
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Gold nanoclusters with bright near-infrared photoluminescence†
Goutam
Pramanik
a,
Jana
Humpolickova
a,
Jan
Valenta
b,
Paromita
Kundu
c,
Sara
Bals
c,
Petr
Bour
 a,
Martin
Dracinsky
a and
Petr
Cigler
*a
a,
Martin
Dracinsky
a and
Petr
Cigler
*a
aInstitute of Organic Chemistry and Biochemistry of the CAS, Flemingovo nam. 2, 166 10 Prague 6, Czech Republic. E-mail: cigler@uochb.cas.cz
bDepartment of Chemical Physics and Optics, Faculty of Mathematics and Physics, Charles University, Ke Karlovu 3, 121 16 Prague 2, Czech Republic
cEMAT, University of Antwerp, Groenenborgerlaan 171, B-2020 Antwerp, Belgium
First published on 8th January 2018
Abstract
The increase in nonradiative pathways with decreasing emission energy reduces the luminescence quantum yield (QY) of near-infrared photoluminescent (NIR PL) metal nanoclusters. Efficient surface ligand chemistry can significantly improve the luminescence QY of NIR PL metal nanoclusters. In contrast to the widely reported but modestly effective thiolate ligand-to-metal core charge transfer, we show that metal-to-ligand charge transfer (MLCT) can be used to greatly enhance the luminescence QY of NIR PL gold nanoclusters (AuNCs). We synthesized water-soluble and colloidally stable NIR PL AuNCs with unprecedentedly high QY (∼25%) upon introduction of triphenylphosphonium moieties into the surface capping layer. By using a combination of spectroscopic and theoretical methods, we provide evidence for gold core-to-ligand charge transfer occurring in AuNCs. We envision that this work can stimulate the development of these unusually bright AuNCs for promising optoelectronic, bioimaging, and other applications.
Introduction
Photoluminescent nanosystems have garnered great interest due to their excellent optical properties, low cost, photostability, convenience, and highly sensitive detection.1 Recently, ultrasmall (<2 nm) photoluminescent gold nanoclusters (AuNCs) have been identified as promising candidates for cell labeling, biosensing,2 photo-therapy applications,3 cancer radiotherapy,4,5 and antimicrobial agents.6 Photoluminescent AuNCs are biocompatible and readily bioconjugable, and they show good photostability and low toxicity.7 Photoluminescent AuNCs can emit from the blue to the near-IR (NIR) spectral region depending on the number of atoms within the cluster.8 In recent years, various approaches have been developed to prepare highly photoluminescent AuNCs with emission in the UV-vis region, for example: (1) engineering the particle surface by using different ligands, such as dendrimers,9 DNA,10,11 peptides, and proteins;12–16 (2) controlling the metal core size;17 (3) aggregation-induced emission;18–21 and (4) rigidification of the Au(I)-thiolate shell.22,23 Recent advances in the synthesis of AuNCs have enabled the development of NIR-photoluminescent (NIR PL) AuNCs functionalized with a variety of thiol-containing ligands, including tiopronin,24 thioctic (lipoic) acid,25–27 polymers,28 11-mercaptoundecanoic acid,29 zwitterionic structures,30 glutathione,31–33 mercaptosuccinic acid,16 and proteins.12,34–36The increase in nonradiative pathways with decreasing energy of emitted light (i.e., the “energy gap law”)37 makes it extremely challenging to prepare AuNCs that emit in the NIR region with high quantum yields (QYs). The photoluminescence QY of AuNCs can be enhanced via charge transfer from the ligands to the metal core (i.e., LMCT) through the Au–S bonds and is parallel with the ligand's capability of donating electron density to the metal core through the S–Au bond (i.e., charge transfer capability of the ligand).38 The luminescence of AuNCs can also be “turned on” upon introduction of sulfur-containing ligands39 or enhanced by either sulfur oxidation at the Au–ligand interface40 or electronic polarization of the bonds between the Au core and thiolate ligands.41 Nevertheless, the currently known NIR-emitting AuNCs achieve relatively low QYs.
One promising approach to a great increase in QY is based on an efficient but experimentally laborious doping of the Au core with a precise number of Ag atoms.3 In contrast, ligand-based PL enhancement approaches are more general and synthetically straightforward and can directly produce water-soluble AuNCs. In this report, we present results from our study of ligand-mediated improvement of the NIR luminescence efficiency of AuNCs. We found that the weak emission of AuNCs stabilized with thioctic acid (TA) and polyethylene glycol (PEG) can be enhanced dramatically by introducing triphenylphosphonium cations in the capping layer. Motivated by this finding, we investigated in detail the photophysical properties of these NIR PL AuNCs. We present experimental evidence suggesting the enhancement of the luminescence QY via charge transfer interactions from the gold core to the triphenylphosphonium cations, i.e. by metal-to-ligand charge transfer (MLCT).
