were applied by Li and Zhang for the removal of water contaminants, such as Cd(II), Pb(II), etc. HR-XPS confirmed that these Fe NPs had a core–shell structure, which resulted in remarkable properties for concurrent sorption and reductive precipitation of metal ions. Such measurements facilitated the identification of the type of element present at the NP surface, the chemical and valence states of these elements and the ratio between the different chemical states of each element. These core/shell Fe/Fe oxide NPs were highly efficient in metal removal.84 Moreover, Sheng et al. used zerovalent Fe NPs immobilized onto diatomite for the sequestration of uranyl (U(VI)) in water. The XPS experiments implied that the diatomite-supported Fe NPs helped to reduce the highly toxic and mobile UO22+ to less toxic and mobile UO2.85 Complementary characterization with EXAFS illustrated that diatomite could act as a scavenger for insoluble products like UO2, therefore enabling more reactive sites to be used for U(VI) reduction. The utility of XPS to acquire information of ligand binding on NPs coated with several types of ligands was demonstrated by Lee and co-workers. The particles studied were CdSe/ZnS quantum dots capped with TOPO, 3-mercaptopropionic acid (MPA) or 1H,1H,2H,2H-perfluorooctanethiol (PFOT). Their analysis with XPS imaging had low sensitivity and limited lateral resolution; however, it provided a statistical, non-destructive method to characterize the ligand–QD binding mode.86 In addition, near ambient pressure (NAP)-XPS was employed to record the changes in the oxidation state of palladium in PdO NPs supported on TiO2 in a temperature range of 30–120 °C. PdO was used to catalyse the oxidation of 2-propanol. Lab-based NAP-XPS instead of synchrotron facilities showed distinct advantages: the instrument is available upon need and it can be integrated permanently with other devices for optimal sample analysis, but there are also some disadvantages: a synchrotron source results in photoelectron peaks with higher intensity, thus obtaining measurements at a higher resolution. However, care needs to be taken because a high-intensity radiation source can destroy certain types of samples. Overall, NAP-XPS is an effective technique to study in situ the steady-state conditions at the solid–gas interface, which are significant in the domains of catalysis, electrochemistry, corrosion and environmental science. The authors mention that additional screening with techniques such as mass spectrometry was needed for a more complete picture of the catalytic process (oxidation of 2-propanol by PdO NPs in this case) in such reactions.87
2.2 Additional techniques for the characterization of the structure, composition and other main NP properties
There are also several other techniques that help in the determination of the structure, composition, size and other basic features of the NPs. Fourier transform infrared spectroscopy (FTIR) is a technique based on the measurement of the absorption of electromagnetic radiation with wavelengths within the mid-infrared region (4000–400 cm−1). If a molecule absorbs IR radiation, the dipole moment is somehow modified and the molecule becomes IR active. A recorded spectrum gives the position of bands related to the strength and nature of bonds, and specific functional groups, providing thus information concerning molecular structures and interactions.88 Feliu and co-workers studied how Pt nanostructures performed on ethanol oxidation, using a combined approach with in situ ATR-FTIR and differential electrochemical mass spectroscopy (DEMS). These techniques helped to probe adsorbates electrochemically and detect volatile reaction products. Their results were in agreement with previous findings, showing that the preferred decomposition products were related to surface structures, with COads formation on (100) domains and acetaldehyde/acetic acid formation on (111) domains.89 In another report, carbon-supported platinum NPs (3–8 nm size) were used for the CO oxidation reaction and this catalytic process was monitored using DRIFTS and quadrupole mass spectrometry (QMS). The FTIR measurements of adsorbed CO confirmed the variations of COad and Oad in different steps of the experiment, in accordance with the results from QMS, while modifications in the CO distributing over various types of Pt surface sites were also noticed. Overall, DRIFTS was regarded as an important tool for the probation of the surface structure of Pt NPs under in situ conditions.90 Shukla et al. published a paper devoted to the FTIR investigation of the surfactant bonding to FePt NPs that were synthesized in the presence of oleic acid and oleylamine. The former ligand was found to bond to FePt NPs in both monodentate and bidentate forms, while oleylamine bonded to FePt molecularly with the NH2 group intact.91 Furthermore, Au/Ag bimetallic NPs stabilized with dodecanethiol and soluble in nonpolar solvents were produced through a two-phase synthetic route in water/toluene mixtures.92 The most important insight derived from XPS and FTIR measurements was that Ag atoms were enriched at the outer part of the alloy clusters in comparison with the Au atoms. In another work, the influence of the Ag NP content on the photocatalytic degradation of oxalic acid adsorbed on TiO2 NPs was evaluated by ATR-FTIR. Various Ag NP amounts were tested, and it was demonstrated that the incorporation of only a small quantity (2%) boosted the photocatalytic performance of TiO2 NPs substantially. AFM and XPS were used to characterize the topography and chemical structure/composition of the composite NP films.93 Tzitzios et al. synthesized Ni NPs with a hexagonal crystal structure in the size range of 13–25 nm via the reduction of nickel stearate in the presence of PEG, oleic acid and oleylamine. FTIR spectra showed the presence of the characteristic groups at the surface of the NPs, such as the –HC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH– arrangement in OAc and OAm, while the binding modes of the ligands onto the NP surface were also examined.94 Copper zinc tin sulpho-selenide (CZTSxSe1−x) nanocrystals were prepared by Haram and colleagues with a hot-injection process. The precursors were dissolved in OAm and heated at T > 200 °C for the synthesis. FTIR measurements showed the adsorption of OAm onto the surface of the particles. Characteristic bands arising from the moieties existing at the molecule of OAm and indicating its successful coordination with the NPs were spotted.95
CH– arrangement in OAc and OAm, while the binding modes of the ligands onto the NP surface were also examined.94 Copper zinc tin sulpho-selenide (CZTSxSe1−x) nanocrystals were prepared by Haram and colleagues with a hot-injection process. The precursors were dissolved in OAm and heated at T > 200 °C for the synthesis. FTIR measurements showed the adsorption of OAm onto the surface of the particles. Characteristic bands arising from the moieties existing at the molecule of OAm and indicating its successful coordination with the NPs were spotted.95
Superparamagnetic ferrite NPs (MFe2O4, M = Ni, Co, Zn, Mn) with high crystallinity and size below 10 nm were synthesized by Sabale et al. with a simple ‘polyol’ method. The observation of tetrahedral (v1) frequency at the FTIR spectrum verified the presence of the spinel ferrite structure. Bands assigned to the –OH and C–O groups denoted the presence of diethylene glycol, thus revealing its successful coating around the ferrite NPs, endowing them a high solubility in water.96 In another report, the presence of Fe–O–P bonds was shown by FTIR measurements for hydrophobic iron nanoparticles which were treated with alkyl phosphonic acid-based ligands in order to turn them into water-soluble iron–iron oxide core–shell NPs.97 FTIR spectroscopy was also employed to characterize multifunctional Fe3O4@C@Ag hybrid NPs, which were prepared with a facile route based on the direct adsorption and spontaneous reduction of Ag ions onto the surface shell of C-coated magnetic NPs. The presence of carboxyl and hydroxyl groups on the NP surface was shown by FTIR. Certain bands attributed to carboxyl vibration implied that carbonyl and other reductive groups were oxidized by Ag ions, which was an indirect sign for the presence of Ag in the products. These hybrid nanostructures displayed a remarkable photocatalytic activity for the photodegradation of neutral red dye under visible light irradiation.98 Duong et al. have shown that FTIR can be successfully used to assess the affinity of polymers bearing phosphate groups as surface ligands for NaYF4:Yb/Er upconversion nanoparticles.99 Trioctylphosphine-stabilized CdS nanorods synthesized by Chen et al. were also characterized by FTIR, revealing C–P stretching peaks related to the aforementioned molecule.100Table 3 presents the IR vibrational assignments of several characteristic functional groups which are involved in nanoparticle synthesis.101
| Vibrational modes | Frequency (cm−1) |
|---|---|
| Methyl C–H asym/sym stretch | 2970–2950/2880–2860 |
| Methyl C–H asym/sym bend | 1470–1430/1380–1370 |
| CC alkenyl stretch |
1680–1620 |
| Aromatic C–H stretch | 3130–3070 |
| O–H hydroxyl group, H-bonded OH stretch | 3570–3200 (broad) |
| C–O stretch, primary alcohol | ∼1050 |
| N–H aliphatic primary amine, NH stretch | 3400–3380, 3345–3325 |
| N–H primary amine, NH bend | 1650–1590 |
| C–N, primary amine, CN stretch | 1090–1020 |
| Carboxylate | 1610–1550/1420–1300 |
| Organic phosphates (PO stretch) |
1350–1250 |
| Aliphatic phosphates (P–O–C stretch) | 1050–990 |
| Sulfonates | 1365–1340/1200–1100 |
| Organic siloxane or silicone (Si–O–Si) | 1095–1075/1055–1020 |
| Organic siloxane or silicone (Si–O–C) | 1100–1080 |
| Thiols (S–H stretch) | 2600–2550 |
| Thiol or thioether, CH2–S– (C–S stretch) | 710–685 |
| Aliphatic chloro-compounds, C–Cl stretch | 800–700 |
| Ammonium ion | 3300–3030/1430–1390 |
Nuclear magnetic resonance (NMR) spectroscopy is another important analytical technique in the quantitative and structural determination of nanoscale materials. It is based on the NMR phenomenon exhibited by nuclei that possess non-zero spin when placed in a strong magnetic field, which causes a small energy difference between the ‘spin-up’ and ‘spin-down’ states. Transitions between these states can be probed by electromagnetic radiation in the radio wave range. NMR is typically used to study the interactions or coordination between the ligand and the surface of diamagnetic or antiferromagnetic NPs. It is, however, not suitable to characterize ferri- or ferromagnetic materials, as the large saturation magnetization of such materials causes variations in a local magnetic field, which lead to shifts of the signal frequency and dramatic decreases in relaxation times. As a result, significant broadening of the signal peaks occurs, making the measurements practically inutile and unable to be interpreted.66
Marbella and Millstone have written a comprehensive review article on the NMR techniques for noble metal NPs. NMR spectroscopy can help toward the routine, straightforward, molecular-scale investigation of NP formation and morphology in situ, in both solution and solid phase. It is particularly useful for analyzing both the formation and final architecture of noble metal NPs. The capping ligands are also typically studied by NMR, and such measurements can yield information on the properties of the particle core (e.g. electronic structure, atomic composition, or compositional architecture). Insights into ligand density, arrangement and dynamics can also be derived.102 Besides facilitating the monitoring of the chemical evolution of ligand precursors and their role in particle growth, NMR is also employed to probe the role of capping ligands for the determination of particle shape. Overall, NMR can screen the chemical conversion of NP precursors in both the solution and solid phase, with high spatial and chemical resolution, under distinct reaction conditions, and for diverse metal identities; this helps in the better comprehension of the reaction mechanisms for NP synthesis. Moreover, NMR is useful for the monitoring of the process and final products of ligand exchange, when the initial capping ligands need to be replaced.102
The 1H NMR chemical shift behaviour is sensitive to the surrounding electronic environment; this includes the electronic structures and bonding environment of the nucleus. Consequently, any changes in the handedness of a molecule can be ‘felt’ by neighboring spin positions and observed as changes in chemical shift. This renders NMR significant to assess the chirality or absence of chirality of small, molecule-like nanoclusters. NMR can also be applied for the direct monitoring of the diffusion of adsorbed gases onto the surface of metal NPs. Finally, NMR is utile for the measurement of the hydrodynamic radius of metal NPs and thus constitutes an important complement to more standard NP sizing techniques, such as TEM and DLS. Similar to DLS, NMR spectra are used to define the NP size via the analysis of particle diffusion. In particular, NMR helps to extract the diffusion coefficient of well-dispersed species in solution diffusing according to Brownian motion only. Then the hydrodynamic size can be calculated through a rearrangement of the Stokes–Einstein equation.102 Finally, a phenomenon known as ‘Knight shift’, which is induced by some metals and can be present upon NMR measurements, is also described in ref. 102. It has to be noted that the particle size, which can be safely analysed by NMR, can exceed by far the 100 nm in the case of polymer-hybrid particles,103,104 whereas metallic NPs have to be at around the size range of 1–5 nm in order to acquire meaningful NMR measurements.
1H solution NMR has been reviewed by Hens and Martins as a tool for the investigation of the surface chemistry of colloidal NPs.105 Diffusion-ordered NMR (DOSY-NMR) offers the possibility to distinguish in situ free ligands from bound ligands, while the distribution of these species can also be quantified. Solution NMR can be employed to identify tightly bound ligands and quantify their surface density of sterically stabilized colloidal NPs.105 Jicsinszky and co-workers studied hydrophilic heptakis(6-deoxy-6-thio)cyclomaltoheptose capped Au NPs with DOSY-NMR. This technique proved to be an effective, reliable and rapid way to investigate the role of the total concentration of gold in solvated metal atom (SMA) solutions as well as of the Au/capping ligand molar ratio on NP sizes. NMR measurements also helped to acquire some basic information on the drug transport and release capabilities of Au NPs. This was achieved through the analysis of the nature of supramolecular aggregation processes and the ability of (Au)n/β-CDSH nanoaggregates to act as hosts for deoxycytidine (DC).106 DOSY-NMR has also been employed to determine the nanoparticle size, e.g. in the case of Au NPs prepared by Canzi et al. This was achieved by analysing the 1H spectrum of the protecting ligands using 2D DOSY NMR, a method that could be facilely adapted also for other metal and semiconductor nanocrystals. Size estimates were acquired by using diffusion coefficient ratios derived from the proton signals from the alkyl thiolate groups bound to Au NPs and a ferrocene internal standard. The authors stated that DOSY NMR was a reliable alternative method to calculate the NP size, being quicker and more cost-effective than TEM.107 Coelho and colleagues used NMR spectroscopy to determine particular intermolecular interactions and mechanisms of drug immobilization and location into surface PEG-modified Au NPs. The authors highlighted the advantages of NMR as a non-destructive, highly reproducible method, sensitive to the structural details of molecules and molecular conjugates, which could be employed for both qualitative and quantitative characterization. Information of size, shape, dynamics, chemical structure, intermolecular interactions, and binding and exchange processes in complex nano-systems could be obtained.108 The combined use of NMR with FTIR, UV-Vis, DLS and TEM could yield significant insights regarding important physicochemical properties of drug delivery systems, which influence their therapeutic efficacy.108
In another report, deuterium (2H) NMR was employed to study the intramolecular ligand dynamics in d15-(PPh3)-capped Au NPs. The authors made use of the ability of NMR to probe ligand structures and surface binding properties on NPs by the in situ analysis of chemical shifts and resonance lines in the solid and liquid states. A specific feature of 2H NMR is its simplicity and the capacity to distinguish the type of dynamics in amorphous and crystalline domains, for organic compounds that are isotopically labelled with deuterons.109 Smith et al. used NMR to investigate the extent of ligand exchange between distinct kinds of thiolated molecules on the surface of Au NPs. In particular, they determined ligand density values for single-moiety ligand shells and then used these data to describe the ligand exchange behaviour with a second, thiolated molecule.110 Triphenylphosphine-capped 1.8 nm Au NPs have been characterized by multinuclear NMR to investigate their surface structure and ligand binding environment. In solution, the ligand exchange kinetic reactions were screened by 1H, 2H and 31P NMR to analyse the exchange process.111 Doyen et al. used UV-Vis and NMR to study the formation of Au NPs by the citrate reduction method. 1D–1H and DOSY-NMR measurements showed that citrate aggregates with Au(I) and Au(0) were formed. That work suggested that citrate, apart from being the reductant and the stabilizing agent for Au NPs, might act as a ‘molecular linker’, which could help in the particle formation.112
The coordination of amine ligands on Ag NPs was evaluated by NMR, SERS and DFT.113 It was found by SERS that the amidine moiety, coming from the silver amidinate precursor, remained bound to the metal surface, whereas the hexadecylamine ligand was in a fast exchange between a surface-bound state and free floating in solution, as revealed by NMR. Solution NMR spectroscopy was a powerful tool for the analysis of short timescale effects. Long-residence-time molecules at the NP surface could not be monitored by this technique due to their very slow tumbling. The SERS analysis of the NPs combined with DFT modelling demonstrated that unexpected organic groups were observed by this latter technique, in contrast with what was shown by solution NMR. SERS is efficient if molecules are in a close contact with the Ag surface, whereas NMR spectroscopy examines molecules in the first and second coordination sphere of the NPs. Despite this, the complementarity of SERS with NMR is beneficial to reveal the molecular environment of the prepared NPs. Amidine hindered the NP aggregation, while hexadecylamine (HDA) helped toward a narrow size distribution of stabilized Ag0 NPs.113 Ag NP/π-conjugated polyelectrolyte systems were investigated by NMR, FTIR and SERS and increased regularity of the high-cis polymers was documented. The IR spectra supported the conclusions drawn from the 1H NMR measurement of the polymer; both techniques consistently illustrated the cis-rich configuration of polymers formed by the solution polymerization in acetonitrile and the cis/trans configuration of the polymers formed by the bulk polymerization.114 Velders and co-workers focused on the use of 1H NMR spectroscopy to determine the NP size in the case of dendrimer-encapsulated Pd NPs. The advantage of NMR in comparison with TEM consisted of its capacity to probe the total population of the NPs, providing more representative information regarding the average NP size. In addition, in situ operation was possible with NMR, and this enabled the monitoring of the changes in the size and the capping ligand environment of the NPs during catalytic reactions.115
Solution NMR spectroscopy has been extensively used also for the characterization of oxide nanoparticle systems. Kahn and co-workers characterized ZnO NPs by 1H and DOSY-NMR. They emphasized on the ability of the latter technique to sort species according to their size, as the diffusion coefficient is inversely proportional to the hydrodynamic radius. Their study, performed on ZnO NPs stabilized by amine molecules, showed that a fast exchange between free and coordinated amine molecules was deduced within the NMR measurement timescale. Overall, the NMR spectra showed that the seemingly simple stabilization of ZnO NPs by amine molecules appeared to be much more complicated than considered beforehand.116 The same group published a study dedicated to the use of NMR techniques for the investigation of the role of amine ligands together with oleic acid on the formation of ZnO NP superlattices in C7D8. Their experiments demonstrated the dependence of the type of ligand adsorbed on the NP surface on the concentration of the colloidal NP solutions. It was suggested that the driving force of the superlattice formation was the presence of ion-paired ammonium carboxylate shells around each particle.117 ϒarger and colleagues investigated phosphonic acid-capped SnO2 NPs with sizes lower than 5 nm, using multinuclear solution and solid-state magic angle spinning (MAS) NMR. The latter technique indicated the absence of acidic protons of the phosphonic acid groups, strongly supporting the formation of P–O–Sn linkages. Insights into the ligand structure and the extent of phosphonic acid protonation upon binding the NP surface were obtained.118 In the case of Ca2SnO4 NPs prepared by the mechanochemical synthetic route, 119Sn MAS-NMR and 119Sn Mössbauer were employed to probe the local environment of Sn nuclei, so as to acquire important insights into the local structural disorder of these NPs. NMR spectroscopy provided information on the magnetic and chemical interactions, while Mössbauer measurements revealed the quadrupolar interactions experienced by the nuclei of 119Sn.119 Magnetite-silica NPs prepared by a two-stage procedure by Bogachev et al. were characterized by NMR relaxometry, AFM and UV-Vis spectroscopy. The aggregation process in the colloidal solutions of Fe3O4–SiO2 NPs was investigated.120 Dextran-coated γ-Fe2O3 NPs were studied by Papavassiliou and colleagues with 57Fe NMR, Mössbauer, TEM and magnetization measurements. The low temperature mechanism of collective magnetic excitation in magnetic NPs, which originated from the fluctuations of the magnetization direction around an energy minimum corresponding to an easy direction of magnetization, was investigated. 10 nm nano-sized samples at low T displayed similar NMR spectra, and thus similar hyperfine fields to the bulk material, implying that the samples had the same magnetic structure.121 Gossuin et al. characterized gadolinium hydroxide and dysprosium oxide NPs using XRD, magnetometry and NMR relaxometry. Nuclear magnetic relaxation dispersion profile represented the evolution of the longitudinal relaxation rate with respect to the magnetic field and provided interesting information about the longitudinal relaxation mechanism.122 Finally, HfO2 and ZrO2 NPs synthesized using the Karlsruhe microwave-plasma process were characterized by several techniques such as 1H MAS NMR, XPS, XRD and electron diffraction. Among these techniques, NMR and XPS helped to identify the chemical composition of the as-prepared NPs. A hydrate surface layer with a hydrogen content of 5–10 wt%, composed of chemisorbed hydroxyl groups and organic precursor fragments, was detected by 1H-MAS NMR.123
Solid-state NMR (SS NMR) spectroscopy is an important characterization tool to investigate the behaviour of solid catalysts and chemical processes occurring at their surface. Such technique may help to resolve not only interactions at the ligand–solvent interface but also result in the acquisition of significant insight into ligand–particle bonding at the hard–soft matter interface.102 For example, 31P is a very sensitive NMR nucleus with 100% natural abundance and high gyromagnetic ratio and it is quite easy to measure the 31P NMR spectra with a good signal to noise ratio even in systems with low ligand concentrations. J-resolved 31P solid-state NMR spectroscopy combined with DFT calculations can provide important information about the structure of heterogenized species and also provide insights into the immobilization of homogeneous metal phosphine catalysts. Gutmann and co-workers have highlighted the crucial role of liquid and partly solid-state NMR techniques for the detection of surface molecules and the discrimination between different binding sites on nanoscale catalysts.124 In particular, 2H MAS NMR has been employed to study chemical reactions such as the hydrogenation of olefins, being capable of detecting reactive intermediates. The authors denoted a weakness of the NMR measurements, which was related to their sensitivity. Solid state 31P NMR was used to characterize phosphinine-stabilised Au NPs and a phosphinine–Au complex, as reported by Mallissery and Gudat.125 NMR spectra showed that in addition to metal-bound intact phosphinine units, several surface-bound species generated by the chemical transformation of the initially supplied ligands were also detected. In another work, two different tripeptides attached on Au NPs were analysed by SS NMR and DFT calculations. Substantial structural differences between CysAlaAla and AlaAlaCys on Au NPs were evidenced through the aforementioned techniques. In particular, the location of the carboxylate moiety relative to the S atom that served to anchor the peptide to the surface played a significant role in determining these structures.126 Novio et al. have used SS NMR and FTIR to characterize the location and dynamics of carbon monoxide coordination on Ru NPs. Two different sets of 2 nm Ru NPs were tested, prepared under a H2 atmosphere, stabilized by either PVP or a bidentate phosphine ligand (dppb). It was demonstrated that CO groups were mobile on the NP surface, while the bulky ancillary ligand dppb slowed down the fluxionality of CO and prevented the exchange at certain positions.127 SS NMR was also employed to characterize 1–2 nm Ru NPs capped by either 1,3,5-triaza-7-phosphaadamantane or PPh3 ligands and exposed to a CO gas atmosphere. That paper presented a new way to analyse interactions and calculate approximate distances between phosphine ligands and CO probe molecules on the surface of Ru NPs employing 31P–13C REDOR NMR.128 Lara et al. decomposed [Ru(COD)(COT)] [(1,5-cyclooctadiene)(1,3,5-cyclooctatriene)ruthenium] and [Pt(CH3)2(COD)] [dimethyl(1,5-cyclooctadiene)platinum(II)] organometallic complexes to produce small core–shell RuPt NPs in the presence of PVP at room temperature. Several characterization techniques were combined for determining the structural composition of the particles, and 13CO was used for adsorption as a probe molecule. FTIR and SS NMR results were in agreement with the coordination of CO to Pt and in this way the presence of a segregated Ru core/Pt shell structure was indicated. Measurements by WAXS, HRTEM, EXAFS and other techniques corroborated these findings.129
Electrically conductive Al-doped ZnO NPs prepared by Avadhut et al. were characterized by SS NMR spectroscopy: a core–shell structure model was proposed for these NPs, which were synthesized with a microwave-assisted polyol method. A combination of different 1D 27Al, 1H, 13C and 2d 27Al{1H} SS NMR techniques helped to gain insight into the particle structure and explain the macroscopically observed conductivities as a function of the NP composition.130 Nanoscale fluorine-doped SnO2 NPs, prepared with a microwave assisted polyol approach were studied by several techniques, including SS NMR. Sn(II) could be distinguished from Sn(IV) using NMR, similar to what Mössbauer spectroscopy can do. Heteronuclear NMR experiments helped to characterize intraparticle interfaces in polycrystalline NPs. The fluorine doped particles showed an increased conductivity, after annealing, in comparison with undoped SnO2 NPs.131 Davidowski and Holland employed SS NMR to characterize mixed phosphonic acid ligand binding and organization on SiO2 NPs. Multinuclear (1H, 29Si, 31P) and multidimensional solid-state NMR techniques were used, while the phosphonic capping ligands were methylphosphonic acid and phenylphosphonic acid. For instance, 31P NMR spectra showed that phosphonic acid functionalized silica NPs displayed three different ligand environments, attributed to physisorbed, monodentate and bi/tridentate.132 The combination of multinuclear SS NMR and DFT calculations has been employed to investigate the structure of NaYF upconverting NPs. A detailed analysis of the crystal lattice and ionic distribution was achieved by these techniques. In particular, 89Y NMR was employed to probe the chemical environment of Y3+ ions in the NaYF4 structure. The presence of a solid solution type cubic structure in which cation sites were randomly occupied was observed.133 Finally, for the characterization of surface species and substrate–surface interactions on metal NPs, the groups of Pruski and Emsley have shown that dynamic nuclear polarization surface enhanced NMR can be a very useful tool for the further increase of the sensitivity of SS NMR.134,135
The Brunauer–Emmett–Teller (BET) technique is also used for the characterisation of nanoscale materials. It is based on the principle of physical adsorption of a gas on a solid surface, and it was named by the initials of the surnames of its developers, Brunauer, Emmett and Teller. It is widely used for the determination of the surface area of nanostructures, being a relatively accurate, rapid and simple method for this purpose.66 Sahoo and co-workers prepared biocompatible ferrofluid containing dye-functionalized Fe3O4 NPs, which can serve as fluorescent markers. Several techniques were used for the characterisation including BET, FTIR and others. The surface area measured by BET was smaller than the estimate obtained from the size distribution and density values of the studied material; this deviation might be caused by the agglomeration of smaller NPs resulting in larger ones, thereby effectively reducing the collective surface area. Such agglomeration risk is probably aggravated considering that the NP samples need to be dried for such measurements: strong hydrogen bonding might occur among the NP surfaces, thus, inducing a certain error.136 In another work, mesoporous polymer microspheres with Au NPs inside their pores were produced, to observe the adsorption behaviour of these NPs, considering their surface functionality and porosity. BET experiments of Au/poly(ethylene glycol dimethacrylate-co-acrylonitrile) composite microspheres, used to measure the microsphere porosity, revealed that the adsorption of Au NPs into the pores kept the pore structure intact and turned it more porous.137 Ma et al. synthesized Fe3O4 NPs by the co-precipitation method of ferrous and ferric species, resulting in a product with high specific surface area (286.9 m2 g−1). This value was much higher than those already reported in the literature for such particles.138
Thermal gravimetric analysis (TGA). While FTIR offers information about the NP–stabiliser interaction and confirmation of the stabiliser type, it does not provide insights into the extent of surface coverage or the mass to mass ratio of NP to stabiliser, which is important to normalize the values of saturation magnetization to purely metallic content, for instance. TGA provides information concerning the mass and composition of the stabilisers. With this technique, a nanomaterial sample is heated and components with different degradation temperatures decompose and vaporise, and a change of mass is recorded. The temperature and the loss of mass are recorded by the TGA device and, taking into account the starting sample mass, the type and quantity of NP organic ligands are determined.139 A method known as microthermogravimetric analysis (μ-TGA) uses the same thermal decomposition principle as TGA, but the mass of the sample investigated is in the order of 1 μg, with mass changes lower than 1 nanogram being able to be detected. In this way, the detection limits of conventional TGA can be improved to a significant extent. Mansfield et al. used μ-TGA to identify the presence and quantity of surface-bound ligand coverage on Au NPs and verify the existence of PEG coating on silica NPs.140 Their results demonstrated that the aforementioned technique is a valid one to determine quantitatively the NPs coatings, while information on the purity and compositional data of the NPs can also be acquired sometimes. The authors highlighted the advantages of TGA, which is a simple and direct technique without any special need for sample preparation, apart from having the sample in dry state. A drawback of conventional TGA is the need to have a few milligrams of the nanomaterial sample, which may raise the cost or lab-scale production feasibility issues. These researchers used a variety of NP systems to illustrate the utility and limitations of μ-TGA and its comparison with conventional TGA. For example, similar results of both techniques were obtained concerning the oxidation temperature and the residual mass measurements in carbon nanotubes. In addition, the ability to identify layer-by-layer coatings on a Au NP core was evidenced by both techniques. The same research group analysed the surface density of PEG on Au NPs by using μ-TGA. The speed and reliability of TGA to determine the fractions of thermally stable and unstable masses of a sample were exploited. Usually, the surface coverage for inorganic particles with combustible ligands can be calculated if particle size and ligand molecular weights are well known. The authors measured the PEG surface densities on Au NPs using both μ-TGA and fluorescence spectroscopy. The lower values for surface densities determined from the latter technique might be attributed to incomplete displacement of the ligands from the Au surfaces.141 In another report, thiol-terminated PEG-coated Au NPs in aqueous solution were studied by TGA and other techniques, aiming to elucidate their structure and hydration. Combining mass density, SANS, SAXS and TGA resulted in the acquisition of precise information on the Au core size and on the capping polymer chains. SANS fits reached their optimal minimizations with a three shell model: the inner one related to the Au core, while the other two are characterized by different polymer–water mixtures with distinct scattering densities. On the other hand, SAXS was principally sensitive to the dimension of the Au core, considering that the contrast in the electron densities between the polymer and the solvent is low. The results of the structural data of the scattering experiments and the volumetric data derived from mass density and TGA measurements were consistent, revealing the complementarity and correctness of this overall characterization approach.142 Jia et al. prepared Au and Pd NPs via a surfactant-free single phase solution route. High-temperature TGA coupled with mass spectroscopy (MS) was used to find the relative amounts of ionic contaminants, since protecting thiolate groups and inorganic contaminants were removed in separate weight loss events. TGA-MS helped to achieve a more accurate determination of the thiolate to Au ratios, revealing a complex composition of the NPs presented therein. TGA-MS could also distinguish between the evaporation of the original thiolate ligands and their oxidized species. The limitations of the above technique include the fact that non-volatile compounds such as Li2O cannot be detected; however, XPS, FTIR and XRD can help toward such detection. In addition, the quantification of the content of certain groups and compounds based on TGA is only precise if their weight losses take place at distinct temperatures. Events that happen at similar temperatures can be separated by optimizing the heating program. However, overlapping events may be identified by MS, but the quantification of the intensities recorded in the MS data is not simple.143
Magnetite NPs with fatty acid (ricinoleic) adsorbed on their surface were investigated with a TGA device coupled with FTIR. The decomposition of ricinoleic acid was studied by TGA under an inert atmosphere, while gas phase FTIR helped to gain information on the decomposition gases released. The impact of the autoxidation of the fatty acids was presented, while an extended reduction of magnetite from carbonaceous residues was also noticed.144 Slight discrepancies between the results from the TGA and XRD experiments on the exact composition of the iron oxides might originate from the formation of oxidized residues in these two different measurements. In another work, Nava-Etzana and co-workers reported the synthesis of BiFeO3 nanostructures by a combustion reaction, in the presence of tartaric acid or glycine as the promoter. The origin of a high purity BiFeO3 nanomaterial together with the formation of certain by-products was described on the basis of metal–ligand interactions. Such high product purity demonstrated by XRD analysis was corroborated with the results from TGA.145 Furthermore, TGA/FTIR and a combination of TG–gas chromatography–mass spectrometry (TG/GC-MS) were employed to characterize the effect of different types of dopants (e.g. SiO2 NPs, multi-walled carbon nanotubes, and montmorillonite) on the thermal decomposition of polypropylene sebacate (PPSeb). It was evidenced through the mass detection analysis of the generated decomposition compounds (aldehydes, alcohols, acids, etc.) that the PPSeb degradation involved mainly β-hydrogen bond scission and also α-hydrogen scission. The insertion of NPs led to the increase of the thermal stability of the polymer.146
Low-energy ion scattering (LEIS) is a modern analytical method that permits the rapid thickness characterization of self-assembled monolayers (SAM), for example, in the case of Au NPs. In this technique, a sample is exposed to low-energy gas ions, and the scattering and subsequent loss of energy of these ions can be related to the elemental composition of the outer layer surface.147 High sensitivity LEIS (HS-LEIS) offers better sensitivity for the investigation of distinct atomic layers with an extensive reduction in surface damage. HS-LEIS illustrated that a complete SAM was formed in the case of C16COOH-functionalized 14 nm Au NPs. The estimated SAM thickness was in good agreement with previous results from simulated electron spectra for the surface analysis of the XPS data. The LEIS thickness values were consistent with the values obtained by AFM, X-ray reflection and sputter depth profiling.147 The high sensitivity of HS-LEIS concerns the top ∼10 nm of the surface atomic layers. This method is fast and rather direct, whereas SESSA simulations require a lengthy analysis of the results for the thickness, but can yield more information on chemical composition. Kauling et al. used HS-LEIS to analyse the outer layer of both functionalized and non-functionalized imidazolium ionic liquids on Au NPs. The description of its operation principle is described therein, together with its capacity to analyse the atomic composition and thickness of the surface of ionic liquids.148 Finally the formation of ruthenium–gold core–shell NPs prepared by the physical vapor deposition method on the TiO2 surface was studied by STM and LEIS, in an article published by Ovari et al. The chemical composition of the NPs was studied by LEIS, and it was found that when Rh was deposited on TiO2 previously covered by Au, Rh atoms impinged to Au clusters moved to subsurface sites; as a consequence, the outermost atomic layer of these clusters remained almost pure Au. STM and LEIS results showed that very limited mixing between Au and Rh in the bimetallic NPs took place (if any).149
UV-Vis spectroscopy (UV-Vis) is another relatively facile and low-cost characterization method that is often used for the study of nanoscale materials. It measures the intensity of light reflected from a sample and compares it to the intensity of light reflected from a reference material. NPs have optical properties that are sensitive to size, shape, concentration, agglomeration state and refractive index near the NP surface, which makes UV-Vis spectroscopy an important tool to identify, characterize and investigate these materials, and evaluate the stability of NP colloidal solutions.150 Gold, silver and copper nanostructure sols exhibit characteristic UV-Vis extinction spectra due to the existence of a LSPR signal in the visible part of the spectrum. In certain cases (e.g. metal chalcogenide NPs and anisotropic gold or silver nanostructures), LSPR bands at the near-infrared (NIR) wavelength region can also appear.151 Besides characterizing the NP optical properties, the size and molar concentration of zerovalent Au, for example, can also be obtained from the UV-Vis measurements. For this calculation, which can also be performed in situ under certain conditions, the position of the LSPR and the extinction at this wavelength, as well as the ratio of extinctions at the wavelength of the LSPR and at 450 nm (ALSPR/A450), are needed.151 The absorbance at 350–400 nm wavelength can also be used to measure the gold colloid concentration, however with an uncertainty up to 20–30% due to a rather slight influence of parameters such as NP size, surface modification and oxidation state. If these factors are taken into account upon calculation, the uncertainty in determining the Au NP concentration can be decreased extensively.151 In fact, the maximum absorbance at the UV-Vis spectra has also been successfully used for the calculation of the concentration of citrate-coated silver NPs.152 Haiss et al. have published a very high profile study on the utility of UV-Vis spectra to determine the size and concentration of Au NPs.153 The colloidal stability of Au NPs can be quantitatively characterized by UV-Vis absorbance spectroscopy, as shown by Pennathur and colleagues. Particle instability parameter (PIP) is a universal technique to quantitatively characterize the stability of plasmonic nanomaterials based on UV-Vis absorbance spectroscopy that does not depend on the colloid system and can fully record the evolution of a given studied system over time. It is a robust and generalizable approach, not only for Au NPs, but also for plasmonic NPs as a whole.154 Another use of UV-Vis spectroscopy involves the ability to detect molecules such as thiamine, by mixing a solution of thiamine in water with a Au NP solution. The presence of thiamine could be detected visually with a color change in the NP solution from red to greenish-grey. Au NPs tested for this application were in the range of 20–30 nm, whereas the limit of detection of thiamine was between 0.5 and 1 μM.155 In another report, Au and Pt NPs prepared by photoreduction synthesis in an aqueous medium containing dodecyltrimethylammonium chloride (DTAC) and PEG were studied by UV-Vis, EXAFS and other techniques (TEM, SAXS). EXAFS confirmed the metallic character of the NPs while SAXS implied that the structure of DTAC and PEG could be fitted with the hard-sphere model having the interaction radius (RHS) and the spherically shaped core–shell structure. The time evolution of the SAXS profiles was consistent with the UV-Vis spectral change during the first 30 min of photoirradiation.156 Behzadi et al. reported the development of a colorimetric sensor array to define the physicochemical properties of NPs dissolved in water with ultra-low concentrations. The effects of several dyes on different types of NPs were probed using variations in the visible spectrum of the dyes. The system should produce unique composite responses to each NP, similar to the well-established colorimetric array that is used to identify toxic chemical vapors.157 The authors prepared four different types of gold nanostructures and they employed their UV-Vis approach to detect and discriminate these particles. Overall, this method can be considered low-cost, non-destructive and quick for the recognition of NP systems and types.
