
Concentration-dependent binding of CdSe quantum dots on the SH3 domain†
David R.
Bell‡
 a,
Seung-Gu
Kang‡
a,
Tien
Huynh
a and
Ruhong
Zhou
*ab
a,
Seung-Gu
Kang‡
a,
Tien
Huynh
a and
Ruhong
Zhou
*ab
aComputational Biology Center, IBM Thomas J Watson Research Center, Yorktown Heights, NY 10598, USA. E-mail: ruhongz@us.ibm.com
bDepartment of Chemistry, Columbia University, New York, NY 10027, USA
First published on 17th November 2017
Abstract
Quantum dots (QDs) are being used increasingly in applications for solar panels, consumer electronics, and biomedical imaging. For biomedical applications, QDs are typically coated with a biocompatible molecule for the system of interest. Experiments have indicated a QD dose-dependent and surface coating-dependent toxicity, with a portion of the toxicity being ascribed to interference with biomolecules. In this work, the interaction of trioctylphosphine oxide (TOPO) coated (CdSe)13 QDs with the SRC homology 3 domain (SH3) protein domain are explored using molecular dynamics simulations. The results of this research agree well with experiments that show that at the lowest concentration, the QDs have little affinity for the native proline-rich motif (PRM) binding site of SH3. At higher concentrations, the QDs aggregate and increasingly prefer the PRM binding site, indicating that the normal SH3 function is impeded. This binding dependence is attributed to changes in the local density of the surface coated TOPO molecules upon aggregation. These results present possible interesting QD toxicity patterns and reveal the interdependence between dose and surface coating effects in QD toxicity.
Introduction
Synthetic nanoparticles are employed in a multitude of applications. With increasing use, it is important to understand the interactions of the nanoparticles with other biological constituents. Carbon nanomaterials, such as graphene and bare carbon nanotubes (CNTs), are highly destructive to cellular membranes1,2 and cause harmful protein adsorption.3–8 Toxicity is lessened in fullerenes and their derivatives,9,10 yet depending on their surface modifications, toxicity can still present. Gadolinium metallofullerenols are a special case, where toxicity contributes to beneficial inhibition of tumour metastasis11,12 as well as aiding uptake of cancer therapeutics.13 To understand these examples, a rigorous study of nanoparticle interactions with biological domains is critical and then they can be used for comprehensive applications.Quantum dots (QDs) are photoluminescent nanoparticles employed in solar panels,14,15 consumer electronics, and biomedical imaging.16–20 QDs have unique optical properties because they are of the same size order as their Bohr radius allowing for generation of an exciton and subsequent photon emission. Because of this distinct yet robust fluorescence, QDs maintain several advantages over organic fluorophores, including a broad ultraviolet light absorption spectrum, narrow emission, and long-term photostability. These properties make QDs an ideal biomedical imaging agent. Cadmium selenide (CdSe) QDs remain one of the most investigated species because of their ease of synthesis and tunability.21 A drawback of using CdSe QDs in biomedical applications is toxicity, which occurs both in vitro and in vivo.22,23 CdSe QD toxicity is suspected to occur through three mechanisms: Cd leaching, generation of reactive oxygen species, and interference with biomolecules.19 Interference with biomolecules is suspected as Cd leaching and reactive oxygen species cannot entirely explain the observed toxicity.19,24
