
Bio-inspired robust non-iridescent structural color with self-adhesive amorphous colloidal particle arrays†
Panmiao
Liu
a,
Jialun
Chen
ab,
Zexi
Zhang
ab,
Zhuoying
Xie
 *ab,
Xin
Du
a and
Zhongze
Gu
*abc
*ab,
Xin
Du
a and
Zhongze
Gu
*abc
aState Key Laboratory of Bioelectronics, School of Biological Science and Medical Engineering, Southeast University, Nanjing, 210096, China. E-mail: zyxie@seu.edu.cn; gu@seu.edu.cn
bNational Demonstration Center for Experimental Biomedical Engineering Education, Southeast University, Nanjing, 210096, China
cSuzhou Key Laboratory of Environment and Biosafety Research Institute of Southeast University in Suzhou, Suzhou, 215123, China
First published on 29th December 2017
Abstract
Here we propose a new method for constructing highly color fast non-iridescent structural color materials by assembling self-adhesive poly-dopamine coated SiO2 nanoparticles (PDA@SiO2) for amorphous colloidal arrays through a “spraying” process. Simply by alkaline vapor treatment, the adhesive forces and fastness of the amorphous colloidal arrays were significantly improved. This was demonstrated by lap shear tests of tape tearing and cohesive failure as well as a series of fastness tests like sandpaper abrasion, finger wiping and ultrasonic cleaning. Besides, the strengthening fastness reaction could occur on different substrates, including glass, metals, polymers and paper, regardless of their chemical and physical properties. Moreover, the structural color of the PDA@SiO2 arrays was bright due to the broadband absorption of PDA, and was tunable according to the size, PDA content and arrangement of the PDA@SiO2 arrays.
Non-iridescent structural colored materials, in which the color arises from coherent-light scattering of amorphous colloidal nanoparticle arrays, have garnered ever-increasing scientific interest during the last decade.1–3 The amorphous structure of colloidal arrays endows the structural materials with angle-independent color like dyes or pigments, while it avoids the problem of chemical/photo bleaching which widely exists in dye/pigment coatings.4 The unique properties of the structural color materials can lead to various applications including building textiles, display boards, print media, colorimetric sensors, and optical devices.5–9 However, the amorphous structure gives rise to a strong incoherent-light scattering across the entire visible spectrum, which causes color whitening problems in human color vision. To address this challenge, black materials10–13 such as carbon nanoparticles/nanotubes and cuttlefish ink have been used to improve the visibility of non-iridescent structural color by doping. However, these materials do not contribute to the fastness of structural color arrays, which is a crucial factor for practical applications. Here we report the construction of highly color fast amorphous colloidal arrays by self-adhesive building blocks, poly-dopamine coated SiO2 nanoparticles (PDA@SiO2). Poly-dopamine (PDA) is a common synthetic melanin produced by the autoxidation of dopamine (DA). Owing to its catechol units, PDA can strongly adhere to most substrates, and this has been applied for the modification of various substrates, and the preparation of functional materials by simple chemistry.14 Besides, PDA can suppress the incoherent-light scattering of amorphous colloidal arrays due to its broad absorption of light.15 For example, Xiao et al.16 and Kohri et al.17,18 respectively utilized PDA and PDA@PS (polystyrene) nanoparticles to fabricate bright and high-visibility non-iridescent structural colors. However, all of these works only focused on improving color visibility and did not develop adhesion properties for structural color materials. In this study, we propose a robust non-iridescent structural color prepared by spraying PDA@SiO2 amorphous colloidal arrays and subsequently treating them with alkaline vapor. The adhesive forces both between particle and particle (P–P) and particle and substrate (P–S) were significantly improved, as demonstrated by lap shear tests of tape tearing and cohesive failure. The tests, including sandpaper abrasion, finger wiping and ultrasonic cleaning, further demonstrated the high color fastness of the PDA@SiO2 amorphous colloidal arrays. Besides, the PDA@SiO2 had higher photo/thermal stability than PDA@polymer and could be mass produced with facile adjustability of light absorption and particle size by changing the thickness of the PDA coating and silica core diameter, respectively. These features of PDA@SiO2 amorphous colloidal arrays promote more extensive practical applications for multiple non-iridescent structural color patterns.
