
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Translocation of silver nanoparticles in the ex vivo human placenta perfusion model characterized by single particle ICP-MS†
Janja
Vidmar
 ab,
Katrin
Loeschner
c,
Manuel
Correia
c,
Erik H.
Larsen
c,
Pius
Manser
d,
Adrian
Wichser
de,
Kailen
Boodhia
f,
Zahraa S.
Al-Ahmady
g,
Jaimé
Ruiz
h,
Didier
Astruc
h and
Tina
Buerki-Thurnherr
*d
ab,
Katrin
Loeschner
c,
Manuel
Correia
c,
Erik H.
Larsen
c,
Pius
Manser
d,
Adrian
Wichser
de,
Kailen
Boodhia
f,
Zahraa S.
Al-Ahmady
g,
Jaimé
Ruiz
h,
Didier
Astruc
h and
Tina
Buerki-Thurnherr
*d
aDepartment of Environmental Sciences, Jožef Stefan Institute, Jamova 39, 1000 Ljubljana, Slovenia
bJožef Stefan International Postgraduate School, Jamova 39, 1000 Ljubljana, Slovenia
cResearch Group for Nano-Bio Science, Division for Food Technology, National Food Institute, Technical University of Denmark, Kemitorvet 201, DK-2800 Kgs. Lyngby, Denmark
dParticles-Biology Interactions, Empa, Swiss Federal Laboratories for Materials Science and Technology, Lerchenfeldstrasse 5, 9014 St. Gallen, Switzerland. E-mail: tina.buerki@empa.ch
eAnalytical Chemistry, Empa, Swiss Federal Laboratories for Materials Science and Technology, Überlandstrasse 129, CH-8600 Dübendorf, Switzerland
fNational Institute for Occupational Health, National Health Laboratory Service, 25 Hospital Street, Constitution Hill, 4788 Johannesburg, South Africa
gFaculty of Biology, Medicine and Health, Division of Pharmacy and Optometry, Nanomedicine Lab, University of Manchester, Oxford Road, M13 9PL Manchester, UK
hISM, UMR CNRS 5255, Univ. Bordeaux, 351 Cours de la Libération, 33405 Talence Cedex, France
First published on 11th June 2018
Abstract
With the extensive use of silver nanoparticles (AgNPs) in various consumer products their potential toxicity is of great concern especially for highly sensitive population groups such as pregnant women and even the developing fetus. To understand if AgNPs are taken up and cross the human placenta, we studied their translocation and accumulation in the human ex vivo placenta perfusion model by single particle ICP-MS (spICP-MS). The impact of different surface modifications on placental transfer was assessed by AgNPs with two different modifications: polyethylene glycol (AgPEG NPs) and sodium carboxylate (AgCOONa NPs). AgNPs and ionic Ag were detected in the fetal circulation in low but not negligible amounts. Slightly higher Ag translocation across the placental barrier for perfusion with AgPEG NPs and higher AgNP accumulation in placental tissue for perfusion with AgCOONa NPs were observed. Since these AgNPs are soluble in water, we tried to distinguish between the translocation of dissolved and particulate Ag. Perfusion with AgNO3 revealed the formation of Ag containing NPs in both circulations over time, of which the amount and their size in the fetal circulation were comparable to those from perfusion experiments with both AgNP types. Although we were not able to clarify whether intact AgNPs and/or Ag precipitates from dissolved Ag cross the placental barrier, our study highlights that uptake of Ag ions and/or dissolution of AgNPs in the tissue followed by re-precipitation in the fetal circulation needs to be considered as an important pathway in studies of AgNP translocation across biological barriers.
Introduction
Silver nanoparticles (AgNPs) are, due to their antibacterial properties, one of the most widely used engineered NPs, and are found in many commercial and medical products (e.g. food packaging, textiles, laundry detergents, prosthetic devices, dietary supplements, cosmetics). Humans may come into contact with AgNPs either accidentally (e.g. through exposure from contaminated drinking water, inhalation or by direct skin contact) or intentionally (e.g. in the case of nanomedical wound dressings or other applications).1 This raises concerns about AgNP accumulation, long-term retention in an organism and subsequent adverse health effects. There is increasing evidence for potential human toxicity of AgNPs2 and possible mechanisms of AgNP toxicity have been described and include their adverse effects on cell membranes, mitochondria and genetic material, causing induction of reactive oxygen species (ROS), oxidative stress, genotoxicity and apoptosis.2–4The growing fetus is particularly vulnerable to harmful compounds even at concentrations that are non-toxic for adults. Although the placenta provides a protective barrier for the entry of harmful substances, previous studies have demonstrated that some NPs can pass the placental barrier and induce pregnancy or fetal complications in pregnant rodents.5–8 For AgNPs, no developmental toxicity has been observed after repeated-dose oral administration in rats.9–11 In contrast, exposure to AgNPs induced teratogenic effects in mice12–14 and aquatic species.15,16 For instance, pulmonary exposure of pregnant mice to AgNPs induced an increased number of resorbed fetuses14 or long-lasting impairment of lung development of the offspring.13 However, results from animal experiments cannot directly be extrapolated to humans due to substantial differences in placental development, structure and function among species.17,18 The most promising and clinically relevant model for studying accumulation and transport of substances across the human placenta is the ex vivo human placental perfusion system developed by Panigel et al.19 Although this model is increasingly being used to study the transfer of different NPs,20,21 no study with AgNPs has been performed so far. The only available data on potential transplacental transfer of AgNPs are from rodent studies, where increased Ag concentrations were detected in fetal organs.11,13,22–24 Recently, the visualization of AgNPs with similar sizes as those of the applied AgNPs in the placenta and fetal tissues has been interpreted as an indicator for placental transfer of these particles.13 However, it is not yet clearly established if Ag transfer occurs in particulate or ionic forms and how different AgNP properties and functionalizations affect placental barrier crossing.
In general, NP toxicity, cellular uptake or transfer across biological barriers depend not only on particle concentration, but also on their physicochemical properties,25 surface modification, agglomeration26 and dissolution.27 Dissolution of AgNPs, also in complex biological systems, has been widely reported in the literature28 but there is a lack of consensus if AgNP-mediated toxicity is driven by the particles per se, the released Ag ions, or their combination.29 Some evidence that the release of Ag ions from NPs is responsible for their toxic effects toward organisms has been provided by several studies showing that AgNPs did not have bactericidal and cytotoxic effects when no dissolution of the NPs occurred28,30 or that dissolved Ag had similar neurotoxic effects as AgNPs.31
A common problem for the reliable quantification and characterization of NPs under realistic exposure concentrations, where transfer rates are expected to be very low, is the insufficient limit of detection (LOD) of many analytical methods (both in terms of NP concentration and size) as well as problems related to matrix interferences and availability of efficient sample preparation methods.8,32 Inductively coupled plasma-mass spectrometry in single particle mode (spICP-MS), established by Degueldre et al.,33 has become a technique of choice for studying metal-containing NPs in various matrices. spICP-MS is capable of providing information on the number-based particle size distribution and quantifying particle number and mass concentration as well as mass concentration of the dissolved fraction of the element in question. In spICP-MS analysis, a highly diluted suspension of NPs (particle mass concentrations usually in the range of ng L−1) and use of short dwell times (from a few hundred microseconds to a few milliseconds) are required. Under ideal conditions, each recorded signal corresponds to a single particle. The frequency of ICP-MS signals can be used to determine the NP number concentration, while the intensity of the signals is related to the mass of the detected element in the NP and thus to its size under the assumption of a spherical particle.34 Furthermore, spICP-MS does not require demanding sample preparation for aqueous suspensions (only dilution), while for NPs in tissue samples, appropriate digestion procedures have to be applied.35,36
The goal of this study was the investigation of AgNP translocation across the human placental barrier in dependence of negative or neutral surface modifications (COONa versus PEG). Since AgNPs can dissolve, one of the aims was to investigate if precipitation of AgNPs from Ag ions occurred. Perfusion experiments were performed using the ex vivo human placenta perfusion model, which for the first time enabled studying translocation and accumulation of AgNPs in a human model.
Experimental section
Nanoparticles
AgNPs (AgPEG and AgCOONa NPs) were provided within the EU FP7 Nanosolutions project (http://www.nanosolutionsfp7.com). The AgNPs were custom-synthesized for the Nanosolutions project to mimic commercial NPs in order to provide a more realistic, real-life safety assessment. A detailed description of the AgNP synthesis and characterization is presented in ESI.† AgNPs were provided as aqueous suspensions with a particle mass concentration of 10 mg mL−1 in 50 mL brown glass vials (as protection from ambient light).Ex vivo human placental perfusion and sample collection
Placentas were obtained from uncomplicated term pregnancies after caesarean section at the Cantonal Hospital or Klinik Stephanshorn in St Gallen. Written informed consent was obtained prior to delivery. The project was approved by the local ethics committee (EKSG 10/078) and performed in accordance with the principles of the Declaration of Helsinki. The placenta perfusion experiments were performed on a human ex vivo placenta perfusion model at the Swiss Federal Laboratories for Materials Science and Technology (Empa) as described previously.37–39 Criteria for a successful perfusions were leak from fetal to maternal circulation of <4 ml h−1 and stable pH (7.2–7.4). The perfusion medium (PM) was M199 tissue culture medium (Sigma, St Louis, MO, USA) diluted 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 with Earl's buffer and supplemented with 1 g L−1 glucose (Sigma, St Louis, MO, USA), 10 g L−1 bovine serum albumin (AppliChem GmbH, Darmstadt, Germany), 10 g L−1 dextran 40 (Sigma, St Louis, MO, USA), 2500 IU L−1 sodium heparin (B. Braun Medical AG, Melsungen, Germany), 250 mg L−1 amoxicillin (GlaxoSmithKline AG, Brentford, UK) and 2.2 g L−1 sodium bicarbonate (Merck, Darmstadt, Germany). Prior to the experiments, AgNPs stock suspensions were gently mixed by inverting the vial, diluted 250-times (AgPEG) or 135-times (AgCOONa) in PM and immediately used for placental perfusion studies. The resulting Ag mass concentrations for AgPEG and AgCOONa NPs in the maternal circulation at the start of the experiment were 12.48 μg mL−1 and 39.26 μg mL−1, respectively, which corresponded to 40 μg mL−1 and 75 μg mL−1 of particle mass concentrations including coating, respectively. The experiments were performed in triplicates (N = 3) for each AgNP type. Maternal and fetal perfusates were sampled in duplicate (experimental subsamples) after 0, 1, 3 and 6 h of perfusion. In order to assess possible precipitation of insoluble silver salts as particles, perfusion with 1 μg mL−1 of ionic Ag (AgNO3) (N = 3) was performed. This concentration was selected as the maximum amount of ionic Ag present in the stock solution of AgNPs. Control perfusion with PM without addition of AgNPs (N = 1) was also conducted to examine the background levels of Ag.