In this research, CdSe QD toxicity on the SRC homology 3 (SH3) protein domain was investigated using all-atom molecular dynamics (MD) simulations. SH3 is a protein binding domain with over 250 examples in the human genome25 and is essential to cell signalling and regulation.25–29 As a result of its widespread use, SH3 is a prime target for investigation of QD toxicity. Several different QD concentrations were explored to understand dose-dependent interactions. The results obtained support the conclusions drawn from the experimental work: that QDs exhibit a dose-dependent and surface coating-dependent toxicity.
Results and discussion
Initial configurations
Fig. 1 illustrates the systems studied in this research. Briefly, the protein structure consisted of the SH3 domain from the c-Crk in complex with a PRM peptide from SOS (PDB ID: 1CKB,30 structure and sequence shown in Fig. 1a and b). The PRM binding site is taken to be the key residues F141, P183, Y186, and W169. The QD studied was a (CdSe)13 nanoparticle (shown in Fig. 1) coated with 10 TOPO molecules bound to surface exposed Cd atoms. A TOPO surface coating was chosen because TOPO is an organometallic solvent widely used in CdSe QD synthesis21,31 and is a default QD coating. TOPO molecules can be exchanged or further modified for solubility or functionality, but TOPO's commonality in CdSe synthesis provides a good starting point for cytotoxicity investigation. Compared to other previously studied nanoparticles such as CNTs3–5,32–34 or fullerene derivatives,11,12,35–38 the TOPO-coated QDs are relatively large and of the same size as the SH3 protein. From literature sources39 and our own simulations, it was found that multiple QDs in solution readily aggregate. Thus, four QD-SH3 systems were studied: a monomer system (M) with the SH3 domain and one QD, a ternary system (Mt) with the SH3 domain, PRM and one QD, a dimer system (D) with the SH3 domain and two QDs aggregated together, and a tetramer system (T) with the SH3 domain and four QDs aggregated together. Note that in the tetramer system, the QDs adopt a tetrahedral configuration to reduce surface area. For each system, five configurations were constructed using equidistant rotation of the SH3 domain, with seven configurations constructed for the tetramer system. Over 200 ns of MD simulation was carried out for each configuration. For simulation details, see the section on Methods.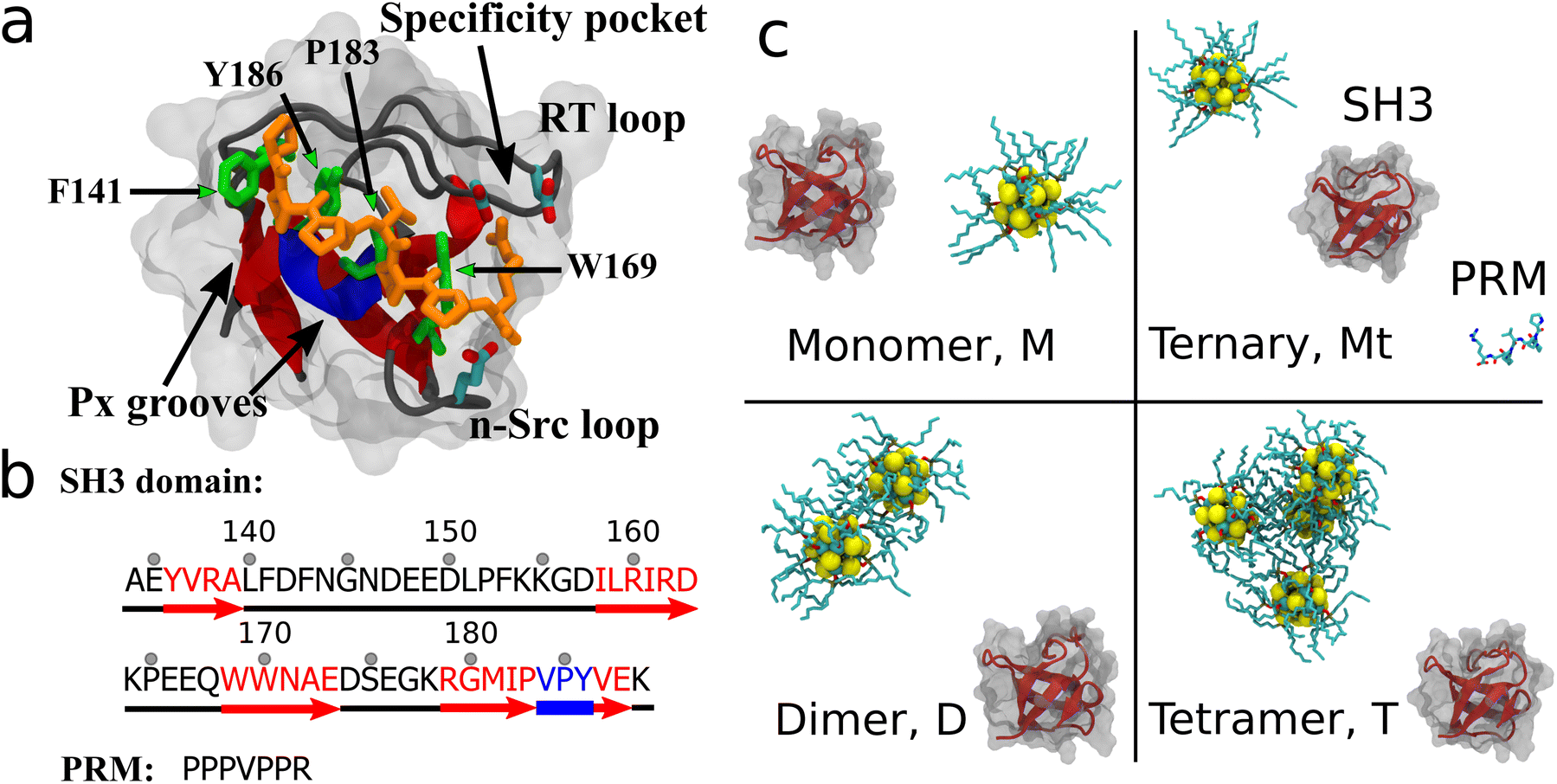 | ||
| Fig. 1 SH3 domain and initial configurations. (a) Native SH3-PRM structure and sequence. PRM is shown in orange with the key PRM binding residues (SH3: F141, W169, P183, Y186) shown in green. (b) SH3 sequence: red arrows are beta strands, blue region is a 310-helix, and the black regions are loops. The RT loop spans residues 140–157 whereas the n-Src loop spans residues 164–168. (c) Initial system configurations: (top left) monomer system M with one QD and the SH3 domain, (top right) ternary system Mt with one QD, the SH3 domain and the native PRM ligand, (bottom left) dimer system D with two aggregated QDs, and (bottom right) tetramer system T with four aggregated QDs. | ||
Simulation stability
As observed from the root mean square deviation (RMSD) of the SH3 backbone over time in Fig. S1 [ESI†], the SH3 molecule remains stable (most RMSD < 2.0 Å, max RMSD = 4.2 Å) in all configurations. There is one simulation (out of five) of the ternary system that does temporarily exceed a 4.0 Å RMSD threshold, but this is not caused by the QD. In this trajectory, the native PRM ligand binds to the SH3 active site and ‘pulls’ on adjacent loop domains in SH3. The overall stability of the SH3 domain in the presence of QDs indicates that the studied QDs do not cause large structural changes. This result contrasts with the effects of carbon allotropes including CNTs and graphene, which have been shown to severely disrupt the protein structure1,2,5,40QD concentration-dependent binding