Results and discussion
To synthesize NIR PL AuNCs, we have utilized a bottom-up approach where the gold precursor is first treated with suitable ligands, followed by the reduction of the gold. Among the various ligands known for the stabilization of AuNCs, multithiol-based ligands (with at least two coordinating groups) provide enhanced colloidal stability to the AuNCs because of the increased number of binding sites between each ligand and the AuNC surface.30 For primary modification of NIR PL AuNCs, we utilized the bidentate thiol anchor TA providing a remarkable colloidal stability to the AuNCs over a broad range of stringent conditions compared to simple monothiol ligands.42 TA also provides a one-phase growth route to nanoparticles with discrete size control in water and offers a carboxylic acid group on the surface of nanoparticles, which can be further functionalized. However, TA-protected AuNCs are unstable in acidic solutions due to the protonation of the carboxylate group. Hence after the preparation of TA-protected AuNCs using a one-step reduction procedure in water,25 we subsequently functionalized them with thiol-terminated PEG (MW 2000), yielding AuNC 1. This led to the improvement of aqueous solubility of AuNCs in acidic pH and a further increase of their colloidal stability. Then we covalently modified 1 with 3-(aminopropyl)triphenylphosphonium (TPP) salts via amidic coupling to surface carboxyl groups, thereby obtaining AuNC 2 (Fig. 1a). We characterized the AuNC structure by high-resolution transmission electron microscopy (HRTEM), inductively coupled plasma optical emission spectrometry (ICP-OES), 31P and 1H NMR, and zeta potential measurements. HRTEM revealed the formation of monodisperse AuNCs (Fig. 2). AuNCs 1 and 2 both showed a narrow metal core size distribution with an average diameter of 1.15 ± 0.2 nm, as judged from the image analysis of more than 100 individual particles. The 31P NMR spectrum of 2 (Fig. S3a†) showed a single peak at δ = 23.7 ppm, which is similar to that of the phosphonium cation in 3-(aminopropyl)triphenylphosphonium bromide (Fig. S3b†). The 31P NMR spectrum of 2 also indicated that the triphenylphosphonium cation was neither degraded nor oxidized during the synthesis. The integration of signal intensities in 1H NMR of 2 (Fig. S6 in ESI†) provided us the molar ratios PEG![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :TA ≈ 5:2 and TA:TPP ≈ 2:1. If we approximate the size of the nanocluster roughly as Au25 (based on the size obtained from HRTEM and on the PL emission maxima), we obtain 18 attachment positions for thiols. Using the above stated ratios, we obtain approximately 10 PEG, 4 TA (dithiol ligand), and 2 TPP moieties attached to each AuNC 2.
:TA ≈ 5:2 and TA:TPP ≈ 2:1. If we approximate the size of the nanocluster roughly as Au25 (based on the size obtained from HRTEM and on the PL emission maxima), we obtain 18 attachment positions for thiols. Using the above stated ratios, we obtain approximately 10 PEG, 4 TA (dithiol ligand), and 2 TPP moieties attached to each AuNC 2.
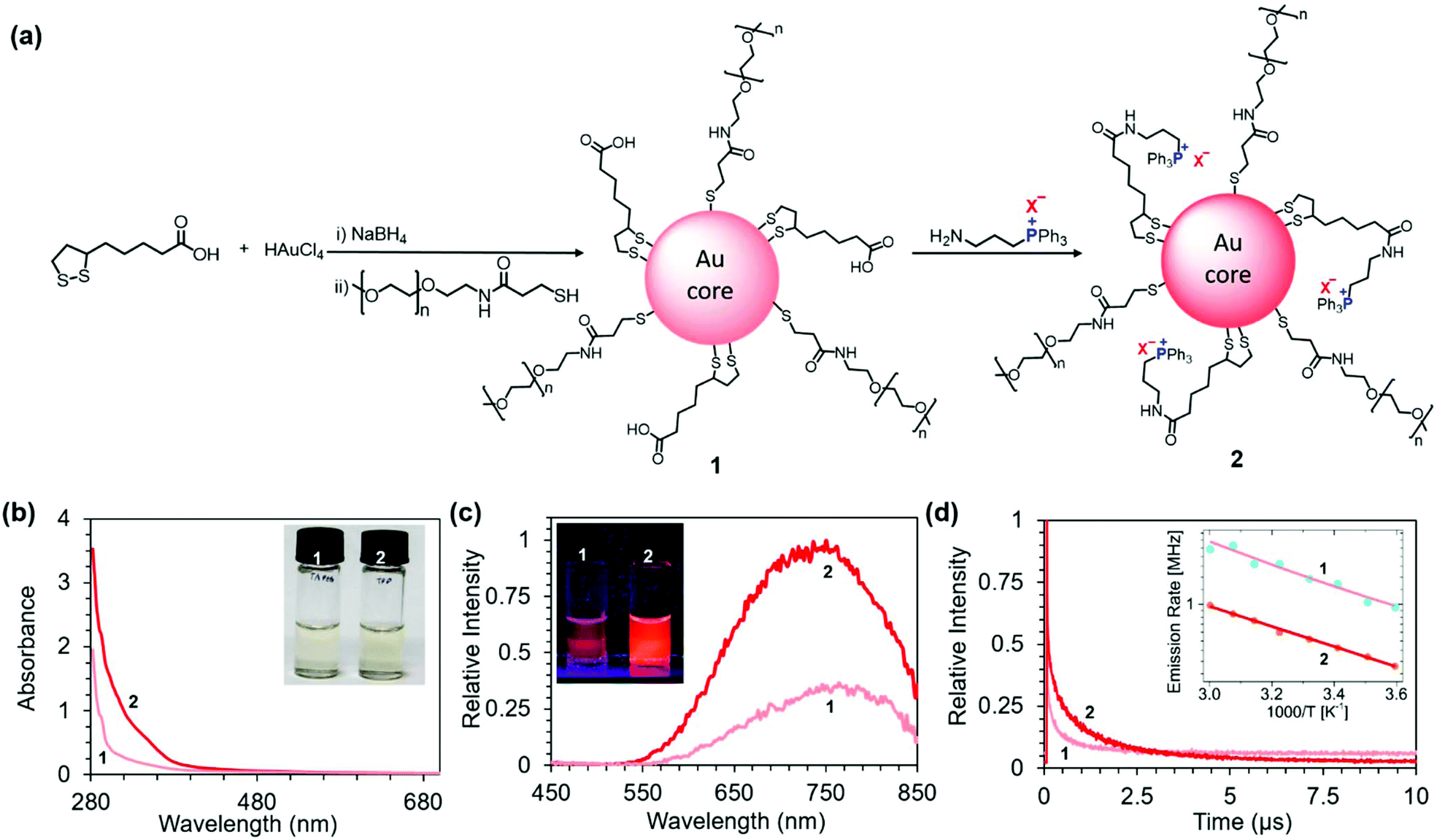 | ||
| Fig. 1 (a) Schematic representation of the structure and preparation of NIR PL AuNCs 1 and 2. X− = Br−, I−, Cl−, or BF4−. (b) Normalized absorption spectra of 200 μg mL−1 aqueous solutions of 1 and 2 (X− = BF4−). Inset: Photograph of the solutions under white light. (c) Normalized emission spectra of 200 μg mL−1 aqueous solutions of 1 and 2 (X− = BF4−) collected upon excitation at 365 nm. Inset: Photograph of the solutions upon UV illumination (365 nm). (d) Time-resolved photoluminescence decay curves for 1 and 2. Inset: Plot of the emission rates (k) of 1 and 2 (X− = BF4−) as a function of reciprocal temperature (1000/T). Fits are based on eqn (2) (note the rate scale is logarithmic with base e). | ||
 | ||
| Fig. 2 Bright field TEM images of 1 (a) and 2 (b) and the corresponding HRTEM images of a single AuNC 1 (c) and 2 (d) showing the typical size around approximately 1 nm. The insets of HRTEM images show the FFTs from the respective particle in the [011] zone where the spots correspond to {111} and {200} lattice planes of Au. | ||
The zeta potentials of 1 and 2 in phosphate buffer (10 mM, pH 7.4) were −10.0 ± 1.8 mV and +1.6 ± 0.3 mV, respectively. The higher zeta potential of 2 compared to 1 is consistent with a charge switch from a part of negatively charged carboxylic groups to positively charged TPP.
