
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA long-chain protic ionic liquid inside silica nanopores: enhanced proton mobility due to efficient self-assembly and decoupled proton transport†
Mounesha N.
Garaga
a,
Vassilios
Dracopoulos
b,
Ulrike
Werner-Zwanziger
a,
Josef W.
Zwanziger
a,
Manuel
Maréchal
c,
Michael
Persson
d,
Lars
Nordstierna
 e and
Anna
Martinelli
*e
e and
Anna
Martinelli
*e
aDepartment of Chemistry, Dalhousie University, Halifax, Canada
bFoundation for Research and Technology (FORTH), Institute of Chemical Engineering Sciences (ICE-HT), GR-26504 Patras, Greece
cUniv. Grenoble Alpes, CNRS, CEA, INAC-SyMMES, 38000 Grenoble, France
dAkzoNobel Pulp & Performance Chemicals AB, Bohus, Sweden
eDepartment of Chemistry, Chalmers University of Technology, SE-41296 Gothenburg, Sweden. E-mail: anna.martinelli@chalmers.se
First published on 25th April 2018
Abstract
We report enhanced protonic and ionic dynamics in an imidazole/protic ionic liquid mixture confined within the nanopores of silica particles. The ionic liquid is 1-octylimidazolium bis(trifluoromethanesulfonyl)imide ([HC8Im][TFSI]), while the silica particles are microsized and characterized by internal well connected nanopores. We demonstrate that the addition of imidazole is crucial to promote a proton motion decoupled from molecular diffusion, which occurs due to the establishment of new N–H⋯N hydrogen bonds and fast proton exchange events in the ionic domains, as evidenced by both infrared and 1H NMR spectroscopy. An additional reason for the decoupled motion of protons is the nanosegregated structure adopted by the liquid imidazole/[HC8Im][TFSI] mixture, with segregated polar and non-polar nano-domains, as clearly shown by WAXS data. This arrangement, promoted by the length of the octyl group and thus by significant chain–chain interactions, reduces the mobility of molecules (Dmol) more than that of protons (DH), which is manifested by DH/Dmol ratios greater than three. Once included into the nanopores of hydrophobic silica microparticles, the nanostructure of the liquid mixture is preserved with slightly larger ionic domains, but effects on the non-polar ones are unclear. This results in a further enhancement of proton motion with localised paths of conduction. These findings demonstrate significant progress in the design of proton conducting materials via tailor-made molecular structures as well as by smart exploitation of confinement effects. Compared to other imidazole-based proton conducting materials that are crystalline up to 90 °C or above, the gel materials that we propose are useful for applications at room temperature, and can thus find applications in e.g. intermediate temperature proton exchange fuel cells.
1. Introduction
Achieving high proton conductivity in solid state materials is one of the current biggest challenges in materials science. Of emerging interest are porous solid materials like zeolites, ordered mesoporous silica and crystalline covalent organic frameworks, impregnated with a suitable proton conducting substance.1,2 Central to these materials is that large pores do not simply cause the flow of charge carriers but can, if smartly designed, also sustain the transport of the hydrogen ion, i.e. the proton.1 Imidazole is a classic example of a proton conducting substance that, by providing both proton-acceptor and proton-donor sites, is able to support fast proton motion via the Grotthuss mechanism, which involves a sequence of proton hopping and molecular reorientations.3 This mechanism of proton conduction is also displayed by other aromatic molecules like triazole or pyrazole, water, and hydrated strong acids like phosphoric or sulphuric acids, which have in common the ability to form an extended hydrogen bonded network. Although water-based systems have the advantage of high proton density and high proton mobility, they are limited to usage at low temperatures due to evaporation and thus dehydration (close to or above 100 °C). By contrast, an amphoteric aromatic molecule can display high proton mobility at much higher temperatures, but have a lower temperature limit defined by the solid-to-liquid transition (which is 90 °C for imidazole).In this context, protic ionic liquids (ILs) have emerged as a new class of potentially good proton conductors, that also provide a negligible volatility and are chemically stable up to temperatures as high as 300 °C. Protic ILs are obtained by proton transfer from a Brønsted acid (AH) to a Brønsted base (B), where the ΔpKa‡ of the acid–base pair determines whether the reaction AH + B → A− + BH+ proceeds to completion. ILs with a ΔpKa value larger than 10 typically result in a low ionic association (or high ionicity), and vice versa.4 Highly ionic ILs display a superior thermal stability, and have exchangeable protons more strongly bound to the cation. This results in higher molar ionic conductivities, as can be revealed by Walden plots, but limits the decoupled motion of the acidic protons. Diffusion NMR experiments have shown that in pure imidazolium-based ionic liquids like 1-ethylimidazolium triflate ([HC2Im+][TfO−]) or 1-ethylimidazolium bis(trifluoromethanesulfonyl)imide ([HC2Im+][TFSI−]) the self-diffusion of the most acidic proton (DH) is not significantly different from that of other aromatic or aliphatic protons (Dcat),5 as would be the case if the Grotthuss mechanism of proton motion occurred. In some ammonium-based ILs, the proton affinity of the acid may be so large that the exchangeable proton appears to diffuse together with the anion.6 Moreover, the correlation observed between the proton affinity of the acid used to produce the protic ILs and the 1H NMR chemical shift of the exchangeable proton enables one to predict the diffusional properties of an IL from its 1H NMR spectrum.6 The diffusional properties of ILs are also correlated with their local structure, as nicely shown for a series of ammonium-based protic ILs by pulsed field gradient (PFG) and heteronuclear (HOESY) NMR experiments.7
Interestingly, protic ILs prepared with non-equimolar amounts of an acid and a base, more precisely in the compositional range of excess base, can show an enhanced proton hopping mechanism as revealed in the case of imidazolium![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :bis(trifluoromethanesulfonyl)imide [(HIm+]:[TFSI−]) mixtures.8,9 Excess proton mobilities with DH/Dcat ratios in the range of 1.1–1.4 could thus be measured between 20 and 120 °C. In our group, we have recently measured an enhanced proton mobility upon addition of imidazole, at low concentrations, to the protic IL [HC2Im+][TFSI−].10 By combining NMR and infrared spectroscopy with MD simulations, we could explain this enhancement by the establishment of new hydrogen bonds between imidazole and both cations and anions. These results give rise to the question whether a selective proton mobility could be further enhanced by choosing a more appropriate molecular structure for the IL, for instance one that provides intrinsic nano-structuration.
:bis(trifluoromethanesulfonyl)imide [(HIm+]:[TFSI−]) mixtures.8,9 Excess proton mobilities with DH/Dcat ratios in the range of 1.1–1.4 could thus be measured between 20 and 120 °C. In our group, we have recently measured an enhanced proton mobility upon addition of imidazole, at low concentrations, to the protic IL [HC2Im+][TFSI−].10 By combining NMR and infrared spectroscopy with MD simulations, we could explain this enhancement by the establishment of new hydrogen bonds between imidazole and both cations and anions. These results give rise to the question whether a selective proton mobility could be further enhanced by choosing a more appropriate molecular structure for the IL, for instance one that provides intrinsic nano-structuration.
To verify this possibility, we have focused this study on the combination of an imidazole with a long-chain protic IL, i.e. 1-octylimidazolium bis(trifluoromethanesulfonyl)imide ([HC8Im+][TFSI−]) that is supposed to provide a close proximity between the cationic head and imidazole in the polar domains and, simultaneously, promote nano-structuration by aggregation of the alkyl chains in the non-polar domains.11–16 We hypothesise that in such a binary mixture the mobility of the cation (Dcat) can be reduced more effectively than that of the exchangeable protons (DH). Moreover, to meet the quest of proton conducting materials in the solid state, we have investigated the diffusional properties of an imidazole/[HC8Im+][TFSI−] mixture confined in silica nanopores, with pores having a diameter of 10 nm and the pore walls being functionalized with trioctylsilyl groups to provide hydrophobicity, and thus reduce interfacial friction. The structural properties and molecular dynamics of the hypothetically proton conducting materials, i.e. the imidazole/[HC8Im+][TFSI−] liquid mixture and the imidazole/[HC8Im+][TFSI−]/silica solid-like gel, were investigated by 1H NMR and vibrational spectroscopy and wide angle X-ray scattering (WAXS), as well as diffusion and solid-state 1H NMR techniques. The results show interesting confinement effects, which depend on the relationship between the size of the nanopores and the nanosegregated structure of the selected IL. The study that we present thus bridges the fields of confined self-organised crystal ILs17 and the nanoconfinement of ILs.18 We find these results relevant not only for the field of fuel cells, that operate upon proton conduction, but also for other electrochemically related fields where the use of ILs as electrolytes is seen as the future solution (for example, batteries or super-capacitors) or for applications based on controlled release from porous structures (e.g. in drug delivery).19
2. Experimental
2.1 Synthesis of the ionic liquids
The Brønsted base 1-octylimidazole (here abbreviated as C8Im) with a purity higher than 98% was purchased in liquid form from IoLiTec (Ionic Liquids Technologies) GmbH, whereas the acid H-bis(trifluoromethanesulfonyl)imide (here abbreviated as HTFSI) with a purity higher than 99% was purchased from Acros Organics. The synthesis of the protic ionic liquid 1-octylimidazolium bis(trifluoromethanesulfonyl)imide (in this paper abbreviated as [HC8Im][TFSI]) was carried out under inert conditions in a glove bag (Atmosbag) purchased from Sigma-Aldrich Co. by mixing equimolar quantities of the base and acid according to the following stoichiometric reaction:| C8Im + HTFSI → HC8Im+ + TFSI− | (1) |
The Karl Fischer titration revealed that water uptake for the protic IL [HC8Im][TFSI] was 0.003% w/w. The molecular structures of the 1-octylimidazolium cation and the TFSI anion are sketched in Fig. 1.