Ag nanostructures have also been extensively studied by UV-Vis spectroscopy. Jha and co-workers investigated the influence of maturing time and concentration of NaBH4 on size with UV-Vis. Their method, under the framework of the Mie theory, was employed to determine the particle size and size distribution. In fact, the LSPR of NPs is affected by size, shape, interparticle interactions, free electron density and surrounding medium, and this helps to obtain a screening of the electron injection and aggregation of NPs. In this way, it was possible to characterize the Ag NP formation kinetics and the final colloidal stability.158 In another work, Ag NPs were prepared via a green synthesis involving the flowers of the Moringa oleifera (MO) plant. This plant acted as a reducing and stabilizing agent, and the resulting particles were studied by FTIR, UV-Vis and other techniques. FTIR experiments demonstrated that proteins in the MO flower extract were adsorbed on Ag NPs, acting as capping agents. It also indicated that retinoic acid, a component of the MO flower extract, acted as a reductant. UV-Vis analysis verified the existence of LSPR in the produced particles and as the concentration of the MO flower extract increased, the absorption spectra showed a blue shift with decreasing NP size.159
Photoluminescence (PL) spectroscopy is another technique used to study nanoscale materials; it monitors the light emitted from atoms or molecules that have absorbed photons. PL is typically useful as the characterization technique for fluorescent nanoparticles, such as quantum dots, as well as metal nanoclusters. Recently, the inherent PL of metallic NPs received remarkable interest. Despite the fact that the quantum efficiency of the emission process is low, this inefficiency can be compensated by the large excitation cross sections at the plasmon resonances. In addition, the PL of metal NPs is free of photobleaching and photoblinking. Thus, PL can be regarded as a better alternative than fluorescent molecules for optical labeling applications. Single-photon and multi-photon excitation PL has been acquired using plasmonic nanostructures of several shapes.160 Gong and co-workers studied the PL behaviour of a single Au nanoflower, a highly branched plasmonic nanostructure. It was demonstrated that the PL measurements of such single Au nanoflower revealed some rather more complex features in comparison with simple nanostructures. Such PL properties of the Au nanoflower were strongly dependent on the excitation wavelength and polarization, and they were further studied in situ. The PL experiments and emission measurements comprised a complementary approach to the optical scattering method, and they are targeted to benefit potential applications in domains such as optical imaging and sensing.160 Andersen et al. illustrated the PL wavelength and polarization engineering by exploiting arrayed Au NPs atop a subwavelength-thin dielectric spacer and optically thick Au film, a configuration that supports gap-surface plasmon resonances.161 On the other hand, quantum dots such as metal chalcogenide NPs have widely been studied by PL. For instance, the extinction and photoluminescence of Cu2−xS, Cu2−xSe and Cu2−xTe NPs have been investigated by Feldmann and co-workers and the tunability and control over those properties have been discussed through the active manipulation over their copper deficiency under oxidative/reductive conditions. It was demonstrated that the presence of NIR LSP resonances in these NPs had a crucial effect on the exciton recombination. For example, the PL of Cu2S nanoclusters was quenched during their gradual transformation to non-stoichiometric nanoclusters (x > 0) under an oxidative environment.162 Metal oxides such as ZnO NPs are also photoluminescent. Saliba et al. synthesized zinc oxide NPs in the presence of branched thermotropic liquid crystals. Three emissions were observed for their particles, depending on the excitation wavelength. The origin of such emissions was attributed to several factors, such as surface defects (e.g. oxygen vacancies).163 Another example of nanoscale materials with photoluminescent properties is cesium lead halide perovskites. Protesescu et al. synthesized cesium lead halide nanocrystals using inexpensive commercial precursors and they studied their photoluminescence properties. The colloidal CsPbX3 (X = Cl, Br, I and mixed Cl/Br, Br/I) NPs were bright (quantum yield = 50–90%), stable, and spectrally narrow and had tunable bandgap energies.164
Dynamic light scattering (DLS) is a widely employed technique to find the size of NPs in colloidal suspensions in the nano- and submicrometer ranges. The NPs dispersed in a colloidal solution are in continuous Brownian motion. DLS measures light scattering as a function of time, which combined with the Stokes–Einstein assumption are used to determine the NP hydrodynamic diameter (i.e. diameter of the NP and the solvent molecules that diffuse at the same rate as the colloid) in solution. In DLS, a relatively low NP concentration is needed so that a multiple scattering effect is avoided.165 Lim et al. have reviewed the characterization of NPs by DLS (Fig. 2) focusing in the case of magnetic particles. They present how various factors such as suspension concentration, particle shape, colloidal stability and surface coating of MNPs influence the size value obtained by DLS measurements. A comparison between the results derived from DLS and other techniques, such as TEM and AFM, is performed and the origins for any discrepancies in the sizing, for either small or larger particles, are discussed, while the working size range for each technique is also given.166 For example, for small-sized NPs, the radius of curvature effect is the principal contributing factor for the large difference observed for the diameter measured by TEM and DLS. Middle-sized Fe3O4 NPs capped with oleic acid and oleylamine seem to have size values that show the best match among DLS and TEM measurements. The authors highlight the use of DLS also for the measurement of the colloidal stability of MNPs. Moreover, DLS has been proven useful to monitor the transient behaviours of β-FeOOH nanorods: these structures self-assemble in a side-by-side fashion to form highly oriented 2-D nanorod arrays, eventually leading to the formation of 3-D layered architectures. Overall, the real-time screening of NPs by DLS provides important insights into their aggregation process, since it measures quantitatively the size of the particle clusters formed. The sensitivity of DLS to large particles is crucial for its excellent diagnostic capability to detect aggregation. Nevertheless, the authors denote that careful analysis is required for the best possible interpretation of the DLS results as they are affected by the factors previously mentioned (shape, coating agents, etc.).166 The advantages of DLS include its quick, easy and precise operation for monomodal suspensions and the fact that it is an ensemble measurement method, yielding a good statistical representation of each NP sample. It is highly sensitive and reproducible for monodisperse, homogeneous samples. A limitation of DLS is the necessary conditions for the particles to be in suspension and undergoing Brownian motion. Large particles scatter much more light and even a small number of large particles can obscure the contribution from smaller particles. Therefore, its resolution for polydisperse, heterogeneous samples is rather low. DLS requires transformative calculations with assumptions that must be taken into account when interpreting the data – particularly with polydisperse samples. Although DLS can sometimes measure anisotropic nanostructures, it generally assumes spherical shaped particles.167,168 Overall, DLS measures the hydrodynamic radius accurately but lacks the resolution to detect small aggregates. However, when coupled with differential centrifugal sedimentation (DCS), for example, it can result in valuable information for core–shell NPs, as in the case of those prepared by Minelli and co-workers: when DCS confirms that the samples are not aggregated, the measurements by DLS can be safely considered as accurate.169 Coleman et al. have compared several methods used to obtain information on particle size distributions. For instance, if ∼1% of larger particles exist in a sample, in comparison with the majority of the particles (e.g. two-fold or three-fold larger than the average size of 99% of the particles), DLS is significantly affected, giving higher values than TEM (e.g. 42 nm for a given silica reference sample compared to 25 nm by TEM). Moreover, DCS, apart from its above-mentioned ability to detect agglomerate clusters, is able to characterize samples with broad size distributions.170
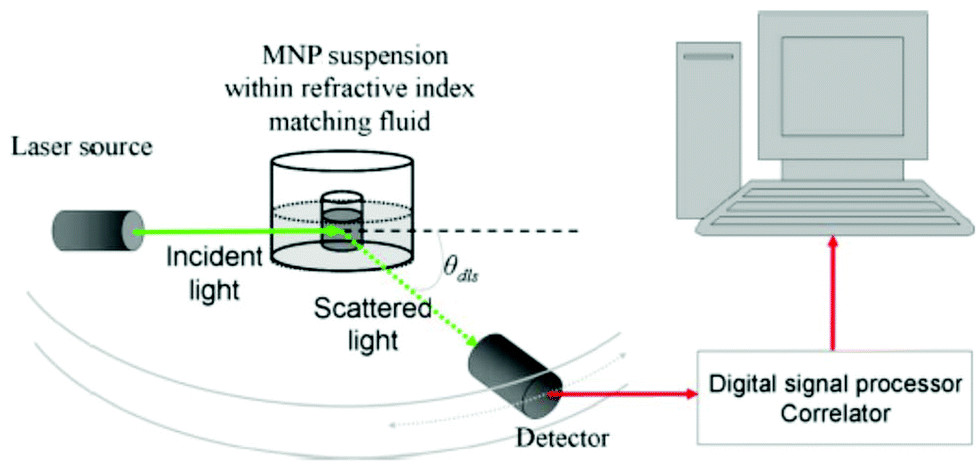 | ||
| Fig. 2 Optical configuration of the typical experimental setup for dynamic light measurements of a nanoparticle suspension. The setup can be operated at multiple angles. Reproduced with permission from ref. 166. Copyright 2013 Springer. | ||
Driskell and co-workers employed DLS to elaborate a fast one-step screening method for the characterization of the specificity of antibody–antigen binding using antibody-conjugated Au NPs. The advantages of DLS detection over the more classic colorimetric technique include better detection limits and higher sensitivity. DLS was used to measure the formation of aggregates produced from virus–antibody binding. The extent of aggregation was employed to assess the interaction between the antibody and the virus. Their novel approach offers an important improvement regarding screening time in comparison with ELISA assays, while giving similarly precise results as the conventional method.171
DLS has also been combined with DOSY- and NOESY-NMR techniques to explore the partitioning behaviour of secondary surfactants added to suspensions of reverse micelles containing either Au or Ag NPs. The critical role of NPs and the surfactant amount on the efficiency of surfactant-assisted NP extraction was investigated. Examples of the surfactants tested were oleylamine, oleic acid and dodecanethiol. The average particle diameters acquired by TEM imaging were lower than those measured by DLS, since the DLS values reflect the outer diameter of the NP-containing AOT reverse micelles together with any related solvent molecules. DLS helped in the monitoring of the irreversible penetration of reverse micelles by specific secondary surfactants.172 Fissan et al. used an aerosol technique, named scanning mobility particle sizer (SMPS), to characterize Au-PVP and Ag-PVP NPs and they compared these results with the ones obtained from techniques such as SEM and DLS. For samples with binary dispersion, DLS failed to provide a correct feedback on the particle size, whereas SEM, SMPS and analytical disk centrifugation (ADC) managed to identify the two different particle size populations. In particular, ADC has a high resolution and can distinguish mixtures if the components cover different size ranges or have distinct densities. ADC is though time-consuming in some cases and it can somewhat underestimate the NP size. Combining SMPS with a nebulizer may result in a method with a higher resolution than ADC.173
Grobelny and co-workers investigated the size and size distribution of polydisperse silver NP colloids using DLS and UV-Vis. Although DLS is more sensitive than UV-Vis, its usual drawback has to do with the difficulty in detecting the presence of smaller NPs; in addition, the UV-Vis spectra did not contain any separate peaks for NPs of different sizes. Therefore, the authors concluded that UV-Vis should not be used for size determination in the case of polydisperse samples. UV-Vis and DLS are low-cost and fast methods, but care is needed when interpreting their results, especially for the aforementioned types of samples, which do not contain a single NP population. Complementary measurements with AFM and TEM/SEM will be certainly needed for polydisperse samples.174 Kestens et al. used numerous techniques (DLS, CLS, SEM, TEM, AFM, and PTA) to measure the size of a ‘standard’ SiO2 nanomaterial sample. Measurements from several researchers working in distinct laboratories were studied. The authors presented the nanomaterial tested as a new reference material with certified values and uncertainties that can be used for assessing the reliability of several particle size analysis methods.175 Murdock et al. characterized a broad range of nanomaterials in solution using DLS and TEM, before assessing their in vitro toxicity. Metal and metal oxide NPs, such as Al, Al2O3, SiO2 and Cu NPs, as well as carbon-based materials such as carbon nanotubes, were tested. DLS measurements showed that depending on the material examined, when the NPs are in solution they do not necessarily retain their nanoscale size.176
Nanoparticle tracking analysis (NTA) is a relatively new, but quickly adopted, technique that can measure NP size, and having a lower concentration detection limit compared to DLS. It utilises the properties of both light scattering and Brownian movement so as to acquire a NP size distribution of samples in liquid dispersion. The details of its operation principle (Fig. 3) and further technical information are provided by Hole et al.177 That paper examined the reproducibility of results acquired by NTA by investigating a wide range of nanoparticle systems and size ranges, in different media. The measurements were performed in 12 distinct laboratories, aiming to obtain a wide database. Examples of the types of nanomaterials tested were Au, SiO2 and polystyrene NPs, dispersed in water or in biological media. An important advantage that NTA offers in comparison with other size measurement techniques is that it is not biased toward larger NPs or aggregates. Furthermore, its confirmed accuracy and reproducibility verified the suitability of NTA to determine the size populations of bimodal samples. The comparison between NTA and DLS was also examined by Jiskoot and colleagues, investigating standard polystyrene beads in the size range of 60–1000 nm.178 Physical mixtures of samples with different NP sizes were also evaluated. It was shown that NTA yielded precise values for the size distribution of both monodisperse and polydisperse samples. The average size values recorded by NTA were slightly smaller and more exact to the nominal ones than those obtained by DLS. Nevertheless, NTA is slower and has a somewhat more difficult operation mode compared to DLS. That study corroborated the above-mentioned findings of other researchers which mention that DLS results are not easily interpreted in the case of polydisperse samples, whereas NTA is able to identify two different sample populations in the same sample.178 Overall, NTA tracks single particles, while DLS studies an ensemble of particles and it is strongly biased to the biggest particles, which are present in the sample. NTA was also studied by Hassellov and co-workers for its capacity to determine the size distributions and concentrations of NPs in liquid samples. Apart from the differences among DLS and NTA, the authors concluded that NTA allows the measurement of large amounts of particles, compared to TEM. Therefore, the statistical confidence is increased and the absence of any particle changes because of the preparation mode of the specimen tested is ensured. Additionally, NTA can potentially use the intensity of light scattered by individual particles to discriminate particles composed of distinct materials within a given size range.179 It is important to note that the sensitivity of NTA is related to the size and composition of the nanomaterials studied. In another report, Ryu et al. prepared CaWO4 and CaMoO4 NPs via the pulsed laser ablation method, and they used several techniques to characterize them, including NTA. The latter technique can dynamically analyse the paths the NPs take under Brownian motion over a suitable time range (e.g. 10–20 s) and visualize deeply sub-micron particles in real time and in a liquid medium. NTA combined with image analysis determined the particle size distribution function of the aforementioned samples. The results for the mean NP size were in accordance with the values derived by TEM and XRD.180
 | ||
| Fig. 3 Schematic of the optical configuration used in NTA. Reprinted with permission from ref. 177. Copyright Springer 2013. | ||
NTA has also been employed to analyse the capping efficiencies of several biomass-derived stabilizers of colloidal Ag suspensions in water. The NTA software identifies and tracks single NPs that undergo Brownian motion and correlates the velocity of the movement with the NP size. For instance, bigger NPs and heavy aggregates move with a slow speed, in comparison with smaller NPs, which have less weight and move faster. It was found that a biorefinery-derived residual syrup acted as an efficient stabilizing agent for silver NPs in solution.181 Another use of NTA, presented by van Leeuwen and co-workers, is the determination of the refractive index which dictates the interaction between light and NPs. Heterogeneous NPs were tested, with sizes <500 nm in suspension, and NTA was capable of discriminating between SiO2 and polystyrene beads on the basis of their different refractive indexes. The authors noted that NTA can overestimate the mean diameter of the beads in comparison with TEM. This was attributed to the uncertainty in the measured diffusion coefficient and to the difference between the hydrodynamic diameter measured by NTA and the physical diameter measured by TEM.182
DCS measures particle size on the basis of their sedimentation rate, which depends upon their size and density. While DLS is a lower resolution analysis method, DCS can be used to detect and resolve peaks down to 2 nm, and differing in size by as little as 2%.183 Minelli and co-workers determined the thickness of immunoglobulin G (IgG) protein on 105 nm polystyrene particles by DCS, DLS and SAXS. While DLS provides precise results for the hydrodynamic size of the particles, comprising their polymeric core and the surrounding protein shell, DCS results are dependent on the density of the particle core and that of the protein shell. On the other hand, as mentioned before, SAXS enables traceable particle size measurements for sufficiently monodisperse particles, and it is a robust tool to identify their size distribution in terms of size and polydispersity, although it relies upon correct modelling for core/shell particles. DCS yielded somewhat larger size than the other two methods. Nevertheless, all techniques showed an increase of the IgG shell thickness with increasing protein concentration during incubation with the NPs, but model refinement was required for their full consistency.184 The same group also published a comparative study of several emerging and established techniques for the characterization of the size of sub-micron particles, evaluating their sizing accuracy and relative resolution. They also demonstrated the variety of the physical principles upon which they are based, aiming to develop a framework in which they can be compared. The particles tested were Stöber silica ones, and it was found that DCS measurements could provide additional information concerning particle porosity that was not accessible to the other techniques. On the other hand, DCS, NTA and SIOS (scanning ion occlusion sensing) were considered to be compact, easy to use and cost-effective. DCS offered a high resolution, which is important for particles with complex structures such as core–shell ones. SMPS had large dynamic range, good resolution and precision. DLS displayed the second highest precision. Shape information could not be provided by SIOS, DCS or NTA, although complementary characterization with TEM could help in this direction.185
Mass spectrometry (MS) has drawn interest as a strong tool for the analytical characterisation of NPs in a reliable way. MS offers invaluable elemental and molecular information on the composition, structure and chemical state of NPs, and their bioconjugation to target biomolecules. Furthermore, it can be used for bioconjugation quantification, as explained by Montoro Bustos et al. in ref. 186. MS is compatible with any type of sample, apart from being a highly sensitive technique. In addition, it is easily coupled with separation techniques to obtain real-time information. In this way, varied and novel insights into the nature of NPs and their final uses and applications can be potentially acquired. Inductively coupled plasma-MS (ICP-MS) is used for the elemental analysis of NPs. It is characterized by robustness, high sensitivity and wide dynamic range, as well as high selectivity and virtual matrix independence. In addition, it is straightforward, usually requiring simple calibration protocols. It allows the reliable quantification and elemental composition characterisation of metallic NPs, and it can determine metallic impurities in non-metallic NPs. Molecular MS techniques, e.g. with electrospray ionisation (ESI) and matrix-assisted laser desorption/ionisation (MALDI), can provide information on the protecting ligands that surround the NPs and also correlate the entire clusters with their chemical composition. Moreover, coupling size-exclusion chromatography with ICP-MS helps to gain information on the size distribution of Au NPs and their elemental characterization. Certain characterization techniques, including capillary electrophoresis, hydrodynamic chromatography, ion mobility spectrometry and field flow fractionation (FFF), also offer useful information about the size and size distribution of NPs. They can be coupled with ICP-MS, for example, FFF-ICP-MS can study the multi-elemental composition and size distribution of natural colloids.
The use of groundbreaking ‘single particle operation mode’ ICP-MS (spICP-MS) has helped to identify the concentration and size distribution of NPs. In that case, highly diluted sample NP suspensions should be used for their characterisation. McLean and colleagues have written a review article on the characterization of thiolate-capped Au NPs by mass spectrometry.187 They reported that apart from characterizing the stabilising ligands and the elemental composition of the NPs, they can also measure the core size and molecular stoichiometry. MS is a formidable tool for elucidating the size distribution of small clusters. It can also observe ligand mixtures with discrete stoichiometry.187 Other techniques, such as NMR spectroscopy, can give population averages, providing only the percentage coverage of different thiolate ligands on an average nanoparticle. For instance, regarding Au NPs, ICP-MS considers the gold core to be of constant mass. This allows the study of the variations in the stoichiometry of distinct ligands on the basis of mass in the following manner: if one characterizes gold NPs containing mixed ligands with ICP-MS, he/she compares ligands of distinct masses and each population of ligands will correspond to a unique mass. This allows the differentiation between the distinct ligands in the cases of NPs capped with more than one ligand.187
ICP-MS can also determine the size distribution and number concentration of NPs in a single, fast analysis. It strongly depends on the matrix of the sample solution. A scheme of the processes involved in the ICP-MS analysis of Au NPs with (A) and without (B) previous Au dissolution is depicted in Fig. 4.188 Regarding its capacity for the size characterisation of Au NPs, Helfrich et al. have published a relevant article.189 They presented an on-line coupling of liquid chromatography or gel electrophoresis with ICP-MS for the size determination and compared the results with other techniques. In particular, they mentioned that DLS is generally expected to give higher values than other techniques because the measured parameter is the hydrodynamic radius of the nanoparticle, but the results obtained by TEM provide information about the diameter of the Au core. Their results illustrated that the performance of on-line GE-ICP-MS is strongly related to the chemical structure of the NP surface composition. Good agreement was found between the different methods used for the size determination of their Au NPs.189 As mentioned before, the possibility to measure the size of Au NPs was also demonstrated using spICP-MS. It has to be noted that for this determination, the chemical composition, density and shape of the NPs are needed to be known. Winchester and co-workers illustrated that precise size measurements by spICP-MS in the range of 20–200 nm can be achieved by operating the ICP-MS instrument in reduced sensitivity modes using a lower extraction voltage, collision cell/KED or higher mass resolution.190 In addition, spICP-MS can detect and quantify the dissolved and nanoparticulate forms of Au at the same time. The detection of Au NPs by the method in discussion is straightforward, but accurate measurement requires careful experimental design and data interpretation. The characterization of complex, polydisperse NP suspension by spICP-MS will require careful experimental design and data interpretation. Pace et al. also used spICP-MS to count and size NPs. They mentioned the above-written advantages of the former method, but they also presented its drawbacks and future challenges. A major hurdle with spICP-MS is the improvement of the size detection limit. For multi-element particles and less ideal systems, spICP-MS may struggle to detect and size particles within the nanoscale range.191 spICP-MS was also employed by Yang and co-workers to analyse Ag and Au NPs in environmental water. The size distribution of these Ag and Au dispersions was in accordance with the TEM results.192
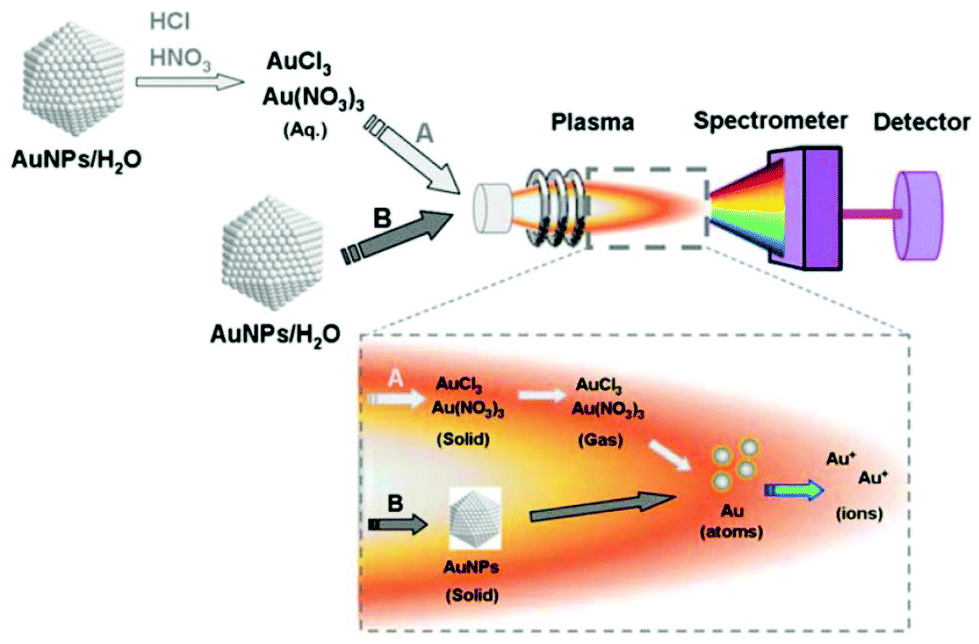 | ||
| Fig. 4 Scheme of the processes involved in the ICP-MS analysis of Au NPs with (A) and without (B) previous gold dissolution. Reprinted with permission from ref. 188. Copyright 2003 Springer. | ||
Arsenic was also determined using ICP-MS by Pereira et al. They detected As(III) and As(V) in environmental and biological samples with the assistance of Cyst-capped thoria (ThO2) NPs. Large amounts of the inorganic As species were successfully removed from polluted water samples.193 In another report, spICP-MS was employed to monitor the detection and characterization of NPs in complex matrices, such as food and biological tissues. NP size, size distribution and particle concentration values were calculated. The size detection limits for four types of NPs studied were 20 nm for Au and Ag NPs, 50 nm for titania NPs and 200 nm for silica NPs. The authors agree with previous reports for the need to combine ICP-MS with separation techniques such as hydrodynamic chromatography and field flow fractionation in order to obtain a more reliable view on the NP features.194 Olesik and Gray have also discussed the use of ICP-MS to calculate the number of particles per litre, for the case of either nanoscale or microscale particles. The main advantages and drawbacks of the method under discussion were the same as mentioned by other researchers. The minimum particle size that can be detected will depend on a number of variables including the sensitivity and the signal due to a dissolved analyte or other continuous signal sources. The minimum quantity of particles per litre of the suspension that is required for detection depends on the equivalent volume of suspension liquid delivered to the ICP in the total measurement time.195
Besides, thiol ligand density was quantified at self-assembled monolayers on Au NPs by ICP-MS. Gold and sulfur concentrations could be determined simultaneously by ICP-MS, and were obtained as ensemble averages of the particle distributions, as shown by Lammerhofer and colleagues.196 The surface coverage of Au NPs was studied quantitatively based on the linear relationship of the gold/sulfur (Au/S) ratio measured by ICP-MS, and the Au NP size measured by TEM. Their method proved to be a valuable tool for the quantification of ligand densities on the surface of Au NPs. spICP-MS was also employed combined with tissue extraction for the quantification and characterization of PVP-capped Au and Ag NPs in environmentally relevant biological tissues.197 The authors described a size detection limit of 20 nm for these Ag and Au NPs, but they noted that this value depends on instrument sensitivity and the ionic background for the metal of interest.
spICP-MS was also employed to characterize TiO2 and Au NPs during water purification, in addition to the Ag NPs. Parameters such as the NP concentration, size, size distribution and dissolved metal element concentration in surface water as well as in purified water were evaluated. Understanding the fate of Ti, Ag and Au during real potable water treatment processes is important since human exposure to these NPs will eventually occur by drinking water. Donovan and co-workers found that lime softening followed by alum coagulation in combination with powdered activated carbon adsorption resulted in the complete removal of Au and Ag NPs and almost complete removal of TiO2 NPs.198 The presence of titania NPs was also investigated in sunscreens, using spICP-MS. The aforementioned parameters were studied (size, size distribution and NP concentration), and the developed method was considered of high throughput, reproducible, low-cost and sensitive.199 The method under discussion has also been applied to detect lanthanide metals doped into the iron cores of superparamagnetic iron oxide NPs in tissue and blood samples. With spICP-MS, more than 10 different NP formulations with distinct physicochemical properties could be directly analysed at the same time. As a proof of concept, their approach was used to study the influence of NP size and surface charge on tumor delivery, biodistribution and blood clearance in vivo.200
Secondary ion mass spectrometry (SIMS) is a mass spectral technique which can be used to obtain molecular chemical information from NPs. It is a surface analysis technique where primary ions, which can be atomic or polyatomic, are used to sputter positively and negatively charged secondary ions. The secondary ions (SIs) originate from the outmost nanometer of the sample.201 SIMS is in particular suitable for the analysis of NPs by virtue of detection sensitivity and lateral (∼100 nm) and depth (∼1 nm) resolution. It is worth mentioning that the secondary ion signature of NPs may be distinct in comparison with the one of bulk materials having the same composition. However, it is necessary to have a well-working methodology to deconvolute the analytical results.202 Blanc et al. used SIMS to analyse the composition of dielectric NPs localized in a silica glass matrix in the core of optical fibers. They performed SIMS imaging at high spatial resolution (NanoSIMS 50L) and their goal was to gain more understanding on the spectroscopic properties of the luminescent ions in these fibers. The authors mentioned that in SIMS the depth resolution is much better than the lateral resolution, which is related to the size of the probe. The partitioning of P, Mg and Er into phase-separated zones was demonstrated, and this indicated that the particle composition was related to the Mg concentration.203 Schweikert and co-workers noted that nano-objects of ‘subcritical assay dimension’ have a SIMS signature that is specific to their physical and chemical features and their environment. A question that arises is how the SIMS response would be influenced in the case of a single layer of NPs with varied composition. The researchers presented an investigation of a single layer of a mixture of Ag and Au NPs. Cluster SIMS was employed to study individual NPs.204
Time of flight secondary ion mass spectrometry (ToF-SIMS) is a material characterisation technique that possesses high chemical sensitivity, high surface sensitivity (upper 2–3 nm probed) and molecular specificity. This method can analyse the nanoparticle drug delivery formulations.205 In fact, ToF-SIMS is extensively used to characterize the nano-zones of larger components, such as electronic devices and thin to ultrathin films of either organic or inorganic nature. The technique under discussion is also utile for the investigation of the surface coating or functional groups of NPs, for example, to analyse peptides coupled to Au NPs and multilayer plasma-deposited organic coatings on Al2O3 NPs. Laus and colleagues noted that SIMS can be destructive while conducting the analysis. Even though the ion dose maximum limit can be adjusted to tackle the molecule destruction issue, the NPs tested may still undergo melting. These authors used SIMS for the depth profiling of certain types of NPs (Au–SiO2 and Ag–SiO2 configurations) and they investigated the depth profiles for melting issues, combining their SIMS study with additional characterization by SEM imaging. In all cases, the interpretation of the SIMS depth profiles illustrated that melting took place, although it is possible that with ultralow energy Cs+ this effect was limited to its minimum.206
Fig. 5 shows a scheme which explains how ToF-SIMS is used to probe NPs.207 NPs are adsorbed on a surface; the bombardment of the primary ions results in the desorption of molecules (NPs or NP conjugates), which then results in the emission of secondary ions from the outermost 1–1.5 nm molecular layers. The secondary ions are fragments of adsorbed molecules: metallic NPs have high secondary ion yields, whereas organic NPs yield chemical-specific fragments that help to determine the surface ligands. Kim et al. mention that when ToF-SIMS is combined with several NP-based signal enhancing strategies, it can probe the functionalization of NPs as well as their locations and interactions in biological systems. NP-based SIMS is important for label-free drug screening because signal-enhancing NPs can be designed to directly measure the enzyme activity. It can also be employed to monitor ligand-exchange processes. The benefit of ToF-SIMS, compared to MALDI-MS (matrix-assisted laser desorption/ionization), is the straightforward analysis of targets without any matrix use. Therefore, ToF-SIMS provides molecular information about functional groups, molecular orientation and conformation as well as denatured species from chemicals and/or from biomolecules. It can also be used to gain information on the core composition of NPs, apart from their surface. The types of NPs usually probed by ToF-SIMS are popular in domains such as biosensing and bio-imaging. Nevertheless, the spatial resolution of ToF-SIMS is limited to only hundreds of nanometers and ToF-SIMS is not particularly sensitive to high mass fragments. For a higher sensitivity and higher spatial resolution for the ability to detect metals in organic matrices, ToF-SIMS can be coupled with laser secondary neutral mass spectrometry (laser-SNMS). High-resolution NanoSIMS can provide monoatomic and diatomic secondary ions with a better sensitivity and spatial resolution than ToF-SIMS.207 Rafati et al. used ToF-SIMS to investigate polymer microspheres for the controlled release of a therapeutic protein from an implantable scaffold. The ability of ToF-SIMS imaging to spatially image the polyvinyl alcohol (PVA) surfactant and protein adsorbed onto the surface of the microspheres was shown for the first time.208 The surfactant layer had a thickness of about 4 nm and it could be readily removed under sputtering with C60, as also confirmed by AFM measurements. Indeed, AFM can act as a complementary technique to ToF-SIMS providing nanometer spatial resolution of the surface topography. Both techniques were able to chemically and physically visualize correspondingly the integrity and pattern of the surfactant across the surface of the NPs. Their work is a good example of what ToF-SIMS imaging can offer, such as the spatial location of the protein, the surfactant and the polymer substrate. Confocal Raman spectroscopy can also be combined with ToF-SIMS to study the bulk distribution of the protein within the microparticles.208 Wiesmann and co-workers also employed ToF-SIMS to detect protein coatings on NP surfaces by ToF-SIMS and advanced electron microscopy techniques. In addition to its other characteristics, this technique can detect all isotopes and offers a simultaneous imaging of the surface distribution of detected molecules and elements. The thicknesses of the different protein coatings of collagen (two different collagen types) were measured by TEM. ToF-SIMS permitted one to distinguish and identify the masses of typical amino acids of the two protein matrixes.209
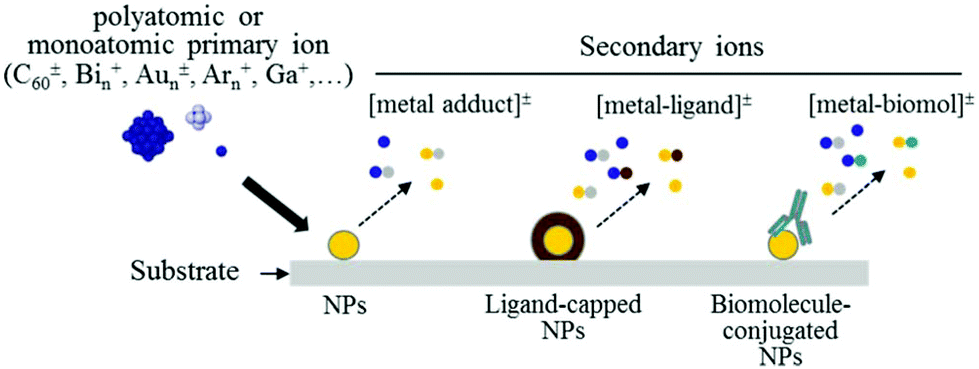 | ||
| Fig. 5 Scheme of probing NPs (NPs) by using ToF-SIMS. Polyatomic or monoatomic bombardment on the surface generates different types of secondary ions from metal NPs that can be encapsulated or conjugated with ligands or biomolecules. Reproduced with permission from ref. 207. Copyright 2015 Wiley-VCH. | ||
Cowin and colleagues employed ToF-SIMS and SEM for an in situ study of 5 nm goat anti-mouse IgG Au NPs in a novel portable vacuum compatible microfluidic device. Characteristic signals of the conjugated Au NPs were successfully spotted through the aperture by EDX in SEM and ToF-SIMS.210 In another report, ToF-SIMS and XPS were used together to study the aging of plasma-mediated coatings with embedded Ag NPs on stainless steel. The variation of film composition (silver release, matrix composition, and thickness) with immersion time in saline solution was analysed. Coating modifications, caused by immersion, were found to depend on the starting Ag content.211
Lee et al. employed and validated an approach combining ToF-SIMS and a confocal laser scanning microscopy (CLSM) imaging method for the cytotoxicity study of ZnO NPs in HaCaT cells. Several compositional and toxicological analysis methods were applied to evaluate the size, shape and other features of the ZnO NPs. Furthermore, their dissolution behaviour and effect on HaCaT cell viability in the presence of various concentrations in water was also studied. Comparative and correlative analyses of the above-mentioned results with ToF-SIMS and CLSM imaging demonstrated a reasonable and acceptable outcome and allowed the consideration of this approach as reliable, quick and sensitive.212 Niehuis and co-workers used ToF-SIMS to study the effect of primary ion parameters (species and energy) on a model system (HfO2 on Si) as well as on Lumidot core–shell NPs. It was indicated that the energy values used in ToF-SIMS caused the melting-up or evaporation of NPs after direct or grazing impact of primary ions. Therefore, although atomic layer-deposited films of HfO2 on Si were well suited for studies on the information depth of ToF-SIMS, experiments on Lumidot NPs implied that the information gained using ALD references cannot be easily transferred to NPs.213
Moreover, with mass spectrometry techniques, the sample needs to undergo ionization and subsequent sorting based on the mass to charge ratio in magnetic and electric fields. The desorption and ionization process can be assisted by ablation with a high energy laser (matrix assisted laser desorption/ionization, MALDI) or a salvo of inert gases (fast atom bombardment). MALDI-ToF MS can characterize very small NPs as it can quantify many particles at a time leading to an improved estimate of dispersity. The size range of the particles that can be analysed is very large and highly sensitive. MALDI-ToF was successfully employed by Hyeon and co-workers to estimate the NP size of spherical Ag NPs in 9-nitroanthracene. The size values matched well with the ones measured by TEM. It was shown that the method under discussion can be used as a generic methodology to estimate with high precision the size and size distribution of NPs with several shapes and sizes.214 MALDI-ToF was also employed to characterize colloidal Pt NPs prepared by Navin et al. The particles analysed were in the 1–4 nm size range and they were stabilized by PVP. Particle sizes determined from mass spectra were found to be in good accordance with those derived from TEM and XRD experiments.215 Zhang et al. used high-performance liquid chromatography coupled with mass spectrometry for the analysis of ultrasmall Pd NPs. Reverse-phase HPLC is expected to offer more accurate determinations of the catalytic, electronic, optical and toxicological properties of metal NPs. Among several separation techniques, HPLC can be considered as an effective approach to isolate different metal NP species. The authors employed RP-HPLC to separate and analyse for the first time water-soluble DMF-Pd NPs. The measurements by MALDI-ToF MS were in agreement with the chemical compositions of the fractions. The aforementioned technique is the most popular MS technique in determining the number of metal atoms of NP fractions. It is further anticipated that RP-HPLC combined with MS can be applied to investigate the growth mechanism of Pd NPs.216
Resonant mass measurement microelectro-mechanical system (RMM-MEMS) is a technique used to detect and count sub-visible and sub-micron particles in a material, and to measure their size and mass and the distributions of these properties. A micro electro-mechanical system (MEMS) sensor, containing a resonating cantilever with a microfluidic channel embedded in its surface, is employed. When a particle with a size between 50 nm–5 μm flows through the fluidic channel, it alters the resonating frequency of the cantilever, which indicates the buoyant mass, and also the dry mass and size of the particle. The information on sample concentration, viscosity, density and volume can also be obtained by the sensor.217 Voevodin and colleagues synthesized Au/Pd bimetallic NPs with a biotemplated approach and deposited them on Au MEMS switch contacts as a NP-based lubricant. The authors of that study noted that since the melting point of NPs is generally lower than that of the bulk materials, NP size and size distribution are important factors for using NPs as MEMS switch lubricants. The bimetallic NPs synthesized by these authors were found to be excellent candidates as surface modifiers/lubricants for MEMS switch lubricants.218
Zeta potential (ζ-potential). The ζ-potential of a sample is a key indicator of the stability of colloidal dispersions. Highly positively or negatively charged particles tend to repel each other, thus forming stable colloidal solutions which show only minor trends to agglomerate. Such highly charged particles are related to pH values which are far from the so-called ‘isoelectric point’ of a solution which refers to the pH value at which the zeta potential is zero. On the other hand, a low value for the ζ-potential of a colloidal NP dispersion causes the flocculation of the colloids and it corresponds to values closer to the isoelectric point of the system. In general, colloids with values for the ζ-potential in the range of ±20–30 mV or higher are considered stable. This property can be tuned through the modification of the surface chemistry, so the stabilisation of the colloidal suspension is obtained via electrostatic repulsion. The ζ-potential is influenced by the concentration of the suspension and composition of the solvent and other additives. Since DLS can also provide indications on the aggregation tendency of a sol, it can be combined with ζ-potential measurements for a more complete characterization.219 Branda et al. employed DLS and ζ-potential studies (which in fact can be carried out in the same device with modern instruments) to analyse the influence of the exposure to growth media on the size and surface charge of silica-based Stöber NPs. These techniques appeared to be valuable tools to investigate the fate of NPs in biological environments. Compared to TEM and SEM, the above-mentioned techniques offer the benefit that the NPs are not exposed to the risk of clustering during sample preparation because of solvent evaporation.220 Dobson and colleagues synthesized and characterized ultra-small superparamagnetic iron oxide NPs thinly coated with SiO2. The authors noted that characterizing the NP surface properties was important for the understanding of properties under physiological conditions and optimizing the conjugation chemistry. Surface charge was characterized by ζ-potential analysis. Acid washes using HNO3 reversed the ζ-potential of the Fe3O4 colloid and removed any remaining ammonium ions, but also caused the material to release Fe2+, converting magnetite to maghemite, with no reduction in particle size.221
pH. The pH is another property frequently measured in colloidal NP solutions. Aroca and co-workers tailored the size and shape of Au NPs in fulvic acid colloidal solution by modifying the pH and concentration of the acid. The reasoning behind the ability to vary the acquired morphology came from the fact that a different pH affected the reaction kinetics.222 The reversible aggregation of Au NPs was induced by pH-dependent modifications in a self-assembled monolayer of disulfide modified poly(L-glutamic acid). The change in the aggregation behaviour with pH took place within minutes and in a narrow range of pH from 4.5 to 5.5.223 In another report, Cappellari et al. synthesized ultrasmall cysteine-coated Au NPs by pH switching of the Au(I)–cysteine polymer. By characterizing their products with several techniques such as XANES and EXAFS, the authors concluded that the pH affected not only the charge state of the polymer, but it also caused a modification in the oxidation state of the metallic centers. The size of the NPs was controlled by the pH value and ultrasmall sizes (∼0.6 nm) appeared for a 4–9 pH range.224 Qin and co-workers synthesized Au NPs by a biosynthetic approach: the products had a tunable shape by simply changing the pH of the reaction solution at room temperature. The structural configuration of moss protein could be induced by pH solutions.225 Hamlett et al. published a study on the pH-dependent adsorption of Au NPs on chemically modified Si3N4 MEMS devices. The maximum adsorption of citrate-passivated Au NPs took place at pH = 5, in agreement with AFM and XPS experiments. The mass adsorption experiments were performed using amino-functionalised Si3N4 ‘flap’ resonators.226 The pH values can also affect the toxicity of nanomaterials, as in the case of Ag NPs reported by Oukarroum et al. The size distribution of their particles depended on the pH of the culture medium. The Ag NP toxicity on the green alga Chlamydomonas acidophila was pH-dependent as shown by the cytotoxicity mediated through the induction of oxidative stress.227
Pavlopoulou et al. monitored the synthesis of Pt NPs using pH-responsive microgel particles. SAXS was employed to study the structure of pH-responsive microgels before and after metal incorporation. The decrease in the microgel radius together with an increase of the fractal dimension f when increasing the solution pH confirmed the pH-responsive character of the microgels. These tertiary amine-based microgels were used as nonreactors for the preparation of Pt NPs.228 Bradu and colleagues published an article on the influence of pH on the catalytic activity and selectivity of Pd–Cu NPs supported on titania in the nitrate reduction reaction. The presence of titania endowed an increased catalytic activity of the nanomaterials studied.229 Gwak et al. studied the physico-chemical changes of ZnO NPs with different sizes and surface chemistries under physiological pH conditions. The ZnO NPs were found to enhance the pH under the physiological pH conditions to a neutral (in the case of the gastric conditions) or basic range (in the case of the intestinal and plasma conditions), showing a dependency on the size and surface chemistry.230 In another report, samarium oxide NPs were synthesized by Yousefi and co-workers through a cathodic electrodeposition approach. The effect of the pH on the morphology of the NPs was studied. With the increase of pH, parameters such as the weight, density and adhesion of the deposit on the electrode were decreased remarkably.231 Engelbrekt et al. synthesized selectively Cu2(OH)3Cl and tenorite CuO NPs with a one-pot protocol and the obtained product was tuned according to the solution pH. In particular, acidic pH values prohibited the formation of NPs, and neutral pH resulted in Cu2(OH)3Cl, whereas CuO NPs were generated in a basic pH environment. The NP morphology was also tuned by controlling the pH.232 Finally, the influence of pH and calcination temperature on the structural and optical properties of Al2O3 NPs was studied by Amirsalari and Shayesteh. It was evidenced that the alumina particles had an optical direct bandgap and the energy gap decreased with increasing calcination temperature and pH of the reaction. The crystalline size of NPs increased according to the pH of the solution.233
Electrophoretic mobility (EPM) is measured to evaluate the surface charge of nanomaterials. The aggregation and disaggregation of iron oxide NPs in relation to NP concentration, pH and natural organic matter were reported.234 Low EPM values were associated with the formation of large aggregates, whereas very high EPM values were observed in the case of very stable NPs for a prolonged time. In another report, DLS and electrophoretic mobility measurements were used to monitor the evolution of silica colloid to silica colloid–polyelectrolyte–iron oxide composites.235 Au NPs, prepared by Merga and co-workers upon the reduction of Au2O3 by H2, were characterized by several techniques, including EPM. Conductivity measurements showed that most of the unreduced Au ions are in solution, but a small fraction resides on the particle. EPM measurements help to obtain the ζ-potential values.236 In fact, Minelli and co-workers compared several techniques in a systematic way for the determination of the ζ-potential of silica NPs in a biological medium. The ζ-potential is directly related to the electrophoretic mobility through the Henry equation and the Smoluchowski or Huckel models. The authors used one ensemble and two particle-by-particle techniques: electrophoretic light scattering (ELS), tunable resistive pulse sensing (TRPS) and zeta particle tracking analysis (z-PTA). Despite differences between the basic measurement principles of the three methods, the results were overall in good agreement.237 Luminescent Au NPs decorated with bifunctional ligands possessing thiol and carboxylic acid functional groups were characterized by electrophoresis, which revealed a monodisperse distribution of NPs. It was suggested that the mercaptoalkanoic acid ligand used to form a Au–S charge transfer complex behaves as a pH-responsive collapsible molecular brush at the surface of the Au NPs.238
Gel permeation chromatography (GPC), also known as size exclusion chromatography, is a highly valuable tool that separates molecules based on their hydrodynamic volume or size. With advanced detection systems coupled to GPC, information about polymers, such as molecular weight (Mw) distribution, average molecular mass, and degree of branching, can be acquired.239 Tadros and colleagues characterized the adsorption of poly(hydroxystearic acid) to TiO2 NPs using GPC. The latter technique was able to resolve and quantify the non-adsorbed molecules by size.240 In another work, GPC was used, together with FTIR and NMR, to characterize a series of succinate linearly linked PLGA-PEG-SA-PEG-PLGA multiblock copolymers which were conjugated with Au NPs. GPC helped to determine the average Mw and Mw distribution of the copolymer samples.241
Differential scanning calorimetry (DSC) is a thermoanalytical technique in which the difference in the amount of heat required to increase the temperature of a sample and a reference is measured. Badia et al. used DSC to detect the phase transitions of C18SH-derivatized Au NPs. These phase transitions could be associated with the reversible disordering of the alkyl chains. Actually, SS NMR measurements show that the chain melting arose from an increased frequency of gauche bonds in the Au-tethered alkanethiol chains. FTIR spectroscopy established that the chain melting starts at the chain terminus and propagates toward the middle of the chain with increasing temperature.242 The melting behaviour of Pb and Sn3.5Ag NPs has also been investigated by DSC studies.243,244 The latter technique has also been used to measure the specific heat capacity and thermal conductivity of PEEK/Ag NPs.245
Inductively coupled plasma optical emission spectrometry (ICP-OES) is a highly sensitive technique that can characterize the core NPs and also their coating ligands. It can reach trace-level concentrations, small changes in concentration can be identified, and multiple elements can be detected at the same time. Therefore, it can provide information on surface species conjugated on Au NPs and quantify the ligand packing density.246 In addition, ICP-OES offers a wide dynamic linear range and it is well reproducible. Magnetic solid phase extraction (MSPE) combined with ICP-OES has been used to identify chromium ions in environmental water samples.247 In addition, trace amounts of Cr, Cu and Pb can also be spotted by the combination of the aforementioned techniques.248
Electrospray differential mobility analysis (ES-DMA) is a rapid technique (analysis timescales on the order of 1–100 min) with sub-nanometer resolution. It can determine the NP concentration, and it is a quick, low-cost technique, with statistically significant results; however it does not offer the atomic-scale resolution of other techniques such as SANS or X-ray crystallography. The size values derived by ES-DMA can match the ones derived from electron microscopy and light scattering techniques.249 A technique belonging to the latter type is elliptically polarized light scattering (EPLS), which is accurate, fast, and non-intrusive and allows in situ function. It can provide information on the size, size distribution, shape and structure of agglomerates.250 Moreover, the thermal lens spectrometry (TLS) technique can be employed to measure the thermal diffusivity of NP solutions, e.g. in the case of 15 nm Au NPs at different pH values at constant NP size and concentration. It provides a reliable alternative to evaluate, with high sensitivity, the thermal diffusivities of semitransparent materials as well as low thermal diffusivities.251
Quartz crystal microbalance (QCM). Compared to ICP and micro-computerized tomography, QCM can be used for the mass measurement of NPs, and it offers the advantages of real-time monitoring, greater sensitivity and lower cost.252 Burg et al. described the use of suspended microchannel resonators as a means to weigh single NPs, single bacterial cells and sub-monolayers of adsorbed proteins in water with sub-femtogram resolution (1 Hz bandwidth).253 In another work, Link and co-workers have shown in a review article the utility of single particle spectroscopy for the characterization of plasmonic NPs with arbitrary size and shape, especially when combined with correlated electron imaging and detailed electromagnetic calculations. They present single nanoparticle spectroscopy performed with several scattering, absorption and extinction methods.254
2.3 Characterization methods for magnetic nanostructures
Magnetic NPs find applications in a broad range of domains, such as magnetic resonance contrast media and as therapeutic agents in cancer treatment. Akbarzadeh et al. have written a review paper on the preparation and physical properties of magnetic NPs as well as their applications, with emphasis on the biomedical ones.255 In this section we focus on the characterization techniques that are employed to evaluate the magnetic properties of such NPs.Superconducting quantum interference device magnetometry (SQUID) is a tool for measuring the magnetic properties of nanoscale materials. Nanomaterials in particular exhibit different properties to those in the bulk state due to their small size and sensitivity to local conditions. As a material decreases in size, it progresses from multi-domain, to single domain and finally to superparamagnetic status. Typical SQUID measurements yield properties such as the magnetization saturation (MS), magnetization remanence (MR) and blocking temperature (TB).88 Apart from NPs, the magnetic response of individual molecules can also be measured by SQUID. In fact, a scanning magnetic microscope including a nanoSQUID has also been developed recently, fabricated on the apex of a sharp quartz. NanoSQUID is considered as a highly promising probe for nanoscale magnetic imaging and spectroscopy. A nanoSQUID sensor requires deep sub-micron Josephson junctions, which are provided by two Dayem nano-bridges (nano-constriction of a superconducting film), fabricated by electron beam lithography or focused ion beam (FIB) with a length and width comparable to the coherence length. The main requirement for a SQUID designed for the detection of magnetic NPs is a very small SQUID area. Ideally, to gain the best coupling factor, the loop size should be comparable to those of the NPs directly coupled to it.256 Regarding the magnetic resonance force microscopy or magneto-optic spin detection, nanoSQUIDs offer the advantage of direct measurement of magnetization changes in small spin systems. The Dayem nanobridges of a nanoSQUID, apart from their easy fabrication by a single nanopatterning step, are also resilient to the magnetic field applied in the plane of the SQUID loop.257 The experimental setup of a nanoSQUID is shown in Fig. 6.