As expected, unlike the UV-vis absorption spectra of larger Au nanoparticles, the spectra of the AuNCs did not display a strong surface plasmon resonance around 520 nm (Fig. 1b). The AuNC solutions are therefore only light yellow in color (inset Fig. 1b). However, the enhanced absorption of 2 compared to 1 in the UV region revealed a strong interaction between the gold core and TPP (Fig. 1b). This enhancement of absorption can be attributed to the charge transfer from the metallic core to the ligands, as described by Sementa et al. for other highly delocalized sterically hindered ligands present on AuNCs.43
Both 1 and 2 showed broad emission with the maxima around 750 nm (Fig. 1c). However, upon introduction of TPP, the PL intensity dramatically increased (Fig. 1c). The observed change in the PL intensity corresponds to the increase in PL QYs from 10% to 25% estimated for 1 and 2 (X− = BF4−), respectively (Fig. S1 and Table S1 in the ESI†). The PL increase is visible to the unaided eye, as documented by photographs of the AuNC solutions upon UV illumination (λex = 365 nm) (Fig. 1c inset). We observed that the PL intensity depends on the Au:P ratio (estimated using ICP OES), with a maximum enhancement at Au:P ≈ 8:1 (Fig. S2 in the ESI†). Considering the approximate size of the nanocluster Au25 (based on the size obtained from HRTEM and on the PL emission maxima), this value suggests ∼3 TPP moieties per one AuNC. This result is fairly consistent with the 2 TPPs per AuNC estimated from 1H NMR (see above).
The time-resolved PL decay profiles of the two sets of AuNCs are shown in Fig. 1d. The PL lifetimes were extracted using a three exponential fit from the PL decay curves using the equation
| I(t) = A1e−t/τ1 + A2e−t/τ2 + A3e−t/τ3 + A4, | (1) |
| Set of τ used for the fit | τ 1 (ns) | τ 2 (ns) | τ 3 (μs) | τ 4 (μs) |
|---|---|---|---|---|
| ∼10 | ∼457 | ∼2.09 | >10 | |
| A 1 | A 2 | A 3 | A 4 | |
| AuNC 1 | 0.79 | 0.12 | 0.03 | 0.06 |
| AuNC 2 | 0.57 | 0.23 | 0.17 | 0.03 |
To further investigate the origin of luminescence enhancement in 2, we carried out temperature-dependent luminescence measurements. The inset of Fig. 1d presents the emission rate k (inverse luminescence decay time, k = τPL−1) as a function of 1000/T. We fit the observed temperature dependence with the equation
| k(T) = k0+k′e−ΔE/(kBT), | (2) |
| k 0 (MHz) | k′ (MHz) | ΔE (meV) | |
|---|---|---|---|
| AuNC 1 | 0.07 | 22 | 92 |
| AuNC 2 | 0.25 | 61 | 144 |
To further investigate the Au core to phosphonium cation charge transfer, we employed X-ray photoelectron spectroscopy (XPS). The binding energy (BE) shift of Au 4f band is influenced by the oxidation state of gold. The BE of Au 4f7/2 transition of both 1 and 2 (Fig. 3) falls between the energies of Au(0) (84 eV) and Au(I) (86 eV) of gold thiolate, suggesting the coexistence of Au(0) and Au(I) in both AuNCs.44,50 However, upon conjugation of TPP, the BE of both Au 4f7/2 and 4f5/2 shifts towards higher values. Because both the AuNCs have a similar size and were stored under similar conditions without exposure to oxidizing agents, we attribute the BE shift of Au 4f band to charge transfer from the Au core to phosphonium cations. The charge transfer makes Au atoms in the cluster more positively charged, which in turn increases their core level binding energy. The XPS result of the Au 4f binding energy shift provides direct experimental evidence for charge transfer from the Au core to the phosphonium cation (Fig. 3).