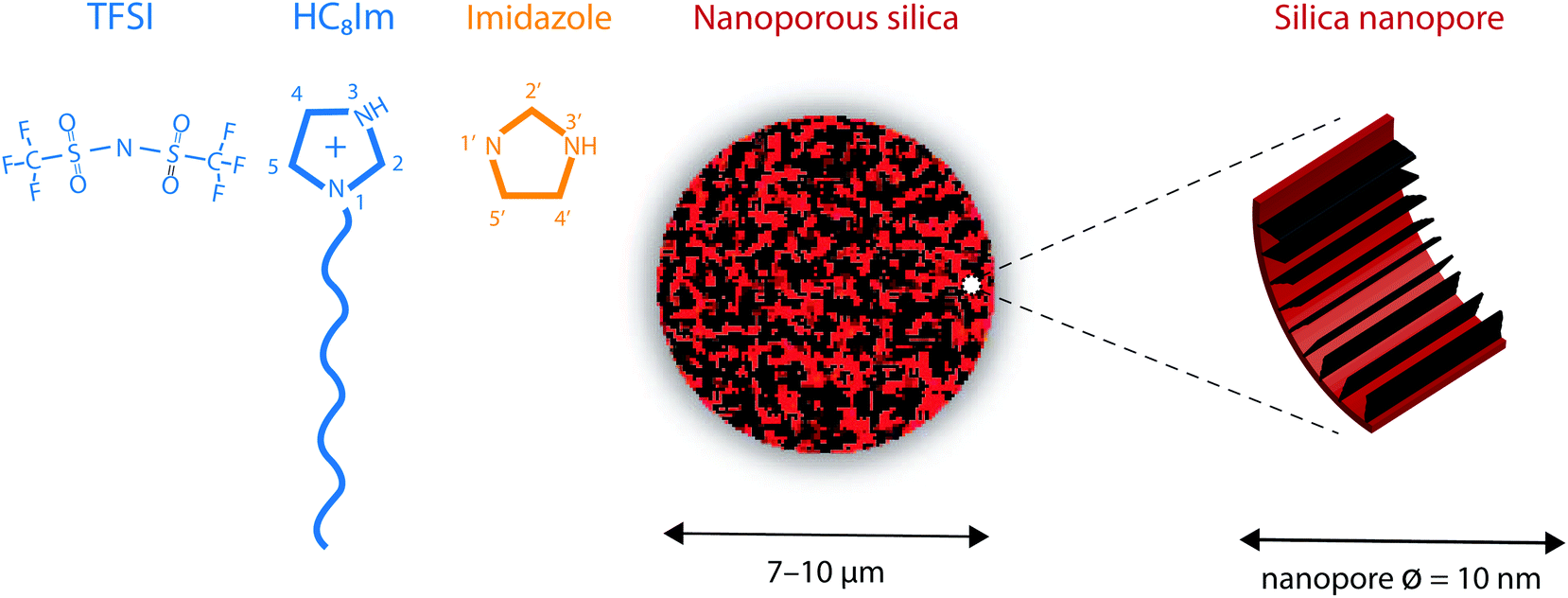 | ||
| Fig. 1 From left to right: Molecular structure of the 1-octylimidazolium cation, the bis(trifluoromethanesulfonyl)imide anion (TFSI) and the neutral imidazole, along with the atom labelling used in this work. A schematic illustration of the nanoporous silica micro-particles and the functionalized pore walls is also provided. | ||
2.2 Synthesis of the nanoporous silica
As already described in ref. 20, the nanoporous silica microparticles were synthesized following an established emulsion solvent evaporation procedure that results in a tunable pore volume and pore size distribution.21 The results from the Brunauer–Emmett–Teller (BET) analysis reveal that the average pore size in these silica particles is 10 nm and that the surface area is 338 m2 g−1.20 The average particle size is estimated to be ∼10 μm, with a narrow size distribution that fulfils the requirement of a dV90/dV10 ratio smaller than 1.5. To provide hydrophobic surfaces, and in this way reduce interfacial friction, the as synthesized silica particles were functionalised with the trioctylsilyl (TOS) groups. New BET analyses revealed that the functionalized silica particles have a surface area reduced to 220 m2 g−1 and can provide a free volume equal to 0.55 cm3 per gram, a value that is crucial to define the degree of filling of the pores by the ionic liquid, given in % (vide infra). In addition, after functionalization, the pore diameter distribution shows a maximum at 8.5 nm, as a consequence of the volume taken up by the TOS groups. A SEM image of this type of nanoporous silica particle, as well as the pore diameter distribution, is given in Fig. SI-1.†Fig. 1 shows a schematic representation of a nanoporous silica microparticle as well as of the hydrophobic/functionalised nanopore wall.2.3 Synthesis of the silica-based gels
The IL filled silica-based gels were prepared according to the following procedure, which is equivalent to the one recently reported in ref. 20 except for the addition of the imidazole to the protic IL [HC8Im][TFSI]. An appropriate amount of imidazole (i.e. 0.03 grams, Mw = 68 g mol−1) was added to 1.30 grams of the protic IL [HC8Im][TFSI] (Mw = 461 g mol−1) under magnetic stirring to obtain an imidazole/IL mixture with a molar fraction of the imidazole, χ, equal to 0.15. This value was chosen from a previous study of imidazole/[HC2Im][TFSI] mixtures that revealed more pronounced proton mobilities at low concentrations of imidazole.10 Part of this solution was kept aside for further characterisation, but a calculated volume of this mixture was added to a weighed amount of the functionalised silica powder to achieve the desired pore filling factor. This is given in percent (%) and represents the fraction of the whole available free space provided by the particles actually filled by the liquid mixture. For example, a 50% pore filling represents silica powder with half of the available empty volume filled by the liquid. A sealed NMR tube containing the functionalized silica powder and the liquid mixture was then centrifuged for the time necessary (about 30 minutes) to obtain a homogeneous gel-like material (see the image in Fig. SI-1†).2.4 Vibrational spectroscopy
Raman spectra of the pure protic IL and the imidazole/IL mixture were collected using the 785 nm line from a near infrared diode laser as the excitation source. The Raman spectrum of the protic IL, however, resulted in a significant luminescence effect, wherefore better quality spectra were also collected with an FT Raman spectrometer using the 1064 nm laser line. Good Raman spectra of the silica gels, on the other hand, could be recorded using the 532 nm laser line, which also provides a suitable wavelength to perform highly confocal mapping experiments. These were performed along vertical lines along the z-axis penetrating the whole volume of silica microparticles, and over x–y surface sectioning particles at their half-diameter height. Scanning steps of 0.5 μm were used to reproduce the contours of the particles and thus monitor the presence of the liquid phase inside the microparticles’ nanopores. For monitoring purposes, the integrated intensity underlying the Raman signal at 742 cm−1 was chosen, which was then converted into a gradient of colours between red (high intensity) and black (low intensity) to visualise the mapping results. The use of the 532 nm line was coupled with a grating of 2400 l mm−1, which provides a spectral resolution better than 1 cm−1.Infrared spectra were collected using a PerkinElmer spectrometer using the attenuated total reflection (ATR) mode and pouring the solutions, or gels, over a single reflectance diamond crystal. For each sample, 32 scans were averaged achieving a nominal spectral resolution of 2 cm−1. The full spectral range 400–4000 cm−1 was investigated. For a more detailed analysis of the 3000–4000 cm−1 spectral region where the N–H stretching modes appear, the software Igor Pro from WaveMetrics was employed to make a peak fitting analysis based on a linear background and Gaussian components.