A QD-SH3 contact analysis reveals that QDs increasingly target the PRM binding site of SH3 with increasing QD concentration as shown in Fig. 2. Here, the system contact ratio (Ct ratio) is defined as the total number of contacts between the QD(s) and the SH3 domain, normalized to the total number of frames. The off-site binding in Fig. 2 is taken to be all residues except for the PRM site residues. For the QD-SH3 systems, off-site binding is dominant for the monomer system as the monomer system was found to have little affinity for the PRM site. Off-site binding is competitive to PRM site binding for the ternary system. Note that the monomer and ternary systems each have one QD, but the difference in contact ratio is attributed to the PRM in the ternary system leading the QD to the PRM binding site. For the dimer and tetramer systems, the QD clearly favored contacting the PRM site. The QD tetramer contacted the PRM binding site the most out of all systems, even surpassing the native PRM ligand (Ct ratio of 0.20 versus 0.12). For the PRM-SH3 contact ratio, the PRM has clear preference for the PRM binding site over off-site binding (Ct ratio of 0.12 versus 0.02), indicating a 6-fold increase in specificity for recognizing the PRM active site of the SH3 domain. This 6-fold increase in specificity is matched or even exceeded by the QD tetramer system (Ct ratio of 0.20 versus 0.03).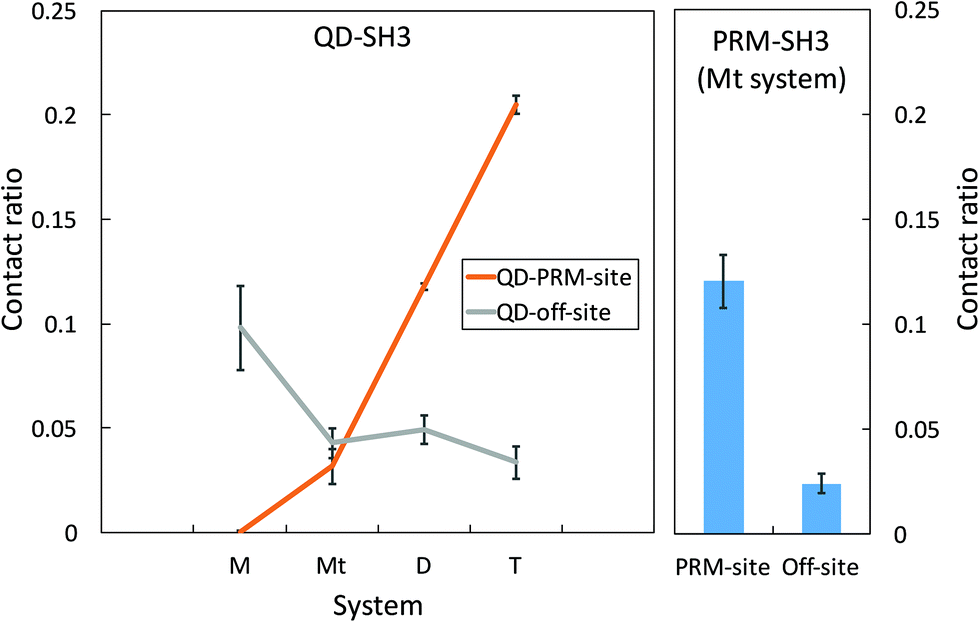 | ||
| Fig. 2 Average QD-SH3 contact ratio for the studied systems (left) with the PRM-SH3 contact ratio for comparison (right). The PRM site of SH3 includes residues F141, W169, P183, and Y186. The off-site of SH3 referenced here includes all residues except PRM site residues. The contact ratio is defined as the number of contacts normalized to the total number of frames. | ||
QD residue – specific contacts
From residue decomposition of the QD contacts (Fig. 3a), there emerge two clear sites of high affinity: the native PRM site with tetramer-dominant affinity (denoted by the arrows) and a distal site at residues Y136, R138, I158, R160, and S175 with monomer-dominant affinity (denoted by the dots). The QD monomer contacts the distal site at approximately twice the rate that the QD tetramer contacts the PRM site (≈0.4 versus ≈0.2). Both the ternary and dimer systems exhibit approximately equal contacts for the distal site, but the dimer system maintains greater contacts with the PRM site. When the QD-SH3 contact ratio is mapped onto the SH3 domain (Fig. 3b), the contact locations become clearly demarcated between the various systems. The monomer system binds almost exclusively to the SH3 distal site, whereas the tetramer system is just the opposite, preferring the native PRM site (which is largely hydrophobic). Interestingly, the distal site contains two central hydrophobic residues surrounded by three charged/hydrophilic residues. Note also the structural differences between the PRM site and distal site surfaces: namely, the PRM site has a broad corrugated surface, whereas the distal site is flat but has three deep pockets well suited to accommodate the TOPO aliphatic chains as discussed in the next section.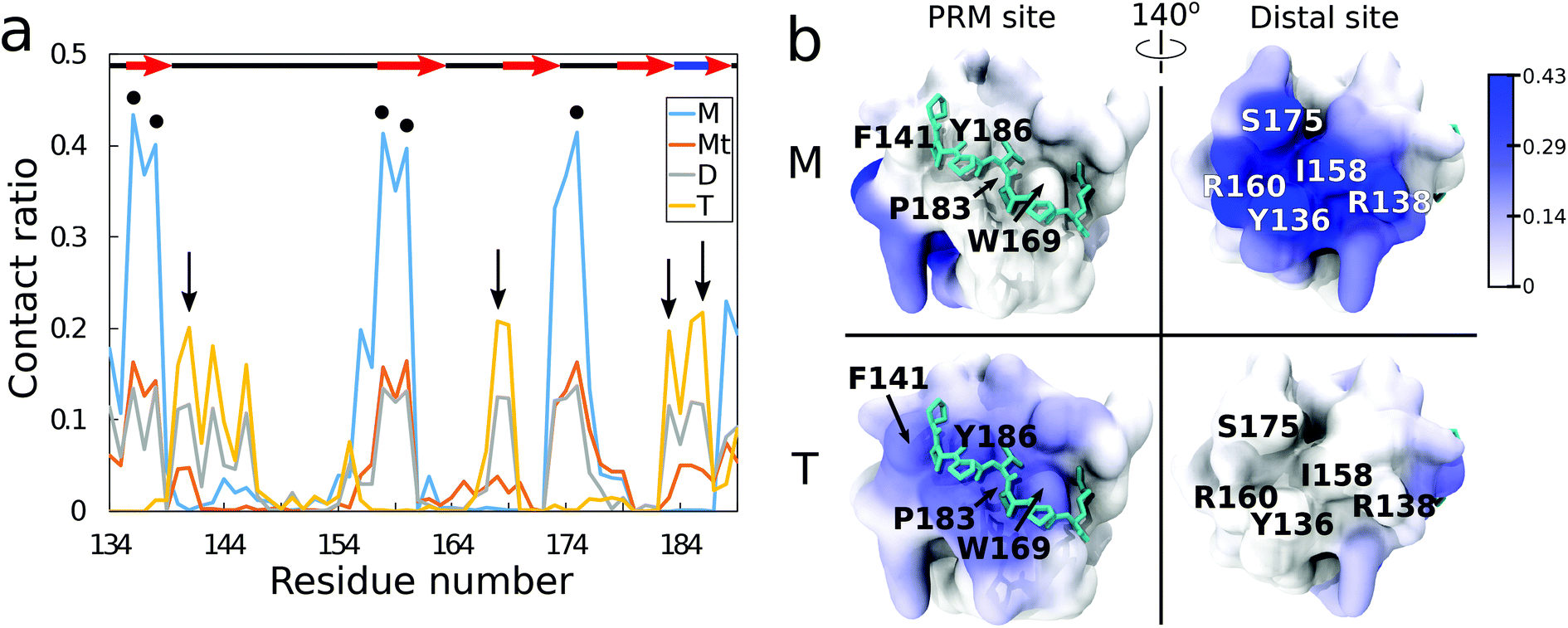 | ||
| Fig. 3 (a) QD contact ratio for all the systems, normalized to number of frames. Secondary structure is shown at the top, where the red arrows indicate beta strands, the blue bold line indicates a 310-helix, and the black lines indicate loop regions. Black arrows indicate the PRM site residues, and, the black dots indicate the distal site residues. Note that the monomer M and ternary Mt systems have little contact with the binding site but favorable contacts with the distal site, whereas the dimer D system has equal contacts between the binding and distal site and the tetramer T system only has a favorable affinity for the binding site. (b) QD contact ratio on the SH3 domain for the monomer M system (top) and the tetramer T system (bottom). High contact ratio residues are shown in dark blue, whereas the zero contact residues are shown in white, with a contact ratio color bar on the top right. The left column shows the PRM active site whereas the right column shows the distal site of high affinity in the monomer M system. | ||
