
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Probing the geometry of copper and silver adatoms on magnetite: quantitative experiment versus theory†
Matthias
Meier
ab,
Zdeněk
Jakub
b,
Jan
Balajka
b,
Jan
Hulva
b,
Roland
Bliem
b,
Pardeep K.
Thakur
 c,
Tien-Lin
Lee
c,
Cesare
Franchini
a,
Michael
Schmid
b,
Ulrike
Diebold
b,
Francesco
Allegretti
d,
David A.
Duncan
*c and
Gareth S.
Parkinson
b
c,
Tien-Lin
Lee
c,
Cesare
Franchini
a,
Michael
Schmid
b,
Ulrike
Diebold
b,
Francesco
Allegretti
d,
David A.
Duncan
*c and
Gareth S.
Parkinson
b
aUniversity of Vienna, Faculty of Physics and Center for Computational Materials Science, 1090 Vienna, Austria
bInstitute of Applied Physics, TU Wien, 1040 Vienna, Austria
cDiamond Light Source, Harwell Science and Innovation Campus, Didcot, OX11 0QX UK. E-mail: david.duncan@diamond.ac.uk
dPhysics Department E20, Technical University of Munich, 85748 Garching, Germany
First published on 15th January 2018
Abstract
Accurately modelling the structure of a catalyst is a fundamental prerequisite for correctly predicting reaction pathways, but a lack of clear experimental benchmarks makes it difficult to determine the optimal theoretical approach. Here, we utilize the normal incidence X-ray standing wave (NIXSW) technique to precisely determine the three dimensional geometry of Ag1 and Cu1 adatoms on Fe3O4(001). Both adatoms occupy bulk-continuation cation sites, but with a markedly different height above the surface (0.43 ± 0.03 Å (Cu1) and 0.96 ± 0.03 Å (Ag1)). HSE-based calculations accurately predict the experimental geometry, but the more common PBE + U and PBEsol + U approaches perform poorly.
Density functional theory (DFT) has become an indispensable tool in modern catalysis research, allowing us to understand long-observed trends in reactivity and unravel complex reaction mechanisms.1 Rapid advances in computational power have fueled efforts to screen, and even predict catalysts from first principles,2–4 but real predictive power requires adsorption energies and reaction barriers to be quantitatively correct. This must begin with an accurate description of the catalyst, but there is little in the way of solid experimental benchmarks5 to test the different exchange–correlation functionals,6–11 van der Waals corrections, both (semi-) empirical12–14 and non-empirical,15,16 and methods beyond DFT.17–23
The emerging field of single-atom catalysis (SAC)24–30 is a case in point. Although, there are reports of highly-active single-atom catalysts,31–37 the field remains controversial38 because such systems are difficult to characterize experimentally. Moreover, the catalytic mechanism is often proposed on the basis of theoretical calculations,31,36,39–43 which utilize an idealized catalyst support with metal adatoms adsorbed at high-symmetry sites on a low index facet. Thus, the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 equivalence of experimental and theoretical data is difficult to establish.
:1 equivalence of experimental and theoretical data is difficult to establish.
FeOx nanocrystallites have been observed as a support material that can anchor single adatoms, with both Pt and Ir adatoms exhibiting catalytic activity.31,33 These two studies utilized the same coprecipitation method to generate the nanocrystallites and, in the work of Lin et al.,33 were observed by X-ray diffraction to be primarily magnetite crystallites. Recently we discovered that the (001) surface of a magnetite single crystal can stabilize ordered arrays of metal adatoms (e.g. Au,44 Pd,45 and Pt46).47 These adatoms were found to be homogenously distributed up to a comparatively high coverage and with high thermal stability, and is therefore a promising model system to provide insight into single atom catalysts supported on FeOx nanocrystallites. It is this remarkable density, stability, and homogeneity of adatom arrays that offers the opportunity to perform a precise structural determination, and test the ability of DFT-based calculations to accurately model these dispersed lone adatoms. To that end, we report a normal incidence X-ray standing waves (NIXSW48) study of two members of this family: Ag1 and Cu1 adatoms on Fe3O4(001). These adatoms were chosen for their nobility, in order to avoid undesired adsorption of the residual gases found in ultra-high vacuum, and thus are used as a comparatively simple benchmark with which to test the performance of theoretical calculations. Both metals were determined to adsorb in a surface tetrahedral cation site, with significantly different adsorption heights (0.96 ± 0.03 Å for Ag and 0.43 ± 0.03 Å for Cu). DFT calculations using the Heyd–Scuseria–Ernzerhof (HSE) functional23 reproduce the geometry well, but the more common Perdew–Burke–Ernzerhof + U (PBE + U) and PBEsol + U approaches perform poorly. Although improved structural agreement can be achieved by constraining the lattice parameter to the experimental value, the failure to meet this experimental benchmark raises concern over this widely used functional in the field of adatoms on metal oxide surfaces.