 | ||
| Fig. 6 Scheme of the experimental setup for the NP magnetization measurements. The variation of the critical current is obtained by averaging the switching current events measured by using a time of flight technique. The resolution of the critical current measurements is about 1 part in 104. The feedback circuit allows the increase of the linear dynamic range of the sensor. The picture shows the holder including the sample and the multiturn feedback coil. Reprinted with permission from ref. 257. Copyright 2013 Springer. | ||
Fiorani and co-workers demonstrated that the latter type of SQUID device is a useful and reliable tool to investigate the magnetic properties of iron oxide NPs.257 Gamarra et al. used SQUID magnetometry and ferromagnetic resonance (FMR) to carry out static and dynamic measurements of a biocompatible ferrofluid based on Fe3O4 NPs. Such measurements were performed as a function of field, temperature and driving frequency. Their magnetization results as a function of the external field showed that for temperatures above TB the hysteresis cycle did not exhibit coercivity, indicating the superparamagnetic behaviour of the material.258 In another report, nickel ferrite NPs synthesized by Malik et al. were investigated by SQUID and Mössbauer. The effect of size on the coercivity and saturation magnetization as derived from the hysteresis loop and the hyperfine parameters obtained from the Mössbauer spectra were reported. The bimodal size distribution was reflected only in the zero-field-cooled–field-cooled (ZFC-FC) measurement done at a very low field, which is also borne out by numerical calculations.259
Vibrating sample magnetometry (VSM) is another method that can be used to record the M–H loops for magnetic nanomaterials and obtain parameters such as MS and MR. The magnetic properties of NPs are studied as a function of magnetic field, temperature and time. The FeCo NPs examined the magnetic properties of superparamagnetic FeCo@SnO2 NPs on graphene–polyaniline. Their enhanced electromagnetic wave absorption properties were investigated. A series of FeCo, FeCo@SnO2 and FeCo@SnO2@graphene@PANI composites were characterized by VSM. The FeCo NPs display strong magnetic dipolar interactions and if an external magnetic field is applied, their magnetic moments would be aligned in the same direction with the field.260
Fabris and colleagues prepared size-controlled magnetite NPs through a direct reduction–precipitation method in the presence of tetramethylammonium hydroxide. All studied samples were found to be superparamagnetic, as evidenced by both zero coercivity and zero remanence on the magnetization loop. The saturation magnetization was a linear function of the NP size.261 Kumari et al. described the concept of first order reversal curves, which is an assembly of partial hysteresis loops originating from the major loop. First order reversal curves (FORC) are efficient for the identification of domain size, composition and interaction in a magnetic system. It is a significant method to obtain a semi-quantitative measure of the effective magnetic particle size. Under certain conditions, FORC may facilitate the revealing of the presence of secondary/minor magnetic contributions, thus helping in a more precise characterization of the magnetic properties. VSM magnetometry was used to obtain such FORC measurements.262
In another work, FeCo NPs with anisotropic long chain structure were prepared by a sputter based gas-condensation method and their magnetic properties were analysed by VSM. A strong exchange coupling interaction between NPs was evidenced in a chain-like sample, while well-dispersed samples showed a distinct magnetic performance. ZFC/FC curves and time dependent remanent magnetic moment measurements helped to gain information on the thermal stability of the studied NPs.263 Apart from the full hysteresis loop properties of the magnetic media, there has been increased interest in the measurement of remanence curves. The measurement of remanence determines only the irreversible component of magnetization and therefore enables the phenomena of switching to be deconvoluted from the hysteresis measurement, which in general includes a reversible component. Two main remanence curves exist: the isothermal remanence and the DC demagnetization curve. The former is measured after the application and removal of a field with the sample initially demagnetized. The DC demagnetization curve is measured after the saturated state by the application of increasing demagnetizing fields. These remanence curves can be obtained by VSM measurements and they can provide the true switching field distribution of the materials.264
Mössbauer spectroscopy is a valuable analytical tool that is based on the recoil-free resonance fluorescence of γ-photons in matter with Mössbauer-active elements, such as Fe. Mössbauer can be used to evaluate the oxidation state, the symmetry and spin state as well as the magnetic ordering of the Fe atoms in a NP sample and thus identify the magnetic phases in a sample. Furthermore, for magnetically ordered materials, Mössbauer spectra recorded as a function of temperature can be used to estimate the magnetic anisotropy energy and quantify the thermal unblocking (superparamagnetism).265 The Mössbauer spectroscopy isomer shift is an important parameter that arises from the nuclear-energy shift that is caused by the coulombic interaction between the nucleus and the electron density at the site of the nucleus. The isomer shift values of Fe2+ and Fe3+ are significantly distinct from each other and Mössbauer spectroscopy has been generally accepted as the method of choice to determine the oxidation number. In the case of doped Fe–ZnO NPs, whilst the Mössbauer isomer shift is related to the charge on the ion in the structure, there is not necessarily a correlation with its oxidation state. The Mössbauer isomer shift cannot act as a necessary determinant of the oxidation number of dopant atoms. In fact, Mössbauer spectroscopy directly probes the charge on the nucleus site. Evidently, this charge is sensitive to the local environment of the atom, both structurally and chemically.266 A schematic diagram of a transmission Mössbauer spectrometer system is depicted in Fig. 7. Oh et al. investigated the magnetic properties of FeCo NPs synthesized by the chemical vapor condensation process and it was found by Mössbauer that the NPs had α-FeOOH, γ-FeOOH and Fe3O4 at their surface. With the complete fit of Mössbauer spectra, the fraction of each phase was quantitatively determined.267 Lange and co-workers mentioned that Mössbauer spectroscopy utilizes hyperfine interactions between nuclei and their surrounding environment. Thus, this method is very sensitive to the surroundings of a given isotope used as a probe. The 57Fe Mössbauer effect can yield information concerning the local chemical and structural environments around the Fe nuclei, permitting the determination of Fe-containing phases as well as the quantitative analysis of their relative proportions. Fitting of the Mössbauer spectra can help to identify parameters such as the magnetic hyperfine field (Hhf) and isomer shift for six-line spectral components, electric quadrupole splitting and isomer shift for quadrupole doublets, and isomer shift for single lines.268
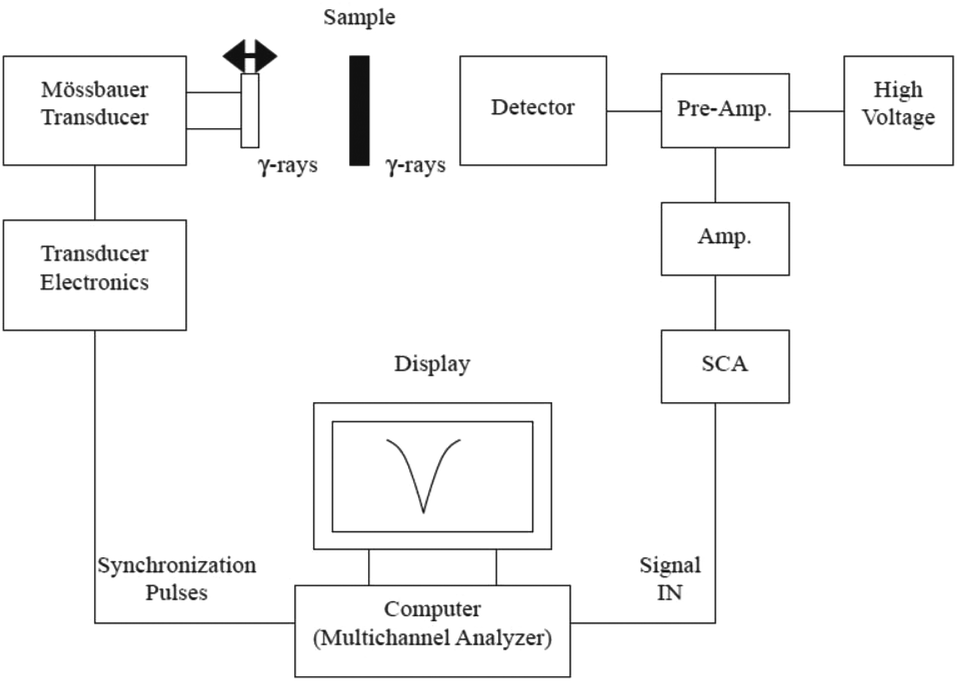 | ||
| Fig. 7 Schematic diagram of a transmission Mössbauer spectrometer system. Reprinted with permission from ref. 267. Copyright 2004 Elsevier. | ||
Tiano et al. employed Mössbauer spectroscopy to probe the nature of metal cation occupancies in MFe2O4 systems (M = Mg, Fe, Co, Ni, Cu or Zn). SQUID and Mössbauer measurements helped to systematically probe their ferrite NPs in an attempt to correlate the magnetic properties with the NP size and composition. Superparamagnetism was found in particles with sizes smaller than 4 nm, whereas the presence of spin canting, uncompensated surface spins and magnetic anisotropy was observed for the majority of the samples. Mössbauer analysis supported the SQUID data, showing that the occupancies of the tetrahedral Fe(A) and octahedral Fe[B] sites were significantly modified, thereby emphasizing the importance of the synthetic method, size and chemical composition.269 In another work, Pankhurst and co-workers used 57Fe Mössbauer spectroscopy to find the composition of magnetite/maghemite mixtures and the stoichiometry of magnetite/maghemite solid solutions. They presented the data on high-purity magnetite and maghemite powders and mixtures thereof, as well as the comparison literature data from nanoparticulate mixtures and solid solutions to demonstrate that there is a linear correlation between the ‘centre of gravity’ parameter δRT (also known as area weighted mean isomer shift at room temperature) and the numerical proportion of iron atoms in the magnetite environment. It has to be noted that XRD cannot distinguish between Fe3O4 and γ-Fe2O3, as their reflections coincide, rendering Mössbauer a successful alternative in this case.270 Still, it has been reported that the Mössbauer spectra of NPs are much more complex compared to the ones of the bulk state.271 Sharma and colleagues synthesized iron oxide NPs by the thermal decomposition of Fe-precursors in Ar and vacuum environments with controlled size distribution and phase composition. Detailed XRD, XANES and Mössbauer experiments demonstrated that the prevailing chemical phase was γ-Fe2O3 in both environments. 57Fe Mössbauer spectroscopy is a powerful tool to characterize iron oxide NPs undergoing superparamagnetic relaxation.272 Rumenapp et al. monitored the aging of magnetite NPs using Mössbauer spectroscopy. The measurements were performed at 4.2 K in order to identify the oxidation state of the iron in the core of the NPs. In Mössbauer spectra, Fe(II) and Fe(III) can be easily distinguished by their different isomer shifts. The authors noticed that the magnetite content of naked magnetic NPs with sizes below about 10 nm decreased rather rapidly after synthesis and use of hydrous solutions or drying in air. However, diethylene glycol provided a resistance to the oxidation of magnetite to maghemite.273 Sundar and co-workers investigated the local structure and magnetic properties of cubic iron oxide NPs formed in zeolite, with the use of Mössbauer spectroscopy. This method was employed to distinguish between the isolated superparamagnetic NPs of iron oxides. The Mössbauer study revealed a strong binding of Fe3O4 NPs in zeolite.274 In another report, Mössbauer measurements helped to study the disordered surface spins in core/shell ferrite NPs. The NPs tested had a nickel ferrite core and a maghemite shell. Their experiments showed that the magnetization temperature dependence of gas-like diluted dispersions of independent NPs is well described by a monodomain ordered core and a surface layer of disordered spins.275
Domracheva et al. performed magnetic resonance and Mössbauer studies on superparamagnetic γ-Fe2O3 NPs encapsulated into liquid-crystalline poly(propylene imine) dendrimers. Mössbauer measurements showed that these NPs were composed of an α-Fe core and a γ-Fe2O3 shell.276 In another report, Siddique et al. investigated the particle size effect on Mössbauer parameters in maghemite NPs. These particles were synthesized by a chemical co-precipitation approach. The presence of a quadrupole doublet indicated the existence of single domain particles. It was evidenced that the internal magnetic field increased with the increase of NP size and the superparamagnetic component remained almost stable. The authors noted that Mössbauer spectroscopy is a very effective and sensitive method to identify the NP size effect and the spin structure in order to analyse the supertransferred hyperfine interactions in nanostructured materials.277 γ-Fe2O3 NPs were also the topic of the study of the Hyeon group and their co-workers: The 57Fe Mössbauer spectra were recorded and a muon spin relaxation study of the magnetodynamics of these monodisperse oleic acid-capped NPs was also carried out. Mössbauer and magnetic susceptibility measurements helped to estimate the magnetic anisotropy constant values. In fact, the relaxation frequencies obtained by Mössbauer spectroscopy and μ-spin relaxation were found to be different and not directly comparable. Mössbauer spectroscopy yielded larger relaxation frequencies than those measured by muon-spin relaxation, a difference which is in agreement with the characteristic times of the two techniques.
The Mössbauer spectra are not fully sensitive to the monodisperse nature of the NPs due to substantial NP interactions, which can appear despite the NP coating by oleic acid.278 CoFe2O4 NPs prepared by a hydrothermal method were also studied by Mössbauer, so as to evaluate their magnetic properties and the cation distribution. Concerning ferrite NPs, in addition to the cation distribution, Mössbauer can provide information on the magnetic domain structure, spin polarization and s-electron density around the Mössbauer probe nuclei. The results of the Mössbauer spectra indicated that these cobalt ferrite NPs had a complete magnetic order. Complementary VSM measurements facilitated a better understanding of the magnetic properties of these materials through the modification of the main magnetic properties (Ms, Hc) with the reaction time and the NP size.27957Fe Mössbauer spectroscopy was used by Tirado and colleagues to investigate iron NPs obtained in situ in conversion ferrite electrodes. Important information was derived concerning parameters such as oxidation state, local environment and magnetic ordering of CoFe2O4 in electrodes cycled vs. lithium.280 Moreover, the thermal reduction of hematite into magnetite was monitored using Mössbauer spectroscopy by Lyubutin and co-workers: the data from ZFC-FC magnetization curves were combined with those from Mössbauer and it was found that the NPs, prepared by the thermal treatment of α-Fe2O3 under inert conditions in octadecene solvent, were strongly coupled by magnetic interactions up to 300 K. Mössbauer spectra illustrated that 95% of the iron was in the magnetite phase while the rest 5% was still in the hematite one.281
Joos et al. studied by Mössbauer iron oxide NPs prepared in diethyleneglycol. They described a protocol to distinguish between maghemite and magnetite using a magnetic field of 0.7 T, at room temperature. This was a remarkable achievement, considering that normally NPs smaller than 15 nm are affected by superparamagnetic relaxation, which hinders their characterization by Mössbauer spectroscopy.282 In another work, β-FeOOH NPs were prepared in a microemulsion system with the use of a non-ionic surfactant. Several characterization techniques were employed to study the properties of the product, and 57Fe Mössbauer spectra showed that the magnetic structure transformed below 150 K, and two kinds of Fe-O octahedra were present in the lattice of the modified β-FeOOH NPs. An approximate Néel temperature (TN) – in a range of 10 degrees – can also be derived from the Mössbauer measurements.283 Another significant feature of Mössbauer spectroscopy is that it does not require the periodic lattice of a crystal, unlike XRD, also knowing that the Mössbauer effect is limited to only a few elements in the periodic table. Mössbauer is powerful in selecting the resonant isotope (e.g.57Fe) in the presence of other atoms in a sample.284 Giersig and co-workers performed the Mössbauer studies of core–shell NPs, and they found out that the magnetic splitting increased with the concentration of maghemite and decreased for magnetite. The Mössbauer spectra of pure γ-Fe2O3, pure Fe3O4 and Fe3O4@γ-Fe2O3 as well as γ-Fe2O3@Fe3O4 core–shell NPs were very different from each other.285 The technique under discussion was also employed to study the biodegradation of magnetic NPs in rat brain, three months after their injection. The presence in the injected ferrofluid of both magnetite NPs and an additional chemical compound containing ferric ion in the high-spin state was evidenced.286
Mazeika et al. studied the effect of the interactions to the properties of ultrasmall CoFe2O4 NPs using Mössbauer spectroscopy: these NPs were prepared by the co-precipitation method and their size was in the range of 1–3 nm. The blocking temperature of the NPs can be determined by several methods, including Mössbauer. In addition, the experimental evidence of the dependence of the Curie temperature on the size of NPs is another interesting task where the application of Mössbauer spectroscopy is valuable.287 Moreover, Gupta and colleagues employed Mössbauer, Raman and XRD to study superparamagnetic ∼9 nm NiFe2O4 NPs prepared by a sol–gel auto-combustion method. Mössbauer measurements recorded at 5 K and under 5 T applied magnetic field demonstrated a mixed spinel structure and canted spin order for the NPs, while a collinear spin order with an inverse spinel structure was observed for larger particles. A prominent central doublet was present at room temperature Mössbauer spectra, showing the superparamagnetic character of the sample at ambient temperature. The measurements from the different techniques concluded that these nickel ferrite NPs consist of a single phase, which is not common with this method of preparation.288 Besides, Ni-substituted Mn0.5Zn0.5Fe2O4 NPs were prepared by Thota et al. with a citrate method. These researchers studied the cation distribution of these NPs and the techniques used were Raman, Mössbauer, XRD and electron spectroscopy. Mössbauer spectroscopy showed the trivalent iron ion distribution between tetrahedral and octahedral sites for all samples with nearly 70–75% of Fe3+ ions sitting on the octahedral sites.289 NiO NPs were also studied with Mössbauer spectroscopy by Bahl and co-workers. These particles were prepared by a combination of chemical precipitation and heating stages. Mössbauer measurements indicated that the nanomaterial was composed of a mixture of ferromagnetic and antiferromagnetic phases. Magnetization measurements yielded larger magnetic moments in comparison with those obtained from the Mössbauer data. This can be explained by interparticle interactions in the samples as well as a difference in the sensitivity of magnetization measurements and Mössbauer spectroscopy to a particle size distribution.290 In addition, Fe3+-doped CeO2 NPs were analysed by XRD, HRTEM and Mössbauer. These particles were prepared by a sol–gel method using ferric nitrate and cerium nitrate as precursors in an alcohol solution. Mössbauer measurements implied the existence of exchange interaction and a sextet pattern observed was assigned to hematite. Magnetic susceptibility also showed the presence of α-Fe2O3, and this was an interesting finding considering that XRD could not confirm the presence of hematite, unlike both of the aforementioned techniques.291
Iron-doped SnO2 NPs were prepared with a hydrothermal route by Diamandescu and colleagues. These particles were characterized by electron magnetic resonance (EMR) and Mössbauer spectroscopies. The EMR data had features attributed to Fe ions in low symmetry crystalline fields and could be related to paramagnetic ions in distorted crystalline positions. Both EMR and Mössbauer studies demonstrated the disordered distribution of iron ions in the bulk and on the surface of SnO2 NPs.292 In another work, Kovalenko and co-workers unraveled the core–shell structure of ligand-capped Sn/SnOx NPs by surface-enhanced SS NMR, Mössbauer and XAS. Oleate or inorganic ligands were employed for the coating of the NPs. XAS and 119Sn Mössbauer spectroscopies were able to identify and quantify amorphous SnO and SnO2 NPs but could not provide insight into the arrangement of these phases within the surface oxide shell. Surface-enhanced SS NMR demonstrated that the outer shell of the NPs was composed exclusively of amorphous SnO2. XRD and TEM showed a crystalline β-Sn core, whereas XAS and Mössbauer measurements detected an interlayer of amorphous SnO and the atomic fraction of each of the three phases. 119Sn NMR signals were not observed due to the low sensitivity of NMR spectroscopy. The combined use of all these techniques resulted in a core/shell 1/shell 2 model of Sn/SnO/SnO2 NPs coated with organic and inorganic ligands, where the only crystalline component was a metallic β-Sn core. In particular, the 119Sn Mössbauer spectroscopy was considered as a highly sensitive tool to determine the oxidation state and chemical environment of tin atoms for several materials, including NPs.293 FeSb2 NPs were prepared by Tremel and co-workers with a wet-chemical approach, and they were analysed by Mössbauer spectroscopy. 57Fe Mössbauer measurements elucidated the remaining iron-containing species during the formation process and determined the purity of the final FeSb2 NPs. Any discrepancies between the XRD and 57Fe Mössbauer data are not surprising due to the fact that these two methods are sensitive to different characteristics, for example, the XRD cannot detect amorphous phases. Mössbauer measurements not only contributed to the comprehension of the formation of the FeSb2 NPs but also provided further proof of the quality of the prepared nanomaterials.294
The surface oxidation of Co NPs prepared by Linderoth and colleagues was analysed by Mössbauer spectroscopy. The structure of CoO formed onto the surface of cobalt particles was considerably well ordered in comparison with the surface oxide formed on iron particles.295 Concas et al. synthesized a cobalt–iron alloy with varying iron content by a sol–gel method followed by thermal treatment under a hydrogen atmosphere. These NPs were embedded in a silica matrix. Mössbauer spectra showed the formation of an ordered component with isomer shift and hyperfine fields characteristic of a Fe–Co alloy only when Fe- and Co-acetate salts were used as precursors, unlike the case of nitrate salts.296 The atomic arrangement in magnetic FePt NPs was analysed by Sakuma and co-workers. XRD and Mössbauer techniques were employed for the analysis of these particles. The order parameter Q was introduced and discussed by the authors, and its value was deduced from Mössbauer measurements. Q denotes the probability of the appearance of the L10-type atomic arrangement. Q is a short-range order parameter, while another parameter named ‘S’ is a long-range order one. The coercivity of the FePt NPs was found to be more dependent on Q than S.297 Another bimetallic NP system is the Fe/Au, and such nanomaterials were studied by XRD, magnetic and Mössbauer experiments by Kauzlarich and colleagues. The authors noted that the XRD pattern that they obtained had a notable simplicity, which was strikingly different from the complexity of the Mössbauer spectra. The latter technique indicated that both uncoated and Au-coated Fe NPs prepared by reduction had three major iron-containing components in their composition. These components were α-Fe, Fe1−xBx alloy and several poorly crystallized iron oxides species.298 FeCu NPs were prepared using an aerosol process by Molins and co-workers, and they were analysed by XRD and Mössbauer. The latter technique played a great role in understanding the processes of formation and decomposition of metastable FeCu alloys.299 In another work, europium sulfide NPs were synthesized with a colloidal approach and they were characterized by Mössbauer spectroscopy. This technique allows a close monitoring of the oxidation state of Eu. The blocking temperature of 20 nm NPs, derived from magnetic measurements, was above 15 K, which is close to the value deduced from the Mössbauer experiments.300
Ferromagnetic resonance (FMR) is a spectroscopic technique that probes the magnetization of ferromagnetic materials, including nanoscale ones. It has similarities with EPR and NMR: for instance FMR probes the sample magnetization that results from the magnetic moments of dipolar-coupled but unpaired electrons, whereas NMR probes the magnetic moments of atomic nuclei that are screened by the atomic or molecular orbitals surrounding such nuclei of non-zero nuclear spin. FMR spectra can provide important information on the average shape and size of catalyst particles, which are composed of ferromagnetic elements (Fe, Ni, Co), and are used for the production of carbon nanotubes. The FMR line width of metal magnetic films is related to the film thickness and depends on the surface anisotropy, defect density and other reasons. The treatment of Si/SiO2/Co substrates in H2 plasma at 350–400 °C resulted in an isotropic FMR spectrum that suggested either the disordered arrangement of catalyst particles or their spherical form on the average. Increasing temperature induced the strong angular dependence of the resonant magnetic field of FMR due to the flattening of the non-spherical and ordered catalyst NPs.301 In fact, the FMR of magnetic NPs differs from the resonance behaviour in bulk materials since the skin depth generally exceeds the particle size, and the multi-magnetic domain structure is excluded from line shape. Increasing the NP size or decreasing the temperature is followed by a shift in the resonance field, an increasingly asymmetric line shape, and an enhanced broadening of the FMR. Surface effects in NMR were revealed at lower temperatures by Murray and co-workers when they studied superparamagnetic cobalt NPs with different crystalline structures and sizes in the range of 4–9 nm by FMR. The comparison of FMR from crystalline magnetic NPs to magnetic NPs with an imperfect structure made it clear that the coherence of the lattice is equally important in describing the anisotropy and hence inhomogeneity of the magnetic properties of the NPs. In total, these authors consider FMR as a sensitive probe of crystallographic imperfection, particle shape and surface composition.302
Morgunov et al. employed FMR spectroscopy to study the magnetic properties of spherical (5–9 nm) Co NPs in a polymer shell. The FMR spectra recorded for cobalt particles did not show any hysteresis, suggesting the existence of the internal field and the presence of remanent magnetization in the NPs. It was found that the saturation magnetization of these NPs was higher than that of the bulk state. In addition, the blocking temperature of the particles was much larger than ambient temperature. The high blocking temperature indicated strong anisotropy, which can be associated with the surface effects in the NPs. Complementary characterization with EPR spectroscopy suggested that the polymer shell interacts with the embedded NPs.303 Stepanov and colleagues investigated Co and Ni NPs implanted in the SiO2 matrix by FMR and TEM methods. FMR signals acquired at room temperature from ensembles of Co and Ni NPs implanted in SiO2 exhibited an out-of-plane uniaxial magnetic anisotropy, typical for thin magnetic films. FMR is in general a suitable method for the evaluation of the magnetic properties of nanogranular media and thin-film systems as it allows the identification of the magnetization value, magnetic anisotropy constants and demagnetization field of a given sample.304 Hochepied and Pileni published a study on the study of the FMR behaviour of nonstoichiometric zinc ferrite NPs doped with Co2+ ions or undoped. FMR measurements on texturated samples (particles subjected to a magnetic field during sample preparation) provided reliable information on the relative thermal variation of the anisotropy constant, and therefore the latter parameter could be evaluated approximately for 3.7 nm zinc ferrite NPs. The FMR spectra of these materials were characterized by an invariant point at a given field, H0. The anisotropy constant varied linearly with temperature and vanished at about ambient temperature.305 The role of dipolar interactions in magnetic NPs was studied by Lezama and co-workers: the FMR measurements of discontinuous multilayers composed of Co80Fe20/Al2O3 were recorded as a function of the angle of the applied magnetic field with respect to the sample at ambient temperature. Angular dependent measurements demonstrate how FMR can be employed to assess interparticle interactions. Overall, FMR can provide significant information not only on ‘bulk’ magnetic properties, but is also useful in evaluating surface magnetic properties and interactions. The g-factor of NPs is one of the parameters that frequency-dependent FMR is able to evaluate. Many of the previously reported FMR studies of NPs had focused on the temperature dependence of the resonance field.306 Dunlop and co-workers have published a study to discuss the 2nd order FMR in NPs; two principal processes in FMR are: the first order absorption of a photon and the creation of a single magnon, which means that the magnon wave-vector should have zero value. Consequently, only the uniform precession magnon (or magnetostatic modes) at the center of the zone can be excited. The second order involves the absorption of a photon, which causes the creation of two magnons of equal and opposite wave-vector. The applications of the 2nd order photon decay of the magnons in FMR include the remagnetization of dilute assemblies of magnetic NPs with high power microwave fields, and the isolation and measurement of magnetic overprints.307 In another report, the magnetic states and FMR in geometrically frustrated arrays of multilayer ferromagnetic NPs ordered on triangular lattices were presented. It was shown that the interlayer coupling resulted in the remarkable splitting of the FMR spectrum. In addition, any magnetizing and remagnetizing of the multilayer systems caused transitions between different ferro-, antiferro- or mixed F/AF interlayer ordering, which were accompanied by dramatic changes in the FMR spectra.308 Lue and colleagues investigated the change from paramagnetic to ferromagnetic resonance for iron NPs produced by the sol–gel method. In ESR (electron spin resonance) the ions are diluted and non-interacting, whereas in FMR the ferromagnetic ions are clustered and interact with each other by the exchange force. Actually both FMR and broadened SPR are relevant to the long-range exchange interaction within the NPs.309 Gamarra et al. used FMR to quantify the amount of superparamagnetic iron oxide NPs in biological materials under both in vitro and in vivo conditions.310 Moreover, FMR was employed to study a phase transition in magnetic field-aligned hematite NPs. Measurements of the temperature dependence of the FMR signal in oriented 9 nm α-Fe2O3 showed anomalies in the intensity, line width and field position in the vicinity of 200 K, implying the occurrence of a phase transition. This transition corresponds to a previously observed Morin transition but having a lower transition temperature than the bulk material. The experiments indicate a transition from a weak ferromagnet to a stronger one at high temperature, whereas in bulk state such transition is from an AF form to a ferromagnetic one.311
Another interesting use of FMR is the determination of the size distribution of NPs. Such possibility was demonstrated by de Biasi and Gondim for the case of γ-Fe2O3 NPs produced by a sol–gel method. By measuring at the temperature range of 10–300 K the relative intensity of the spectrum due to superparamagnetic particles, and the anisotropy field of the spectrum due to ferromagnetic NPs, the size distribution of the particles was obtained. The overall shape of the FMR spectrum of randomly oriented NPs reflected the magnetic anisotropy of the particles. Their work showed that FMR can be used to acquire the size distribution not only in ferrimagnetic precipitates, but also for randomly oriented particles, since the standard deviation of the particle size distribution is nearly the same as the one derived by TEM. The size value measured by TEM is around 40% larger than the corresponding value measured by FMR, because of the presence of a disordered layer in the surface of the particles that makes the ‘magnetic size’ of the NPs to be smaller than the physical size. Depending on the application, the ‘magnetic size’ of the NPs may be more important than the physical size. There are other ways to obtain the size distribution of small magnetic NPs from magnetic measurements, such as from hysteresis loops and from ZFC/FC curves. The FMR method is most suitable when the magnetocrystalline anisotropy is relatively small and the particles are approximately spherical. In that case, a quick and quite precise estimation of the size distribution of the magnetic NPs can be achieved.312 In another work, ∼10 nm maghemite NPs in a PMMA polymer matrix were studied by FMR and DSC. Iron hydroxide gel was used as a precursor for the NP synthesis, and the FMR experiments exposed the temperature range of a superparamagnetic regime (60–290 K) and the blocking temperature, TB ∼ 60 K. The significance of the dipole–dipole interaction for a high concentration of maghemite and temperatures above 220 K was demonstrated.313
Owens studied the ferromagnetic resonance of magnetic field oriented magnetite NPs in frozen ferrofluids: it was shown that by freezing magnetic NPs suspended in a fluid in a magnetic field it is possible to determine the orientational dependence of the FMR spectrum and abstract parameters such as the g value and magnetic anisotropy constant, K. Comparing the data with the FMR measurements in the bulk material indicated that the magnetic phase transition at 136 K does not happen in the NPs until a lower temperature in the range of 25 K.314 In another report, the ferromagnetic resonance of magnetostatically coupled shifted chains of NPs in an oblique magnetic field was published. The resonance field is what routinely measured in FMR measurements with a rotating applied magnetic field, and it permits the characterization of the system with regard to its physical parameters. In that study, this could be useful to characterize, inter alia, the magnetostatic interaction between the chains and to investigate the critical shift as a function of the applied field (restricted to a dimer).315 FePt–Au NPs were also the subject of a study with FMR. The author of that study notes that a relatively small amount of material is capable of providing a good signal to noise ratio. It was shown that the experimental spectra noticed in partially ordered FePt–Au films arise mainly in the low anisotropy disordered phase.316 Vargas et al. characterized by FMR the order–disorder transformation in FePt NPs. These particles were studied in both as-made and annealed forms. The as-prepared particles were synthesized in phenyl ether, they crystallized in the low magnetic anisotropy fcc phase and their diameter was in the range of 2–4 nm. The aim of that study was to evaluate, by means of FMR, the dynamical response of as-made and thermally treated FePt NPs. FMR helped to estimate the magnetic anisotropy in a collection of FePt NPs annealed at various temperatures.317
FMR spectroscopy was also employed to investigate magnetic nickel NPs that are generated through the thermal decomposition of the layered lithium–aluminum double hydroxide with intercalated nickel–EDTA complexes. The Curie temperature (TC) of the resulting NPs measured using FMR spectroscopy was close to the corresponding one for bulk nickel. A numerical simulation of the FMR spectra of these systems was carried out, and the information on the size and shape of Ni NPs was acquired, being consistent with the data obtained through other methods. In addition, insights into the early generation stages of a ferromagnetic phase were gained.318 In another work, Romero and colleagues analysed the surface and frustration evidence in Co–Ni–B NPs through FMR experiments. These particles were amorphous and the measurements were performed as a function of temperature. The FMR measurements provided microscopic information on the internal magnetic order of the particles, which may be hidden by interparticle interactions in magnetization measurements.319 Furthermore, the ferromagnetic resonance in Ni–Zn ferrite NPs in different aggregation states was studied by Ammar and co-workers. These particles were prepared through force hydrolysis in polyol using acetate salts of the corresponding metals as precursors. The products ranged from isolated particles with a size around 5 nm to 20 nm clusters. In FMR experiments, where the absorption is measured by the microwave field, the time window is smaller than in SQUID experiments and thus it shows an ordered magnetic structure for considerably higher temperatures. Any inconsistency in the results derived by the aforementioned techniques is attributed to their different timescales. For instance, at certain temperatures a given sample can appear to be ferromagnetic with one technique, whereas the other technique could characterize it as superparamagnetic.320
X-ray magnetic circular dichroism (XMCD) is a technique which is utilized as a local probe for the study of the site symmetry and the magnetic moments of transition metal ions in ferro- and ferrimagnetic materials. XMCD uses the differential absorption of left and right circularly polarized light in a magnetic field. The external magnetic field is applied along the X-ray propagation vector and the measurement is recorded at the L2,3 edges of the transition elements. For example, XMCD and XAS were employed to study the effects of the size of γ-Fe2O3 NPs on their chemical and magnetic structures. XMCD allows the separation and quantification of the magnetic contributions of FeA3+ and FeB3+ ions to the magnetization. In the case of a phosphate-modified surface (for particles coated with phosphoric acid), XMCD experiment results implied that the surface disordered spins could be mainly FeB3+ spins. XMCD signals recorded for 2.7 and 8 nm particles and acquired by decreasing values of the external applied field helped to detect a greater disorder of FeB3+ spins with respect to the field direction than for FeA3+ spins. The existence of a preferential spin canting of FeB3+ spins at the surface was evidenced, and overall these results were in agreement with a core–shell model of the magnetic structure previously proposed for the particles.321 The same group published another work on maghemite NPs measured by XMCD: the experiments were carried out at the L2,3 edges of iron to analyse the site-specific magnetic contribution of ions in the NPs of γ-Fe2O3. The site-specificity of XMCD renders it a robust tool to analyse the magnetic contributions of the different atoms in the NPs of spinel oxides. In that study, XMCD experiments helped to investigate the magnetic order on tetrahedral and octahedral sites in those NPs at liquid He temperature as a function of the external magnetic field. From such measurements on a single size of particles, it was not possible to conclude whether this magnetic disorder was a surface effect or a core effect.322
Cai et al. analysed the orbital and spin moments of Fe3O4 NPs with size in the range of 5 to 11 nm using the XMCD method. Unlike magnetometry, XMCD is element-specific. Their results implied that while the magnetic moment in the larger NPs appears somewhat to the corresponding one in Fe3O4 single crystal, it may be reduced by a number of factors associated with the nanostructuration: preparation method, particle ligand environment, and NP shape and size.323 Manna and co-workers published a study on the structural and magnetic deconvolution of FePt/FeOx-NPs using XMCD. XAS fit parameters represent the ‘real’ chemical material contribution, whereas the XMCD fit parameters represent the magnetic contribution of each component of the material. The potential of XAS/XMCD techniques for an accurate structural and magnetic characterization with a high spatial resolution at the nanoscale was shown in this work. A core–shell-like structure seemed to be a suitable term to describe this type of structure – and not a ‘dimer-like’ one.324 Takahashi et al. employed XMCD to study the orbital magnetic moment and coercivity of SiO2-coated FePt NPs. In XMCD, one can eliminate the extrinsic magnetic signals, such as those from oxidized Fe and those from the diamagnetic SiO2 coating.325 FeRh NPs were also studied with XMCD, by Chaudret et al. XMCD constitutes a valuable tool to unravel the role of each element in the overall magnetic behaviour of bimetallic NPs. Powerful sum rules permit the direct identification from the experimental spectra not only the value of spin and orbital contributions to the total magnetic moment, but also its orientation. It was observed that the spin and orbital moments induced on Rh could be strongly influenced by the chemical composition of NPs and by their synthesis process.326 Moreover, XMCD spectra were recorded at the L2,3 edges of Co, Cu, Ag and Au and at the K edges of Co and Cu for a series of multilayer systems of partially self-assembled Co NPs, both coated with Al2O3 and with different metals (Cu, Ag and Au). Because of its element selectivity and high sensitivity, XMCD proved useful to provide information regarding the orbital and spin moment components of the Co and the capping metals independently. Direct evidence of the hybridization of the interatomic 3d–nd and the Co intra-atomic 3d–4p bands was acquired through the XMCD measurements. These experiments resulted in the acquisition of the values for the spin and orbital moments averaged over the core and surface of the particle, and the number of holes nh in the empty exchange split nd subbands.327