In Fig. S2 (ESI†), the average root mean square fluctuations (RMSFs) of the residue atoms are shown, together with the Ct ratio. Excluding terminal residues, all residues have RMSF values less than 2.5 Å, confirming that the SH3 domain remains stable during the simulations. There is little relationship between high RMSF values and high Ct ratios, with the highest RMSF values obtained from the RT loop and n-Src loop regions. This reiterates that the QDs do not cause large changes in SH3 structure. In terms of nanotoxicity, this result points towards disruption in the native biomolecular interactions as a possible cause for toxicity, and this is discussed further in the next sections.
QD binding free energy surfaces and binding modes
QD-SH3 binding transitions from the distal site centric space in the monomer system to the PRM site centric space in the tetramer system as shown through the [images] presented in Fig. 4. Distal site binding occurs at a QD key residue distance of approximately 1.0 nm. At this location, the QDs typically bind with a contact area of 7–10 nm2. The PRM site well is located at a QD key residue distance of 0.25 nm and a broad contact area well of 5–15 nm2. For comparison, the PRM binding surface is given in Fig. S3 (ESI†) and it shows a PRM site well similar to that of the QD systems, but with a smaller contacting area (7 nm2) given the smaller size of the PRM ligand. The monomer system binding free energy surface shows only one well at the distal site whereas the tetramer system shows only one well at the PRM site. In the dimer and ternary systems, the binding surface shows both distal site and PRM site wells, with the ternary system preferring the distal site and the dimer system preferring the PRM site. From the binding modes (Fig. 4b, d, f and h), several trends were observed and are discussed next.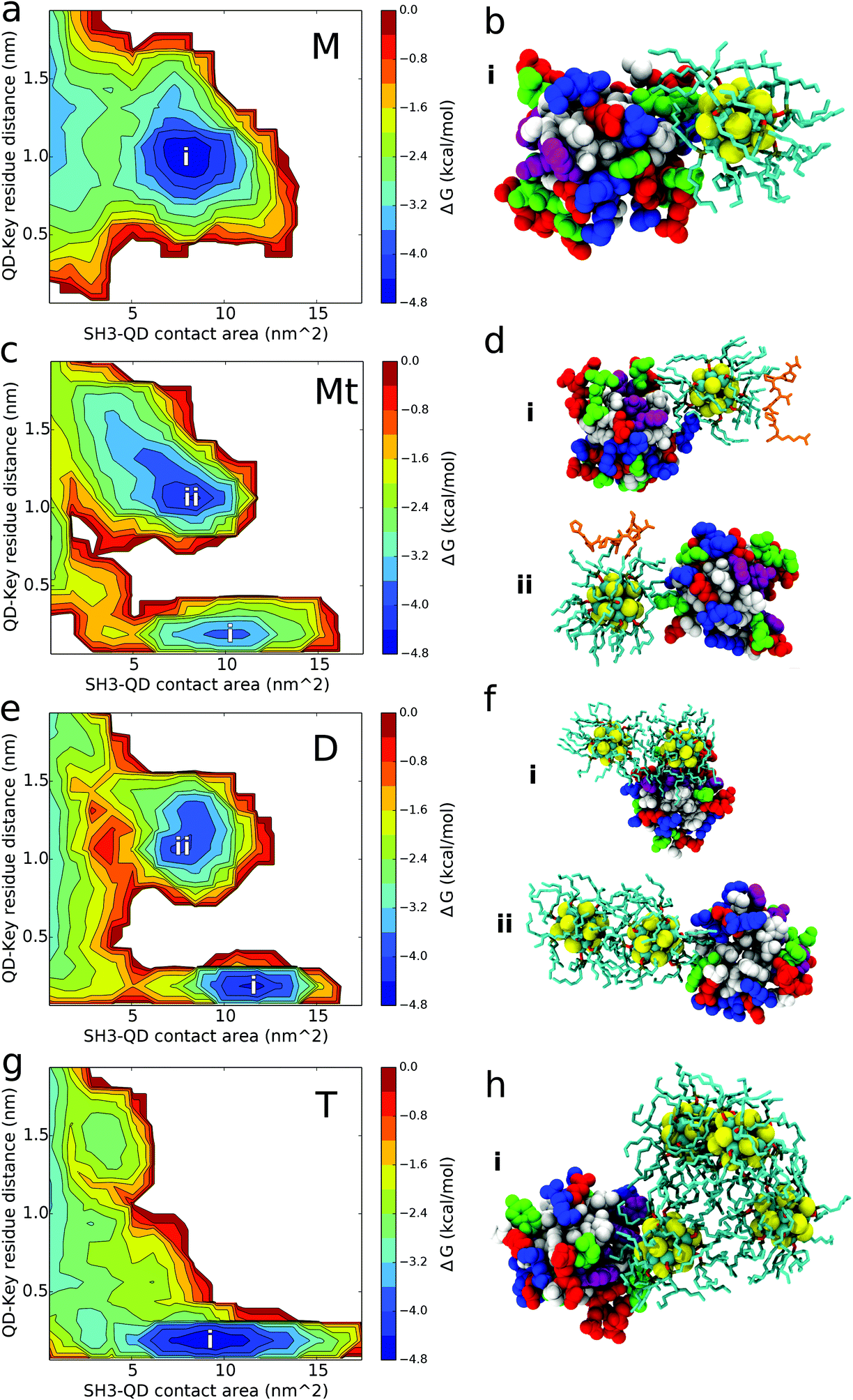 | ||
| Fig. 4 Binding free energy surfaces and structures. (a, c, e, g) Binding free energy surfaces of (a) monomer system M, (c) ternary system Mt, (e) dimer system D, and (g) tetramer system T. (b, d, f, h) Characteristic binding modes of binding site well (blue area) for (b) monomer system M, (d) ternary system Mt, (f) dimer system D, and (h) tetramer system T. For the ternary and dimer systems, two free energy wells are observed and the respective binding mode structures are labeled accordingly (i, ii). | ||
The QD monomer binds strongly to the SH3 distal site because of these combined effects: two positively charged arginines at the distal site form favorable electrostatic interactions with the exposed Se atoms of the QD core while the deep hydrophobic pockets of the distal site nicely accommodate the aliphatic TOPO chains. The long, positively charged side chains of arginine residues R138 and R160 of the SH3 distal site are able to form beneficial electrostatic interactions with the partially negatively charged Se atoms of the QD core [see Table S1 (ESI†) for QD core charges]. Cadmium atoms in the QD core are positively charged and will act to repel other positive charges; however, most of the Cd atoms are covalently bound to the TOPO chains, allowing the more exposed Se atoms to dominate the electrostatic interactions. The absence of PRM site binding for the monomer system can be explained by the lack of positively charged residues at the PRM site for forming favorable electrostatic interactions. Instead, the PRM site is largely hydrophobic which, although well suited to interact with TOPO chains, is unable to balance the exposed charges on the QD core. In one of the five configurations for the