
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Metallic impurities in black phosphorus nanoflakes prepared by different synthetic routes†
Carmen C.
Mayorga-Martinez
 a,
Zdeněk
Sofer
b,
David
Sedmidubský
b,
Jan
Luxa
b,
Bahareh
Kherzi
a and
Martin
Pumera
*ab
a,
Zdeněk
Sofer
b,
David
Sedmidubský
b,
Jan
Luxa
b,
Bahareh
Kherzi
a and
Martin
Pumera
*ab
aDivision of Chemistry & Biological Chemistry, School of Physical Mathematical Science, Nanyang Technological University, Singapore 637371, Singapore. E-mail: pumera.research@gmail.com
bDepartment of Inorganic Chemistry, University of Chemistry and Technology Prague Technická 5, 166 28 Prague 6, Czech Republic
First published on 5th January 2018
Abstract
The allotropes of elementary phosphorus materials, such as black phosphorus and its single layer form phosphorene have recently regained scientific attention due to their outstanding properties suitable for applications in electronics, optical devices, and energy applications. As with many other materials, there is a fundamental question of the presence of impurities in these materials and their influence on the properties. Such impurities are expected to dramatically influence the electronic and electrochemical properties of black phosphorus in a way similar to the way they do in the case of graphene. Here, we synthesize black phosphorus from commercially available red phosphorus via different techniques. We utilize high-pressure conversion of red phosphorus to black phosphorus and vapor phase growth of black phosphorus using a gold/tin alloy-like solvent for red phosphorus. We show that both methods lead to black phosphorus containing a large number of metallic impurities. We show that the classical tin-based method for preparing black phosphorus adds a significant amount of tin and that the metallic impurities present have an observable effect on the electrochemical properties of black phosphorus.
Introduction
Black phosphorus has attracted scientific attention as this material is an elemental solid and individual layers are held together by weak van der Waals interactions to create a crystal with orthorhombic symmetry.1–3 Black phosphorus can therefore be exfoliated to few or single layered phosphorene, in a similar manner of graphite to graphene or bulk MoS2 to its single layer form. Black phosphorus was first prepared more than 100 years ago using a high hydrostatic pressure of 1.2 GPa at 200 °C within 5–30 min.4 More than sixty years ago the first “low pressure” synthesis procedure was developed based on the catalytic conversion of white phosphorus with mercury5 and recently also a new method using Sn–Au alloys as well as pure Sn in the presence of SnI4 based on the vapour transport growth method was developed.6–8 Black phosphorus remained a scientific curiosity for almost a hundred years, until recently when it was reported to have potential applications in photocatalysis9–11 semiconductors,12 rechargeable batteries13 and (bio)sensing.14–16 There are several methods that are used for the preparation of black phosphorus, such as high pressure or Au/Sn alloy based vapour phase growth using red phosphorus as a precursor. White, black and red phosphorus are allotropes of elemental phosphorus. Each allotrope exhibits different crystal structures: white phosphorus displays a cubic structure, while red phosphorus is amorphous and black phosphorus has an orthorhombic structure. Single layer black phosphorus has recently been termed phosphorene to show its relationship with 2D graphene (although in the case of BP the suffix “-ene” has no nomenclature support). Despite the fact that BP does not show aromaticity or single atom thickness as graphene, it has something else in common with graphene as we report below. It was believed for many years that graphene is a pure material and it was found only later that the graphene contains metallic impurities, either originating from graphite as a source material,17,18 from the catalyst in the case of CVD graphene19,20 or from the chemicals (and impurities in these chemicals) used for graphene preparation.21,22 It was shown that these metallic impurities dramatically influence its catalytic21,23 and electronic properties.20 Graphene, in many ways, followed the previous example of carbon nanotubes.12,22 In a similar way, it was demonstrated that MoS2 contains many impurities24–29 which originate from the natural mineral of MoS2 or from the synthetic precursors of MoS2. These impurities influence the electronic and catalytic properties of MoS2.30,31 Having these examples of layered and 2D materials in mind and given the explosive growth of black phosphorus applications in many areas, from electronic, to optical to electrochemical applications, one would be curious to know whether there are any impurities in black phosphorus and whether they have any influence on its properties.Here we prepare black phosphorus (BP) by two fundamentally different routes, high pressure conversion (HPC) or vapour phase growth (VPG) using Au/Sn as a solvent (Scheme 1). In both cases, highly pure red phosphorus is used as a precursor. We show that indeed, there is a significant amount of metallic impurities in black phosphorus, and we demonstrate that the impurities influence its electrochemical properties. These findings are of immense conceptual importance when discussing the fabrication of black phosphorus derived devices.