:2 with Earl's buffer and supplemented with 1 g L−1 glucose (Sigma, St Louis, MO, USA), 10 g L−1 bovine serum albumin (AppliChem GmbH, Darmstadt, Germany), 10 g L−1 dextran 40 (Sigma, St Louis, MO, USA), 2500 IU L−1 sodium heparin (B. Braun Medical AG, Melsungen, Germany), 250 mg L−1 amoxicillin (GlaxoSmithKline AG, Brentford, UK) and 2.2 g L−1 sodium bicarbonate (Merck, Darmstadt, Germany). Prior to the experiments, AgNPs stock suspensions were gently mixed by inverting the vial, diluted 250-times (AgPEG) or 135-times (AgCOONa) in PM and immediately used for placental perfusion studies. The resulting Ag mass concentrations for AgPEG and AgCOONa NPs in the maternal circulation at the start of the experiment were 12.48 μg mL−1 and 39.26 μg mL−1, respectively, which corresponded to 40 μg mL−1 and 75 μg mL−1 of particle mass concentrations including coating, respectively. The experiments were performed in triplicates (N = 3) for each AgNP type. Maternal and fetal perfusates were sampled in duplicate (experimental subsamples) after 0, 1, 3 and 6 h of perfusion. In order to assess possible precipitation of insoluble silver salts as particles, perfusion with 1 μg mL−1 of ionic Ag (AgNO3) (N = 3) was performed. This concentration was selected as the maximum amount of ionic Ag present in the stock solution of AgNPs. Control perfusion with PM without addition of AgNPs (N = 1) was also conducted to examine the background levels of Ag.
In order to investigate the degree of AgNPs accumulation, placental tissue samples from the villous chorion were collected before (0 h) and after each perfusion experiment (after 6 h) and homogenized by mixing 2 g of tissue in 2 mL of perfusion buffer (dilution factor of 2). All samples for spICP-MS analysis, i.e. perfusates and tissue homogenates, were immediately delivered to the Technical University of Denmark at 4 °C and analyzed by spICP-MS within 3 days. A part of the replicates (N = 1 for AgPEG NPs and N = 2 for AgNO3) were performed one year later.
DLS measurements
Determination of hydrodynamic size and size distribution of AgNPs in UPW (resistivity 18.2 mΩ cm−1), obtained from a Millipore Element apparatus (Millipore, Milford, MA, USA) and PM was performed by DLS (Zetasizer, Malvern Instruments, UK). The measurements were carried out using dust-free disposable cuvettes (DTS0012, Malvern Instruments, UK). The measurement position was fixed at the center of the cuvette (4.65 mm from the cuvette wall). The detection angle was 173° and the measurement was performed at a temperature of 25 °C, which was achieved following equilibration for 1–2 minutes. Five consecutive measurements were performed using a minimum of 11 runs of 10 s each. The results obtained by DLS of the measured samples, given as Z-average (Zave) and polydispersity index, were determined by averaging the results of the five repeated measurements (N = 5) for one diluted AgNPs suspension.Enzymatic tissue digestion
For the enzymatic treatment of tissue, the enzyme solution was prepared by diluting 3 mg mL−1 of Proteinase K (commercial protease from Engyodontium album) and 0.5% (m/v) sodium dodecyl sulphate (SDS, ReagentPlus with ≥98.5% purity) in enzyme buffer, consisting of 50 mM ammonium bicarbonate buffer (with 99% purity) and 200 μg mL−1 sodium azide (with ≥98% purity), all purchased from Sigma-Aldrich (St Louis, MO, USA) at pH 7.4 (PHM 240 pH/ION meter from Radiometer, Copenhagen, Denmark). To allow double determinations, each homogenated sample was divided into two subsamples before enzymatic treatment. A mass of 0.2 g of tissue homogenate (corresponding to 0.1 g of wet tissue) was weighed into plastic tubes with round bottom (Karter Scientific Labware Manufacturing, Lake Charles, LA, USA). After the addition of 2.0 mL of the enzyme solution (60 μg of enzyme per mg of wet tissue) and 300 μL of UPW, samples were shortly vortexed with a MS2 minishaker (IKA Works, Inc., Wilmington, NC, USA) until no sample adhered to the tube walls. The samples were placed in the water bath at 37 °C (HetoHMT 200 RS thermostat) and magnetically stirred with the use of Telesystem HP15 magnetic stirrer (Holm & Halby, Allerød, Denmark) for 1 h. At the end of enzymatic treatment, the completely digested samples were placed in ice water to slow down the enzymatic digestion process and appropriately diluted with UPW prior to spICP-MS measurements.spICP-MS analysis
A Thermo Scientific iCAP Q ICP-MS instrument (Thermo Fisher Scientific GmbH, Bremen, Germany) was used for single particle measurement of the samples from all the perfusion experiments with the following practical exceptions. For the samples from one of the perfusion experiments with AgCOONa NPs an Agilent 8800 Triple Quadrupole ICP-MS instrument (Agilent Technologies, California, USA) was applied and an Agilent 7900 ICP-MS instrument was used for measuring the samples from one perfusion experiment with AgPEG NPs and two perfusion experiments with AgNO3. Instrument tuning was performed prior to analysis by using a tuning solution according to the manufacturer's recommendation. Settings for all three instruments are given in Table S1.†Accurate sample flow rate was determined daily by weighing. Quantification of particle size (particle diameter) was based on a calibration curve constructed from a blank (UPW) and four concentration levels of certified standard solutions of Ag (1000 μg mL−1, PlasmaCAL, SCP Science, Baie D'Urfé, QC, Canada) ranging from 0.2 to 2.0 ng mL−1. For each sample, the isotope 107Ag was detected using 3 ms dwell time for a total measurement time of 180–600 s (equivalent to 60000–200000 data points).
The recorded signal intensity data were plotted (in cps) versus number of “events”, to create a signal distribution histogram using Microsoft Excel (Microsoft, WA, USA). The threshold between signals from instrumental noise/dissolved Ag and particulate Ag was set to a value that gives a lowest detectable particle diameter of 25 nm. This value corresponded to the maximum of dissolved Ag concentration that was found in the PM samples from the maternal side at time 0 h (i.e., up to 68 ng L−1 in diluted samples). Lower diameters (down to 13 nm) could be detected in fetal perfusates and maternal perfusates collected at longer perfusion times. However, the application of the same threshold for all samples ensured that a direct comparison of particle mass and number concentrations as well as median particle diameters between samples was possible. The Ag mass fraction below the selected threshold was used to estimate the mass concentration of dissolved Ag and AgNPs smaller than 25 nm.
The transport efficiency of the liquid NP samples through the sample introduction system was determined according to the “particle size” method40 with the use of reference gold nanoparticle suspension (RM8012), obtained from the National Institute for Science and Technology (NIST, Gaithersburg, MD, USA) with a known average particle diameter (27.6 ± 2.1 nm as determined by TEM) and gold mass concentration (48.17 ± 0.33 μg g−1). This suspension, diluted 107-times with UPW, in combination with a calibration curve made of dissolved Au standards (0, 0.5 and 1 ng mL−1) (1000 μg mL−1 Au standard, PlasmaCAL from SCP Science, Baie D'Urfé, QC, Canada) was used to determine the transport efficiency of the NPs. With the use of AuNPs, a calibration curve that relates the most common signal intensity to the average particle mass was created. Then, a corresponding calibration curve from the dissolved Au standards was constructed by multiplying dissolved Au standard concentration by the sample flow rate and the dwell time to calculate the total Au mass. Finally, the ratio of the slope from the calibration curve made from the ionic Au standards and the slope from the calibration curve created from the AuNPs was used to determine the transport efficiency. To assess the accuracy of the transport efficiency determined by the “particle size” method with the use of AuNPs, the “particle frequency” method40 using 40 nm AgPEG NPs (NanoXact Silver Nanospheres, purchased from Nanocomposix, San Diego, CA, USA) was applied. There was no significant difference in the so determined transport efficiencies. For the validation of the spICP-MS method, the particle number concentration of 40 nm AgPEG NPs (Nanocomposix) was determined and compared to the expected particle number concentration (calculated from the size provided by TEM and Ag mass concentration provided by ICP-MS), where 95 ± 1% (N = 2) recovery was achieved. Complete ionization of AgNPs was confirmed by the comparable Ag concentrations observed for the digested (i.e., dissolved) AgCOONa NPs (determined by conventional ICP-MS) and undigested AgCOONa NPs (determined by spICP-MS) (Fig. S1†).
To prevent AgNPs from sticking to the ICP-MS tubings and to avoid a memory effect during ICP-MS measurements, the following washing procedure was applied after each sample: (1) 1% (v/v) HCl (34–37%), 1% (v/v) HNO3 (67–69%), both of PlasmaPURE quality, and 0.1% (m/v) Triton X-100 (Merck, Darmstadt, Germany) for 1 min at 1.5 mL min−1, (2) UPW for 30 s at 1 mL min−1. Following the analysis of each sample, UPW was aspirated into the ICP-MS for additional rinsing and to ensure that no carryover from the previous measurement was detectable.