Full experimental details can be found in the ESI.† The as-prepared surface exhibits a sharp (√2 × √2)R45° low energy electron diffraction (LEED) pattern (not shown), and scanning tunneling microscopy (STM) images reveal rows of surface Feoct atoms running in the [110] directions (see Fig. 1A and B). Here, Feoct refers to atoms with octahedral coordination to oxygen in bulk Fe3O4. The surface Feoct rows exhibit a characteristic distortion due to an ordered array of subsurface cation vacancies and interstitials,47 the so-called subsurface cation vacancy (SCV) reconstruction.
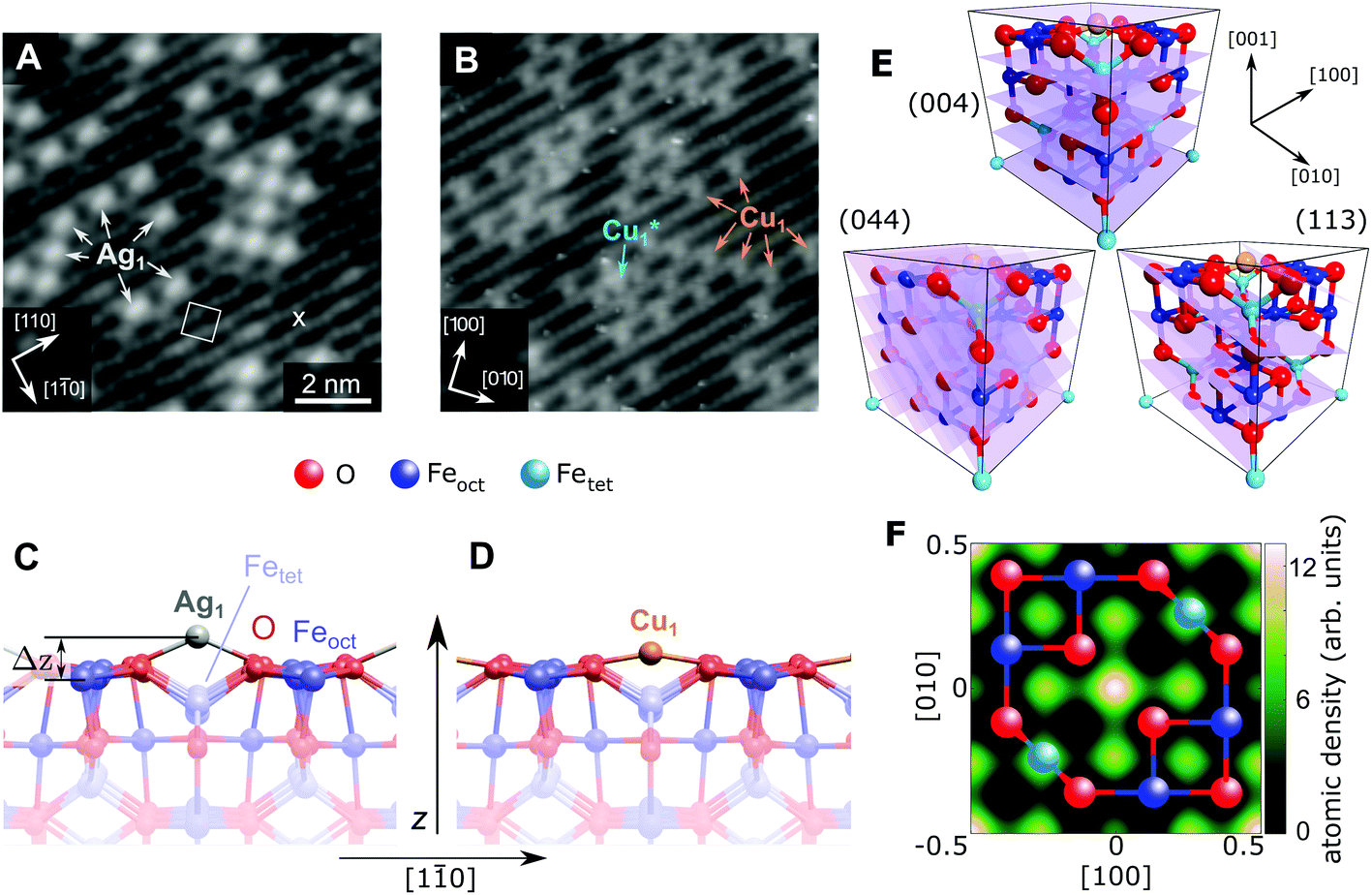 | ||
| Fig. 1 STM image of (A) 0.27 ML Ag and (B) 0.41 ML Cu on the Fe3O4(001) surface (Vsample = +1.2 V/+2.0 V, respectively, Itunnel = 0.3 nA). All Ag1 adatoms occupy the “narrow” site marked by an × in the figure and the (√2 × √2)R45° unit cell is indicated by a white square. The stable majority site (Cu1) and metastable minority site (Cu1*) are labelled. (C, D) Side view of the optimum Ag/Fe3O4(001) and Cu/Fe3O4(001) structures determined by HSE, with a height above the relaxed Feoct surface atoms (Δz) of 1.12 Å and 0.59 Å, respectively. (E) Unit cell of Fe3O4(001) with the experimentally determined site of the Cu1 adatom. The (004), (113) and (044) planes utilized in the NIXSW experiments are indicated. (F) A 2D atomic density map of the Cu adatom obtained from the NIXSW measurements, as described in the ESI,† overlaid with a ball-and-stick model representing an idealized FeoctO2 bulk termination. The adsorption site can be clearly identified at the center and corners of the map, corresponding to an oxygen bridge site. | ||
