
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMembrane-containing virus particles exhibit the mechanics of a composite material for genome protection†
S.
Azinas
 ab,
F.
Bano
b,
I.
Torca
c,
D. H.
Bamford
d,
G. A.
Schwartz
e,
J.
Esnaola
c,
H. M.
Oksanen
d,
R. P.
Richter
*bf and
N. G.
Abrescia
*ag
ab,
F.
Bano
b,
I.
Torca
c,
D. H.
Bamford
d,
G. A.
Schwartz
e,
J.
Esnaola
c,
H. M.
Oksanen
d,
R. P.
Richter
*bf and
N. G.
Abrescia
*ag
aMolecular recognition and host–pathogen interactions programme, CIC bioGUNE, CIBERehd, Derio, Spain
bBiosurfaces Lab, CIC biomaGUNE, San Sebastian, Spain
cMechanical and Industrial Production Department, Mondragon University, Arrasate-Mondragón, Spain
dMolecular and Integrative Biosciences Research Programme, Faculty of Biological and Environmental Sciences, Viikki Biocenter, University of Helsinki, Finland
eCentro de Física de Materiales, (CSIC-UPV/EHU) & Donostia International Physics Center, San Sebastian, Spain
fSchool of Biomedical Sciences, Faculty of Biological Sciences, School of Physics and Astronomy, Faculty of Mathematics and Physical Sciences, and Astbury Centre for Structural Molecular Biology University of Leeds, Leeds, UK. E-mail: R.Richter@leeds.ac.uk; Tel: +44 113 3431969
gIKERBASQUE, Basque Foundation for Science, Bilbao, Spain. E-mail: nabrescia@cicbiogune.es; Fax: +34 946572502; Tel: +34 946572523
First published on 16th April 2018
Abstract
The protection of the viral genome during extracellular transport is an absolute requirement for virus survival and replication. In addition to the almost universal proteinaceous capsids, certain viruses add a membrane layer that encloses their double-stranded (ds) DNA genome within the protein shell. Using the membrane-containing enterobacterial virus PRD1 as a prototype, and a combination of nanoindentation assays by atomic force microscopy and finite element modelling, we show that PRD1 provides a greater stability against mechanical stress than that achieved by the majority of dsDNA icosahedral viruses that lack a membrane. We propose that the combination of a stiff and brittle proteinaceous shell coupled with a soft and compliant membrane vesicle yields a tough composite nanomaterial well-suited to protect the viral DNA during extracellular transport.
1. Introduction
Virions (infectious virus particles) are nano-sized carriers of information whose objective is to infect a host cell and generate progeny that can, in turn, repeat the infection cycle. When traveling outside of the host cell, virions may encounter harsh environmental conditions. Knowledge of the deformability (stiffness, k), the energy required for mechanical failure (toughness, T), and the limits to fatigue for virions has been acquired through studies using atomic force microscopy (AFM), but mainly for viruses that do not have an internal membrane.1–4 The mechanical properties of these virions ultimately recapitulate the molecular interactions across the viral icosahedral shell.5,6 There is no comparable information available for icosahedral virions in which an internal membrane vesicle surrounds the densely packaged DNA and, to our knowledge, such viruses with RNA genome are yet to be identified. Defining how a membrane and capsid provide protection to the genome within will provide new insights into the function and properties of biological materials, thereby inspiring the design of novel nanostructures. PRD1, the prototype for icosahedral dsDNA viruses with an internal membrane, is a large and complex enterobacterial virus (family Tectiviridae; ∼65 nm mean diameter and triangulation number pseudo-T = 25, Fig. 1a). Its crystal structure revealed the molecular interactions across the major capsid proteins (MCPs) P3 (395 residues) and with the membrane.7,8 P3 trimers (capsomers) are arranged in the group of nine (GON) layout in the virion facets (Fig. S1a†). The minor capsid protein P30 cements the edges of the icosahedron. Eleven of the twelve vertices are capped by peripentonal P3 trimers and by spike complexes7–11 (Fig. 1a and S1a†). The internal membrane enclosing the densely packaged dsDNA is composed of a roughly equal mass of lipids and membrane proteins.8 The twelfth vertex is the unique, membrane-embedded portal used for DNA packaging into the procapsid and later for DNA ejection via the formation of a proteo-lipidic tube.12,13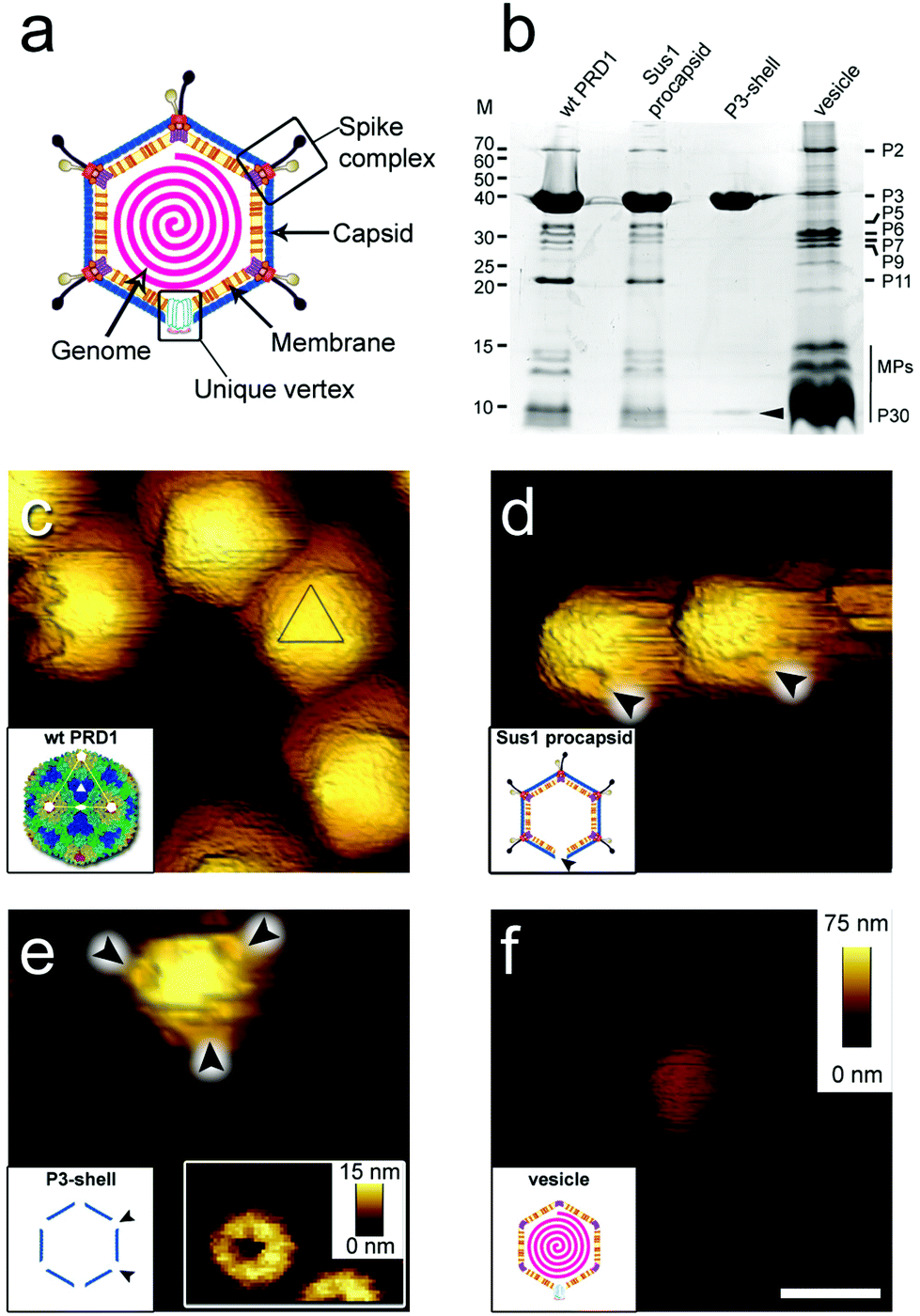 | ||
| Fig. 1 (a) Schematic of PRD1 highlighting the main structural features of the virus particle. (b) Protein composition of wt PRD1 and subviral particles analysed by SDS-PAGE and Coomassie staining. Left: Molecular masses (M; kDa). Right: Positions of the most abundant proteins; the vertical black line refers collectively to membrane proteins (MPs, including P14, P16, P18, P20, P22, P31 and P32). Cementing protein P30 identified in the P3-shell by mass spectrometry is indicated by a black arrowhead. (c–f) Atomic force micrographs of wt PRD1 and subviral particles with scale bar (100 nm) and z range (75 nm) indicated in (f). (c) Wt PRD1 featuring different particle orientations. Inset: Crystal structure of wt PRD1.7 Yellow, green, cyan and blue denote the P3 pseudo-hexameric capsomers composing the icosahedral asymmetric unit; white lines delineate a virus facet; white pentagons, triangles, and ovals indicate icosahedral 5-, 3- and 2-fold symmetry axes, respectively; (d) Sus1 procapsids (no DNA within). Arrowheads indicate the visible depression due to the missing packaging portal in the unique vertex. Inset: Schematic of Sus1 procapsid; (e) P3-shell. Arrowheads highlight the depressions visible at all vertices due to the absence of the peripentonal capsomers and vertex complexes. Right inset: The star-shaped vertex depression at higher resolution (z range: 15 nm). Left inset: Schematic of P3-shell. Arrowheads indicate some of the de-capped vertices. (f) DNA-filled vesicle. Inset: Schematic of vesicle represented as an icosahedron for clarity and consistency with the wt PRD1 schematic representation – this shape, however, might not be retained in the purified membrane-vesicle. | ||
Guided by the available genetic, biochemical, and structural information, we chose to utilize (i) wild type PRD1 (wt PRD1), (ii) a PRD1 mutant that forms procapsids devoid of DNA (Sus1 procapsid), (iii) the icosahedral P3-shell composed of MCP P3 and the minor capsid protein P30 (Fig. 1b and Table S1†), but lacking the pentons and peripentonal capsomers (P3-shell), and (iv) proteo-lipidic membrane vesicles enclosing a complete genome (vesicle; Fig. 1b). We used AFM to examine the mechanical responses of these particles in an aqueous environment by assessing their stiffness and yield behaviour under an applied force and used finite element modelling, where possible, to aid the analysis.