Prior to spICP-MS analysis, two (analytical) subsamples were taken from each experimental subsample of perfusate and diluted with ultrapure water (UPW). The list of analyzed samples together with suitable dilution factors and total measurement times, which were determined experimentally, are presented in Table S2.† For the sake of counting statistics, a minimum number of 400 particles were detected per sample and the ratio between detected particles and number of dwells restricted to a maximum of 20% to limit the probability of multiple particle events during one dwell to 10%. The results for the translocation experiments are expressed as Ag mass concentration in perfusates normalized to the Ag mass concentration measured on the maternal side at time 0 h (M0). Ag mass concentrations determined in placental tissues, collected before the start of perfusion, were subtracted from the Ag values determined in tissues after 6 h of perfusion. Results were expressed as Ag mass concentrations in the tissues that were normalized to the applied Ag dose, measured in M0 samples by conventional ICP-MS, taking into account the total mass of the placenta tissue and the total volume of perfusate. Since there was not enough MO sample (maternal perfusate after 0 min of perfusion) left from one of the perfusion with AgCOONa NPs for conventional ICP-MS analysis, M15 sample, collected shortly after the beginning of perfusion (15 min), was used to estimate the starting concentration. This might lead to a slight underestimation of the tissue concentration, however, the error is likely smaller than the biological variations observed between the individual experiments.
Behavior of AgNPs in perfusion medium
For studying aggregation of AgNPs in PM by DLS and spICP-MS, AgPEG and AgCOONa NPs stock suspensions were diluted in UPW or PM 5- and 10-times, respectively, (in case of DLS analysis) and 125- and 1000-times, respectively (in case of spICP-MS analysis). In order to assess if Ag ions could precipitate as NPs in PM, different concentrations of ionic Ag (100 μg mL−1, 10 μg mL−1 and 1 μg mL−1) were prepared in PM and their size distributions measured by DLS immediately after their preparation and 4 days later after storage at room temperature (RT). spICP-MS was applied for detecting possible formation of Ag precipitates in PM in the lower concentration range (4, 2 and 0.2 μg mL−1), over a period of 3 days at RT. Prior to spICP-MS analysis (3 min measurement time), samples containing 4, 2 and 0.2 μg mL−1 ionic Ag were diluted 4000, 2000 and 200-times, respectively. PM was diluted in the same way and measured after each sample.Statistical analysis
Results based on repeated measurements are given as mean ± one standard deviation (STD). The number of repetitions N is stated in parentheses. Data (comparison of quantiles of particle size distributions at different perfusion times, repeatability and reproducibility for sizing and quantification of AgNPs) were statistically analyzed by using one-way Analysis of Variance (ANOVA) in Microsoft Office Excel 2010 (Microsoft, Redmond, WA, USA). A p-value ≤ 0.01 was considered statistically significant.Results and discussion
Characterization of AgNPs
The basic characteristics for the stock suspensions of both AgNP types, including total Ag mass concentration, mass fraction of dissolved Ag, mass fraction of surface modification, primary particle size and zeta potential are presented in Table 1. Besides revealing primary particle size, TEM analysis further showed that AgPEG suspension contained three populations -Ag spheres of 2–8 nm, Ag spheres close to 7–15 nm, and a third minor population of large and irregular particles in the 200–1000 nm size range. The AgCOONa NP suspensions comprised, besides the primary particles, large assemblies of 100–500 nm containing multiple individual Ag spheres of 5–15 nm (Fig. S2†).| AgNP characteristic | AgPEG | AgCOONa |
|---|---|---|
| Results for Ag mass concentration (N = 3), mass fraction of dissolved Ag (N = 2) and zeta potential (N = 5) are presented as mean ± STD. | ||
| Ag mass concentration (mg mL−1) | 3.12 ± 0.06 | 5.30 ± 0.01 |
| Mass fraction of dissolved Ag (w/w %) | 4.9 ± 0.1 | 0.11 ± 0.01 |
| Mass fraction of surface modification (w/w %) | 64.1 | 16.3 |
| Primary particle diameter (nm) | 2–15 | 5–15 |
| Zeta potential at pH 5 in UPW (mV) | −10.7 ± 0.4 | −32.5 ± 1.1 |
All NPs have been tested for endotoxin contamination and were endotoxin free (<0.5 EU mL−1 in Chromogenic LAL assay).
Stability of AgNPs in perfusion medium
In order to assess if AgNPs aggregated/agglomerated in PM, hydrodynamic size and size distribution of AgNPs in UPW and PM were determined by DLS (Fig. 1). Size distributions obtained for AgPEG and AgCOONa NPs in UPW were in agreement with the TEM results (Fig. S2†). For AgPEG NPs in PM, a slightly broader size distribution was observed in comparison to UPW. Size distribution for pure PM (peaks around 10 nm and 500 nm – data not shown) indicated that the size distributions of AgNPs in PM can be attributed to the AgNPs and not to scattering entities in the PM. For AgCOOH NPs in PM, the size distribution shifted to significantly larger sizes with the peak maximum at 1000 nm due to agglomeration/aggregation of the NPs.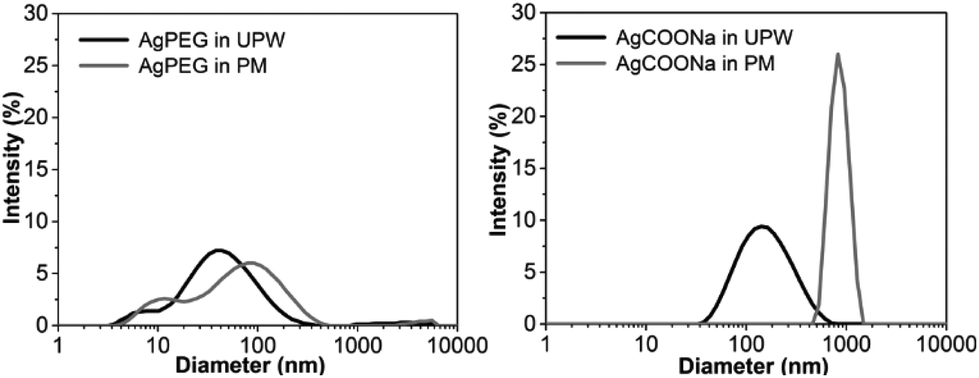 | ||
| Fig. 1 DLS hydrodynamic size measurements of AgNPs dispersed in ultrapure water or perfusion medium. Intensity-based size distributions of AgPEG (left) and AgCOONa NPs (right), dispersed in ultrapure water (UPW) or perfusion medium (PM) at Ag mass concentration of approximately 0.3 mg mL−1. The data is representative of five (N = 5) consecutive DLS measurements for each AgNPs type and media. | ||
Agglomeration/aggregation of AgCOONa NPs in PM was further confirmed by spICP-MS measurements (Table 2 and Fig. 2). While size distributions and mass concentrations of AgPEG NPs were similar in UPW and PM, AgCOONa NPs aggregated/agglomerated in PM resulting in bigger median diameters, higher mass concentration of AgNPs >25 nm and lower mass concentration of AgNPs ≤25 nm in comparison to UPW.
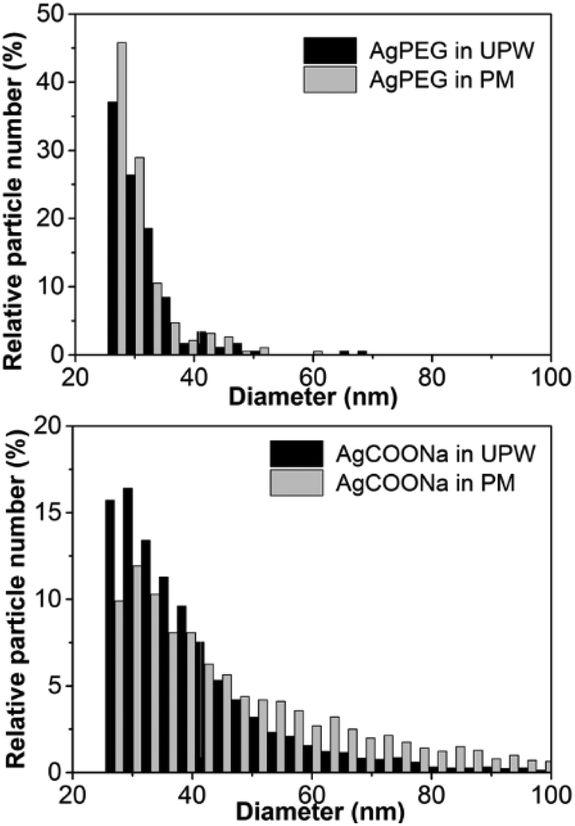 | ||
| Fig. 2 Size distributions of AgPEG NPs and AgCOONa NPs, dispersed in ultrapure water or perfusion medium. The minimum detectable size by spICP-MS was 25 nm. In case of AgCOONa NPs in perfusion medium, NPs up to 270 nm nm were detected. Size distributions are representative of two replicates for each medium and each AgNP type. | ||