Fig. 1 shows STM images of the surface following the deposition of 0.27 ML Ag (a) and 0.41 ML Cu (b). Isolated adatoms appear as bright protrusions between the surface Feoct rows, with apparent heights relative to the surface Feoct rows of 1.1 ± 0.3 Å and 0.6 ± 0.2 Å for Ag1 and Cu1 respectively. As observed previously,49 adatom adsorption occurs almost exclusively at the site marked by an × in Fig. 1A, i.e., where the separation of the Feoct rows appears narrowest with a sample bias of 1–1.5 V.47 Approximately 10% of the Cu adatoms occupy an alternative adsorption site, Cu1*, after room temperature deposition. This site is metastable, and can be converted into regular Cu1 by annealing at 550 K. For Ag, small clusters begin to form at a coverage of ≈0.5 ML.51 This aggregation is irreversible, but does not affect the position of the majority adatom species measured by the NIXSW method.
Fig. 1C and D show the minimum-energy configuration for Cu1 and Ag1 adatoms on the Fe3O4(001) surface, as determined by HSE-based calculations. The favoured adsorption site for both adatoms is twofold coordinated to surface oxygen atoms between the surface Feoct rows. Specifically, the adatoms bind to the two oxygen atoms without a subsurface Fetet (i.e. tetrahedrally coordinated Fe) neighbour, where the next Fetet atom would reside if the bulk structure were continued outward. The calculations predict that the Ag1 adatom protrudes further from the surface than the Cu1 (Δz = 1.12 Å and Δz = 0.59 Å, respectively), which corresponds remarkably well to the STM apparent heights in the bias range Vsample = 1–1.5 V.