The building blocks, monodispersed PDA@SiO2, were synthesized by polymerizing DA onto spherical silica nanoparticles in 1 mM (pH 7–7.5) Tris-buffer (Fig. 1a). Fig. 1b shows the transmission electron microscopy (TEM) image of the PDA@SiO2 particles with an average diameter of 295 nm. The strong bands at 1355 and 1579 cm−1 in the Raman spectra indicate that the PDA was coated on the SiO2 nanoparticles (Fig. S1b†). The thickness of the PDA shell could be adjusted by the initial concentration of DA. As shown in Fig. S1c,† the thickness of the PDA shell increased with the concentration of DA, which resulted in the increase of the PDA@SiO2 diameter (Fig. S1d†). The corresponding color of the PDA@SiO2 suspension gradually became dark with the increment of PDA shell thickness (Fig. S1e†). Non-iridescent structural color was fabricated by spraying an aqueous suspension of monodispersed PDA@SiO2 onto the substrate using a spraying–drying method (Fig. 1c and Movie S1†).19,20Fig. 1d shows bright green films with a thickness of 16 μm on glass, fabricated from monodispersed PDA@SiO2 particles with average diameters of 232 nm (Fig. S1a†). The color in ambient light was invariable when changing viewing angle. The scanning electron microscopy (SEM) images and two-dimensional Fourier analyses showed that the film was formed by amorphous PDA@SiO2 arrays without long-range order (Fig. S2e†). The bright non-iridescent structural color arose from the scattering of coherent-light and absorption of scattered incoherent-light by the amorphous PDA@SiO2 arrays. In the reflection spectra, a broad reflection peak emerged at 520 nm and its position had a low-dependence on view angle (Fig. S2a and b†). Furthermore, the PDA coated on the SiO2 nanoparticles absorbed incoherent-light scattering and improved the visibility of the non-iridescent structural color (Fig. S3†). As shown in Fig. 1e and f, the amorphous PDA@SiO2 arrays exhibited 80–93% absorptivity, while amorphous SiO2 arrays only had an absorptivity of 30–60%.
 | ||
| Fig. 1 (a) Schematic of the synthesis of PDA@SiO2 and (b) TEM image of PDA@SiO2 particles of 295 nm size (comprising 288 nm silica cores and 7 nm PDA shells). (c) Schematic of the fabrication of amorphous PDA@SiO2 arrays. (d) Array images of 232 nm PDA@SiO2 particles at various viewing angles. TEM transmission, absorption, and reflection of (e) PDA@SiO2 arrays and (f) SiO2 arrays with 16 μm thickness detected at normal incidence. | ||
The self-adhesion of the amorphous colloidal nanoparticle arrays was inspired by mussels. Mussels attach to the surface of substrates by the byssus which is distally tipped by a flared adhesive plaque.21,22 The plaque is assembled from proteins stockpiled in the foot of the mussel. The adhesive protein subtypes of Mytilus edulis foot protein, Mefp-3 and Mefp-5 are firstly squirted onto the surface of the substrate by the foot and localized near the interface between the adhesive pad and the substrate. Mefp-3 and Mefp-5 have the highest 3,4-dihydroxyphenylalanine (DOPA) contents of 20 and 30 mol% respectively,23,24 which are believed to contribute to adhesion (Fig. 2a).25 Because PDA has similar catechol units to DOPA, PDA has been confirmed to achieve strong adhesion with most substrates. Here we reinforced the PDA@SiO2 arrays and the adhesion to substrates via ammonia vapor treatment because the PDA shells are further oxidized and cross-linked in an alkaline atmosphere (Fig. 2b). To confirm the high color fastness of the arrays, we investigated the damage resistance performance of PDA@SiO2 arrays treated with ammonia vapor (PDA@SiO2-AM) as well as that of bare SiO2 arrays and untreated PDA@SiO2 arrays. Fig. 2c shows the samples on glass substrates before and after fastness tests, including sandpaper abrasion (Fig. S4†), finger wiping and ultrasonic cleaning. After 10 cycles of abrasion by sandpaper, the retained area was zero for SiO2 arrays, 45% for PDA@SiO2 arrays and 100% for PDA@SiO2-AM arrays. Fig. S5† shows the SEM images of these samples after abrasion. Only some traces along the abrasion direction were observed in the PDA@SiO2-AM arrays, but a little peeling and a lot of peeling were presented respectively in PDA@SiO2 arrays and SiO2 arrays. Besides, the color of the PDA@SiO2-AM arrays was unchanged after sandpaper abrasion, which was also revealed by the reflection spectra in Fig. S5.