2. Experimental
2.1 PRD1 specimen production
Wt PRD1 and mutant PRD1 and mutant sus1 [amber mutation in gene IX] were propagated on Salmonella enterica serovar Typhimurium strain DS88 or on suppressor strains PSA(pLM2) or DB7156(pLM2).14–18 Cells were grown in Luria–Bertani (LB) medium at 37 °C.For the production of wt and mutant phage particles, DS88 cells were infected using a multiplicity of infection of 8–10. For mutant particle production, infected cells were collected 15 min after infection (Sorvall rotor F12, 5000 rpm, 10 min, 22 °C) and transferred to a fresh pre-warmed medium. Virus particles were purified by polyethylene glycol–NaCl precipitation and rate zonal ultracentrifugation in sucrose (Sorvall rotor AH629), as previously described.19 For AFM, wt PRD1 and Sus1 procapsids were further purified by equilibrium ultracentrifugation in sucrose (Sorvall rotor AH629). The particles were concentrated by differential centrifugation (Sorvall rotor T647.5, 32![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm, 2 h, 5 °C). A buffer containing 20 mM potassium phosphate pH 7.2 and 1 mM MgCl2 was used for purification and resuspension.
000 rpm, 2 h, 5 °C). A buffer containing 20 mM potassium phosphate pH 7.2 and 1 mM MgCl2 was used for purification and resuspension.
For P3-shell preparation, the rate zonal purified Sus1 mutant particles (2 mg ml−1 in 20 mM Tris-HCl, pH 7.2, 1 mM MgCl2) were treated with 1% (w/v) sodium dodecyl sulfate (SDS) for 15 min at 25 °C.20 P3 shells were isolated by rate zonal centrifugation in a linear 5–20% (w/v) sucrose gradient using the Tris buffer (Sorvall rotor AH629, 24000 rpm, 1 h 45 min, 20 °C). The particles were concentrated by centrifugation (Sorvall rotor T865, 34000 rpm, 4 h, 5 °C) and resuspended in Tris buffer.
For membrane vesicle preparation, the rate zonal purified Sus607 particles devoid of the major membrane protein P11 (1 mg ml−1 in 20 mM Tris-HCl, pH 7.2) were treated with 2.5 M GuHCl21 and membrane vesicles were purified by equilibrium centrifugation in a linear 20–70% (w/v) sucrose gradient (Sorvall rotor TH641, 22000 rpm, 16–18 h, 20 °C). Protein concentrations were determined by Bradford assay.22 Particles were analyzed using sodium dodecyl sulfate polyacrylamide gel electrophoresis23 (SDS-PAGE; 16% acrylamide; Fig. 1b).
Virus particles were stored at 4 °C for no more than 4 weeks. During this time, more than 95% of the wt PRD1 particles remained intact and without the loss of their genome as visualized by cryo-electron microscopy (cryo-EM) 2D imaging [a JEM-2200FS (JEOL) transmission electron microscope equipped with an UltraScan 4000 SP 4k × 4k camera (GATAN)] (Fig. S2†). Sus1 procapsid, P3-shell particles and vesicles were similarly stored and also used within this time frame (Fig. S2†).
2.2 Protein identification by mass spectrometry
Silver stained24 protein bands were cut out of the polyacrylamide gel and “in-gel” digested. Cysteine bonds were reduced with 0.045 M dithiothreitol (Sigma-Aldrich, USA) for 20 min at 37 °C and alkylated with 0.1 M iodoacetamide (Fluka, Sigma-Aldrich, USA) at room temperature. Samples were digested by adding 0.75 μg trypsin (Sequencing Grade Modified Trypsin, V5111, Promega) overnight at 37 °C. After digestion, peptides were purified with C18 microspin columns (Harvard Apparatus) according to the manufacturer's protocol. The dried peptides were reconstituted in 30 μl of buffer A [0.1% trifluoroacetic acid (TFA) in 1% acetonitrile (ACN)].Liquid chromatography coupled to tandem mass spectrometry (LC-MS/MS) analysis was carried out on an EASY-nLC1000 (Thermo Fisher Scientific, Germany) connected to a Velos Pro-Orbitrap Elite hybrid mass spectrometer (Thermo Fisher Scientific) with a nano-electrospray ion source (Thermo Fisher Scientific). The LC-MS/MS samples were separated using a two-column setup consisting of a 2-cm C18-Pepmap trap column (Thermo Fisher Scientific), followed by a 15-cm C18-Pepmap analytical column (Thermo Fisher Scientific). The linear separation gradient consisted of 5% buffer B in 5 min, 35% buffer B in 60 min, 80% buffer B in 5 min and 100% buffer B in 10 min at a flow rate of 0.3 μl min−1 (buffer B: 0.1% TFA acid in 98% acetonitrile). Six microliters of sample were injected per LC-MS/MS run and analyzed. Full MS scan was acquired with a resolution of 60000 in the normal mass range in an orbitrap analyzer and followed with CID-MS2 top 20 most intense precursor ions within the ion trap (energy 35). Data were acquired using LTQ Tune software.
The acquired MS2 scans were searched against the enterobacteria phage PRD1 (NCBI) protein database using the Sequest search algorithms in Thermo Proteome Discoverer. The allowed mass error for the precursor ions was 15 ppm and for the fragment 0.8 Da. A static residue modification parameter was set for carbamidomethyl +57021 Da (C) of the cysteine residue. Methionine oxidation was set as dynamic modification +15995 Da (M). Only full-tryptic peptides were allowed for the maximum of one missed cleavage.