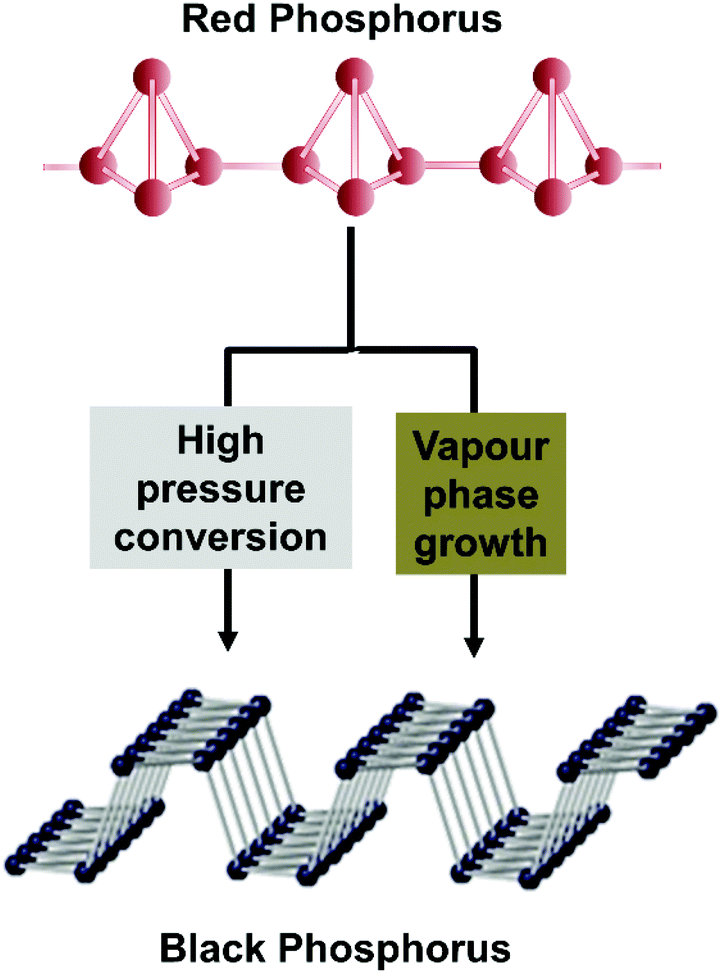 | ||
| Scheme 1 Crystal structure and synthesis methods used for the production of black phosphorus from red phosphorus. | ||
Experimental section
Materials
Red phosphorus (99.999%) and tin (99.999%) were obtained from Sigma-Aldrich (Czech Republic). Iodine (99.9%), carbon disulfide (99.99%) and chloroform (99.9%) were obtained from Penta, Czech Republic. Gold (99.99%) was obtained from Safina, Czech Republic.For the first method, 10 g of red phosphorus was wrapped in graphite foil and was exposed to high pressure (6 GPa) and temperature (600 °C) conditions using uniaxial pressing apparatus of 1′′ size, followed by slowly cooling (100 °C min−1) to room temperature. Finally, the resulting ceramic piece (20 mm × 5 mm) was mechanically removed from the graphite foil. For the preparation of BP using the second method, we followed the method reported by Lange et al. in 20076 and modified by our group.8 500 mg of Au/Sn alloy (prepared by melting stoichiometric amounts of tin and gold under high vacuum), 15 mg SnI4 and 720 mg of red phosphorus were placed inside a quartz ampoule, followed by sealing of the ampoule using an oxygen/hydrogen torch. Subsequently, the ampoule was heated at 400 °C for 1 hour using a muffle furnace and kept in these conditions for two more hours. After that the temperature was increased until 600 °C for 24 h. Finally, the muffle furnace was cooled to room temperature overnight, and then the resulting plates (up to 5 mm × 2 mm) were washed with CS2 to remove the remaining white phosphorus32 and SnI433 formed during the synthesis. For the preparation of SnI4, iodine and tin in chloroform was placed under reflux followed by the purification of SnI4 obtained by recrystallization from chloroform. After the materials were synthesized, a solution of 5 mg mL−1 of each material was prepared and subjected to sonication for 6 hours to obtain a good nanosheet dispersion.
where
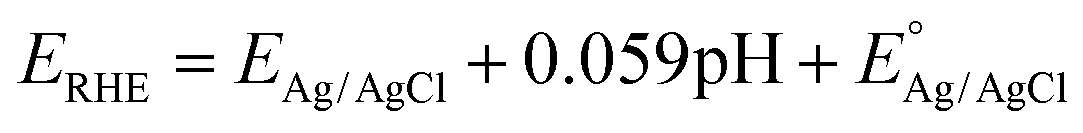
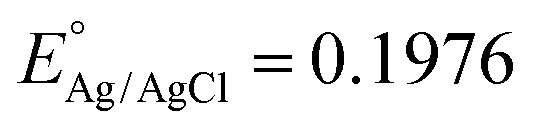 at 25 °C
at 25 °C
Results and discussion
After black phosphorus materials were synthesized using high purity chemical reagents by high pressure conversion and vapor phase growth synthesis (Scheme 1), we characterized the resulting black phosphorus materials with a variety of methods, including scanning electron microscopy (SEM), transmission electron microscopy (TEM), X-ray diffraction (XRD), X-ray photoelectron spectroscopy (XPS) and inductively coupled plasma mass spectrometry (ICP-MS).The morphology of black phosphorus prepared by high pressure conversion (BP HPC) and vapor phase growth (BP VPG) synthesis was studied by SEM (Fig. 1). The layered morphology is clearly visible only for black phosphorus prepared by vapour phase growth. The layered morphology was not clearly observed for black phosphorus prepared by high pressure synthesis, which is also indicated by the broadening of the X-ray diffraction patterns due to its nanocrystalline structure as discussed below. For comparison, the SEM image of amorphous red phosphorus used as a starting material is also shown in Fig. 1.
 | ||
| Fig. 1 The SEM images at different magnifications of red phosphorus (RP), black phosphorus prepared by high-pressure conversion (BP HPC) and vapour phase growth (BP VPG) synthesis. | ||
Once we confirmed that the BP materials were successfully synthesized, the materials were mechanically exfoliated by mild ultrasonic treatment for 6 hours. We used TEM to further investigate the crystallinity of the exfoliated BP nanoflakes. High-resolution TEM (HR-TEM) images and selective area electron diffraction (SAED) patterns show the orthorhombic crystalline nature of the investigated black phosphorus materials (see Fig. 2).