2.5 Liquid state 1H NMR spectroscopy
All NMR measurements were carried out on a Bruker Avance 600 spectrometer, equipped with a Bruker Diff30 probe with a 5 mm 1H/2H RF double coil insert and connected to a 40 A gradient amplifier. The relaxation times were obtained by standard inversion recovery (T1) and CPMG (T2) pulse sequences. All NMR signals, in both liquid and gel-like samples, revealed strict mono-exponential behaviour for the relaxation recovery. Regarding the NMR self-diffusion studies of the liquid samples, the standard stimulated echo sequence was used with a gradient pulse length, d, equal to 1 ms and diffusion time, D, equal to 100 ms. In each experiment, the gradient strength, g, was ramped uniformly in 16 steps from 0.1 to 12 T m−1. The experiments were carried out at 30 °C, collecting data at each gradient value with a sum of 8 acquisitions, and using a 7 s recycle delay time to ensure complete longitudinal relaxation of all signals. For the gel-like samples (i.e. the liquid mixture inside the nanopores of silica), the stimulated echo sequence using bipolar pulses was used to suppress the influence of internal background gradients. Due to fast spin–spin relaxation, a gradient pulse length d equal to 0.37 ms was used, while the diffusion time D was set to 300 ms. In each experiment, the gradient strength g was ramped uniformly in 16 steps from 0.1 to 17 T m−1. The experiments were carried out at 30 °C, collecting data at each gradient value with a sum of 16 acquisitions, and using a 7 s recycle delay time to ensure complete longitudinal relaxation of all signals. The gradient strength was calibrated with the self-diffusion coefficient of HDO traces in a standard sample of pure D2O. For all samples, a strict one-component attenuation of the logarithmic NMR signal intensity against k was noted, where k is equal to (γgd)2·(D − d/3) and the 1H gyromagnetic ratio is denoted γ.2.6 Solid-state NMR spectroscopy
One-dimensional (1D) 1H and two-dimensional (2D) 29Si{1H} HETCOR NMR experiments were performed on a 400 MHz Bruker Avance DSX NMR spectrometer operating at a magnetic field of 9.4 T (with 1H and 29Si Larmor frequencies of 400.24 and 79.51 MHz, respectively) using a 7 mm double-resonance probe head. 1H NMR spectra were acquired over 8 scans with 5 seconds of recycling delay at room temperature (RT) and variable temperature (VT) from 290 K to 335 K.22 The sample was spun at 6 kHz. The 1H 90° pulse was calibrated to 12.5 μs. 29Si{1H} CPMAS NMR spectra were collected over 1024 scans with 2 seconds of recycling delay at a 5 kHz spinning speed, where RAMP shape was used to transfer polarization with 8 ms of contact time. The 2D spectra were collected under the same CP conditions. The signals were accumulated over 256 scans for each of 76 increments with 2.5 seconds of relaxation delay. The 2D NMR experiments were performed at room temperature. A saturation recovery and CPMG pulse sequences were used to measure 1H T1 and T2 relaxations as a function of temperature (290–335 K) while spinning the sample at 6 kHz. The 1H chemical shifts were externally referenced to those of isopropanol.2.7 X-ray scattering
Wide- and small-angle X-ray scattering (WAXS and SAXS) experiments were performed on a Mat:Nordic instrument purchased from SAXSLAB, a Xenocs company, and available at the Chalmers Materials Analysis Laboratory (CMAL) at the Chalmers University of Technology. The X-ray source was generated using a Rigaku 003+ high brilliance microfocus Cu-radiation source, while the detector was a Pilatus 300K. Samples were inserted into thin capillaries with a wall diameter of 0.15 mm, and silver behenate was used as the standard to calibrate the q-range. All experiments were performed at room temperature. To collect WAXS and SAXS data, an accumulation time of 8 and 12 minutes, respectively, was used. Since the diffraction peaks of interest in this paper fell within the q-range covered by the WAXS setup, only these data will be presented and discussed.2.8 Surface analysis
Nitrogen sorption analyses were performed using a TriStar 3000 instrument from Micromeritics Instrument Corporation. Prior to the measurements, the materials were outgassed overnight at 120 °C. The pore size distribution was calculated according to the BJH (Barrett–Joyner–Halenda) method,23 whereas the surface area was obtained using the BET (Brunauer–Emmett–Teller) method.24 The pore volume was calculated using a single point adsorption value at the relative pressure (p/p0) of 0.990. The obtained pore size distribution is shown in Fig. SI-1.†3. Results and discussion
3.1. Chemistry and nanostructure of the protic ionic liquid
Upon complete proton transfer from the acid (HTFSI) to the base (C8Im), a new N–H bond should be created in the cation, on site N3 according to the atom labelling given in Fig. 1. The formation of the N–H bond was straightforwardly demonstrated by infrared spectroscopy in the high frequency range where the N–H stretching modes typically appear, as shown in Fig. 2. The new N–H stretching mode appears at about 3265 cm−1, with a broad shape indicative of a wide distribution of the N–H bond lengths and thus of the establishment of hydrogen bonds with various strengths. This can be compared to the infrared spectrum of the analogous aprotic IL (i.e. [C1C8Im][TFSI]) that is featureless in the frequency region above 3200 cm−1. That the proton has been transferred to the base and is now residing on the 1-octylimidazolium cation is also demonstrated by 1H NMR spectroscopy, through the (N3)H proton resonance that appears at 11.81 ppm (Table S1† and Fig. 3), as will be discussed in more detail.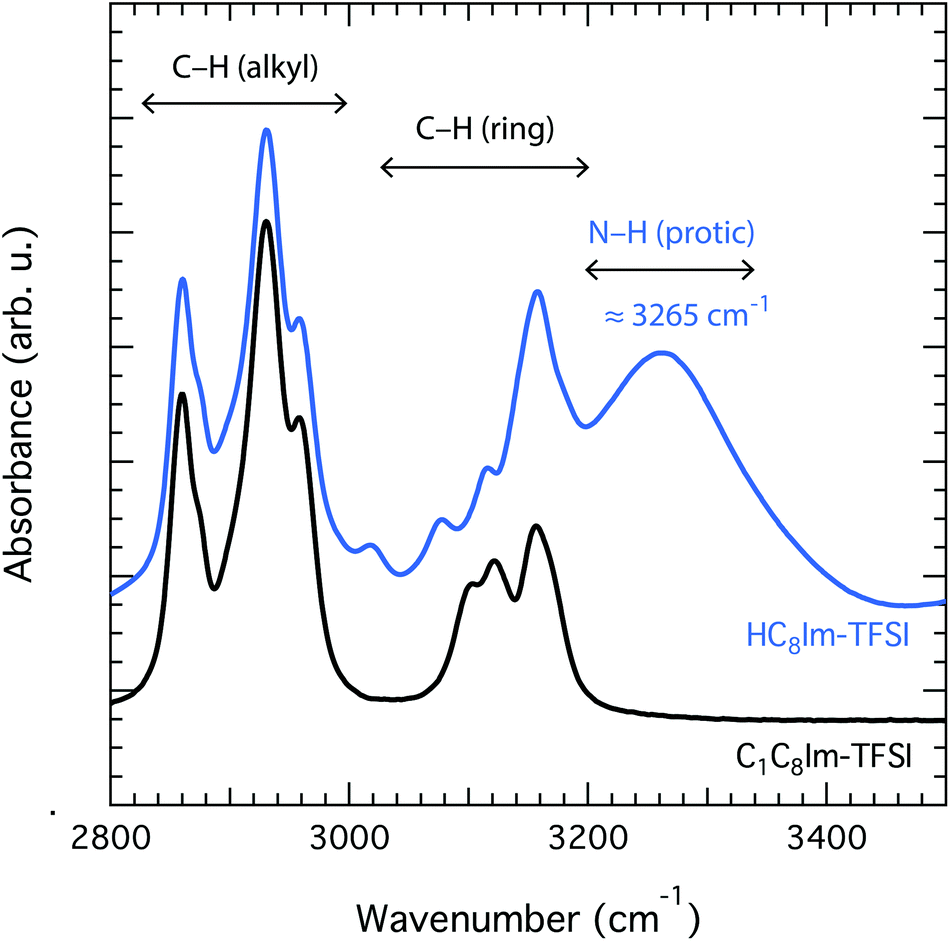 | ||
| Fig. 2 Infrared spectra collected for the aprotic IL [C1C8Im][TFSI] (black) and the protic IL [HC8Im][TFSI] (blue). The broad vibrational mode peaked at 3265 cm−1 is assigned to the N–H stretching that appears after protonation at site N3. | ||
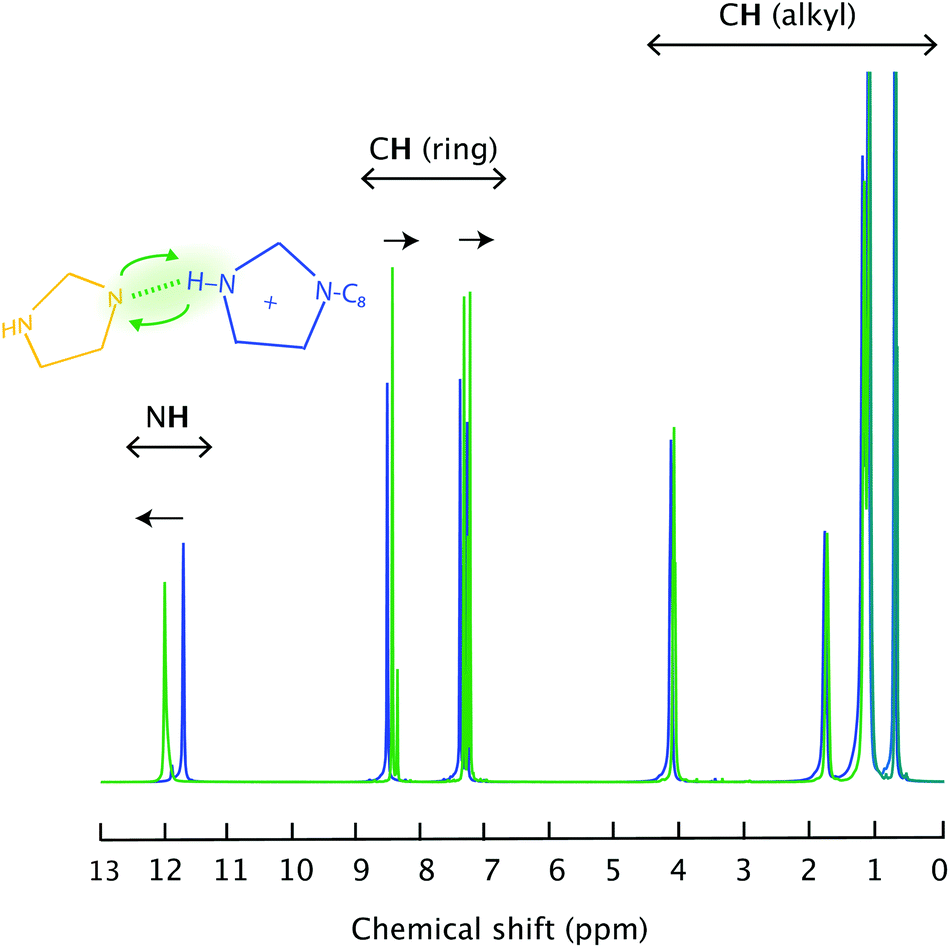 | ||
| Fig. 3 Liquid state 1H NMR spectra recorded for the pure IL [HC8Im][TFSI] (blue) and the imidazole/[HC8Im][TFSI] mixture (green). | ||
Because [HC8Im][TFSI] is a new tailor made IL, there has been no reported information on its structural properties. We can thus report the first X-ray scattering study of a long-chain imidazolium-based protic IL. Our wide angle X-ray scattering (WAXS) data reveal three clear diffraction peaks centred at 3.4, 8.6 and 13.4 nm−1, indicated as q1 (also known as the pre-peak), q2 and q3 in Fig. 4. In accordance with previous X-ray scattering experiments and MD simulations performed on aprotic 1-methyl-3-alkyl-imidazolium-based ILs,11–16 these peaks are associated with a local structure with segregated polar (cationic heads and anions) and non-polar (alkyl chains) domains, with characteristic sizes in the nanometer range. For the pure IL [HC8Im][TFSI] investigated in this study, the polar and non-polar correlation lengths, calculated using the Bragg's diffraction rule as di = 2π/qi, are 1.8 and 0.7 nm respectively. These values are in full agreement with those found for [C1C8Im][TFSI], i.e. the aprotic IL is analogous to [HC8Im][TFSI], although the intensity of peak q1 is markedly enhanced, see Fig. SI-2.† This enhancement is attributed to the protonation of the imidazolium ring, which increases the polarity of the ionic domains thus promoting the segregation from the non-polar regions and increasing the contrast of scattering length densities between these domains.