2.3 Surface preparation and immobilization of PRD1 particles
Freshly cleaved surfaces (∼1 cm2) of mica (Ted-Pella Inc., CA, USA) were functionalized with 3-(2,2-aminoethylamino)-ethylaminopropyltrimethoxysilane (APTES; Sigma-Aldrich) by adapting a previously described procedure.25 Briefly, mica was left to incubate with 30 μL of APTES in a 4 l gas incubator for 2 h. A small amount of water (10 μl) was deliberately added to generate a root-mean-square surface roughness of 1.7 to 2 nm, which proved beneficial to particle attachment. Following the APTES functionalization, surfaces were covered with 100 μl Hepes buffer (10 mM HEPES, pH 7.4, 150 mM NaCl, 2 mM MgCl2) containing ∼10 μg virus particles (wt PRD1, Sus1 procapsid, or P3-shell). PRD1 vesicles were attached to freshly cleaved mica instead of APTES-coated mica. Virus particles were allowed to adhere for 30 min, and excess particles were removed by washing with buffer.2.4 AFM imaging and nanoindentation
All PRD1 particles were found to attach as a monolayer to the prepared mica surfaces that could be readily visualized by AFM. Imaging and nanoindentation measurements were carried out using a MultiMode 8 AFM with a Nanoscope V controller (Bruker, CA, USA) at room temperature in Hepes buffer. Wt PRD1, Sus1 procapsids, and P3-shells were analysed with sharpened triangular Si3N4 cantilevers with a nominal spring constant of 0.7 N m−1 (ScanAsyst-Liquid; Bruker). For PRD1 vesicles, Si3N4 cantilevers with a nominal spring constant of 0.1 N m−1 (AC40; Bruker) were used. The real spring constants of the cantilevers were measured using the thermal noise method26 as implemented in the NanoScope software and were found to be close to the nominal values. Imaging was performed in Peak Force Tapping mode, with a typical tapping amplitude of 40 nm and a driving frequency of 2 kHz. For wt PRD1, Sus1 procapsids, and vesicles, the peak force was usually 100 pN; for P3-shells, it was 80 pN. Under these conditions, the image quality was sufficient to reliably identify the virus particles (and in some cases, their orientation and surface sub-features), and in general, intact PRD1 particles did not degrade or break upon repeated imaging. On already degraded particles, gradual further deterioration was observed, and particles were also found to detach from the surface. Images were plane fitted when required, using Gwyddion software (http://gwyddion.net/) without the application of noise filtering or sharpening.Nano-indentation measurements were performed at individually selected particles using the ‘point-and-shoot’ function within the NanoScope software. Briefly, the area of interest was first imaged to localize the virus particle; the ‘point-and-shoot’ function was then activated and force curves were taken at the particle centre. subsequently, the area was imaged once more to verify successful nano-indentation. The accuracy of localization of the particle centre was found to be limited by piezo drifts and estimated to be within 5 nm. Force vs. distance (F/z) curves were acquired at a constant approach velocity of 200 nm s−1. The approach and retract distances were 100 nm, corresponding to a total time of 1 s per complete approach and retract cycle. The maximal load was 4 nN, except for P3-shells, where the maximal load was lowered to 2 nN. F/z curves were analysed using the NanoScope software.
2.5 Force curve analysis
To avoid including particles that might have been displaced or changed orientation upon indentation, force curves where force levels dropped and remained below 500 pN over distances of 20 nm and more before hard-wall contact were also discarded from analysis. A representative force curve for each PRD1 particle type with AFM micrographs before and after indentation is shown in Fig. S3,† and additional force curves illustrating sample to sample variations are shown in Fig. S4.†
R3 ≈ π/6h3, we obtain T ≈ 6/πFyεy/h2.
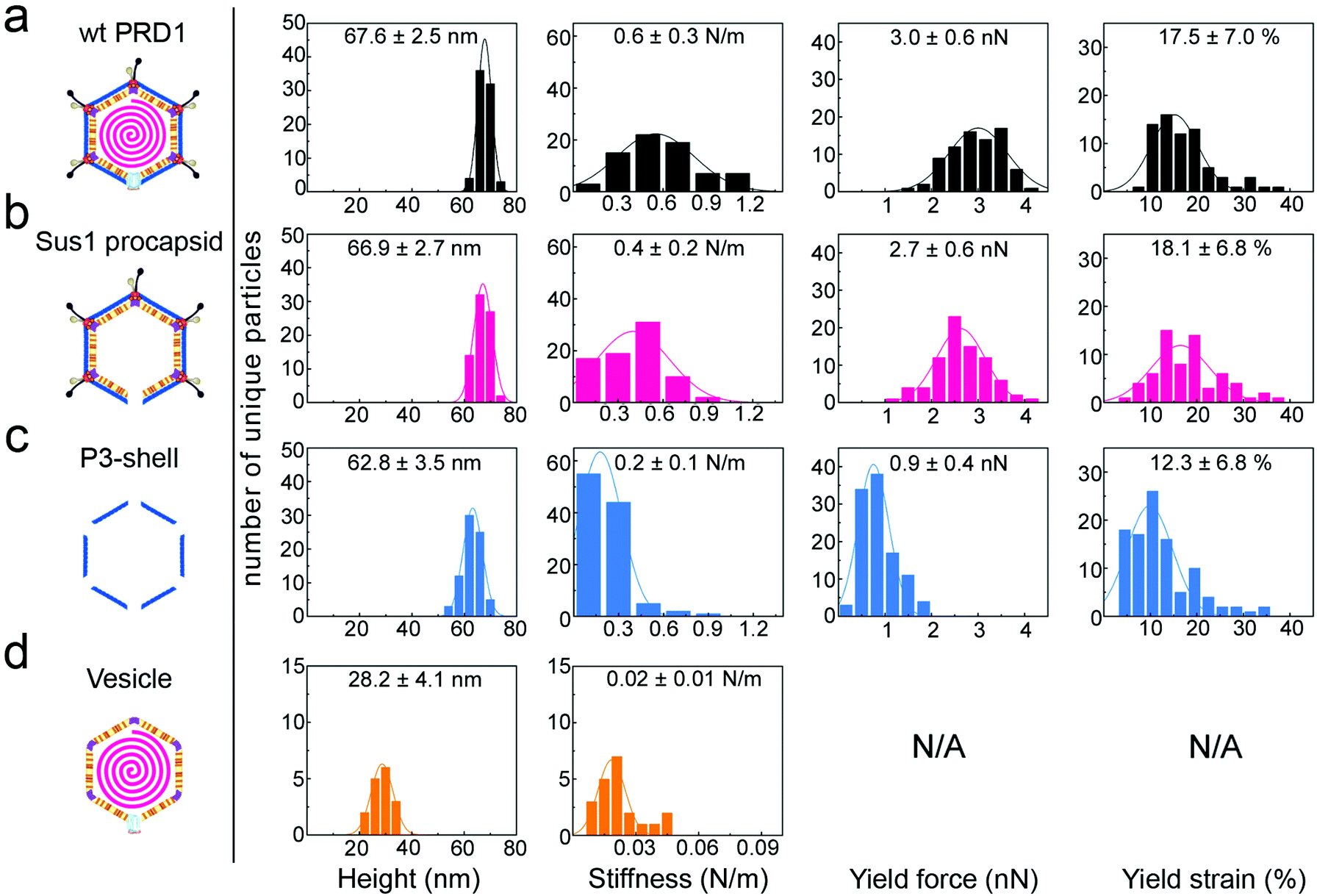 | ||
| Fig. 2 Histograms of various properties of the four viral particle types, reporting mean values and standard deviations for each. y-Axis: number of unique particles (75 for wt PRD1 and Sus1 procapsid, 103 for P3-shell and 21 for vesicles). x-Axis: property denoted below each column. Specimen heights closely match the particle dimensions previously obtained by averaging techniques, except for the vesicle, which is slightly flattened upon immobilization. Yield force and strain are not shown for vesicles, as these do not have a defined yield point. | ||
2.6 Finite-element analyses