To quantitatively benchmark the adatom geometry, we performed NIXSW experiments48,49 at beamline I09, Diamond Light Source. NIXSW exploits the standing wavefield generated by the interference between incident and reflected photon beams at a specific Bragg condition of the substrate. As the incident photon energy is varied near such a condition, the standing wavefield moves relative to the Bragg planes. Since the standing wave also extends beyond the substrate surface, the photoemission intensity from an adatom core level (Ag 3d or Cu 2p, in this case), excited by the X-rays, varies with the photon energy. Maximum (minimum) intensity is observed when the antinode (node) of the standing wavefield coincides with an adatom. An analysis of the NIXSW profile yields two parameters: the coherent position (Phkl) and coherent fraction (fhkl).48,49 These are, colloquially, the mean position and level of order of the adatom between the Bragg planes. In total we exploited the (004), (113), and (044) reflections (schematically shown in Fig. 1E), occurring at hν = 2960, 2450 and 4180 eV respectively. Note that at exact normal incidence, the wavelength of the light will be twice the spacing between the planes. Atomic density maps (e.g.Fig. 1F), reconstructed from three reflections using a Fourier expansion described in the ESI,† directly (and unambigiously) identify the three dimensional adsorption site as the surface oxygen bridge site, confirming the site inferred from the STM/calculated data. Here we focus primarily on the (004) data, which specifically probes the vertical positions of the adatoms. Details of the complete NIXSW analysis are included in the ESI.†
Fig. 2 shows the fitted (004) NIXSW profiles following the deposition of 0.4 ML of Cu and Ag at room temperature. The photon energy scale is plotted relative to the Bragg energy, defined by the X-ray reflectivity curve shown in the lower curve. Clearly, the maximum photoemission intensity occurs at significantly different energies for the Ag 3d5/2 and Cu 2p3/2 data, indicating a significant difference in their position with respect to the Fe3O4(004) planes. Specifically, P004 values of 0.71 ± 0.02 (Cu1) and 0.96 ± 0.01 (Ag1) were obtained, which corresponds to heights of HCu = 0.43 ± 0.03 Å (Cu1) and HAg = 0.96 ± 0.03 Å (Ag1) above an idealized FeoctO2 bulk termination. The corresponding f004 values are 0.71 ± 0.03 (Cu1) and 0.66 ± 0.03 (Ag1). Annealing the Fe3O4(001)/Cu surface to 550 K resulted in a dramatic increase in the coherent fraction to 0.93 ± 0.03, consistent with the conversion of metastable Cu1* species (Fig. 1B) into regular Cu1 adatoms. The relatively low coherent fraction of Ag1 is attributable to clustering.50 Since the clusters are likely three dimensional, and thus have an equal occupation of all sites within the projected layer spacings, their contribution to the NIXSW simply lowers the coherent fraction without altering P004.
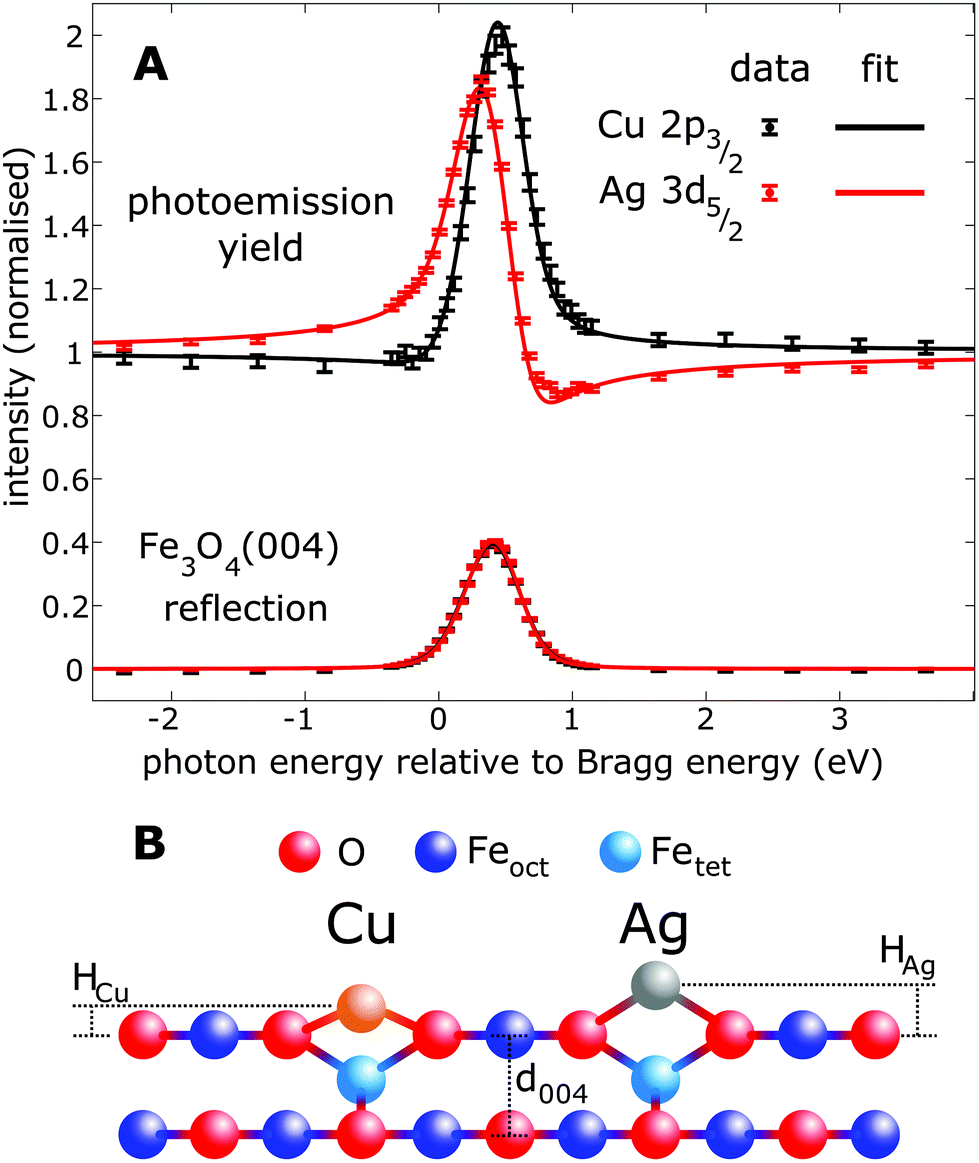 | ||
| Fig. 2 Results of the fitting of the (A) NIXSW data from the (004) reflection of Fe3O4. (B) Schematic of the apparent heights (Had) with respect to a bulk-like terminated Fe3O4(001) surface. The difference in height is 0.52 ± 0.04 Å, with absolute HAg and HCu values of 0.96 ± 0.03 Å and 0.43 ± 0.03 Å, respectively. | ||
