
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
2-Formylpyrrole natural products: origin, structural diversity, bioactivity and synthesis
James M.
Wood
a,
Daniel P.
Furkert
 *a and
Margaret A.
Brimble
*ab
*a and
Margaret A.
Brimble
*ab
aSchool of Chemical Sciences, University of Auckland, 23 Symonds St, Auckland, 1142, New Zealand. E-mail: m.brimble@auckland.ac.nz; Web: http://www.brimble.chem.auckland.ac.nz
bMaurice Wilkins Centre for Molecular Biodiscovery, University of Auckland, Private Bag 92019, Auckland 1142, New Zealand
First published on 24th July 2018
Abstract
Covering: up to April 2018
2-Formylpyrroles are ubiquitous in nature, arising from the non-enzymatic Maillard reactions of amines and sugars. Often confused for secondary metabolites, these Maillard products display interesting biological activities including hepatoprotective, immunostimulatory, antiproliferative and antioxidant effects. This review presents all 2-formylpyrrole natural products reported to date and identifies structural sub-classes for their categorisation. The origin, biological activity and chemical syntheses of these natural products are discussed herein.
 James M. Wood | James Wood graduated from the University of Auckland with BSc (Hons) in 2014, before undertaking a PhD with Distinguished Professor Margaret Brimble and Dr Daniel Furkert. His doctoral research concerned the synthesis of 2-formylpyrrole natural products using a Maillard-type reaction. He is currently a research associate at the University of Bristol with Professor John Bower. Areas of interest include the development of new synthetic methods to access bioactive heterocycles and their application in total synthesis and drug discovery. |
 Daniel P. Furkert | Daniel Furkert is a Senior Research Fellow at The University of Auckland, leading Professor Brimble's natural product synthesis and medicinal chemistry group in the School of Chemical Sciences. After obtaining his undergraduate and doctoral degrees at The University of Auckland, he carried out postdoctoral fellowships in the total synthesis of azaspiracid at Oregon State University (USA), and medicinal chemistry of novel opioid receptor ligands at the University of Bath (UK). He returned to New Zealand take up his current role in 2010, where his current interests include asymmetric synthesis and medicinal chemistry applications of natural products, novel chemical reactivity and drug discovery. |