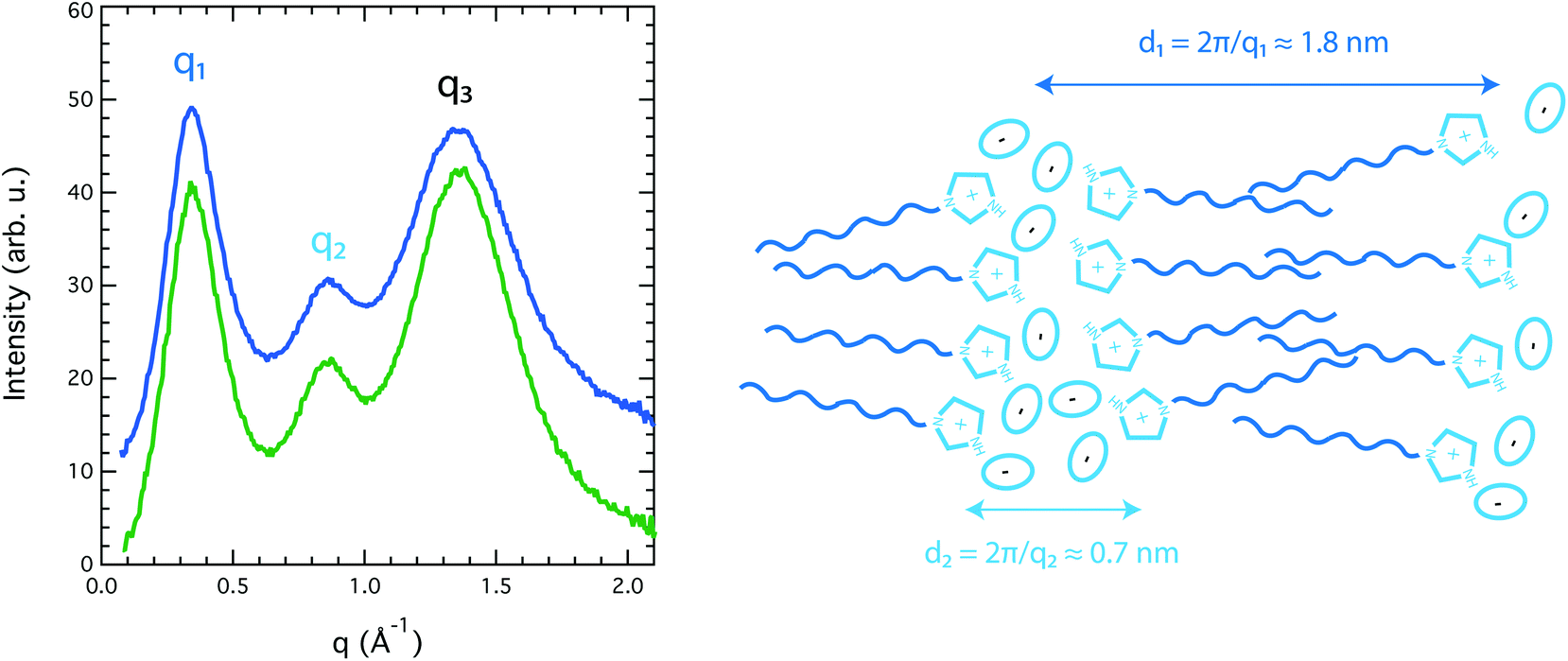 | ||
| Fig. 4 Wide angle X-ray scattering intensity collected for the pure protic IL [HC8Im][TFSI] (blue) and the imidazole/[HC8Im][TFSI] mixture (green). The pre-peak attributed to the non-polar domains and the second peak attributed to the polar and ionic domains are indicated as q1 and q2, respectively. An illustration of the nano-segregated structure that gives rise to the observed scattering pattern, along with the associated correlation lengths, is also provided. | ||
Upon addition of an imidazole to [HC8Im][TFSI], specific hydrogen bonds are formed that primarily involve the (N3)H and the N1′ sites as hydrogen bond donors and acceptors, respectively. This is particularly clear in the liquid state 1H NMR spectrum that reveals a down-field shift of the merged (N)H chemical shift from 11.81 to 12.04 ppm, Fig. 3. Concomitantly, the chemical shift of aromatic and aliphatic protons shifts slightly upfield, see also Table S1.† These features reflect the proximity of the imidazole to the imidazolium ring as well as a fast proton exchange (on a time scale shorter than 1 ms), since only one narrow (N)H feature is observed in the 1H NMR spectrum, rather than two as expected from two non-interacting molecular species (note that the (N3′)H chemical shift of neat imidazole is expected to appear at 13.5 ppm). In addition, the high ppm value observed for the merged (N)H signal reflects the acidic character of the (N)H protons. In fact, by adding an imidazole the average N3–H bond becomes longer, as manifested in the infrared spectrum by a small but detectable red shift of the N3–H stretching mode to 3261 cm−1, Fig. SI-3.†
3.2. Diffusional properties in the liquid mixture
The narrow and distinct proton resonances shown in Fig. 3 are suitable for performing 1H diffusion experiments and thus obtain the self-diffusion coefficient, D, of individual hydrogen nuclei. These experiments reveal homogeneous D values in the case of the neat IL [HC8Im][TFSI], with insignificant differences between the aliphatic, aromatic or (N)H protons, as shown in Table S1.† In the imidazole/[HC8Im][TFSI] mixture, however, all diffusion values increase, and remain similar for aliphatic and aromatic protons. This increase in mobility is attributed to a decrease in viscosity, as previously also discussed for imidazole/[HC2HIm][TFSI] mixtures.10 Hydrogen atoms at position C2′ show a higher D value, which can be explained, invoking the Stokes–Einstein relation D = (kBT)/(cηr),§ by the smaller size of the imidazole compared to the bulkier octylimidazolium cation.The most interesting result, however, is that the self-diffusion coefficient of the exchangeable (N3,3′)H protons is the highest,¶ indicating that they move faster than both imidazolium and imidazole. The implication of such an observation can only be discussed through a detailed analysis of all possible dynamical contributions. In a complex system like the one investigated here, three potential proton carriers must be considered, i.e. the octylimidazolium cation, the neutral imidazole, and the free proton H+, whereby the self-diffusion value measured from the (N3,3′)H resonance (DNH) is assumed to contain contributions from both the vehicular (Dcat and DIm) and the Grotthuss (DH) mechanisms of proton motion (eqn (2)):
| DNH = χ1·DH + χ2·DIm + χ3·Dcat | (2) |
Based on the observation that in the pure protic IL DH ≈ Dcat, the addition of imidazole appears to be a crucial approach to enhance proton mobility. This is a result of the amphoteric nature of the imidazole, which is able to create hydrogen bonded molecular networks by acting as both a hydrogen bond donor and acceptor. This property has already been discussed for imidazole added polymers25 (in which DH ≈ 2.5·Dmol was measured), liquids and liquid crystals,25,26 as well as for imidazole-containing complex compounds.27,28 A proton diffusivity greater than that of the parent molecule has also been reported for imidazole:HTFSI mixtures at non-equimolar ratios (DH ≈ 1.2·Dmol),9 and pure imidazole (DH ≈ 2·Dmol).29
From the above analysis, it turns out that the contribution of the Grotthuss mechanism of proton transport is more enhanced in the imidazole/[HC8Im][TFSI] mixture (DH ≈ 4·Dcat) than in the imidazole/[HC2Im][TFSI] mixture previously investigated by us,10 in which mobile protons were observed to diffuse about two times faster than the cations (DH ≈ 2·Dcat). This is explained by a more pronounced nanosegregation in [HC8Im][TFSI] than in [HC2Im][TFSI], due to the longer chain (octyl, C8, versus ethyl, C2), yet the same chemical environment in the polar domains where imidazole and exchangeable protons reside. Hence, in both cases the decoupled proton motion is strictly related to the nature and strength of the hydrogen bonded network formed by the imidazole and imidazolium, whilst the molecular mobility is markedly reduced in [HC8Im][TFSI] due to stronger chain–chain interactions. That the characteristic nanostructure with segregated polar and non-polar nanodomains is preserved even upon addition of imidazole is shown in Fig. 4, where the diffraction peaks q1, q2 and q3 observed for the imidazole/[HC8Im][TFSI] mixture perfectly match in position with those of the neat [HC8Im][TFSI] IL.**
This type of nanostructure-related proton motion is similar to that discussed in ref. 26 and 27, which, however, report the cases of non-ionic systems that require temperatures above 110 °C for good proton conductivities. With respect to a selective ionic motion, that is the enhancement of the mobility of either the cation or the anion compared to its counterion, reports are available on ILs that exhibit liquid-crystal like nanostructures. For instance, at temperatures below the liquid-crystal phase transition, i.e. below ∼80 °C, a long-chain fan-shaped tri-nonylammonium-based ionic liquid displays diffusion values for the anion twice as large as those of the cations.30 On the other hand, in [C1C2Im][C8OS] the 1-ethyl-3-methylimidazolium cations diffuse two to three times faster than the octylsulfate anions in the rigid-gel phase, which sets in at temperatures between 20 and 25 °C depending on the level of hydration.31 These experimental findings are in great accordance with the molecular dynamics simulations recently provided by Erbas et al.,32 who have demonstrated the pivotal role of nanomorphologies for enhanced counterion mobilities in ILs.