| AgNP type | Medium | Expected Ag mass conc.a (μg mL−1) | Mass conc. of AgNPs >25 nm (μg mL−1) | Mass conc. of AgNPs ≤25 nm + Ag ions (μg mL−1) | Mass fraction of AgNPs >25 nmb (%) | Ag mass recoveryc (%) | Median particle diameter (nm) |
|---|---|---|---|---|---|---|---|
| a Based on ICP-MS analysis of digested stock suspensions. b Based on the total Ag mass concentration determined by spICP-MS (≤25 nm + >25 nm). c Ag mass concentration (≤25 nm + >25 nm) determined by spICP-MS in comparison to the Ag mass concentration determined by conventional ICP-MS after acid digestion. | |||||||
| AgPEG | UPW | 12.5 | 0.141 ± 0.004 | 9.05 ± 0.24 | 1.5 | 73.6 | 27.7 ± 0.3 |
| PM | 0.121 ± 0.004 | 8.98 ± 0.14 | 1.3 | 72.9 | 27.4 ± 0.3 | ||
| AgCOONa | UPW | 39.3 | 13.5 ± 0.3 | 17.0 ± 0.7 | 44 | 77.6 | 34.3 ± 0.3 |
| PM | 29.1 ± 2.0 | 7.65 ± 0.31 | 79 | 93.6 | 40.8 ± 0.4 | ||
Translocation of Ag across the human placental barrier
40 μg ml−1 AgPEG NPs (12.5 μg ml−1 Ag) or 74 μg ml−1 AgCOONa (39 μg ml−1 Ag) were added to the maternal circulation in an ex vivo placenta perfusion model, and changes of Ag mass concentrations of the fraction >25 nm and the fraction ≤25 nm (AgNPs ≤25 nm and dissolved Ag) were followed over the time of perfusion in maternal and fetal circulations by spICP-MS (Fig. 3). AgPEG NPs and AgCOONa NPs were selected as AgNPs with neutral and negatively charged surface modifications, respectively, since studies showed that cellular uptake and translocation of negatively charged NPs is less likely to occur compared to neutral or positively charged NPs.20,41–43 The concentrations of AgNPs were chosen as doses that were non-toxic to BeWo trophoblast cells after 6 h of exposure according to an MTS viability assay (Fig. S8†) but still relatively high to be able to detect particle uptake and/or translocation and to mimic a potential worst-case exposure scenario.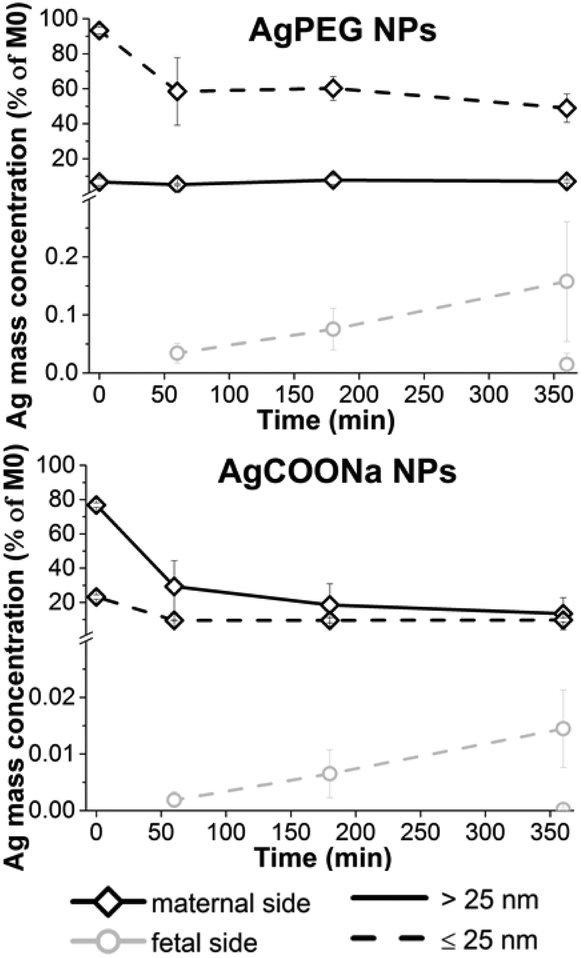 | ||
| Fig. 3 Perfusion profiles of AgPEG and AgCOONa NPs during ex vivo human placental perfusion. Ag mass fraction >25 nm (continuous line) and ≤25 nm (dashed line) were measured on the maternal (black color) and fetal side (gray color) over a time period of 6 h by spICP-MS. Missing points present non-detectable concentrations (<LOD). Data represent the mean ± STD of the independent experiments (N = 3 for AgPEG NPs, N = 3 for AgCOONa NP). | ||
After perfusion with AgCOONa or AgPEG NPs, Ag mass fraction > and ≤25 nm in the fetal circulation increased with time of perfusion but overall, only very low amounts of the maternally applied Ag doses were detected on the fetal side (Fig. 3). The mass percentage of Ag fraction >25 nm that crossed the placental barrier after 6 h of perfusion was 0.0148 ± 0.0192% for AgPEG NPs (N = 3) and 0.0002 ± 0.0002% for AgCOONa NPs (N = 3). However, the highest placental translocation was not observed for the AgNPs > 25 nm but for the small AgNPs and/or Ag ions (fraction ≤25 nm) with 0.1578 ± 0.1032% for AgPEG NPs and 0.0151 ± 0.0077% for AgCOONa NPs. On the maternal side, the Ag fraction >25 nm remained constant, while the Ag fraction ≤25 nm decreased by 44% during 6 h of perfusion in case of AgPEG NPs. In the case of AgCOONa NPs, a strong decrease of both fractions was observed on the maternal side during the 6 h of perfusion (63% decrease for Ag fraction >25 nm and 13% decrease for Ag fraction ≤25 nm).
In addition to quantifying the Ag mass concentrations of different size fractions, spICP-MS analysis was also applied to obtain information about the (number-based) particle size distribution on the maternal side over increasing time of perfusion and on the fetal side at the end of perfusion (Fig. 4). For quantitative comparison of the particle size distribution the 2.5, 50 and 97.5% percentiles of the size distributions were calculated.
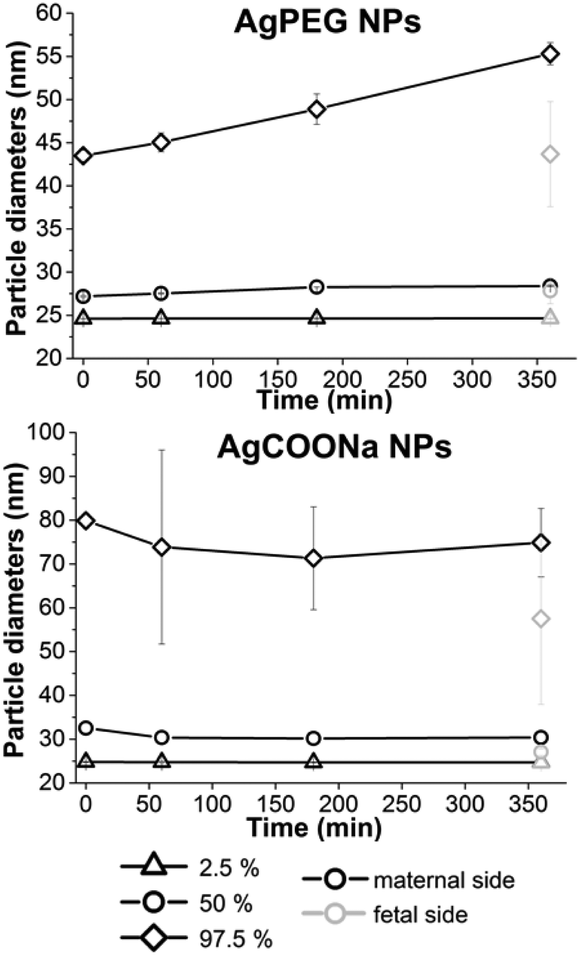 | ||
| Fig. 4 Size distributions of AgNPs formed in perfusion with AgPEG and AgCOONa NPs over time and determined by spICP-MS. Missing points present non-detectable concentrations (<LOD). Data represent the mean ± STD of the independent experiments (N = 3 for AgPEG NPs, N = 3 for AgCOONa NP). | ||
For perfusion experiments with AgCOONa NPs, smaller particles were present in the fetal circulation at the end of perfusion (median particle diameter (MPD) 27.4 ± 0.6 nm) as compared to the maternal side at the beginning of perfusion (MPD 32.8 ± 0.5 nm). In contrast, there was no significant difference in the particle size distribution of AgPEG NPs between maternal (MPD 27.5 ± 0.3 nm) and fetal side (MPD 28.7 ± 1.1 nm). In case of AgPEG NPs, a statistically significant shift (p < 0.01) for all three percentile values of particle size distribution towards larger particles occurred with time on the maternal side (MPD 29.0 ± 0.1 nm after 6 h of perfusion) and reached a size similar to the one observed on the fetal side at the end of the perfusion. For AgCOONa NPs a statistically significant decrease of the particle size with perfusion time was observed (significant only for 50% percentile value) to a size of 30.8 ± 0.1 nm (MPD) on the maternal side after 6 h of perfusion.
To confirm the results obtained by spICP-MS total Ag mass concentrations on the maternal and fetal side for varying time of perfusion were also determined by conventional ICP-MS analysis and compared to the results obtained by spICP-MS (Ag mass fraction > + ≤25 nm, the latter includes also the dissolved Ag fraction) (Fig. S1†). There was no significant difference (t-test, p < 0.01) between the Ag concentrations determined by both methods for the perfusion studies with AgCOONa NPs. However, in case of AgPEG NPs, about 3-times higher Ag concentrations were detected in the maternal perfusates at time 0 h by ICP-MS compared to spICP-MS. This indicates unforeseen Ag mass losses during the spICP-MS measurements. We suspect that the surface properties (PEG coating) led to partial adhesion to the tubings of the sample introduction system of the ICP-MS. However, the recoveries were more favorable (73%) for AgPEG NPs freshly spiked to PM (Table 2). Nevertheless, reduced spICP-MS recoveries of AgPEG NPs do not have an impact on the obtained results, as relative changes were investigated. The percentage of total Ag that crossed the placental barrier (measured on the fetal side after 6 h of perfusion) was 0.0239 ± 0.0197% for AgCOONa and 0.0620 ± 0.0454% for AgPEG NPs when determined by conventional ICP-MS and 0.0152 ± 0.0080% for AgCOONa and 0.0615 ± 0.0482% for AgPEG NPs when determined by spICP-MS.