 Margaret A. Brimble | Margaret Brimble is a Fellow of the Royal Society (London) and a Distinguished Professor at the University of Auckland where her research program focuses on the synthesis of bioactive natural products and peptides. She has published >460 papers, 65 reviews, holds 30 patents, won the 2018 RSC Sosnovsky Award, 2016 Marsden Medal, 2012 Rutherford Medal (NZ top science award), the 2010 RSC Natural Products Award, named an IUPAC Distinguished Women in Chemistry (2015) and L'Oreal-UNESCO Women in Science laureate for Asia-Pacific, and conferred the Queen's Honour CNZM. She is Past-President of IUPAC Organic and Biomolecular Division III, co-Founder of cancer vaccine company SapVax, Associate Editor for Organic and Biomolecular Chemistry, and Past-President of the International Society of Heterocyclic Chemistry. |
1. Introduction
5-Hydroxymethylpyrrole-2-carbaldehydes (Fig. 1), sometimes referred to as 2-formylpyrroles or pyrralines, have been isolated from a large array of natural sources,1,2 as well as traditional medicine preparations3 and cooked foods.4,5 Their presence in various thermally-processed products reveals their likely origin — the non-enzymatic Maillard reaction — and to date no enzymatic biosynthesis has been proposed for these compounds. Regardless of their non-metabolic origin, these compounds exhibit a range of valuable bioactivities including hepatoprotective,6 immunostimulatory,7 antiproliferative8 and antioxidant effects.1,9,10 Furthermore, certain members of this compound family possess highly unique and complex structures, providing attractive targets for total synthesis and the development of novel synthetic methodologies.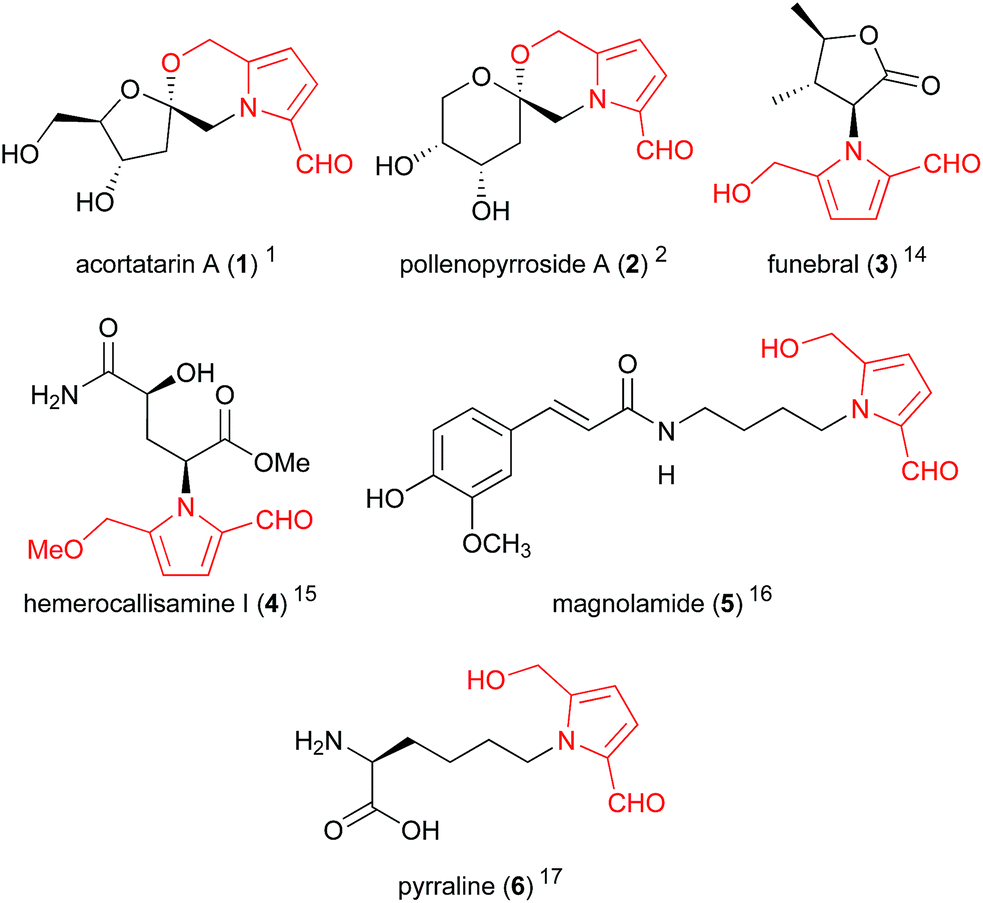 | ||
| Fig. 1 Representative 2-formylpyrroles (5-hydroxymethylpyrrole-2-carbaldehyde system highlighted in red). | ||
This review aims to highlight the interesting biological properties of 2-formylpyrroles and the synthetic methods developed to access them. One particular 2-formylpyrrole, the advanced glycation end-product pyrraline (6), will not be discussed here as it has been reviewed recently.11 Acortatarin A (1) was reviewed previously by Butler and Aponick,12 while a recent review by Shahzad and co-workers13 provides a detailed account of the synthetic approaches to acortatarin A (1) and related pyrrolomorpholine spiroketals. These compounds will be discussed herein within the wider context of 2-formylpyrrole natural products.
2. Non-enzymatic origin
It has been established that 2-formylpyrroles form in biological systems,18 yet no enzymatic biosynthesis has been proposed for the 5-hydroxymethylpyrrole-2-carbaldehyde ring system. Rather, it is accepted that these compounds arise from the condensation of amines with sugars in what are commonly referred to as Maillard or browning reactions.19,20 This hypothesis stems from the first reports of 2-formylpyrroles in which these compounds were isolated directly from reaction mixtures of reducing sugars and amines in 1970.21,22 Kato and Fujimaki characterised pyrroles 13–15 as the reaction products of D-glucose (7) and methylamine (10), ethylamine (11) or butylamine (12) respectively (Scheme 1).21 This reaction was reproducible with hexoses D-galactose (8) and D-fructose (9), albeit with reduced yields.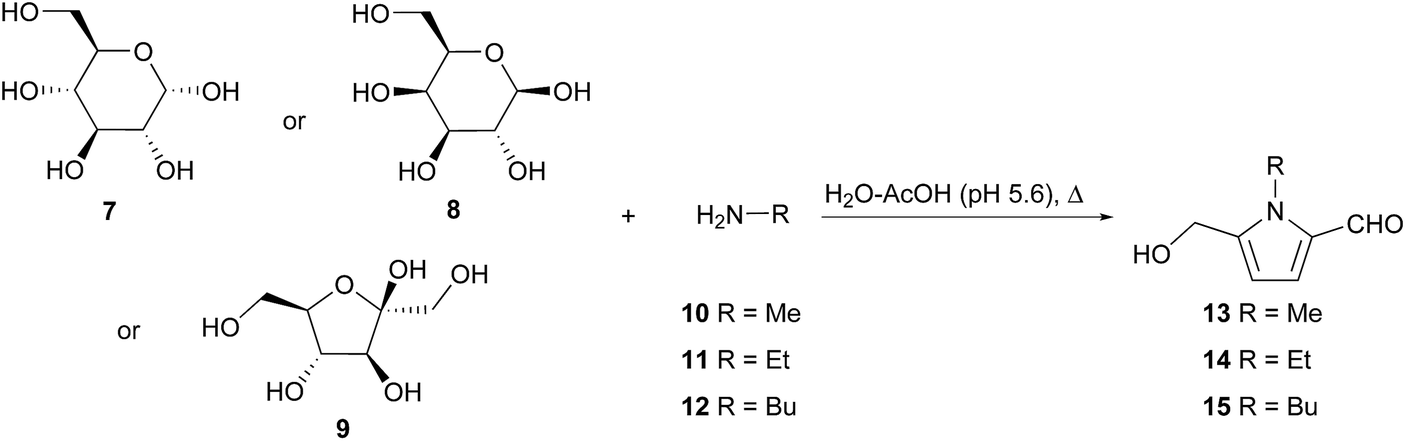 | ||