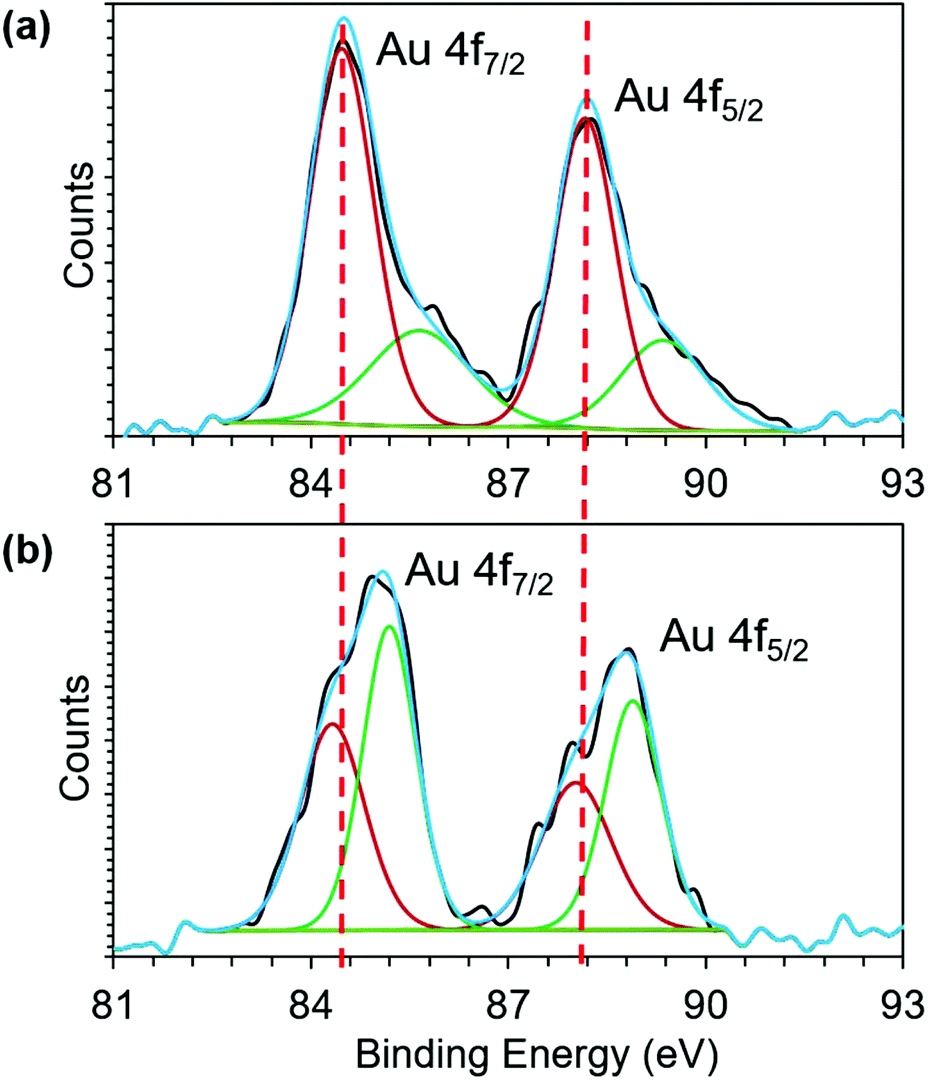 | ||
| Fig. 3 Recorded XPS spectra of the Au 4f peaks (black line) of (a) 1 and (b) 2. The fitted curve (blue) shows that the spectra comprise two doublets of Au(0) (dark red) and Au(I) (green). | ||
Because charge transfer involving the triphenylphosphonium cation is also influenced by the counteranion,51 we investigated the effect of phosphonium salt counteranions on the emission properties of 2. We prepared AuNC 2 variants bearing a set of four different counteranions (BF4−, Cl−, Br−, and I−) by reactions of the corresponding triphenylphosphonium salts. The anions differ in oxidizability,51 forming a series (BF4− > Cl− > Br− > I−) ranging from the nonoxidizable BF4− anion to the most oxidizable I−. As Fig. 4b shows, the PL intensity depends on the counteranion. Decreases in the emission intensity of 2 follow the increasing oxidizability of the counteranions. This trend can be interpreted according to the proposed mechanism of PL enhancement (Fig. 4a). The charge transfer from the counteranion to the phosphonium cation decreases in the order I− > Br− > Cl− > BF4− and competes with the gold core-to-phosphonium ion charge transfer (i.e., MLCT). The more readily oxidizable the counteranion, the more charge transfer takes place from the counteranion to phosphonium cation and the less gold core-to-phosphonium cation charge transfer occurs (and vice versa). These observations further support the involvement of the MLCT mechanism in the enhancement of the PL emission of 2.