Accumulation of Ag in the placental tissue
Although placental Ag transfer was very low, Ag mass concentration in the maternal circulation was declining, in particular for AgCOONa NPs (Fig. 3). In order to investigate the degree of Ag accumulation in the placenta, tissue samples were enzymatically treated to release AgNPs and dissolved Ag from the tissue. Ag mass fraction > and ≤25 nm were measured in placental tissues after 6 h of perfusion with AgPEG NPs and AgCOONa NPs (Fig. 5).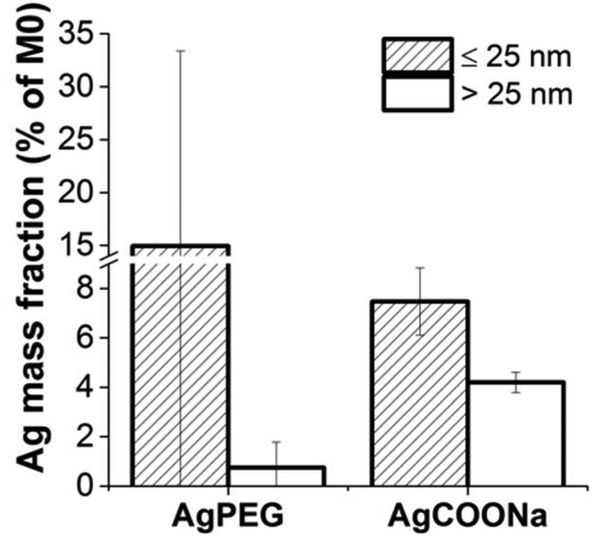 | ||
| Fig. 5 Ag mass concentrations in placental tissues from perfusion experiment with AgPEG and AgCOONa NPs. Ag mass fraction > and ≤25 nm were determined by spICP-MS in enzymatically treated tissue samples. Ag mass concentrations in placenta collected after 6 h of perfusion are normalized to the applied Ag concentration measured in M0 samples (initial concentrations in maternal perfusates) by conventional ICP-MS. Data represent the mean ± STD of the independent experiments (N = 3 for AgPEG NPs, N = 3 for AgCOONa NPs). | ||
For AgCOONa NPs, a much higher Ag fraction >25 nm of the applied dose accumulate in the placenta (4.2%) as compared to AgPEG NPs (0.75%), while the mass concentration of the Ag fraction ≤25 nm was 7.5% for AgCOONa NP and 15% for AgPEG. Overall, Ag mass fraction ≤25 nm represented 95% and 64% of the total internalized Ag mass concentration (≤25 nm + >25 nm) for AgPEG and AgCOONa NPs, respectively. The amount of Ag determined in the placental tissue was therefore much higher in comparison to the translocated Ag (below 0.02% of Ag NPs >25 nm for both AgNP types), suggesting that the transfer of Ag to the fetal circulation was substantially hindered by the placental barrier. Strong accumulation of Ag fraction >25 nm in the placental tissue after the perfusion with AgCOONa NPs was reflected also through the decrease of the Ag concentration on the maternal side over time (Fig. 3). A possible explanation for the high Ag concentrations in placenta may be the presence of high levels of the metal-binding protein metallothionein in this tissue.44 The strong accumulation of Ag in the placental tissue is of potential threat to the developing fetus if it interferes with proper placental development and function or the release of mediators (e.g. inflammatory, angiogenic or endocrine factors) from placental tissue. Such indirect toxicity has been previously described in rodent and human in vitro placental transfer studies for CoCr NPs.25,45,46 Although we did not directly investigate the impact of Ag on placental tissue viability and functionality, the maintenance of barrier integrity throughout the perfusion experiments suggests the absence of major acute toxicity to the placenta. However, further studies would be required to understand the long-term consequences of high Ag tissue levels on placental function. In addition, even if placental transfer of Ag is low, direct teratogenic effects may occur from translocated Ag in particular for a chronic exposure scenario since the fetus is particularly vulnerable to toxic compounds already at low levels. Previous studies with pregnant rodents revealed that AgNPs may cause adverse effects on embryo's health.13,47 Moreover, there is recent evidence that AgNPs cause developmental neurotoxicity in human embryonic neural precursor/stem cells in vitro.48,49
When comparing the sizes of AgNPs determined on the maternal and fetal side after 6 h of perfusion with AgCOONa NPs with those in placental tissue (Fig. 6), we observed larger Ag particles (aggregates/agglomerates) on the maternal side and in the placental tissue compared to the smaller particles on the fetal side. In case of AgPEG NPs, there was no difference in MPDs on the maternal side, in placental tissue and on the fetal side.
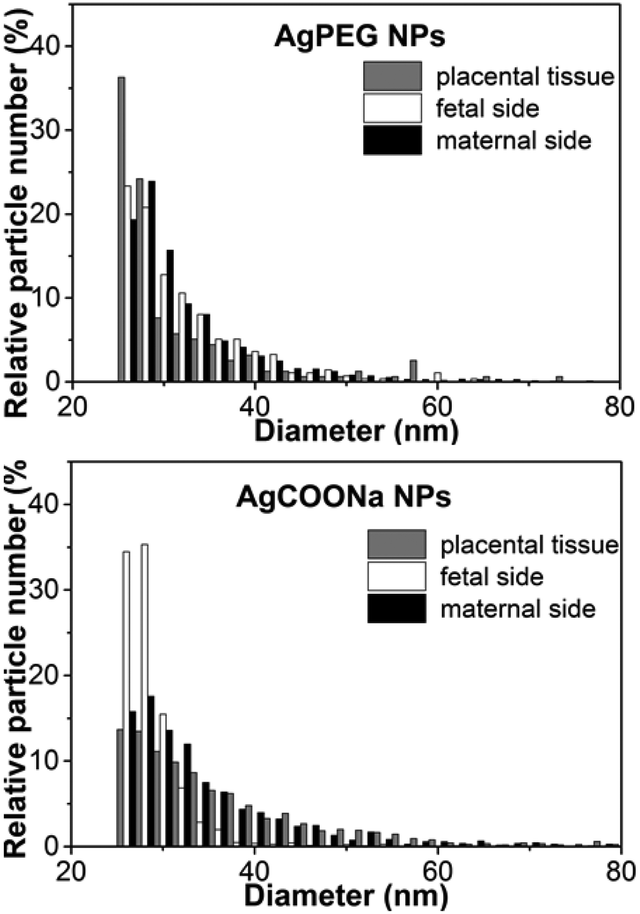 | ||
| Fig. 6 Size distributions of AgPEG and AgCOONa NPs on maternal, fetal side and in placental tissue. Size distributions were determined by spICP-MS after 6 h of perfusion and are representative of three independent perfusion experiments (N = 3 for AgPEG NPs, AgCOONa NPs and AgNO3). | ||
The presence and localization of AgNPs in placental tissue after 6 h of perfusion with AgPEG and AgCOONa NPs was also assessed by CytoViva dark field microscopy (Fig. S4†). The technique has the ability to detect a broad range of differently sized NPs (from small monodispersed NPs50 up to large NP or NP agglomerates/aggregates51) and to distinguish between NP types if spectral profiles are sufficiently distinct.50 Particulates were observed in all perfused tissues and were mainly localized in the syncytiotrophoblast layer. Particulates were neither detected in control samples of placental tissue perfused with perfusion medium only nor in placental tissue sections taken before perfusion with AgNPs. To further verify the specificity of the Ag signals, scans were also performed on AgNP suspensions alone to generate spectral libraries, which were then mapped onto the scans of the NP-perfused tissues using the image classification algorithm, spectral angle mapper (Fig. S5 and S6†). Positive signals of particulate Ag were identified but an unambiguous identification of all AgNP signals was not possible since the spectral profile of the particles changed in the tissue as compared to the spectral library of the NPs in stock solution.
Translocation of dissolved Ag and formation of Ag containing particles
Since AgNPs are soluble, one objective of the study was to distinguish between the translocation and uptake of dissolved and particulate Ag in the ex vivo placenta perfusion model. In our experiments with AgNPs, dissolved Ag was present on the maternal or fetal side either due to particle dissolution in PM during the perfusion experiment or due to the dissolved Ag fraction that was already present in the AgNP suspensions (4.9% for AgPEG and 0.11% for AgCOONa NP suspensions – Table 1) or both. Loza et al.28 have shown that the predominant species formed from dissolved Ag in biological media in the presence of chloride ions is colloidally dispersed nanoparticulate AgCl, whereas in tissues ionic Ag typically deposits as selenium and/or sulfur containing particles.52 We therefore assumed that AgCl precipitation from dissolved Ag would occur in the used PM. In order to study potential de novo formation of Ag containing NPs from ionic Ag during the perfusion experiment, DLS and spICP-MS analyses for different concentrations of ionic Ag in PM were performed. AgNP precipitation in PM (additional peak in the size distribution with a peak maximum at around 100–200 nm) was only detectable by DLS when the ionic Ag concentration was 10 μg mL−1 or higher (Fig. S7†). Even after prolonged incubation (4 days at RT), no Ag particle formation was evident at relevant concentrations of dissolved Ag (1 μg mL−1) (data not shown), most probably because DLS is not sensitive enough to detect NP formation at such small concentrations. No Ag-containing NPs could be detected even by the more sensitive spICP-MS method in PM spiked with 2 or 4 μg mL−1 ionic Ag (2000- and 4000-times diluted prior to analysis, respectively) (data not shown). In PM spiked with 0.2 μg mL−1 a very low number of NPs (92) was detected. A similar number (108) was, however, observed in the corresponding blank sample (200-times diluted PM) indicating that less diluted PM has a stronger ability to wash out AgNPs, adhered to ICP-MS tubes from previous measurements. Therefore, the particles detected in a freshly prepared 0.2 μg mL−1 ionic Ag sample solution are most probably the consequence of the particles wash-out rather than formation of AgNPs in PM. No Ag-containing NPs could be detected after 3 days incubation at RT either (data not shown). In summary, at