| Scheme 1 Synthesis of 2-formylpyrroles from amine and hexose reducing sugar mixtures by Kato and Fujimaki. | ||
It is widely proposed that 2-formylpyrroles 26 arise from the condensation of amines with 3-deoxy-D-glucosone (3-DG) (19) (Scheme 2).23 3-DG (19) is an intermediate in many Maillard reaction pathways and is a degradation product of Amadori compounds, which in turn form by condensation of glucose (7) with amines.20,24,25 A number of plausible mechanisms exist for the formation of 2-formylpyrroles 26 from 3-DG (19). It has been suggested that this process is initiated by further dehydration of 3-DG (19) to enone 20 (Scheme 2).23 Keto–enol tautomerisation, followed by conjugate addition of an amine affords hemiaminal 22, which undergoes elimination of water. 5-exo-Trig cyclisation gives rise to another hemiaminal species 25, which undergoes elimination of water to form the aromatic pyrrole ring system.26
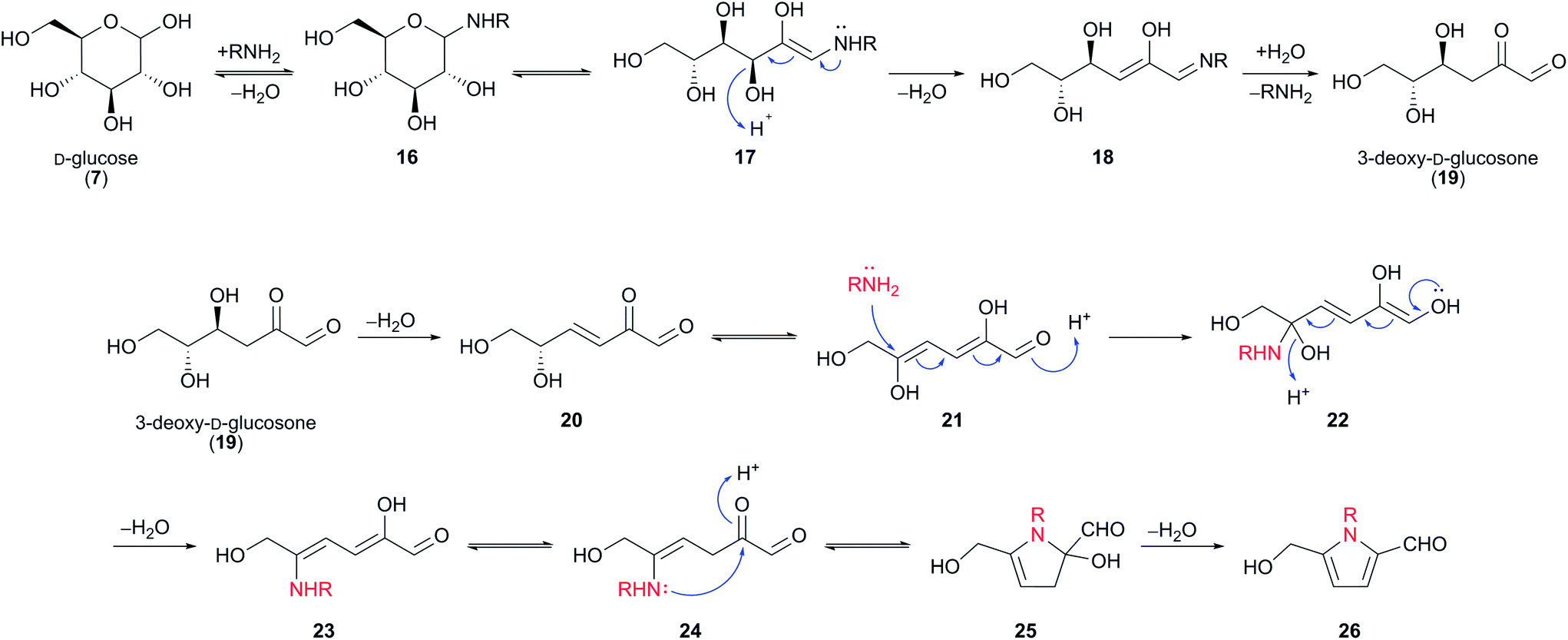 | ||
| Scheme 2 Proposed mechanism for the formation of 2-formylpyrroles 26 from amines and D-glucose (7), via 3-deoxy-D-glucosone (19). | ||
This non-enzymatic synthesis from sugars is quite distinct from most characterised biosynthetic pathways leading to pyrroles, which involve amino acid (glycine, proline, serine, threonine, and tryptophan) and dicarboxylic acid (malonate, oxaloacetate, and succinate) precursors.27 It should however be noted that a unique pyrrole biosynthesis from fructose-6-phosphate (27) was identified by Lautru and co-workers in 2012, operative in Streptomyces ambofaciens (Scheme 3).28 4-Acetamidopyrrole-2-carboxylate (36) was established as a biosynthetic precursor to congocidine (37), a DNA minor groove binder of the pyrrolamide family of natural products. The proposed biosynthetic pathway for 4-acetamidopyrrole-2-carboxylate (36) involves enzymes and precursors from carbohydrate metabolism. The conversion of 4-aminopentose 32 into pyrroline 33 was attributed to Cgc10, an enzyme that resembles a glycosyltransferase. Spontaneous dehydration and subsequent oxidation by a putative alcohol dehydrogenase, Cgc17, delivers 2-formylpyrrole metabolite 35en route to 4-acetamidopyrrole-2-carboxylate (36). This biosynthetic pathway was later extended to the pyrrolamides distamycin and disgocidine in Streptomyces netropsis DSM40846.29 It is therefore evident that biosynthetic pathways exist for the conversion of carbohydrates into 2-formyl and 2-carboxylpyrroles, and it remains to be seen whether similar biosynthetic pathways will be characterised for 5-hydroxymethylpyrrole-2-carbaldehydes.