3.3. Enhanced dynamics inside the nanopores
Whether the structural properties and molecular dynamics of the liquid mixture are affected by nanoconfinement was a key question in this work. The 1H NMR and infrared spectroscopic studies as well as the WAXS experiments were therefore repeated for a gel prepared by filling the nanopores of the functionalised silica microparticles with the imidazole/[HC8Im][TFSI] mixture. Particular attention was thus paid to verify that the liquid mixture effectively penetrated the nanopores. This was verified by using confocal Raman spectroscopy, and collecting Raman spectra while scanning a large X–Y surface area containing the silica microparticles. To do so in the most precise way, a single layer of particles was isolated over a thin glass slide. Moreover, to avoid artefacts due to surface roughness, the confocal volume from where the Raman scattered light is collected was positioned at half-height with respect to the particles’ diameter (in the z-direction), see Fig. 5A for the details of this experimental setup. The presence, or absence, of the IL within the scanned area was verified by monitoring the integrated area of the vibrational mode at 742 cm−1, the strongest Raman signature in TFSI containing ILs, Fig. 5B. The analysis of the collected data confirms that the liquid mixture resides inside the silica particles, which is clear from the reproduction of spherical shapes with distinct contours and a characteristic diameter of ∼10 μm, Fig. 5C. Complementary 2D 29Si{1H} solid state HETCOR NMR experiments confirm that TOS groups are anchored to the silica walls, as shown in Fig. SI-5,† but chainIL–chainTOS cross correlations could not be observed due to the high flexibility of the octyl groups. However, a self-orientation of the octyl chains of imidazolium cations towards those anchored to the silica walls is expected, as already evidenced for a similar system based on shorter, i.e. butyl, chains.33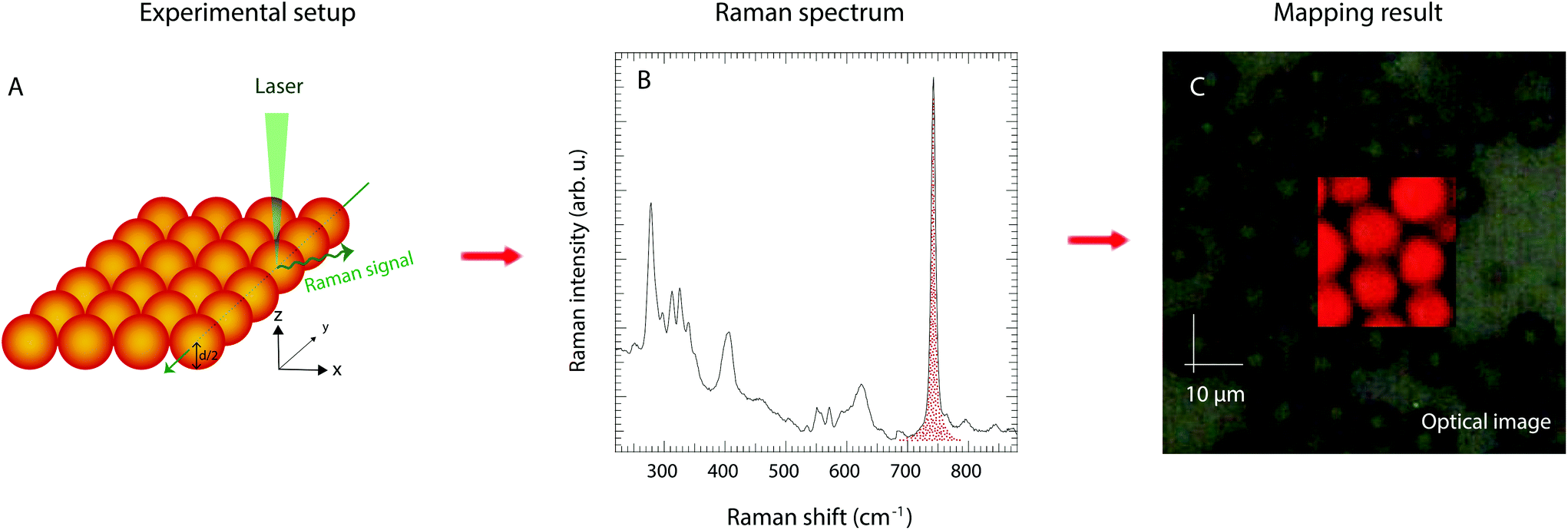 | ||
| Fig. 5 Surface mapping experiments by confocal Raman spectroscopy performed over silica particles whose nanopores were 100% filled with the imidazole/PIL mixture. A: Experimental setup with the confocal point of the laser positioned at half-height with respect to the particles diameter. B: Integration of the most intense Raman signal in TFSI containing compounds, i.e. the expansion–contraction mode of the TFSI anion at 742 cm−1. C: Results from visualising the Raman intensity as in (B) on a colour scale, where red indicates strong intensity and black indicates low intensity. The net red spherical shapes reproduced in (C) confirm that the liquid mixture has penetrated the particles and fills the particles’ nanopores. | ||
One striking property of the liquid mixture confined in the nanopores is the higher diffusion value measured for all H nuclei compared to the bulk case, as shown in Table S1.† This is an unusual observation given that molecular and ionic mobilities in nanodomains are usually reduced by physical obstruction and/or liquid–wall interactions.20,33–37 This dynamics retardation can be a limit for the practical use of gels, especially in gel electrolytes whose functionality is based on a high and selective ionic mobility. Hence, with the aim to counteract the reduced dynamics, different strategies have been explored, which include e.g. surface modification of the walls in the host matrix or the selection of specific size domains that change the phase behaviour of the guest liquid.33,38–42 Nevertheless, ionic mobilities higher in the gel than in the bulk state have been reported in a few cases only, i.e. in directional nanochannels and/or carbon nanotubes,43,44 but never in nanoporous silica gels. By using the measured self-diffusion values and implementing eqn (2) as done above for the liquid mixture, we found that inside the nanopores the protons move about seven times faster than in the imidazolium cations (DH ≈ 7·Dcat), see also Table 1. This is, to the best of our knowledge, the greatest enhancement of proton mobility ever reported for nanoporous materials in the anhydrous state. Highly mobile protons inside the nanopores were also evidenced by solid state 1H NMR spectra recorded at increasing temperatures, from 290 to 335 K, as summarised in Fig. 6.†† Upon increasing the temperature, we observe a chemical shift down-field for the merged (N)H resonance, from 11 to 12 ppm, and a concomitant narrowing of the line, with a FWHM varying from 313 to 206 Hz in the temperature range investigated. By comparison, the chemical shift and line width of other proton resonances, for instance the one at position C6, are not affected in the same way (the chemical shift change remains below 0.1 ppm and the line width decreases by only 15 Hz). Similar shift and line width effects with increased temperature had been observed in other imidazole-based proton conductors, indicating dynamic hydrogen bonds.45 Hence, the high-resolution solid state NMR experiments shown in Fig. 6 reveal that the hydrogen bond type interactions established in the imidazole/[HC8Im][TFSI] mixture are retained also inside the nanopores of silica.