Table 1 shows a selection of the computational results for the Fe3O4(001)/Cu1 and Fe3O4(001)/Ag1 systems. Full details of the various calculations are contained within the ESI,† but briefly, we utilized the Vienna ab initio Simulation Package (VASP)51,52 with the following functionals: PBE,9 PBE + U,9,53,54 PBEsol + U,11 and HSE.23 The surface calculations utilized an asymmetric surface slab with 5 fixed and 2 relaxed layers FeoctO2 layers including the SCV reconstruction.47 Initially we followed the standard procedure for calculating Fe3O4 surfaces, using a theoretical lattice parameter obtained by relaxing the bulk unit cell with the relevant functional. PBE + U and HSE overestimate the lattice by 0.75% and 0.18%, respectively, whereas PBE and PBEsol + U underestimate it by 0.01% and 0.61%, respectively. All calculations except the PBE find that the addition of the metal adatom reduces the total magnetic moment from 60μB to 59μB in both cases, indicating a charge state of +1 for the adatoms.
| Method | HSE | PBE + U | PBE + U | PBE | PBEsol + U | NIXSW |
|---|---|---|---|---|---|---|
| Lattice param. a (Å) | 8.411 | 8.459 | 8.396 | 8.390 | 8.345 | 8.396 |
| a–aexpt (%) | +0.18 | +0.75 | 0 | −0.01 | −0.61 | 0 |
| H Ag (Å) | 0.88 | 0.78 | 0.89 | 0.75 | 0.64 | 0.96 ± 0.03 |
| ΔzAg (Å) | 1.12 | 1.00 | 1.05 | 0.96 | 0.85 | — |
| Ag–O bond length (Å) | 2.11 | 2.09 | 2.09 | 2.06 | 2.02 | — |
| E ad (eV) | −1.99 | −2.19 | −1.93 | −2.30 | −2.63 | — |
| H Cu (Å) | 0.37 | 0.33 | 0.41 | 0.38 | 0.31 | 0.43 ± 0.03 |
| ΔzCu (Å) | 0.59 | 0.53 | 0.55 | 0.56 | 0.50 | — |
| Cu–O bond length (Å) | 1.86 | 1.85 | 1.84 | 1.84 | 1.82 | — |
| E ad (eV) | −3.46 | −3.76 | −3.60 | −3.85 | −4.17 | |
To directly compare the theoretical geometry to the NIXSW results, the adatom height must be calculated with respect to the bulk lattice. Thus we must convert the theoretical adatom height with respect to the bottom fixed FeoctO2 layer in the DFT slab, zad, to Had (ad = Ag, Cu) by:
| Had = zad − n·d004, | (1) |
Interestingly, the predicted charge state (+1 – supported by X-ray photoelectron spectroscopy, see ESI†) and Cu–O and Ag–O bond lengths (1.86 Å and 2.11 Å, respectively) are similar to the bulk compounds Cu2O and Ag2O (1.8481 ± 0.0004 Å and 2.043 ± 0.002 Å, respectively),56 where the cations bind linearly to O2− anions. Thus, the adatom geometry can be understood as the metal adopting its favored bond length to oxygen, with the constraint that the surrounding Fe3O4 lattice precludes the ideal linear geometry. It is then straightforward to understand why the Had predicted by PBE + U is too low (HAg = 0.78 Å, HCu = 0.33 Å). Although the Cu–O and Ag–O bond lengths are similar to HSE, the overestimation of the lattice parameter leads to a widening of the relevant O–O distance, and the adatoms sink towards the substrate. Simply rescaling the calculation to the experimental lattice makes matters worse, because this also reduces the Ag–O/Cu–O bondlengths, and thus the adatom height. If instead, utilising the PBE + U functional, the substrate lattice parameter is constrained to the experimental value (8.396 Å) at the outset of the calculation, the Ag–O and Cu–O bondlengths are unaffected, and Had values closer to the experiment are obtained. The local structure of the adatoms provides a very good approximation to the HSE results, though it must be noted that constraining the lattice in this way leads to a small expansion in the z direction in the relaxed layers. Altering the U(eff) parameter did not provide a more accurate modelling of the geometric structure (see ESI/Fig. S5†). Given