To test how the membrane vesicle affects the stiffness and stability in a composite system of a proteinaceous capsid shell with an underneath membrane vesicle, modelling of the mechanical response of PRD1 was performed using a continuum-mechanics finite-element analysis (FEA) with the software ABAQUS 6.14 (ABAQUS, Fremont, CA) (Fig. S6 and S7†). In this approach, the proteinaceous capsid and the proteo-lipidic vesicle were represented as mechanically isotropic spherical shells, i.e., neglecting the structural details of each shell (see section 3.3). This method can effectively deal with the architectural complexity introduced by the presence of the vesicle whilst the number of adjustable parameters is kept small.3. Results and discussion
3.1 Imaging PRD1 assemblies at the single particle level
When immobilized, the wt particles oriented with a 3-fold, 2-fold or 5-fold symmetry axis normal to the supporting surface (Fig. 1c). The preferred orientation (>67% of the particles) was with a facet down (i.e., a 3-fold axis normal to the surface). The average height was 67.8 ± 2.5 nm (mean ± s.d.) for wt PRD1, 66.9 ± 2.7 nm for the Sus1 procapsid (n = 75 each; Fig. 2a and b, and 62.8 ± 3.5 nm for the P3-shell (n = 103; Fig. 2c), in good agreement with electron-microscopy and X-ray data7,28,29 (Fig. S1b†). Strikingly, we observed on the Sus1 procapsid a mild circular depression, ∼13 nm in diameter at ∼8% of the visualized vertices (n = 103) (Fig. 1d and S8†). This percentage, combined with the estimated dimensions, defines at the single-molecule level that this feature is the unique vertex that lacks the external part of the packaging portal.12 This, however, was not detected on wt PRD1. Indeed, the structural difference in the capsid context between the ‘wild type’ unique vertex and the remaining 11 vertices is much smaller than that between the unique vertex and the other 11 vertices in the procapsid,12 and apparently, it is too small to be resolved with our AFM set-up. P3-shells showed holes, ∼25 nm wide, at each vertex (Fig. 1e) consistent with the missing peripentonal P3 capsomers, spike complexes, and internal vesicles28,29 (Fig. S1b†). The forces needed to be lowered from ∼100 to ∼80 pN to enable imaging of P3-shells indicating that these particles are more sensitive to breakage compared to wt PRD1. By contrast, vesicles displayed a featureless surface and a height of 28.2 ± 4.1 nm (Fig. 1f and 2d). This height is less than the diameter of DNA-containing vesicles (∼35 nm),8 indicating that vesicles readily deform when immobilized.3.2 Stiffness of PRD1 particles
The AFM force vs. distance (F/z) curves (Fig. S3 and S4†) generated by the nanoindentation testing of individual particles quantified the resistance to small elastic deformation (stiffness, k), as well as the force and strain at the point of mechanical failure (yield force, Fy; yield strain, εy) (Fig. 2 and 3a–c).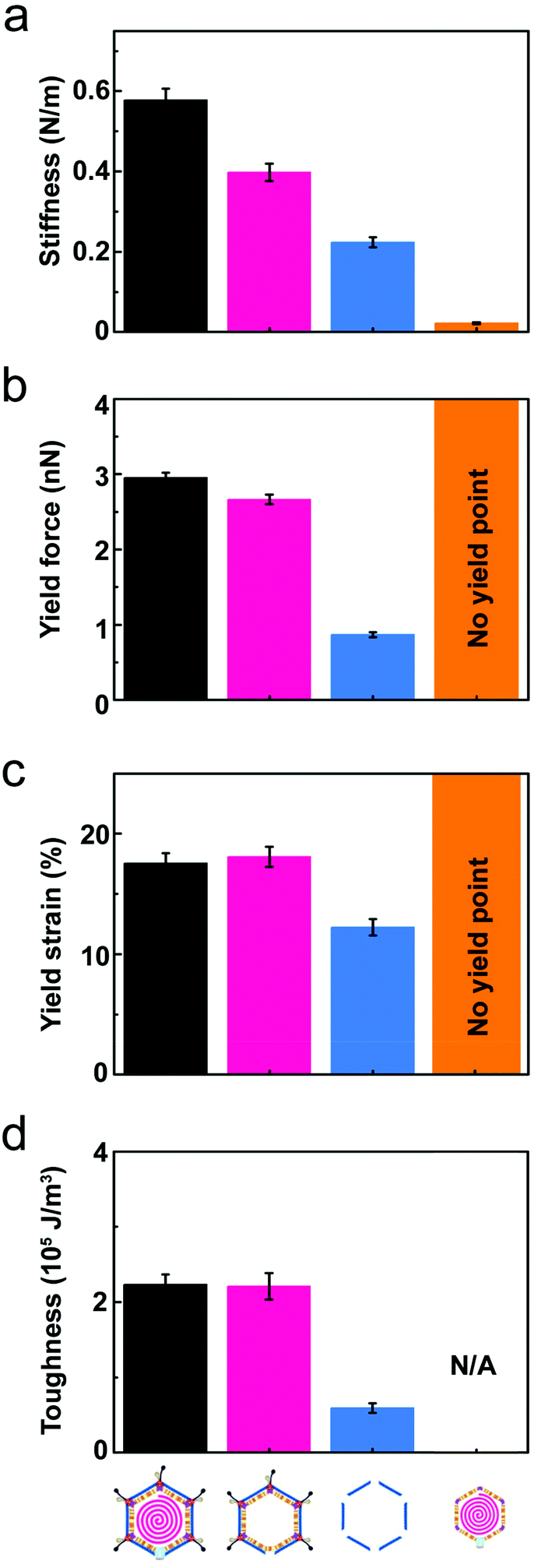 | ||
| Fig. 3 Comparison of the average mechanical properties across the different PRD1-derived particles. In all panels: black bar, PRD1 wt; pink bar, Sus1 procapsid; blue bar, P3-shell; orange bar, vesicle. (a) Stiffness (k); (b) yield force (Fy); (c) yield strain (εy); (d) toughness (T). All panels report the mean values and standard errors of the mean that were derived from the data shown in Fig. 2 (for the vesicle, calculation of toughness is not possible because there is no defined yield point). | ||
The F/z curves typically exhibited a relatively small non-linear regime at the smallest indentation forces (F < 200 pN), which is likely to represent the Hertzian deformation of the capsid shell.27 This was followed by an extended linear regime, justifying the quantification of elastic properties in terms of the stiffness k. For quantitative stiffness analysis, we considered this linear regime for strains up to 10%, which was well below the yield point. For quasi-spherical shells such as the Sus1 procapsid and the P3-shell, this linear regime can be associated with shell bending.27
Wt PRD1 possessed the greatest stiffness (0.57 ± 0.03 N m−1, mean ± s.e.m.), followed by the Sus1 procapsid (0.39 ± 0.02 N m−1) and the P3-shell (0.22 ± 0.01 N m−1; Fig. 3a). Most likely, the enhanced resistance of wt PRD1 to elastic deformation arises from the pressure exerted by the DNA.8,30 PRD1 genome packaging produces a radial expansion of the internal membrane (e.g., the radius of the outer leaflet increases by ∼6%), which presses the vesicle closer to the capsid.12,28,30
The DNA-filled vesicle displayed the least stiffness (0.022 ± 0.002 N m−1; Fig. 3a), indicating that its direct effect on virion stiffness is marginal but that it contributes to virion stiffness by transmitting pressure from the DNA to the capsid. The volume inside the rigid capsid is reduced upon indentation accentuating the effect of DNA pressure on the stiffness of wt PRD1. In contrast, the soft membrane can stretch upon indentation, thus reducing any effect of DNA pressure on the vesicle stiffness.