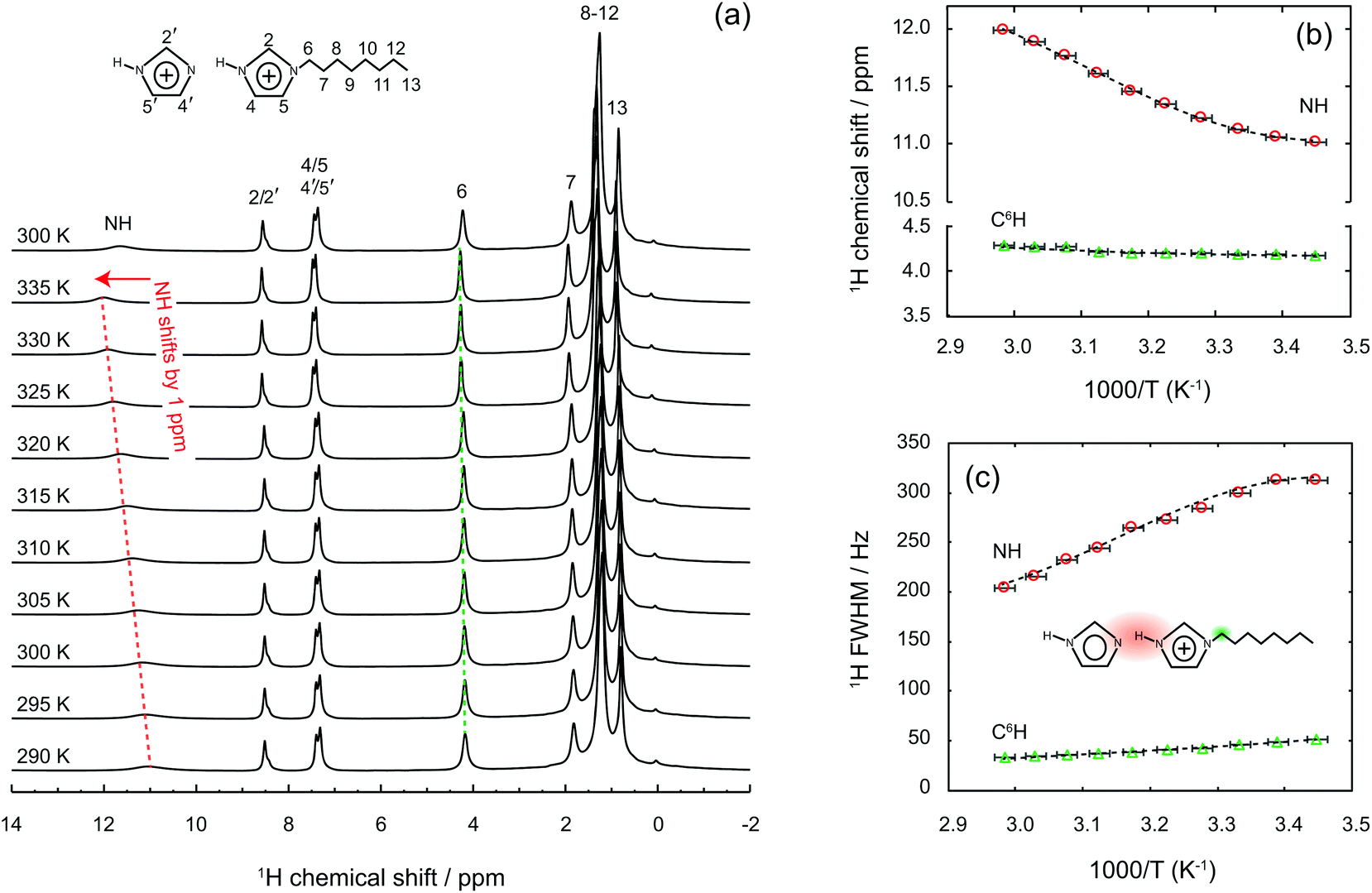 | ||
| Fig. 6 Temperature dependent 1H solid-state NMR spectra of a gel collected at a magnetic field of 9.4 T and a spinning frequency of 6 kHz, from 290 K to 335 K. Dotted lines are just a guide to the eye. | ||
| Sample | State (at 30 °C) | D cat (10–11 m2 s−1) | D Im (10–11 m2 s−1) | D NH (10–11 m2 s−1) | D H/Dcat |
|---|---|---|---|---|---|
| a This value was deduced from assuming the same DNH/DIm and DIm/Dcat ratios as in the liquid Im/[HC8Im][TFSI] mixture. | |||||
| [HC8Im][TFSI] | Liquid | 0.80 | — | 0.79 | 1 |
| 0.15Im/0.85[HC8Im][TFSI] | Liquid | 0.86 | 1.24 | 1.35 | 3.7 |
| 0.15Im/0.85[HC8Im][TFSI]/C8-SiO2 | Gel | 1.45 | 2.2a | 2.4 | 7.0 |
An explanation for the observed molecular dynamics was sought in the local structure adopted by the liquid in the confined state, which can a priori be captured from the WAXS data. Fig. 7 compares the WAXS intensities measured for the imidazole/[HC8Im][TFSI] mixture in the bulk and inside the nanopores. Distinct diffraction features are observed also for the confined liquid, albeit shifted compared to the bulk case. The peak at position q3 appears shifted to higher q-values, but this is an apparent effect due to the stronger contribution of the silica particles with their characteristic peak at ∼1.6 Å−1. The peak at position q2, on the other hand, is shifted to lower q-values, from 0.86 to 0.78 Å−1, which corresponds to a correlation length d2 of 0.8 nm. Determining the position and intensity of peak q1 was a major challenge, due to the strong interference in the low-q range from the structural features of the silica microparticles, i.e. the 10 nm large nanopores and the pore wall thickness of ∼6 nm. Thus, to best describe the overall collected intensity in this range a power-law contribution, as observed in the WAXS intensity of bare silica microparticles (C8-SiO2), was included. For the liquid-filled silica gel, a power of −2.6 was found (i.e. I ∝ q−2.6), which indicates smooth pore wall surfaces and a planar-like (2D) distribution of the ionic liquid, most likely as a consequence of a bilayer forming towards the TOS groups. A power law contribution decays strongly with q and we found it unable to fit, on its own, the experimental data of the gel for q-values between 0.2 and 0.5 Å−1, see Fig. SI-7A.† By contrast, the fit is more consistent if a contribution at a q-value between 0.33 and 0.35 Å−1, attributable to the non-polar domains, is considered, as shown in Fig. SI-7B.† It is very important to mention, however, that during the peak fit procedure both the intensity and position of peak q1 change slightly when small variations are imposed on the power-law contribution. This makes conclusive statements on the value of d1 difficult. In all cases, though, the intensity of peak q1 is found to be less intense, when compared to that of q2, in the nanopores than in the bulk (Fig. SI-7C†), which suggests a certain loss of the long-range order. This could be an effect of the concave shape of the pores or of a loose packing of the molecules. Additional structural data, in the lower q-range, supporting this argument can be found in Fig. SI-8 of the ESI.†
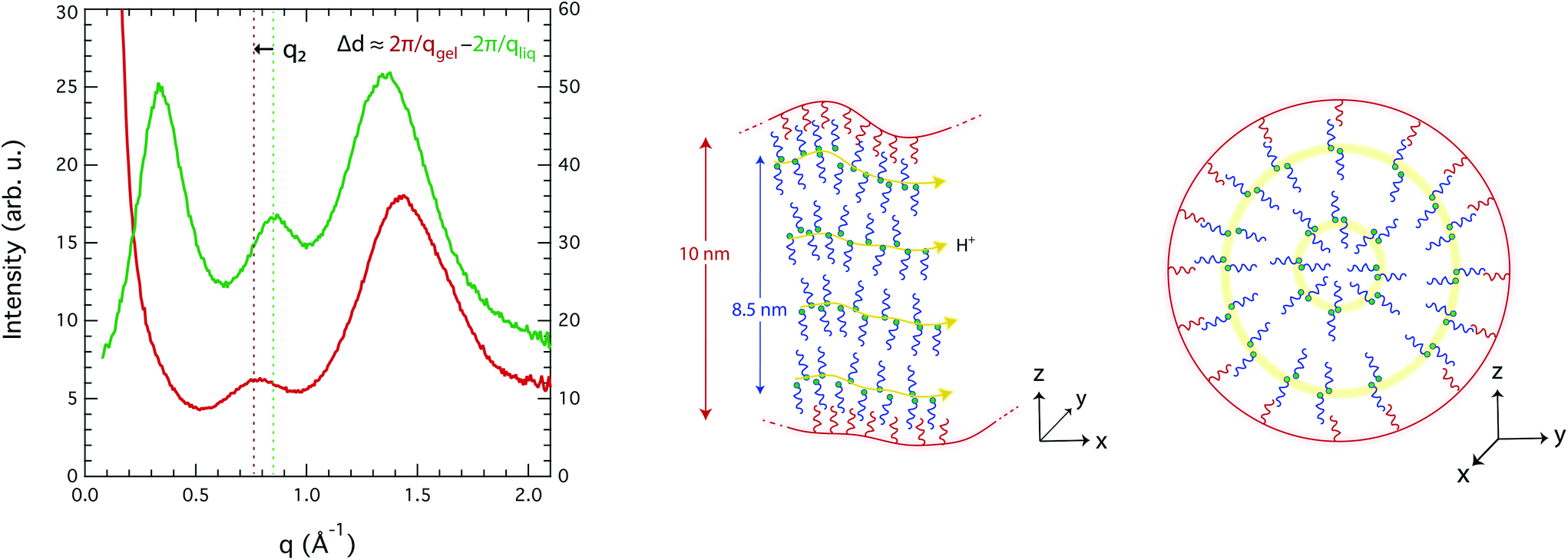 | ||
| Fig. 7 Left: Comparison of the WAXS pattern recorded for the Im/[HC8Im][TFSI] mixture (green trace, right axis) and the gel obtained by confining the Im/[HC8Im][TFSI] mixture inside nanoporous silica microparticles (red trace, left axis). Right: Schematic picture of the plausible nanostructuration of the liquid mixture inside the nanopores of silica, with perspectives showing the molecular organisation along the pores (left) and across a pore section (right). | ||
Since these are the first experimentally measured X-ray diffraction data for an IL in nanopores, it is difficult to use the literature data as a benchmark for discussion. Nevertheless, density fluctuations and order/disorder effects due to nanoconfinement have recently been discussed in the context of MD simulations, e.g. by Ori et al.46 and by Shi et al.47 Accordingly, we hypothesize that a density change inside the nanopores of this work could arise from a mismatch between the real space available and the structural parameters of the liquid, i.e. from the fact that the diameter of the pore is different from an integer number of bilayers (diampore ≠ n·(d1 + d2)). A schematic representation of the local structure plausibly adopted inside the nanopores is given in Fig. 7, from different axial perspectives. This arrangement is in accordance with the tendency of long-chain ILs to form bilayer structures at confined interfaces, as Smith et al. have reported.48 The figure also shows that due to an efficient self-assembly, preferred paths for the transport of protons are created, which is in good agreement with the enhanced DNH/Dcat ratios observed in the gel phase and already discussed above.