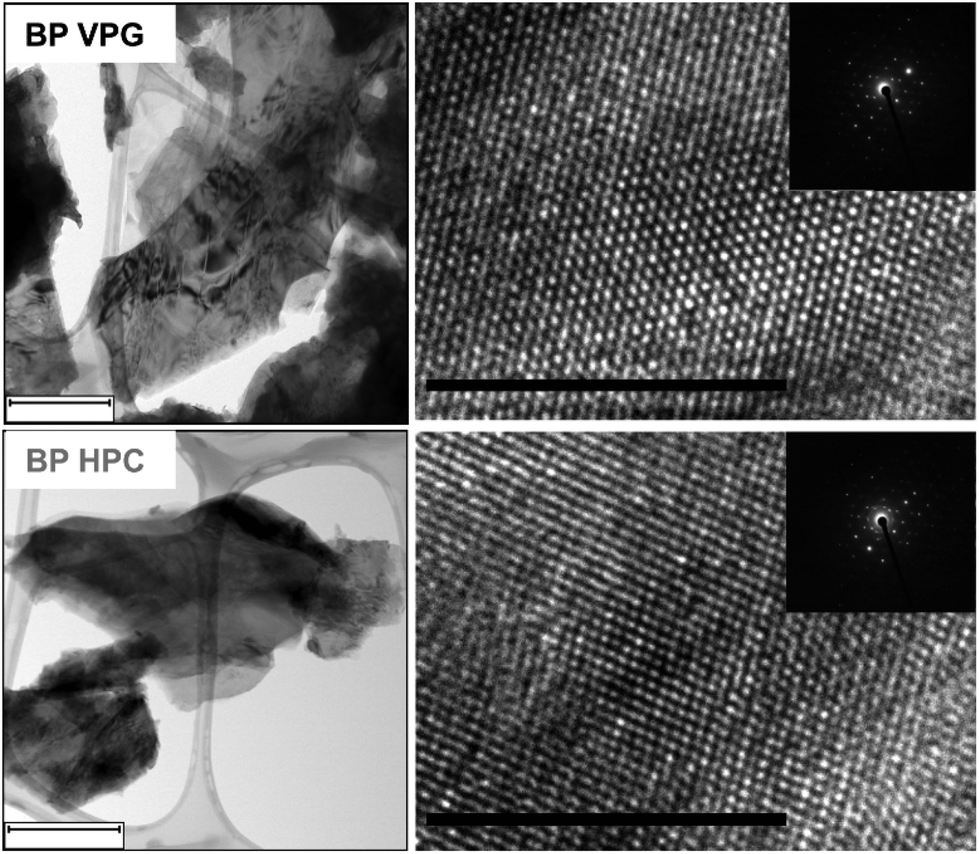 | ||
| Fig. 2 The TEM images (left) and HR-TEM images (right) with the corresponding SAED (insert) of BP HPC and BP VPG nanoflakes. The scale bar corresponds to 500 nm (TEM images) and 10 nm (HR-TEM). | ||
The phase composition of BP HPC and BP VPG was proved by X-ray diffraction. Both the black phosphorus samples prepared by vapour growth and high pressure synthesis show an orthorhombic structure (Cmca space group; PDF card # 01-076-1957). Vapour phase growth phosphorus (BP VPG) shows strong preferential orientation along the (0k0) direction and an extremely narrow diffraction pattern. This indicates the high crystallinity of the material obtained by vapour phase growth and is in good agreement with the observed layered structure by SEM. It was observed that the black phosphorus samples prepared by high pressure synthesis show the significant broadening of all diffraction patterns and only minimal preferential orientation originating from the layered structure. This shows the nanocrystalline character of black phosphorus prepared by high pressure synthesis, in the range of tenths of nanometers corresponding to tenths to hundreds of BP layers. For comparison the diffractogram of amorphous red phosphorus used as a starting material is also shown. The red phosphorus is clearly amorphous. The observed broad diffraction patterns are typical of an amorphous material and correspond to the average inter-atomic distances on short range ordering (chemical bonds in the glass state material; e.g. P4 tetrahedra forming amorphous red phosphorus). The X-ray diffractograms are shown in Fig. 3A.