3.3 Modelling of PRD1's shell elasticities
To confirm the above findings on PRD1's elastic properties, we confronted the experimental data with continuum-mechanics finite-element computational modelling (see Experimental and Fig. S6†). In this simulation, the particle was placed between a rigid plane and a rigid indenter (Fig. 4a). The displacement of the rigid plane was constrained, and the indenter apex was modelled as a sphere of 10-nm radius positioned coaxially with the virus-derived particle in the direction normal to the plane. For indentation, the indenter applied a force in the direction normal to the plane. The contact between all bodies was assumed to be hard in the direction normal to the contact (i.e., interpenetration was not allowed) and frictionless in the tangential direction (i.e., relative displacement and rotation were allowed with no constraint).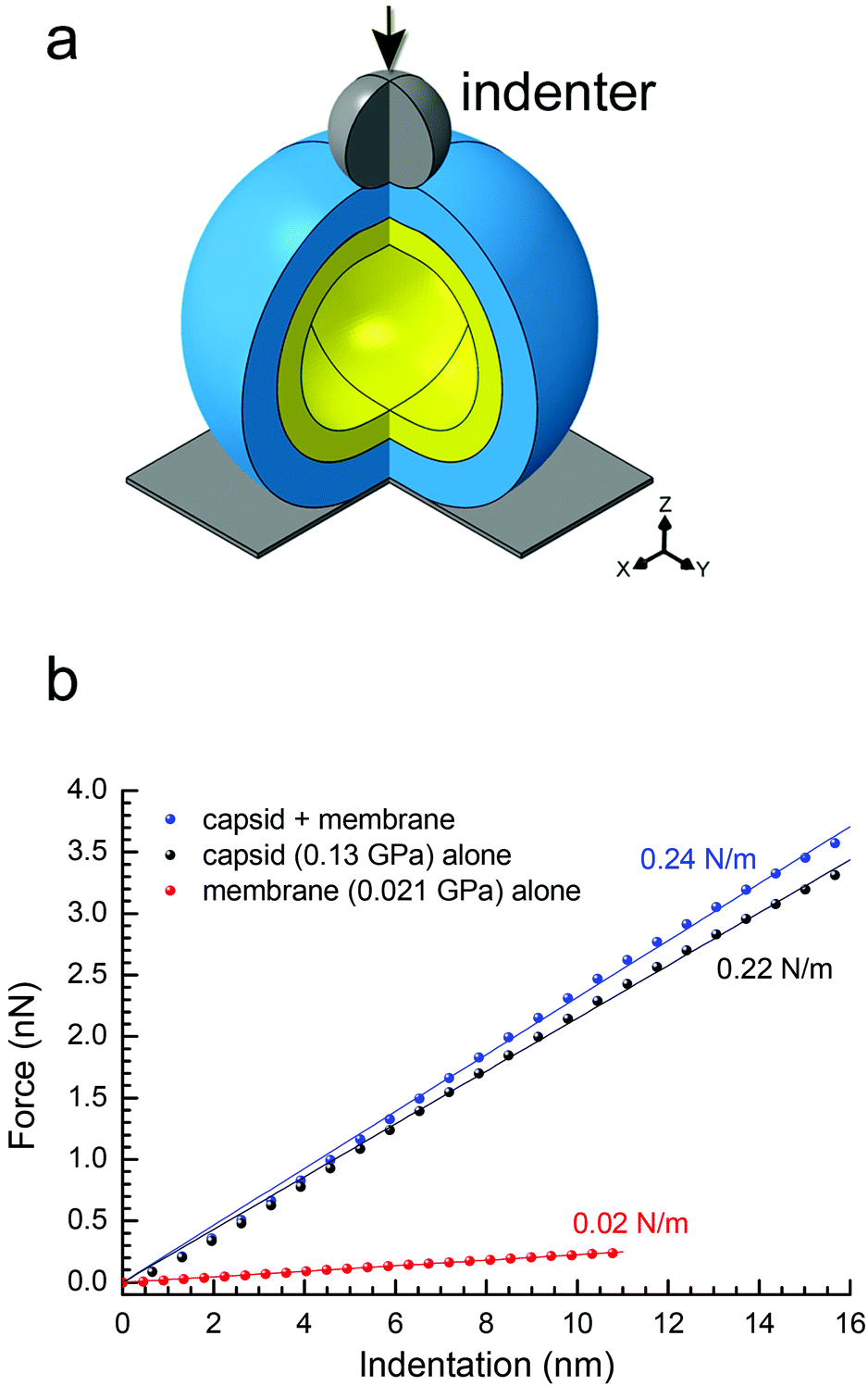 | ||
| Fig. 4 (a) Schematic of the spherical two-shell model (outer protein shell in blue, inner vesicle shell in yellow) for the finite-element modelling; three-quarters of the spheres are shown with the indenter apex represented as a grey sphere with the direction of the applied force as indicated by the black arrow; in the bottom right corner, the Cartesian coordinate system; (b) predicted curves of force vs. indentation for a protein shell (with E = 0.13 GPa, representing the P3-shell; black dots), a vesicle shell (with E = 0.021 GPa, representing the proteo-lipidic vesicle; red dots) and a composite of these protein and vesicle shells (blue dots). In matching colours: lines are linear fits through the origin, and texts the stiffness values corresponding to the slopes. The stiffness values of protein and vesicle shells match the experimental data for P3-shell and vesicle (Fig. 3a), respectively, confirming the correct choice of Young's modulus values; the stiffness of the composite is well approximated by the sum of the stiffness values of the constituent shells and thus only marginally larger than the stiffness of the protein shell alone. | ||
To extract material properties from the experimental data, we first considered the proteinaceous capsid individually. For the capsid, we estimated an outer radius R = 33.2 nm and a thickness d = 8 nm from the rotationally averaged electron density maps of the PRD1 virion.28,30 By treating the capsid as isotropic and linearly elastic, its properties can be described by two independent parameters: Young's modulus E and the Poisson ratio ν. By neglecting a non-linear regime at very small strains (ε < 3%), the predicted relationship between force and particle indentation δ was approximately linear and scaled with the Young's modulus E for indentations up to 2d (ε up to 24%; Fig. S6a†). Our results were rather insensitive to the Poisson ratio (Fig. S7a–c†), and we thus fixed ν = 0.4.
These features are consistent with previous computations by others and with the predictions of thin-shell theory,4,31,32 and thus validate the numerical model. The linear relationship between force and indentation is also consistent with our experimental data (Fig. S3b, e, h, and S4a–d†), where we note that the strain regime of ε < 10% used for the analysis of experimental data lies well within the range over which theory predicts a linear response. This implies that stiffness analysis is far away from any buckling transition.33 It justifies the use of linear elasticity in our simple theoretical model, and also the use of stiffness k = F/δ to characterize the shell's elastic properties. With F/E ∼ δ and k = F/δ, it is also clear that k ∼ E, and a linear fit to the data in Fig. S6a† gives k ≈ 1.65 nm × E. With this equation, we estimate E = 0.13 GPa for the P3-shell from the experimentally determined mean stiffness value for this particle (k = 0.22 N m−1; Fig. 3a).
A recent computational modelling study on a smaller non-membrane-containing virus has shown that capsid proteins can dynamically re-structure upon capsid indentation with appreciable effects on capsid elasticity compared to an idealized homogeneous shell.34 Our experimental data do not allow deconvoluting these effects. However, the extended linear regime in the F/z curves observed experimentally for the P3-shell and the Sus1 procapsid (Fig. S4c and d†) indicates that the bending elasticity of the PRD1 capsid shell remains appropriately described by a Young's modulus (where this would effectively include the possible re-structuration effects).
The elasticity of the proteolipidic membrane was estimated analogous to that of the proteinaceous capsid, assuming an outer radius identical to the inner radius of the capsid (R = 25.2 nm) for direct contact of the two shells, and a membrane thickness d = 5.5 nm from a rotationally averaged electron density map.30 Fig. S6b† shows the dependence of F/E on vesicle deformation for these geometrical parameters, from which k ≈ 1.07 nm × E can be derived. Assuming to a first approximation that the stiffness of the membrane shell is similar to the experimentally accessible value for the genome-containing vesicle (k = 0.022 N m−1; Fig. 3a), we can estimate E = 0.021 GPa.
To predict the elastic behaviour of the composite capsid-membrane system, we modelled a system of two concentric shells with the inner shell adopting the geometry and Young's modulus of the membrane and the outer shell adopting the geometry and Young's modulus of the P3-shell (Fig. 4a). The stiffness of this composite system was k = 0.24 N m−1, that is the presence of the vesicle enhanced the stiffness only marginally, by about 10%, compared to the P3-shell alone. More generally, the stiffness values shown in Fig. 4b exemplify that the stiffness of the composite (0.24 N m−1) is well approximated by the sum of the stiffness values of the constituent shells (0.22 N m−1 + 0.02 N m−1). The small enhancement in stiffness was virtually independent of the Poisson ratios of both the capsid and membrane (Fig. S7d†).
The above modelling exercise provides reasonable predictions about the trends that can be expected based on the experimental AFM-derived shell elastic mechanical properties. Here, we have operated with the P3-shell as a reference system because experimental data for this single-shell system were readily available. Using the above-identified stiffness relationship, we can now also estimate the Young's modulus of the complete capsid shell from the experimentally determined stiffness values of the Sus1 procapsid and the membrane. The closure of 11 of 12 vertices by additional proteins enhances the elasticity of the Sus1 procapsid shell compared to the P3-shell, whilst a further enhancement of the capsid elasticity by the unique vertex – missing in the Sus1 procapsid – is likely to be marginal. kcapsid ≈ kcapsid+membrane − kmembrane ≈ 0.39 N m−1 − 0.02 N m−1 = 0.37 N m−1 (Fig. 3a) and E ≈ k/1.65 nm (Fig. S5a†) give E ≈ 0.22 GPa, a value comparable to that for some non-enveloped and enveloped viral capsids.35
Indeed, the two-shell modelling confirmed that the stiffness is only marginally affected, by less than 10%, by an internal soft membrane contacting the capsid (Fig. 4b). The reduced stiffness of the P3-shell (E = 0.13 GPa) is likely due to the absence of the stabilizing pentons and peripentonal capsomers.