In another work, Prado et al. probed using XMCD the magnetic anisotropy of cyanide-bridged core and core–shell coordination NPs. XMCD allows the determination of the relative orientation of the magnetic moments throughout the core–shell NPs. This method is particularly useful for core–shell nanostructures, in which three different magnetic ions are present. In comparison with SQUID measurements for Co-containing NPs, the XMCD magnetization curve reaches the maximum magnetization more gradually. This difference might be due to the sensitivity of the XMCD technique to the surface.328 Hochepied et al. used XAS and XMCD to measure at the Fe and Co L2,3 edges of mixed cobalt–zinc ferrite NPs. Such measurements allowed the identification of their magnetic structure and cationic distribution. The advantage of XMCD compared to neutron diffraction is that the former method can also be used for particles that are not well crystallized, and for particles with relatively small size. Elements can be easily separated by the values of their L2,3 edges. Furthermore, XMCD is sensitive to the site symmetry of the absorbing ions, and to the orientation and amplitude of the local magnetic moments. Isotropic spectra are sensitive to the ratio between octahedral and tetrahedral site occupancy, whereas XMCD signals are sensitive to the ratio of magnetic moments of the two sites.329 The complementarity of the information extracted from isotropic and from XMCD spectra was confirmed. Any discrepancies between the XMCD results and magnetization curves could be assigned to the sample preparation, since for XMCD measurements a powder of particles is inserted into layers, resulting in strong interactions between the NPs and radical shape effects, while in SQUID experiments, the particles were dispersed in a polymer matrix.329 In another study, Pd NPs prepared under a high purity atmosphere showed ferromagnetic properties and were characterized by XMCD. This technique proved useful for the evaluation of the electronic and magnetic states of Pd NPs. The researchers who authored that study claim that this was the first observation of the inherent ferromagnetic moment in Pd NPs achieved by performing XMCD measurements.330 Besides, Yamamoto et al. published an XMCD study of polymer-protected Au NPs. This was considered as a direct observation of the spontaneous spin polarization of Au NPs using the technique under discussion. The magnetization assessed by XMCD was in accordance with the values derived by DC magnetization. The origin of spin polarization observed was assigned to an interaction between the protecting polymer and the NPs, although this assumption was not that clear.331
XMCD has also provided evidence of the ZnO NP ferromagnetic behaviour. The size of the particles was 20 nm, and three different surfactants were used: trioctylphosphine, dodecylamine and dodecanethiol. The occurrence of ferromagnetic-like (FML) property up to room temperature was shown. The Zn K-edge XMCD measurements revealed the co-existence of two distinct magnetic contributions: a paramagnetic response from the core of the NP, and a ferromagnetic-like contribution stemming from the interface formed between the ZnO core of the NP and the organic molecule.332 In another work, Kataoka et al. used XMCD to investigate the origin of room temperature ferromagnetism in Fe-doped ZnO NPs. These particles were prepared by a chemical pyrophoric reaction approach. The XMCD spectral line shape of the Zn0.9Fe0.1O NPs was different from that of Fe metal, implying that the magnetism in this sample was not due to metallic Fe clusters, but assigned to the ionic Fe atoms with localized 3d electrons. The XAS results indicated that iron ions were mainly in the trivalent state, together with a small amount of Fe2+. Room-temperature ferromagnetism for these NPs was primarily attributed to the antiferromagnetic coupling between unequal amounts of Fe3+ ions occupying two sets of non-equivalent positions in the region of the XMCD probing depth of 2–3 nm.333
Magnetic susceptibility. Measuring the magnetic susceptibility of a nanomaterial is another way to measure its magnetic properties. The susceptibility indicates whether a material is attracted into or repelled out of a magnetic field, which has implications for practical applications. It is expressed as the ratio of the magnetization to the applied magnetizing field intensity. Herrera et al. reported that poly(N-isopropylacrylamide) [pNIPAM]-coated magnetic NPs showed aggregation through AC susceptibility measurements, which was not evident from DLS experiments. SANS measurements supported the above information derived from AC susceptibility.334 Usually, magnetic susceptibility measurements are performed over a range of temperatures, rather than frequencies, because of the limited available frequencies of most susceptometers. Broadband alternating current magnetic susceptibility measurements were employed to characterize magnetic NPs in natural materials.335 Rinaldi and co-workers carried out AC susceptibility measurements of cobalt ferrite NPs to determine the viscosity of mineral oil. Oleic acid was used a capping ligand for the suspended NPs in the oil and an excellent agreement was found between the nanoscale and macroscale viscosities.336 Lima and colleagues were able to evaluate the size and size dispersity of magnetic NPs in polymeric templates through susceptibility measurements. They also managed to evaluate the magnetocrystalline anisotropy values of magnetite NPs considering the field dependence of the susceptibility peak. NP size parameters acquired from the analysis by the dynamic susceptibility data were in accordance with the values obtained from the fitting of the TEM data.337
In another work, Enpuku and co-workers used AC susceptibility measurements to detect magnetic Fe3O4 NPs with a weight down to 7 ng. To achieve this, an excitation field was applied to the particles, and the resulting signal field from the particles was detected with a pickup coil. An advantage of the susceptibility measurement is that the magnetic signal is determined only by the total weight of the particles and is nearly independent of the size of each particle. A disadvantage of the susceptibility measurement is that the magnetic signal must be measured in the presence of an excitation field, while the signal can be measured in the absence of the excitation field, in the case of relaxation, and remanence measurements for the detection of NPs.338 Magnetic susceptibility measurements were also employed to quantify PVA-coated Fe3O4 NPs in granular sludge. The authors of that study mentioned that compared to other analytical methods, magnetic susceptibility did not require any sample preparation and enabled the straightforward quantification of engineered magnetic NPs in both water phase and granular sludge. Their approach allowed the development of a calibration and correlation of the measured magnetic susceptibility with the iron concentration of the NPs. The Fe concentration for the calibration was identified by ICP-OES. In fact, measuring the magnetic susceptibility with magnetic susceptibility balance (MBS) offers a simple, quick and high accuracy method to determine the concentration of added magnetic NPs without special sample preparation in complex matrices.339 It was observed in another work that if Ni0.6Zn0.4Fe2−xCrxO4 (x = 0–0.5) ferrite NPs were randomly orientated, the overall susceptibility was decreased by decreasing temperature. The temperature dependence of the real and imaginary parts of the effective magnetic susceptibility was measured. Fitting the experimental data of susceptibility with a Néel–Brown model yields unphysical high values for relaxation time and implies the presence of strong interactions between ferrite NPs.340 In another report, the temperature variation of the low-field magnetic susceptibility for antiferromagnetic NPs of ferritin and ferrihydrite in the superparamagnetic regime was studied. The authors of that study managed to show why the temperature variation of the low-field susceptibility in antiferromagnetic NPs, such as the above mentioned ones, deviates from the Curie law variation even without invoking the interparticle interaction.341 In addition, FePt NPs produced in the presence of polyol and PVP were found to possess a high magnetic susceptibility to alternate AC fields at around ambient temperature for biomedical applications, such as magnetic sensing devices for diagnostics and magnetic hyperthermia. The AC magnetic susceptibility reached its maximum value at a temperature near the blocking temperature, and the blocking temperature of the FePt NPs was required to be adjusted at approximately room temperature to ameliorate biomedical performances. Crystallite size and blocking temperature were increased with higher synthesis reaction temperature, resulting in the enhancement of magnetic susceptibility in the range of 300–350 K.342
Magnetophoretic mobility arises from the motion of an electrically neutral body in a viscous medium when exposed to an inhomogeneous magnetic field. It is defined as the ratio of a particle-field interaction parameter to the particle friction coefficient. The particle mobility is a significant factor in predicting the separation when a mixture of particles of different mobilities is exposed to an external field.343 Superparamagnetic iron oxide NPs (SPIONs) were used by Lee et al. in an approach based on the concept that such particles can act as a magnetophoretic mobility switch. More specifically, these particles undergo aggregation only in the presence of target analytes. These authors developed a new LSPR detection technique based on the programmed assembly of SPIONs and the respective change in mobility under external magnetic fields. They noticed a substantial improvement in LSPR response, permitting a selective detection of target molecules without the need to immobilize receptors on the sensor surface. The sensing performance could be tuned by modifying the concentrations of the reactants and the NP sizes.344 Yang and co-workers published a study on the magnetophoretic mobility and superparamagnetism of core–shell iron oxide NPs with dual targeting and imaging functionality. The efficiency of magnetic targeting depends mainly on the magnetophoretic mobility, a parameter that can be increased only by increasing the size of the magnetic NPs. Preliminary in vivo investigation confirmed the suitability of utilizing these NPs in yielding distinctive magnetic resonance imaging of the brain tumor in a rat model.345 The calculated magnetophoretic mobility of a range of magnetic compounds has identified FeCo to be an alternative for magnetite in vitro biological cell applications. In a simple model, the magnetophoretic mobility of a magnetic NP is deduced for a spherical magnetic carrier, which moves slowly in a liquid medium under the effect of an applied inhomogeneous magnetic field. The NPs tested were capped by oleic acid, their size was in the range of 1–11 nm and the stoichiometric (Fe50Co50) alloy was the best one from the magnetophoretic mobility point of view.346 Bharti et al. published a report on the magnetophoretic assembly of flexible NPs/lipid microfilaments. In the presence of a uniform magnetic field, the magnetophoretic attraction of the particles combined with interparticle dipole–dipole attraction drives the microfilament assembly. The magnetophoretic assembly is guided by the distribution of the external magnetic field. In this way, the aggregation of lipid-coated sticky iron oxide NPs into unusually thick and flexible microfilaments takes place.347 In another work, Bakuzis and co-workers reported a mass magnetophoretic experiment applied for the separation of biocompatible magnetic NPs with the potential to magnetohyperthermia. These researchers performed a mass magnetophoretic experiment to segregate NPs according to their diameter and size dispersion. The analysis of HRTEM images proved that with a few hours of exposure to the gradient field, the mean diameter and size dispersion of the NPs near the surface of the fluid showed a significant change.348
Magnetophoretic separation is one of the most promising approaches for harvesting microalgae since the utilization of iron oxide NPs are both technically and economically competent to remove the suspended cells from the surrounding media. Toh et al. investigated the real-time kinetic behaviour of the magnetophoretic separation of Chlorella sp. and the bio-interaction between the Chlorella sp. and surface-functionalized iron oxide NPs under low gradient magnetic separation. The reliability of magnetophoretic separation for microalgal biomass collection was demonstrated, and this method could be employed as an effective downstream process for biofuel production.349 Faraudo and colleagues published an article on the simulation of magnetophoretic separation processes in dispersions of superparamagnetic NPs in the non-cooperative regime. The magnetophoretic separation process of a mixture containing NPs with different sizes and magnetic responses was studied. It was demonstrated that the homogeneous magnetophoretic conditions created by a closed type separator (high magnetic field over almost the whole sample and constant magnetic gradient) enhance the separation process, resulting in a better control over the process, and decreasing the expected separation time when compared to the open-type version of the separator.350 In addition, non-magnetic particles were also affected by the application of a magnetic field gradient in magnetic media. The so-called negative magnetophoresis was used to separate such non-magnetic NPs based on their size.351
Superparamagnetic relaxometry (SPMR) is a technique that combined the use of sensitive magnetic sensors and the superparamagnetic properties of Fe3O4 NPs. It is an emerging technology with applications in various fields, including cancer research where the functionalization of NPs with biomarkers permits the specific binding to cancer cells. In magnetorelaxometry, the magnetic moments of the NPs are aligned by a magnetizing field pulse of amplitude of a few mT and length of some seconds, and after abruptly switching off the field, the decay of the net magnetic moment of the sample is recorded. The magnetic flux density from the sample's net magnetic moment is obtained using high-sensitivity magnetic field sensors, such as SQUIDs (Fig. 8) and fluxgates.352 Similar to AC susceptibility, magnetorelaxometry provides information on the relaxation times (magnetization dynamics) for MNPs in a carrier liquid or for immobilized magnetic NPs.353 Flynn and colleagues reported that the SPRM technology can be employed to specifically determine different types of Ab and cancer cell lines through incubation measurements. Superparamagnetic NPs were conjugated to biomarkers and they could be detected through SPMR measurements, ensuring high contrast in vivo. Unbound NPs did not give any SPMR signal, falling in the measurement time window. Overall, their experiments demonstrated that SPMR is an ideal approach for cancer detection and treatment monitoring.352 Ludwig et al. published a comparative study on the characterisation of magnetic core–shell NPs by fluxgate magnetorelaxometry, AC susceptibility, TEM and photocorrelation spectroscopy (PCS). The samples tested were commercial Fe3O4 NPs with polyacrylic acid shells. There was good agreement between the hydrodynamic size determined from the magnetorelaxometry and AC susceptibility measurements and that obtained from PCS. This suggests that, although clustering occurred, magnetic interactions were negligible and that the models were applicable. In comparison with other methods, magnetorelaxometry is very rapid, it can be performed in opaque media and its signal is less dominated by bigger particles than in AC susceptibility or PCS. It can also be used to characterize both the core properties and the hydrodynamic size distribution of magnetic NPs and clusters, respectively. In magnetorelaxometry, the magnetic moments of the NPs are aligned by an external magnetic field of the order of 1–2 mT for typically 1–2 s, and the decay of the net magnetic moment of the sample is recorded after abruptly switching off the magnetizing field. Compared to magnetorelaxometry utilizing sensitive SQUID sensors, the differential fluxgate magnetorelaxometry setup has the advantages that the measurements can be carried out without magnetic shielding and that the whole magnetization–relaxation cycle can be recorded.354 In a magnetorelaxometry experiment, the relaxation of the superparamagnetic NPs can happen via the Brownian and Néel mechanisms. The magnetorelaxometry-derived size obtained by these authors was found to be slightly larger than that determined from TEM imaging.354
 | ||
| Fig. 8 MRX experimental setup. (1) LiHe Dewar; (2) optional superconducting quantum interference detector (SQUID) magnetometer channel (SQUID sensor not shown); (3) seven-channel second-order SQUID gradiometers (SQUID sensors not shown); (4) Helmholtz coil; and (5) manually controlled non-magnetic 3D stage with optical readout. Reprinted with permission from ref. 352. Copyright 2015 DeGruyter. | ||
Magnetite NPs were characterized by Adolphi et al. by SQUID-relaxometry and magnetic needle biopsy. They found that the magnetization detected by SQUID-relaxometry was 0.33% of that detected by susceptometry, implying that the sensitivity of SQUID-relaxometry could be significantly improved through better control of the NP size. These researchers developed SQUID relaxometry as a highly sensitive platform for detecting and localizing superparamagnetic magnetite NPs specifically bound to cell–surface antigens (or other disease targets) in vivo. Both relaxometry and susceptometry can be used together in a complementary way, in order to quantitatively analyse nanoparticle–cell binding experiments and to evaluate the results obtained by the moment superposition model analysis.355
In fact, both magnetic relaxometry and MRI can be used to detect and locate targeted magnetic NPs, noninvasively and without ionizing radiation. Magnetic relaxometry has specificity (only NPs are detected) and linear dependence of the relaxometry signal on the number of NPs present. Relaxometry is well suited for therapeutic monitoring applications where the quick and precise measurement of a high concentration of magnetic NPs is needed. SQUID-detected magnetorelaxometry has been reported to offer accurate quantification over a wider range of NP concentrations compared to MRI. In addition, the former technique can be more rapid and cheaper than the MRI.356 Urbano-Bojorge et al. evaluated and compared alternating gradient field magnetometry and relaxometry as effective tools to assess the biodistribution of the magnetic NPs and to detect them on ex vivo tissue. To do this, a standard dose of the Fe-oxide core and dextran coated magnetic NPs were injected in the retro-orbital sinus on mice. The relaxometry time and magnetometry data were consistent with the distribution of magnetic NPs and specific uptake in the reticuloendothelial system.357 Another comparison between fluxgate and SQUID magnetorelaxometry techniques for the characterization of magnetic core–shell NPs was reported by Schilling and colleagues. They mention that the advantages of using fluxgate magnetometers are that they are easier to operate since they do not need cryogenic cooling and since they are less susceptible to magnetic disturbances.
In addition, the fluxgate method measures the absolute value of the corresponding vector component of the magnetic field, and not just flux/field changes as SQUID magnetometers; therefore the complete magnetization–relaxation process can be recorded and zero signal is defined. Whereas fluxgate approach acts as a compact, user-friendly and affordable tool for the standard magnetic characterization, SQUID relaxometry shows a high sensitivity performance.358 Peng et al. published a study on engineered water-soluble two-dimensional magnetic nanocomposites, aiming for high magnetorelaxometry properties. In terms of MRI activity, the relaxometric properties of nanoparticulate contrast agents were structure-related and highly dependent on the interaction between water protons and the core magnetic NPs within the magnetic nanocomposites. These researchers used hydrophobic Mn-doped ferrite NPs and they turned them into hydrophilic colloids through a one-step direct solvent evaporation method, involving aqueous graphene oxide solution as a phase transfer agent. The resultant unique two-dimensional magnetic nanocomposite construct showed improved water accessibility and water retention in between the aggregated hydrophobic Mn-doped ferrite samples. Thus, it resulted in enhanced relaxometric properties.359
2.4 Microscopy techniques for NP characterization
Transmission electron microscopy (TEM) is a microscopy technique that exploits the interaction between a uniform current density electron beam (i.e. the energies are usually within a range of 60 to 150 keV) and a thin sample. When the electron beam reaches the sample, part of the electrons are transmitted, while the rest are elastically or inelastically scattered.360 The magnitude of the interaction depends on several factors, such as size, sample density and elemental composition. The final image is built with the information acquired from the transmitted electrons. As it is clear from the previous sections, size and morphology define the unique set of physical properties, such as optical,361 magnetic,362 electronic363 and catalytic,364 of NPs, as well as their interaction with biological systems.365,366 TEM is the most common technique to analyse nanoparticle size and shape, since it provides not only direct images of the sample but also the most accurate estimation of the nanoparticle homogeneity. Nevertheless, some limitations have to be considered when using this technique, such as the difficulty in quantifying a large number of particles or misleading images due to orientation effects. When characterizing very homogeneous samples, other techniques that analyse larger amounts of NPs can provide more reliable results, such as SAXS for larger and spherical NPs,367 or XRD by exploiting the bordering of the XRD reflections and the Scherrer formula.368 However, a previous analysis has to be performed to ensure sample homogeneity.Nanoparticle properties not only depend on their size and morphology but also other factors, like interparticle distance. For instance, when two metal NPs are brought in close proximity, their plasmons couple, red-shifting their plasmon band and changing their colour. Therefore, TEM has been used to characterize the nanoparticle aggregation for different biomedical applications, including (1) sensing and diagnostics, where the aggregation depends on the presence of a biomarker or analyte;369,370 (2) therapy, where the aggregation causes an increase of the nanoparticle therapeutic effect;371 and (3) imaging, where the aggregation improves the response signal.372 In order to obtain reliable results, extra care should be taken for sample preparation, since an inadequate protocol can result in sample alteration or artefact creation,373e.g. aggregation during the drying of the colloid suspension. Thus, TEM is usually combined with other techniques that can measure larger numbers of particles, and require less sample preparation, such as UV-Vis and DLS.374,375 In recent years strong control over the nanoparticle assembly has been achieved, and a controlled NP self-assembly can lead to well-defined NP superlattices. The systematic assembly of different nanocrystals yields new multifunctional structures that combine the features of the individual building blocks, as well as the rise of new and exciting properties.376 TEM has been one of the techniques used to characterize the formation of different super-lattice nanocomposites, which can be isostructural to several atomic crystal systems (Fig. 9).377 These new three-dimensional arrays are made of different NPs (e.g. quantum dots, metals and magnetic NPs), and their final structure and composition can be controlled by tailoring the colloid surface charge377or directional bonding with DNA.378
 | ||
| Fig. 9 The depicted superlattices are assembled from a, 13.4 nm γ-Fe2O3 and 5.0 nm Au; b, 7.6 nm PbSe and 5.0 nm Au; c, 6.2 nm PbSe and 3.0 nm Pd; d, 6.7 nm PbS and 3.0 nm Pd; e, 6.2 nm PbSe and 3.0 nm Pd; f, 5.8 nm PbSe and 3.0 nm Pd; g, 7.2 nm PbSe and 4.2 nm Ag; h, 6.2 nm PbSe and 3.0 nm Pd; i, 7.2 nm PbSe and 5.0 nm Au; j, 5.8 nm PbSe and 3.0 nm Pd; k, 7.2 nm PbSe and 4.2 nm Ag; and l, 6.2 nm PbSe and 3.0 nm Pd NPs. Scale bars: a–c, e, f, i–l, 20 nm; d, g, h, 10 nm. The lattice projection is labelled in each panel above the scale bar. Reprinted with permission from ref. 377. Copyright 2006 Nature Publishing Group. | ||
In the last few years, the scientific community has started to view NPs as dynamic systems, where their structure and properties can evolve over time as they interact with their surroundings.379 Therefore, it is important to characterize their dynamic transformations in order to optimize their performance in many applications. For instance, sunlight has been reported to aggregate Ag NPs and decrease their cytotoxicity. TEM imaging showed that nanobridges were formed between the NPs upon sunlight exposure.380 These morphological changes combined with surface sulfidation affected the nanoparticle dissolution rate, which caused the toxicity to decrease. Furthermore, TEM and DLS have been used to study the biodegradation of the nanoparticle polymeric coating by bacteria. The loss of the particle coating caused colloidal aggregation, which affected their mobility and cytotoxicity.381
Furthermore, traditional TEM cannot be used to study the growth of NPs in solution (this topic is further discussed in the Liquid-TEM section of this review). Nevertheless, it can be used to characterize the formation of colloids from solid precursors. For example, TEM has been used to image the growth dynamics of copper NPs.382 These were synthesized in a heating holder by reducing copper phyllosilicate platelets with hydrogen. The in situ visualization allowed the characterization of the phase transformation of copper as the reaction was progressing. Another use of NPs concerns the field of therapeutic carriers, since they can enhance the efficiency of drugs by improving their stability and cellular uptake.383 Two main techniques are used to study the interaction between NPs and cells: TEM and CLSM.384 Both techniques are complementary and frequently used together, since TEM provides higher resolution than any other imaging technique while CLSM allows the live cell imaging and fluorescent labelling of different cell components. NPs are internalized through endocytosis after interacting with cell membrane receptors, such as scavenger receptors.385 However, in order to increase their therapeutic effect, NPs need to escape from the vesicles and be released into the cytoplasm.386 Thus, TEM has been used to assess the location of NPs within a cell. For instance, it was used to study the Au NP shape and size requirements for higher cellular uptake and later vesicle escape (Fig. 10).387
 | ||
| Fig. 10 Representative TEM images of U87 cells after treatment with NP-siRNA constructs indicate that larger constructs can distribute in the cytoplasm. U87 cells were treated with 0.5 nM of (a) 13 nm spheres, (b) 50 nm spheres, and (c) 40 nm stars for 24 h. The images in the boxes (lower panel) indicate zoomed-in views. The yellow arrows indicate NPs distributed outside vesicles; the orange arrows indicate locally disrupted vesicle membranes. Reprinted with permission from ref. 387. Copyright 2017 American Chemical Society. | ||
As mentioned in an earlier section of this review, the aggregation of NPs can change their physical properties. Therefore, TEM has been applied to characterize the dispersion of NPs after their internalization. For example, Au NPs grafted with PEG were well dispersed, and in low proportions within the intracellular vesicles of macrophages, while non-grafted Au NPs mostly accumulated as aggregates in the vesicles.388 An additional advantage of TEM is that it allows the assessment of the changes of subcellular structures caused by the NPs. For instance, apoptosis-related vacuoles were observed in melanoma cells after magnetic field hyperthermia treatment with iron oxide NPs was applied.389 This observation helped to understand the cell death pathways in response to magnetic field hyperthermia. Finally, TEM has also been employed to define the degree of penetration of NPs through different tissues, such as TiO2 NPs through the skin for sunscreen applications.390
High-resolution TEM (HRTEM) is an imaging mode of transmission electron microscopy that uses phase-contrast imaging, where both transmitted and scattered electrons are combined to produce the image.391 In comparison with traditional TEM imaging, HRTEM requires a larger objective aperture in order to employ the scattered electrons. Phase-contrast imaging is the technique with the highest resolution ever developed and allows the detection of the arrays of atoms in crystalline structures. HRTEM provides important information on the nanoparticle structure; in particular, while conventional electron microscopies can provide the statistical assessment of NP morphology, they do not have enough resolution to image the single particle crystal structure. Thus, HRTEM has become the most common technique to characterize the internal structure of NPs.
For instance, HRTEM has been used to study the effect of ligands in the final structure of Pt NPs grown by organometallic synthesis.392 Similarly, it has been employed to observe that the Pd nanoclusters (sizes between 1 and 1.5 nm) synthesized by a different organometallic protocol are a mixture of four different structured crystals with comparable energy levels,393 see Fig. 11 and 12.
 | ||
| Fig. 11 HREM images of Pd particles with fcc structure. (a) and (b) are in a 〈1 1 0〉 orientation and (c) is in a 〈1 0 0〉 orientation, while (d) corresponds to a particle with a hexagonal profile, which corresponds to a distorted 〈1 1 0〉 orientation. The corresponding FFT is included in each case. Reprinted with permission from ref. 393. Copyright 2001 Elsevier. | ||
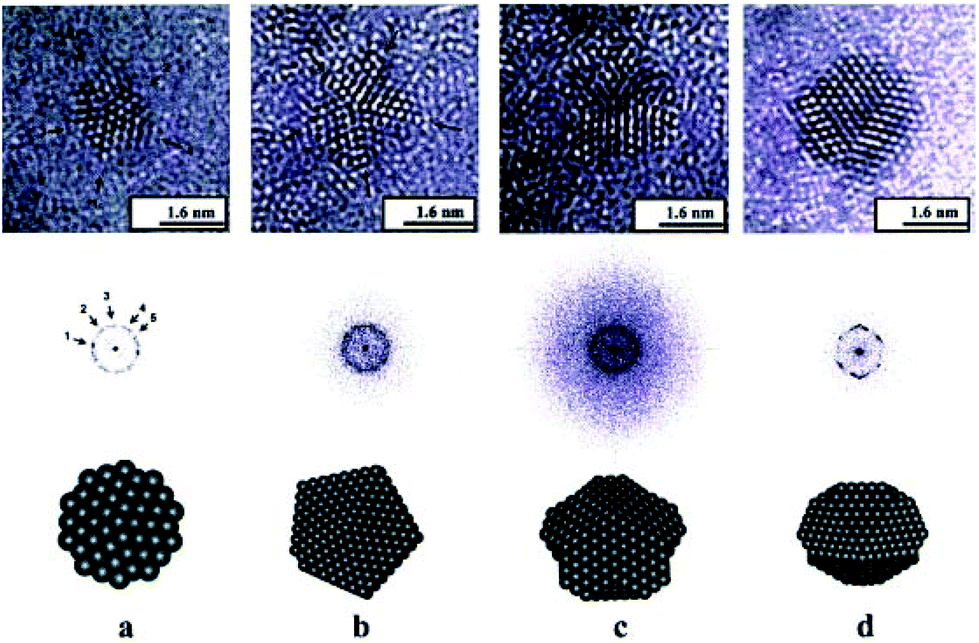 | ||
| Fig. 12 Sequence of HRTEM images for decahedral Pd particles showing different orientations with respect to the one five-fold axis parallel to the electron beam. A model shows in each case the orientation the corresponding FFT is included in the figure. Reprinted with permission from ref. 393. Copyright 2001 Elsevier. | ||
Furthermore, HRTEM can distinguish between single crystal and polycrystalline anisotropic Au NPs that present similar optical properties.394 HRTEM also allows the characterization of structural transitions, such as the thermal transition from disordered face-centred cubic to ordered L10 in iron–platinum NPs.395 This thermal-induced event yields NPs with enhanced coercivity and larger magnetocrystalline anisotropy, which are necessary qualities to build permanent magnets.
The imaging of single crystals also offers the opportunity to identify structural defects that may explain the unusual properties. For instance, it had been reported that the lattice constant of CeO2 NPs increased with decreasing particle size.396 Nevertheless, no explanation had been found for such abnormal behaviour. A later study using HRTEM showed that these changes were not caused by either disclinations (line defects) or volume expansions in the high angle boundaries.397 Combining these results with the ones from Raman spectroscopy, the authors concluded that the lattice expansion was the result of an increased number of point defects at smaller particle sizes.398 Even though HRTEM is a powerful technique, it is worth mentioning that the characterization of NPs is not always feasible by this technique. Due to the random orientation of the crystals relative to the electron source, there may be directions where the atoms are not well aligned, resulting in complex images that cannot be directly used to define the structure.398
Insights regarding NP growth and structure-related properties can also be gained through HRTEM observations. For instance, Zhang et al. studied the formation of CuO NPs by in situ HRTEM. They observed that the leading mechanism was coalescence,399 which was much faster than others, such as nanocrystal reshaping. Furthermore, they witnessed that if the colloids were aligned before merging, the resulting NPs were single crystals. Furthermore, HRTEM has been used to clarify the effect of substrates on the properties of metal NPs. For instance, it was observed that Cu NPs deposited on graphite substrates presented a distinctive melting behaviour and selective superheating, in comparison with NPs supported on CuO2.400 Based on the HRTEM images and molecular dynamics, the authors of the study attributed the distinctive behaviour to the absorption of a thin layer of carbon on the NP surface, which improved their thermal stability.
Liquid TEM. As mentioned earlier a fundamental component of TEM is the vacuum system, which prevents the damage of the filament and decreases the electron beam scattering. Traditional TEM imaging has been solely used on solid and dried samples, since the evaporation of liquids could compromise the vacuum. Thus, the characterization of solid–liquid systems at the nanoscale has been neglected for many decades. Early attempts to characterize liquid samples date back to the 1930s, when L. Marton imaged biological samples trapped between aluminum thin foils.401 Nevertheless, the technical challenge of preserving the vacuum and avoiding the liquid evaporation prevented any significant advancement until recent years, when the nanofabrication of sealed liquid cells was developed. In 2003, Frances M. Ross and collaborators developed a TEM liquid cell using epoxy-sealed silicon nitride (SiN) membranes.402 These membranes were electron transparent and confined the liquid sample, preserving the microscope vacuum. Ross et al. were able to image the growth of Cu nanoclusters with 5 nm spatial resolution and a time resolution of 30 images per second. Since then, several modifications have been introduced to the liquid TEM grid. For instance, better cell sealing was achieved by replacing the original SiO2 spacers with softer indium thin films.403 SiN window grids were fabricated by binding commercially available SiN wafers with polymer O-rings.404 The polymers greatly simplified the cell fabrication, decreasing the assembly times down to 10 to 15 min, and allowing the re-use of the wet cells. SiN TEM grids are currently commercially available with electrochemistry and heating packages, making them the most popular option among the different TEM liquid cells. However, these grids suffer from lower image resolution, due to the SiN membrane and liquid layer thickness, which contribute to scatter the electron beam (Fig. 13).405 An alternative to SiN membrane grids is imaging solid–liquid systems in low vapor pressure ionic liquids (ILs).406 The solids are dispersed in the ILs and imaged without sealing, since ILs do not evaporate. The absence of cell membrane provides lower electron scattering and better image resolution. Nevertheless, working with ILs is technically demanding and the number of systems that can be imaged is very limited, since ILs react with a wide range of components. Recently, a new kind of liquid cell has been developed by enclosing liquid samples between two thin graphene sheets.407 In these, the thickness of both liquid layer and sealing membrane is highly minimized, decreasing the electron beam scattering and achieving images with atomic resolution. Furthermore, the van der Waals forces between the graphene layers keep the system assembled and no further sealing step is required. Although graphene membrane cells have become a hot topic, they still present some limitations that must be considered before using them. For example, low operation voltages are required (80 kV) in order to minimize the electron knock-off effects on the graphene atoms. Most of the imaging techniques only provide information at a single time point, usually after the nanoparticle growth has finished. Therefore, they can characterize the final nanoparticle structure but not the growing mechanism. Liquid TEM allows the tracking of the nanoparticle trajectory while this is growing, providing direct observation of the nanoparticle evolution. For instance, liquid TEM has been used to study the growth mechanism of platinum NPs, which can follow two different growing mechanisms, i.e. monomer attachment or coalescence, and still yield the same final nanoparticle size.408
 | ||
| Fig. 13 (A) O-ring sealed in situ wet cell design. (B) Sealless in situ liquid TEM setup utilizing low vapor ionic liquids. (C) Illustration of an in situ liquid cell formed by atomic thin graphene membranes. (D) Atomic resolution images obtained with C, showing the Pt nanocrystal growth procedure. Reprinted with permission from ref. 405. Copyright 2015 Royal Society of Chemistry. | ||
Interestingly, graphene liquid cell resolution is high enough to study the facet-dependence interaction between NPs. Alivisatos’ group observed that the platinum nanoparticle growth through the coalescence mechanism is facet-specific, where the attachment is favoured on the lowest energy surfaces.407 Other interesting mechanistic observations imaged through liquid TEM include the oscillatory growth dynamics of bismuth NPs, where both Ostwald and anti-Ostwald ripening occur,409 the Kirkendall effect on the synthesis of hollow bismuth oxide NPs410 and the galvanic replacement on the formation of hollow palladium NPs.411 Furthermore, in situ imaging can be used to calculate the redox reaction rates on the growth of heterocomplexes, such as core–shell gold–palladium NPs.412
NPs within fluids are under constant movement. In addition to Brownian motion, several other parameters can contribute to their movement, such as chemical-induced changes of the environment or liquid flow. Liquid TEM has been used to characterize some of them. For instance, the groups of Alivisatos and Dahmen recorded the movement of inorganic NPs during fluid evaporation.413 Nevertheless, the most exciting application of recording nanoparticle motion is the 3D reconstruction of the colloid. Park et al. imaged the free movement of platinum NPs in liquid in order to reconstruct their structure at the near-atomic scale.414 Nanoparticle assembly and superlattice formation are emerging as important fields of research within nanoscience because they can present different properties in comparison with the individual NPs and bulk materials. The fundamental understanding of their formation mechanisms requires characterizing not only the final structure but also the assembly process. In this direction, liquid TEM has been used for the direct observation of Pt NP superlattice formation, which includes an initial amorphous agglomerate condensation and a subsequent array crystallization.415
Cryo-electron microscopy (cryo-TEM) is a subclass of TEM that allows the visualization of the near-unaltered samples in their frozen-native environment by vitrifying them at cryogenic temperatures.416 Very recently, the 2017 Nobel Prize of Chemistry was awarded to Dubochet, Frank and Henderson for the development of cryo-electron microscopy for the high resolution structure determination of biomolecules in solution. Liquid nitrogen is usually employed to freeze the samples. This technique is commonly used in molecular biology and colloid chemistry due to the lack of factors (i.e. staining and sample's preservation in non-physiological environments) that can alter the conformation or assembly of the sample's molecules. The liquid samples are usually vitrified by commercial automated plunge-freezers, which freeze water solutions by decreasing their temperature extremely fast, so the water molecules cannot reorganize in long-range ordered crystal lattices. This results in an amorphous state that is similar to the native liquid.417 Plunge-freezers accomplish this amorphous state in four steps: (1) placing the liquid sample in the carbon-coated copper grid, (2) removing the excess of liquid in order to produce a thin film, (3) plunge-freezing the grid into the liquid N2 and (4) storing the vitrified sample in a storage box that contains liquid N2.