experimentally ionic Ag concentrations of 0.2 to 4 μg mL−1 no formation of Ag-containing NPs was observed. The minimum detectable particle size in all experiments with dissolved Ag was higher than in ex vivo perfusion experiments (approximately 47 nm) due to the lower degree of dilution of samples prior to spICP-MS analysis. A possible formation of particles smaller than this size cannot be excluded. By performing a perfusion experiment with dissolved Ag, we wanted to assess if the dissolved Ag was able to cross the placental barrier and if any Ag-containing NPs could form from the dissolved Ag in the presence of PM and placental tissue.28,53,54 After perfusion with 1 μg mL−1 AgNO3 (Fig. 7A), an increase of the Ag mass fraction ≤25 nm was measured in the fetal circulation over time by spICP-MS. Interestingly, Ag particles >25 nm appeared in the maternal circulation (already at time 0 h) and their concentration increased gradually throughout the course of a perfusion experiment. A significant number of particles was formed on the maternal ((2.89 ± 1.23) × 107 particles per mL) and fetal side ((1.10 ± 0.37) × 105 particles per mL) after 6 h of perfusion (N = 3). The mass concentration of AgNP >25 nm on the fetal side was 0.0165 ± 0.0070 μg L−1 after 6 h of perfusion (Fig. 7A). This value was in the same concentration range as the mass concentration of AgNPs >25 nm that was observed on the fetal side after perfusion with AgNPs (0.819 ± 1.074 μg L−1 for AgPEG (N = 3) and 0.081 ± 0.071 μg L−1 for AgCOONa (N = 3)) (Fig. 3). The size distribution of the newly precipitated particles on the fetal side after 6 h (MPD 26.1 ± 0.4 nm) was similar to the size distribution of particles detected on the fetal side after AgPEG (MPD 28.7 ± 1.1 nm) or AgCOONa NPs (MPD 27.4 ± 0.6 nm) were added (Fig. S3A†). Moreover, the particle size on the maternal side increased with time of perfusion and larger particles were observed on the maternal side (MPD 31.9 ± 1.5 nm) compared to the fetal side (MPD 26.1 ± 0.4 nm) after 6 h of perfusion (Fig. 7B and Fig. S3B†). Finally, spICP-MS analysis revealed that predominantly Ag ions and/or small AgNPs were taken up by the placental tissue (18% for fraction ≤25 nm, 0.02% for AgNPs >25 nm) (Fig. 7C) and particulates similar to the ones observed after perfusion with AgNPs (Fig. S4†) were observed in placental tissue sections by CytoViva dark field microscopy (Fig. 7D).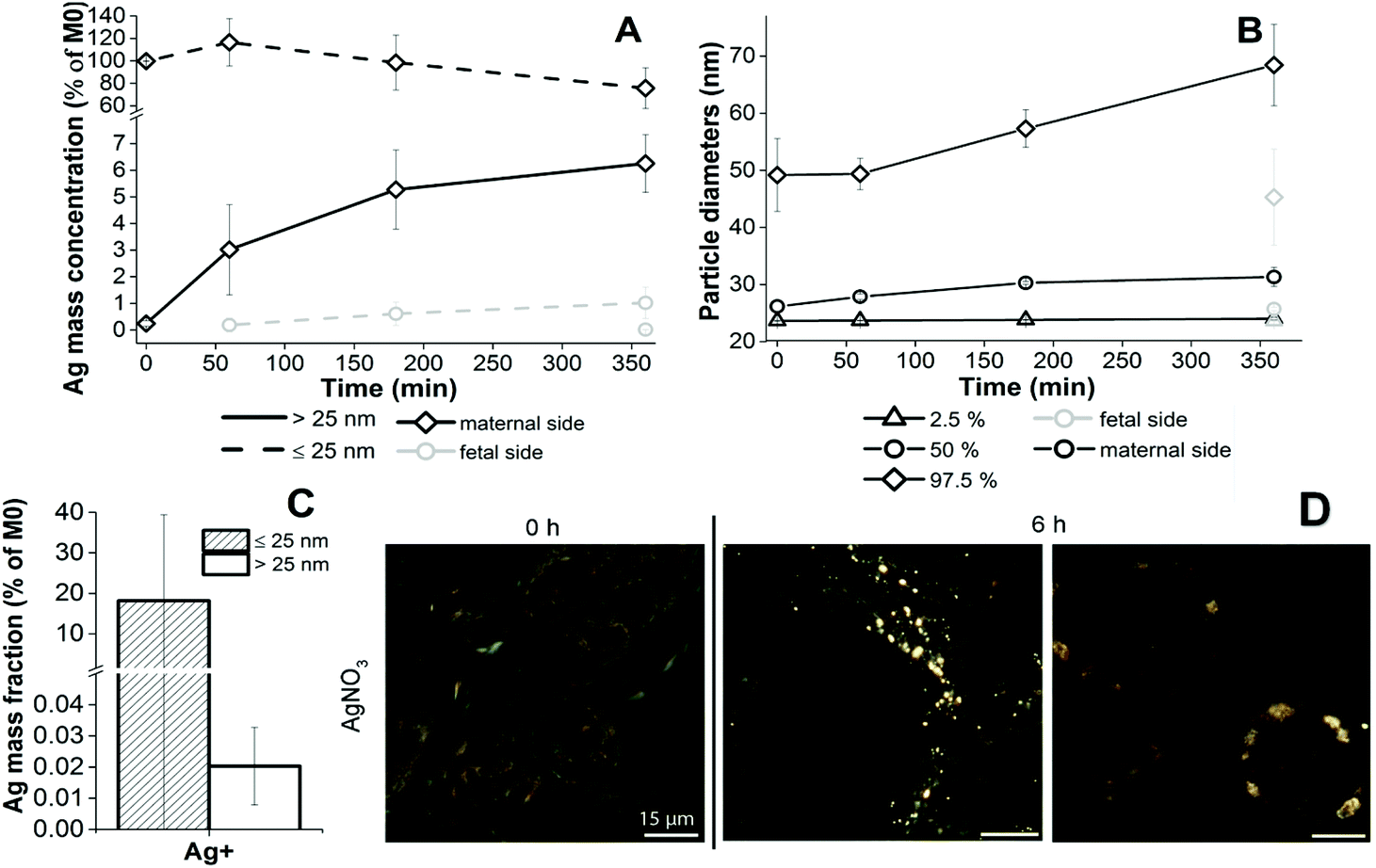 | ||
| Fig. 7 Characteristics of AgNO3 in perfusates and placental tissues during the perfusion experiment. (A) Perfusion profiles of dissolved Ag during ex vivo human placental perfusion determined by spICP-MS. Ag mass fraction >25 nm (continuous line) and ≤25 nm (dashed line) were measured on the maternal (black color) and fetal side (gray color) over a time period of 6 h. Missing points present non-detectable concentrations (<LOD). Data represent the mean ± STD of three independent perfusion experiments (N = 3). (B) Size distributions of AgNPs formed in perfusion with dissolved Ag over time, determined by spICP-MS. Missing points present non-detectable concentrations (<LOD). Data represent the mean ± STD of three independent perfusion experiments (N = 3). (C) Ag mass concentrations in placental tissues from a perfusion experiment with AgNO3. Ag mass fraction >and ≤25 nm were determined by spICP-MS in enzymatically treated tissue samples. Ag mass concentrations in placenta collected after 6 h of perfusion are normalized to the applied Ag concentration measured in M0 samples (initial concentrations in maternal perfusates) by conventional ICP-MS. Data represent the mean ± STD of three independent perfusion experiments (N = 3). (D) Dark-field images of placental tissue perfused with AgNO3. Dark-field images assessed by CytoViva dark field microscopy were captured at 60× magnification of placental tissue before perfusion (0 h) or after 6 h of perfusion. Scale bar represents 15 μm. | ||
The concentrations and size distributions of the particles detected on the fetal side after the addition of dissolved Ag were comparable to those from the AgNP perfusion experiments. The exact composition of the formed NPs could not be determined with spICP-MS, because only one element per particle could be detected. Since no particle formation was observed in PM, which contained relevant ionic Ag concentrations, the formation of NPs cannot be explained with interaction of Ag ions and PM only, but occurred most probably due to the presence of proteins in the placental tissue. For instance, amino acids containing thiol groups (i.e. cysteine) have a high affinity for ionic Ag, forming Ag(I)–sulfide complexes.55 Bolea et al.56 substantiated this by showing that association of dissolved Ag released from AgNPs with the proteins is accelerated in the presence of cells.
Consequently, the observed presence of NPs in the fetal circulation in ex vivo perfusion experiments with AgNPs could therefore be either the result of the translocation of the pristine AgNPs (scenario 1) or be caused by the translocation and subsequent reduction or reaction with available ligands of dissolved Ag (originating from AgNPs dissolution) (scenario 2). With the use of the spICP-MS method that provides information only about particle size and not about the complete particle composition we were not able to unambiguously confirm the exact mechanism. Assuming the first scenario, quantitatively different degrees of Ag translocation of AgPEG NPs and AgCOONa NPs across the placenta could be explained by their different size distributions in PM as a result of different surface modifications: AgCOONa NPs formed large agglomerates/aggregates in the micrometer size range in PM. In contrast, the size distribution of AgPEG NPs was not significantly changed in PM (Table 2, Fig. 1). The higher degree of agglomeration/aggregation observed for AgCOONa NPs in PM could be attributed to the carboxylate groups which become less ionized at high ionic strength in PM. The repulsive forces between AgCOONa NPs are thereby weakened, leading to particle aggregation. PEG coating, on the other hand, provides strong steric stabilization for the AgPEG NPs even in solution with high ionic strength. Since agglomeration of NPs is expected to significantly reduce placental transfer,20,37 a higher dose of AgCOONa NPs compared to AgPEG NPs was applied to the maternal circulation. However, even at higher initial concentrations in the PM, the percentage of AgNPs in fetal perfusates was lower for AgCOONa than for AgPEG NPs. To conclude, the influence of NP surface charge/modification on translocation could not be studied as a single factor as it also affected the agglomeration of NPs in perfusion medium and consequently the size distribution.
NP size has been shown to be a key determinant in placental transfer with lower transfer for larger NPs.20 Further evidence that size is the key determinant for AgNP translocation was reflected in a more efficient translocation of small AgNPs/ionic silver (Ag fraction ≤25 nm) across the placental barrier than larger AgNPs. Moreover, the comparison of particle size distributions on the maternal and fetal side after 6 h of perfusion showed that smaller AgCOONa were present on the fetal side (up to 96 nm) in comparison to the maternal side (NPs/aggregates up to 230 nm) and the particles accumulated in placental tissue (up to 140 nm). Assuming the second scenario (translocation of dissolved Ag followed by Ag reduction or reaction with available ligands), the higher translocation of Ag in case of AgPEG NPs in comparison to AgCOONa NPs could be explained with the larger mass fraction of dissolved Ag of 4.9% in comparison to 0.1% in the NP formulations and/or potentially higher solubility of AgPEG NPs.