3.4 Mechanical stability of PRD1 particles
The presence of the genome and the ensuing particle stiffening had no appreciable effect on the mechanical stability of the virion: the yield force and yield strain of wt PRD1 (Fy = 3.0 ± 0.1 nN, εy = 17.5 ± 0.8%; mean ± s.e.m.) and the Sus1 procapsid (Fy = 2.7 ± 0.1 nN, εy = 18.1 ± 1.4%) were similar (Fig. 3b and c). This is in contrast to other phages, such as phage λ, where the DNA augments both stiffness and mechanical stability.36 As for the P3-shell, it was much more sensitive to breakage than wt PRD1 or the Sus1 procapsid (Fy = 0.9 ± 0.1 nN, εy = 12.3 ± 0.7%; Fig. 3b and c).The mechanical failure of the PRD1 particles upon indentation was frequently accompanied by the loss of capsomers from the capsid shell (Fig. S3a–c,† insets). In addition, F/z curves of wt PRD1, the Sus1 procapsid, and the P3-shell revealed slip events coincident with the occurrence of micro-fractures during force loading likely reflecting the local displacements of MCPs (Fig. S5†). While all three particle populations presented a similar density of micro-fractures (average 1.3 per nN of applied compressive force), wt PRD1 and the procapsid withstood more of these fractures before yielding.
Previous studies on binary component viral systems – genome encapsulated by a protein shell – have highlighted the role of DNA or RNA in contributing to the capsid stiffness where the mechanical reinforcement is achieved by the genome anchoring the protein shell from the interior.35 In other cases, such as the herpes simplex virus type 1 (HSV-1) nucleocapsid, the stiffness and yield force remain practically the same whether the particle is fully packaged or devoid of the genome, and stabilizing viral proteins appear to be responsible for this assembly type.37,38 In PRD1, the Sus1 procapsid and the mature particle display similar yield forces whereas the relative increase in the stiffness of the wt PRD1 can be attributed to the presence of DNA. The packaging of the genome leads to an expansion of the membrane-vesicle increasing the membrane's interactions with the capsid proteins.12,28–30
3.5 Toughness analysis of PRD1 and other icosahedral dsDNA viruses
The resistance to material fracture is typically expressed as toughness T, here defined as the amount of energy per unit volume that can be absorbed before mechanical failure. The toughness of PRD1, 2.2 × 105 J m−3, is relatively high compared to that reported for other icosahedral dsDNA viruses (see Fig. 3d and 5, and Table S2†). Thus, PRD1 together with human adenovirus is one of the toughest among all icosahedral dsDNA viruses.7,39–41 Adenovirus shares a common MCP fold and assembly mechanism with PRD1; however, whilst it lacks a membrane, it possesses a complex set of cementing proteins stabilizing the structure – about 300 in total (composed of sixty copies of each of the proteins IIIa, V, VI, VIII and IX) instead of the mere 60 copies of the single protein species P30 guiding the assembly in PRD1.7,42Altogether, these comparisons highlight that the layered complex of the capsid and membrane vesicle relieves the genome from a stabilizing role and endows PRD1 with remarkably high mechanical stability.
To explore how the PRD1 vesicle and capsid together influence toughness, we compared the toughness of the PRD1-derived particles: the Sus1 procapsid, the P3-shell, and the vesicle. The P3-shell (T = 0.54 × 105 J m−3) was more susceptible to mechanical failure than the Sus1 procapsid (T = 2.1 × 105 J m−3; Fig. 3d). The fact that the P3-shell's toughness was ∼4-fold less but its stiffness was only 2-fold less revealed its rather stiff, brittle nature (Fig. 3a). The P3-shell lattice is held together by the (C-I type and C-II type) interactions established by the GON within each facet and along the facets via the MCPs C-termini and by the P30 proteins7 (Fig. S1a†). The relative ease of breakage of this lattice might facilitate morphological corrections as the capsomers assemble on the vesicle mould during the procapsid formation. Closing the icosahedral vertices with the peripentonal capsomers and penton proteins (Fig. S1a†) and plugging the unique vertex with the portal complex produce a stable procapsid that can withstand dsDNA translocation powered by the packaging ATPase P9.12
More generally, the brittle nature of the capsid is not only manifested in the small yield force of the P3-shell but also in the above-mentioned microfractures. The DNA-filled vesicles, on the other hand, did not show a clear yield point and typically recovered their original shape even after strains exceeding 60%, which indicated that they were ductile and very soft (Fig. 3a).
Thus, comparative analysis with other dsDNA viruses indicates that the toughness of PRD1 is superior to most other dsDNA viruses with comparable capsid organization but lacking the membrane, and is only rivalled by adenovirus – a non-membrane-containing virus – which is exceptionally rich in cementing proteins stabilizing the capsid (Fig. 5).
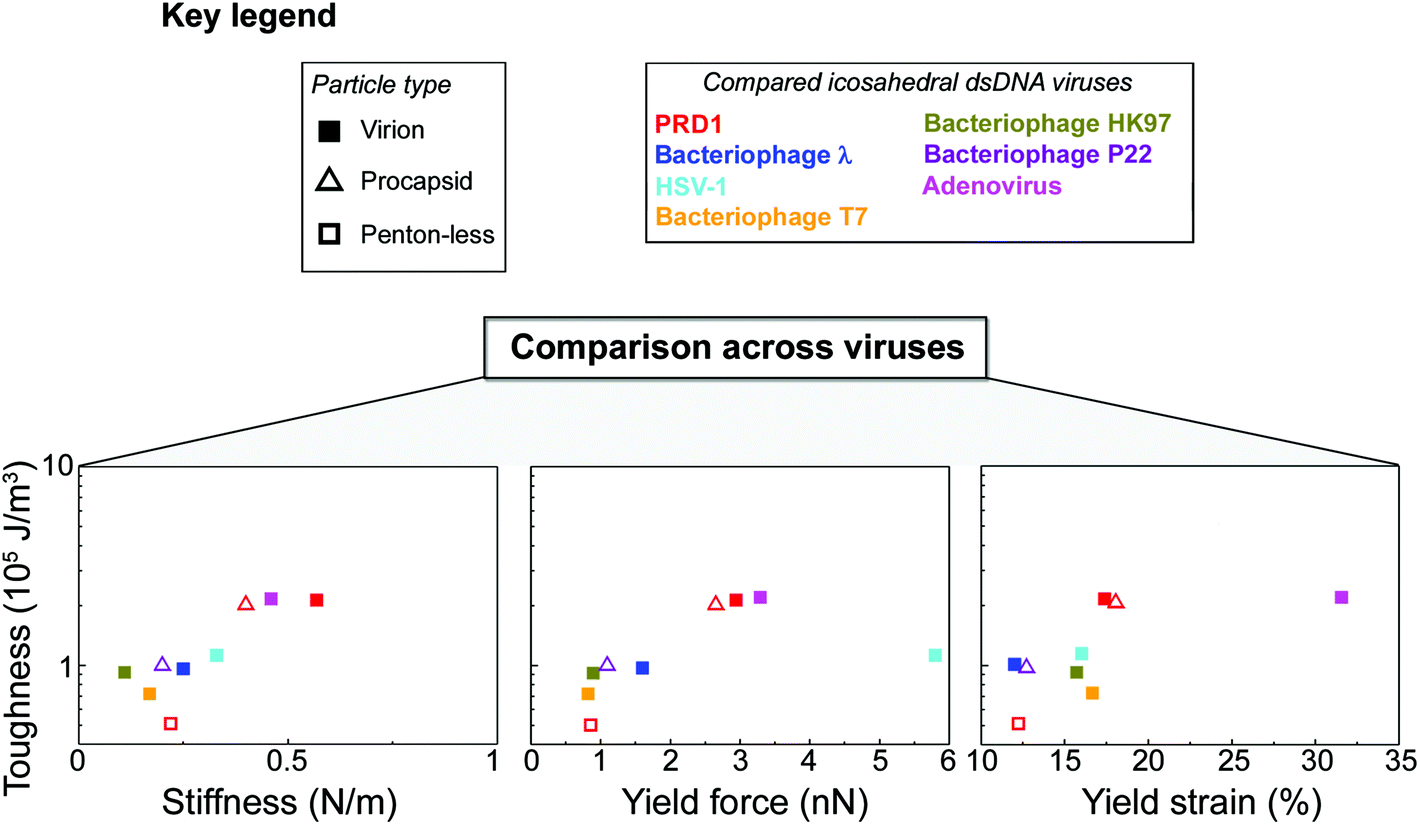 | ||
| Fig. 5 PRD1's mechanical properties compared to other dsDNA viruses. Key for the viruses and particle types shown in the other panels (in the case of HSV-1 virion, we refer to its nucleocapsid). Comparison across viruses via Ashby plots of toughness (y-axis in log scale) vs. stiffness, yield force and yield strain (x-axis), respectively (see the Table S2† for references). | ||
3.6 Composite material model of PRD1