Before liquid TEM became commercially available, cryo-TEM was one of the two most common techniques used to visualize the nanoparticle growth (the other one involved arresting the NPs at intermediate reaction stages, and performing normal TEM characterization).418 Cryo-TEM has been used to study complex growth mechanisms, such as the aggregative growth of zeolite crystals, where several amorphous aggregates are formed before they rearrange into the final crystals.419 Other studied systems include the formation of biphasic particles,420 made of silica and polystyrene, and the “popcorn”-like growth of gold nanorods.421 The latter is a good example to highlight the strengths and weaknesses of this technique. On the one hand, it can provide direct images of the NPs while growing in their native environment. On the other hand, the concentration of the particles usually is too low (in the nanomolar range) to provide statistical information. In addition to the NP growth, cryo-TEM has been used to visualize the molecular templates, such as block copolymers and CTAB, that direct the growth of lanthanide-based NPs422 and Au nanorods,423 respectively. The morphology and volume transitions of thermoresponsive core–shell NPs have also been imaged by cryo-TEM, where the morphology of the thermosensitive shell is preserved after the plunge freezing, and clearly visible without staining.424 Lastly, it is worth mentioning that cryo-TEM can achieve sub-nanomolar resolutions. For instance, the Au (200) planes of 15 nm Au NPs have been imaged with structural resolutions below 0.2 nm.425
Cryo-TEM has been used to study complex nanoparticle aggregation mechanisms, such as the kinetic manipulation of block copolymer nanostructures (Fig. 14)426 or the assembly of binary NP superlattices using protein cages.427 Furthermore, cryo-TEM imaging is usually required to confirm the unusual assembly behaviours, since the fast plunge freezing avoids the particle rearrangement during the sample preparation and visualization. As an example, conventional TEM showed that cellulose NPs laterally self-assemble into flat objects. Nevertheless, cryo-TEM imaging was required to confirm that these assemblies were not drying or staining artefacts.428 In addition to qualitative characterization, cryo-TEM can also be used to quantify the thermodynamic forces involved in the formation of assemblies. Even though several theoretical models have been developed to explain the contribution of these forces,429 there is very limited amount of experimental data available. The formation free energy of quantum dot nanostructures was calculated from cryo-TEM images.430 The free energy was later separated into the entropic and enthalpic contributions, exploiting the variation of the assemblies with temperature.
 | ||
| Fig. 14 TEM images of directed gold nanoparticle assembly in the charged polyacrylic acid region. (A and B) Bright-field images. Dark stripes are concentrated gold nanoparticle areas. The insert shows the proposed structures. Yellow dots denote gold NPs. (C) High-resolution TEM (HRTEM) imaging of the lattice structure of gold single crystals. (D and E) High-angle annular dark-field (HAADF) imaging of periodic gold stripes. Gold particles appear as bright stripes. (F) TEM image of periodic gold stripes when polyamine functionalized gold particles are used as counterions. Reprinted with permission from ref. 426. Copyright 2007 AAAS. | ||
Electron diffraction (ED), also known as selected area electron diffraction (SAED), is another important microscopy tool for the study of the crystal structure of NPs. Experiments are usually performed in a TEM, or a scanning electron microscope (SEM) as electron backscatter diffraction. In these instruments, electrons are accelerated by an electrostatic potential in order to gain the desired energy and determine their wavelength before they interact with the sample to be studied. The periodic structure of a crystalline solid acts as a diffraction grating, scattering the electrons in a predictable manner. Working back from the observed diffraction pattern, it may be possible to deduce the structure of the crystal producing the diffraction pattern. Buffat discussed the use of electron diffraction and HRTEM to investigate multiply-twinned structures and dynamical events in metal NPs. The author noted that measuring the shrinkage of the lattice space by XRD may be complicated, as instrumental parameters, reflection broadening due to a very small NP size and matrix effects can lead to unclear XRD results. With ED, particles are lying rather free on a substrate, lower material quantity is needed for the measurement, and correlation with direct images of the crystals is possible. However, the study of size effects in Au and Pt by ED requires a careful interpretation of its results due to the complex multiply-twinned or icosahedral-like structure that appears in NPs to lower the total free energy.431
The SAED technique is limited by the fact that many NPs contribute to the diffraction pattern because of the relatively large size of the illuminated area, making their individual study difficult. In the more modern ‘nanodiffraction’ technique, the area of the sample which contributes to the diffraction pattern is limited by the size of the electron probe, which in a field emission TEM can be as small as 0.1 nm. In a paper concerning decahedral Au NPs, the ‘nanodiffraction’ approach was employed enabling the study of single NPs, but it was demonstrated that the beam convergence produced a loss of symmetry from 10- to 5-fold in the diffraction pattern of the NPs.432 Schamp and Jesser used ED to calculate interplanar spacings and other lattice parameters of Au NPs. An improved calculation of such parameters allows a more precise determination of the anisotropic strains in the gold NPs.433 In another work, the origin of the ‘forbidden’ reflections present in the [111] and [112] electron diffraction patterns of triangular-flat-thin Au NPs was explained.434 Regarding another noble metal, Ag, Smyslov and co-workers combined SAXS, ED and microscopy experiments to determine the size and phase composition of Ag NPs in a gel film of bacterial cellulose. In that report, SAXS provided a reliable estimate of the size of the NPs in the moisture-containing composite; ED and electron microscopy permitted the performance of phase analysis, obtain images of NPs and visualize their arrangement in the composite matrix.435
Bismuth NPs have also been studied by electron diffraction: the authors of that study noted that the diffraction pattern is produced from the whole ensemble of the NPs, and if there are populations of NPs with two different sizes, the larger size NPs will contribute more to the diffraction intensity than the smaller ones.436 Fe-based NPs (alloys and oxides) have often been investigated with electron diffraction. Sato and Hirotsu used ED to study the order–disorder transformation in L10-FePd NPs. The disappearance of the long-range atomic order in such 10 nm NPs was examined by ED using a specimen heating stage attached to a TEM, for an in situ annealing. A particle size dependence of the order–disorder transformation temperature of 10 nm sized FePd isolated NPs was evidenced. Compared to the bulk alloy, such temperature was lower by around 80 K for 13.5 nm FePd NPs.437 The same group employed ED to determine the long-range order (LRO) parameters of two-dimensional epitaxially-grown dispersed monocrystalline 10 nm L10-FePd NPs. In that case, the very small volume of the 2D sample would hinder the applicability of the usual XRD measurements for the calculation of the LRO parameters.438 Similarly, the LRO parameters of L10-FePt NPS were also determined through the use of ED. It is reported that when using transmission electron diffraction with fast electrons, the scattering power of atoms for electrons is about 104 times larger than that for X-rays.439 Thus, ED has a great advantage in acquiring superstructure reflections for these bimetallic NPs with ordered structure and 2D dispersion. Nevertheless, the dynamical scattering effect complicates the analysis of ED intensity. Still, the LRO parameters of such FePt NPs can be calculated with accuracy by ED taking into account the multiple scattering effect.439
Li et al. analysed the structure of CoFe–Fe3O4 core–shell NPs by electron imaging and diffraction. These researchers employed both ED and HRTEM to find out if the core is composed of CoFe2O4. HRTEM images can provide significant information on the real-space structure, but only the NPs oriented along specific directions and the lattice planes that are large enough to be resolved by TEM can give rise to lattice fringes in the image. Electron diffraction patterns recorded from a large number of NPs have a unique advantage, i.e. all of the lattice planes are represented in the diffraction pattern. HRTEM, ED and EDS microanalysis helped in the combination for determining the structure and composition of such core–shell NPs.440 Langguth and co-workers combined ED and XRD for the structural characterization of iron oxide/hydroxide NPs in 9 different parenteral drugs for the treatment of iron deficiency anaemia. While XRD permits a higher resolution of small d-distances because of the low wavelength of about λ = 0.154 nm, the combination of STEM with diffraction analyses allows the selective investigation of crystalline areas in the sample.441 Finally, electron imaging and diffraction were used in a complementary way to determine the crystalline planes and directions of the surface facets and edges of hematite NPs as well as to calculate their Miller indices.442
In scanning transmission electron microscopy (STEM), the electron beam is focused to a fine spot that is then scanned over the sample in a raster, unlike conventional TEM. The rastering of the beam across the sample makes STEM appropriate for techniques such as Z-contrast annular dark-field imaging (explained below) and spectroscopic mapping by energy dispersive X-ray (EDX) spectroscopy or electron energy loss spectroscopy (EELS). Using EDX or EELS spectroscopy in the STEM it is possible to obtain elemental maps that show features down to the atomic scale. For the proper operation of STEM, the experimental determination of the absolute cross section is very challenging, as electron donors of high dynamical range are required. This has hampered the application of the STEM-based technique to a broad range of particle sizes, as one would wish. Nevertheless, mass information can be acquired through STEM-based mass measurements if a known mass standard can be established. For the characterization of the 3D morphology of NPs, STEM electron tomography (analysed later in this review) is a very powerful technique and has been successfully employed for embedded and stable NPs. The main restriction of the method is the time needed to take full tomographs and this might exclude many electron beam sensitive samples from analysis. To tackle that difficulty, Palmer and co-workers developed a ‘single-shot’ approach to a three-dimensional measurement problem, using Au NPs as the model system.443 Haigh and co-workers published a paper on the investigation of the limitations and optimisation of EDX tomography within a STEM, focusing on the application of the technique to characterize the 3D elemental distribution of bimetallic AgAu NPs. A key question they worked on for EDX tomography was whether the characteristic X-ray intensity generated in the STEM meets the requirements for the constraints of a particular sample and detector geometry.444 Ag NPs exposed to light and humic substances were investigated by a combination of high resolution STEM, EELS and UV-Vis techniques. This multimethod approached facilitated the acquiring of information on NP morphology, surface chemistry transformations and corona formation. Despite the signal loss, probably by dissolution, that was noticed, there was no direct evidence of oxidation from the STEM-EELS.445 The Palmer group has reported that not many applications of quantitative STEM exist in nanomaterial characterization. Therefore, they demonstrated a new approach to quantify the imaging contrast in STEM using size-selected clusters. The nanoclusters used consisted of Pd (Z = 46) and Au (Z = 79).446 Finally, Deiana et al. used STEM-EDX to investigate the core–shell structure of bimetallic Pd–Hg NPs, which proved to be a crystalline core–shell structure, with a Pd core and a Pd–Hg ordered alloy shell. The ordered shell was considered to be responsible for the high oxygen reduction selectivity to H2O2.447
High-angle annular dark-field imaging (HAADF-STEM). Annular dark-field imaging is a method of mapping samples in a STEM. These images are formed by collecting scattered electrons with an annular dark-field detector. An annular dark-field image formed only by very high angle, incoherently scattered electrons (Rutherford scattered from the nucleus of the atoms) – as opposed to Bragg scattered electrons – is highly sensitive to variations in the atomic number of atoms in the sample (Z-contrast images). This technique is also known as high-angle annular dark-field imaging (HAADF). HAADF-STEM is a valuable tool to observe local atomic structures and has been successfully applied to the imaging of various material interfaces. Akita et al. used this technique to observe Au NPs supported on CeO2 for the first time to investigate the mechanism of the cyclic structural change according to the switching on and off of the electron beam. The sequential HAADF-STEM observations can directly detect the atomic process of the structural change as well as the detailed behaviour of Au atoms at the perimeter edge. HAADF-STEM images can also represent the correct atomic column positions of Au and Ce as maximum intensity positions without any artifact under their observation conditions. This is a difference from the usual HRTEM images, where intensive image simulations are essential to decide atomic positions due to the significant occurrence of artifacts, especially at surfaces or interfaces.448 HAADF-STEM has straightforward interpretability, although multislice simulation is often required in order to take into account the strong dynamical screening effect if quantitative structure information is needed.
The technique under discussion has applications in tomography (discussed below), size mass/thickness measurement at the atomic scale, structure characterization and composition measurement. HAADF-STEM uses a sharply focused beam to scan across the specimen, and the annular dark-field (ADF) detector collects only the scattered electrons.449 The observation of Au NPs on TiO2 was also achieved using HAADF-STEM. This technique together with HRTEM is indispensable for the direct observation of the atomic structure of heterointerfaces. HAADF-STEM can resolve atomic configurations directly without image simulations considering the defocus value and the thickness of the samples, although it is hard to image the light atoms, such as oxygen.450 In the study by Haruta and co-workers, the distance between the Au and Ti layers at the interface is estimated from the HAADF-STEM image, which is essential to evaluate the status of oxygen layers affecting the catalytic activity. The oxygen columns on the TiO2 surface and in the bulk TiO2 region were not detected in the HAADF-STEM image, because oxygen atoms are light compared with titanium, and the signal-to-noise ratio was not high enough. STEM images are easily distorted during image acquisition by the sample drift or mechanical and electronic vibrations. Although the atomic columns are detected in the HAADF-STEM image, it is hard to detect the local displacement of each atom. The complementary combination of HAADF-STEM imaging and first-principles calculations should be a promising approach to elucidate the atomic and electronic structure at the interfaces.450
Li et al. mention that HAADF-STEM is appealing to probe the 3D-structure of NPs because its intensity is strongly dependent not only on the atomic number Z of the observed atoms but also on the number of atoms in a column. They combined quantitative HAADF-STEM analysis with molecular-dynamics-based model structure search procedures, and realistic image contrast simulations in order to identify not only the size and shape but also the structure and orientation of soft-landed Au nanoclusters.451 Badonneau et al. studied by HAADF-STEM the Au and Ag NPs embedded in dielectric capping. The authors illustrated that this method is a convenient tool for revealing the morphology of buried NPs, and highlighting the influence of the NP size and the dielectric-capping layer on the aspect ratio and optical response of the NPs. The information on the long-range order and the random (or not) orientation of the NPs can be derived. In comparison with cross-sectional bright-field TEM, the HAADF-STEM data represent a statistical average over 103 NPs. The morphological parameters derived from a HAADF-STEM analysis can be used to simulate accurately the absorption spectra obtained in the visible range by spectroscopic ellipsometry measurements of the sandwiched Ag NPs, thus confirming the validity of the HAADF-STEM analysis. The HAADF-STEM analysis helps to reveal the average shape (in-plane diameter and height) of the individual embedded NPs, with no need for cross-section preparation or Z-contrast tomography measurements, which require a big number of projections to be collected over a wide tilt range.452 In another report, microscopy techniques, including HAADF-STEM, were employed to characterize bimetallic Cu–Au NPs with size in the range of 1–7 nm. The researchers noted that the HAADF-STEM imaging provides thickness contrast, which is linearly proportional to specimen thickness, and atomic number contrast, which is proportional to the atomic number Z. The compositional sensitivity of HAADF images allows the investigation of heterogeneous materials with components of very different atomic numbers present. In that paper, the HAADF-STEM imaging of a cubo-octahedral particle supported a mixed Cu–Au configuration.453 Calvino and colleagues focused on the characterization of Au catalysts supported on a Ce–Tb–Zr mixed oxide. In general, HAADF-STEM operates well when metal NPs are dispersed within light support materials, such as zeolites or alumina, for which large differences between metal and support element atomic numbers contribute to a high contrast in the images. The quantitative 3D HAADF-STEM tomographic analysis of nanometer-sized noble metal particles supported on oxides of high atomic number (Ce, Tb and Zr) was proved to be feasible.454
Quantitative HAADF imaging at the atomic level can also be used to measure the number of atoms contained in a NP or a cluster. For instance, bimetallic 8 nm FePd NPs were studied by HAADF-STEM to determine their chemical composition. Particularly, HAADF was used to identify the chemical variations of a population of NPs, i.e. measure the statistical dispersion in chemical composition.455 Filippousi et al. studied with HAADF-STEM the polyhedral iron oxide core–shell NPs in a biodegradable polymeric matrix, and they found out that the NPs consisted of well-defined polyhedral structures with multiple facets.456
Aberration-corrected electron microscopy. The performance of electron microscopes may be limited by spherical aberration, a feature of all round lenses that causes image distortion and limits the resolution. The relatively recent development of aberration correctors for the objective lens resulted in a radical improvement in the resolution limits of HAADF-STEM microscopes. The Palmer group used aberration-corrected electron microscopy and atomistic computer simulations to demonstrate the hierarchy of metastability in the deposited, size-selected Au nanoclusters.457 They have also investigated the atomic structure of the Au55(PPh3)12Cl6 Schmid cluster by using aberration-corrected STEM combined with the multislice simulation of STEM images. The combination of size-fractionation by the STEM mass balance method and atomic structure determination in the aberration-correction regime might be able to reveal the isomeric structures of other types of NPs too.458 The use of chromatic aberration correction is in general expected to allow a much larger fraction of the incident electrons to be used to record high spatial resolution images than by using energy filtering.459 In another report, aberration-corrected STEM was used to probe, one cluster at a time, the atomic structure of a statistical ensemble of 79 Au clusters as a function of irradiation time. Each cluster contained 923 ± 23 atoms.460 Midgley and co-workers used high resolution aberration-corrected electron microscopy and 3D electron tomography to localize Au NPs supported on TiO2. The aberration correctors in the HAADF-STEM imaging helped to gain insights into the atomic level structure critical to understanding the reactivity properties of nanocatalysts.461 Rellinghaus and colleagues used aberration-corrected HRTEM for the quantitative measurement of the surface self-diffusion on Au NPs.462 In another report, aberration-corrected STEM provided the direct atomic-resolution imaging of surface migration, coalescence and atomic rearrangement of Au clusters on a Y:ZrO2 support.463
Bimetallic NPs, such as Au/Pd NPs, have also been investigated by aberration-corrected STEM. Ferrer et al. used this technique to study the atomic structure of three-layer Au/Pd NPs, in combination with theoretical simulations and single particle diffraction. The authors note that the aberration corrector offers the possibility to study atomic structures at a resolution lower than 0.1 nm, enabling the acquiring of more detailed information.464 Esparza et al. also used the technique under discussion for Au–Pd core–shell NPs and they observed the presence of Au NPs with preferential surfaces enriched with Pd atoms. These NPs were synthesized using Au NPs as core seeds and the final Au–Pd particles reached an average size of 5.5 nm.465 Ricolleau and co-workers performed aberration-corrected electron microscopy measurements and revealed in an unambiguous way the existence of long-range chemical orders in Au–Pd NPs. These ordered Au–Pd NPs may offer a new class of advanced nanocatalysts for various chemical reactions.466 Jose-Yacaman and co-workers combined aberration-corrected STEM with spectral and chemical analysis STEM-EDS and STEM-EELS to identify and better understand the interface structure of Pd–Au NPs. The atomistic structure and alloying of Pd–Au–Pd tri-layer NPs were investigated.467 In another report, Co/Au and Pd/Au NPs were deposited on grids aiming to study the coalescence of the different metals. The as-synthesized materials (Co/Au) were sintered by thermal treatment or by strong beam irradiation and the subsequent characterization was performed in situ in an aberration corrected STEM.468
Jian and Palmer investigated the variation of the core atomic structure of thiolated (AuxAg1−x)321±55 nanoclusters with composition using aberration-corrected HAADF-STEM. Fig. 15 demonstrates a comprehensive set of high-resolution HAADF-STEM images of AuAg alloy clusters with their respective simulated images (for bare Au309 clusters).469 Cu–Au core/shell clusters synthesized through cluster-beam synthesis were also analysed by aberration-corrected STEM. Insights were obtained into the growth kinetics of the bimetallic clusters leading to the controlled, selective and efficient production of different metastable but practical core/shell NP morphologies.470 Furthermore, Herzing et al. showed that aberration-corrected STEM-EDX can provide important high spatial resolution compositional information on (i) alloy homogeneity and phase segregation effects within individual NPs, (ii) particle-size composition correlations, (iii) the detection of trace amounts of the alloying element and (iv) metal component distribution in extremely highly dispersed catalyst systems for the case of Au–Ag and Au–Pd bimetallic NP systems.471 The disclinations in those decahedral Pd nanostructures with D5h symmetry were studied by aberration-corrected HRTEM. These researchers mentioned that the advantage of the aforementioned technique is to minimize the possibility of image artefacts that might confuse the geometric phase analysis of the NPs.472 The coalescence and sintering of small (<3 nm) Pt NPs under the influence of the electron beam was also studied by aberration-corrected HAADF-STEM in real time. The authors of that study showed that this technique allows single atomic columns to be clearly identified within each nanoparticle. Such measurements are significant in order to understand how particle size influences mass transport in nanoscale materials.473 Hashimoto et al. used aberration-corrected scanning confocal electron microscopy for the 3D analysis of Pt NPs on carbon nanohorn aggregate supports. In comparison with HAADF-STEM, the confocal electron microscopy improves the depth resolution because in the former method such resolution is limited by the lateral size of the objects and the illumination angle. It is expected that a continuous improvement in aberration correction will enable the use of larger convergence and collection angles, or image-processing techniques, such as the deconvolution method may result in depth resolution values close to those theoretically predicted. In this way, such approach can become a routine one for structural NP characterization.474 The same researchers used the aberration-corrected TEM for the in situ observation of Pt NPs on graphene layers. The structural changes and motions at the Pt colloids under high temperature were also characterized by the assistance of EELS measurements. The ability of single atom detection even at high temperature by aberration-corrected TEM facilitates the understanding of the interactions between catalytic NPs or atoms and graphene on an atomic scale, resulting in the development of more efficient catalyst–graphene composites.475
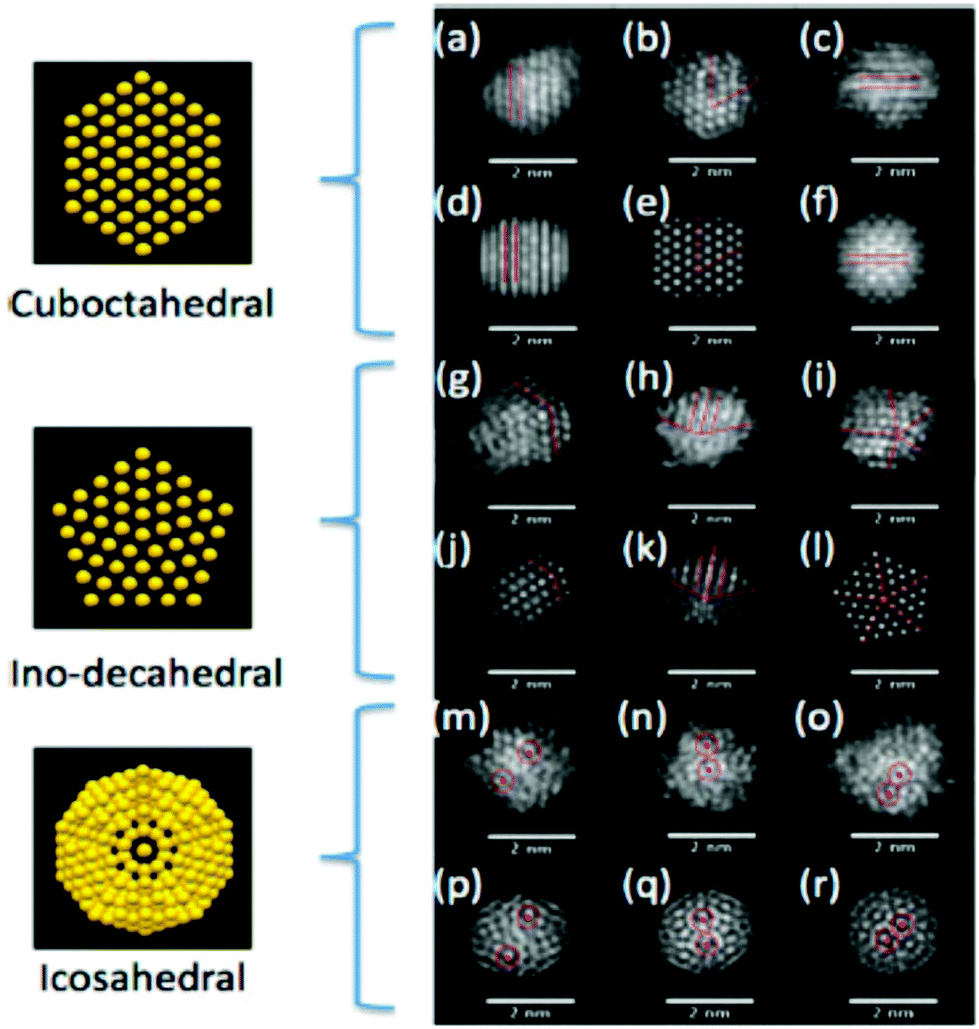 | ||
| Fig. 15 Typical HAADF-STEM images of thiolated (AuxAg1−x)312±55 clusters. (a)–(c), (g)–(i) and (m)–(o) are clusters assigned to cubooctahedral, ino-decahedral or icosahedral structures, based on (d)–(f), (j)–(l) and (p)–(r), the corresponding simulated images (for bare Au309 clusters, which is the closest full shell size of cuboctahedral, icosahedral and ino-decahedral). Reprinted with permission from ref. 469. Copyright 2015 American Chemical Society. | ||
Pt/γ-Al2O3 NPs (Pt clusters on an alumina support) were investigated by Sinkler et al. through a combined approach using aberration-corrected TEM (AC-TEM) and in situ XAFS. In comparison with STEM, aberration-corrected TEM uses a broad coherent electron beam and thus can offer advantages relative to STEM for the structure determination of fine clusters; this is because of the reduced tendency of the structures to deteriorate under the electron beam upon using AC-TEM. The complementarity of AC-TEM with the XAFS measurements is assured because it provides an ensemble-averaged view of the structures.476 Ling and Zhang used aberration-corrected STEM (AC-STEM) to map the reactions of Cr(VI) in Fe NPs. Fig. 16 provides STEM-EDS elemental mapping of Fe(Kα), Cr(Kα), O(Kα) and corresponding color overlays of the spent iron NPs after 24 h of reactions with hexavalent chromium.477 Ortalan et al. demonstrated the use of AC-STEM for the study of supported Rh–Ir clusters, combined with dynamic multislice image simulations, so as to identify individual atoms, map the full structure and determine changes in the positions of metal atoms in sequential images. The outmost goal of their approach was to help the development of new and improved catalysts and other functional nanostructures. The combination of AC-STEM with the simulations provided the critical experimental input required to determine the full 3D-structure of a nanocluster composed of Rh and Ir atoms.
 | ||
| Fig. 16 Aberration-corrected STEM-EDS elemental mapping of nZVI reactions with Cr(VI): (a) Fe, (b) O, (c) Cr and (d) Fe + O + Cr overlay. Reprinted with permission from ref. 477. Copyright Royal Society of Chemistry 2014. | ||
The resolution achieved went down to the atomic scale and the authors confirmed the capacity of AC-STEM to provide information on the size, shape, number and types of atoms in a nanocluster, as well as how they change with processing conditions and under the influence of reactants.478 Lead chalcogenide NPs were also investigated using aberration-corrected STEM; in that study, PbSe and PbTe NPs were chemically synthesized and a combination of electron diffraction, EDXS, EELS and AC-STEM was employed to acquire information related to their morphology, crystal structure and composition.479 The results obtained implied the presence of a NP surface rich in Pb and poor in chalcogen, with no oxygen, and a clear C signal that might be attributed either to the supporting film or to the presence of carbon in the capping layer as well.
Electron energy loss spectroscopy (EELS) is a family of techniques that measure the change in kinetic energy of electrons after their interaction with a material. The sample tested is exposed to a beam of electrons with a known, narrow range of kinetic energies. Some of the electrons will undergo inelastic scattering, which means that they lose energy and have their paths slightly and randomly deflected. The amount of energy loss can be measured via an electron spectrometer and interpreted in terms of what caused the energy loss.
EELS is typically used to identify the atomic structure and chemical properties of a sample, including: the type and quantity of atoms present, chemical state of atoms and collective interactions of atoms with their neighbors. Schaffer et al. compared energy-filtering TEM (EF-TEM) and STEM-EELS for the plasmon mapping of Au NPs in a monochromated TEM. They found out that the STEM EELS approach provides higher energy resolution, and thus allows the accurate mapping of peak positions, whereas the EFTEM technique provides spatially highly resolved information over large fields of view in a comparably short acquisition time. It is thus the ideal technique to monitor long distance effects as encountered in coupled systems.480
McComb and co-workers demonstrate the use of EELS-STEM as a powerful tool for the study of LSPR in silver NPs. Plasmon modes were highly sensitive to changes in local geometry and could be affected by electron beam damage, although special care in specimen preparation techniques could minimize such damage. Experimental data were in good agreement with theoretical predictions.481 Collins et al. noted that it is possible to combine EELS with electron tomography in order to image surface plasmon resonances qualitatively at the nanoscale in a 3D mode. The eigenmode tomography enables EELS to analyse not a particular electron-induced response, but the underlying geometric modes characteristic of particle surface plasmons. The precise optical analysis of single particles, particle ensembles and plasmonic devices is possible. Fig. 17 shows an EELS analysis of a plan-view silver right bipyramid that highlights many key issues in 2D imaging.482 In another report, Wei et al. noted that the high spatial and energy resolution EELS-STEM approach could be used to study several coupling interactions of a plethora of metal–semiconductor nanocomposite systems. In particular these researchers observed a strong exciton–plasmon coupling between ZnO nanowires and Ag NPs by the monochromated EELS-STEM technique.483
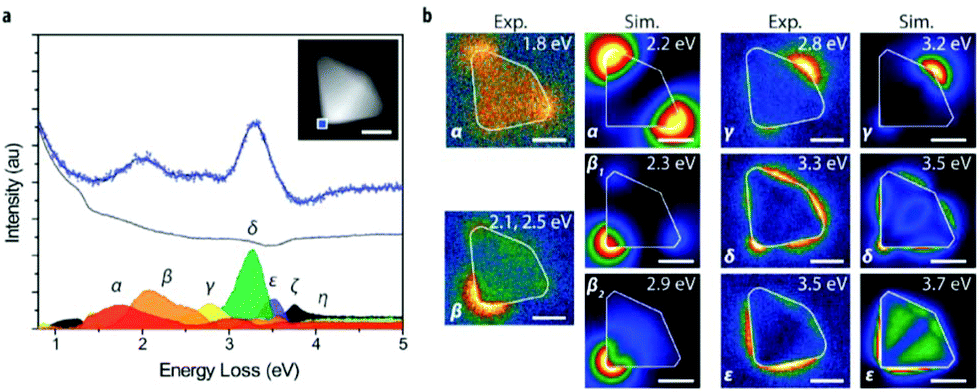 | ||
| Fig. 17 Plan-view EELS spectrum imaging of a silver right bipyramid. (a) Non-negative matrix factorization of EELS for a selected area (blue square, inset). The decomposition is shown for a 4.3 nm × 4.3 nm (9 pixel × 9 pixel) region. Blue dots represent the summed raw spectra, the black line is the sum of all decomposition components, the gray line corresponds to the spectral signature of the zero loss peak, and each of the remaining components corresponds to a spatial map that exhibits a dominant contribution matching a surface plasmon mode of the bipyramid (α–ε), the bulk plasmon (ζ), or the MoO3 substrate band edge (η). (b) NMF component maps (Exp.) and simulated energy loss probability maps (Sim.). Intensities are plotted on a normalized scale for each map. Subscripts on β denote fully resolved peaks in simulated spectra represented in the single experimental non-negative matrix factorization component β. Energies refer to peak maxima in the respective experimental and simulated spectra. Scale bars are 25 nm. Reprinted with permission from ref. 482. Copyright 2015 American Chemical Society. | ||
Ni NPs encapsulated in carbon, prepared by Rojas and colleagues, were characterized by TEM, EELS and EFTEM.484 The combined analysis indicated that the Ni nanocrystallites were surrounded by amorphous C, which provided some protection to the metallic Ni from oxidation. The EELS technique records core level absorption edges in an analogous way to XAS but provides information at a microscopic level. In another report, the location and role of Al in Al-modified titania NPs were determined using low-temperature heat capacity, EELS and XRD. EELS measurements confirmed that Al entered the TiO2 lattice but it also indicated that the short-ranged structure around the Al atoms shifted from a TiO2-like environment toward an Al2O3-like environment, as the dopant concentration increased. XRD showed that the long-range order of the NPs decreased as the dopant concentration increased but retained a basic TiO2-like structure. The heat capacity data showed that lattice vacancies increased significantly with the addition of the Al dopant, suggesting that the Al3+ cations entered the titania lattice and created vacancies due to the charge difference between Al3+ and Ti4+.485 Crozier and colleagues used monochromated EELS-STEM to measure bandgap states in individual non-stoichiometric praseodymium-ceria NPs. The authors of that study reported that the combination of EELS and AC-STEM offers new opportunities for the local nanoscale probing of bandgap states, and correlation with structure and chemistry at the 0.1 nm level. EELS allows the width and energy position of the state to be determined with respect to the top of the valence band, while optical observations of chemically-induced color changes are employed to provide further information on the energy shift of the inter-band state when the Pr oxidation state is changed. In that paper, high spatial and energy resolution monochromated EELS helped to detect a state within the bandgap of ∼30 nm NPs composed of PrxCe1−xO2−δ. That inter-band state was associated with Pr4+ 4f levels. The ultra-high resolution STEM-EELS permits inter-band states to be probed with high spatial resolution and should be applicable to other systems where nanocharacterization is necessary, such as grain boundaries, dislocations and precipitates.486
De La Fuente and co-workers used spatially-resolved EELS to analyse the antibody distribution on biofunctionalized core–shell Fe3O4 NPs. Spatially resolved EELS-STEM analysis was performed on such biofunctionalized NPs on the basis of its suitability to gain insight not only into the morphology and chemical composition of the NP surface, but also into the direct visualization and spatial localization of the organic biomolecules. In that report, the authors showed that their functional moieties (i.e. the antibodies) were located only in specific areas of the NP surface, namely those in which N was detected. Both biochemistry techniques and TEM studies provided complementary information for the evaluation and understanding of the validity of their functionalization protocol.487 Another interesting ability of quantitative EELS measurements is to distinguish the core from the shell in NPs with such configuration, for example in the case of the MnOx/MnOy and FeOx/MnOx core/shell, in a study published by Peiro and colleagues. The EELS data obtained from spectrum lines across several NPs showed that the Mn oxidation state was 3+ at the outer part of the NPs (where no Fe signal was found) and decreased moderately towards the centre of the NPs. Importantly, it appears that the power of the quantitative EELS technique to resolve core/shell structures is sufficient even in cases where HRTEM or HAADF cannot distinguish them.488
TEM and relevant techniques give a two-dimensional (2D) projection of three-dimensional (3D) objects. To tackle this problem, 3D electron microscopy or so-called ‘electron tomography’ (ET) has been developed. Apart from 3D structural information, the chemical composition can be analysed in 3D by combining the concepts of tomography with analytical TEM techniques. In this way, electron tomography is now considered as an important tool for the comprehension of the relation between the properties and structure of NPs. Nowadays, ET can provide quantitative 3D information down to the atomic scale. In addition to NPs, ET can also be employed for the study of NP assemblies. With ET, typically a tilt of photos (snapshots) is acquired by tilting the sample in TEM over a large tilt angle range. Using a mathematical algorithm, the tilt series is combined into a 3D reconstruction of the original object. In this manner, several different 3D images of the NP are obtained, together with a video that is a sum of all projections. An example of such reconstruction is shown in Fig. 18. In that image, a reconstruction of the structure of Au nanorods is shown. Such experiments allow the study of the influence of the synthesis method on the final shape of Au nanorods.489
 | ||
| Fig. 18 Atomic scale reconstruction of Au nanorods. (a, b) Orthogonal slices through the atomic scale reconstruction of Au nanorods prepared using different surfactants. The side facets of these rods can be clearly recognized. (c) Strain measurement along the major axis of the nanorod. Reprinted with permission from ref. 489. Copyright Wiley-VCH 2014. | ||
Fig. 19 shows a schematic summary of the function mode of the ET. In the paper containing that figure, Meurig Thomas et al. have described the concept of compressed sensing (CS) that can be allied to the ET aiming to use the resultant CS-ET approach, especially for particles of organic or biological materials, which are particularly prone to damage by the electron beam. The aim of compressed sensing is to obtain a signal from fewer measurements than would normally be required.490 van Aken and co-workers used ET to study the growth of 1D-CuPcF16 nanostructures onto Au NPs. To understand this growth, it is necessary to know the shape of the 1D nanostructure and its geometrical relationship with the Au particle. The experimental results from the combination of tomography and HRTEM provided a detailed 3D insight into the structural properties of the 1D self-organization of CuPcF16 molecules onto the Au NPs, which resulted in the proposed growth model of the 1D nanostructures.491 The benefits of ET for the characterization of the precise morphology of core–shell Au@Ag NPs and its implications on their plasmonic properties were also analysed by Coronado and colleagues. In their paper, it is noted that ET provides more statistically significant information on core–shell systems compared to other commonly used techniques. Bright-field TEM (BF-TEM) imaging may easily lead to artifacts upon 3D reconstruction, whereas HAADF-STEM matches much better with the requirements needed for tomographic applications (e.g. minimal diffraction or phase contrast).492
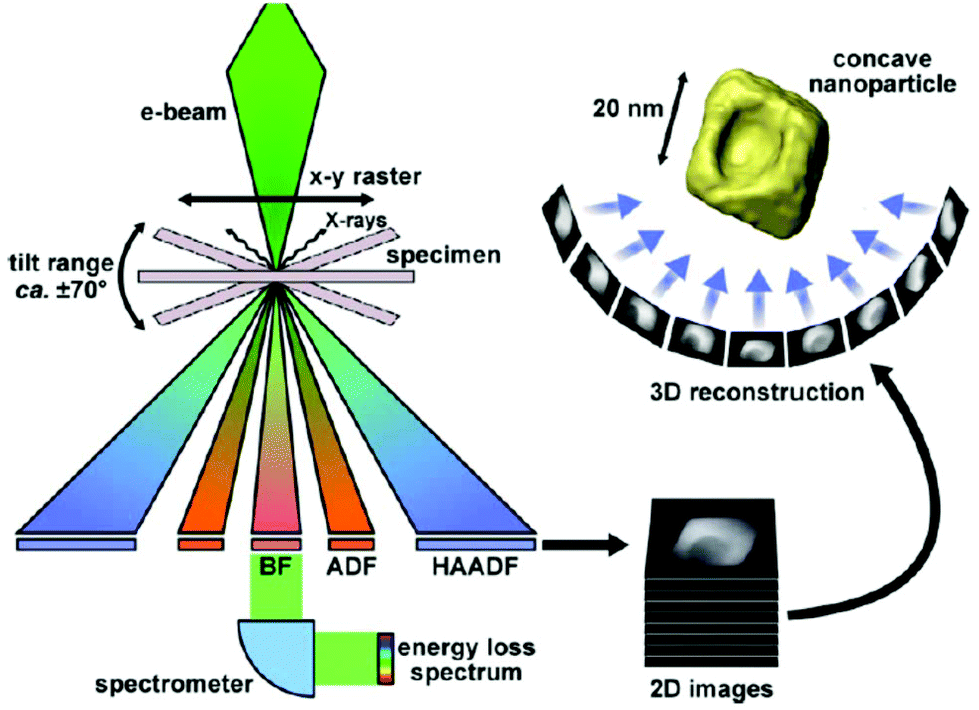 | ||
| Fig. 19 The essence of ET: an angular series of 2D projection images is recorded by tilting the specimen in the (scanning) transmission electron microscope. The ‘tilt-series’ of images are then back-projected into space to obtain a 3D reconstruction. A variety of signals may be recorded, including HAADF signals. The bright-field detector can be removed to allow the transmitted electrons to pass through to a spectrometer and form an energy-loss spectrum. Reproduced with permission from ref. 490. Copyright 2013 Elsevier. | ||
Other noble metals such as Pd have also been investigated using HAADF-STEM-electron tomography. Pd NPs with complex shapes were the subject of the study of Berhault and colleagues. Such 3D approach is expected to yield quantitative information, such as angle measurements and facets indexing, deduced from the acquired tomogram. Shapes such as pentagonal rods and bipyramids were among the Pd nanostructures monitored in that study.493 Florea et al. used ET to study the selective deposition of Pd NPs inside the bimodal porosity of β-SiC. The spatial distribution and connection of the porous network of the medium surface area β-SiC synthesized through the gas–solid reaction were investigated by ET. The obtained results illustrated the unique character of the ET to shed light on the morphology, internal structure, and spatial distribution of a nanoscale material. Such information is crucial for the field of catalysis, for instance.494 In another report, Blacher and co-workers monitored the localization of Pd NPs within their silica support, in two heterogeneous catalysts prepared by the sol–gel method, with different metal loadings. By using ET, it was found that the presence of artifacts was associated with an overestimation of the size of the Pd NPs. The resolution of the tomograms could be roughly estimated as the ratio of the thickness of the sample to the number of projections used for the reconstruction. The Pd NPs were located deep inside the silica skeleton. It was found that the dispersion manner of the Pd NPs also partially depended on their loading amount.495 ET has also been employed for the characterization of Pt nanostructures, such as nanodendrites496 and small NPs entrapped in zeolite. The quantitative and qualitative location of the latter particles was achieved through ET. H2PtCl6 was used as the Pt source for the impregnation of ultrastable (USY) zeolite support and 3–4 nm Pt NPs did not show any preferential location in mesopores or at the surface of the crystals. The size distribution as well as the distances between Pt NPs were also identified. A comparison of the size values with those obtained by EXAFS was also included in that work.497