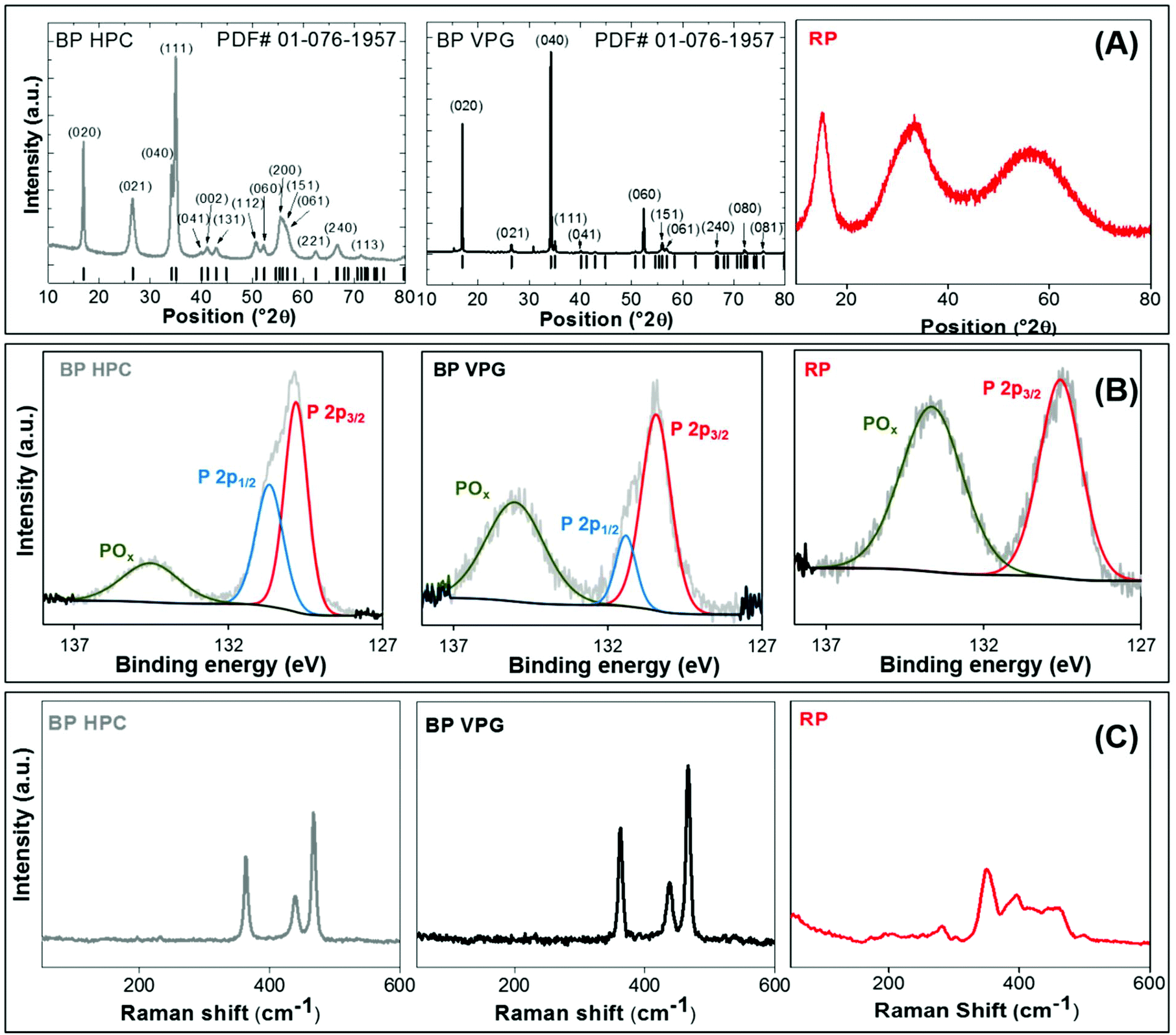 | ||
| Fig. 3 X-ray diffractogram (A), high resolution XPS spectrum of the P 2p core level (B) and Raman spectra (C) of black phosphorus prepared by vapour phase growth (BP VPG), black phosphorus produced by high pressure conversion (BP HPC), and commercial amorphous red phosphorus (RP) used as a precursor for the synthesis. | ||
Furthermore, the prepared BP materials mechanically exfoliated by mild ultrasonic treatment for 6 hours were characterized by X-ray photoelectron spectroscopy (XPS) in order to obtain the detailed information on the chemical composition of their structure and the chemical bonds present. The BP VPG, BP HPC, as well as the precursor RP as the reference were investigated. Fig. 3B shows the high resolution XPS spectrum of the P 2p core level for all the elemental phosphorus allotropes. The characteristic binding energies of two features in the regions of P 2p that correspond to P–P bonding were observed for both black phosphorus materials (VPG and VPC). While for the commercial red phosphorus only one feature was obtained, and the data are in good agreement with the literature.1 The surface oxidation features were observed in all the materials at 134 ± 0.6 eV, being more evident in RP. The oxidative feature observed on the materials is due to low stability and easy passivation of the phosphorus allotropes in water solution.34 Moreover, energy-dispersive X-ray (EDX) spectra were recorded for both BP materials (VPG and HPC) immediately after the synthesis and before being subjected to mechanical exfoliation. The EDX analysis for the commercial RP before dissolving in water by sonication was performed as well (see Fig. S1 in the ESI†). These measurements confirm the absence of oxygen in the commercial red phosphorus; oxygen was introduced during the dissolution by sonication.