monomer system, a QD is located 15 Å directly opposite from the PRM site, yet the QD still bypasses the active site for contacts with the distal site. It is proposed that the monomer QD disfavors the PRM site because of a low “effective” density of the TOPO chains (explained below), allowing electrostatic charge from the QD core to have a larger influence in binding. This is supported by analysis of QD core-SH3 contacts, which reveal that the monomer QD core contacts the SH3 domain the most out of all systems [shown in Fig. S4; ESI†]. Given that the QD core has a partial charge, it is evident that the QD core protein electrostatic interactions play a larger role in monomer QD binding, which explains the monomer's affinity for the positively charged distal site and its aversion for the uncharged PRM site.
For the ternary system, Fig. 4 shows the affinity of the QD for both the distal and PRM site, with the QD preferring the distal site. The slight contact affinity of the QD for the PRM site is because the PRM “leads” the QD to the active site. As observed in Fig. 4d, the QD can bind both the PRM and SH3 domain simultaneously, and from our trajectories the QD only contacts the PRM site when the PRM is bound to the QD and directly “leads” the QD to the PRM site. Another aspect to consider is that when the PRM is bound to the QD, the PRM arginine forms charged interactions with the Se atoms of the QD core. This decreases the availability of the QD Se atoms to form favourable interactions with the arginines at the SH3 distal site. Regardless of the dominant mechanism, the effect of PRM on QD binding is robustly supported by comparison between the ternary and monomer systems. In the presence of only the QD monomer, the QD does not contact the PRM site at all; however, in the presence of the PRM (ternary system), the QD does contact the PRM site, indicating that the PRM causes the QD to contact the PRM site. Interestingly, from computing the PRM Ct ratios, the PRM has a stronger preference to interact with the QD over the SH3 domain (Fig. S5; ESI†). This favourable PRM-QD behaviour is driven by hydrophobic interactions with the TOPO chains and charged interactions with the QD core. Six of the seven PRM residues are hydrophobic which interact favourably with the aliphatic TOPO chains of the QD. The seventh PRM residue, a positively charged arginine, is observed to form electrostatic interactions with the negatively charged Selenium atoms of the QD core. This arginine leads the PRM to have a high contact ratio with the QD core (Ct ratio and binding mode shown in Fig. S4; ESI†).
The aggregated dimer and tetramer QD systems exhibit favorable binding affinity for the PRM active site of the SH3 domain. The tetramer system binds to the PRM site as a broad, hydrophobic surface interacting with the large hydrophobic PRM site. The tetramer can bind to the PRM active site as a monomer (1 QD binds at a time), dimer (2 QDs), or trimer (3 QDs), but not 4 QDs at a time given the steric nature of the tetrahedral configuration. For dimer system D, the QD dimer only binds to the PRM site as a dimer, whereas it binds to the distal site using a monomer interface as shown in Fig. 4. For the dimer and tetramer systems, the distribution of QD multiplicity in binding key residue distance and binding contact area is given in Fig. S6.† These distributions reiterate that in the dimer system, the QD dimer binds as a dimer to the PRM site and as a monomer to the distal site. For the tetramer case, there is substantial overlap in QD multiplicity over key-residue distances and contact area distributions (Fig. S6; ESI†). This indicates that once the QD concentration increases to form a tetramer, the aggregated QDs are able to robustly bind the PRM active site without preferring a particular tetramer interface (monomer, dimer, or trimer).