Ricolleau and colleagues published a paper on the comparison of ET and HRTEM slicing methods as tools to measure the thickness of CoPt NPs deposited on a substrate. Regarding thickness measurements, ET presents several advantages over HRTEM, although the precision of the experiments obtained by these two techniques is similar.498 ET is a more direct (in what concerns the recording of the thickness value) and readily statistical approach, since more particles can be analysed at once. Concerning imaging modes, the bright-field one is associated with the presence of artifacts, as mentioned before. The HAADF-STEM mode avoids the occurrence of diffraction contrast problems, but BF-TEM involves short exposure time for each image, less distortion due to a residual specimen drift, limited sample contamination and small image distortion due to electrical instability.498
Iron oxide NPs have also been extensively studied by ET: for example, Midgley and colleagues applied the compressed sensing-ET approach in order to reveal the morphology of iron oxide NPs with reactive concave surfaces in great detail, and with fewer artifacts in comparison with the use of more ‘classic’ reconstruction algorithms. The reduction of missing wedge and star artifacts allows the simpler and more objective segmentation of tomograms, leading to a greater reliability of the 3D quantitative analysis of nanostructures. Only a few projections are enough for the reconstruction of decent tomograms using CS-ET, thus showing the ability of this technique for rapid data acquisition.499 In another report, magnetic NP composites with a Fe3O4 core and a hydroxyapatite coating were synthesized using the precipitation method followed by hydrothermal treatment. The combination of energy-filtered TEM (EF-TEM) and 3D-reconstructured electron tomography demonstrated that the nanocomposites consisted only of needle-like hydroxyapatite nanocrystals coating the magnetite spherical NPs which had internal nanopores.500 The capacity of the compressed sensing anisotropic total variation algebraic reconstruction technique (CSATV-ET) to improve the quality and accuracy of tomograms using fewer datasets when compared to more ‘common’ reconstruction techniques (e.g. SIRT and BP) was illustrated by Monsegue et al. also for the case of hematite NPs.501
Scanning electron microscopy (SEM) is a widely used method for the high-resolution imaging of surfaces that can be employed to also characterize nanoscale materials. SEM uses electrons for imaging, much as a light microscope uses visible light. Mazzaglia et al. combined field-emission SEM (FE-SEM) and XPS measurements to study supramolecular colloidal systems of Au NPs/amphiphilic cyclodextrin. These two techniques provided important information on the morphology and nature of the interaction of (thiohexyl carbon chain) SC6NH2 and (thiohexadecyl carbon chain) SC16NH2 with Au NPs onto the silicon surface.502 Sinclair and co-workers have reported that SEM and NanoSIMS can be employed to locate Au NPs in cells. SEM analysis illustrated its superiority compared to NanoSIMS for the analysis of inorganic NPs in complex biological systems. NanoSIMS has a lower spatial resolution of around 50 nm while SEM is able to achieve resolutions down to 1 nm. The particles tested were Raman-active Au–core NPs and NanoSIMS resulted in somewhat blurred images in certain cases due to its limited resolution. However, NanoSIMS has the unique capability to differentiate between isotopes, although this is not relevant for the case of Au NPs.503High-resolution SEM (HRSEM) was used by Goldstein et al. for the imaging of Au NPs in cells and tissues. The straightforward visualization of metallic NPs is assured with this technique, and the sample preparation is fast and easy. However, in case of biological specimens, the need to decrease charging artefacts might make metal coating necessary, thus increasing the risks of radiation damage for the samples. The advantage of HRSEM, compared to other imaging techniques, is the capacity to scale down and study the arrangements of nanometric elements in their wider context. It allows the study of the specific spatial arrangement of NPs and thus the examination of possible interactions between them. The results of that study indicated the potential of HRSEM as a relatively simple tool to qualitatively screen the factors that enhance Au NPs penetration, through the skin barrier. It can be considered as a powerful and diverse tool for the study of the interactions between biological systems and metallic nanostructures.504 In another report, SEM and AFM measurements were compared for the same set of NPs, that is, SiO2 and Au NPs on mica or silicon substrates. For example, AFM observations enabled the measurement of the height of a nano-object with sub-nanometric accuracy, but the lateral measurements (along the X and Y axes) had large errors because of the tip/sample convolution. In contrast to the AFM, SEM cannot provide any metrological information on the height of the NPs; however, modern SEM can give decent measurements of their lateral dimensions. In fact, the measurements of nearly spherical SiO2 NPs by using both techniques gave similar results, showing the coherency and complementarity of both instruments.505
SEM can be operated in the transmission mode, i.e. through the technique called ‘transmission in scanning electron microscope’ (T-SEM) (see Fig. 20). In the transmission mode, advanced NP analysis can be carried out by gaining in-depth information as well as analysis of ensembles of NPs. In a paper by Rades et al., the combination of complementary techniques as SEM, T-SEM, EDX and scanning Auger microscopy (SAM) was proven to be a powerful strategy for comprehensive morphological and chemical evaluation of the properties of individual silica and titania NPs. On the other hand, methods such as SAXS, DLS, XPS, XRD and BET would be suitable to characterize only the ensembles of the NPs, and not single particles. T-SEM allows a quick examination of the NP shape, though its lateral resolution is limited to NP sizes down to 5–10 nm. TEM provides images with better quality, but T-SEM can be easily combined with EDX for a fast check of the NP size and elemental composition.506 Hodoroaba et al. proved that T-SEM imaging provides a size distribution that is slightly broader than that obtained by TEM. For small SiO2 NPs, the precise delimitation of the particles in the T-SEM mode is definitely constrained by the lower spatial resolution achieved compared to that of conventional TEM. In addition, with the T-SEM, the surface layer of the particles might not be always easily detected.507 The same author noted in another paper that the conventional SEM imaging mode could not detect the NPs on the back side of the support film that was required for the observations. Therefore, an explicit knowledge of the T-SEM operator is needed for the measurements. The authors observed that the obtained SiO2 NP size distributions by SEM and TSEM in their work and for various conditions agreed well with each other, within the associated measurement uncertainties.508 In another report, 3D reconstruction by focused ion beam (FIB) cutting and SEM imaging were combined to comprehend the evolution of pore volume, pore shape and other parameters during the two-step sintering of ZnO NPs. In this way, the sintering process at the nanoscale for such particles can be better understood.509 Ni- and Cu-co-doped zinc oxide NPs prepared by the co-precipitation method were investigated by Ashokkumar and Muthukumaran by microstructure, optical and FTIR measurements. The depicted shape by SEM was in good agreement with the mathematical determinations from XRD, whereas FTIR provided important information on chemical bonding.510
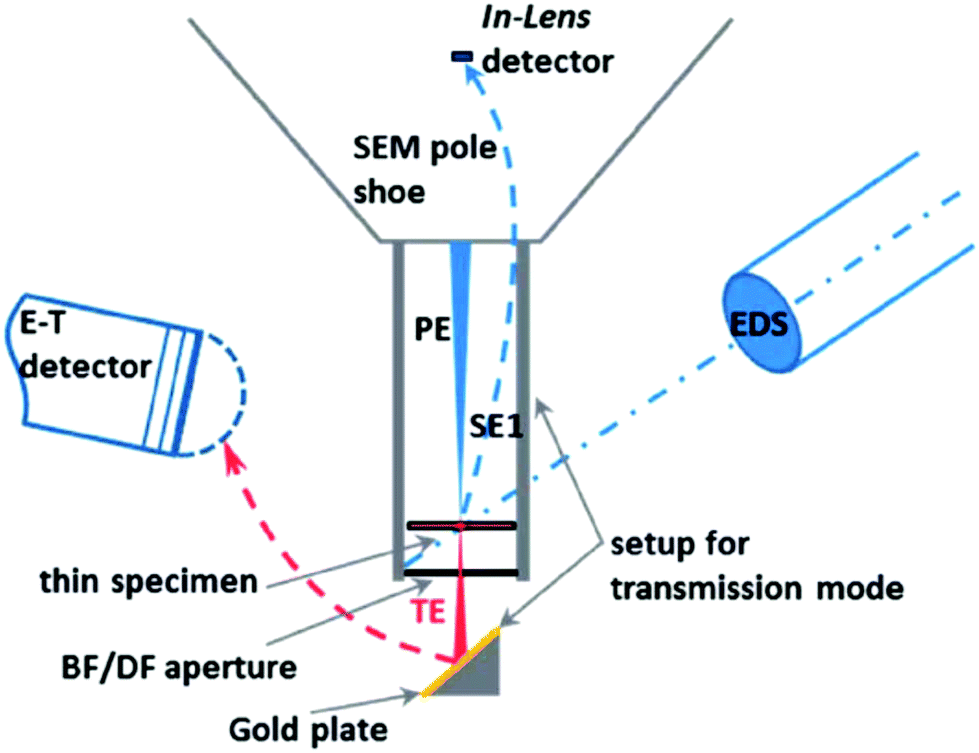 | ||
| Fig. 20 Scheme of a SEM/EDS system operating in the transmission mode with the Zeiss single-unit transmission setup (PE: primary electrons; SE1: secondary electrons emitted at the point of impact of the PE on the sample; TE: transmitted electrons; BF: bright field; DF: dark field; E–T: Everhart–Thornley detector). Reprinted with permission from ref. 506. Copyright Royal Society of Chemistry 2014. | ||
Electron backscatter diffraction (EBSD) is a microstructural–crystallographic characterisation technique for the study of crystalline or polycrystalline materials, including nanoscale ones. This technique aims at the comprehension of the structure, crystal orientation, and phase of the materials in SEM. Normally, EBSD is employed to examine microstructures, revealing texture, defects, grain morphology and deformation. High resolution and non-destructive analysis of these parameters can be achieved. In EBSD, an electron beam hits a sample that is tilted at an angle of typically 70° towards the detector. The detector, usually a phosphor screen, which captures the inelastically backscattered electrons from the sample surface, is able to make a diffraction pattern.511 EBSD can effectively improve the statistics of the analysis of NPs compared to TEM, thus giving a better overview of a larger ensemble of NPs. The heteroepitaxial relationship of Au NPs with an average size of 60–80 nm on (001) ϒ1Ba2Cu3O7−δ has been investigated by EBSD. In that case the small size of Au NPs compared to the spatial resolution of the EBSD caused a certain challenge on the orientation analysis.512
In another report, EBSD was used to directly measure the crystallographic orientation of embedded Y2BaCuO5 and Y2Ba4CuMOx NPs in melt-textured YBCO, with a spatial resolution of around 40 nm. The researchers of that study aimed to explore how the behaviour of the superconducting matrix was modified upon embedding a certain quantity of NPs. The interactions between these NPs and the surrounding YBCO matrix were studied. A novel finding of that work concerned the observation of twin boundaries within the melt-textured YBCO samples through the use of EBSD. The EBSD analysis showed that the addition of depleted uranium oxide had a remarkable effect on the resulting microstructure of the melt-processed YBCO samples.513 The same group found out that a homogeneous YBCO matrix can be formed, even though a large number of embedded particles are present.514 Small et al. reported that the primary cause of the reduced EBSD pattern quality from NPs is an increase in the diffuse background contribution or noise resulting from electron penetration through the small particles into thick, amorphous mounting substrates and not the loss of the coherent scattering intensity. It was suggested that designing an EBSD sample holder that accommodates particles mounted on thin film substrates would help to decrease radically the background produced as a result of electron interactions with the mounting substrate. This would lead to an increase in pattern quality, extending the application of EBSD phase identification analysis to relatively low-Z, low-ρ particles as small as 120 nm in size.515
Atomic force microscopy (AFM) is a microscopy technique capable of creating three-dimensional images of surfaces at high magnification. It was initially developed by Gerard Binning and Heinrich Rohrer at IBM in 1986.516 AFM is based on measuring the interacting forces between a fine probe and the sample. The probe is a sharp tip and is coupled to the end of a cantilever, which is made of silicon or silicon nitride. When the AFM scans the sample, the cantilever gets deflected as a result of the attractive or repulsive forces between the tip and the sample surface (Fig. 21A). The bending is quantified by a laser beam that reflects on the cantilever back side. The forces are finally calculated by combining the information from the laser variation and the known cantilever stiffness. AFM can scan under three different modes depending on the degree of proximity between the probe and the sample, i.e. contact, non-contact and tapping mode (also known as intermediate or oscillating mode).517 The latter is the most common when characterizing NPs. However, it is very sensitive to the free amplitude of the oscillating tip.518 In addition, other parameters, such as tip curvature radius, and surface energy and elasticity of the nanoparticle, influence the final topological values. Nevertheless, these factors can be minimized by plotting the particle height against the free amplitude of the oscillating probe, providing more reliable results.518 Alternatively, non-contact is preferred when the sample is very sensitive and can be influenced by the tip–sample forces.517
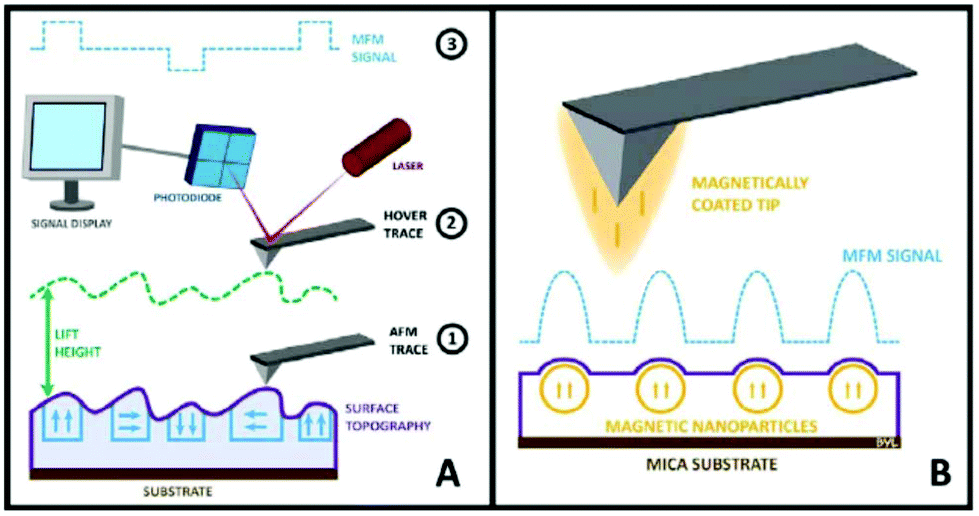 | ||
| Fig. 21 Schematic of AFM and MFM imaging techniques. (A) (1) An AFM tip scans the surface of a sample to produce a topographical trace, (2) the cantilever is raised to a user-defined height away from the sample surface and the retrace follows the original topographical pattern from the first step; (3) during the retrace, the magnetic signal is scanned and recorded for the sample. In all cases, the signals are recorded via the reflection of a laser beam off the back of the cantilever and onto a photodiode, where changes in cantilever deflection are detected. (B) In the case of using magnetic force microscopy to scan magnetic NPs on mica substrates, a magnetically coated tip is used to scan the sample surface and an MFM signal is obtained as it interacts magnetically with the sample and its magnetic domains or NPs. Reprinted with permission from ref. 536. Copyright 2016 United Scientific Group. | ||
AFM has the advantage that it does not require any surface modification or coating prior to imaging. Thus, the topological analysis of small NPs (≤6 nm), such as ion-doped Y2O3, has been performed by AFM without any special treatment.519 Low density materials, which present poor contrast in electron microscopy, have also been characterized. For instance, AFM was used to understand the formation mechanism of uniform patchy and hollow rectangular nanoplatelets made of polymer mixtures.520 Side-by-side comparison between AFM and electron microscopies, i.e. SEM and TEM, showed that AFM provided comparable results when analysing NP sizes.521–523 AFM has the advantage that images the sample in three dimensions and allows the characterization of the nanoparticle height. Furthermore, it has similar resolution to SEM and TEM, while costing much less and occupying smaller laboratory space. Nevertheless, AFM displays slower scanning times than any electron microscope. Alternatively, spectroscopic techniques, such as DLS and photon correlation spectroscopy (PCS), have also been used to characterize the nanoparticle size. DLS and AFM provided similar results when the sample analysed was monodisperse and uniform.523 However, only AFM could properly characterize NPs with bimodal distribution sizes.524 The study of alumina nanopowder formation in the solvent by ultrasonic treatment, and posterior sedimentation to a thin film, showed that correlated information could be obtained by PCS and AFM, even though the former analysed liquid samples and the latter solid ones.525 The combination of different technique strengths, such as the high magnification of HRTEM and height measurements of AFM, has helped to understand longstanding problems in nanoscience, like the role of the dendrimer template on the growth of Pt NPs.526 AFM and XRD were jointly used to characterize Ag NP films, where both techniques provided complementary information.527 In particular, AFM allowed the characterization of the grain size and the nanoparticle coverage of the surface, while XRD identified the preferential growth direction of the particles. Interestingly, at higher NP coverage, AFM showed that the film was made of larger particle grains. Nevertheless, XRD indicated that the crystal size remained the same. This apparent contradiction suggested that the larger particles were formed by coalescence of different crystals, yielding larger polycrystalline grains. It is worth mentioning that a densely packed nanoparticle film can be challenging to characterize by AFM, since part of the particles are hidden by their neighbours. Therefore, several algorithms have been developed to estimate the nanoparticle size from the visible part of the image. These algorithms can be applied to densely packed spherical528 and non-spherical particles.529
The catalytic activity of Rh NPs in the polymerization of phenylacetylene was characterized by AFM and TEM.530 Both techniques were able to track the formation of poly-phenylacetylene fibers around the NPs. However, only TEM could solve the pitch of the polymer helical structure. AFM has also been used to characterize different NP-based metal substrates for SERS sensing (Fig. 22).531,532 Different parameters, such as NP composition, size, shape and surface properties, were correlated to the measured enhanced factors, and near single molecule detection limit was achieved for one of the substrates.533 In addition, AFM was further employed to study the SERS phenomenon at the single-NP level.534 Lastly, AFM and Kelvin probe force microscopy could be combined to generate three-dimensional maps of nanoparticle surface potential distributions.535 These were obtained by monitoring the corrosion behaviour of individual iron and stainless steel NPs under a sulfuric acid environment (Fig. 23).
 | ||
| Fig. 22 AFM images of a nickel plate before (upper) and after (lower) the deposition of silver colloidal NPs. Reproduced with permission from ref. 531. Copyright 2007 Elsevier. | ||
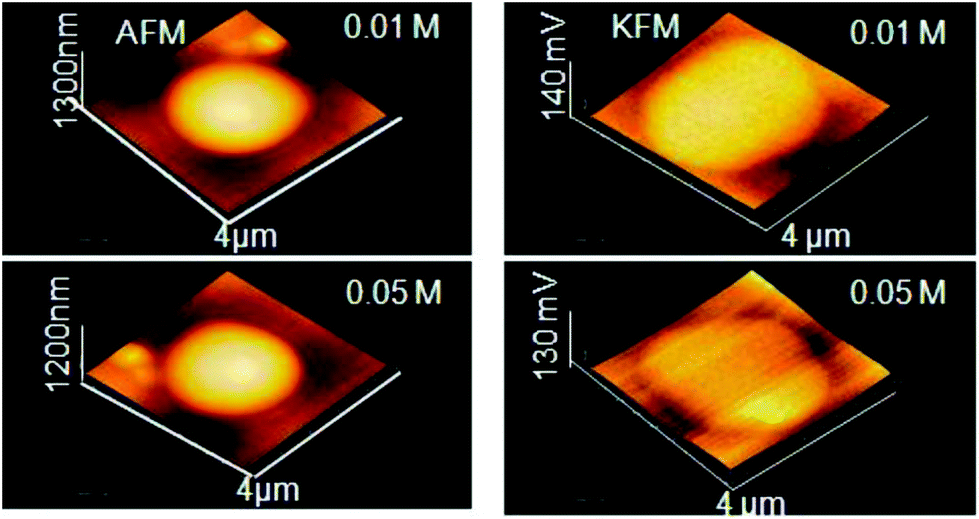 | ||
| Fig. 23 AFM (left) and KFM (right) images of pure iron nanoparticles in different concentrations of H2SO4. Reprinted with permission from ref. 535. Copyright 2014 ESG. | ||
Magnetic force microscopy (MFM) is a type of scanning probe microscopy where a magnetic probe is used to raster-scan the sample surface, of which its magnetic field interacts with the magnetic tip to offer insight into the magnetic properties (Fig. 21B). MFM has the ability to separate the magnetic interactions from the other tip sample forces (such as van der Waals, and other forces recorded in AFM). The most common measurement method is called the ‘two-pass technique’ in which the sample is scanned twice, once to produce an AFM image, and a second time to produce an MFM one. The MFM advantages include its non-destructive character, lack of surface preparation or NP modification, and no use of labels or tags. MFM allows the determination of the magnetic moment of a single NP and study of how this measurement changes with the NP size as well as probe distance from the sample, something which bulk magnetic analysis is not capable of. The development of MFM operating in liquid brings excellent possibilities of the studies of magnetic NPs under biologically relevant conditions, such as in the interior of cells. In addition, MFM can operate under ambient conditions, at varying temperatures and in a ultra-high vacuum environment. Furthermore, it can provide a resolution down to less than 10 nm.536 Neves et al. published a paper on how MFM can be used to discriminate between magnetic and non-magnetic NPs. MFM can detect and localize the magnetic fields arising from nanoscopic magnetic domains, such as magnetic NPs. Unfortunately, there are cases where MFM can give a strong response even on non-magnetic NPs or under circumstances where no magnetic interaction would be expected, potentially giving rise to misleading results.537
The magnetic field from NPs and consequently the phase shift that is detected in MFM depend very strongly on the particle diameter. MFM is sensitive to magnetic fields coming from magnetic NPs with diameters around 40–60 nm.537 On the other hand, the evaluation by MFM of small ferromagnetic or superparamagnetic NPs is particularly challenging: in this case, the formation of the MFM contrast takes place under conditions of strong interaction of the probe field and the particle magnetic moment, which complicates the interpretation of the experimental results. The application of an external magnetic field can result in the redistribution of the magnetic contrast from low-coercive Co NPs, as reported by Mironov et al., making it possible to distinguish between the contribution of the magnetic and van der Waals interactions to the generation of a phase contrast of the MFM images. Therefore, the observation of MFM contrast from superparamagnetic Co NPs smaller than 10 nm is still possible under magnetic moment stabilization in a strong external magnetic field.538
Asenjo and co-workers employed MFM for the study of superparamagnetic versus blocked states in aggregated of Fe3−xO4 NPs. Two distinct magnetic behaviours were observed depending on the particle size. Aggregates of NPs of about 11 and 49 nm in size were investigated. For the former sample, a homogeneous attractive tip–sample interaction was observed, displayed as a uniform dark contrast on the MFM images, arising from the coherent rotation of the spins within the aggregate as they align along the tip stray-field. This reflected the predominant superparamagnetic character of these small NPs within the characteristic acquisition time of the MFM technique and at zero applied field. For the sample with the 49 nm NP aggregates, dark/bright contrast associated with the existence of magnetic domains and magnetization–polarization prevailed in the MFM images all along the magnetic cycle. This happened due to the fact that the net magnetization of these large particles remained blocked during the acquisition time of the MFM images, even at zero applied field.539 Athanassiou and colleagues have published a study on a quantitative, high spatially resolved MFM imaging of samples based on 11 nm diameter superparamagnetic iron oxide NPs in air at room temperature, characterizing magnetic textures down to the single particle level. Energy loss imaging in the tapping mode can provide high compositional sensitivity and magnetic features as small as a few tens of nanometers lying under the surface were pointed out by MFM, whereas topographical imaging alone would not be able to detect them.540
2.5 Focus on NP size – distinct examples of characterization with different techniques
In this section we provide some examples of the literature where different methods are used at given samples to characterize their size, as this property is one of the most basic ones for NPs and it deserves special attention. Akbari et al. used TEM, PCS, BET and XRD to evaluate the size and size distribution of alumina NPs. The NP size was found to be in the range of 5–95 nm. XRD and TEM size values were in agreement for these particles. The authors of that study mention that PCS is well suited for the measurement of narrow particle size distribution in the range of 1–500 nm, but for systems where agglomeration occurs, comparison with other methods is recommended. The size value deduced from BET was also in accordance with the ones derived by TEM and XRD, as expected for particles with a spherical shape, but the recorded PCS value was higher.541 Gollwitzer et al. compared several techniques for the size measurement of silica NPs dispersed in water and in the cell culture medium. The techniques used were DLS, CLS (centrifugal liquid sedimentation), SAXS and PTA (particle tracking analysis). PTA is practically the same as NTA, but PTA is a more generic term which covers a larger range of particle sizes. The DLS results in the cell culture medium differed to a significant extent from the other methods, due to the presence of agglomerates, which diminish the DLS accuracy. The particle agglomeration caused by the cell culture medium resulted in a significant size increase in PTA, whereas the NP size value remained stable for SAXS and CLS measurements. SAXS offered highly precise values while CLS yielded detailed size distributions from which further information on the agglomeration state can be derived.542 Minelli and co-workers used tunable resistive pulse sensing (TRPS), DCS and DLS to measure the size of silica NPs in serum. Also in this case, DLS precision was not sufficient because of the presence of agglomerates. DCS and TRPS values were quite similar, though. The agglomeration measured by DCS was more significant than that observed by TRPS. In fact, in contrast to DLS and DCS, TRPS performs particle-by-particle measurements, providing a statistical distribution of the data across a NP sample rather than average results. The researchers authoring that study confirmed that TRPS is a sensitive and high resolution technique in the characterization of NPs in biological media.543 In another report, a certified reference material, ERM-FD100, composed of SiO2 NPs with a nominal equivalent spherical diameter of 20 nm, was characterized by researchers from 34 laboratories using DLS, CLS, EM (TEM/SEM), SAXS and ELS. Participants from both the industry and academic institutions showed that a good agreement for the results by different methods was confirmed. The good comparability of results enabled the certification of the colloidal SiO2 materials for NP size analysis.544 The size measurement uncertainties of near-monodisperse, near-spherical NPs composed of reference gold and polystyrene materials were compared in a paper by Mast and colleagues. PTA proved to be a precise and non-biased method for the determination of the modal hydrodynamic diameter in the range of 30–200 nm. TEM was accurate and non-biased for the measurement of the mean area-equivalent circular diameter in the size range between 8 and 200 nm of the investigated near-monomodal near-spherical materials. Therefore, PTA was found to be a good alternative to TEM for measuring the NP size, with the exception of 8.9 nm Au NPs, because that sample had a size below the detection limit of the former technique.545 Carney et al. described a 2D analytical ultracentrifugation approach for the determination of the size, density and molecular weight distributions of gold-based NPs. The extracted values for the sedimentation and diffusion coefficients from the analytical ultracentrifugation helped to find the above-mentioned parameters.546 Shard and co-workers used PTA to quantify the IgG protein adsorption to gold NPs. In the low protein coverage regime, the measured amount of protein depended upon the technique: NTA and DLS gave similar values that correlated well with the plasmon frequency shift. DCS analysis underestimated the protein shell thicknesses in that regime. DLS and NTA measurements resulted in larger diameters for the citrate-capped Au NPs than those provided by the supplier, with the DCS method giving smaller diameters. DLS and NTA assess NP size from the analysis of Brownian motion and should be expected to result in identical diameters for a monodisperse sample. It was noted that DCS was more precise than either DLS or NTA and less prone to artifacts. Apart from aggregates, DLS is also sensitive to impurities and NTA is statistically limited by the number of significant observations of the position that can be made on a single particle before it moves from the field of view.547 In another report, the combination of techniques such as WAXS and DLS that are sensitive to different characteristics of colloidal particles (Au, Ni(OH)2), such as crystalline core and overall size, permitted the estimation of the thickness of polymeric stabilizing layers. WAXS was efficient for the determination of the size and shape of dispersed colloidal particles.548 Finally, in a study concerning Ag NPs, quasielastic light scattering was employed for size measurement, and it was reported that it achieved rapid measurements, with a slightly higher size in comparison with TEM.5493. Summary and outlook
This review described the role of several different techniques for the characterization of nanoscale materials. Through this comprehensive summary of NP characterization methods, we demonstrated the uses of each one of them, emphasizing on their advantages and limitations, as well as on explaining how they can be effectively combined and how they can complement each other. The acquisition of a full picture of the variety of features that are associated with a nanomaterial requires typically the use of numerous techniques, often needing to use more than one of them for evaluating well and completely even a single property. By presenting the role of each technique in a comparative way, our review will act as a robust guide, helping the scientific community to understand better the discussed topic. In this way, researchers will be helped for the choice of the most suitable techniques for their characterization, together with the ability to assess their use in a more precise manner.Of course, there are challenges in the scientific community for the further improvement of the accuracy and resolution of many techniques. Therefore, we finally hope that a careful reading of this review will help to identify which valuable techniques merit efforts for further technical improvements.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge funding from the EPSRC (EP/M018016/1). NTKT thanks the AOARD grant (FA2386-17-1-4042). Dr Torsten Gutmann (Technical University of Darmstadt, Germany) is thanked by the authors for his feedback on the NMR technique.References
- N. T. K. Thanh, N. Maclean and S. Mahiddine, Chem. Rev., 2013, 114, 7610 CrossRef PubMed.
- K. M. Koczkur, S. Mourdikoudis, L. Polavarapu and S. E. Skrabalak, Dalton Trans., 2015, 44, 17883 RSC.
- Y. Wang, D. Wan, S. Xie, X. Xia, C. Z. Huang and Y. Xia, ACS Nano, 2013, 7, 4586 CrossRef PubMed.
- F. Kim, S. Connor, H. Song, T. Kuykendall and P. Yang, Angew. Chem., Int. Ed., 2004, 43, 3673 CrossRef PubMed.
- C. Minelli , talk on ‘Measuring nanoparticle properties: are we high and dry or all at sea?’ at ‘Nanoparticle Characterisation – Challenges for the Community’ event – IOP (Institute of Physics), book of abstracts, July 2016, London.
- P. Dobson , talk on ‘NPs: What do we need to know and can we measure everything we need to?’ at ‘Nanoparticle Characterisation – Challenges for the Community’ event – IOP (Institute of Physics), book of abstracts, July 2016, London.
- S. Upadhyay, K. Parekh and B. Pandey, J. Alloys Compd., 2016, 678, 478 CrossRef.
- W. Yan, V. Petkov, S. M. Mahurin, S. H. Overbury and S. Dai, Catal. Commun., 2005, 6, 404 CrossRef.
- W. Li, R. Zamani, P. Rivera Gil, B. Pelaz, M. Ibanez, D. Cadavid, A. Shavel, R. A. Alvarez-Puebla, W. J. Parak, J. Arbiol and A. Cabot, J. Am. Chem. Soc., 2013, 135, 7098 CrossRef PubMed.
- A. J. Pugsley, C. L. Bull, A. Sella, G. Sankar and P. F. McMillan, J. Solid State Chem., 2011, 184, 2345 CrossRef.
- X. Chen, Q. Cai, W. Wang, G. Mo, L. Jiang, K. Zhang, Z. Chen, Z. Wu and Z. Wu, Chem. Mater., 2008, 20, 2757 CrossRef.
- J. M. Ramallo-Lopez, L. Giovanetti, A. F. Craievich, F. C. Vicentin, M. Marin-Almazo, M. Jose-Yacaman and F. G. Requejo, Physica B, 2007, 389, 150 CrossRef.
- M. A. Newton, S. G. Fiddy, G. Guilera, B. Jyoti and J. Evans, Chem. Commun., 2005, 118 RSC.
- V. V. Srabionyan, V. V. Pryadchenko, A. A. Kurzin, S. V. Belenov, L. A. Avakyan, V. E. Guterman and L. A. Bugaev, Phys. Solid State, 2016, 58, 752 CrossRef.
- F. Klasovsky, J. Hohmeyer, A. Bruckner, M. Bonifer, J. Arras, M. Steffan, M. Lucas, J. Radnik, C. Roth and P. Claus, J. Phys. Chem. C, 2008, 112, 19555 CrossRef.
- T. Liu, L. Guo, Y. Tao, T. D. Hu, Y. N. Xie and J. Zhang, Nanostruct. Mater., 1999, 11, 1329 CrossRef.
- V. Krishnan, R. K. Selvan, C. O. Augustin, A. Gedanken and H. Bertagnolli, J. Phys. Chem. C, 2007, 111, 16724 CrossRef.
- C. Schmitz-Antoniak, Reg. Prog. Phys., 2015, 78, 062501 CrossRef PubMed.
- E. M. Moroz, Russ. Chem. Rev., 2011, 80, 293 CrossRef.
- J. A. Gomes, M. H. Sousa, G. J. da Silva, F. A. Tourinho, J. Mestnik-Filho, R. Itri, G. de Azevedo and J. Depeyrot, J. Magn. Magn. Mater., 2006, 300, e213 CrossRef.
- Z. Wu, L. Guo, H. Li, Q. Yang, Q. Li and H. Zhu, Mater. Sci. Eng., A, 2000, 286, 179 CrossRef.
- U. Wongpratat, S. Maensiri and E. Swatsitang, Appl. Surf. Sci., 2016, 380, 60 CrossRef.
- K. Zhang, Z. Zhao, Z. Wu and Y. Zhou, Nanoscale Res. Lett., 2015, 10, 37 CrossRef PubMed.
- Y. Tan, D. Sun, H. Yu, B. Yang, Y. Gong, S. Yan, Z. Chen, Q. Cai and Z. Wu, CrystEngComm, 2014, 16, 9657 RSC.
- J. Rockenberger, L. Troger, A. Kornowski, T. Vossmeyer, A. Eychmuller, J. Feldhaus and H. Weller, J. Phys. Chem. C, 1997, 101, 2691 CrossRef.
- G. Rafeletos, S. Norager and P. O'Brien, J. Mater. Chem., 2001, 11, 2542 RSC.
- C. I. Pearce, V. S. Coker, J. M. Charnock, R. A. D. Pattrick, J. F. W. Mosselmans, N. Law, T. J. Beveridge and J. R. Lloyd, Nanotechnology, 2008, 19, 155603 CrossRef PubMed.
- M. Dubiel, S. Brunsch, W. Seifert, H. Hofmeister and G. L. Tan, Eur. Phys. J. D, 2001, 16, 229 CrossRef.
- M. Heinz, V. V. Srabionyan, A. L. Bugaev, V. V. Pryadchenko, E. V. Ishenko, L. A. Avakyan, Y. V. Zubvichus, J. Ilhemann, J. Meinertz, E. Pippel, M. Dubiel and L. A. Bugaev, J. Alloys Compd., 2016, 681, 307 CrossRef.
- V. V. Srabionyan, A. L. Bugaev, V. V. Pryadchenko, A. V. Makhiboroda, E. B. Rusakova, L. A. Avakyan, R. Schneider, M. Dubiel and L. A. Bugaev, J. Non-Cryst. Solids, 2013, 382, 24 CrossRef.
- M. Dubiel, X. C. Yang and S. Brunsch, Phys. Scr., 2015, T115, 729 Search PubMed.
- M. Dubiel, S. Brunsch and L. Trogen, J. Synchrotron Radiat., 2001, 8, 539 CrossRef PubMed.
- A. Kafizas, S. A. Parry, A. V. Chadwick, C. J. Carmalt and P. Parkin, Phys. Chem. Chem. Phys., 2013, 15, 8254 RSC.
- H. Modrow, X-Ray Methods for the Characterization of NPs, in Nanofabrication Towards Biomedical Applications: Techniques, Tools, Applications and Impact, ed. C. S. S. R. Kumar, J. Hormes and C. Leuschner, Wiley-VCH, 2005, ch. 7 Search PubMed.
- B. Ingham, Crystallogr. Rev., 2015, 21, 229 CrossRef.
- A. M. Beale and B. M. Weckhuysen, Phys. Chem. Chem. Phys., 2010, 12, 5562 RSC.
- A. I. Frenkel, J. Synchrotron Radiat., 1999, 6, 293 CrossRef PubMed.
- Y. Sun, A. I. Frenkel, R. Isserof, C. Shonbrun, M. Forman, K. Shin, T. Koga, H. White, L. Zhang, Y. Zhu, M. H. Rafailovich and J. C. Sokolov, Langmuir, 2006, 22, 807 CrossRef PubMed.
- G. Cheng, J. D. Carter and T. Guo, Chem. Phys. Lett., 2004, 400, 122 CrossRef.
- A. Sharma, M. Varshney, J. Park, T.-K. Ha, K.-H. Chae and H.-J. Shin, RSC Adv., 2015, 5, 21762 RSC.
- A. Kabelitz, A. Guilherme, M. Joester, U. Reinholz, M. Radtke, R. Bienert, K. Schulz, R. Schmack, R. Kraenhert and F. Emmerling, CrystEngComm, 2015, 17, 8463 RSC.
- J. Leveneur, G. I. N. Waterhouse, J. Kennedy, J. B. Metson and D. R. G. Mitchell, J. Phys. Chem. C, 2011, 115, 20978 CrossRef.
- X. Chen, J. Schroder, S. Hauschild, S. Rosenfeldt, M. Dulle and S. Forster, Langmuir, 2015, 31, 11678 CrossRef PubMed.
- C. J. Kim, K. Sondergeld, M. Mazurowski, M. Gallei, M. Rehahn, T. Spehr, H. Frielinghaus and B. Stuhn, Colloid Polym. Sci., 2013, 291, 2087 CrossRef.
- T. Schindler, M. Schmiele, T. Schmutzler, T. Kassar, D. Segets, W. Peukert, A. Radulescu, A. Kriele, R. Gilles and T. Unruh, Langmuir, 2015, 31, 10130 CrossRef PubMed.
- W. Wang, X. Chen, Q. Cai, G. Mo, L. S. Jiang, K. Zhang, Z. J. Chen, Z. H. Wu and W. Pan, Eur. Phys. J. B, 2008, 65, 57 CrossRef.
- T. Li, A. J. Senesi and B. Lee, Chem. Rev., 2016, 116, 11128 CrossRef PubMed.
- M. Singh, I. Sinha, A. K. Singh and R. K. Mandal, J. Nanopart. Res., 2011, 13, 4387 CrossRef.
- L. Bulavin, N. Kutsevol, V. Chumachenko, D. Soloviov, A. Kuklin and A. Marynin, Nanoscale Res. Lett., 2016, 11, 35 CrossRef PubMed.
- M. Singh, I. Sinha, A. K. Singh and R. K. Mandal, Colloids Surf., A, 2011, 384, 668 CrossRef.
- Y. Zhao, K. Saijo, M. Takenaka, S. Koizumi and T. Hashimoto, Polymer, 2009, 50, 2696 CrossRef.
- M. Harada, N. Tamura and M. Takenaka, J. Phys. Chem. C, 2011, 115, 14081 CrossRef.
- Y.-C. Liang, Y.-W. Juan, K.-T. Lu, U.-S. Jeng, S.-A. Chen, W.-T. Chuang, C.-J. Su, C.-L. Liu, C.-W. Pao, J.-F. Lee, H.-S. Sheu and J.-M. Chen, J. Phys. Chem. C, 2012, 116, 26649 CrossRef.
- A. P. LaGrow, B. Ingham, M. F. Toney and R. D. Tilley, J. Phys. Chem. C, 2013, 117, 16709 CrossRef.
- A. Ulyanenkov, J. Chrost, P. Siffalovic, L. Chitu, E. Majkova, K. Erlacher, H. Guerault, G. Maier, M. Cornejo, B. Ziberi and F. Frost, Phys. Status Solidi A, 2011, 208, 2619 CrossRef.
- D. J. Tobler, S. Shaw and L. G. Benning, Geochim. Cosmochim. Acta, 2009, 73, 5377 CrossRef.
- A. Tarasov, V. Goertz, E. Goodilin and H. Nirschl, J. Phys. Chem. C, 2013, 117, 12800 CrossRef.
- A. Turkovic, P. Dubcek, M. Rakic, M. Loncaric, B. Etlinger and S. Bernstorff, Vacuum, 2012, 86, 750 CrossRef.
- Z. H. Chen, C. Kim, X.-b. Zeng, S. H. Hwang, J. Jang and G. Ungar, Langmuir, 2012, 28, 15350 CrossRef PubMed.
- B. L. Caetano, C. V. Santilli, S. H. Pulcinelli and V. Briois, Phase Transitions, 2011, 84, 714 CrossRef.
- N. Krins, J. D. Bass, B. Julian-Lopez, P. Evrar, C. Boissiere, L. Nicole, C. Sanchez, H. Amenitsch and D. Grosso, J. Mater. Chem., 2011, 21, 1139 RSC.
- F. Meneau, G. Sankar, N. Morgante, S. Cristol, C. R. A. Catlow, J. M. Thomas and G. N. Greaves, Nucl. Instrum. Methods Phys. Res., Sect. B, 2003, 199, 499 CrossRef.
- S.-H. Wang, Y.-S. Sun, A. S.-T. Chiang, H.-F. Hung, M.-C. Chen and K. Wood, J. Phys. Chem. C, 2011, 115, 11941 CrossRef.
- L. Broussous, C. V. Santilli, S. H. Pulcinelli and A. F. Craievich, J. Phys. Chem. B, 2002, 106, 2855 CrossRef.
- B. L. Caetano, F. Meneau, C. V. Santilli, S. H. Pulcinelli, M. Magnani and V. Briois, Chem. Mater., 2014, 26, 2677 CrossRef.
- L. T. Lu, Ph.D. thesis, Water-dispersible Magnetic NPs for Biomedical applications: Synthesis and Characterisation, University of Liverpool, 2011 Search PubMed.
- D. D. Sharma, P. K. Santra, S. Mukherjee and A. Nag, Chem. Mater., 2013, 25, 1222 CrossRef.
- A. Shard, J. Phys. Chem. C, 2012, 116, 16806 CrossRef.
- L. Caprile, A. Cossaro, E. Falletta, C. D. Pina, O. Cavalleri, R. Rolandi, S. Terreni, R. Ferrando, M. Rossi, L. Floreano and M. Canepa, Nanoscale, 2012, 4, 7727 RSC.
- N. A. Belsey, A. G. Shard and C. Minelli, Biointerphases, 2015, 10, 019012 CrossRef PubMed.
- M. Y. Smirnov, A. V. Kalinkin, A. V. Bukhtiyarov, I. P. Prosvirin and V. I. Bukhtiyarov, J. Phys. Chem. C, 2016, 120, 10419 CrossRef.
- I. Tunc, U. K. Demirok, S. Suzer, M. A. Correa-Duartea and L. M. Liz-Marzan, J. Phys. Chem. B, 2005, 109, 24182 CrossRef PubMed.
- C. Battocchio, F. Porcaro, S. Mukherjee, E. Magnano, S. Nappini, I. Fratoddi, M. Quintiliani, M. V. Russo and G. Polzonetti, J. Phys. Chem. C, 2014, 118, 8159 CrossRef.
- Y.-C. Wang, M. H. Engelhard, D. R. Baer and D. G. Castner, Anal. Chem., 2016, 88, 3917 CrossRef PubMed.
- N. Maiti, S. Thomas, A. Debnath and S. Kapoor, RSC Adv., 2016, 6, 56406 RSC.
- M. Ramstedt and P. Franklyn, Surf. Interface Anal., 2010, 42, 855 CrossRef.
- M. Muniz-Miranda, S. Caporali, P. Marsili and E. Giorgetti, Mater. Chem. Phys., 2015, 167, 188 CrossRef.
- G. Hota, S. B. Idage and K. C. Khilar, Colloids Surf., A, 2007, 293, 5 CrossRef.
- P. Prieto, V. Nistor, K. Nouneh, M. Oyama, M. Abd-Lefdil and R. Diaz, Appl. Surf. Sci., 2012, 258, 8807 CrossRef.
- A. Kalinkin, A. M. Sorokin, M. Y. Smirnov and V. I. Bukhtiyarov, Kinet. Catal., 2014, 55, 371 CrossRef.
- N. Chakroune, G. Viau, S. Ammar, L. Poul, D. Veautier, M. M. Chehimi, C. Magneney, F. Villain and F. Fievet, Langmuir, 2005, 21, 6788 CrossRef PubMed.
- T. Ashida, K. Miura, T. Nomoto, S. Yagi, H. Sumida, G. Kutluk, K. Soda, H. Namatame and M. Taniguchi, Surf. Sci., 2007, 601, 3898 CrossRef.
- F. Bernardi, J. D. Scholten, G. H. Fecher, J. Dupont and J. Morais, Chem. Phys. Lett., 2009, 479, 113 CrossRef.
- X.-Q. Li and W.-X. Zhang, J. Phys. Chem. C, 2007, 111, 6939 CrossRef.
- G. Sheng, P. Yang, Y. Tang, Q. Hu, H. Li, X. Ren, B. Hu, X. Wang and Y. Huang, Appl. Catal., B, 2016, 193, 189 CrossRef.
- J. W. Kim, B. Son, H. Yu, H. M. Park and Y.-S. Lee, Surf. Interface Anal., 2014, 46, 193 CrossRef.
- A. Jurgensen, N. Heutz, H. Raschke, K. Merz and R. Hergenroder, Anal. Chem., 2015, 87, 7848 CrossRef PubMed.
- C. Blanco-Andujar, Ph.D. Thesis, Sodium Carbonate Mediated Synthesis of Iron Oxide NPs to Improve Magnetic Hyperthermia Efficiency and Induce Apoptosis, University College London, 2014 Search PubMed.
- C. Buso-Rogero, S. Brimaud, J. Solla-Gullon, F. J. Vidal-Iglesias, E. Herrero, R. J. Behm and J. M. Feliu, J. Electroanal. Chem., 2016, 763, 116 CrossRef.
- S. K. Cheah, V. P. Bernardet, A. A. Franco, O. Lemaire and P. Gelin, Phys. Chem. Chem. Phys., 2016, 18, 15278 RSC.
- N. Shukla, C. Liu, P. M. Jones and D. Weller, J. Magn. Magn. Mater., 2003, 266, 178 CrossRef.
- S. W. Han, Y. Kim and K. Kim, J. Colloid Interface Sci., 1998, 208, 272 CrossRef PubMed.
- I. Tunc, Mater. Chem. Phys., 2014, 144, 444 CrossRef.
- V. Tzitzios, G. Basina, M. Gjoka, V. Alexandrakis, V. Georgakilas, D. Niarchos, N. Boukos and D. Petridis, Nanotechnology, 2006, 17, 3750 CrossRef.
- Y. A. Jadhav, P. R. Thakur and S. K. Haram, Data Brief, 2016, 8, 1072 CrossRef PubMed.