† Furthermore, during the finger-wipe test, 200 wipes removed SiO2 arrays completely, while it removed only 20% of the PDA@SiO2 arrays and almost none of the PDA@SiO2-AM arrays. During the ultrasonic cleaning test, which was conducted with a power of 90 W at a distance of 2 cm from the bottom of the ultrasonic instrument, the retained area was 10% for SiO2 arrays, 35% for PDA@SiO2 arrays and 65% for PDA@SiO2-AM arrays after 30 minutes. The curves of retained area for these tests are shown in Fig. 2d. The shedding speed of the SiO2 arrays was always faster than that of the PDA@SiO2 arrays, which was faster than that of the PDA@SiO2-AM arrays for all fastness tests. As a result, compared to the initial SiO2 arrays, the PDA@SiO2 arrays exhibited increased resistance to abrasion (45%), wiping (20%) and ultrasonic treatment (25%). However the PDA@SiO2-AM arrays presented remarkably improved resistance to abrasion (100%), wiping (100%) and ultrasonic treatment (55%). To further investigate the adhesion energy of the PDA@SiO2-AM arrays, the tensile stress derived from tape tearing was examined by lap shear testing, as shown in Fig. 2e. The measured lap shear force of the SiO2 arrays was close to 0.001 N (line a), while that for PDA@SiO2 arrays was 0.32 N (line b). The residue of a thin layer of particles on the tape shown in Fig. S6† revealed that the lap shear forces by tape tearing for SiO2 arrays and PDA@SiO2 arrays were primarily attributed to the P–P adhesion. However, the PDA@SiO2-AM arrays were almost completely peeled off from the substrates and stuck on the tape during the tape tearing process because of the high P–P adhesion. Thus the stable tension values of around 0.55 N for the PDA@SiO2-AM sample (line c) were mostly attributed to P–S forces. To measure the P–P force of PDA@SiO2-AM, we stuck the PDA@SiO2-AM arrays on the surface of tape. As expected, the arrays torn from the P–P interface presented an average of 0.88 N tension (line d), close to threefold that of the PDA@SiO2 sample. Further, the lap shear test of vertical uniaxial tension was used to measure cohesive failure tension using a 1 cm × 1 cm area tape cured onto the arrays, as illustrated in Fig. 2f. The displacement-force curves displayed that the PDA@SiO2 samples suffered cohesive failure at a tension of 7.2 N between the particles and tape surface, which was double that of the SiO2 samples (3.97 N). The samples of PDA@SiO2-AM fractured between the substrates and tape surface after 13 N tension but the cohesion between the arrays and tape surface was still tight (Fig. S7†). This indicated that the cohesive failure tension of PDA@SiO2-AM between particles and particles exceeded 13 N, which was approximately fourfold higher than that for the SiO2 samples. Therefore, the mechanical strength and adhesion were highly improved by the PDA shells and the binding was further reinforced by alkaline treatment.26
 | ||