the importance of the lattice parameter and metal–oxygen bonding in general, one might expect that PBEsol + U, specifically designed to correct for the disfavor of density overlapping of PBE, should perform well. Such calculations do indeed yield a lattice parameter closer to experiment than PBE + U, but the Ag adatom height is dramatically underestimated because the Ag–O bond length is also significantly reduced, therefore constraining the lattice constant to the experimental value resulted in a greater disagreement with experiment. This concomitant overbinding of adsorbates by PBEsol has been observed previously.21 A similar overbinding is observed in the PBE calculations (without U), due to highly reduced degree of localization of the Fe 3d electrons. Thus PBE and PBEsol are clearly not a suitable choice for SAC studies.
Conclusions
In summary, we demonstrate via direct NIXSW imaging, that Cu and Ag adatoms occupy a bulk-continuation cation site on the Fe3O4(001) surface. Furthermore the NIXSW data indicates their height above the surface differs significantly by 0.52 ± 0.04 Å. Successful theoretical modelling of this quantitative experimental result was found to be dependent not only on the choice of functional, but also on the bulk lattice parameter.Large deviations from the lattice parameter are known to affect calculations of phonon and magnetic properties, but the values obtained here would not normally be considered problematic, especially for adsorption studies. However, the PBE + U functional only obtains quantitative agreement with experiment when the lattice parameter is within 0.2% of the experimental value (8.396 Å). Despite this ability to “shoe-horn” the PBE + U calculations into more accurately modelling the experimental results, it is clear that, by predicting an underbinding of the atoms in the substrate and a relative overbinding between the substrate and the adatom, PBE + U fails at this experimental benchmark. Thus the use of the popular PBE + U functional must be questioned, certainly in its application to metal adatoms supported on magnetite, potentially to single metal adatoms supported on other metal oxides surfaces as well, and possibly even to metal nanoclusters supported on metal oxides in general.
Conflicts of interest
The authors have no conflicts to declare.Acknowledgements
The authors gratefully acknowledge funding through projects from the Austrian Science Fund FWF (START-Prize Y 847-N20 (MM, JH & GSP); Special Research Project ‘Functional Surfaces and Interfaces’, FOXSI F4505-N16 and F4507-N16 (MS & UD)), the European Research Council (UD & JB: ERC-2011-ADG_20110209 Advanced Grant ‘OxideSurfaces’), and the Doctoral College TU-D (ZJ) and Solids4fun (W1243: RB). The computational results presented have been achieved using the Vienna Scientific Cluster (VSC). We also thank Diamond Light Source for the award of beam time.Notes and references
- A. J. Medford, A. Vojvodic, J. S. Hummelshøj, J. Voss, F. Abild-Pedersen, F. Studt, T. Bligaard, A. Nilsson and J. K. Nørskov, J. Catal., 2015, 328, 36–42 CrossRef CAS.
- J. K. Nørskov, T. Bligaard, J. Rossmeisl and C. H. Christensen, Nat. Chem., 2009, 1, 37–46 CrossRef PubMed.
- Z. W. Seh, J. Kibsgaard, C. F. Dickens, I. Chorkendorff, J. K. Nørskov and T. F. Jaramillo, Science, 2017, 355, 146 CrossRef PubMed.
- F. Calle-Vallejo, D. Loffreda, T. M. KoperMarc and P. Sautet, Nat. Chem., 2015, 7, 403–410 CrossRef CAS PubMed.