Later Raman spectroscopy was performed for the BP materials evaluated in this study, see Fig. 3C. The Raman spectra of BP VPG show the characteristic intensities of A1g, B2g, and A2g at 360 cm−1, 440 cm−1 and 465 cm−1 respectively, while for the BP HPC the intensities were at 362.5 cm−1 (A1g), 440 cm−1 (B2g) and 465 cm−1 (A2g).35–38 The Raman spectrum of RP shows a typical peak of amorphous red phosphorus.39
The metal impurities were quantified using inductively coupled plasma mass spectrometry (ICP-MS). Interestingly, the ICP-MS results of the starting material commercial red phosphorus (with a nominal purity of 99.999%) show the content of some metal impurities such as Cr (41 ppm), Fe (51 ppm), Ni (317 ppm), Cu (147 ppm), Zn (121 ppm), Sn (77 ppm) and Pb (44 ppm). As we can see in Table 1, the amount of Fe, Cu and Zn impurities increased significantly after RP was exposed to high pressure and vapour phase growth methods to obtain BP.
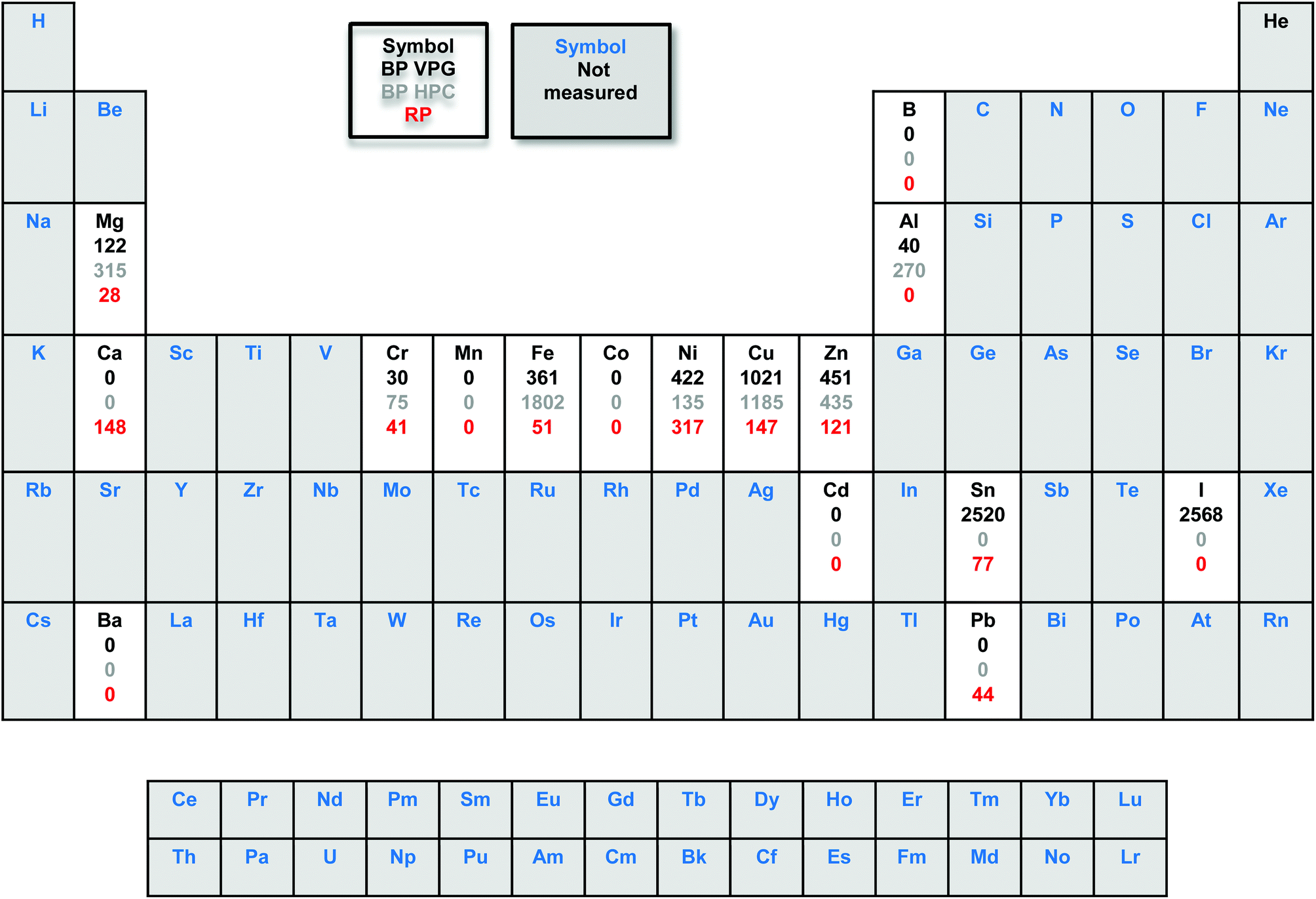
|
However, the Ni content increased for the BP VPG and decreased for BP HPC. Although we used reagents of high purity, the resulting materials have a high content of metallic impurities such as Fe, Ni, Cu, Zn and Sn; also the presence of iodine was observed. Particularly, in the BP vapor phase growth the Sn and I impurities came from Sn and SnI4 used during the synthesis and remained even after washing with CS2. In the case of BP prepared by high pressure conversion the metal impurities came from the high pressure system used for the synthesis and packing in graphite foil used as a vessel for such synthesis.