| Fig. 2 (a) Photograph of a mussel attached to a stone surface and the illustrated mechanism of mussel adhesion.22 (b) Schematic of PDA@SiO2 arrays treated by ammonia vapor. (c) Photograph of SiO2, PDA@SiO2 and PDA@SiO2-AM samples on glass substrates before and after robustness tests of sandpaper abrasion, finger-wiping and ultrasonic tape-exposure. (d) Retained area ratios during the tests of sandpaper abrasion, finger-wiping and ultrasonic tape-exposure for SiO2 (square), PDA@SiO2 (circle) and PDA@SiO2-AM (triangle) arrays on glass substrates. (e) Schematic of the lap shear test by tape tearing and the obtained displacement-force curves for SiO2 arrays (P–P force; line a), PDA@SiO2 arrays (P–P force; line b), PDA@SiO2-AM arrays (P–S force; line c) and PDA@SiO2-AM arrays (P–P force; line d). (f) Schematic of the lap shear test of tensile cohesive failure and the obtained displacement-force curves for SiO2 arrays, PDA@SiO2 arrays and PDA@SiO2-AM arrays. The dashed areas show the cohesive failure positions on the curves. | ||
In order to investigate the adhesion mechanism after ammonia vapor treatment on a molecular scale, attenuated total reflection-infrared (ATR-IR) spectroscopy and Raman spectroscopy were conducted for the SiO2 arrays, PDA@SiO2 arrays and PDA@SiO2-AM arrays. Fig. 3a reveals that the absorption of aromatic ring vibration (νC![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C) peaks from the PDA structure are located at 1541 cm−1 in the PDA@SiO2 spectrum, while at 1458 cm−1 for the PDA@SiO2-AM arrays.27 The significant displacement of this νCC peak to lower wavenumber for the PDA@SiO2 arrays can probably be attributed to the crosslinking of aromatic rings after alkaline treatment, which produces a conjugate effect and decreases the absorption energy and absorption frequency (Fig. 3c). The Raman spectra further confirmed that the PDA was distributed in the PDA@SiO2 and PDA@SiO2-AM arrays from the PDA characteristic bands appearing in the range of 1379 and 1539 cm−1, as shown in Fig. S9.†28 Among them, the aromatic ring characteristic bands of PDA@SiO2 also exhibited a lower wavenumber shift from 1410 cm−1 to 1379 cm−1 after ammonia vapor treatment because of the conjugate effect (Fig. 3b). Moreover, the decreased broad absorbance of νOH in the spectral region 3600–2800 cm−1 together with the increased H-bond absorbance peak at 2980 cm−1 (Fig. 3d) demonstrated that more H-bonds composed from surface bound H-bonds and intramolecular H-bonds with phenolic hydroxyl and phenol carbonyl oxygens were generated for the PDA@SiO2 arrays after alkaline treatment (Fig. 3e).29 Besides, due to the involvement of PDA, the intensity of the SiO2 characteristic peaks gradually decreased from SiO2 arrays to PDA@SiO2 arrays and PDA@SiO2-AM arrays (Fig. S8†). Hence, the decrement from the PDA@SiO2 arrays to the PDA@SiO2-AM arrays also provided evidence for the greater self-polymerization degree of the PDA@SiO2 arrays after ammonia vapor treatment. These results demonstrated that the ammonia vapor provided a humid alkaline environment which accelerated the oxidative self-polymerization among PDA shells and promoted more covalent and non-covalent bonding.30,31 On the other hand, it also contributed to the binding between PDA@SiO2 particles and substrates from the formation of more hydrogen bonds between the OH groups of the catechol and the O atoms in the glass.32
C) peaks from the PDA structure are located at 1541 cm−1 in the PDA@SiO2 spectrum, while at 1458 cm−1 for the PDA@SiO2-AM arrays.27 The significant displacement of this νCC peak to lower wavenumber for the PDA@SiO2 arrays can probably be attributed to the crosslinking of aromatic rings after alkaline treatment, which produces a conjugate effect and decreases the absorption energy and absorption frequency (Fig. 3c). The Raman spectra further confirmed that the PDA was distributed in the PDA@SiO2 and PDA@SiO2-AM arrays from the PDA characteristic bands appearing in the range of 1379 and 1539 cm−1, as shown in Fig. S9.