- T. Bligaard, R. M. Bullock, C. T. Campbell, J. G. Chen, B. C. Gates, R. J. Gorte, C. W. Jones, W. D. Jones, J. R. Kitchin and S. L. Scott, ACS Catal., 2016, 6, 2590–2602 CrossRef CAS.
- Y. Zhang and W. Yang, Phys. Rev. Lett., 1998, 80, 890–890 CrossRef CAS.
- A. D. Becke, Phys. Rev. A, 1988, 38, 3098–3100 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37, 785–789 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S.-C. Qi, J.-i. Hayashi and L. Zhang, RSC Adv., 2016, 6, 77375–77395 RSC.
- J. P. Perdew, A. Ruzsinszky, G. I. Csonka, O. A. Vydrov, G. E. Scuseria, L. A. Constantin, X. Zhou and K. Burke, Phys. Rev. Lett., 2008, 100, 136406 CrossRef PubMed.
- S. Grimme, J. Comput. Chem., 2006, 27, 1787–1799 CrossRef CAS PubMed.
- A. Tkatchenko and M. Scheffler, Phys. Rev. Lett., 2009, 102, 073005 CrossRef PubMed.
- J. Klimeš and A. Michaelides, J. Chem. Phys., 2012, 137, 120901 CrossRef PubMed.
- M. Dion, H. Rydberg, E. Schröder, D. C. Langreth and B. I. Lundqvist, Phys. Rev. Lett., 2004, 92, 246401 CrossRef CAS PubMed.
- J. Klimeš, D. R. Bowler and A. Michaelides, Phys. Rev. B: Condens. Matter Mater. Phys., 2011, 83, 195131 CrossRef.
- V. I. Anisimov, J. Zaanen and O. K. Andersen, Phys. Rev. B: Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS.
- J. F. Dobson and B. P. Dinte, Phys. Rev. Lett., 1996, 76, 1780–1783 CrossRef CAS PubMed.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2003, 118, 8207–8215 CrossRef CAS.
- J. P. Perdew, M. Ernzerhof and K. Burke, J. Chem. Phys., 1996, 105, 9982–9985 CrossRef CAS.
- L. Schimka, J. Harl, A. Stroppa, A. Grüneis, M. Marsman, F. Mittendorfer and G. Kresse, Nat. Mater., 2010, 9, 741–744 CrossRef CAS PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652 CrossRef CAS.
- J. Heyd, G. E. Scuseria and M. Ernzerhof, J. Chem. Phys., 2006, 124, 219906 CrossRef.
- X.-F. Yang, A. Wang, B. Qiao, J. Li, J. Liu and T. Zhang, Acc. Chem. Res., 2013, 46, 1740–1748 CrossRef CAS PubMed.
- S. Liang, C. Hao and Y. Shi, ChemCatChem, 2015, 7, 2559–2567 CrossRef CAS.
- J. Liu, ACS Catal., 2017, 7, 34 CrossRef CAS.
- G. Kyriakou, M. B. Boucher, A. D. Jewell, E. A. Lewis, T. J. Lawton, A. E. Baber, H. L. Tierney, M. Flytzani-Stephanopoulos and E. C. H. Sykes, Science, 2012, 335, 1209–1212 CrossRef CAS PubMed.
- Q. Fu, H. Saltsburg and M. Flytzani-Stephanopoulos, Science, 2003, 301, 935–938 CrossRef CAS PubMed.
- M. Flytzani-Stephanopoulos and B. C. Gates, Annu. Rev. Chem. Biomol. Eng., 2012, 3, 545–574 CrossRef CAS PubMed.
- F. Dvořák, M. Farnesi Camellone, A. Tovt, N.-D. Tran, F. R. Negreiros, M. Vorokhta, T. Skála, I. Matolínová, J. Mysliveček, V. Matolín and S. Fabris, Nat. Commun., 2016, 7, 10801 CrossRef PubMed.
- B. Qiao, A. Wang, X. Yang, L. F. Allard, Z. Jiang, Y. Cui, J. Liu, J. Li and T. Zhang, Nat. Chem., 2011, 3, 634–641 CrossRef CAS PubMed.
- T. K. Ghosh and N. N. Nair, ChemCatChem, 2013, 5, 1811–1821 CrossRef CAS.
- J. Lin, A. Wang, B. Qiao, X. Liu, X. Yang, X. Wang, J. Liang, J. Li, J. Liu and T. Zhang, J. Am. Chem. Soc., 2013, 135, 15314–15317 CrossRef CAS PubMed.