HSI analysis indicated that the NPs detected in placental tissue were (at least to some extent) the pristine AgNPs, which means that complete dissolution of the AgNPs on the maternal side can be excluded. The finding does, however, not allow to conclude that pristine AgNPs translocated to the fetal side.
In summary, our study demonstrates that the derivatised AgNPs underwent complex reactions in the conducted translocation studies due to their readily soluble nature and their chemical reactivity. In order to distinguish between translocation of intact AgNPs and that of dissolved Ag/ions originating from the NPs further studies are needed, where other methods for determining chemical composition of translocated AgNPs (e.g. energy dispersive X-ray spectroscopy52,57) are required. Another possibility would be isotope labeling in combination with conventional ICP-MS, which however can suffer from the risk of artificial and/or false-positive results associated with labeled materials (e.g. loss of label and changes in NP properties and biointeractions). Moreover, distinguishing between particulate and ionic silver would require additional separation techniques such as filtration or centrifugation. These additional steps could result in retention of the ionic Ag-macromolecule fraction to the filter or co-precipitation in centrifugation due to a reaction of ionic Ag with macromolecules in biological environments. This could be problematic in particular for this study where we deal with very low amounts of translocated Ag. The use of two stable Ag isotopes could be another promising approach to monitor the transformation of AgNPs and dissolved Ag in biological systems by spICP-MS.58 However, using an enriched Ag isotope label in conjunction with spICP-MS is not yet possible since only one isotope can be detected by most ICP-MS instruments (some quadrupole ICP-MS instruments are able to switch between two masses during high time resolution measurements but quantitative mass analysis is hardly possible with this technique because important peak information is lost during the mass switching time and signal intensities will suffer from spectral intensity skew error59).
Comparison of translocation data with other studies
So far, potential AgNP translocation across the placenta has only been investigated in pregnant rodents. The translocation levels of Ag, measured by ICP-MS in the tissues of pups as the total Ag mass concentrations and expressed as the percentages of the administered dose, ranged from 0.008–0.009%,23 0.036%,11 0.085–0.147%,22 0.097%59 to 0.2–0.5%,24 while the percentage of Ag accumulated in placental tissue varied between 0.2%13,23 and 0.04–1.3%.24 In our study Ag translocation across the human placental barrier (0.015–0.062%, calculated as Ag mass fraction > and ≤25 nm) was in the same range as those observed in rodent studies, while the percentage of accumulated Ag in the placental tissue was about 10-times higher (12–16%). Although significant biological differences in placental structure and function between humans and rodents may contribute to the observed differences, a direct comparison is challenging due to additional physico-chemical differences in the NP types (size, surface modifications), applied doses and routes of administration, among others. For instance, the higher observed accumulation of Ag in human placental tissue is most probably related to the different doses of AgNPs at the placental barrier. In in vivo experiments where AgNPs were intravenously or orally dosed to the rodents, NPs were not only distributed to the placenta but also to other maternal organs, mainly the liver and kidneys.52 Therefore, biodistribution studies in rodents will still be highly valuable to provide an estimate of the expected doses at the placental barrier while human ex vivo placenta perfusion studies can deliver important mechanistic insights on NP translocation and accumulation at the intact human placental barrier.Conclusions
Different surface properties of the selected AgNPs (neutral and negatively charged coatings), which can affect their solubility and size through aggregation/agglomeration in PM, most probably influence different translocation and accumulation behavior of Ag. The results further demonstrate that particles detected on the fetal side are not necessarily the result of translocated pristine AgNPs, but could also be Ag precipitates formed from dissolved Ag that crosses the placental barrier. The detected Ag-containing particles in the fetal circulation, which can result from translocation of intact AgNPs or from re-precipitated Ag, were in the same concentration and size range, which hampered the identification of any translocation mechanism of AgNPs. Therefore, careful assessment of particle dissolution and their possible re-precipitation under physiological conditions as well as the inclusion of ionic controls should be an integral part of future translocation studies with soluble NPs. Such measure would enable accurate interpretation of the results and help to identify strategies towards prevention of placental transfer of nanomaterials. Although translocation was low for both NP types the small amount of translocated ions/NPs may be sufficient to adversely affect fetal health, which should be carefully addressed in future studies.Our study further demonstrates that spICP-MS is a promising technique to study the translocation of NPs. In contrast to conventional ICP-MS, which was used in other published studies dealing with translocation,23,24,60 spICP-MS additionally provides information on the NP size distribution. This allows studying NP changes like dissolution/aggregation and, in this way, helps identifying mechanisms of transport. Theoretically, spICP-MS can even distinguish between NPs and the dissolved fraction of the analysed element if the size of the studied NPs does not overlap with the minimum detectable size.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
We thank Agilent for providing the Agilent 8800 instrument. We thank Prof. Bengt Fadeel and Dr Audrey Gallud, Karolinska Institute, for endotoxin testing of AgPEG and AgCOONa NPs used in this study. The research leading to these results has received funding from the European Union's Seventh Framework Programme (FP7/2007–2013) under grant agreement no 309329 (NANOSOLUTIONS). This work was further supported by the Ad futura scholarship (Slovene Human Resources and Scholarship Fund) that financed the research visit of Janja Vidmar at the Technical University of Denmark.References
- S. Marin, G. M. Vlasceanu, R. E. Tiplea, I. R. Bucur, M. Lemnaru, M. M. Marin and A. M. Grumezescu, Curr. Top. Med. Chem., 2015, 15(16), 1596–1604 CrossRef PubMed; M. E. Quadros and L. C. Marr, J. Air Waste Manage. Assoc., 2010, 60(7), 770–781 CrossRef.
- M. Ahamed, M. S. AlSalhi and M. K. J. Siddiqui, Clin. Chim. Acta, 2010, 411, 1841–1848 CrossRef PubMed.
- M. C. Stensberg, Q. Wei, E. S. Mclamore and D. Marshall, Nanomedicine, 2012, 6, 879–898 CrossRef PubMed.
- S. Kim and D. Y. Ryu, J. Appl. Toxicol., 2013, 33, 78–89 CrossRef PubMed.
- K. Yamashita, Y. Yoshioka, K. Higashisaka, K. Mimura, Y. Morishita, M. Nozaki, T. Yoshida, T. Ogura, H. Nabeshi, K. Nagano, Y. Abe, H. Kamada, Y. Monobe, T. Imazawa, H. Aoshima, K. Shishido, Y. Kawai, T. Mayumi, S.-I. Tsunoda, N. Itoh, T. Yoshikawa, I. Yanagihara, S. Saito and Y. Tsutsumi, Nat. Nanotechnol., 2011, 6, 321–328 CrossRef PubMed.
- A. Pietroiusti, M. Massimiani, I. Fenoglio, M. Colonna, F. Valentini, G. Palleschi, A. Camaioni, A. Magrini, G. Siracusa, A. Bergamaschi, A. Sgambato and L. Campagnolo, ACS Nano, 2011, 5, 4624–4633 CrossRef PubMed.
- L. Campagnolo, M. Massimiani, G. Palmieri, R. Bernardini, C. Sacchetti, A. Bergamaschi, L. Vecchione, A. Magrini, M. Bottini and A. Pietroiusti, Part. Fibre Toxicol., 2013, 10, 21 CrossRef PubMed.
- K. S. Hougaard, L. Campagnolo, P. Chavatte-Palmer, A. Tarrade, D. Rousseau-Ralliard, S. Valentino, M. V. D. Z. Park, W. H. de Jong, G. Wolterink, A. H. Piersma, B. L. Ross, G. R. Hutchison, J. S. Hansen, U. Vogel, P. Jackson, R. Slama, A. Pietroiusti and F. R. Cassee, Reprod. Toxicol., 2015, 56, 118–140 CrossRef PubMed.
- J.-S. Hong, S. Kim, S. H. Lee, E. Jo, B. Lee, J. Yoon, I.-C. Eom, H.-M. Kim, P. Kim, K. Choi, M. Y. Lee, Y.-R. Seo, Y. Kim, Y. Lee, J. Choi and K. Park, Nanotoxicology, 2014, 8, 349–362 CrossRef PubMed.
- W. J. Yu, J. M. Son, J. Lee, S. H. Kim, I. C. Lee, H. S. Baek, I. S. Shin, C. Moon, S. H. Kim and J. C. Kim, Nanotoxicology, 2013, 1, 1–7 Search PubMed.
- M. Charehsaz, K. S. Hougaard, H. Sipahi, A. I. D. Ekici, C. Kaspar, M. Culha, U. U. Bucurgat and A. Aydin, Daru, J. Pharm. Sci., 2016, 24, 1–13 CrossRef PubMed.
- N. A. Philbrook, L. M. Winn, A. R. M. N. Afrooz, N. B. Saleh and V. K. Walker, Toxicol. Appl. Pharmacol., 2011, 257, 429–436 CrossRef PubMed.
- E. Paul, M.-L. Franco-Montoya, E. Paineau, B. Angeletti, S. Vibhushan, A. Ridoux, A. Tiendrebeogo, M. Salome, B. Hesse, D. Vantelon, J. Rose, F. Canouï-Poitrine, J. Boczkowski, S. Lanone, C. Delacourt and J.-C. Pairon, Nanotoxicology, 2017, 11, 484–495 CrossRef PubMed.
- L. Campagnolo, M. Massimiani, L. Vecchione, D. Piccirilli, N. Toschi, A. Magrini, E. Bonanno, M. Scimeca, L. Castagnozzi, G. Buonanno, L. Stabile, F. Cubadda, F. Aureli, P. H. Fokkens, W. G. Kreyling, F. R. Cassee and A. Pietroiusti, Nanotoxicology, 2017, 1–36 Search PubMed.
- C. M. Powers, E. D. Levin, F. J. Seidler and T. A. Slotkin, Neurotoxicol. Teratol., 2012, 33, 329–332 CrossRef PubMed.
- Y. Wu, Q. Zhou, H. Li, W. Liu, T. Wang and G. Jiang, Aquat. Toxicol., 2010, 100, 160–167 CrossRef PubMed.