†28 Among them, the aromatic ring characteristic bands of PDA@SiO2 also exhibited a lower wavenumber shift from 1410 cm−1 to 1379 cm−1 after ammonia vapor treatment because of the conjugate effect (Fig. 3b). Moreover, the decreased broad absorbance of νOH in the spectral region 3600–2800 cm−1 together with the increased H-bond absorbance peak at 2980 cm−1 (Fig. 3d) demonstrated that more H-bonds composed from surface bound H-bonds and intramolecular H-bonds with phenolic hydroxyl and phenol carbonyl oxygens were generated for the PDA@SiO2 arrays after alkaline treatment (Fig. 3e).29 Besides, due to the involvement of PDA, the intensity of the SiO2 characteristic peaks gradually decreased from SiO2 arrays to PDA@SiO2 arrays and PDA@SiO2-AM arrays (Fig. S8†). Hence, the decrement from the PDA@SiO2 arrays to the PDA@SiO2-AM arrays also provided evidence for the greater self-polymerization degree of the PDA@SiO2 arrays after ammonia vapor treatment. These results demonstrated that the ammonia vapor provided a humid alkaline environment which accelerated the oxidative self-polymerization among PDA shells and promoted more covalent and non-covalent bonding.30,31 On the other hand, it also contributed to the binding between PDA@SiO2 particles and substrates from the formation of more hydrogen bonds between the OH groups of the catechol and the O atoms in the glass.32
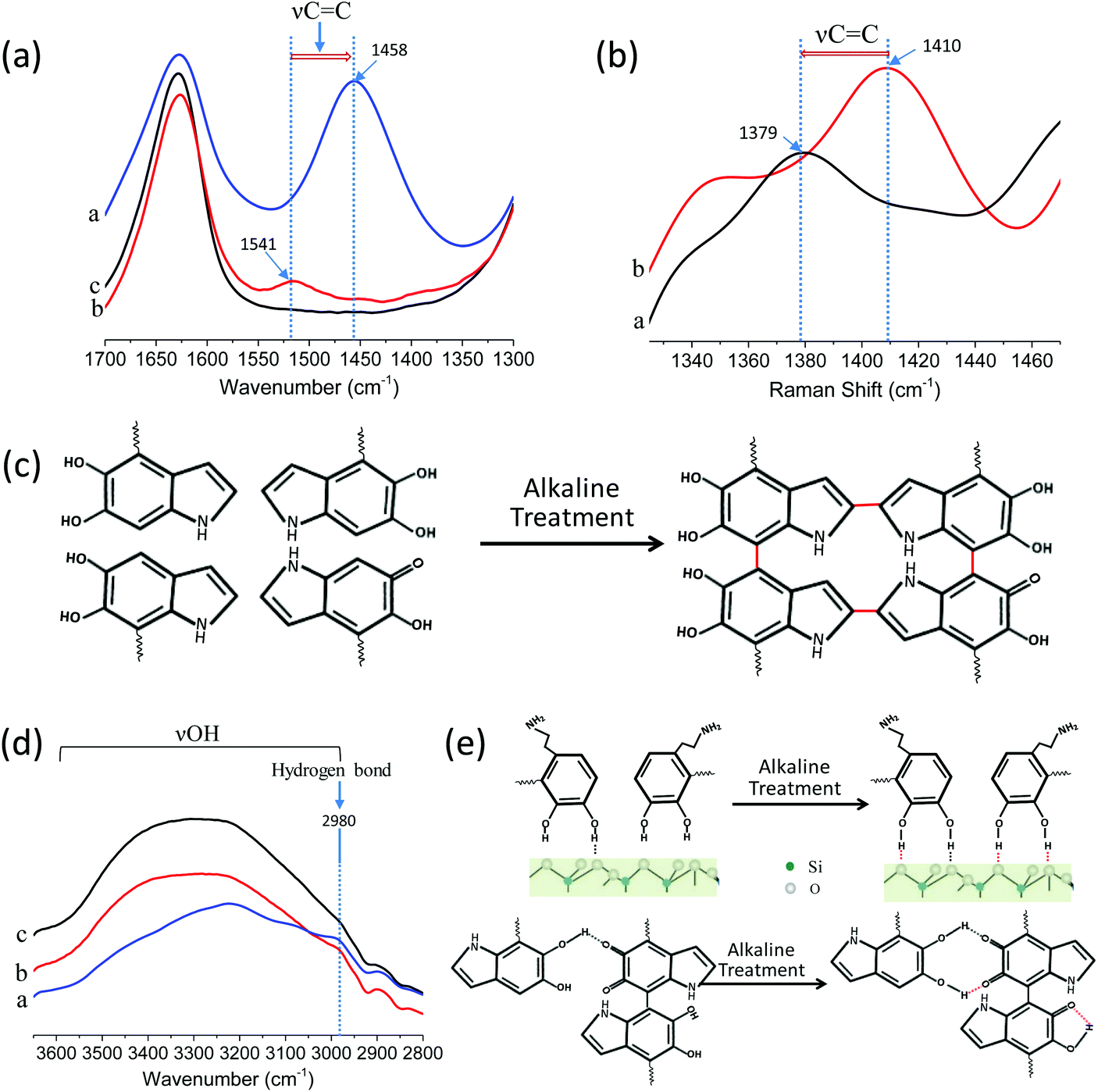 | ||
| Fig. 3 (a) ATR-IR spectrum of PDA@SiO2-AM (line a), PDA@SiO2 (line b) and SiO2 (line c) arrays in 1700–1300 cm−1 region. The aromatic ring (νCC) of PDA@SiO2-AM located at 1458 cm−1 and PDA@SiO2 arrays for 1541 cm−1. (b) Raman spectra of PDA@SiO2-AM (line a) and PDA@SiO2 (line b) arrays range from 1335 cm−1 to 1470 cm−1. The characterization spectra of aromatic ring (νCC) for PDA@SiO2-AM is 1379 cm−1 and for PDA@SiO2 arrays is 1410 cm−1. (c) Schematic of cross-linking for PDA@SiO2 after treatment of ammonium vapor and the red line is the generated covalent bond among PDA moleculars. (d) ATR-IR spectrum of PDA@SiO2-AM (line a), PDA@SiO2 (line b) and SiO2 (line c) arrays in 3600–2800 cm−1 region and hydrogen bond located at 2980 cm−1. (e) Schematic of H-bond generation for PDA@SiO2 after treatment of ammonium vapor and the red dashed line is the generated H-bond between the PDA moleculars or PDA moleculars-substrates. | ||
To better understand the performance of the self-adhered non-iridescent structural color material, we investigated the color fastness of the PDA@SiO2-AM arrays prepared on various substrates, including plastic, metal and paper. As shown in Movies S4–S7,† the fastness of the colloidal nanoparticle arrays on poly(methylmethacrylate) (PMMA), aluminum (Al) foil and paper, respectively, were examined by sandpaper abrasion. As expected, the PDA@SiO2-AM arrays exhibited remarkably increased fastness on these substrates, as presented in Fig. 4a. The changing curves of retained area ratio on the PMMA substrate showed that for the SiO2 arrays and PDA@SiO2 arrays this decreased to 44% and 80% original area, respectively, at a constant speed, while the PDA@SiO2-AM arrays remained constant during the whole abrasion process (Fig. S10a†). For the substrate of Al foil, the SiO2 arrays were removed completely after only five abrasion cycles and the PDA@SiO2 arrays and PDA@SiO2-AM arrays were reduced to 26% and 70% after 10 abrasion cycles, respectively (Fig. S10b†). However, the situation was different on paper substrates, on which the nanoparticle arrays were protected by their porous structures, resulting in all samples on paper dropping off only a small amount after 10 cycles of sandpaper abrasion (Fig. S10c†). Furthermore, finger wipe tests showed that the PDA@SiO2-AM patterns on these substrates did not display any damage after 200 wipes (Fig. S12 and Movie S2†). These results indicated that the PDA@SiO2-AM arrays showed a remarkable resistance to abrasion on various substrates due to the self-adhesive