- S. Sabale, V. Jadhav, V. Khot, X. Zhu, M. Xin and H. Chen, J. Mater. Sci.: Mater. Med., 2015, 26, 127 CrossRef PubMed.
- K. Gharbi, F. Salles, P. Mathieu, C. Amiens, V. Colliere, Y. Coppel, K. Philippot, L. Fontaine, V. Montembault, L. Samia Smiri and D. Ciuculescu-Pradines, New J. Chem., 2017, 41, 11898 RSC.
- H. Liang, H. Niu, P. Li, Z. Tao, C. Mao, J. Song and S. Zhang, Mater. Res. Bull., 2013, 48, 2415 CrossRef.
- H. T. T. Duong, Y. Chen, S. A. Tawfik, S. Wen, M. Parviz, O. Shimoni and D. Jin, RSC Adv., 2018, 8, 4842 RSC.
- S. Chen, X. Zhang, Q. Zhang and W. Tan, Nanoscale Res. Lett., 2009, 4, 1159 CrossRef PubMed.
- J. Coates, Interpretation of Infrared Spectra, a Practical Approach, in Encyclopedia of Analytical Chemistry, ed. R. A. Meyers, John Wiley & Sons Ltd, 2006 Search PubMed.
- L. E. Marbella and J. E. Millstone, Chem. Mater., 2015, 27, 2721 CrossRef.
- D. Scheid, D. Stock, T. Winter, T. Gutmann, C. Dietz and M. Gallei, J. Mater. Chem. C, 2016, 4, 2187 RSC.
- S. Vowinkel, S. Paul, T. Gutmann and M. Gallei, Nanomaterials, 2017, 7, 390 CrossRef PubMed.
- Z. Hens and J. C. Martins, Chem. Mater., 2013, 25, 1211 CrossRef.
- G. Uccello-Barretta, C. Evangelisti, F. Balzano, L. Vanni, F. Aiello and L. Jicsinszky, Carbohydr. Res., 2011, 346, 753 CrossRef PubMed.
- G. Canzi, A. A. Mrse and C. P. Kubiak, J. Phys. Chem. C, 2011, 115, 7972 CrossRef.
- S. C. Coelho, M. Rangel, M. C. Pereira, M. A. N. Coelho and G. Ivanova, Phys. Chem. Chem. Phys., 2015, 17, 18971 RSC.
- R. Sharma, R. E. Taylor and L.-S. Bouchard, J. Phys. Chem. C, 2011, 115, 3297 CrossRef.
- A. M. Smith, L. E. Marbella, K. A. Johnson, M. Hartmann, S. E. Crawford, L. M. Kozycz, D. S. Seferos and J. E. Millstone, Anal. Chem., 2015, 87, 2771 CrossRef PubMed.
- R. Sharma, G. P. Holland, V. C. Solomon, H. Zimmermann, S. Schiffenhaus, S. A. Amin, D. A. Buttry and J. L. Yarger, J. Phys. Chem. C, 2009, 113, 16387 CrossRef.
- M. Doyen, K. Bartik and G. Bruylants, J. Colloid Interface Sci., 2013, 399, 1 CrossRef PubMed.
- J. Cure, Y. Coppel, T. Dammak, P. F. Fazzini, A. Mlayah, B. Chaudret and P. Fau, Langmuir, 2015, 31, 1362 CrossRef PubMed.
- T. Faukner, L. Slany, I. Sloufova, J. Vohlidal and J. Zednik, Macromol. Res., 2016, 24, 441 CrossRef.
- M. V. Gomez, J. Guerra, V. S. Myers, R. M. Crooks and A. H. Velders, J. Am. Chem. Soc., 2009, 131, 14634 CrossRef PubMed.
- Y. Coppel, G. Spataro, C. Pages, B. Chaudret, A. Maisonnat and M. L. Kahn, Chem. – Eur. J., 2012, 18, 5384 CrossRef PubMed.
- Y. Coppel, G. Spataro, V. Colliere, B. Chaudret, C. Mingotaud, A. Maisonnat and M. L. Kahn, Eur. J. Inorg. Chem., 2012, 2691 CrossRef.
- G. Holland, R. Sharma, J. O. Agola, S. Amin, V. C. Solomon, P. Singh, D. A. Buttry and J. L. Yarger, Chem. Mater., 2007, 19, 2519 CrossRef.
- V. Sepelak and K. D. Becker, Chem. Mater., 2009, 21, 2518 CrossRef.
- Y. Y. Bogachev, J. S. Chernenco, K. G. Gareev, I. E. Kononova, L. B. Matyushkin, V. A. Moshnikov and S. S. Nalimova, Appl. Magn. Reson., 2014, 45, 329 CrossRef.
- M. Fardis, A. P. Douvalis, D. Tsitrouli, I. Rabias, D. Stamopoulos, T. Kehagias, E. Karakosta, G. Diamantopoulos, T. Bakas and G. Papavassiloou, J. Phys.: Condens. Matter, 2012, 24, 156001 CrossRef PubMed.
- Y. Gossuin, A. Hocq, Q. L. Vuong, S. Disch, R. P. Hermann and P. Gillis, Nanotechnology, 2008, 19, 475102 CrossRef PubMed.
- M. Forker and P. de la Presa, Phys. Rev. B: Condens. Matter, 2008, 77, 054108 CrossRef.
- T. Gutmann, A. Grunberg, N. Rothermel, M. Werner, M. Srour, S. Abdulhussain, S. Tan, Y. Xu, H. Breitzke and G. Buntkowsky, Solid State Nucl. Magn. Reson., 2013, 55–56, 1 CrossRef PubMed.
- S. K. Mallissery and D. Gudat, Dalton Trans., 2010, 39, 4280 RSC.
- I. Karki, H. Wang, N. R. Geise, B. W. Wilson, J. P. Lewis and T. Gullion, J. Phys. Chem. B, 2015, 119, 11998 CrossRef PubMed.
- F. Novio, K. Philippot and B. Chaudret, Catal. Lett., 2010, 140, 1 CrossRef.
- T. Gutmann, E. Bonnefille, H. Breitzke, P.-J. Debouttiere, K. Philippot, R. Poteau, G. Buntkowsky and B. Chaudret, Phys. Chem. Chem. Phys., 2013, 15, 17383 RSC.
- P. Lara, M.-J. Casanove, P. Lecante, P.-F. Fazzini, K. Philippot and B. Chaudret, J. Mater. Chem., 2012, 22, 3578 RSC.
- Y. S. Avadhut, J. Weber, E. Hammarberg, C. Feldmann and J. S. auf der Gunne, Phys. Chem. Chem. Phys., 2012, 14, 11610 RSC.
- Y. S. Avadhut, J. Weber, E. Hammarberg, C. Feldmann, I. Schellenberg, R. Pottgen and J. Schmedt auf der Gunne, Chem. Mater., 2011, 23, 1526 CrossRef.
- S. K. Davidowksi and G. P. Holland, Langmuir, 2016, 32, 3253 CrossRef PubMed.
- A. A. Arnold, V. Terskikh, Q. Y. Li, R. Naccache, I. Marcotte and J. A. Capobianco, J. Phys. Chem. C, 2013, 117, 25733 CrossRef.
- F. A. Perras, J. D. Padmos, R. L. Johnson, L.-L. Wang, T. J. Schwartz, T. Kobayashi, J. H. Horton, J. A. Dumesic, B. H. Shanks, D. D. Johnson and M. Pruski, J. Am. Chem. Soc., 2017, 139, 2702 CrossRef PubMed.
- A. J. Rossini, A. Zagdoun, M. Lelli, A. Lesage, C. Coperet and L. Emsley, Acc. Chem. Res., 2013, 46, 1942 CrossRef PubMed.
- Y. Sahoo, A. Goodarzi, M. T. Swihart, T. Y. Ohulchanskyy, N. Kaur, E. P. Furlani and P. N. Prasad, J. Phys. Chem. B, 2005, 119, 3879 CrossRef PubMed.
- Y.-J. Kim, J.-W. Kim, J.-E. Lee, J.-H. Ryu, J. Kim, I.-S. Chang and K.-D. Suh, J. Polym. Sci., Part A: Polym. Chem., 2004, 42, 5627 CrossRef.
- J. Ma, L. Wang, Y. Wu, X. Dong and Q. Ma, Mater. Trans., 2014, 55, 1900 CrossRef.
- L. A. Wormell Green, Ph.D Thesis, Synthesis and Characterization of FePt Magnetic NPs, University College London, 2014 Search PubMed.
- E. Mansfield, K. M. Tyner, C. M. Poling and J. L. Blacklock, J. Anal. Chem., 2014, 86, 1478 Search PubMed.
- K. B. Sebby and E. Mansfield, Anal. Bioanal. Chem., 2015, 407, 2913 CrossRef PubMed.
- M. Maccarini, G. Briganti, S. Rucareanu, X.-D. Lui, R. Sinibaldi, M. Sztucki and R. B. Lennox, J. Phys. Chem. C, 2010, 114, 6937 CrossRef.
- W. Jia, J. McLachlan, J. Xu, S. M. Tadayyon, P. R. Norton and S. H. Eichhorn, Can. J. Chem., 2006, 84, 998 CrossRef.
- M. Rudolph, J. Erler and U. A. Peuker, Colloids Surf., A, 2012, 397, 16 CrossRef.
- J. L. Ortiz-Quinonez, D. Diaz, I. Zumeta-Dube, H. Arriola-Santamaria, I. Betancourt, P. Santiago-Jacinto and N. Nava-Etzana, Inorg. Chem., 2013, 52, 10306 CrossRef PubMed.
- K. Chrissafis, E. Roumeli, K. M. Paraskevopoulos, N. Nianias and D. N. Bikiaris, J. Anal. Appl. Pyrolysis, 2012, 96, 92 CrossRef.
- A. Rafati, R. ter Veen and D. G. Castner, Surf. Interface Anal., 2013, 45, 1737 CrossRef PubMed.
- A. Kauling, G. Ebeling, J. Morais, A. Padua, T. Grehl, H. H. Brongersma and J. Dupont, Langmuir, 2013, 29, 14301 CrossRef PubMed.
- L. Ovari, A. Berko, N. Balazs, Z. Majzik and J. Kiss, Langmuir, 2010, 26, 2167 CrossRef PubMed.
- UV/Vis/IR Spectroscopy Analysis of NPs, September 2012, NanoComposix (Nanocomposix.com).
- T. Hendel, M. Wuithschick, F. Kettemann, A. Birnbaum, K. Rademann and J. Polte, Anal. Chem., 2014, 86, 11115 CrossRef PubMed.
- D. Paramelle, A. Sadovoy, S. Gorelik, P. Free, J. Hobley and D. G. Fernig, Analyst, 2014, 139, 4855 RSC.
- W. Haiss, N. T. K. Thanh, J. Aveyard and D. G. Fernig, Anal. Chem., 2007, 79, 4215 CrossRef PubMed.
- T. R. Ray, B. Lettiere, J. de Rutte and S. Pennathur, Langmuir, 2015, 31, 3577 CrossRef PubMed.
- M. Zielinski, M.Sc. Thesis, Determination of thiamine in solution by UV-Visible spectrophotometry: the effect of interaction with gold NPs, Ypsilanti, Michigan, 2014 Search PubMed.
- M. Harada, K. Saijo and N. Sakamoto, Colloids Surf., A, 2009, 349, 176 CrossRef.
- S. Behzadi, F. Ghasemi, M. Ghalkhani, A. A. Ashkarran, S. M. Akbari, S. Pakpour, M. R. Hormozi-Nezhad, Z. Jamshidi, S. Mirsadeghi, R. Dinarvand, F. Atyabi and M. Mahmoudi, Nanoscale, 2015, 7, 5134 RSC.
- R. Desai, V. Mankad, S. K. Gupta and P. K. Jha, Nanosci. Nanotechnol. Lett., 2012, 4, 30 CrossRef.
- M. R. Bindhu, V. Sathe and M. Umadevi, Spectrochim. Acta, Part A, 2013, 115, 409 CrossRef PubMed.
- T. Zhang, G. Lu, H. Shen, K. Shi, Y. Jiang, D. Xu and Q. Gong, Sci. Rep., 2014, 4, 3867 CrossRef PubMed.
- S. K. H. Andersen, A. Pors and S. I. Bozhevolnyi, ACS Photonics, 2015, 2, 432 CrossRef.
- I. Kriegel, C. Jiang, J. Rodriguez-Fernandez, R. D. Schaller, D. V. Talapin, E. da Como and J. Feldmann, J. Am. Chem. Soc., 2012, 134, 1583 CrossRef PubMed.
- S. Saliba, Y. Coppel, C. Mingotaud, J.-D. Marty and M. L. Kahn, Chem. – Eur. J., 2012, 18, 8084 CrossRef PubMed.
- L. Protesescu, S. Yakunin, M. I. Bodnarchuk, F. Krieg, R. Caputo, C. H. Hendon, R. X. Yang, A. Walsh and M. V. Kovalenko, Nano Lett., 2015, 15, 3692 CrossRef PubMed.
- H. Kato, Size Determination of NPs by Dynamic Light Scattering, in Nanomaterials: Processing and Characterization with Lasers, ed. S. C. Singh, H. Zeng, C. Guo and W. Cai, Wiley-VCH, 2012, ch. 8 Search PubMed.
- J. Lim, S. P. Yeap, H. X. Che and S. C. Low, Nanoscale Res. Lett., 2013, 8, 381 CrossRef PubMed.
- Nanoparticle characterization instrumentation, Australian Government, National Measurement Institute – Nanometrology Section. Powerpoint presentation, November 2012, Website: measurement.gov.au.
- M. Wolfgang , powerpoint presentation: Nanoparticle Size Analysis: A Survey and Review, in Nanomedicines Alliance, October 2015.
- N. A. Belsey, A. G. Shard and C. Minelli, poster presentation: Shell thickness determination of core-shell NPs, in Euramet.
- V. A. Coleman, A. K. Jamting, H. J. Catchpoole, M. Roy and J. Herrmann, Proc. SPIE, 8105, 2011, 810504, DOI:10.1117/12.894297.
- Y. H. Lai, S. Koo, S. H. Oh, E. A. Driskell and J. D. Driskell, Anal. Methods, 2015, 7, 7249 RSC.
- C. Dumas and C. J. Meledandri, Langmuir, 2015, 31, 7193 CrossRef PubMed.
- H. Fissan, S. Ristig, H. Kaminski, C. Asbach and M. Epple, Anal. Methods, 2014, 6, 7324 RSC.
- E. Tomaszewska, K. Soliwoda, K. Kadziola, B. Tkacz-Szczesna, G. Celichowski, M. Cichomski, W. Smaja and J. Grobelny, J. Nanomater., 2013, 313081 Search PubMed.
- V. Kestens, G. Roebben, J. Herrmann, A. Jamting, V. Coleman, C. Minelli, C. Clifford, P.-J. de Temmerman, J. Mast, L. Junjie, F. Babick, H. Colfen and H. Emons, J. Nanopart. Res., 2016, 18, 171 CrossRef PubMed.
- R. C. Murdock, L. Braydich-Stolle, A. M. Schrand, J. M. Schlager and S. M. Hussain, Toxicol. Sci., 2008, 101, 239 CrossRef PubMed.
- P. Hole, K. Sillence, C. Hannell, C. M. Maguire, M. Roesslein, G. Suarez, S. Capracotta, Z. Magdolenova, L. Horev-Azaria, A. Dybowska, L. Cooke, A. Haase, S. Contal, S. Mano, A. Vennemann, J.-J. Sauvain, K. C. Staunton, S. Anguissola, A. Luch, M. Dusinska, R. Korenstein, A. C. Gutleb, M. Wiemann, A. Prina-Mello, M. Riediker and P. Wick, J. Nanopart. Res., 2013, 15, 2101 CrossRef PubMed.
- V. Filipe, A. Hawe and W. Jiskoot, Pharm. Res., 2010, 27, 796 CrossRef PubMed.
- J. A. Gallego-Urrea, J. Tuoriniemi and M. Hassellov, Trends Anal. Chem., 2011, 30, 473 CrossRef.
- J. H. Ryu, S. Y. Bang, J.-W. Yoon, C. S. Lim and K. B. Shim, Appl. Surf. Sci., 2007, 253, 8408 CrossRef.
- R. Luque, M. Ojeda, A. Garcia, C. Lastres, R. Campos, A. Pineda, A. A. Romero and A. Yepez, RSC Adv., 2013, 3, 7119 RSC.
- E. van der Pol, F. A. W. Coumans, A. Sturk, R. Nieuwland and T. G. van Leeuwen, Nano Lett., 2014, 14, 6195 CrossRef PubMed.
- analytikLTD – Technical note. http://www.analytic.co.uk.
- C. Minelli, R. Garcia-Diez, A. E. Sikora, C. Gollwitzer, M. Krumrey and A. G. Shard, Surf. Interface Anal., 2014, 46, 663 CrossRef.
- N. C. Bell, C. Minelli, J. Tompkins, M. M. Stevens and A. G. Shard, Langmuir, 2012, 28, 10860 CrossRef PubMed.
- A. R. Montoro Bustos, J. Ruiz Encinar and A. Sanz-Medel, Anal. Bioanal. Chem., 2013, 405, 5637 CrossRef PubMed.
- K. M. Harkness, D. E. Cliffel and J. A. McLean, Analyst, 2010, 135, 868 RSC.
- R. Allabashi, W. Stach, A. de la Escosura-Muniz, L. Liste-Calleja and A. Merkoci, J. Nanopart. Res., 2009, 11, 2003 CrossRef.
- A. Helfrich, W. Bruchert and J. Bettmer, J. Anal. At. Spectrom., 2006, 21, 431 RSC.
- J. Liu, K. E. Murphy, R. I. MacCuspie and M. R. Winchester, Anal. Chem., 2014, 86, 3405 CrossRef PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361 CrossRef PubMed.
- Y. Yang, C.-L. Long, H.-P. Li, Q. Wang and Z.-G. Yang, Sci. Total Environ., 2016, 563–564, 996 CrossRef PubMed.
- F. J. Pereira, M. D. Vazquez, L. Deban and A. J. Aller, Anal. Methods, 2015, 7, 598 RSC.
- R. Peters, Z. Herrera-Rivera, A. Undas, M. van der Lee, H. Marvin, H. Bouwmeester and S. Weigel, J. Anal. At. Spectrom., 2015, 30, 1274 RSC.
- J. W. Olesik and P. J. Gray, J. Anal. At. Spectrom., 2012, 27, 1143 RSC.
- H. Hinterwirth, S. Kappel, T. Waitz, T. Prohaska, W. Lindner and M. Lammerhofer, ACS Nano, 2013, 7, 1129 CrossRef PubMed.
- E. P. Gray, J. G. Coleman, A. J. Bednar, A. J. Kennedy, J. F. Ranville and C. P. Higgins, Environ. Sci. Technol., 2013, 47, 14315 CrossRef PubMed.
- A. R. Donovan, C. D. Adams, Y. Ma, C. Stephan, T. Eicholz and H. Shi, Chemosphere, 2016, 144, 148 CrossRef PubMed.
- Y. Dan, H. Shi, C. Stephan and X. Liang, Microchem. J., 2015, 122, 119 CrossRef.
- S. H. Crayton, D. R. Elias, A. A. Zaki, Z. Cheng and A. Tsourkas, Biomaterials, 2012, 33, 1509 CrossRef PubMed.
- S. R. Rajagopal Achary, Ph.D. thesis, Characterization of individual NPs and applications of NPs in mass spectrometry, Texas A&M University, 2010 Search PubMed.
- C.-K. Liang, M. J. Eller, S. V. Verkhoturov and E. A. Schweikert, J. Am. Soc. Mass Spectrom., 2015, 26, 1259 CrossRef PubMed.
- W. Blanc, C. Guillermier and B. Dussardier, Opt. Mater. Express, 2012, 2, 1504 CrossRef.
- S. Rajagopalachary, S. V. Verkhoturov and E. A. Schweikert, Surf. Interface Anal., 2011, 43, 547 CrossRef.
- D. Scurr , oral presentation: ‘Challenges and Opportunities in Nanoparticle Analysis using ToF-SIMS?’ at ‘Nanoparticle Characterisation – Challenges for the Community’ event – IOP (Institute of Physics), book of abstracts, July 2016, London.
- L. Yang, M. P. Seah, I. S. Gilmore, R. J. H. Morris, M. G. Dowsett, L. Boarino, K. Sparnacci and M. Laus, J. Phys. Chem. C, 2013, 117, 16042 CrossRef.
- Y.-P. Kim, H. K. Shon, S. K. Shin and T. G. Lee, Mass Spectrom. Rev., 2015, 34, 237 CrossRef PubMed.
- A. Rafati, A. Boussahel, K. M. Shakesheff, A. G. Shard, C. J. Roberts, X. Chen, D. J. Scurr, S. Rigby-Singleton, P. Whiteside, M. R. Alexander and M. C. Davies, J. Controlled Release, 2012, 162, 321 CrossRef PubMed.
- J. Neunzehn, F. Draude, U. Golla-Schindler, H. F. Arlinghaus and H.-P. Wiesmann, Surf. Interface Anal., 2013, 45, 1340 CrossRef.
- L. Yang, Z. Zhu, X.-Y. Yu, E. Rodek, L. Saraf, T. Thevuthasan and J. P. Cowin, Surf. Interface Anal., 2014, 46, 224 CrossRef.
- S. Zanna, C. Saulou, M. Mercier-Bonin, B. Despax, P. Raynaud, A. Seyeux and P. Marcus, Appl. Surf. Sci., 2010, 256, 6499 CrossRef.
- P.-L. Lee, B.-C. Chen, G. Gollavelli, S.-Y. Shen, Y.-S. Yin, S.-L. Lei, C.-L. Jhang, W.-R. Lee and Y.-C. Ling, J. Hazard. Mater., 2014, 277, 3 CrossRef PubMed.
- R. Kersting, D. Breitenstein, B. Hagenhoff, M. Fartmann, D. Heller, T. Grehl, P. Bruner and E. Niehuis, Surf. Interface Anal., 2013, 45, 503 CrossRef.
- B. H. Kim, H. Chang, M. J. Hackett, J. Park, P. Seo and T. Hyeon, Bull. Korean Chem. Soc., 2014, 35, 961 CrossRef.
- J. K. Navin, M. E. Grass, G. A. Somorjai and A. L. Marsh, Anal. Chem., 2009, 81, 6295 CrossRef.
- L. Zhang, Z. Li, Y. Zhang, M. C. Paau, Q. Hu, X. Gong, S. Shuang, C. Dong, X. Peng and M. M. F. Choi, Talanta, 2015, 131, 632 CrossRef PubMed.
- Malvern Instruments. Website: https://www.malvern.com/en/products/product-range/archimedes.
- S. T. Patton, J. M. Slocik, A. Campbell, J. Hu, R. R. Naik and A. A. Voevodin, Nanotechnology, 2008, 19, 405705 CrossRef PubMed.
- F. Baldassarre, M. Cacciola and G. Ciccarella, J. Nanopart. Res., 2015, 17, 377 CrossRef.
- F. Branda, B. Silvestri, A. Costantini and G. Luciani, J. Sol-Gel Sci. Technol., 2015, 73, 54 CrossRef.
- A. Bumb, M. W. Brechbiel, P. L. Choyke, L. Fugger, A. Eggeman, D. Prabhakaran, J. Hutchinson and P. J. Dobson, Nanotechnology, 2008, 19, 335601 CrossRef PubMed.
- D. S. dos Santos Jr., R. A. Alvarez-Puebla, O. N. Oliveira Jr. and R. F. Aroca, J. Mater. Chem., 2005, 15, 3045 RSC.
- Y.-Y. Shim and V. K. Gupta, J. Colloid Interface Sci., 2007, 316, 977 CrossRef PubMed.
- P. S. Cappellari, D. Buceta, G. M. Morales, C. A. Barbero, M. S. Moreno, L. J. Giovanetti, J. M. Ramallo-Lopez, F. G. Requejo, A. F. Craievich and G. A. Planes, J. Colloid Interface Sci., 2015, 441, 17 CrossRef PubMed.
- X. Zhang, X. He, K. Wang, F. Ren and Z. Qin, Nanotechnology, 2011, 22, 355603 CrossRef PubMed.
- C. A. E. Hamlett, P. T. Docker, M. C. L. Ward, P. D. Prewett, K. Critchley, S. D. Evans and J. A. Preece, J. Exp. Nanosci., 2009, 4, 147 CrossRef.
- A. Oukarroum, M. Samadani and D. Dewez, Water, Air, Soil Pollut., 2014, 225, 2038 CrossRef.
- E. Pavlopoulou, G. Portale, K. E. Christodoulakis, M. Vamvakaki, W. Bras and S. H. Anastasiadis, Macromolecules, 2010, 43, 9828 CrossRef.
- F. Papa, I. Balint, C. Negrila, E.-A. Olaru, I. Zgura and C. Bradu, Ind. Eng. Chem. Res., 2014, 53, 19094 CrossRef.
- G.-H. Gwak, W.-J. Lee, S.-M. Paek and J.-M. Oh, Colloids Surf., B, 2015, 127, 137 CrossRef PubMed.
- T. Yousefi, M. T. Mostaedi, M. Ghasemi and A. Ghadirifar, Synth. React. Inorg. Met., 2016, 46, 137 CrossRef.
- C. Engelbrekt, P. Malcho, J. Andersen, L. Zhang, K. Stahl, B. Li, J. Hu and J. Zhang, J. Nanopart. Res., 2014, 16, 2562 CrossRef.
- A. Amirsalari and S. F. Shayesteh, Superlattices Microstruct., 2015, 82, 507 CrossRef.
- M. Baalousha, Sci. Total Environ., 2009, 407, 2093 CrossRef PubMed.
- H. X. Che, S. P. Yeap, A. L. Ahmad and J. Lim, Chem. Eng. J., 2014, 243, 68 CrossRef.
- G. Merga, N. Saucedo, L. C. Cass, J. Puthussery and D. Meisel, J. Phys. Chem. C, 2010, 114, 14811 CrossRef.
- A. Sikora, D. Bartczak, D. Geiβler, V. Kestens, G. Roebben, Y. Ramaye, Z. Varga, M. Palmai, A. G. Shard, H. Goenaga-Infante and C. Minelli, Anal. Methods, 2015, 7, 9835 RSC.
- C.-W. Lee, C. Takagi, T. Truong, Y.-C. Chen and A. Ostafin, J. Phys. Chem. C, 2010, 114, 12459 CrossRef.
- A. K. Mohammad and J. Reineke, Quantitative Nanoparticle Organ Disposition by Gel Permeation Chromatography, in Nanotoxicity – Methods and Protocols, ed. J. Reineke, Springer New York Heidelberg Dordrecht London, 2012, ch. 23 Search PubMed.
- B. J. Naden, L. M. Kessell, P. F. Luckham and T. F. Tadros, Colloids Surf., A, 2015, 478, 36 CrossRef.
- M. Gajendiran, S. M. J. Yousuf, V. Elangovan and S. Balasubramanian, J. Mater. Chem. B, 2014, 2, 418 RSC.
- A. Badia, L. Cuccia, L. Demers, F. Morin and R. B. Lennox, J. Am. Chem. Soc., 1997, 119, 2682 CrossRef.
- Y. Li, S. C. Ng and Z. P. Lu, Philos. Mag. Lett., 1998, 78, 37 CrossRef.
- C. Zou, Y. Gao, B. Yang and Q. Zhai, J. Mater. Sci.: Mater. Electron., 2010, 21, 868 CrossRef.
- L. Riviere, N. Causse, A. Lonjon, E. Dantras and C. Lacabanne, Polym. Degrad. Stab., 2016, 127, 98 CrossRef.
- S. Elzey, D.-H. Tsai, S. A. Rabb, L. L. Yu, M. R. Winchester and V. A. Hackley, Anal. Bioanal. Chem., 2012, 403, 145 CrossRef PubMed.
- C. Cui, M. He, B. Chen and B. Hu, Anal. Methods, 2014, 6, 8577 RSC.
- J. S. Suleiman, B. Hu, H. Peng and C. Huang, Talanta, 2009, 77, 1579 CrossRef PubMed.
- R. Anumolu and L. F. Pease III, Rapid Nanoparticle Characterization, in Nanotechnology and Nanomaterials – The Delivery of NPs, ed. A. A. Hashim, ch. 17, 2012, http://www.intechopen.com Search PubMed.
- M. Kozan, Ph.D. thesis, Characterization of colloidal nanoparticle aggregates using light scattering techniques, University of Kentucky, 2007 Search PubMed.
- J. L. Jimenez-Perez, J. F. Sanchez-Ramirez, Z. N. Correa-Pacheco, A. Cruz-Orea, E. Chigo Anota and F. Sanchez-Sinencio, Int. J. Thermophys., 2013, 34, 955 CrossRef.
- T.-H. Wu, S.-C. Liao, Y.-F. Chen, Y.-Y. Huang, Y.-S. Wei, S.-J. Tu and K.-S. Chen, Appl. Surf. Sci., 2013, 274, 1066 Search PubMed.
- T. P. Burg, M. Godin, S. M. Knudsen, W. Shen, G. Carlson, J. S. Foster, K. Babcock and S. R. Manalis, Nature, 2007, 446, 955 CrossRef PubMed.
- J. Olson, S. Dominguez-Medina, A. Hoggard, L.-Y. Wang, W.-S. Chang and S. Link, Chem. Soc. Rev., 2015, 44, 40 RSC.
- A. Akbarzadeh, M. Samiei and S. Davaran, Nanoscale Res. Lett., 2012, 7, 144 CrossRef PubMed.
- R. Russo, E. Esposito, C. Granata, A. Vettoliere, M. Russo, C. Cannas, D. Peddis and D. Fiorani, Phys. Procedia, 2012, 36, 293 CrossRef.
- C. Granata, R. Russo, E. Esposito, A. Vettoliere, M. Russo, A. Musinu, D. Peddis and D. Fiorani, Eur. Phys. J. B, 2013, 86, 272 CrossRef.
- L. F. Gamarra, W. M. Pontuschka, J. B. Mamani, D. R. Cornejo, T. R. Oliveira, E. D. Vieira, A. J. Costa-Filho and E. Amaro Jr., J. Phys.: Condens. Matter, 2009, 21, 115104 CrossRef.
- R. Malik, S. Annapoorni, S. Lamba, V. Raghavendra Reddy, A. Gupta, P. Sharma and A. Inoue, J. Magn. Magn. Mater., 2010, 322, 3742 CrossRef.
- Y. Wang, W. Zhang, C. Luo, X. Wu and G. Yan, Ceram. Int., 2016, 42, 12496 CrossRef.
- A. L. Andrade, M. A. Valente, J. M. F. Ferreira and J. D. Fabris, J. Magn. Magn. Mater., 2012, 324, 1753 CrossRef.
- M. Kumari, M. Widdrat, E. Tompa, R. Uebe, D. Schuler, M. Posfai, D. Faivre and A. M. Hirt, J. Appl. Phys., 2014, 116, 124304 CrossRef.
- J. Liu, K. Wu and J.-P. Wang, AIP Adv., 2016, 6, 056126 CrossRef.
- Lake Shore Cryotronics, Inc. http://www.lakeshore.com.
- http://www.nanomag-project.eu/Mössbauer-spectroscopy.html .
- J. Xiao, A. Kuc, S. Pokhrel, L. Madler, R. Pottgen, F. Winter, T. Frauenheim and T. Heine, Chem. – Eur. J., 2013, 19, 3287 CrossRef PubMed.
- S.-J. Oh, C.-J. Choi, S.-J. Kwon, S.-H. Jin, B.-K. Kim and J.-S. Park, J. Magn. Magn. Mater., 2004, 280, 147 CrossRef.
- M. Bystrzejewski, A. Grabias, J. Borysiuk, A. Huczko and H. Lange, J. Appl. Phys., 2008, 104, 054307 CrossRef.
- A. L. Tiano, G. C. Papaefthymiou, C. S. Lewis, J. Han, C. Zhang, Q. Li, C. Shi, A. M. Milinda Abeykoon, S. J. L. Billinge, E. Stach, J. Thomas, K. Guerrero, P. Munyaco, R. B. Scorzelli, P. Burnham, A. J. Viescas and S. S. Wonghenko, Chem. Mater., 2015, 27, 3572 CrossRef.
- J. Fock, L. K. Bogart, D. Gonzalez-Alonso, J. I. Espeso, M. F. Hansen, M. Varon, C. Frandsen and Q. A. Pankhurst, J. Phys. D: Appl. Phys., 2017, 50, 265005 CrossRef.
- B. Kalska-Szostko, U. Wykowska and D. Satula, Appl. Surf. Sci., 2014, 306, 7 CrossRef.
- Sarveena, J. M. Vargas, D. K. Shukla, C. T. Meneses, P. Mendoza Zelis, M. Singh and S. K. Sharma, Phys. Chem. Chem. Phys., 2016, 18, 9561 RSC.
- C. Rumenapp, F. E. Wagner and B. Gleich, J. Magn. Magn. Mater., 2015, 380, 241 CrossRef.
- L. H. Singh, R. Govindaraj, G. Amarendra and C. S. Sundar, Appl. Phys. Lett., 2013, 103, 193104 CrossRef.
- E. C. Sousa, H. R. Rechenberg, J. Depeyrot, J. A. Gomes, R. Aquino, F. A. Tourinho, V. Dupuis and R. Perzynski, J. Appl. Phys., 2009, 106, 093901 CrossRef.
- N. E. Domracheva, A. V. Pyataev, R. A. Manapov and M. S. Gruzdev, ChemPhysChem, 2011, 12, 3009 CrossRef PubMed.
- M. Siddique, E. Ahmed and N. M. Butt, Physica B, 2010, 405, 3964 CrossRef.
- L. Rebbouh, R. P. Hermann, F. Grandjean, T. Hyeon, K. An, A. Amato and G. J. Long, Phys. Rev. B: Condens. Matter, 2007, 76, 174422 CrossRef.
- M. Liu, M. Lu, L. Wang, S. Xu, J. Zhao and H. Li, J. Mater. Sci., 2016, 51, 5487 CrossRef.
- J.-C. Jumas, M. Womes, R. Alcantara, P. Lavela and J. L. Tirado, Hyperfine Interact., 2008, 183, 1 CrossRef.
- I. S. Lyubutin, E. A. Alkaev, Y. V. Korzhetskiy, C. R. Lin and R. K. Chiang, Hyperfine Interact., 2009, 189, 21 CrossRef.
- A. Joos, C. Rumenapp, F. E. Wagner and B. Gleich, J. Magn. Magn. Mater., 2016, 399, 123 CrossRef.
- Y. X. Yang, M. L. Liu, H. Zhu, Y. R. Chen, G. J. Mu, X. N. Liu and Y. Q. Jia, J. Magn. Magn. Mater., 2008, 320, L132 CrossRef.
- C. E. Johnson and J. A. Johnson, Hyperfine Interact., 2012, 204, 47 CrossRef.
- B. Kalska-Szostko, M. Cydzik, D. Satula and M. Giersig, Acta Phys. Pol., A, 2011, 119, 15 CrossRef.
- D. M. Polikarpov, R. R. Gabbasov, V. M. Cherepanov, M. A. Chuev, V. A. Korshunov, M. P. Nikitin, S. M. Deyev and V. Y. Panchenko, IEEE Trans. Magn., 2013, 49, 436 Search PubMed.
- K. Mazeika, A. Mikalauskaite and A. Jagminas, J. Magn. Magn. Mater., 2015, 389, 21 CrossRef.
- A. Ahlawat, V. G. Sathe, V. R. Reddy and A. Gupta, J. Magn. Magn. Mater., 2011, 323, 2049 CrossRef.
- S. Thota, S. C. Kashyap, S. K. Sharma and V. R. Reddy, Mater. Sci. Eng., B, 2016, 206, 69 CrossRef.
- C. R. H. Bahl, M. F. Hansen, T. Pedersen, S. Saadi, K. H. Nielsen, B. Lebech and S. Morup, J. Phys.: Condens. Matter, 2006, 18, 4161 CrossRef PubMed.
- S. Acharya, A. Bandyopadhyay, S. Modak, S. Mukherjee, D. Das and P. K. Chakrabarti, J. Magn. Magn. Mater., 2009, 321, 2701 CrossRef.
- M. N. Grecu, S. G. Costantinescu, D. Ghica, D. Tarabasanu-Mihaila and L. Diamandescu, Hyperfine Interact., 2012, 205, 111 CrossRef.
- L. Protesescu, A. J. Rossini, D. Kriegner, M. Valla, A. de Kergommeaux, M. Walter, K. V. Kravchyk, M. Nachtegaal, J. Stangl, B. Malaman, P. Reiss, A. Lesage, L. Emsley, C. Coperet and M. V. Kovalenko, ACS Nano, 2014, 8, 2639 CrossRef PubMed.
- C. S. Birkel, G. Kieslich, D. Bessas, T. Claudio, R. Branscheid, U. Kolb, M. Panthofer, R. P. Hermann and W. Tremel, Inorg. Chem., 2011, 50, 11807 CrossRef PubMed.
- F. Bodker, S. Morup, S. W. Charles and S. Linderoth, J. Magn. Magn. Mater., 1999, 196–197, 18 CrossRef.
- C. Concas, F. Congiu, G. Ennas, G. Piccaluga and G. Spano, J. Non-Cryst. Solids, 2003, 330, 234 CrossRef.
- H. Sakuma, T. Taniyama, K. Ishii, Y. Kitamoto and Y. Yamazaki, J. Magn. Magn. Mater., 2006, 300, 284 CrossRef.
- S.-J. Cho, A. M. Shahin, G. J. Long, J. E. Davies, K. Liu, F. Grandjean and S. M. Kauzlarich, Chem. Mater., 2006, 18, 960 CrossRef.
- E. A. Shafranovsky, Y. I. Petrov, Ll. Casas and E. Molins, J. Nanopart. Res., 2011, 13, 4913 CrossRef.
- C. E. Johnson, L. Costa, J. A. Johnson, D. E. Brown, S. Somarajan, W. He and J. H. Dickerson, J. Phys. D: Appl. Phys., 2014, 47, 075001 CrossRef.
- E.-S. M. Durai and Kh. A. Abdullin, J. Magn. Magn. Mater., 2009, 321, L69 CrossRef.
- M. R. Diehl, J.-Y. Yu, J. R. Heath, G. A. Held, H. Doyle, S. Sun and C. B. Murray, J. Phys. Chem. B, 2001, 105, 7913 CrossRef.
- R. B. Morgunov, A. I. Dmitriev, G. I. Dzhardimalieva, A. D. Pomogailo, A. S. Rozenberg, Y. Tanimoto, M. Leonowicz and E. Sowka, Phys. Solid State, 2007, 49, 1507 CrossRef.
- I. S. Edelman, E. A. Petrakovskaja, D. A. Petrov, S. M. Zharkov, R. I. Khaibullin, V. I. Nuzhdin and A. L. Stepanov, Appl. Magn. Reson., 2011, 40, 363 CrossRef.
- J. F. Hochepied and M. P. Pileni, J. Magn. Magn. Mater., 2001, 231, 45 CrossRef.
- D. S. Schmool, R. Rocha, J. B. Sousa, J. A. M. Santos, G. N. Kakazei, J. S. Garitaonandia and L. Lezama, J. Appl. Phys., 2007, 101, 103907 CrossRef.
- D. Walton, H. Boehnel and D. J. Dunlop, Phys. Status Solidi, 2001, 201, 3257 CrossRef.
- V. L. Mironov, E. V. Skorohodov and J. A. Blackman, J. Appl. Phys., 2014, 115, 184301 CrossRef.
- C. T. Hseih, W. L. Huang and J. T. Lue, J. Phys. Chem. Solids, 2002, 63, 733 CrossRef.
- L. F. Gamarra, A. J. da Costa-Filho, J. B. Mamani, R. de Cassia Ruiz, L. F. Pavon, T. T. Sibov, E. D. Vieira, A. C. Silva, W. M. Pontuschka and E. Amaro Jr., Int. J. Nanomed., 2010, 5, 203 CrossRef.
- F. J. Owens, J. Magn. Magn. Mater., 2009, 321, 2386 CrossRef.
- R. S. de Biasi and E. C. Gondim, Solid State Commun., 2006, 138, 271 CrossRef.
- J. Typek, N. Guskos, A. Szymczyk and D. Petridis, J. Non-Cryst. Solids, 2008, 354, 4256 CrossRef.
- F. J. Owens, J. Phys. Chem. Solids, 2003, 64, 2289 CrossRef.
- R. Bastardis, J.-L. Dejardin, F. Vernay and H. Kachkachi, J. Appl. Phys., 2016, 119, 174302 CrossRef.
- A. Butera, Eur. Phys. J. B, 2006, 52, 297 CrossRef.
- J. M. Vargas, R. D. Zysler and A. Butera, Appl. Surf. Sci., 2007, 254, 274 CrossRef.
- M. M. Yulikov, I. S. Abornev, O. N. Mart'yanov, V. F. Yudanov, V. P. Isupov, L. E. Chupakhina, K. A. Tarasov and R. P. Mitrofanova, Kinet. Catal., 2004, 45, 735 CrossRef.
- E. de Biasi, R. D. Zysler, C. A. Ramos and H. Romero, J. Magn. Magn. Mater., 2005, 294, e87 CrossRef.
- R. Valenzuela, F. Herbst and S. Ammar, J. Magn. Magn. Mater., 2012, 324, 3398 CrossRef.
- S. Brice-Profeta, M.-A. Arrio, E. Tronc, N. Menguy, I. Letard, C. Cartier dit Moulin, M. Nogues, C. Chaneac, J.-P. Jolivet and Ph. Sainctavit, J. Magn. Magn. Mater., 2005, 288, 354 CrossRef.