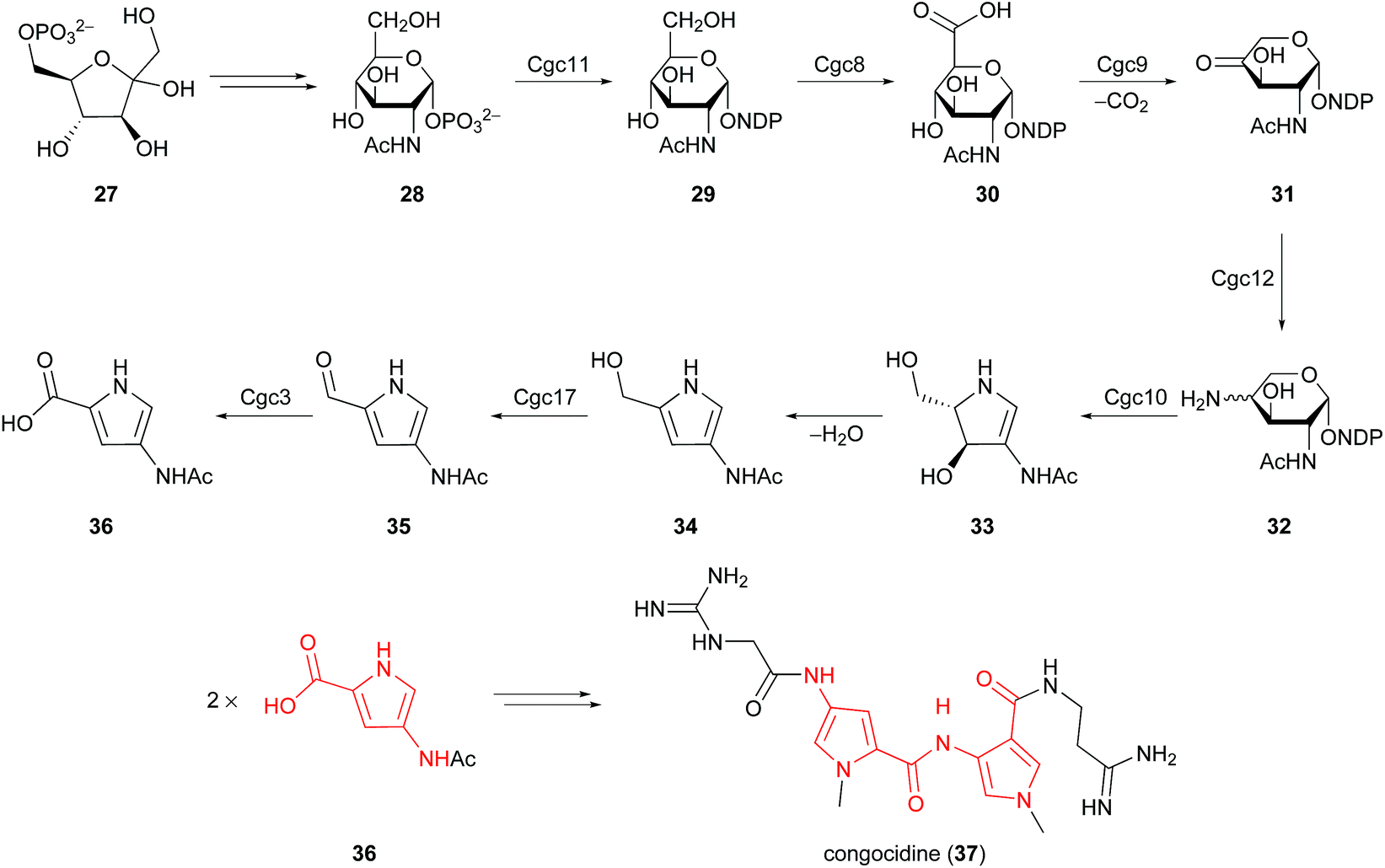 | ||
| Scheme 3 Proposed biosynthesis of 4-acetamidopyrrole-2-carboxylate (36) from fructose-6-phosphate (27) in Streptomyces ambofaciens. | ||
3. Structural diversity and bioactivity
2-Formylpyrroles appear frequently in the natural products isolation literature. The sources from which these compounds have been isolated are of limited significance, as no evidence exists for a biosynthetic pathway for these compounds. As such, details of isolation will be minimal, primarily serving to illustrate the ubiquity of 2-formylpyrroles in nature. This section instead highlights compounds with interesting biological activities and provides an overview of the structural diversity across this compound family. Compounds have been organised into three broad categories according to the type of amine from which they arise: amino acid, biogenic amine or amino sugar.3.1. 2-Formylpyrroles derived from amino acids
Compounds which incorporate proteinogenic amino acids represent a major sub-class of 2-formylpyrroles (Fig. 2). Their prevalence reflects the ubiquity of the amino acids from which they arise. As an example