4. Conclusions
To conclude, we have demonstrated a strategy for materials’ design that results in an enhanced proton mobility decoupled from molecular diffusion. One important factor that enables this achievement is the intrinsic nanostructuration of long-chain ionic liquids,12 with separated polar and non-polar nanodomains. This property is here combined with the protonation of the aromatic head, which provides localised and acidic protic sites. Nevertheless, protons diffuse independently of the parent molecules only upon addition of an imidazole, an amphoteric molecule that resides in the polar domains and promotes the formation of an extended hydrogen bonded network. This finding is per se of highest relevance, primarily in the field of fuel cells, since it highlights the possibility of selective proton motion under anhydrous conditions, as required for use in next-generation devices. In addition, the liquid mixture reported here can provide a high proton mobility over a wide temperature range, that bridges over the upper limit of water-based electrolytes (i.e. the dehydration temperature of 80 °C) and the lower limit of aromatic proton conductors like imidazole (i.e. the temperature for the solid–liquid transition of 90 °C or above). In fact, the imidazole/[HC8Im][TFSI] mixture is fluid at room temperature and is expected to be chemically stable up to at least 200 °C. The inclusion of imidazole/[HC8Im][TFSI] into the nanopores of silica microparticles revealed a further enhancement in ionic and protonic mobilities, which is a rare observation. The faster dynamics that we observed are attributed partly to weak surface interactions, thanks to the anchored TOS groups, and partly to confinement effects that have an influence on the nanostructure of the confined liquid. Here, the concave shape of the pore walls may also play a role. A better understanding of the structure and dynamics of ILs in the nanodomains is also relevant to the fields of batteries and supercapacitors, which foresee the use of electrolytes inside nanoporous electrodes. Based on these results, future studies will focus on confining a longer-chain ionic liquid inside silica particles with smaller pores, with the aim to further localise the protonic charges.Conflicts of interest
There is no conflict to declare.Acknowledgements
Funding from the Knut and Alice Wallenberg Foundation and from the Area of Advance Nanoscience and Nanotechnology is acknowledged. The authors M. N. Garaga, U. Werner-Zwanziger and J. W. Zwanziger gratefully acknowledge funding from the NSERC (Grant RGPIN 261987) and the NSF (Grant DMR-1407404). All the authors thank Milene Zezzi do Valle Gomez for performing the BET experiments, Dr Katarina Logg for technical help while performing the X-ray diffraction measurements, and Dr Charlotte Hamngren for taking SEM images of the nanoporous silica microparticles.References
- H. Xu, S. Tao and D. Jiang, Proton conduction in crystalline and porous covalent organic frameworks, Nat. Mater.– Lett., 2016, 15, 722–727 CrossRef PubMed.
- R. Marschall, M. Sharifi and M. Wark, Proton conductivity of imidazole functionalized ordered mesoporous silica: Influence of type of anchorage, chain length and humidity, Microporous Mesoporous Mater., 2009, 123, 21–29 CrossRef.
- K. D. Kreuer, S. J. Paddison, E. Spohr and M. Schuster, Transport in proton conductors for fuel-cell applications: simulations, elementary reactions, and phenomenology, Chem. Rev., 2004, 104(10), 4637–4678 CrossRef PubMed.
- M. Yoshizawa, W. Xu and C. A. Angell, Ionic Liquids by proton transfer: vapor pressure, conductivity, and the relevance of ΔpKa from aqueous solutions, J. Am. Chem. Soc., 2003, 125(50), 15411–15419 CrossRef PubMed.
- A. Yaghini, L. Nordstierna and A. Martinelli, Ionic Liquids by proton transfer: vapor pressure, conductivity, and the relevance of ΔpKa from aqueous solutions, Phys. Chem. Chem. Phys., 2014, 16(20), 9266–9275 RSC.
- S. K. Davidowski, F. Thompson, W. Huang, M. Hasani, S. A. Amin, C. A. Angell and J. L. Yarger, NMR characterization of ionicity and transport properties for a series of diethylmethylamine based protic ionic liquids, J. Phys. Chem. B, 2016, 120(18), 4279–4285 CrossRef PubMed.
- P. Judeinstein, C. Iojoiu, J.-Y. Sanchez and B. Ancian, Proton conducting ionic liquid organization as probed by NMR: Self-diffusion coefficients and heteronuclear correlations, J. Phys. Chem. B, 2008, 112(12), 3680–3683 CrossRef PubMed.
- M. L. Hoarfrost, M. Tyagi, R. A. Segalman and J. A. Reimer, Proton hopping and long-range transport in the protic ionic liquid [Im][TFSI], probed by pulsed-field gradient NMR and quasi-elastic neutron scattering, J. Phys. Chem. B, 2012, 116(28), 8201–8209 CrossRef PubMed.
- A. Noda, M. A. B. Hasan Susan, K. Kudo, S. Mitsushima, K. Hayamizu and M. Watanabe, Brønsted acid-base ionic liquids as proton-conducting nonaqueous electrolytes, J. Phys. Chem. B, 2003, 107(17), 4024–4033 CrossRef.
- N. Yaghini, V. Gomez-Gonzalez, L. M. Varela and A. Martinelli, Structural origin of the proton mobility in a protic ionic liquid/imidazole mixture. Insights from computational and experimental results, Phys. Chem. Chem. Phys., 2016, 18(33), 23195–23206 RSC.
- J. N. A. Canongia Lopes and A. A. H. Padua, Nanostructural organization in ionic liquids, J. Phys. Chem. B, 2006, 110(7), 3330–3335 CrossRef PubMed.
- A. Martinelli, M. Marechal, A. Ostlund and J. Cambedouzou, Insights into the interplay between molecular structure and diffusional motion in 1-alkyl-3-methylimidazolium ionic liquids: A combined PFG NMR and X-ray scattering study, Phys. Chem. Chem. Phys., 2013, 15(15), 5510–5517 RSC.
- K. Shimizu, C. E. S. Bernardes, A. Triolo and J. N. Canongia Lopes, Nano-segregation in ionic liquids: Scorpions and vanishing chains, Phys. Chem. Chem. Phys., 2013, 15(38), 16256–16262 RSC.
- O. Russina, A. Triolo, L. Gontrani and R. Caminiti, Mesoscopic structural heterogeneities in room-temperature ionic liquids, J. Phys. Chem. Lett., 2012, 3(1), 27–33 CrossRef.
- A. Triolo, O. Russina, H.-J. Bleif and E. Di Cola, Nanoscale segregation in room temperature ionic liquids, J. Phys. Chem. B, 2007, 111(18), 4641–4644 CrossRef PubMed.
- N. M. Garaga, M. Nayeri and A. Martinelli, Effect of the alkyl chain length in 1-alkyl-3-methylimidazolium ionic liquids on inter-molecular interactions and rotational dynamics: A combined vibrational and NMR spectroscopic study, J. Mol. Liq., 2015, 210(Part B), 169–177 CrossRef.
- B. Soberats, M. Yoshio, T. Ichikawa, S. Taguchi, H. Ohno and T. Kato, 3D anhydrous proton-transporting nanochannels formed by self-assembly of liquid crystals composed of a sulfobetaine and a sulfonic acid, J. Am. Chem. Soc., 2013, 135(41), 15286–15289 CrossRef PubMed.
- R. Futamura, T. Iiyama, Y. Takasaki, Y. Gogotsi, M. J. Biggs, M. Salanne, J. Ségalini and P. Simon, Partial breaking of the coulombic ordering of ionic liquids confined in carbon nanopores, Nat. Mater., 2017, 16(12), 1225–1232 CrossRef PubMed.
- A. S. Amarasekara, Acidic ionic liquids, Chem. Rev., 2016, 116(10), 6133–6183 CrossRef PubMed.
- M. N. Garaga, M. Persson, N. Yaghini and A. Martinelli, Local coordination and dynamics of a protic ammonium based ionic liquid immobilized in nano-porous silica micro-particles probed by Raman and NMR spectroscopy, Soft Matter, 2016, 12(9), 2583–2592 RSC.
- N. Andersson, B. Kronberg, R. Corkery and P. Alberius, Combined emulsion and solvent evaporation (ESE) synthesis route to well-ordered mesoporous materials, Langmuir, 2007, 23(3), 1459–1464 CrossRef PubMed.
- A. Bielecki and D. P. Burum, Temperature dependence of 207 Pb MAS spectra of solid lead nitrate. An accurate, sensitive thermometer for variable-temperature MAS, J. Magn. Reson., Ser. A, 1995, 116(2), 215–220 CrossRef.