- S. Brice-Profeta, M.-A. Arrio, E. Tronc, I. Letard, Ch. Cartier dit Moulin and Ph. Sainctavit, Phys. Scr., 2005, T115, 626 CrossRef.
- Y. P. Cai, K. Chesnel, M. Trevino, A. Westover, R. G. Harrison, J. M. Hancock, S. Turley, A. Scherz, A. Reid, B. Wu, C. Graves, T. Wang, T. Liu and H. Durr, J. Appl. Phys., 2014, 115, 17B537 CrossRef.
- D. Nolle, E. Goering, T. Tietze, G. Schutz, A. Figuerola and L. Manna, New J. Phys., 2009, 11, 033034 CrossRef.
- ϒ. Takahashi, T. Kadono, S. Yamamoto, V. R. Singh, V. K. Verma, K. Ishigami, G. Shibata, T. Harano, Y. Takeda, T. Okane, Y. Saitoh, H. Yamagami, M. Takano and A. Fujimori, Phys. Rev. B: Condens. Matter, 2014, 90, 024423 CrossRef.
- A. Smekhova, D. Ciuculescu, P. Lecante, F. Wilhelm, C. Amiens, A. Rogalev and B. Chaudret, IEEE Trans. Magn., 2008, 44, 2776 Search PubMed.
- J. Bartolome, L. M. Garcia, F. Bartolome, F. Luis, R. Lopez-Ruiz, F. Petroff, C. Deranlot, F. Wilhelm, A. Rogalev, P. Bencok, N. B. Brookes, L. Ruiz and J. M. Gonzalez-Calbet, Phys. Rev. B: Condens. Matter, 2008, 77, 184420 CrossRef.
- Y. Prado, M.-A. Arrio, F. Volatron, E. Otero, C. Cartier dit Moulin, P. Sainctavit, L. Catala and T. Mallah, Chem. – Eur. J., 2013, 19, 6685 CrossRef PubMed.
- J. F. Hochepied, Ph. Sainctavit and M. P. Pileni, J. Magn. Magn. Mater., 2001, 231, 315 CrossRef.
- Y. Oba, H. Okamoto, T. Sato, T. Shinohara, J. Suzuki, T. Nakamura, T. Muro and H. Osawa, J. Phys. D: Appl. Phys., 2008, 41, 134204 CrossRef.
- Y. Yamamoto, T. Miura, T. Teranishi, M. Suzuki, N. Kawamura, H. Miyagawa, T. Nakamura, K. Kobayashi and H. Hori, J. Magn. Magn. Mater., 2004, 272–276, e1183 CrossRef.
- C. Guglieri, M. A. Laguna-Marco, M. A. Garcia, N. Carmona, E. Cespedes, M. Garcia-Hernandez, A. Espinosa and J. Chaboy, J. Phys. Chem. C, 2012, 116, 6608 CrossRef.
- T. Kataoka, M. Kobayashi, G. S. Song, Y. Sakamoto, A. Fujimori, F.-H. Chang, H.-J. Lin, D. J. Huang, C. T. Chen, S. K. Mandal, T. K. Nath, D. Karmakar and I. Dasgupta, Jpn. J. Appl. Phys., 2009, 48, 04C200 CrossRef.
- A. P. Herrera, C. Barrera, Y. Zayas and C. Rinaldi, J. Colloid Interface Sci., 2010, 342, 540 CrossRef PubMed.
- K. Kodama, J. Geophys. Res.: Solid Earth, 2012, 118, 1 Search PubMed.
- V. L. Calero-DdelC, D. I. Santiago-Quinonez and C. Rinaldi, Soft Matter, 2011, 7, 4497 RSC.
- A. F. R. Rodriguez, A. C. Oliveira, P. C. Morais, D. Rabelo and E. C. D. Lima, J. Appl. Phys., 2003, 93, 6963 CrossRef.
- T. Q. Yang, M. Abe, K. Horiguchi and K. Enpuku, Physica C, 2004, 412–414, 1496 CrossRef.
- M. P. Herrling, K. L. Fetsch, M. Delay, F. Blauert, M. Wagner, M. Franzreb, H. Gorn and S. Lackner, Sci. Total Environ., 2015, 537, 43 CrossRef PubMed.
- A. Ghasemi, J. Cluster Sci., 2016, 27, 979 CrossRef.
- M. S. Seehra and A. Punnoose, Phys. Rev. B: Condens. Matter, 2001, 64, 132410 CrossRef.
- Y. Kitamoto and J.-S. He, Electrochim. Acta, 2009, 54, 5969 CrossRef.
- M. Zborowski, L. R. Moore, P. S. Williams and J. J. Chalmers, Sep. Sci. Technol., 2002, 37, 3611 CrossRef.
- K. S. Lee, M. Lee, K. M. Byun and I. S. Lee, J. Mater. Chem., 2011, 21, 5156 RSC.
- F. Yu, L. Zhang, Y. Huang, K. Sun, A. E. David and V. C. Yang, Biomaterials, 2010, 31, 5842 CrossRef PubMed.
- A. Hutten, D. Sudfeld, I. Ennen, G. Reiss, K. Wojczykowski and P. Jutzi, J. Magn. Magn. Mater., 2005, 293, 93 CrossRef.
- B. Bharti, A.-L. Fameau and O. D. Velev, Faraday Discuss., 2015, 181, 437 RSC.
- M. S. Carriao, K. Skeff Neto and A. F. Bakuzis, J. Phys. D: Appl. Phys., 2014, 47, 025003 CrossRef.
- P. Y. Toh, B. W. Ng, C. H. Chong, A. L. Ahmad, J.-W. Yang, C. J. C. Derek and J. K. Lim, RSC Adv., 2014, 4, 4114 RSC.
- J. S. Andreu, P. Barbero, J. Camacho and J. Faraudo, J. Nanomater., 2012, 2012, 678581 CrossRef.
- S.-E. K. Fateen and M. Magdy, Chem. Eng. Res. Des., 2015, 95, 69 CrossRef.
- L. P. de Haro, T. Karaulanov, E. C. Vreeland, B. Anderson, H. J. Hathaway, D. L. Huber, A. N. Matlashov, C. P. Nettles, A. D. Price, T. C. Monson and E. R. Flynn, Biomed. Eng.-Biomed. Tech., 2015, 60, 445 Search PubMed.
- http://www.nanomag-project.eu/magnetorelaxometry.html .
- F. Ludwig, E. Heim and M. Schilling, J. Magn. Magn. Mater., 2009, 321, 1644 CrossRef.
- N. L. Adolphi, D. L. Huber, J. E. Jaetao, H. C. Bryant, D. M. Lovato, D. L. Fegan, E. L. Venturini, T. C. Monson, T. E. Tessier, H. J. Hathaway, C. Bergemann, R. S. Larson and E. R. Flynn, J. Magn. Magn. Mater., 2009, 321, 1459 CrossRef PubMed.
- N. L. Adolphi, K. S. Butler, D. M. Lovato, T. E. Tessier, J. E. Trujillo, H. J. Hathaway, D. L. Fegan, T. C. Monson, T. E. Stevens, D. L. Huber, J. Ramu, M. L. Milne, S. A. Altobelli, H. C. Bryant, R. S. Larson and E. R. Flynn, Contrast Media Mol. Imaging, 2012, 7, 308 CrossRef PubMed.
- A. L. Urbano-Bojorge, N. Feliz-Gonzalez, T. Fernandez, F. del Pozo-Guerrero, M. Ramos and J. J. Serrano-Olmedo, J. Nano Res., 2015, 31, 129 Search PubMed.
- F. Ludwig, E. Heim, D. Eberbeck, K. Schwarz, L. Trahms and M. Schilling, IEEE Trans. Magn., 2009, 45, 4857 Search PubMed.
- E. Peng, F. Wang, B. Zheng, S. F. Y. Li and J. M. Xue, Nanoscale, 2015, 7, 7819 RSC.
- L. Reimer and H. Kohl, Transmission Electron Microscopy Physics of Image Formation, Springer, New York, 2009, vol. 51, pp. 1–15 Search PubMed.
- M. C. Daniel and D. Astruc, Chem. Rev., 2004, 104, 293 CrossRef PubMed.
- Q. Pankhurst, J. Connolly, S. K. Jones and J. Dobson, J. Phys. D: Appl. Phys., 2003, 36, R167–R181 CrossRef.
- J. T. Nurmi, P. G. Tratnyek, V. Sarathy, D. R. Baer, J. E. Amonette, K. Pecher, C. Wang, J. C. Linehan, D. W. Matson, R. L. Penn and M. D. Driessen, Environ. Sci. Technol., 2005, 39, 1221 CrossRef PubMed.
- D. Astruc, F. Lu and J. R. Aranzaes, Angew. Chem., Int. Ed., 2005, 44, 7852 CrossRef PubMed.
- Y. W. Jun, J. W. Seo and J. Cheon, Acc. Chem. Res., 2008, 41, 179 CrossRef PubMed.
- Y. Pan, S. Neuss, A. Leifert, M. Fischler, F. Wen, U. Simon, G. Schmid, W. Brandau and W. Jahnen-Dechent, Small, 2007, 3, 1941 CrossRef PubMed.
- H. Borchert, E. V. Shevchenko, A. Robert, I. Mekis, A. Kornowski, G. Grübel and H. Weller, Langmuir, 2015, 21, 1931 CrossRef PubMed.
- L. E. Alexander and H. P. Klug, J. Appl. Phys., 1950, 21, 137 CrossRef.
- D. Vilela, M. C. González and A. Escarpa, Anal. Chim. Acta, 2012, 751, 24 CrossRef PubMed.
- N. T. K. Thanh and Z. Rosenzweig, Anal. Chem., 2002, 74, 1624 CrossRef PubMed.
- J. Nam, N. Won, H. Jin, H. Chung and S. Kim, J. Am. Chem. Soc., 2009, 131, 13639 CrossRef PubMed.
- S. D. Perrault and W. C. W. Chan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 11194 CrossRef PubMed.
- D. Mahl, J. Diendorf, W. Meyer-Zaika and M. Epple, Colloids Surf., A, 2011, 377, 386 CrossRef.
- A. Albanese and W. C. W. Chan, ACS Nano, 2011, 5, 5478 CrossRef PubMed.
- R. M. Pallares, S. L. Kong, T. H. Ru, N. T. K. Thanh, Y. Lu and X. Su, Chem. Commun., 2015, 51, 14524 RSC.
- A. Courty, A. Mermet, P. A. Albouy, E. Duval and M. P. Pileni, Nat. Mater., 2005, 4, 395 CrossRef PubMed.
- E. V. Shevchenko, D. V. Talapin, N. A. Kotov, S. O'Brien and C. B. Murray, Nature, 2006, 439, 55 CrossRef PubMed.
- M. R. Jones, R. J. Macfarlane, B. Lee, J. Zhang, K. L. Young, A. J. Senesi and C. A. Mirkin, Nat. Mater., 2010, 9, 913 CrossRef PubMed.
- M. R. Wiesner, G. V. Lowry, E. Casman, P. M. Bertsch, C. W. Matson, R. T. Di Giulio, J. Liu and M. F. Hochella, ACS Nano, 2011, 5, 8466 CrossRef PubMed.
- Y. Cheng, L. Yin, S. Lin, M. Wiesner, E. Bernhardt and J. Liu, J. Phys. Chem. C, 2011, 115, 4425 CrossRef.
- T. L. Kirschling, P. L. Golas, J. M. Unrine, K. Matyjaszewski, K. B. Gregory, G. V. Lowry and R. D. Tilton, Environ. Sci. Technol., 2011, 45, 5253 CrossRef PubMed.
- R. Van Den Berg, C. F. Elkjaer, C. J. Gommes, I. Chorkendorff, J. Sehested, P. E. De Jongh, K. P. De Jong and S. Helveg, J. Am. Chem. Soc., 2016, 138, 3433 CrossRef PubMed.
- R. A. Petros and J. M. DeSimone, Nat. Rev. Drug Discovery, 2010, 9, 615 CrossRef PubMed.
- A. Ostrowski, D. Nordmeyer, A. Boreham, C. Holzhausen, L. Mundhenk, C. Graf, M. C. Meinke, A. Vogt, S. Hadam, J. Lademann, E. Ruhl, U. Alexiev and A. D. Gruber, Beilstein J. Nanotechnol., 2015, 6, 263 CrossRef PubMed.
- J. I. Cutler, E. Auyeung and C. A. Mirkin, J. Am. Chem. Soc., 2012, 134, 1376 CrossRef PubMed.
- J. Panyam, W.-Z. Zhou, S. Prabha, S. K. Sahoo and V. Labhasetwar, FASEB J., 2002, 16, 1217 CrossRef PubMed.
- J. Yue, T. J. Feliciano, W. Li, A. Lee and T. W. Odom, Bioconjugate Chem., 2017, 28, 1791 CrossRef PubMed.
- C. D. Walkey, J. B. Olsen, H. Guo, A. Emili and W. C. W. Chan, J. Am. Chem. Soc., 2012, 134, 2139 CrossRef PubMed.
- C. Blanco-Andujar, D. Ortega, P. Southern, S. Nesbitt, N. T. K. Thanh and Q. Pankhurst, Nanomedicine, 2016, 11, 121 CrossRef PubMed.
- E. Gontier, M.-D. Ynsa, T. Bíró, J. Hunyadi, B. Kiss, K. Gáspár, T. Pinheiro, J.-N. Silva, P. Filipe, J. Stachura, W. Dabros, T. Reinert, T. Butz, P. Moretto and J.-E. Surlève-Bazeille, Nanotoxicology, 2008, 2, 218 CrossRef.
- D. B. Williams and C. B. Carter, in High-resolution TEM in Transmission Electron Microscopy, Springer US, Boston, MA, 2nd edn, 2009, pp. 483–509 Search PubMed.
- M. R. Axet, K. Philippot, B. Chaudret, M. Cabié, S. Giorgio and C. R. Henry, Small, 2011, 7, 235 CrossRef PubMed.
- M. José-Yacamán, M. Marín-Almazo and J. Ascencio, J. Mol. Catal. A: Chem., 2001, 173, 61 CrossRef.
- R. M. Pallares, Y. Wang, S. H. Lim, N. T. K. Thanh and X. Su, Nanomedicine, 2016, 11, 2845 Search PubMed.
- O. Dmitrieva, B. Rellinghaus, J. Kästner and G. Dumpich, J. Cryst. Growth, 2007, 303, 645 CrossRef.
- S. Tsunekawa, K. Ishikawa, Z. Q. Li, Y. Kawazoe and A. Kasuya, Phys. Rev. Lett., 2000, 85, 3440 CrossRef PubMed.
- F. Zhang, S. W. Chan, J. E. Spanier, E. Apak, Q. Jin, R. D. Robinson and I. P. Herman, Appl. Phys. Lett., 2002, 80, 127 CrossRef.
- J.-Y. Ascencio, Electron Microscopy of Nanostructured and Ancient Materials, in Handbook of Nanostructured Materials and Nanotechnology, Academic Press, Cambridge, MA, 1999, vol. 2, p. 393 Search PubMed.
- W. J. Zhang and D. E. Miser, J. Nanopart. Res., 2006, 8, 1027 CrossRef.
- C. Chen, Z. Hu, Y. Li, L. Liu, H. Mori and Z. Wang, Sci. Rep., 2016, 6, 19545 CrossRef PubMed.
- L. Marton, Bull. Acad. R. Med. Belg., 1934, 20, 439 Search PubMed.
- M. J. Williamson, R. M. Tromp, P. M. Vereecken, R. Hull and F. M. Ross, Nat. Mater., 2003, 2, 532 CrossRef PubMed.
- H. Zheng, U. M. Mirsaidov, L. W. Wang and P. Matsudaira, Nano Lett., 2012, 12, 5644 CrossRef PubMed.
- R. Franks, S. Morefield, J. Wen, D. Liao, J. Alvarado, M. Strano and C. Marsh, J. Nanosci. Nanotechnol., 2008, 8, 4404 CrossRef PubMed.
- X. Chen, C. Li and H. Cao, Nanoscale, 2015, 7, 4811 RSC.
- H. G. Liao, Y. Shao, C. Wang, Y. Lin, Y. X. Jiang and S. G. Sun, Mater. Lett., 2014, 116, 299 CrossRef.
- J. M. Yuk, J. Park, P. Ercius, K. Kim, D. J. Hellebusch, M. F. Crommie, J. Y. Lee, A. Zettl and A. P. Alivisatos, Science, 2012, 336, 61 CrossRef PubMed.
- H. M. Zheng, R. K. Smith, Y. W. Jun, C. Kisielowski, U. Dahmen and A. P. Alivisatos, Science, 2009, 324, 1309 CrossRef PubMed.
- H. L. Xin and H. Zheng, Nano Lett., 2012, 12, 1470 CrossRef PubMed.
- K. Y. Niu, J. Park, H. Zheng and A. P. Alivisatos, Nano Lett., 2013, 13, 5715 CrossRef PubMed.
- E. Sutter, K. Jungjohann, S. Bliznakov, A. Courty, E. Maisonhaute, S. Tenney and P. Sutter, Nat. Commun., 2014, 5, 4946 CrossRef PubMed.
- E. A. Sutter and P. W. Sutter, J. Am. Chem. Soc., 2014, 136, 16865 CrossRef PubMed.
- H. Zheng, S. A. Claridge, A. M. Minor, A. P. Alivisatos and U. Dahmen, Nano Lett., 2009, 9, 2460 CrossRef PubMed.
- J. Park, H. Elmlund, P. Ercius, J. M. Yuk, D. T. Limmer, Q. Chen, K. Kim, S. H. Han, D. A. Weitz, A. Zettl and A. P. Alivisatos, Science, 2015, 349, 290 CrossRef PubMed.
- J. Park, H. Zheng, W. C. Lee, P. L. Geissler, E. Rabani and A. P. Alivisatos, ACS Nano, 2012, 6, 2078 CrossRef PubMed.
- D. Danino, Curr. Opin. Colloid Interface Sci., 2012, 17, 316 CrossRef.
- C. A. Angell, Annu. Rev. Phys. Chem., 2004, 55, 559 CrossRef PubMed.
- D. A. Zweifel and A. Wei, Chem. Mater., 2005, 17, 4256 CrossRef PubMed.
- S. Kumar, Z. Wang, R. L. Penn and M. Tsapatsis, J. Am. Chem. Soc., 2008, 130, 17284 CrossRef PubMed.
- J.-C. Taveau, D. Nguyen, A. Perro, S. Ravaine, E. Duguet and O. Lambert, Soft Matter, 2008, 4, 311 RSC.
- J. A. Edgar, A. M. McDonagh and M. B. Cortie, ACS Nano, 2012, 6, 1116 CrossRef PubMed.
- F. Bouyer, C. Gérardin, F. Fajula, J. L. Putaux and T. Chopin, Colloids Surf., A, 2003, 217, 179 CrossRef.
- R. M. Pallares, X. Su, S. H. Lim and N. T. K. Thanh, J. Mater. Chem. C, 2016, 4, 53 RSC.
- Y. Lu, S. Proch, M. Schrinner, M. Drechsler, R. Kempe and M. Ballauff, J. Mater. Chem., 2009, 19, 3955 RSC.
- O. Balmes, J.-O. Malm, N. Pettersson, G. Karlsson and J.-O. Bovin, Microsc. Microanal., 2006, 12, 145 CrossRef PubMed.
- H. Cui, Z. Chen, S. Zhong, K. L. Wooley and D. J. Pochan, Science, 2007, 317, 647 CrossRef PubMed.
- M. A. Kostiainen, P. Hiekkataipale, A. Laiho, V. Lemieux, J. Seitsonen, J. Ruokolainen and P. Ceci, Nat. Nanotechnol., 2012, 8, 52 CrossRef PubMed.
- S. Elazzouzi-Hafraoui, Y. Nishiyama, J. L. Putaux, L. Heux, F. Dubreuil and C. Rochas, Biomacromolecules, 2008, 9, 57 CrossRef PubMed.
- L. Filion, M. Marechal, B. Van Oorschot, D. Pelt, F. Smallenburg and M. Dijkstra, Phys. Rev. Lett., 2009, 103, 188302 CrossRef PubMed.
- J. van Rijssel, B. H. Erné, J. D. Meeldijk, M. Casavola, D. Vanmaekelbergh, A. Meijerink and A. P. Philipse, Phys. Chem. Chem. Phys., 2011, 13, 12770 RSC.
- P. A. Buffat, Mater. Chem. Phys., 2003, 81, 368 CrossRef.
- L. D. Romeu and J. Reyes-Gasga, Microsc. Microanal., 2011, 17, 279 CrossRef PubMed.
- C. T. Schamp and W. A. Jesser, Ultramicroscopy, 2005, 103, 165 CrossRef PubMed.
- J. Reyes-Gasga, A. Gomez-Rodriguez and M. Jose-Yacaman, Ultramicroscopy, 2008, 108, 929 CrossRef PubMed.
- V. V. Volkov, V. V. Klechkovskaya, E. V. Shtykova, K. A. Dembo, N. A. Arkharova, G. I. Ivakin and R. Y. Smyslov, Crystallogr. Rep., 2009, 54, 169 CrossRef.
- A. R. Esmail, A. Bugayev and H. E. Elsayed-Ali, J. Phys. Chem. C, 2013, 117, 9035 CrossRef.
- K. Sato and Y. Hirotsu, Mater. Trans., 2006, 47, 59 CrossRef.
- K. Sato, Y. Hirotsu, H. Mori, Z. Wang and T. Hirayama, J. Appl. Phys., 2005, 97, 084301 CrossRef.
- K. Sato and Y. Hirotsu, Mater. Trans., 2003, 44, 1518 CrossRef.
- J. Li, H. Zeng, S. Sun, J. P. Liu and Z. L. Wang, J. Phys. Chem. B, 2004, 108, 14005 CrossRef.
- S. Futterer, I. Andrusenko, U. Kolb, W. Hofmeister and P. Langguth, J. Pharm. Biomed., 2013, 86, 151 CrossRef PubMed.
- S. Li, Y. Zhang, C. Esling, J. Muller, J.-S. Lecomte, G. W. Qin, X. Zhao and L. Zuo, J. Appl. Crystallogr., 2009, 42, 519 CrossRef.
- N. P. Young, Z. Y. Li, Y. Chen, S. Palomba, M. Di Vece and R. E. Palmer, Phys. Rev. Lett., 2008, 101, 246103 CrossRef PubMed.
- T. J. A. Slater, A. Jannsen, P. H. C. Camargo, M. G. Burke, N. J. Zaluzec and S. J. Haigh, Ultramicroscopy, 2016, 162, 619 CrossRef PubMed.
- I. Romer, Z. W. Wang, R. C. Merrifield, R. E. Palmer and J. Lead, Environ. Sci. Technol., 2016, 50, 2183 CrossRef PubMed.
- Z. W. Wang, Z. Y. Li, S. J. Park, A. Abdela, D. Tang and R. E. Palmer, Phys. Rev. B: Condens. Matter, 2011, 84, 073408 CrossRef.
- D. Deiana, A. Verdaguer-Casadevall, P. Malacrida, I. E. L. Stephens, I. Chorkendorff, J. B. Wagner and T. W. Hansen, ChemCatChem, 2015, 7, 3748 CrossRef.
- T. Akita, S. Tanaka, K. Tanaka, M. Haruta and M. Kohyama, J. Mater. Sci., 2011, 46, 4384 CrossRef.
- D. S. He, Z. Y. Li and J. Yuan, Micron, 2015, 74, 47 CrossRef PubMed.
- T. Akita, K. Tanaka, M. Kohyama and M. Haruta, Surf. Interface Anal., 2008, 40, 1760 CrossRef.
- Z. Y. Li, N. P. Young, M. Di Vece, S. Palomba, R. E. Palmer, A. L. Bleloch, B. C. Curley, R. L. Johnston, J. Jiang and J. Yuan, Nature, 2008, 451, 46 CrossRef PubMed.
- D. Babonneau, D. Lantiat, S. Camelio, J. Toudert, L. Simonot, F. Pailloux, M.-F. Denanot and T. Girardeau, Eur. Phys. J.: Appl. Phys., 2008, 44, 3 CrossRef.
- D. T. Tran, I. P. Jones, J. A. Preece, R. L. Johnston and C. R. van den Brom, J. Nanopart. Res., 2011, 13, 4229 CrossRef.
- J. C. Gonzalez, J. C. Hernandez, M. Lopez-Haro, E. del Rio, J. J. Delgado, A. B. Hungria, S. Trasobares, S. Bernal, P. A. Midgley and J. J. Calvino, Angew. Chem., Int. Ed., 2009, 48, 5313 CrossRef PubMed.
- T. Epicier, K. Sato, F. Tournus and T. Konno, J. Nanopart. Res., 2012, 14, 1106 CrossRef.
- M. Filippousi, T. Altantzis, G. Stefanou, M. Betsiou, D. N. Bikiaris, M. Angelakeris, E. Pavlidou, D. Zamboulis and G. Van Tendeloo, RSC Adv., 2013, 3, 24367 RSC.
- D. M. Wells, G. Rossi, R. Ferrando and R. E. Palmer, Nanoscale, 2015, 7, 6498 RSC.
- N. Jian, C. Stapefeldt, K.-J. Hu, M. Froba and R. E. Palmer, Nanoscale, 2015, 7, 885 RSC.
- J.-P. Baudoin, J. R. Jinschek, C. B. Boothroyd, R. E. Dunin-Borkowski and N. de Jonge, Microsc. Microanal., 2013, 19, 814 CrossRef PubMed.
- Z. W. Wang and R. E. Palmer, Phys. Rev. Lett., 2012, 108, 245502 CrossRef PubMed.
- J. C. Hernandez-Garrido, K. Yoshida, P. L. Gai, E. D. Boyes, C. H. Christensen and P. A. Midgley, Catal. Today, 2011, 160, 165 CrossRef.
- A. Surrey, D. Pohl, L. Schultz and B. Rellinghaus, Nano Lett., 2012, 12, 6071 CrossRef PubMed.
- J. Li, Z. Wang, C. Chen and S. Huang, Sci. Rep., 2014, 4, 5521 CrossRef PubMed.
- D. Ferrer, D. A. Blom, L. F. Allard, S. Mejia, E. Perez-Tijerina and M. Jose-Yacaman, J. Mater. Chem., 2008, 18, 2442 RSC.
- R. Esparza, O. Tellez-Vazquez, G. Rodriguez-Ortiz, A. Angeles-Pascual, S. Velumani and R. Perez, J. Phys. Chem. C, 2014, 118, 22383 CrossRef.
- J. Nelayah, N. T. Nguyen, D. Alloyeau, G. Y. Wang and C. Ricolleau, Nanoscale, 2014, 6, 10423 RSC.
- F. L. Deepak, G. Casillas-Garcia, R. Esparza, H. Barron and M. Jose-Yacaman, J. Cryst. Growth, 2011, 325, 60 CrossRef PubMed.
- A. Mayoral, F. Leonard Deepak, R. Esparza, G. Casillas, C. Magen, E. Perez-Tijerina and M. Jose-Yacaman, Micron, 2012, 43, 557 CrossRef.
- N. Jian and R. E. Palmer, J. Phys. Chem. C, 2015, 119, 11114 CrossRef.
- F. Yin, Z. W. Wang and R. E. Palmer, J. Am. Chem. Soc., 2011, 133, 10325 CrossRef PubMed.
- A. A. Herzing, M. Watanabe, J. K. Edwards, M. Conte, Z.-R. Tang, G. J. Hutchings and C. J. Kiely, Faraday Discuss., 2008, 138, 337 RSC.
- W. Ji, W. Qi, X. Li, S. Zhao, S. Tang, H. Peng and S. Li, Mater. Lett., 2015, 152, 283 CrossRef.
- M. A. Asoro, D. Kovar, Y. Shao-Horn, L. F. Allard and P. J. Ferreira, Nanotechnology, 2010, 21, 025701 CrossRef PubMed.
- A. Hashimoto, P. Wang, M. Shimojo, K. Mitsuishi, P. D. Nellist, A. I. Kirkland and M. Takeguchi, Appl. Phys. Lett., 2012, 101, 253108 CrossRef.
- A. Hashimoto and M. Takeguchi, J. Electron Microsc., 2012, 61, 409 Search PubMed.
- W. Sinkler, S. I. Sanchez, S. A. Bradley, J. Wen, B. Mishra, S. D. Kelly and S. R. Bare, ChemCatChem, 2015, 7, 3779 CrossRef.
- L. Ling and W.-X. Zhang, Anal. Methods, 2014, 6, 3211 RSC.
- V. Ortalan, A. Uzun, B. C. Gates and N. D. Browning, Nat. Nanotechnol., 2010, 5, 843 CrossRef PubMed.
- D. I. Garcia-Gutierrez, D. F. Garcia-Guttierez, L. M. De Leon-Covian, M. T. Trevino-Gonzalez, M. A. Garza-Navarro, I. E. Moreno-Cortez and R. F. Cienfuegos-Pelaes, J. Phys. Chem. C, 2014, 118, 22291 CrossRef.
- B. Schaffer, W. Grogger, G. Kothleitner and F. Hofer, Ultramicroscopy, 2010, 110, 1087 CrossRef.
- A. L. Koh, K. Bao, I. Khan, W. E. Smith, G. Kothleitner, P. Nordlander, S. A. Maier and D. W. McComb, ACS Nano, 2009, 3, 3015 CrossRef PubMed.
- S. M. Collins, E. Ringe, M. Duchamp, Z. Saghi, R. E. Dunin-Borkowski and P. A. Midgley, ACS Photonics, 2015, 2, 1628 CrossRef.
- J. Wei, N. Jiang, J. Xu, X. Bai and J. Liu, Nano Lett., 2015, 15, 5926 CrossRef PubMed.
- T. C. Rojas, M. J. Sayagues, A. Caballero, Y. Koltypin, A. Gedanken, L. Ponsonnet, B. Vacher, J. M. Martin and A. Fernandez, J. Mater. Chem., 2000, 10, 715 RSC.
- J. M. Schliesser, R. E. Olsen, D. B. Enfield and B. F. Woodfield, J. Phys. Chem. C, 2015, 119, 17867 CrossRef.
- W. J. Bowman, K. March, C. A. Hernandez and P. A. Crozier, Ultramicroscopy, 2016, 167, 5 CrossRef PubMed.
- R. Arenal, L. de Matteis, L. Custardoy, A. Mayoral, M. Tence, V. Grazu, J. M. De La Fuente, C. Marquina and M. R. Ibarra, ACS Nano, 2017, 7, 4006 CrossRef PubMed.
- S. Estrade, Ll. Yedra, A. Lopez-Ortega, M. Estrader, G. Salazar-Alvarez, M. D. Baro, J. Nogues and F. Peiro, Micron, 2012, 43, 30 CrossRef PubMed.
- S. Bals, B. Goris, L. M. Liz-Marzan and G. Van Tendeloo, Angew. Chem., Int. Ed., 2014, 53, 10600 CrossRef PubMed.
- J. Meurig Thomas, R. Leary, P. A. Midgley and D. J. Holland, J. Colloid Interface Sci., 2013, 392, 7 CrossRef PubMed.
- N. Y. Jin-Phillipp, T. N. Krauss and P. A. van Aken, ACS Nano, 2012, 6, 4039 CrossRef PubMed.
- J. C. Hernandez-Garrido, M. S. Moreno, C. Ducati, L. A. Perez, P. A. Midgley and E. A. Coronado, Nanoscale, 2014, 6, 12696 RSC.
- S. Benlekbir, T. Epicier, M. Bausach, M. Aouine and G. Berhault, Philos. Mag. Lett., 2009, 89, 145 CrossRef.
- I. Florea, M. Houlle, O. Ersen, L. Roiban, A. Deneuve, I. Janowska, P. Nguyen, C. Pham and C. Pham-Huu, J. Phys. Chem. C, 2009, 113, 17711 CrossRef.
- C. J. Gommes, K. de Jong, J.-P. Pirard and S. Blacher, Langmuir, 2005, 21, 12378 CrossRef PubMed.
- S. Mourdikoudis, M. Chirea, T. Altantzis, I. Pastoriza-Santos, J. Perez-Juste, F. Silva, S. Bals and L. M. Liz-Marzan, Nanoscale, 2013, 5, 4776 RSC.
- J. Zecevic, A. M. J. van der Eerden, H. Friedrich, P. E. de Jongh and K. P. de Jong, Microporous Mesoporous Mater., 2012, 164, 99 CrossRef.
- D. Alloyeau, C. Ricolleau, T. Oikawa, C. Langlois, Y. Le Bouar and A. Loiseau, Ultramicroscopy, 2009, 109, 788 CrossRef PubMed.
- Z. Saghi, D. J. Holland, R. Leary, A. Falqui, G. Bertoni, A. J. Sederman, L. F. Gladden and P. A. Midgley, Nano Lett., 2011, 11, 4666 CrossRef PubMed.
- M. Okuda, M. Takeguchi, O. O. Ruairc, M. Tagaya, Y. Zhu, A. Hashimoto, N. Hanagata, W. Schmitt and T. Ikoma, J. Electron Microsc., 2010, 59, 173 CrossRef PubMed.
- N. Monsegue, X. Jin, T. Echigo, G. Wang and M. Murayama, Microsc. Microanal., 2012, 18, 1362 CrossRef PubMed.
- A. Mazzaglia, L. M. Scolaro, A. Mezzi, S. Kaciulis, T. De Caro, G. M. Ingo and G. Padeletti, J. Phys. Chem. C, 2009, 113, 12772 CrossRef.
- P. J. Kempen, C. Hitzman, L. S. Sasportas, S. S. Gambhir and R. Sinclair, Mater. Res. Soc. Symp. Proc., 2013, 1569, 157 CrossRef PubMed.
- A. Goldstein, Y. Soroka, M. Frusic-Zlotkin, I. Popov and R. Kohen, J. Microsc., 2014, 256, 237 CrossRef PubMed.
- A. Delvalee, N. Feltin, S. Ducourtieux, M. Trabelsi and J. F. Hochepied, Meas. Sci. Technol., 2015, 26, 085601 CrossRef.
- S. Rades, V.-D. Hodoroaba, T. Salge, T. Wirth, M. P. Lobera, R. H. Labrador, K. Natte, T. Behnke, T. Gross and W. E. S. Unger, RSC Adv., 2014, 4, 49577 RSC.
- V.-D. Hodoroaba, S. Raddes and W. E. S. Unger, Surf. Interface Anal., 2014, 46, 945 CrossRef.
- V.-D. Hodoroaba, C. Motzkus, T. Mace and S. Vaslin-Reimann, Microsc. Microanal., 2014, 20, 602 CrossRef PubMed.
- B. Chen, Z. Xia and K. Lu, J. Eur. Ceram. Soc., 2013, 33, 2499 CrossRef.
- M. Ashokkumar and S. Muthukumaran, Opt. Mater., 2014, 37, 671 CrossRef.
- A. Vilalta-Clemente, G. Naresh-Kumar, M. Nouf-Allehiani, P. Gamarra, M. A. di Forte-Poisson, C. Trager-Cowand and A. J. Wilkinson, Acta Mater., 2017, 125, 125 CrossRef.
- A. Bochmann, C. Katzer, F. Schmidl and S. Teichert, J. Appl. Phys., 2015, 117, 215306 CrossRef.
- A. Koblischka-Veneva, M. R. Koblischka, N. H. Babu, D. A. Cardwell, L. Shlyk and G. Krabbes, Supercond. Sci. Technol., 2006, 19, S562 CrossRef.
- A. Koblischka-Veneva, M. R. Koblischka, N. H. Babu, D. A. Cardwell and F. Mucklich, Physica C, 2006, 445, 379 CrossRef.
- J. A. Small, J. R. Michael and D. S. Bright, J. Microsc., 2002, 206, 170 CrossRef PubMed.
- G. Binnig and C. F. Quate, Phys. Rev. Lett., 1986, 56, 930 CrossRef PubMed.
- A. Vilalta-Clemente and K. Gloystein, Principles of Atomic Force Microscopy (AFM), in Proceedings of Physics of Advanced Materials Winter School, 2008 Search PubMed.
- Á. Mechler, J. Kopniczky, J. Kokavecz, A. Hoel, C. G. Granqvist and P. Heszler, Phys. Rev. B: Condens. Matter, 2005, 72, 125407 CrossRef.
- D. Patel, C. Perry and S. Kennedy, Proceedings of SPIE, Optical Components and Materials IV, San Jose, CA, 2007, vol. 6469, p. 64690A Search PubMed.
- H. Qiu, Y. Gao, C. E. Boott, O. E. C. Gould, R. L. Harniman, M. J. Miles, S. E. D. Webb, M. A. Winnik and I. Manners, Science, 2016, 352, 697 CrossRef PubMed.
- A. Pitto-Barry, L. M. A. Perdigao, M. Walker, J. Lawrence, G. Costantini, P. J. Sadler and N. P. E. Barry, Dalton Trans., 2015, 44, 20308 RSC.
- F. Hubenthal, D. Blázquez Sánchez and F. Träger, Appl. Sci., 2012, 2, 566 CrossRef.
- M. Oćwieja, M. Morga and Z. Adamczyk, J. Nanopart. Res., 2013, 15, 1460 CrossRef PubMed.
- C. M. Hoo, N. Starostin, P. West and M. L. Mecartney, J. Nanopart. Res., 2008, 10, 89 CrossRef.
- J. R. Viguié, J. Sukmanowski, B. Nölting and F. X. Royer, Colloids Surf., A, 2007, 302, 269 CrossRef.
- Y. Gu, H. Xie, J. Gao, D. Liu, C. T. Williams, C. J. Murphy and H. J. Ploehn, Langmuir, 2005, 21, 3122 CrossRef PubMed.
- J. E. De Andrade, R. Machado, M. A. Macêdo and F. G. C. Cunha, Polimeros, 2013, 23, 19 Search PubMed.
- L. Fekete, K. Kůsová, V. Petrák and I. Kratochvílová, J. Nanopart. Res., 2012, 14, 1062 CrossRef.
- A. A. Akhmadeev and M. K. Salakhov, J. Phys.: Conf. Ser., 2014, 560, 12005 CrossRef.
- M. Kopaczyńska, J. H. Fuhrhop, A. M. Trzeciak, J. J. Ziółkowski and R. Choukroun, New J. Chem., 2008, 32, 1509 RSC.
- M. Muniz-Miranda, B. Pergolese, A. Bigotto, A. Giusti and M. Innocenti, Mater. Sci. Eng., C, 2007, 27, 1295 CrossRef.
- L. H. Lu, G. Y. Sun, H. J. Zhang, H. S. Wang, S. Q. Xi, J. Q. Hu, Z. Q. Tian and R. Chen, J. Mater. Chem., 2004, 14, 1005 RSC.
- M. Fan and A. G. Brolo, Phys. Chem. Chem. Phys., 2009, 11, 7381 RSC.
- S. Nie and S. R. Emory, Science, 1997, 275, 1102 CrossRef PubMed.
- X. J. Raj and T. Nishimura, Int. J. Electrochem. Sci., 2014, 9, 2090 Search PubMed.
- G. Cordova, B. Y. Lee and Z. Leonenko, NanoWorld J., 2016, 2, 10 Search PubMed.
- C. S. Neves, P. Quaresma, P. V. Baptista, P. A. Carvalho, J. P. Araujo, E. Pereira and P. Eaton, Nanotechnology, 2010, 21, 305706 CrossRef PubMed.
- V. L. Mironov, D. S. Nikitushkin, D. S. Petrov, A. B. Shubin and P. A. Zhdan, J. Surf. Invest., 2007, 1, 348 CrossRef.
- C. Moya, O. Iglesias-Freire, X. Battle, A. Labarta and A. Asenjo, Nanoscale, 2015, 7, 17764 RSC.
- B. Torre, G. Bertoni, D. Fragouli, A. Falqui, M. Salerno, A. Diaspro, R. Cingolani and A. Athanassiou, Sci. Rep., 2011, 1, 202 CrossRef PubMed.
- B. Akbari, M. P. Tavandashti and M. Zandrahimi, Iran. J. Mater. Sci. Eng., 2011, 8, 48 Search PubMed.
- C. Gollwitzer, D. Bartczak, H. Goenaga-Infante, V. Kestens, M. Krumrey, C. Minelli, M. Palmai, Y. Ramaye, G. Roebben, A. Sikora and Z. Varga, Anal. Methods, 2016, 8, 5272 RSC.
- A. Sikora, A. G. Shard and C. Minelli, Langmuir, 2016, 32, 2216 CrossRef PubMed.
- A. Braun, V. Kestens, K. Franks, G. Roebben, A. Lamberty and T. P. J. Linsinger, J. Nanopart. Res., 2012, 14, 1021 CrossRef.
- P.-J. de Temmerman, E. Verleysen, J. Lammertyn and J. Mast, J. Nanopart. Res., 2014, 16, 2628 CrossRef.
- R. P. Carney, J. Y. Kim, H. Qian, R. Jin, H. Mehenni, F. Stellacci and O. M. Bakr, Nat. Commun., 2011, 2, 335 CrossRef PubMed.
- N. C. Bell, C. Minelli and A. G. Shard, Anal. Methods, 2013, 5, 4591 RSC.
- S. J. S. Qazi, A. R. Rennie, J. K. Cockcroft and M. Vickers, J. Colloid Interface Sci., 2009, 338, 105 CrossRef PubMed.
- A. Ruiz-Baltazar, A. Escobedo, U. Pal, R. Perez and G. Rosas, MRS Proc., 2010, 1275, 27 Search PubMed.
| This journal is © The Royal Society of Chemistry 2018 |