Subsequently, we were interested in investigating if the metallic impurities had an influence on the electrochemical properties of the materials. It is known that some of the transition metals show the inherent electrochemical signature originating from their oxidation/reduction.39 These include Ni, which is present as an impurity in BP, see Table 1. We investigated whether Ni impurities influence the voltammetric signature of black phosphorus. Fig. 4 shows the CV recorded in alkali media of GC electrodes modified with 5 μg of BP VGP, BP HPC and RP materials; NiO NPs were also tested for comparison. After 20 continuous voltammetric scans, the oxidative and reductive peaks were observed in the BP VPG and BP HPC materials. Fig. 4 shows the 20th scan, where the oxidation peak intensity constantly reaching an almost stable contour. The potentials of these peaks corresponding to Ni(OH)2/NiOOH were observed, as confirmed by comparison with NiO NPs (Fig. 4, blue line).22,40
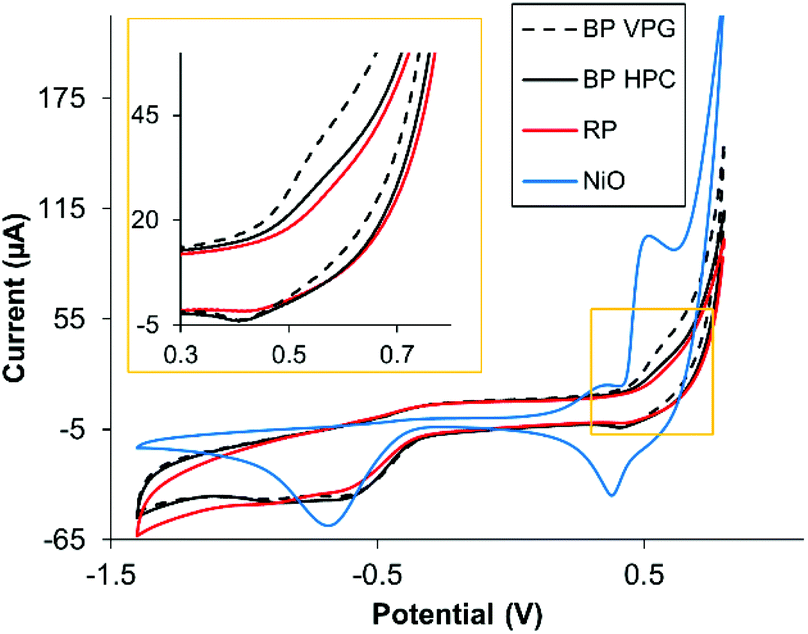 | ||
| Fig. 4 Overlay of the 20th CV scan of NiO NPs and BP VPG, BP HPC and RP in 0.1 M NaOH, scan rate: 0.1 V s−1, GC electrodes (3 mm of diameter) were modified with the materials under study. | ||
In addition, some of the transition metals exhibit a catalytic effect on the hydrogen evolution reaction whilst black phosphorus is not particularly catalytic for hydrogen evolution.3 Differences between different BP materials in their HER activities may serve as an indicator of impurities. We observed that the BP HPC and BP VPG show different hydrogen evolution reactions (HERs) (Fig. 5A); BP HPC shows higher HER electrocatalysis than BP VPG. Interestingly, the BP obtained using the HPC method shows a higher content of Fe impurity than the BP obtained by the VGP method (Table 1). Therefore, we also evaluated the HER performance of Fe3O4 which catalyzes HER. We therefore show the direct relationship between the Fe based impurities in BP and its HER catalysis. Moreover, a slight improvement in the HER of RP is observed, this performance was reported before, where RP was used for visible light driven photocatalytic H2 formation from water.9
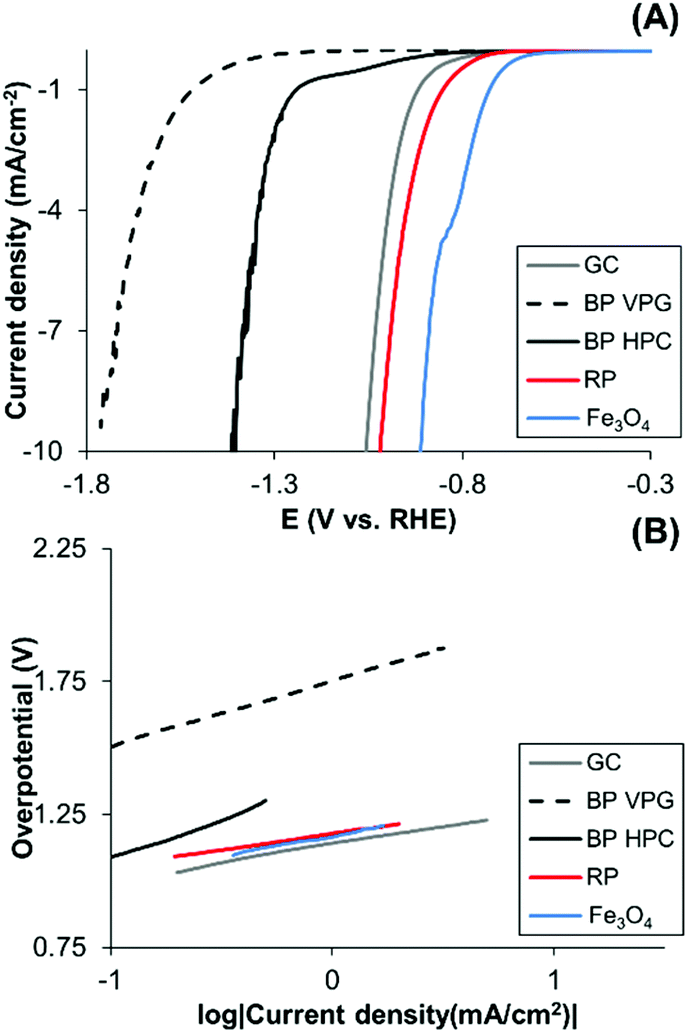 | ||
| Fig. 5 Hydrogen evolution reaction (HER) performed in 0.5 M H2SO4 solution. Linear sweep voltammetry (A) and Tafel plot (B) of BP VPG, BP HPC, RP GC bare and Fe3O4. Glassy carbon electrodes (3 mm of diameter) were modified with the materials under study. | ||
Moreover, to elucidate the electrochemical mechanisms, Tafel analysis is executed (Fig. 5B). The mechanisms of the hydrogen evolution on black phosphorus materials, red phosphorus and Fe3O4 surfaces are in agreement with the Volmer adsorption step.