- E. P. Barrett, L. G. Joyner and P. P. Halenda, The determination of pore volume and area distributions in porous substances. 1. Computations from nitrogen isotherms, J. Am. Chem. Soc., 1951, 73(1), 373–380 CrossRef.
- S. Brunauer, P. H. Emmett and E. Teller, Absorption of gases in multimolecular layers, J. Am. Chem. Soc., 1938, 60(2), 309–319 CrossRef.
- K. D. Kreuer, A. Fuchs, M. Ise, M. Spaeth and J. Maier, Imidazole and pyrazole-based proton conducting polymers and liquids, Electrochim. Acta, 1998, 43(10–11), 1281–1288 CrossRef.
- T. Liang, Y. Wu, S. Tan, X. Yang and B. Wei, Enhancing proton conduction via doping of supramolecular liquid crystals (4-alkoxybenzoic acids) with imidazole polymers and liquids, Chem. Phys. Lett., 2015, 637, 22–25 CrossRef.
- S. Tan, B. Wei, T. Liang, X. Yang and Y. Wu, Anhydrous proton conduction in liquid crystals containing benzimidazole moieties, Chem. Phys. Lett., 2016, 6, 34038–34042 Search PubMed.
- D. Basak, C. Versek, J. A. Harvey, S. Christensen, J. Hillen, S. M. Auerbach, M. T. Tuominen and D. Venkataraman, Enhanced anhydrous proton conduction in binary mixtures of 1H-imidazole–1H-1,2,3-triazole based compounds, J. Mater. Chem., 2012, 22, 20410–20417 RSC.
- W.-Q. Deng, V. Molinero and W. A. Goddard III, Fluorinated imidazoles as proton carriers for water-free fuel cell membranes, J. Am. Chem. Soc., 2004, 126(48), 15644–15645 CrossRef PubMed.
- A. E. Erise, T. Ichikawa, M. Yoshio, H. Ohno, S. V. Dvinskikh, T. Kato and I. Furó, Ion conductive behaviour in a confined nanostructure: NMR observation of self-diffusion in a liquid-crystalline bicontinuous cubic phase, Chem. Commun., 2009, 46(5), 728–730 Search PubMed.
- O. Cabeza, L. Segade, M. Domínguez-Pérez, E. Rilo, D. Ausín, A. Martinelli, N. Yaghini, B. Gollas, M. Kriechbaum, O. Russina, A. Triolo, E. López-Lago and L. M. Varela, Mesostructure and physical properties of aqueous mixtures of 1-ethyl-3-methyl imidazolium octyl sulfate doped with magnesium sulfate in the liquid and mesomorphic states, Phys. Chem. Chem. Phys., 2018, 20(13), 8724–8736 RSC.
- A. Erbas and M. Olvera de la Cruz, Morphology-enhanced conductivity in dry ionic liquids, Phys. Chem. Chem. Phys., 2016, 18(9), 6441–6450 RSC.
- M. N. Garaga, L. Aguilera, N. Yaghini, A. Matic, M. Persson and A. Martinelli, Achieving enhanced ionic mobility in nanoporous silica by controlled surface interactions, Phys. Chem. Chem. Phys., 2017, 19(8), 5727–5736 RSC.
- M. Nayeri, M. Aronson, D. Bernin, B. F. Chmelka and A. Martinelli, Surface effects on the structure and mobility of the ionic liquid C6C1ImTFSI in silica gels, Soft Matter, 2014, 10(30), 5618–5627 RSC.
- E. D. Hazelbaker, R. Guillet-Nicolas, M. Thommes, F. Kleitz and S. Vasenkov, Influence of confinement in mesoporous silica on diffusion of a mixture of carbon dioxide and an imidazolium-based ionic liquid by high field diffusion NMR, Microporous Mesoporous Mater., 2015, 206, 177–183 CrossRef.
- C. Iacob, J. Sangoro, W. Kipnusu and F. Kremer, Rotational and translational diffusion of ionic liquids in silica nanopores, in Dynamics in geometrical confinement, Advances in Dielectrics, Springer International Publishing, Switzerland, 2014, pp. 151–163 Search PubMed.
- P. Huber, Soft matter in hard confinement: phase transition thermodynamics, structure, texture, diffusion and flow in nanoporous media, J. Phys. Condens. Matter, 2015, 27(10), 103102 CrossRef PubMed.
- Y. Daiko, Proton conduction in nanopores of sol-gel derived porous glasses and thin films, J. Sol-Gel Sci. Technol., 2014, 70(2), 172–179 CrossRef.
- Y. Daiko, H. Katayama, T. Araki, A. Mineshige, M. Kobune, S. Yusa, T. Yazawa, K. Nagasaka, S. Murata, S. Matsumoto, A. Koiwai and I. Konomi, In situ oxidation of alkanethiol groups and proton transfer in nanopores of sodium borosilicate glasses, J. Phys. Chem. C, 2009, 113(5), 1891–1895 Search PubMed.
- S. Khatua, A. K. Bar and S. Konar, Tuning proton conductivity by interstitial guest change in size- adjustable nanopores of a Cu-MOF: a potential platform for versatile proton carriers, Chem. – Eur. J., 2016, 22(45), 16277–16285 CrossRef PubMed.
- A. Guyomard-Lack, B. Said, N. Dupre, A. Galarneau and J. Le Bideau, Enhancement of lithium transport by controlling the mesoporosity of silica monoliths filled by ionic liquids, New J. Chem., 2016, 40, 4269–4276 RSC.
- T. Sato, T. Marinaga, S. Marukane, T. Narutomi, T. Igarashi, Y. Kawano, K. Ohno, T. Fukuda and Y. Tsujii, Novel solid-state polymer electrolyte of colloidal crystal decorated with ionic-liquid polymer brush, Adv. Mater., 2011, 23(42), 4868–4872 CrossRef PubMed.
- Q. Berrod, F. Ferdeghini, P. Judeinstein, N. Genevaz, R. Ramos, A. Fournier, J. Dijon, J. Ollivier, S. Rols, D. Yu, R. A. Mole and J.-M. Zanotti, Enhanced ionic liquid mobility induced by confinement in 1D CNT membranes, Nanoscale, 2016, 8(15), 7845–7848 RSC.
- S. M. Chathoth, E. Mamontov, S. Dai, X. Wang, P. F. Fulvio and D. J. Wesolowski, Fast diffusion in a room temperature ionic liquid confined in mesoporous carbon, Europhys. Lett., 2012, 97(6), 66004 CrossRef.
- G. R. Goward, M. F. H. Schuster, D. Sebastiani, I. Schnell and H. W. Spiess, High-resolution solid-state NMR studies of imidazole-based proton conductors: Structure motifs and chemical exchange from 1H NMR, J. Phys. Chem. B, 2002, 106(36), 9322–9334 CrossRef.
- G. Ori, F. Villemot, L. Viau, A. Vioux and B. Coasne, Ionic liquid confined in silica nanopores: molecular dynamics in the isobaric-isothermal ensemble, Mol. Phys., 2014, 12(9–10), 1350–1361 CrossRef.
- W. Shi and D. R. Luebje, Enhanced gas absorption in the ionic liquid 1-n-hexyl-3-methylimidazolium bis(trifluoromethylsulfonyl)amide ([hmim][Tf2N]) confined in silica slit pores: A molecular simulation study, Langmuir, 2013, 29(18), 5563–5572 CrossRef PubMed.
- A. M. Smith, K. R. J. Lovelock, N. N. Gosvami, P. Licence, A. Dolan, T. Welton and S. Perkin, Monolayer to bilayer structural transition in confined pyrrolidinium-based ionic liquids, J. Phys. Chem. Lett., 2013, 4(3), 378–382 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional and complementary data achieved by wide angle X-ray scattering, vibrational (infrared and Raman) spectroscopy, and solid-state NMR. A comprehensive table with observed 1H NMR chemical shifts as well as self-diffusion values for the [HC8Im][TFSI] IL, the imidazole/[HC8Im][TFSI] mixture and the imidazole/[HC8Im][TFSI]/C8-SiO2 gel. See DOI: 10.1039/C8NR02031K |
| ‡ Where ΔpKa = pKa(BH+) − pKa(AH). |
| § Where kB is the Boltzmann's constant, T is the absolute temperature in K, c is a frictional factor, η is the viscosity, and r is the hydrodynamic radius. |
| ¶ This has been verified by repeating the PFG NMR experiments and varying both the gradient strength, G, and the diffusion time, Δ, to confirm that the D values for the (N)H protons are systematically and significantly higher than those of imidazole. |
| || We approximate to the scenario where 0.15 cationic protons are set in faster motion by the presence of the imidazole and the establishment of stronger hydrogen bonds, while the remaining 0.70 cations thus move as protonated molecular species. |
| ** The WAXS pattern of the imidazole/[HC8Im][TFSI] liquid also confirms the proximity of the imidazole to imidazolium cations. This is deduced from the slightly reduced intensity of peak q1, mainly related to intermolecular interactions, and the slight increase of peak q3, mainly related to intramolecular interactions, two effects that can co-exist only if imidazole resides in the ionic phase close to the cations. |
| †† Fig. SI-6† shows broad lines for the liquid-state NMR spectra of the gel, which motivates the use of MAS solid-state NMR methods to achieve higher spectral resolution. |
| This journal is © The Royal Society of Chemistry 2018 |