property of PDA@SiO2. PDA@SiO2 relied on different binding mechanisms to adhere to these substrates. On inorganic surfaces, PDA@SiO2 formed coordination bonds, whereas on organic surfaces, oxidized PDA@SiO2 was capable of adhering via covalent bond formation.33,34 Thus bonds between PDA@SiO2 and organic substrates were stronger than those of PDA@SiO2-inorganic substrates. As demonstrated in Fig. 2d, Fig. S10a and S10b,† the shedding speeds of PDA@SiO2 arrays on glass and Al substrates were faster than those on the PMMA substrate. Furthermore, on inorganic surfaces PDA@SiO2 arrays formed H-bonding and/or multivalent chelating interactions, which strongly depend on the surface wettability.33 Therefore Al foil (with a water contact angle of 96.5 ± 0.2°, Fig. S11b†) presented a faster shedding speed than glass substrates (with a water contact angle of 36.3 ± 0.1°, Fig. S11a†) for the PDA@SiO2-AM arrays, as shown in Fig. 2d and Fig. S10b.† Due to the nanoparticle protective effect of a rough porous structure,35 PDA@SiO2-AM arrays on wettable porous paper presented inconspicuous shedding after 10 cycles of abrasion.
 | ||
| Fig. 4 (a) SiO2, PDA@SiO2 and PDA@SiO2-AM arrays after 10 cycles treatment with sandpaper abrasion on PMMA, Al foil and paper substrates, respectively. The dashed areas show the original areas of the samples. (b) PDA@SiO2 arrays on glass treated for different times with 20 wt% ammonia vapor. (c) PDA@SiO2 arrays with particle sizes of 172, 193, 204, 215, 232 and 252 nm (top line from left to right) and 264, 273, 286, 291, 309 and 323 nm (bottom line from left to right). (d) PDA@SiO2 arrays with original DA concentrations of 0, 1, 5, 10, 20 mM (formed with 275 nm silica core particles and 2 nm, 7 nm, 12 nm, 20 nm coatings). (e) The multi-color patterns of PDA@SiO2-AM arrays on glass, PMMA, Al foil and paper. | ||
Since the alkaline treatment caused further polymerization of PDA, its influence on color was investigated. As shown in Fig. 4b, the appearance of the PDA@SiO2 pattern as well as the color analysis (Fig. S13†) was unchanged with the increase of ammonia vapor treatment time. Since the color multiplicity is necessary for structural color patterns, multiple structural colors were achieved by changing the size of the silica core, PDA content and arrangement of PDA@SiO2. Firstly, the non-iridescent structural colors with hue ranging from blue to purple-red were obtained by varying the size of PDA@SiO2, as shown in Fig. 4c. The hue of such structural color was consistent with the dominant reflection wavelength in the reflection spectrum, which linearly increased with the diameter of PDA@SiO2 (Fig. S14a and b†). Secondly, the luminance of the color was tuned by the content of PDA in PDA@SiO2 (Fig. 4d). With the increase of PDA content, the film sprayed from 295 nm PDA@SiO2 particles gradually revealed a red color from coherent-light scattering accompanied with the decrease of reflectance in the whole visible region (Fig. S15a and b†). Since the content of PDA was related to the DA concentration during the growth of the PDA shell on SiO2, the luminance declined with the increase of DA concentration (Fig. S15c†). Finally, the saturation of color was slightly tuned by the arrangement of PDA@SiO2. A higher concentration of colloid suspension tended to form a more ordered arrangement, which improved the saturation (Fig. S16†). However the thickness of the sprayed coating was negligible to the non-iridescent structural color when it appeared a certain color (Fig. S17†). Fig. 4e exhibits multi-color PDA@SiO2-AM patterns of the Chinese-style painting, Peking Opera, on PMMA, glass, paper and Al foil. The vivid color patterns illustrate that the high fastness and bright non-iridescent structural color presents a good alternative for future color painting nanomaterials.