The hydrophobic binding of the tetramer QD systems to the PRM site is attributed to sequestration of the charged QD cores and increasing pressure to expose the TOPO chains, causing a high “effective” density or surface coverage of hydrophobic TOPO chains. Upon aggregation, the QD cores form charged electrostatic interactions with the other QD cores, leading them to become buried inside the oligomer. Meanwhile, the TOPO chains are pushed outward to accommodate the QD core sequestration. Thus, with the QD tetramer system, the exposed surface of the QDs is thought to be more hydrophobic given the increased exposure of the TOPO chains. This larger, more exposed hydrophobic surface than the QD monomer and dimer can more effectively bind the PRM site, and do so with less dependence on the number of QDs contacting the PRM site in comparison to the dimer system. In addition, the enhanced hydrophobic surface of the tetramer is thought to strongly shield electrostatic interactions with the QD core, explaining the loss of binding with the charged distal site.
The dimer system spans the gap between the tetramer and monomer binding behavior. In the QD dimer, the two QD cores are slightly pressed together because of favorable electrostatic interactions. This causes some TOPO chains to be pressed outward, but there is less pressure than the tetramer system. This means that the two ends (i.e., the monomer interface) of the QD dimer are reasoned to still have a similar hydrophobic TOPO distribution to QD monomers, allowing electrostatic interactions with the QD core. This explains why only the monomer interface of the QD dimer binds to the charged distal site. Conversely, the dimer interface of the QD dimer is more hydrophobic than the monomer interface because of the slightly higher density of TOPO molecules. This hydrophobic dimer interface binds to the hydrophobic PRM site. Furthermore, the dimer system binds with a higher contact area well than the tetramer because: (1) it can more easily adapt to the binding surface, and (2) the tetramer well includes monomer, dimer, and trimer QD interfaces whereas the dimer system PRM site well only contains binding from the dimer QD interface.
It is important to note that QDs used in experiments vary widely in size (and curvature), and that the size (curvature) of the QDs will also affect biomolecular interactions. It was speculated that the larger QDs with the same TOPO surface coating distribution would interact more strongly with the PRM binding site of SH3. This is a result of larger QDs having a lower curvature, and thus, a higher “effective” density of TOPO surface coating molecules, which was found to cause preferential targeting of the PRM binding site. If the density of TOPO surface coating molecules were reduced on larger QDs by removal of TOPO molecules, then at a certain point it was anticipated that electrostatic interactions with the QD core would once again dominate interactions with SH3, leading to distal site binding.
An important result from this work is that QD binding and interference with biomolecules is heavily modulated by surface coating characteristics including composition and density, providing evidence for surface coating dependent toxicity. It was found that increasing the concentration of QDs affects the distribution of the surface coating, and thus, affinity for the active site, proposing an intriguing argument that concentration dependent toxicity can, in effect, be surface coating “induced” toxicity. The TOPO chains used here (and which remain popular in experiments) were profoundly hydrophobic, enabling aggregation and interaction with the hydrophobic PRM binding site of SH3. TOPO molecules are also unique because of their three aliphatic chains, which can both fan out across the QD surface and interdigitate upon QD aggregation and subsequent higher TOPO density. Another unique aspect of the system studied is that the PRM site of the SH3 domain contains three aromatic residues: F141, W169, and Y186. Thus, it is proposed that for the SH3 system studied, a QD surface coating of aromatic ligands would form strong interactions with the PRM site resulting in interference of native SH3 interactions. Ultimately, all QD surface coatings will need to be addressed individually to understand their behaviour with biological molecules. However, in general, it is suspected that a hydrophilic surface coating ligand, such as polyethylene glycol (PEG), will decrease aggregation and affect the binding affinity of the QD to the hydrophobic protein sites. This agrees well with an experiment that shows decreased toxicity when the QDs are coated in PEG.41
Conclusions
In this work, TOPO coated (CdSe)13 QD toxicity and the effect of concentration on the SH3 protein domain was investigated. At low concentrations, QD specificity for the SH3 binding site is low, instead preferring a distal site. This corresponds to the SH3 domain retaining function/activity with PRM binding. With increasing concentration, there is increasing preference for the QD to contact the PRM binding site of the SH3 domain. With increasing QD concentration, the QDs readily aggregate, causing sequestration of QD cores while pushing the surface coated TOPO molecules outward. This increased outward pressure causes a high local density of hydrophobic TOPO molecules on the surface, resulting in a broad, hydrophobic surface capable of forming strong, yet non-specific interactions with the hydrophobic PRM binding site of SH3. The large binding affinity for the PRM binding site with high QD concentrations indicates that the SH3 domain will not be able to bind PRM implying loss of SH3 function. This proposed loss of function indicates that at high concentrations, the studied TOPO coated (CdSe)13 QDs are toxic. However, it was observed that there was a substantial interdependence between QD concentration and surface coating behaviour. These results reiterate that QD toxicity is a complex landscape dependent on a multitude of variables including choice of QD surface coating and concentration.Methods
The SH3 protein structure used in this work was taken from a crystallized c-Crk, N-terminal SH3 domain in complex with the proline-rich SOS peptide (PPPVPPR), PDB ID: 1CKB.30 The QDs used consisted of (CdSe)13 nanoparticles, with ten trioctylphosphine oxide (TOPO) covalently bound to surface exposed Cd ions. Four initial system setups were used: a “monomer” system with 1 QD and SH3, a “ternary” system with 1QD, PRM and SH3, a system with two aggregated QDs (e.g., QD dimer) and SH3, and a system with four aggregated QDs (e.g., QD tetramer) and SH3. All the macromolecules (SH3, QD, PRM, but not the QD dimer and QD tetramer) were initially separated by 15 Å. QD dimers and tetramers were simulated because of the QDs’ tendency to aggregate in solution and dimers and tetramers were built by simulating separated QDs in solution, with aggregation occurring within 200 ns. For each configuration setup, five different systems were generated by rotating the SH3 protein 72° about a vertical axis and additionally, two simulation runs were added for the QD tetramer configuration. In total, 32 systems were simulated. Each system was immersed in a roughly 87 × 87 × 87 Å box of TIP3P water molecules. Sodium chloride ions were added to all systems to achieve 0.1 M salt concentration. The CHARMM36 force field42 was used for protein parameters, and the QD parameters were found from the density functional theory (DFT) calculations43 with TOPO parameters from the CHARMM22 lipid force field.44,45 van der Waals interactions were cut off at 12 Å, whereas long range electrostatics were treated using the particle mesh Ewald46 method. Systems were energy minimized for 15![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps with fixed protein atoms, followed by 5000 steps of energy minimization with all the atoms unrestrained. Following minimization, systems were equilibrated for 500000 steps with a 0.5 fs time step. Production MD simulations were run for 200 ns with a 2 fs time step. All simulations were run in the NPT ensemble, with a pressure of 1 atm and temperature of 310 K. In total, over 6.5 μs of all-atom MD was simulated. Simulations were run using NAMD247 on both an IBM Blue Gene/Q48 supercomputer as well as an IBM Power 8 cluster.