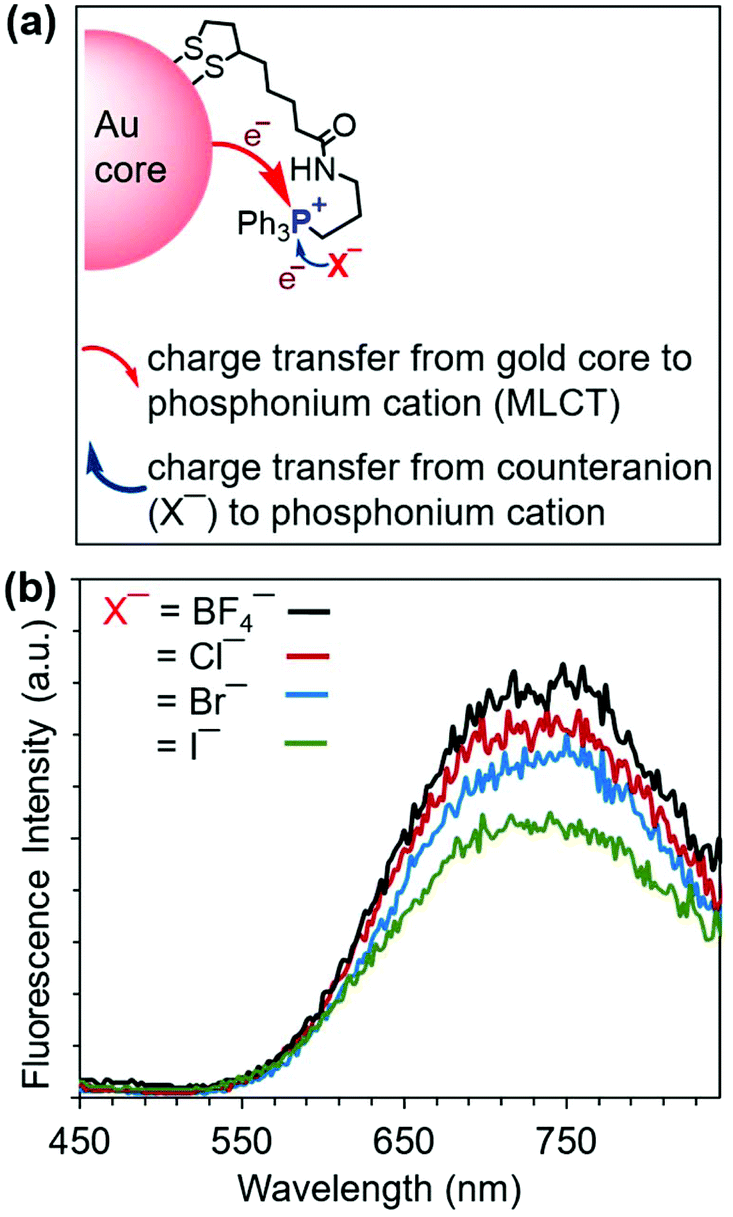 | ||
| Fig. 4 (a) Proposed luminescence enhancement mechanism in 2. (b) The relative photoluminescence spectra of 2 with different counteranions collected upon excitation at 365 nm. | ||
Because the intrinsic hydrophobicity of the TPP moiety containing three phenyl groups can be (partially) responsible for the enhancement of QY due to the increased local hydrophobicity in 2, we performed two different experiments investigating this potential environmental effect. First, we introduced a model hydrophobic moiety 2-aminoanthracene to 1, providing 3 (Scheme S1 in the ESI†). The characteristic intrinsic fluorescence of 2-aminoanthracene in the blue region of the spectra indicates the presence of the moiety on 3. However, in contrast to 1 and 2, the AuNC 3 did not show any PL peak in the wavelength range of 600–850 nm (same concentration of all the AuNCs; Fig. S4 in the ESI†), which would be expected upon an increase of local hydrophobicity. Second, we recorded the PL spectra of 2 in binary mixtures of water and dimethyl sulfoxide (DMSO) with increasing volume fractions of DMSO. DMSO is a known disruptor of hydrogen-bond network in water52 and its presence can strongly modulate the local polarity around fluorophores and influence their QYs.53 Nevertheless, we did not observe any change of PL spectra of 2 with an increasing concentration of DMSO (either in intensity or as a solvatochromic shift; Fig. S5 in the ESI†). Our results suggest that the polarity effects of the TPP do not contribute to the enhancement of the PL of AuNCs.
To analyze further the chemical structure and electronic behaviour of the AuNCs we used 1H NMR spectroscopy. The 1H NMR spectrum of 1 (Fig. S6†) shows that the signals corresponding to the TA are missing. However, after conjugation of TPP with the carboxylic acid group on the surface of 1, the TA signals reappear. The disappearance of TA signals in 1 may be explained by the interaction of TA with a paramagnetic gold core, leading to extreme signal broadening due to the paramagnetic relaxation enhancement. It is likely that the carboxylate group also interacts with the nanoparticle surface, because even the signals of hydrogen atoms close to the carboxylate group are missing in the spectrum (Fig. S6†). The reappearance of the signals after TPP addition indicates that the structure of the interacting radical (gold core) is different. Signals of hydrogen atoms distant from the sulphur atoms are broad but in the same positions as for free TA and signals of hydrogen atoms close to sulphur (and AuNC) are significantly broadened and shifted from their original positions. These spectral patterns indicate that TA interacts with a paramagnetic centre (gold core), but the nature of the radical (gold core) has changed upon TPP introduction leading to a suppression of paramagnetic relaxation and relative line narrowing, most likely because of the MLCT effect.
We have also simulated the possibility of electronic charge transfer from AuNC to TPP under electronic excitations by the time dependent density functional theory (TDDFT). From the emission spectra (Fig. 1c), we suppose that our AuNCs are closely related to the Au25 cluster. The Au25 cluster is based on a centered icosahedral Au13 core, which is capped by an exterior shell composed of twelve Au atoms.54 For simplicity, we approximated our particles with the Au13 core bearing one TPP molecule. The Au13 AuNC interacted strongly with TPP and a substantial amount of electronic charge was transferred from the Au13 nanocluster to the TPP. We found the frontier orbitals (HOMO/LUMO) centered at the phosphorus atom and gold cluster, respectively (Fig. 5). This implies an extensive charge transfer during the excitation, large transition dipole moments and hence strong fluorescence intensities.