of this prevalence, phenylalanine-derived lactone 38 has been isolated from flue-cured tobacco,30,31 fruit (Celastrus orbiculatus Thunb. (Celastraceae),32Morus alba33), fungi (Xylaria nigripes34) and actinobacteria (Jishengella endophytica 161111,35Streptomyces albospinus RLe7 (ref. 36)).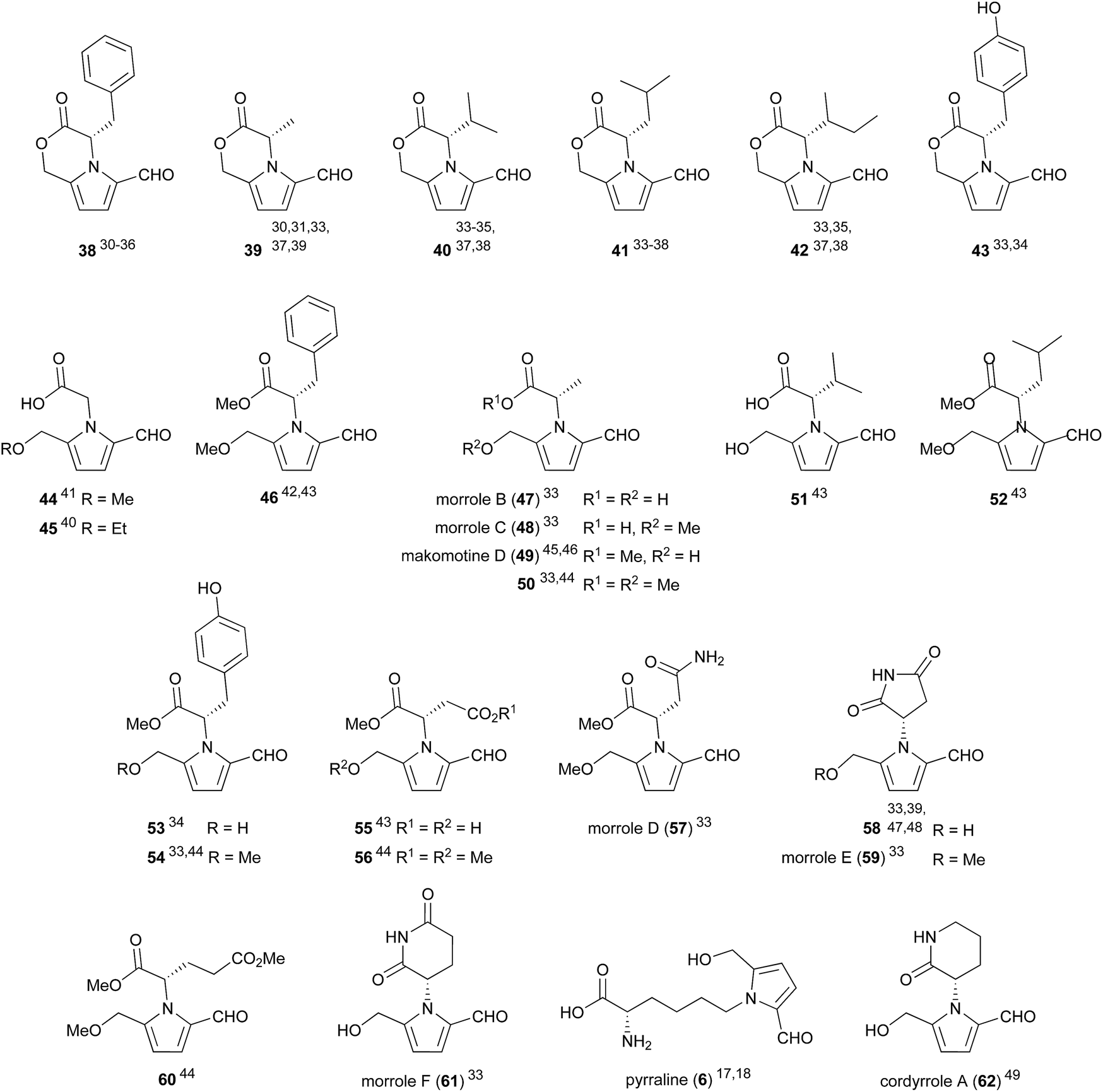 | ||
| Fig. 2 2-Formylpyrroles derived from proteinogenic amino acids. | ||
Some notable compounds within this 2-formylpyrrole sub-class are tyrosine derivative 43 and alanine derivative 50 which inhibit porcine pancreatic lipase by 70% and 40% respectively at 100 μM.33 In a separate report, cordyrrole A (62) was also shown to inhibit porcine pancreatic lipase at 100 μM.49 Pancreatic lipase is a target of interest for the development of anti-obesity treatments. Makomotine D (49) has been shown to induce quinone reductase (QR) activity thus enhancing carcinogen detoxification, with a concentration of 43.1 μM required to double QR activity.46 In the same report, makomotine D (49) was shown to possess hydroxyl radical scavenging ability with an ED50 (concentration for hydroxyl radical scavenging by 50%) of 16.7 μM.