Conclusion
In summary in this study, we have demonstrated that different synthetic routes (high pressure conversion and vapor phase growth) to black phosphorus introduce or retain different metal impurities. We found that there are impurities in the resulting black phosphorus, which originate from the impurities in the starting red phosphorus and from the agents used for the synthesis of black phosphorus, such as Sn alloy. We demonstrated that metallic impurities, in this case Ni and Fe based, influence the electrochemical response of black phosphorus. Both synthetic methods retain the impurity from the red phosphorus (precursor), Ni, that influences the voltammetric signature of black phosphorus. Fe impurities are introduced during both synthetic methods. The high pressure conversion route introduces a higher amount of Fe impurities that results in a larger catalytic influence upon HER. This study serves as a warning to researchers working in the field of black phosphorus and phosphorene as metal impurities in general influence not only the electrochemical properties but may also influence the electronic and other properties of black phosphorus-derived materials.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. P. thanks the Tier 1 fund (01/13). Z. S., D. S. and J. L. were supported by the Czech Science Foundation (GACR No. 15-09001S) and by the specific university research (MSMT No. 20-SVV/2017). This work was performed with the financial support from the Neuron Foundation for science support. This work was supported by the project Advanced Functional Nanorobots (Reg. No. CZ.02.1.01/0.0/0.0/15_003/0000444 financed by the EFRR).Notes and references
- N. B. Goodman, L. Ley and D. W. Bullett, Phys. Rev. B: Condens. Matter Mater. Phys., 1983, 27, 7440–7450 CrossRef CAS.
- S. H. Aldave, M. N. Yogeesh, W. Zhu, J. Kim, S. S. Sonde, A. P. Nayak and D. Akinwande, 2D Mater., 2016, 3, 014007 CrossRef.
- Z. Sofer, D. Sedmidubsky, S. Huber, J. Luxa, D. Bousa, C. Boothroyd and M. Pumera, Angew. Chem., Int. Ed., 2016, 55, 3382–3386 CrossRef CAS PubMed.
- P. W. Bridgman, J. Am. Chem. Soc., 1914, 36, 1344–1363 CrossRef CAS.
- H. Krebs, H. Weitz and K. H. Worms, Z. Anorg. Allg. Chem., 1955, 280, 119–133 CrossRef CAS.
- S. Lange, P. Schmidt and T. Nilges, Inorg. Chem., 2007, 46, 4028–4035 CrossRef CAS PubMed.
- M. Kopf, N. Eckstein, D. Pfister, C. Grotz, I. Kruger, M. Greiwe, T. Hansen, H. Kohlmann and T. J. Nilges, Cryst. Growth, 2014, 405, 6–10 CrossRef.
- C. C. Mayorga-Martinez, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2015, 127, 14525–14528 CrossRef.
- F. Wang, W. K. H. Ng, J. C. Yu, H. Zhu, C. Li, L. Zhang, Z. Liu and Q. Li, Appl. Catal., B, 2012, 111–112, 409–414 CrossRef CAS.
- Z. Shen, Z. Hu, W. Wang, S.-F. Lee, D. K. L. Chan, Y. Li, T. Gu and J. C. Yu, Nanoscale, 2014, 6, 14163–14167 RSC.
- H. Wang, X. Yang, W. Shao, S. Chen, J. Xie, X. Zhang, J. Wang and Y. Xie, J. Am. Chem. Soc., 2015, 137, 11376–11382 CrossRef CAS PubMed.
- (a) G. Fasol, J. Phys. C: Solid State Phys., 1985, 18, 1729–1741 CrossRef CAS; (b) D. Li, A. E. D. R. Castillo, H. Jussila, G. Ye, Z. Ren, J. Baib, X. Chen, H. Lipsanen, Z. Suna and F. Bonaccorsoc, Appl. Mater. Today, 2016, 4, 17 CrossRef.
- C.-M. Park and H.-J. Sohn, Adv. Mater., 2007, 19, 2465–2468 CrossRef CAS.
- C. C. Mayorga-Martinez, N. M. Latiff, A. Y. S. Eng, Z. Sofer and M. Pumera, Anal. Chem., 2016, 88, 10074–10079 CrossRef CAS PubMed.