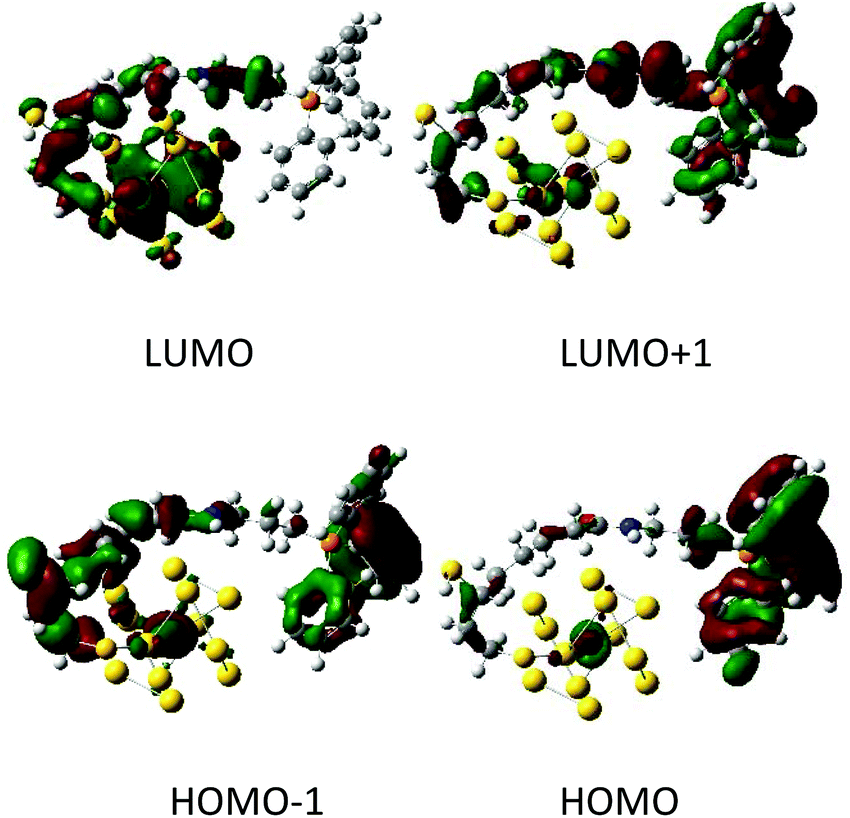 | ||
| Fig. 5 Examples of orbitals in the Au13-TPP model calculated at the BP86/6-31G**/MWB60 level. The red and green colors correspond to the positive and negative lobes. | ||
Finally, to demonstrate a broader applicability of our PL enhancement method, we prepared another NIR PL AuNC 4 stabilized with mercaptosuccinic acid and PEG. Then we conjugated it with TPP salt via amidic coupling to obtain 5 (Scheme S2 in ESI†). We observed a similar enhancement of absorption and PL upon conjugation of TPP to the case of 2 (Fig. S8†). This experimental result shows that the method developed in this study can be extended to other thiolate-protected NIR emitting AuNCs for PL enhancement.
By using a simple synthetic procedure, we were able to prepare high QY colloidally stable AuNCs emitting in the NIR region. In comparison with other reported methods3 of substitution our two step synthetic method in water (without the need for an organic solvent) at room temperature uses very simple chemistry, which could be appealing for broader research community interested in long term in vitro and in vivo imaging of cells and tissues with a high signal-to-background ratio by avoiding auto fluorescence, reduced light scattering, and high tissue penetration. Most importantly, our method produces water soluble AuNCs, which does not require ligand exchange for biological applications in an aqueous environment. By using state-of-the-art characterization techniques such as HRTEM, XPS, UV-vis absorption, temperature dependent fluorescence lifetime, NMR spectroscopy, counteranion dependent emission enhancement, in combination with DFT modelling, we were able to provide further insight into the mechanism of PL enhancement. Our results indicate a gold-to-phosphonium ion charge transfer, i.e., metal-to-ligand (MLCT) responsible for enhancement of PL. In contrast to commonly observed PL enhancement by ligand-to-metal charge transfer (LMCT) via the S–Au bond from the electron-rich groups (e.g., carboxylic, and amino groups) present in the ligand (e.g., glutathione),38,55 our findings of MLCT mediated that PL enhancement offers an alternative route to achieve high QY NIR PL AuNCs.
Conclusions
We discovered unprecedentedly high enhancement of luminescence in AuNCs explicable by efficient charge transfer from the gold nanocluster to triphenylphosphonium cations present in the ligand-capping layer. Our findings provide a design principle useful for the synthesis of high-QY NIR PL AuNCs for bioimaging applications. We believe that our work will stimulate further experimental and theoretical research on photoluminescent gold nanoclusters for broad applications, including light-harvesting systems that involve charge transfer. Moreover, our results show that in addition to the size and composition of the Au core and ligand attachment chemistry, the electronic properties of surface ligands play an extremely important role in PL behavior of AuNCs. We envision that our simple synthetic protocol can be extended to fabricate highly photoluminescent NIR emitting AuNCs protected with custom-designed ligands with different functionalities.Experimental section
Preparation of AuNC 1
In a typical reaction, 1.7 μL of 470 mg mL−1 HAuCl4·3H2O was added to 3.9 mL deionized water containing TA (1.3 mg, 6.3 μmol) and 10 μL of 2 M NaOH solution. The mixture was stirred at room temperature for 15 min, then 80 μL NaBH4 (1.9 mg mL−1 freshly prepared stock solution) was added, followed by further stirring of the reaction mixture overnight. The solution was purified by applying three cycles of centrifugation/filtration using a membrane filtration device (Millipore) with a molecular weight cut-off 3 kDa. Next, thiol-terminated polyethylene glycol (MW 2000; 2.6 mg; 1.3 μmol) was added to the solution, and the mixture was stirred overnight. The dispersion was purified by applying three cycles of centrifugation/filtration using a membrane filtration device (Millipore) with a molecular weight cut-off of 3 kDa.Preparation of AuNC 2
AuNC 1 solution (4 mL, 0.2 mg mL−1) and 3-(aminopropyl)triphenylphosphonium bromide (or the respective TPP salt with BF4−, Cl−, or I−) (2 mg, ∼5 μmol) were mixed together, and the pH was adjusted to the range of 4.5–6.0 with 1 M HCl. The reaction was started by adding excess N-(3-dimethylaminopropyl)-N′-ethylcarbodiimide hydrochloride (EDC·HCl) (10 mg, 52 μmol) and was stirred overnight. The dispersion was purified by applying three cycles of centrifugation/filtration using a membrane filtration device (Millipore) with a molecular weight cut-off of 3 kDa to obtain AuNC 2.Characterization
UV-vis absorption and photoluminescence spectra of AuNCs were recorded using a TECAN M1000 microplate reader. Phosphorus and gold contents were estimated using an optical emission spectrometer (OES) with radial observation of inductively coupled plasma (ICP) (SPECTRO Arcos – SPECTRO Analytical Instruments, Kleve, Germany). NMR spectra were recorded on a Bruker Avance III 500 spectrometer (499.88 MHz for 1H and 125.71 MHz for 13C) equipped with a 5 mm PFG cryoprobe. All 1H and 13C spectra were acquired for samples in CD3CN and referenced to the solvent signal (1.94 ppm in 1H). Signals of all hydrogen atoms of TA were assigned by using a combination of 1D and 2D (H,H-COSY, H,C-HSQC and H,C-HMBC) techniques. HRTEM was performed with an FEI Osiris instrument operated at 200 kV. The samples were prepared by drop-casting on an ultrathin film C grid. Zeta potential experiments were carried out on a Zetasizer Nano ZSP (Malvern Instruments, UK) with a 633 nm laser at 25 °C. For zeta potential measurements, the universal dip cell in disposable cuvettes was used. 10 μL of AuNC solution (200 μg mL−1) was mixed with 1 mL phosphate buffer solution (10 mM, pH 7.4) and the solution was used for the measurement. For zeta potential measurements, the average value of at least three data points is reported.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the GACR project Nr. 18-12533S. J. V. acknowledges funding from the Ministry of Education, Youth and Sports of the Czech Republic via the V4+Japan project No. 8F15001 (cofinanced by the International Visegrad Fund). P. B. acknowledges GACR project No. 16-05935S and Ministry of Education, Youth and Sports of the Czech Republic project No. LTC17012.References
- C. Dong, Z. Liu, J. Liu, C. Wu, F. Neumann, H. Wang, M. Schäfer-Korting, B. Kleuser, J. Chang, W. Li, N. Ma and R. Haag, Adv. Healthcare Mater., 2016, 5, 2214–2226 CrossRef CAS PubMed.