000 steps with fixed protein atoms, followed by 5000 steps of energy minimization with all the atoms unrestrained. Following minimization, systems were equilibrated for 500000 steps with a 0.5 fs time step. Production MD simulations were run for 200 ns with a 2 fs time step. All simulations were run in the NPT ensemble, with a pressure of 1 atm and temperature of 310 K. In total, over 6.5 μs of all-atom MD was simulated. Simulations were run using NAMD247 on both an IBM Blue Gene/Q48 supercomputer as well as an IBM Power 8 cluster.
The contact ratio was computed for all four systems studied, with a contact distance of 4.5 Å. The contact ratio is defined as the number of frames the QD contacted the SH3 domain, divided by the total number of frames. For the aggregated dimer and tetramer systems, contact was calculated between the QD group and the SH3, not individually. Binding free energy surfaces were computed using a similar method to that given by Zhou et al.49 by creating a two dimensional probability histogram P(S,D) of surface contact area (S) and key-residue distance (D). The PMF was computed from W(S,D) = −RTln(P(S,D)) assuming a uniform reference distribution. The key binding site residues are F141, W169, P183, and Y186.
Conflicts of interest
There are no conflicts to declare.References
- Y. Tu, M. Lv, P. Xiu, T. Huynh, M. Zhang, M. Castelli, Z. Liu, Q. Huang, C. Fan, H. Fang and R. Zhou, Nat. Nanotechnol., 2013, 8, 594–601 CrossRef CAS PubMed.
- R. Zhou and H. Gao, Wiley Interdiscip. Rev.: Nanomed. Nanobiotechnol., 2014, 6, 452–474 CrossRef CAS PubMed.
- J. Gao, L. Wang, S.-G. Kang, L. Zhao, M. Ji, C. Chen, Y. Zhao, R. Zhou and J. Li, Nanoscale, 2014, 6, 12828–12837 RSC.
- R. El-Sayed, K. Bhattacharya, Z. Gu, Z. Yang, J. K. Weber, H. Li, K. Leifer, Y. Zhao, M. S. Toprak, R. Zhou and B. Fadeel, Sci. Rep., 2016, 6, 21316 CrossRef CAS PubMed.
- G. Zuo, S.-G. Kang, P. Xiu, Y. Zhao and R. Zhou, Small, 2013, 9, 1546–1556 CrossRef CAS PubMed.
- C. Ge, J. Du, L. Zhao, L. Wang, Y. Liu, D. Li, Y. Yang, R. Zhou, Y. Zhao, Z. Chai and C. Chen, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16968–16973 CrossRef CAS PubMed.
- Y. Liu, Y. Zhao, B. Sun and C. Chen, Acc. Chem. Res., 2013, 46, 702–713 CrossRef CAS PubMed.
- A. B. Seabra, A. J. Paula, R. de Lima, O. L. Alves and N. Durán, Chem. Res. Toxicol., 2014, 27, 159–168 CrossRef CAS PubMed.
- H. J. Johnston, G. R. Hutchison, F. M. Christensen, K. Aschberger and V. Stone, Toxicol. Sci., 2010, 114, 162–182 CrossRef CAS PubMed.
- K. Aschberger, H. J. Johnston, V. Stone, R. J. Aitken, C. L. Tran, S. M. Hankin, S. A. K. Peters and F. M. Christensen, Regul. Toxicol. Pharmacol., 2010, 58, 455–473 CrossRef CAS PubMed.
- S. G. Kang, R. Araya-Secchi, D. Q. Wang, B. Wang, T. Huynh and R. H. Zhou, Sci. Rep., 2014, 4, 8 Search PubMed.
- S. G. Kang, G. Q. Zhou, P. Yang, Y. Liu, B. Y. Sun, T. Huynh, H. Meng, L. N. Zhao, G. M. Xing, C. Y. Chen, Y. L. Zhao and R. H. Zhou, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 15431–15436 CrossRef CAS PubMed.
- X.-J. Liang, H. Meng, Y. Wang, H. He, J. Meng, J. Lu, P. C. Wang, Y. Zhao, X. Gao, B. Sun, C. Chen, G. Xing, D. Shen, M. M. Gottesman, Y. Wu, J.-J. Yin, L. Jia and I. Pastan, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 7449–7454 CrossRef CAS PubMed.
- P. V. Kamat, J. Phys. Chem. Lett., 2013, 4, 908–918 CrossRef CAS PubMed.
- G. H. Carey, A. L. Abdelhady, Z. Ning, S. M. Thon, O. M. Bakr and E. H. Sargent, Chem. Rev., 2015, 115, 12732–12763 CrossRef CAS PubMed.