Conclusions
In conclusion, we prepared a robust non-iridescent structural color by spraying PDA@SiO2 amorphous colloidal arrays. By simple treatment with alkaline vapor, the colloidal arrays presented remarkably higher fastness than bare SiO2 and PDA@SiO2 arrays without treatment, as demonstrated by lap shear tests of tape tearing and cohesive failure, and fastness tests of sandpaper abrasion, finger wiping and ultrasonic cleaning. Besides, the strengthening fastness reaction for the PDA@SiO2 arrays was compatible with various substrates, including glass, plastic, metal and paper. Moreover, the structural color of the PDA@SiO2 arrays was bright due to the broadband absorption of PDA, and was tunable according to the size, PDA content and arrangement of PDA@SiO2. This study provided a facile and universal approach for constructing fast non-iridescent structural color and has practical applications in optical display, painting and cosmetics. Due to the good biocompatibility of both SiO2 and PDA, this kind of structural color also has potential applications for biomedical materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by the research Fund of the National Key Research and Development Program of China (No. 2017YFA0205700), the National Natural Science Foundation (No. 21327902, 21635001, 51628102), the Natural Science Foundation of Jiangsu Province (Grant BK20150024), and the Fundamental Research Funds for the Central Universities.Notes and references
- J. D. Forster, H. Noh, S. F. Liew, V. Saranathan, C. F. Schreck, L. Yang, J.-G. Park, R. O. Prum, S. G. J. Mochrie, C. S. O'Hern, H. Cao and E. R. Dufresne, Adv. Mater., 2010, 22, 2939 CrossRef PubMed.
- S. Magkiriadou, J.-G. Park, Y.-S. Kim and V. N. Manoharan, Opt. Mater. Express, 2012, 2, 1343 CrossRef CAS.
- J.-G. Park, S.-H. Kim, S. Magkiriadou, T. M. Choi, Y.-S. Kim and V. N. Manoharan, Angew. Chem., Int. Ed., 2014, 53, 2899 CrossRef CAS PubMed.
- Y. Takeoka, M. Honda, T. Seki, M. Ishii and H. Nakamura, ACS Appl. Mater. Interfaces, 2009, 1, 982 Search PubMed.
- M. Kuang, J. Wan, B. Bao, F. Li, L. Wang, L. Jiang and Y. Song, Adv. Opt. Mater., 2014, 2, 34–38 CrossRef.
- D. Ge, E. Lee, L. Yang, Y. Cho, M. Li, D. S. Gianola and S. Yang, Adv. Mater., 2015, 27, 2489–2495 CrossRef CAS PubMed.
- J. Hou, H. Zhang, B. Su, M. Li, Q. Yang, L. Jiang and Y. Song, Chem. - Asian J., 2014, 53, 5791–5795 CAS.
- Y. Ohtsuka, T. Seki and Y. Takeoka, Angew. Chem., Int. Ed., 2015, 127, 15588 CrossRef.
- M. Xiao, Y. Li, J. Zhao, Z. Wang, M. Gao, N. C. Gianneschi, A. Dhinojwala and M. D. Shawkey, Chem. Mater., 2016, 28, 5516 CrossRef CAS.
- Y. Takeoka, S. Yoshioka, M. Teshima, A. Takano, M. Harun-Ur-Rashid and T. Seki, Sci. Rep., 2013, 3, 2371 CrossRef PubMed.
- K. Katagiri, Y. Tanaka, K. Uemura, K. Inumaru, T. Seki and Y. Takeoka, NPG Asia Mater., 2017, 9, e355 CrossRef CAS.