- L.-Y. Chen, C.-W. Wang, Z. Yuan and H.-T. Chang, Anal. Chem., 2015, 87, 216–229 CrossRef CAS PubMed.
- S. Wang, X. Meng, A. Das, T. Li, Y. Song, T. Cao, X. Zhu, M. Zhu and R. Jin, Angew. Chem., Int. Ed., 2014, 53, 2376–2380 CrossRef CAS PubMed.
- N. Goswami, Z. Luo, X. Yuan, D. T. Leong and J. Xie, Mater. Horiz., 2017, 4, 817–831 RSC.
- X.-D. Zhang, Z. Luo, J. Chen, X. Shen, S. Song, Y. Sun, S. Fan, F. Fan, D. T. Leong and J. Xie, Adv. Mater., 2014, 26, 4565–4568 CrossRef CAS PubMed.
- K. Zheng, M. I. Setyawati, D. T. Leong and J. Xie, ACS Nano, 2017, 11, 6904–6910 CrossRef CAS PubMed.
- Y. Ju-Nam, Y.-S. Chen, J. J. Ojeda, D. W. Allen, N. A. Cross, P. H. E. Gardiner and N. Bricklebank, RSC Adv., 2012, 2, 10345–10351 RSC.
- J. Zheng, C. Zhang and R. M. Dickson, Phys. Rev. Lett., 2004, 93, 077402 CrossRef PubMed.
- J. Zheng, J. T. Petty and R. M. Dickson, J. Am. Chem. Soc., 2003, 125, 7780–7781 CrossRef CAS PubMed.
- R. Zhou, M. Shi, X. Chen, M. Wang and H. Chen, Chem. – Eur. J., 2009, 15, 4944–4951 CrossRef CAS PubMed.
- W.-Y. Chen, G.-Y. Lan and H.-T. Chang, Anal. Chem., 2011, 83, 9450–9455 CrossRef CAS PubMed.
- H. Kawasaki, K. Hamaguchi, I. Osaka and R. Arakawa, Adv. Funct. Mater., 2011, 21, 3508–3515 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, J. Am. Chem. Soc., 2009, 131, 888–889 CrossRef CAS PubMed.
- T.-H. Chen and W.-L. Tseng, Small, 2012, 8, 1912–1919 CrossRef CAS PubMed.
- P. L. Xavier, K. Chaudhari, A. Baksi and T. Pradeep, Nano Rev., 2012, 3, 14767 CrossRef CAS PubMed.
- M. A. H. Muhammed, S. Ramesh, S. S. Sinha, S. K. Pal and T. Pradeep, Nano Res., 2010, 1, 333–340 CrossRef.
- J. Zheng, P. R. Nicovich and R. M. Dickson, Annu. Rev. Phys. Chem., 2007, 58, 409–431 CrossRef CAS PubMed.
- Z. Luo, X. Yuan, Y. Yu, Q. Zhang, D. T. Leong, J. Y. Lee and J. Xie, J. Am. Chem. Soc., 2012, 134, 16662–16670 CrossRef CAS PubMed.
- N. Goswami, F. Lin, Y. Liu, D. T. Leong and J. Xie, Chem. Mater., 2016, 28, 4009–4016 CrossRef CAS.
- N. Goswami, Q. Yao, Z. Luo, J. Li, T. Chen and J. Xie, J. Phys. Chem. Lett., 2016, 7, 962–975 CrossRef CAS PubMed.
- A. Yahia-Ammar, D. Sierra, F. Mérola, N. Hildebrandt and X. Le Guével, ACS Nano, 2016, 10, 2591–2599 CrossRef CAS PubMed.
- K. Pyo, V. D. Thanthirige, K. Kwak, P. Pandurangan, G. Ramakrishna and D. Lee, J. Am. Chem. Soc., 2015, 137, 8244–8250 CrossRef CAS PubMed.
- H.-H. Deng, X.-Q. Shi, F.-F. Wang, H.-P. Peng, A.-L. Liu, X.-H. Xia and W. Chen, Chem. Mater., 2017, 29, 1362–1369 CrossRef CAS.