Our findings indicate that the capsid is stiff and brittle, whereas the membrane vesicle is ductile and soft. Might the hierarchical combination of these contrasting material properties be responsible for the high toughness of PRD1 particles? Quantifying accurately the contribution of the membrane vesicle to the overall toughness displayed by the PRD1 particle remains a challenge. To our knowledge, no PRD1 intact capsid particles can be biochemically or genetically produced that would allow the AFM probing, and yet the resulting information might be still limited for an exhaustive modelling of the yield behaviour of the full capsid as a brittle material. However, in nature, other macroscopic systems use similar arrangements to create tough materials. One such illustrative example is the coat protecting bird egg, in which a stiff, yet brittle, calcified shell is bonded to a soft and compliant proteinaceous inner skin43 (Fig. 6a).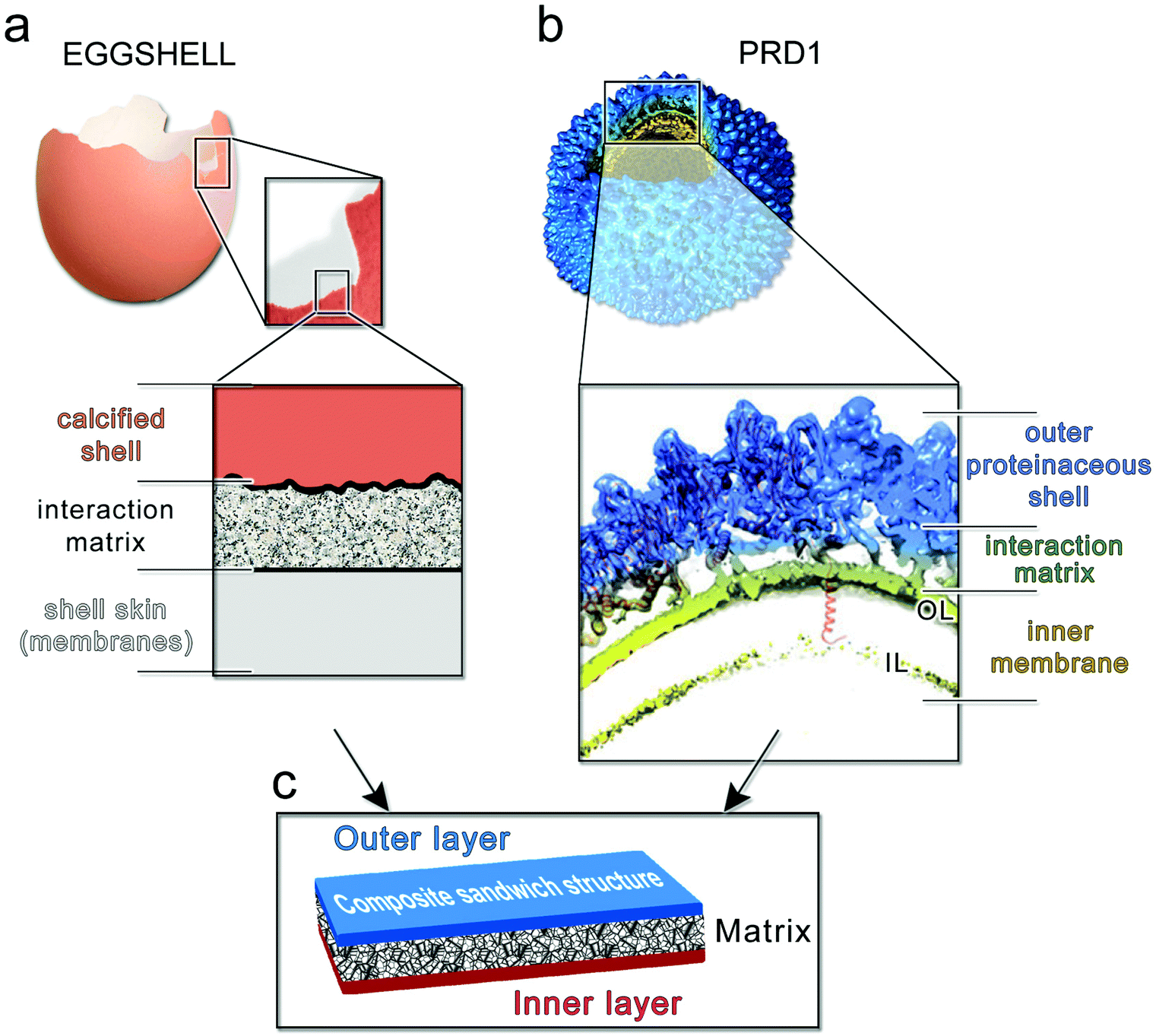 | ||
| Fig. 6 (a) Multilayered structure of an avian eggshell; (b) cryo-electron density of wt PRD1 with an octant removed displayed in Chimera45 to show (inset) the layered/composite structure of the virion with the proteinaceous shell in blue, the membrane-vesicle in yellow-lime (OL: outer leaflet; IL: inner leaflet) and with the horizontal black lines marking the distinct layers and the interacting matrix region between the capsid and the membrane. As cartoon representation in red fitted in density the P3 MCPs with N-terminal α-helices interacting with the OL and the P16 transmembrane protein crossing the membrane vesicle. The dsDNA has been removed from within the membrane vesicle for clarity; (c) schematic of a composite sandwich material to which (a) and (b) recapitulate. | ||
Altogether, these layers constitute a tough composite material that protects the progeny against mechanical stress while carrying out other essential functions. Analogously, the PRD1 capsid and vesicle are bonded to protect the integrity of the virion and its genome (Fig. 6b). Specifically, the connection between the capsid and membrane vesicle is made of polypeptide stretches: the capsid shell – composed of 240 copies of interacting trimers of the MCP P3 and glued at the icosahedral edges by the cementing protein P30 – anchors the membrane through several N-termini of MCPs P3; this connectivity is further augmented by the anchoring N-terminal transmembrane helix of protein P16 located at the base of the peripentonal MCPs P37 (inset in Fig. 6b).
Moreover, it is conceivable that during force loading onto the capsid, these connectors will act as nano-staples increasing the capability of the system of absorbing energy before mechanical failure (an additional energetic cost would be necessary to disrupt protein–membrane interactions44).
4. Conclusions
Composites have evolved in nature driven by selection for the efficient use of materials, adaptability, and multi-functionality. Viruses can be seen as composite biological entities where nucleic acids, proteins and lipids assemble to produce functional particles for infection. We propose that, as in a composite sandwich material (Fig. 6c), an interfacial protein/polypeptide matrix in PRD1 generates a tight connection that mechanically couples the capsid and the membrane (Fig. 6b). The flexibility of this matrix and possibly also the fluidity of the membrane facilitate the displacement of the two shells relative to each other, and thus assist in maintaining the capsomers in place whilst allowing for the correction of small scale defects in the (re-)assembly process.In summary, we here presented the nanomechanical characterization of a virus that features a membrane inside its capsid. The combination of a stiff, yet brittle, proteinaceous capsid with a soft proteolipidic vesicle in PRD1 facilitates multiple stages of the virus life cycle, including virus assembly, mechanical protection for the extracellular virion, and the vesicle-to-tube transformation during DNA ejection.13 From a broader perspective, it appears that the evolution of membrane-containing viruses has yielded, at the nanometer scale, composite properties comparable to those known for macroscopic natural materials, where the capsid and vesicle are bonded into a tough composite that protects the integrity of the virus and its genome.
Our findings provide both foundational quantitative information and inspiration that can encourage the engineering of tough nanoscale devices and particles capable of protecting fragile cargos.
Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank Sari Korhonen and Helin Veskiväli at the Instruct Centre for Virus and Macromolecular Complex Production (ICVIR; 2009–2017), University of Helsinki, for skilled technical assistance in virus production and purification (the current Biocomplex). We are grateful to the Abrescia Lab's members Eva S. Cunha, Diego Charro and Hani Y. Boshra for discussions throughout the study and Isaac Sántos-Pérez for help in virus production and electron microscopy imaging. David I. Stuart at the University of Oxford is also acknowledged for the permission to adapt original images for use in Fig. S1a.† We also thank MINECO for the Severo Ochoa Excellence Accreditation to the CIC bioGUNE (SEV-2016-0644) and the University of Helsinki and Academy of Finland (grant 1306833) for the support to Biomolecular Complex Purification (Biocomplex), part of Instruct-FI, used in this study. This research was supported by Bruker – Spain (to S. A. and N. G. A. A.), by the Academy of Finland (Academy Professor funding grants 283072 and 255342 to D. H. B.), by the European Research Council (starting grant FP7-ERC-2012-StG-306435 to R. P. R.), and by the Spanish Ministry of Economy and Competitiveness (MINECO/FEDER; MAT2014-54867-R to R. P. R.; MAT2015-63704-P to G. A. S.; BFU2015-64541-R to N. G. A. A.).Notes and references
- M. Hernando-Perez, S. Lambert, E. Nakatani-Webster, C. E. Catalano and P. J. de Pablo, Nat. Commun., 2014, 5, 4520, DOI:10.1038/ncomms5520.