- Y. T. Yew, Z. Sofer, C. C. Mayorga-Martinez and M. Pumera, Mater. Chem. Front., 2017, 1, 1130–1136 RSC.
- V. Kumar, J. R. Brent, M. Shorie, H. Kaur, G. Chadha, A. G. Thomas, E. A. Lewis, A. P. Rooney, L. Nguyen, X. L. Zhong, M. G. Burke, S. J. Haigh, A. Walton, P. D. McNaughter, A. A. Tedstone, N. Savjani, C. A. Muryn, P. O'Brien, A. K. Ganguli, D. J. Lewis and P. Sabherwal, ACS Appl. Mater. Interfaces, 2016, 8, 22860–22868 CAS.
- A. Ambrosi, C. K. Chua, B. Khezri, Z. Sofer, R. D. Webster and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 12899–12904 CrossRef CAS PubMed.
- A. Ambrosi, S. Y. Chee, B. Khezri, R. D. Webster, Z. Sofer and M. Pumera, Angew. Chem., Int. Ed., 2012, 51, 500–503 CrossRef CAS PubMed.
- A. Ambrosi and M. Pumera, Nanoscale, 2014, 6, 472–476 RSC.
- G. Lupina, J. Kitzmann, I. Costina, M. Lukosius, C. Wenger, A. Wolff, S. Vaziri, M. Östling, I. Pasternak, A. Krajewska, W. Strupinski, S. Kataria, A. Gahoi, M. C. Lemme, G. Ruhl, G. Zoth, O. Luxenhofer and W. Mehr, ACS Nano, 2015, 9, 4776–4785 CrossRef CAS PubMed.
- C. H. A. Wong, Z. Sofer, M. Kubešová, J. Kucera, S. Matejková and M. Pumera, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 13774–13779 CrossRef CAS PubMed.
- R. J. Toh, A. Ambrosi and M. Pumera, Chem. – Eur. J., 2012, 18, 11593–11596 CrossRef CAS PubMed.
- C. H. A. Wong, C. K. Chua, B. Khezri, R. D. Webster and M. Pumera, Angew. Chem., Int. Ed., 2013, 52, 8685–8688 CrossRef CAS PubMed.
- C. E. Banks, A. Crossley, C. Salter, S. J. Wilkins and R. G. Compton, Angew. Chem., Int. Ed., 2006, 45, 2533–2537 CrossRef CAS PubMed.
- W. Z. Teo and M. Pumera, ChemPhysChem, 2014, 15, 3819–3023 CrossRef CAS PubMed.
- L. Wang and M. Pumera, Chem. Commun., 2014, 50, 12662–12664 RSC.
- L. Wang, A. Ambrosi and M. Pumera, Anal. Chem., 2013, 85, 6195–6197 CrossRef CAS PubMed.
- L. Wang, A. Ambrosi and M. Pumera, Electrochem. Commun., 2013, 26, 71–73 CrossRef CAS.
- M. Pumera, A. Ambrosi and E. L. K. Chng, Chem. Sci., 2012, 3, 3347–3355 RSC.
- R. Addou, S. McDonnell, D. Barrera, Z. Guo, A. Azcatl, J. Wang, H. Zhu, C. L. Hinkle, M. Quevedo-Lopez, H. N. Alshareef, L. Colombo, J. W. P. Hsu and R. M. Wallace, ACS Nano, 2015, 9, 9124–9133 CrossRef CAS PubMed.
- R. J. Toh, Z. Sofer, J. Luxa and M. Pumera, ChemCatChem, 2017, 9, 1168–1171 CrossRef CAS.
- A. D. F. Toy, Comprehensive Inorganic Chemistry, The chemistry of phosphorus, Pergamon Press, Oxford, 1975, ch. 20, pp. 398–542 Search PubMed.
- M. E. Dorfman and J. H. Hildebra, J. Am. Chem. Soc., 1927, 49, 729–737 CrossRef CAS.
- Y. Huang, J. Qiao, K. He, S. Bliznakov, E. Sutter, X. Chen, D. Luo, F. Meng, D. Su, J. Decker, W. Ji, R. S. Ruoff and P. Sutter, Chem. Mater., 2016, 28, 8330–8339 CrossRef CAS.
- V. Eswaraiah, Q. Zeng, Y. Long and Z. Liu, Small, 2016, 12, 3480–3502 CrossRef CAS PubMed.
- S. Bagheria, N. Mansouria and E. Aghaie, Int. J. Hydrogen Energy, 2016, 41, 4085–4095 CrossRef.
- S. Lin, S. Liu, Z. Yang, Y. Li, T. W. Ng, Z. Xu, Q. Bao, J. Hao, C.-S. Lee, C. Surya, F. Yan and S. P. Lau, Adv. Funct. Mater., 2016, 26, 864–871 CrossRef CAS.
- D. J. Olego, J. A. Baumann, M. A. Kuck, R. Schachter, C. G. Michel and P. M. Raccah, Solid State Commun., 1984, 52, 311–314 CrossRef CAS.
- A. Ambrosi, A. Bonanni, Z. Sofer and M. Pumera, Nanoscale, 2013, 5, 2379–2387 RSC.
- W. Z. Teo and M. Pumera, ChemElectroChem, 2014, 1, 249–253 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c7nr05718k |
| This journal is © The Royal Society of Chemistry 2018 |