- Y. Zhang, B. Dong, A. Chen, X. Liu, L. Shi and J. Zi, Adv. Mater., 2015, 27, 4719 CrossRef CAS PubMed.
- F. Li, B. Tang, J. Xiu and S. Zhang, Molecules, 2016, 21, 547 CrossRef PubMed.
- H. Lee, S. M. Dellatore, W. M. Miller and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- M. d'Ischia, K. Wakamatsu, A. Napolitano, S. Briganti, J. Garcia-Borron, D. Kovacs, P. Meredith, A. Pezzella, M. Picardo, T. Sarna, J. D. Simon and S. Ito, Pigm. Cell Melanoma Res., 2013, 26, 616–633 CrossRef PubMed.
- M. Xiao, Y. Li, M. C. Allen, D. D. Deheyn, X. Yue, J. Zhao, N. C. Gianneschi, M. D. Shawkey and A. Dhinojwala, ACS Nano, 2015, 9, 5454 CrossRef CAS PubMed.
- M. Kohri, Y. Nannichi, T. Taniguchi and K. Kishikawa, J. Mater. Chem. C, 2015, 3, 720 RSC.
- A. Kawamura, M. Kohri, G. Morimoto, Y. Nannichi, T. Taniguchi and K. Kishikawa, Sci. Rep., 2016, 6, 33984 CrossRef CAS PubMed.
- Y. Takeoka, S. Yoshioka, A. Takano, S. Arai, K. Nueangnoraj, H. Nishihara, M. Teshima, Y. Ohtsuka and T. Seki, Angew. Chem., Int. Ed., 2013, 52, 7261 CrossRef CAS PubMed.
- L. Cui, Y. Zhang, J. Wang, Y. Ren, Y. Song and L. Jiang, Macromol. Rapid Commun., 2009, 30, 598 CrossRef CAS PubMed.
- J. H. Waite, Int. J. Adhes. Adhes., 1987, 7, 9 CrossRef CAS.
- J. H. Waite and A. Moule, Integr. Comp. Biol., 2002, 42, 1172 CrossRef CAS PubMed.
- B. P. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99 CrossRef CAS PubMed.
- V. V. Papov, T. V. Diamond, K. Biemann and J. H. Waite, J. Biol. Chem., 1995, 270, 20183 CrossRef CAS PubMed.
- E. W. Danner, Y. Kan, M. U. Hammer, J. N. Israelachvili, J. H. Waite and X. Qin, Biochemistry, 2012, 51, 6511 CrossRef CAS PubMed.
- H. Lee, B. P. Lee and P. B. Messersmith, Nature, 2007, 448, 338 CrossRef CAS PubMed.
- D. R. Dreyer, D. J. Miller, B. D. Freeman, D. R. Paul and C. W. Bielawski, Langmuir, 2012, 28, 6428 CrossRef CAS PubMed.
- D. Yan, P. Xu, Q. Xiang, H. Mou, J. Xu, W. Wen, X. Li and Y. Zhang, J. Mater. Chem. A, 2016, 4, 3487 CAS.
- W. Wei, L. Petrone, Y. Tan, H. Cai, J. N. Israelachvili, A. Miserez and J. Herbert Waite, Adv. Funct. Mater., 2016, 26, 3496 CrossRef CAS PubMed.
- S. Hong, Y. S. Na, S. Choi, I. T. Song, W. Y. Kim and H. Lee, Adv. Funct. Mater., 2012, 22, 4711 CrossRef CAS.
- X. L. Wang, Q. Ye, J. X. Liu, X. J. Liu and F. Zhou, J. Colloid Interface Sci., 2010, 351, 261 CrossRef CAS PubMed.
- J. L. Dalsin, B.-H. Hu, B. P. Lee and P. B. Messersmith, J. Am. Chem. Soc., 2003, 125, 4253 CrossRef CAS PubMed.
- H. Lee, N. F. Scherer and P. B. Messersmith, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 12999 CrossRef CAS PubMed.
- A. M. Baty, P. K. Leavitt, C. A. Siedlecki, B. J. Tyler, P. A. Suci, R. E. Marchant and G. G. Geesey, Langmuir, 1997, 13, 5702 CrossRef CAS.
- Y. Lu, S. Sathasivam, J. Song, C. R. Crick, C. J. Carmalt and I. P. Parkin, Science, 2015, 347, 1132 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr08056e |
| This journal is © The Royal Society of Chemistry 2018 |