A small number of 2-formylpyrroles comprising unnatural amino acids have also been reported (Fig. 3). Funebral (3) and its Schiff base funebrine (63) were isolated from Quararibea funebris, a flowering tree native to Mexico.14,50 The plant is used in traditional medicine as a cough remedy, antipyretic, and to control menstrual disorders and psychopathic fears,51 although the bioactivity of funebral (3) and funebrine (63) has yet to be evaluated. γ-Hydroxyisoleucine, the amino acid incorporated in these two compounds, was isolated alongside funebrine (63).50
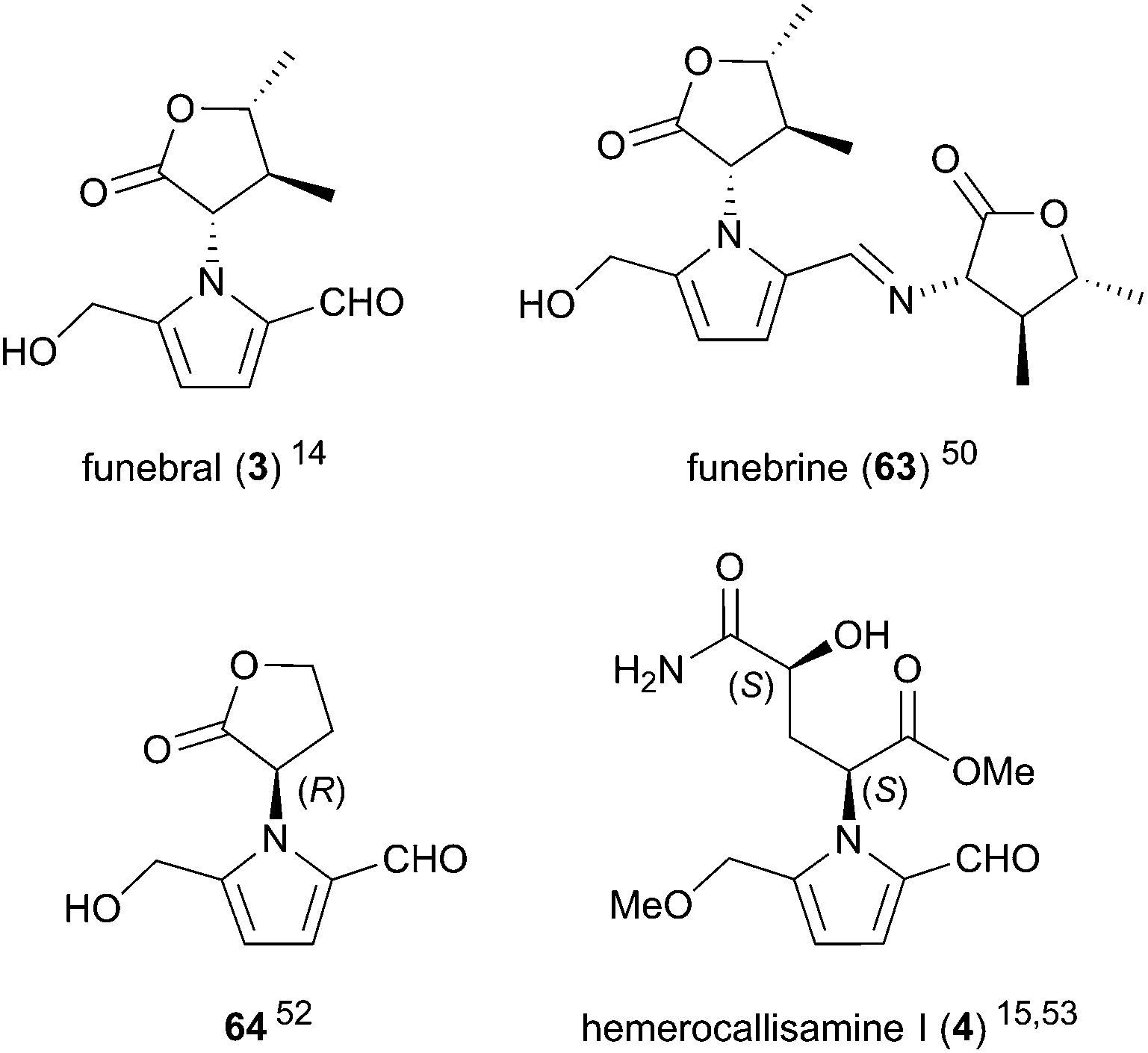 | ||
| Fig. 3 2-Formylpyrroles derived from non-proteinogenic amino acids. | ||
Homoserine lactone derivative 64 was isolated from 10 day-old Pisum sativum seedlings in 1987.52 The absolute configuration of lactone 64, which corresponds to D-homoserine lactone, was confirmed by total synthesis. The unusual D-homoserine moiety was proposed to arise from opine biosynthetic pathways. Pyrrole 64 was found to inhibit trigonelline-induced cell cycle arrest in G2 phase with an ED50 of 0.5 μM, signifying the first chemically characterised substance to override hormonally induced cellular arrest in complex tissues. Notably, the synthesised (S)-enantiomer of pyrrole 64 was shown to be inactive.
Hemerocallisamine I (4) was isolated from Hemerocallis fulva var. kwanso, H. flava, and H. minor (daylily flowers) by Matsuda and co-workers in 2014 and again in 2016.15,53 Originally assigned as (2R,4R)-4 by X-ray crystal structure, the structure of hemerocallisamine I was later revised to the enantiomer (2S,4S)-4 by total synthesis,54 suggesting hemerocallisamine I (4) arises from L-γ-hydroxyglutamine metabolites in Hemerocallis. A variety of L-glutamine-based metabolites have been isolated from Hemerocallis,55,56 some of which inhibit αβ42 aggregation and accelerate neurite outgrowth in PC12 cells, signifying their potential as novel therapies for Alzheimer's disease.53 Hemerocallisamine I (4), however, was shown to lack any such neurological activity.