- G. Wang, T. Huang, R. W. Murray, L. Menard and R. G. Nuzzo, J. Am. Chem. Soc., 2005, 127, 812–813 CrossRef CAS PubMed.
- L. Shang, N. Azadfar, F. Stockmar, W. Send, V. Trouillet, M. Bruns, D. Gerthsen and G. U. Nienhaus, Small, 2011, 7, 2614–2620 CrossRef CAS PubMed.
- C.-A. J. Lin, T.-Y. Yang, C.-H. Lee, S. H. Huang, R. A. Sperling, M. Zanella, J. K. Li, J.-L. Shen, H.-H. Wang, H.-I. Yeh, W. J. Parak and W. H. Chang, ACS Nano, 2009, 3, 395–401 CrossRef CAS PubMed.
- D. Mishra, F. Aldeek, E. Lochner, G. Palui, B. Zeng, S. Mackowski and H. Mattoussi, Langmuir, 2016, 32, 6445–6458 CrossRef CAS PubMed.
- X. Huang, Y. Luo, Z. Li, B. Li, H. Zhang, L. Li, I. Majeed, P. Zou and B. Tan, J. Phys. Chem. C, 2011, 115, 16753–16763 CAS.
- H.-C. Chang, Y.-F. Chang, N.-C. Fan and J. A. Ho, ACS Appl. Mater. Interfaces, 2014, 6, 18824–18831 CAS.
- F. Aldeek, M. A. H. Muhammed, G. Palui, N. Zhan and H. Mattoussi, ACS Nano, 2013, 7, 2509–2521 CrossRef CAS PubMed.
- K. G. Stamplecoskie and P. V. Kamat, J. Am. Chem. Soc., 2014, 136, 11093–11099 CrossRef CAS PubMed.
- Z. Wu, C. Gayathri, R. R. Gil and R. Jin, J. Am. Chem. Soc., 2009, 131, 6535–6542 CrossRef CAS PubMed.
- Y. Negishi, Y. Takasugi, S. Sato, H. Yao, K. Kimura and T. Tsukuda, J. Am. Chem. Soc., 2004, 126, 6518–6519 CrossRef CAS PubMed.
- X. Wen, P. Yu, Y.-R. Toh, A.-C. Hsu, Y.-C. Lee and J. Tang, J. Phys. Chem. C, 2012, 116, 19032–19038 CAS.
- X. Wen, P. Yu, Y.-R. Toh and J. Tang, J. Phys. Chem. C, 2012, 116, 11830–11836 CAS.
- P.-C. Chen, C.-K. Chiang and H.-T. Chang, J. Nanopart. Res., 2012, 15, 1336 CrossRef.
- J. Michl and V. Bonacic-Koutecky, Electronic Aspects of Organic Photochemistry, John Wiley & Sons, Inc., New York, 1990 Search PubMed.
- Z. Wu and R. Jin, Nano Lett., 2010, 10, 2568–2573 CrossRef CAS PubMed.
- S. E. Crawford, C. M. Andolina, A. M. Smith, L. E. Marbella, K. A. Johnston, P. J. Straney, M. J. Hartmann and J. E. Millstone, J. Am. Chem. Soc., 2015, 137, 14423–14429 CrossRef CAS PubMed.
- J. Jiang, C. V. Conroy, M. M. Kvetny, G. J. Lake, J. W. Padelford, T. Ahuja and G. Wang, J. Phys. Chem. C, 2014, 118, 20680–20687 CAS.
- G. Wang, R. Guo, G. Kalyuzhny, J.-P. Choi and R. W. Murray, J. Phys. Chem. B, 2006, 110, 20282–20289 CrossRef CAS PubMed.
- E. Oh, K. Susumu, R. Goswami and H. Mattoussi, Langmuir, 2010, 26, 7604–7613 CrossRef CAS PubMed.
- L. Sementa, G. Barcaro, A. Dass, M. Stener and A. Fortunelli, Chem. Commun., 2015, 51, 7935–7938 RSC.
- C. Zhou, C. Sun, M. Yu, Y. Qin, J. Wang, M. Kim and J. Zheng, J. Phys. Chem. C, 2010, 114, 7727–7732 CAS.
- A. Vogler and H. Kunkely, Coord. Chem. Rev., 2001, 219–221, 489–507 CrossRef CAS.
- V. W.-W. Yam and E. C.-C. Cheng, Chem. Soc. Rev., 2008, 37, 1806–1813 RSC.
- T. Huang and R. W. Murray, J. Phys. Chem. B, 2001, 105, 12498–12502 CrossRef CAS.
- E. Sakuda, Y. Ando, A. Ito and N. Kitamura, Inorg. Chem., 2011, 50, 1603–1613 CrossRef CAS PubMed.
- E. A. Medlycott and G. S. Hanan, Chem. Soc. Rev., 2005, 34, 133–142 RSC.
- Y. Negishi, K. Nobusada and T. Tsukuda, J. Am. Chem. Soc., 2005, 127, 5261–5270 CrossRef CAS PubMed.
- C. Imrie, T. A. Modro and C. C. P. Wagener, J. Chem. Soc., Perkin Trans. 2, 1994, 1379–1382 RSC.
- K.-I. Oh, K. Rajesh, J. F. Stanton and C. R. Baiz, Angew. Chem., Int. Ed., 2017, 56, 11375–11379 CrossRef CAS PubMed.
- J. R. Lakowicz, Principles of Fluorescence Spectroscopy, Springer, Boston, MA, 3rd edn, 2006 Search PubMed.
- M. Zhu, C. M. Aikens, F. J. Hollander, G. C. Schatz and R. Jin, J. Am. Chem. Soc., 2008, 130, 5883–5885 CrossRef CAS PubMed.
- K. G. Stamplecoskie, Y.-S. Chen and P. V. Kamat, J. Phys. Chem. C, 2014, 118, 1370–1376 CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr06050e |
| This journal is © The Royal Society of Chemistry 2018 |