- T. I. Ala-Kokko, P. Myllynen and K. Vähäkangas, Int. J. Obstet. Anesth., 2000, 9, 26–38 CrossRef.
- A. C. Enders and T. N. Blankenship, J. Physiol., 1999, 38, 3–15 Search PubMed.
- M. Panigel, M. Pascaud and J. L. Brun, J. Physiol., 1967, 59, 277 Search PubMed.
- C. Muoth, L. Aengenheister, M. Kucki, P. Wick and T. Buerki-Thurnherr, Nanomedicine, 2016, 11, 941–957 CrossRef PubMed.
- T. Buerki-Thurnherr, U. Von Mandach and P. Wick, Swiss Med. Wkly., 2012, 142, 1–9 Search PubMed.
- E. A. Melnik, Y. P. Buzulukov, V. F. Demin, V. A. Demin, I. V. Gmoshinski, N. V. Tyshko and V. A. Tutelyan, Acta Nat., 2013, 5, 107–115 Search PubMed.
- C. A. Austin, T. H. Umbreit, K. M. Brown, D. S. Barber, B. J. Dair, S. Francke-Carroll, A. Feswick, M. A. Saint-Louis, H. Hikawa, K. N. Siebein and P. L. Goering, Nanotoxicology, 2011, 6, 1–11 Search PubMed.
- T. R. Fennell, N. P. Mortensen, S. R. Black, R. W. Snyder, K. E. Levine, E. Poitras, J. M. Harrington, C. J. Wingard, N. A. Holland, W. Pathmasiri and S. C. J. Sumner, J. Appl. Toxicol., 2017, 37, 530–544 CrossRef PubMed.
- J. S. Refuerzo, B. Godin, K. Bishop, S. Srinivasan, S. K. Shah, S. Amra, S. M. Ramin and M. Ferrari, Am. J. Obstet. Gynecol., 2011, 204, 546.e5–546.e9 CrossRef PubMed.
- A. Bruinink, J. Wang and P. Wick, Arch. Toxicol., 2015, 89, 659–675 CrossRef PubMed.
- A. E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klaessig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef PubMed.
- K. Loza, J. Diendorf, C. Sengstock, L. Ruiz-Gonzalez, J. M. Gonzalez-Calbet, M. Vallet-Regi, M. Köller and M. Epple, J. Mater. Chem. B, 2014, 2, 1634 RSC.
- A. R. Gliga, S. Skoglund, I. O. Wallinder, B. Fadeel and H. L. Karlsson, Part. Fibre Toxicol., 2014, 11, 11 CrossRef PubMed.
- Z. Xiu, Q. Zhang, H. L. Puppala, V. L. Colvin and P. J. J. Alvarez, Nano Lett., 2012, 12, 4271–4275 CrossRef PubMed.
- N. Hadrup, K. Loeschner, A. Mortensen, A. K. Sharma, K. Qvortrup, E. H. Larsen and H. R. Lam, NeuroToxicology, 2012, 33, 416–423 CrossRef PubMed.
- H. M. Braakhuis, S. K. Kloet, S. Kezic, F. Kuper, M. V. D. Z. Park, S. Bellmann, M. van der Zande, S. Le Gac, P. Krystek, R. J. B. Peters, I. M. C. M. Rietjens and H. Bouwmeester, Arch. Toxicol., 2015, 89, 1469–1495 CrossRef PubMed.
- C. Degueldre and P. Y. Favarger, Colloids Surf., A, 2003, 217, 137–142 CrossRef.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, E. P. Gray, C. P. Higgins and J. F. Ranville, Environ. Sci. Technol., 2012, 46, 12272–12280 CrossRef PubMed.
- E. P. Gray, J. G. Coleman, A. J. Bednar, A. J. Kennedy, J. F. Ranville and C. P. Higgins, Environ. Sci. Technol., 2013, 47, 14315–14323 CrossRef PubMed.
- K. Loeschner, M. S. J. Brabrand, J. J. Sloth and E. H. Larsen, Anal. Bioanal. Chem., 2014, 406, 3845–3851 CrossRef PubMed.
- P. Wick, A. Malek, P. Manser, D. Meili, X. Maeder-Althaus, L. Diener, P. A. Diener, A. Zisch, H. F. Krug and U. Von Mandach, Environ. Health Perspect., 2010, 118, 432–436 CrossRef PubMed.
- S. Grafmueller, P. Manser, L. Diener, L. Maurizi, P.-A. Diener, H. Hofmann, W. Jochum, H. F. Krug, T. Buerki-Thurnherr, U. von Mandach and P. Wick, Sci. Technol. Adv. Mater., 2015, 16, 44602 CrossRef PubMed.
- S. Grafmüller, P. Manser, H. F. Krug, P. Wick and U. von Mandach, J. Visualized Exp., 2013, 1–7 Search PubMed.
- H. E. Pace, N. J. Rogers, C. Jarolimek, V. A. Coleman, C. P. Higgins and J. F. Ranville, Anal. Chem., 2011, 83, 9361–9369 CrossRef PubMed.
- K. R. Di Bona, Y. Xu, P. A. Ramirez, J. DeLaine, C. Parker, Y. Bao and J. F. Rasco, Reprod. Toxicol., 2014, 50, 36–42 CrossRef PubMed.
- H. Yang, C. Sun, Z. Fan, X. Tian, L. Yan, L. Du, Y. Liu, C. Chen, X. Liang, G. J. Anderson, J. A. Keelan, Y. Zhao and G. Nie, Sci. Rep., 2012, 2, 847 CrossRef PubMed.
- F. Tian, D. Razansky, G. G. Estrada, M. Semmler-Behnke, A. Beyerle, W. Kreyling, V. Ntziachristos and T. Stoeger, Inhalation Toxicol., 2009, 21, 92–96 CrossRef PubMed.
- M. P. Waalkes, A. M. Poisner, G. W. Wood and C. D. Klaassen, Toxicol. Appl. Pharmacol., 1984, 74, 179–184 CrossRef PubMed.
- G. Bhabra, A. Sood, B. Fisher, L. Cartwright, M. Saunders, W. H. Evans, A. Surprenant, G. Lopez-Castejon, S. Mann, S. A. Davis, L. A. Hails, E. Ingham, P. Verkade, J. Lane, K. Heesom, R. Newson and C. P. Case, Nat. Nanotechnol., 2009, 4, 876–883 CrossRef PubMed.
- A. Sood, S. Salih, D. Roh, L. Lacharme-Lora, M. Parry, B. Hardiman, R. Keehan, R. Grummer, E. Winterhager, P. J. Gokhale, P. W. Andrews, C. Abbott, K. Forbes, M. Westwood, J. D. Aplin, E. Ingham, I. Papageorgiou, M. Berry and J. Liu, Nat. Nanotechnol., 2011, 6, 824–833 CrossRef PubMed.
- X.-F. Zhang, J.-H. Park, Y.-J. Choi, M.-H. Kang, S. Gurunathan and J.-H. Kim, Int. J. Nanomed., 2015, 10, 7057–7071 Search PubMed.
- E. Söderstjerna, F. Johansson, B. Klefbohm and U. Englund Johansson, PLoS One, 2013, 8, 1–13 CrossRef PubMed.
- F. Liu, M. Mahmood, Y. Xu, F. Watanabe, A. S. Biris, D. K. Hansen, A. Inselman, D. Casciano, T. A. Patterson, M. G. Paule, W. Slikker and C. Wang, Front. Neurosci., 2015, 9, 1–9 CrossRef PubMed.
- M. Mortimer, A. Gogos, N. Bartolome, A. Kahru, T. D. Bucheli and V. I. Slaveykova, Environ. Sci. Technol., 2014, 48, 8760–8767 CrossRef PubMed.
- K. Sarlo, K. L. Blackburn, E. D. Clark, J. Grothaus, J. Chaney, S. Neu, J. Flood, D. Abbott, C. Bohne, K. Casey, C. Fryer and M. Kuhn, Toxicology, 2009, 263, 117–126 CrossRef PubMed.
- K. Loeschner, N. Hadrup, K. Qvortrup, A. Larsen, X. Gao, U. Vogel, A. Mortensen, H. Lam and E. H. Larsen, Part. Fibre Toxicol., 2011, 8, 18 CrossRef PubMed.
- M. van der Zande, R. J. Vandebriel, E. Van Doren, E. Kramer, Z. Herrera Rivera, C. S. Serrano-Rojero, E. R. Gremmer, J. Mast, R. J. B. Peters, P. C. H. Hollman, P. J. M. Hendriksen, H. J. P. Marvin, A. A. C. M. Peijnenburg and H. Bouwmeester, ACS Nano, 2012, 6, 7427–7442 CrossRef PubMed.
- J. Liu, Z. Wang, F. D. Liu, A. B. Kane and R. H. Hurt, ACS Nano, 2012, 6, 9887–9899 CrossRef PubMed.
- J. Liu, D. A. Sonshine, S. Shervani and R. H. Hurt, ACS Nano, 2010, 4, 6903–6913 CrossRef PubMed.
- E. Bolea, J. Jiménez-Lamana, F. Laborda, I. Abad-Álvaro, C. Bladé, L. Arola and J. R. Castillo, Analyst, 2014, 139, 914–922 RSC.
- S. Juling, G. Bachler, N. von Götz, D. Lichtenstein, L. Böhmert, A. Niedzwiecka, S. Selve, A. Braeuning and A. Lampen, Food Chem. Toxicol., 2016, 97, 327–335 CrossRef PubMed.
- S. Yu, Y. Yin, X. Zhou, L. Dong and J. Liu, Environ. Sci.: Nano, 2016, 3, 883–893 RSC.
- A. Praetorius, A. Gundlach-Graham, E. Goldberg, W. Fabienke, J. Navratilova, A. Gondikas, R. Kaegi, D. Günther, T. Hofmann and F. von der Kammer, Environ. Sci.: Nano, 2017, 4, 307–314 RSC.
- Y. Lee, J. Choi, P. Kim, K. Choi, S. Kim, W. Shon and K. Park, Toxicol. Res., 2012, 28, 139–141 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr02096e |
| This journal is © The Royal Society of Chemistry 2018 |