3.2. 2-Formylpyrroles derived from biogenic amines
Amino acid decarboxylation products, commonly referred to as biogenic amines, can arise from both the enzymatic decarboxylation of amino acids, as well as the Strecker degradation of amino acids with reducing sugars.20,57 The most commonly encountered 2-formylpyrroles within this sub-class are those arising from γ-aminobutyric acid (GABA), the decarboxylation product of glutamic acid (pyrroles 65–75, Fig. 4). 4-(2-Formyl-5-(hydroxymethyl)-1H-pyrrol-1-yl)butanoic acid (PBA) (65) was first isolated from kako-bushi-matsu, a thermally processed Aconitum japonicum root product used in oriental medicine for its analgesic, diuretic and cardiac effects.58 PBA (65) has been isolated no fewer than eleven times,4–7,34,41,58–62 and its methyl ether 66 no fewer than eight,6–8,34,38,46,63,64 from both plant and fungal sources. The yield of PBA (65) from kako-bushi-matsu, which is prepared by autoclaving Aconitum roots at 110 °C, was determined to be 10-fold greater than the yield of PBA (65) from Aconitum roots dried at 50–55 °C.58 This observation is consistent with the proposal that PBA is the product of Maillard-type reactions.5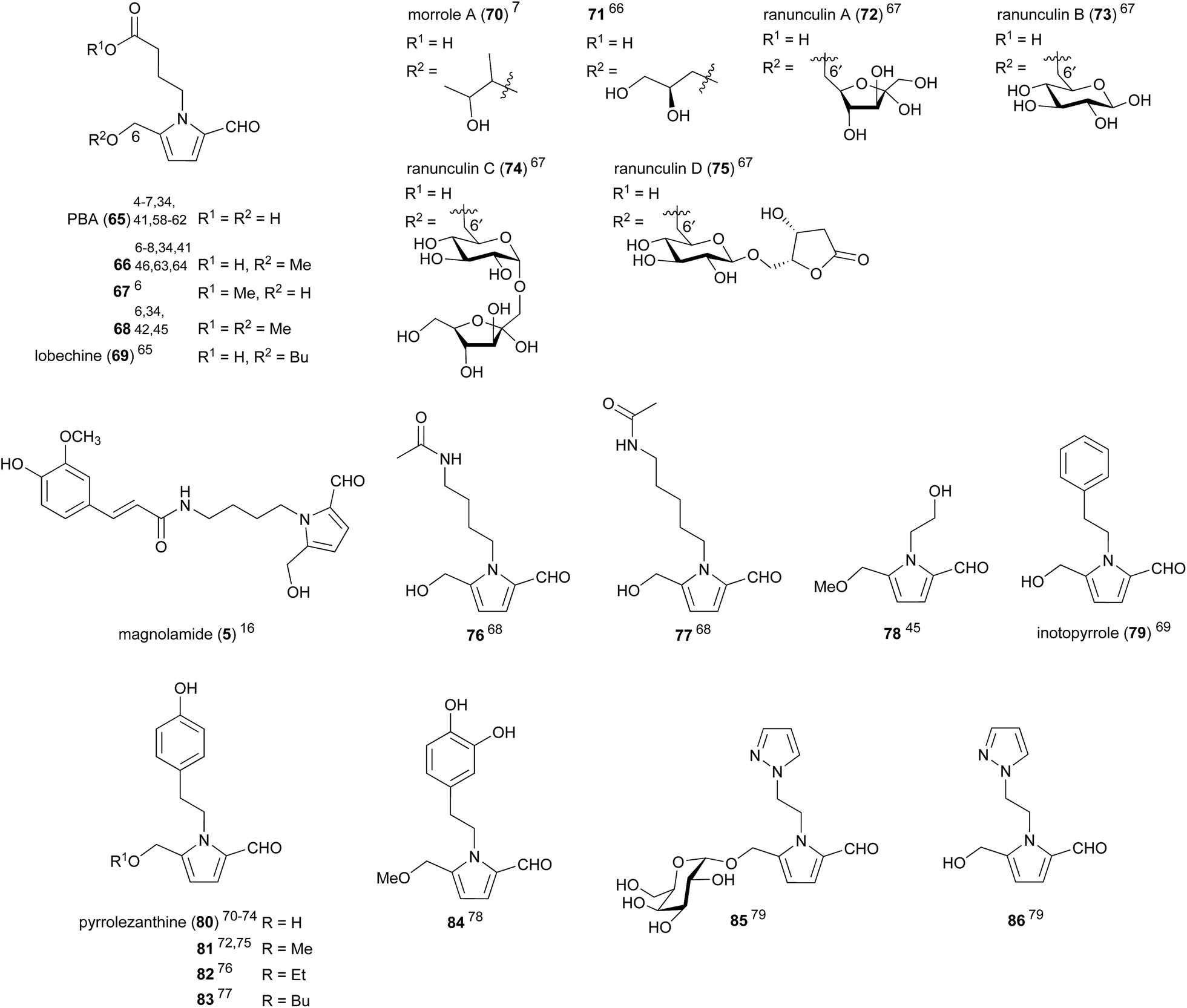 | ||
| Fig. 4 2-Formylpyrroles derived from biogenic amines. | ||
PBA (65) and its derivatives possess an impressive array of biological activities. A 50 μg dose of PBA (65) caused a significant increase in the peripheral blood flow (90.3 ± 18.2 mL/30 min/100 g) of mice.58 Free acids PBA (65) and methyl ether 66 were both shown to exhibit hepatoprotective effects at 0.1 μM (64.4 ± 3.9% and 65.8 ± 5.6% cell viability respectively).6 The methyl esters 67 and 68 also exhibited hepatoprotective effects to a lesser degree, suggesting the importance of the free acid for high hepatoprotective activity. In another report, PBA (65) and methyl ether 66 were shown to possess immunostimulatory activity.7 Incubation of RAW 264.7 macrophage cells with PBA (65) resulted in a significant increase in phagocytotic activity, while methyl ether 66 caused a smaller increase in activity and morrole A (70) had no effect. It was supposed that the 5-hydroxymethyl moiety was important for macrophage stimulatory activity as increasingly large substitution at this position corresponded with decreased activity.