- A. Llauró, B. Schwarz, R. Koliyatt, P. J. de Pablo and T. Douglas, ACS Nano, 2016, 10, 8465–8473 CrossRef PubMed.
- C. Carrasco, A. Carreira, I. A. Schaap, P. A. Serena, J. Gómez-Herrero, M. G. Mateu and P. J. de Pablo, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 13706–13711 CrossRef CAS PubMed.
- J. P. Michel, I. L. Ivanovska, M. M. Gibbons, W. S. Klug, C. M. Knobler, G. J. Wuite and C. F. Schmidt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 6184–6189 CrossRef CAS PubMed.
- C. Carrasco, A. Luque, M. Hernando-Pérez, R. Miranda, J. L. Carrascosa, P. A. Serena, M. de Ridder, A. Raman, J. Gómez-Herrero, I. A. Schaap, D. Reguera and P. J. de Pablo, Biophys. J., 2011, 100, 1100–1108 CrossRef CAS PubMed.
- E. K. Dimitriadis, F. Horkay, J. Maresca, B. Kachar and R. S. Chadwick, Biophys. J., 2002, 82, 2798–2810 CrossRef CAS PubMed.
- N. G. Abrescia, J. J. Cockburn, J. M. Grimes, G. C. Sutton, J. M. Diprose, S. J. Butcher, S. D. Fuller, C. San Martín, R. M. Burnett, D. I. Stuart and D. H. Bamford, Nature, 2004, 432, 68–74 CrossRef CAS PubMed.
- J. J. Cockburn, N. G. Abrescia, J. M. Grimes, G. C. Sutton, J. M. Diprose, J. M. Benevides, G. J. Thomas, J. K. Bamford, D. H. Bamford and D. I. Stuart, Nature, 2004, 432, 122–125 CrossRef CAS PubMed.
- J. T. Huiskonen, V. Manole and S. J. Butcher, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 6666–6671 CrossRef CAS PubMed.
- S. T. Jaatinen, S. J. Viitanen, D. H. Bamford and J. K. Bamford, J. Virol., 2004, 78, 9790–9797 CrossRef CAS PubMed.
- L. Xu, S. D. Benson, S. J. Butcher, D. H. Bamford and R. M. Burnett, Structure, 2003, 11, 309–322 CrossRef CAS PubMed.
- C. Hong, H. M. Oksanen, X. Liu, J. Jakana, D. H. Bamford and W. Chiu, PLoS Biol., 2014, 12, 1002024 Search PubMed.
- B. Peralta, D. Gil-Carton, D. Castaño-Díez, A. Bertin, C. Boulogne, H. M. Oksanen, D. H. Bamford and N. G. Abrescia, PLoS Biol., 2013, 9, 1001667 Search PubMed.
- R. H. Olsen, J. S. Siak and R. H. Gray, J. Virol., 1974, 14, 689–699 CAS.
- L. Mindich, D. H. Bamford, T. McGraw and G. Mackenzie, J. Virol., 1982, 44, 1021–1030 CAS.
- L. Mindich, J. Cohen and M. Weisburd, J. Bacteriol., 1976, 126, 177–182 CAS.
- J. K. Bamford and D. H. Bamford, Virology, 1990, 177, 445–451 CrossRef CAS PubMed.
- F. Winston, D. Botstein and J. H. Miller, J. Bacteriol., 1979, 137, 433–439 CAS.
- J. K. Bamford and D. H. Bamford, Virology, 1991, 181, 348–352 CrossRef CAS PubMed.
- C. Luo, S. Butcher and D. H. Bamford, Virology, 1993, 194, 564–569 CrossRef CAS PubMed.
- D. H. Bamford and L. Mindich, J. Virol., 1982, 44, 1031–1038 CAS.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS PubMed.
- V. M. Olkkonen and D. H. Bamford, Virology, 1989, 171, 229–238 CrossRef CAS PubMed.
- K. L. O'Connell and J. T. Stults, Electrophoresis, 1997, 18, 349–359 CrossRef PubMed.
- N. Crampton, W. A. Bonass, J. Kirkham and N. H. Thomson, Langmuir, 2005, 21, 7884–7891 CrossRef CAS PubMed.
- H. J. Butt and M. Jaschke, Nanotechnology, 1995, 6, 1 CrossRef.
- O. Koronova, J. Snijder, Y. Kholodov, K. A. Marx, G. J. Wuite, W. H. Roos and V. Barsegov, PLoS Comput. Biol., 2016, 12, e1004729 Search PubMed.
- C. San Martín, J. T. Huiskonen, J. K. Bamford, S. J. Butcher, S. D. Fuller, D. H. Bamford and R. M. Burnett, Nat. Struct. Mol. Biol., 2002, 9, 756–7630 Search PubMed.
- C. San Martın, R. M. Burnett, F. de Haas, R. Heinkel, T. Rutten, S. D. Fuller, S. J. Butcher and D. H. Bamford, Structure, 2001, 9, 917–930 CrossRef.
- I. Sántos-Pérez, H. M. Oksanen, D. H. Bamford, F. M. Goñi, D. Reguera and N. G. Abrescia, Biochim. Biophys. Acta, 2017, 1861, 664–672 CrossRef PubMed.
- I. L. Ivanovska, P. J. de Pablo, B. Ibarra, G. Sgalari, F. C. MacKintosh, J. L. Carrascosa, C. F. Schmidt and G. J. Wuite, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 7600–7605 CrossRef CAS PubMed.
- L. Pauchard and S. Rica, Philos. Mag. B, 1998, 78, 225–233 CrossRef CAS.
- O. Koronova, J. Snijder, M. Brasch, J. Cornelissen, R. I. Dima, K. A. Marx, G. J. Wuite, W. H. Roos and V. Barsegov, Biophys. J., 2013, 105, 1893–1903 CrossRef PubMed.
- O. Koronova, F. Maksudov, K. A. Marx and V. Barsegov, J. Phys.: Condens. Matter, 2018, 30, 044006 CrossRef PubMed.
- M. G. Mateu, Virus Res., 2012, 168, 1–22 CrossRef CAS PubMed.
- I. Ivanovska, G. Wuite, B. Jönsson and A. Evilevitch, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 9603–9608 CrossRef CAS PubMed.
- W. H. Roos, K. Radtke, E. Kniesmeijer, H. Geertsema, B. Sodeik and G. J. Wuite, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 9673–9678 CrossRef CAS PubMed.
- U. Sae-Ueng, T. Liu, C. E. Catalano, J. B. Huffman, F. L. Homa and A. Evilevitch, Nucleic Acids Res., 2014, 42, 9096–9107 CrossRef CAS PubMed.
- H. Liu, L. Jin, S. B. Koh, I. Atanasov, S. Schein, L. Wu and Z. H. Zhou, Science, 2010, 329, 1038–1043 CrossRef CAS PubMed.
- V. S. Reddy, S. K. Natchiar, P. L. Stewart and G. R. Nemerow, Science, 2010, 329, 1071–1075 CrossRef CAS PubMed.
- A. Ortega-Esteban, A. J. Pérez-Berná, R. Menéndez-Conejero, S. J. Flint, C. San Martín and P. J. de Pablo, Sci. Rep., 2013, 3, 1434 CrossRef CAS PubMed.
- V. S. Reddy and G. R. Nemerow, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 11715–11720 CrossRef CAS PubMed.
- J. Du, M. T. Hincke, M. Rose-Martel, C. Hennequet-Antier, A. Brionne, L. A. Cogburn, Y. Nys and J. Gautron, BMC Genomics, 2015, 16, 792, DOI:10.1186/s12864-015-2013-3.
- A. G. Lee, Biochim. Biophys. Acta, Biomembr., 2003, 1612, 1–40 CrossRef CAS.
- E. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt, E. C. Meng and T. E. Ferrin, J. Comput. Chem., 2004, 25, 1605–1612 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nr00196k |
| This journal is © The Royal Society of Chemistry 2018 |