- S. Sun, G. Zhang, N. Gauquelin, N. Chen, J. Zhou, S. Yang, W. Chen, X. Meng, D. Geng, M. N. Banis, R. Li, S. Ye, S. Knights, G. A. Botton, T.-K. Sham and X. Sun, Sci. Rep., 2013, 3, 1775 CrossRef.
- J. Lin, B. Qiao, N. Li, L. Li, X. Sun, J. Liu, X. Wang and T. Zhang, Chem. Commun., 2015, 51, 7911–7914 RSC.
- J.-X. Liang, X.-F. Yang, A. Wang, T. Zhang and J. Li, Catal. Sci. Technol., 2016, 6, 6886–6892 CAS.
- M. Yang, J. Liu, S. Lee, B. Zugic, J. Huang, L. F. Allard and M. Flytzani-Stephanopoulos, J. Am. Chem. Soc., 2015, 137, 3470–3473 CrossRef CAS PubMed.
- K. Ding, A. Gulec, A. M. Johnson, N. M. Schweitzer, G. D. Stucky, L. D. Marks and P. C. Stair, Science, 2015, 350, 189–192 CrossRef CAS PubMed.
- J.-X. Liang, J. Lin, X.-F. Yang, A.-Q. Wang, B.-T. Qiao, J. Liu, T. Zhang and J. Li, J. Phys. Chem. C, 2014, 118, 21945–21951 CAS.
- C. K. Narula, L. F. Allard, G. M. Stocks and M. Moses-DeBusk, Sci. Rep., 2014, 4, 7238 CrossRef CAS PubMed.
- Y. Tang, Y.-G. Wang and J. Li, J. Phys. Chem. C, 2017, 121, 11281–11289 CAS.
- Y. Tang, S. Zhao, B. Long, J.-C. Liu and J. Li, J. Phys. Chem. C, 2016, 120, 17514–17526 CAS.
- F. Li, Y. Li, X. C. Zeng and Z. Chen, ACS Catal., 2015, 5, 544–552 CrossRef CAS.
- Z. Novotný, G. Argentero, Z. Wang, M. Schmid, U. Diebold and G. S. Parkinson, Phys. Rev. Lett., 2012, 108, 216103 CrossRef PubMed.
- G. S. Parkinson, Z. Novotny, G. Argentero, M. Schmid, J. Pavelec, R. Kosak, P. Blaha and U. Diebold, Nat. Mater., 2013, 12, 724–728 CrossRef CAS PubMed.
- R. Bliem, J. E. S. van der Hoeven, J. Hulva, J. Pavelec, O. Gamba, P. E. de Jongh, M. Schmid, P. Blaha, U. Diebold and G. S. Parkinson, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 8921–8926 CrossRef CAS PubMed.
- R. Bliem, E. McDermott, P. Ferstl, M. Setvin, O. Gamba, J. Pavelec, M. A. Schneider, M. Schmid, U. Diebold, P. Blaha, L. Hammer and G. S. Parkinson, Science, 2014, 346, 1215–1218 CrossRef CAS PubMed.
- D. P. Woodruff, Rep. Prog. Phys., 2005, 68, 743 CrossRef CAS.
- J. Zegenhagen, Surf. Sci. Rep., 1993, 18, 202–271 CrossRef.
- R. Bliem, R. Kosak, L. Perneczky, Z. Novotny, O. Gamba, D. Fobes, Z. Mao, M. Schmid, P. Blaha, U. Diebold and G. S. Parkinson, ACS Nano, 2014, 8, 7531 CrossRef CAS PubMed.
- G. Kresse and J. Hafner, Phys. Rev. B: Condens. Matter Mater. Phys., 1993, 48, 13115–13118 CrossRef CAS.
- G. Kresse and J. Furthmüller, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS.
- A. Kiejna, T. Ossowski and T. Pabisiak, Phys. Rev. B: Condens. Matter Mater. Phys., 2012, 85, 125414 CrossRef.
- I. Bernal-Villamil and S. Gallego, J. Phys.: Condens. Matter, 2015, 27, 293202 CrossRef CAS PubMed.
- J. C. Sharp, Y. X. Yao and C. T. Campbell, J. Phys. Chem. C, 2013, 117, 24932–24936 CAS.
- T. Suzuki, J. Phys. Soc. Jpn., 1960, 15, 2018–2024 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, as well as further details on the results and analyses. See DOI: 10.1039/c7nr07319d |
| This journal is © The Royal Society of Chemistry 2018 |