- T. Jamieson, R. Bakhshi, D. Petrova, R. Pocock, M. Imani and A. M. Seifalian, Biomaterials, 2007, 28, 4717–4732 CrossRef CAS PubMed.
- W. W. Yu, E. Chang, R. Drezek and V. L. Colvin, Biochem. Biophys. Res. Commun., 2006, 348, 781–786 CrossRef CAS PubMed.
- S. J. Rosenthal, J. C. Chang, O. Kovtun, J. R. McBride and I. D. Tomlinson, Chem. Biol., 2011, 18, 10–24 CrossRef CAS PubMed.
- K. D. Wegner and N. Hildebrandt, Chem. Soc. Rev., 2015, 44, 4792–4834 RSC.
- K. Sun, M. Vasudev, H.-S. Jung, J. Yang, A. Kar, Y. Li, K. Reinhardt, P. Snee, M. A. Stroscio and M. Dutta, Microelectron. J., 2009, 40, 644–649 CrossRef CAS.
- Z. A. Peng and X. Peng, J. Am. Chem. Soc., 2001, 123, 183–184 CrossRef CAS PubMed.
- R. Hardman, Environ. Health Perspect., 2006, 114, 165–172 CrossRef PubMed.
- K. M. Tsoi, Q. Dai, B. A. Alman and W. C. W. Chan, Acc. Chem. Res., 2013, 46, 662–671 CrossRef CAS PubMed.
- S. T. Stern, B. S. Zolnik, C. B. McLeland, J. Clogston, J. Zheng and S. E. McNeil, Toxicol. Sci., 2008, 106, 140–152 CrossRef CAS PubMed.
- T. Pawson and P. Nash, Science, 2003, 300, 445–452 CrossRef CAS PubMed.
- T. Pawson, Cell, 2004, 116, 191–203 CrossRef CAS PubMed.
- T. Pawson and P. Nash, Genes Dev., 2000, 14, 1027–1047 CAS.
- J. D. Scott and T. Pawson, Science, 2009, 326, 1220–1224 CrossRef CAS PubMed.
- B. J. Mayer, J. Cell Sci., 2001, 114, 1253–1263 CAS.
- X. Wu, B. Knudsen, S. M. Feller, J. Zheng, A. Sali, D. Cowburn, H. Hanafusa and J. Kuriyan, Structure, 1995, 3, 215–226 CrossRef CAS PubMed.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- G. Zuo, Q. Huang, G. Wei, R. Zhou and H. Fang, ACS Nano, 2010, 4, 7508–7514 CrossRef CAS PubMed.
- B. Shi, G. Zuo, P. Xiu and R. Zhou, J. Phys. Chem. B, 2013, 117, 3541–3547 CrossRef CAS PubMed.
- Y. Zhang, C. A. Jimenez-Cruz, J. Wang, B. Zhou, Z. Yang and R. Zhou, Sci. Rep., 2014, 4, 7229 CrossRef CAS PubMed.
- S.-G. Kang, T. Huynh and R. Zhou, Nanoscale, 2013, 5, 2703–2712 RSC.
- S.-G. Kang, T. Huynh and R. Zhou, Sci. Rep., 2012, 2, 957 CrossRef PubMed.
- Y. Pan, L. Wang, S.-G. Kang, Y. Lu, Z. Yang, T. Huynh, C. Chen, R. Zhou, M. Guo and Y. Zhao, ACS Nano, 2015, 9, 6826–6836 CrossRef CAS PubMed.
- X. Yin, L. Zhao, S.-G. Kang, J. Pan, Y. Song, M. Zhang, G. Xing, F. Wang, J. Li and R. Zhou, Nanoscale, 2013, 5, 7341–7348 RSC.
- B. O. Dabbousi, J. Rodriguez-Viejo, F. V. Mikulec, J. R. Heine, H. Mattoussi, R. Ober, K. F. Jensen and M. G. Bawendi, J. Phys. Chem. B, 1997, 101, 9463–9475 CrossRef CAS.
- B. Luan, T. Huynh, L. Zhao and R. Zhou, ACS Nano, 2015, 9, 663–669 CrossRef CAS PubMed.
- M. A. Walling, J. A. Novak and J. R. E. Shepard, Int. J. Mol. Sci., 2009, 10, 441–491 CrossRef CAS PubMed.
- R. B. Best, X. Zhu, J. Shim, P. E. M. Lopes, J. Mittal, M. Feig and A. D. MacKerell, J. Chem. Theory Comput., 2012, 8, 3257–3273 CrossRef CAS PubMed.
- Y. Gao, B. Zhou, S. G. Kang, M. S. Xin, P. Yang, X. Dai, Z. G. Wang and R. H. Zhou, RSC Adv., 2014, 4, 27146–27151 RSC.
- M. Schlenkrich, J. Brickmann, A. D. MacKerell and M. Karplus, in Biological Membranes: A Molecular Perspective from Computation and Experiment, ed. K. M. Merz and B. Roux, Birkhäuser Boston, Boston, MA, 1996, pp. 31–81, DOI:10.1007/978-1-4684-8580-6_2.
- S. E. Feller, D. Yin, R. W. Pastor and A. D. MacKerell, Biophys. J., 1997, 73, 2269–2279 CrossRef CAS PubMed.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092 CrossRef CAS.
- J. C. Phillips, R. Braun, W. Wang, J. Gumbart, E. Tajkhorshid, E. Villa, C. Chipot, R. D. Skeel, L. Kalé and K. Schulten, J. Comput. Chem., 2005, 26, 1781–1802 CrossRef CAS PubMed.
- S. Kumar, C. Huang, G. Zheng, E. Bohm, A. Bhatele, J. C. Phillips, H. Yu and L. V. Kale, IBM J. Res. Dev., 2008, 52, 177–188 CrossRef CAS.
- R. Zhou, B. J. Berne and R. Germain, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 14931–14936 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr06148j |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2018 |