PBA methyl ether 66 was found to inhibit rat lens aldose reductase with an IC50 of 39.71 ± 1.77 μM.63 Aldose reductase is the first enzyme in the polyol pathway of glucose metabolism. Increased flux through this pathway can cause diabetic complications including retinopathy, neuropathy, nephropathy and cataracts. Methyl ether 66 also exhibits moderate antiproliferative activity, inhibiting four different human tumour cell lines; A459, SK-OV-3, SK-MEL-2 and HCT-15, with an IC50 range of 21.52 ± 1.82 μM to 40.74 ± 2.41 μM.8 The cancer chemopreventative properties of PBA methyl ether 66 were investigated and it was found to exhibit hydroxyl radical scavenging activity.46 PBA methyl ester 66 was also shown to induce quinone reductase (QR) with a concentration of 2.4 μM required to double QR activity.
Ranunculins A–D (72–75) were recently isolated from the roots of Ranunculus ternatus Thunb, a plant used in traditional Chinese medicine.67 While no bioactivity was investigated for these four GABA-derived pyrroles, they contain an unprecedented 6-6′ ether link between the pyrrole hydroxymethyl substituent, putatively arising from C-6 of 3-DG (19), and the C-6 carbons of fructose (ranunculin A (72)) or glucose (ranunculins B–D (73–75)). This tail–tail ether connectivity has not been observed in any other natural products, whereas head–head (e.g. trehalose: 1,1 connected), head–tail (e.g. isomaltose: 1,6 connected) and head-body (e.g. sucrose: 1,2 connected) connectivities are all well-known. Attempts to synthesise these natural products by 6-6′ ether formation between PBA (65) and the appropriate sugar derivatives were unsuccessful.
Magnolamide (5), which contains trans-feruloyl and putrescine moieties, was shown to possess antioxidant activity, inhibiting Cu2+/O2-induced LDL lipid peroxidation with an IC50 of 9.7 ± 2.8 μM.9 This was comparable to the positive control resveratrol (IC50 13.1 ± 2.6 μM). Putrescine and cadaverine-derived pyrroles 76 and 77 showed weak activity against human cancer cell lines HeLa, K-562 and L-929, with IC50 values ranging between 8.9–20.2 μg mL−1, but were inactive against the microbes Bacillus subtilis, Staphylococcus aureus, Escherichia coli, and Candida albicans (IC50 > 10 μg mL−1).68
Tyramine derivative pyrrolezanthine (80) was found to exhibit moderate cytotoxicity against lung cancer A-549 and human colon cancer SW480 cell lines with IC50 values of 38.3 and 33.7 μM respectively.74 Pyrrolezanthine (80) was also demonstrated to possess anti-inflammatory activity, inhibiting NO production in RAW 264.7 macrophage cells with an IC50 of 58.8 μM.73 Pyrrolezanthine butyl ether 83, which was isolated from the butanol soluble fraction of Reynoutria ciliinervis (Nakai) Moldenke extract, was found to possess antifungal activity against Sclerotinia sclerotiorum with a MIC of 31.2 μg mL−1.77
Pyrroles 85 and 86 were isolated from watermelon (Citrullus lanatus) seeds in 2015.79 They were the first compounds containing both pyrazole and pyrrole rings to be isolated from a natural source and are likely derived from β-(1-pyrazolyl)alanine, a non-proteinogenic amino acid produced in watermelon.
3.3. N-Unsubstituted 2-formylpyrroles derived from amino sugars
A small sub-class of 2-formylpyrroles exist with no N-substituents (Fig. 5). These compounds plausibly arise from the self-condensation of hexosamines. Interestingly, O-D-galactopyranoside 94 appears to arise either from a disaccharide precursor, or from the glycosylation of 2-formylpyrrole 88. Of these N-unsubstituted 2-formylpyrroles, only ilanepyrrolal (92) was shown to exhibit biological activity. Ilanepyrrolal (92), which was isolated from rice fermented with the endophytic fungus Annulohypoxylon ilanense (Xylariaceae), was demonstrated to inhibit Mycobacterium tuberculosis growth with a MIC value of 76.8 mM.80 | ||
| Fig. 5 N-Unsubstituted 2-formylpyrroles. | ||
3.4. Pyrrolomorpholine spiroketals derived from amino sugars
Another small group of amino sugar derivatives, referred to as pyrrolomorpholine spiroketals (Fig. 6), are thought to arise from the intermolecular condensation of 3-DG (19) with 1-amino-1,3-dideoxy-D-fructose (95) (acortatarin A (1), pollenopyrroside A (2), shensongine B (97) and shensongine A (98)) or 1-amino-1-deoxy-D-fructose (100) (acortatarin B (101) and shensongine C (102)).5 3-DG (19) is a deamination product of Amadori compounds, while 1-amino-1-deoxyhexoses are the products of Strecker degradation. Both processes are associated with Maillard pathways. The generation of pyrrolomorpholine spiroketals by a Maillard pathway was proposed by Jiang and Peterson, who isolated acortatarin A (1) and a [6,6]-pyrrolomorpholine spiroketal, the stereochemistry of which was not determined, from bread.4,5 These compounds were present in higher concentrations in the bread crust, which is exposed to the highest temperatures during baking, supporting this Maillard hypothesis.4 There are regioisomeric [5,6]-spiroketals and [6,6]-spiroketals within this group of compounds depending on which hydroxy group of 1-amino-1,3-dideoxy-D-fructose (95) is engaged in spirocyclisation. For each regioisomer there are two possible configurations at the spiroketal center giving rise to pairs of spiroketal anomers.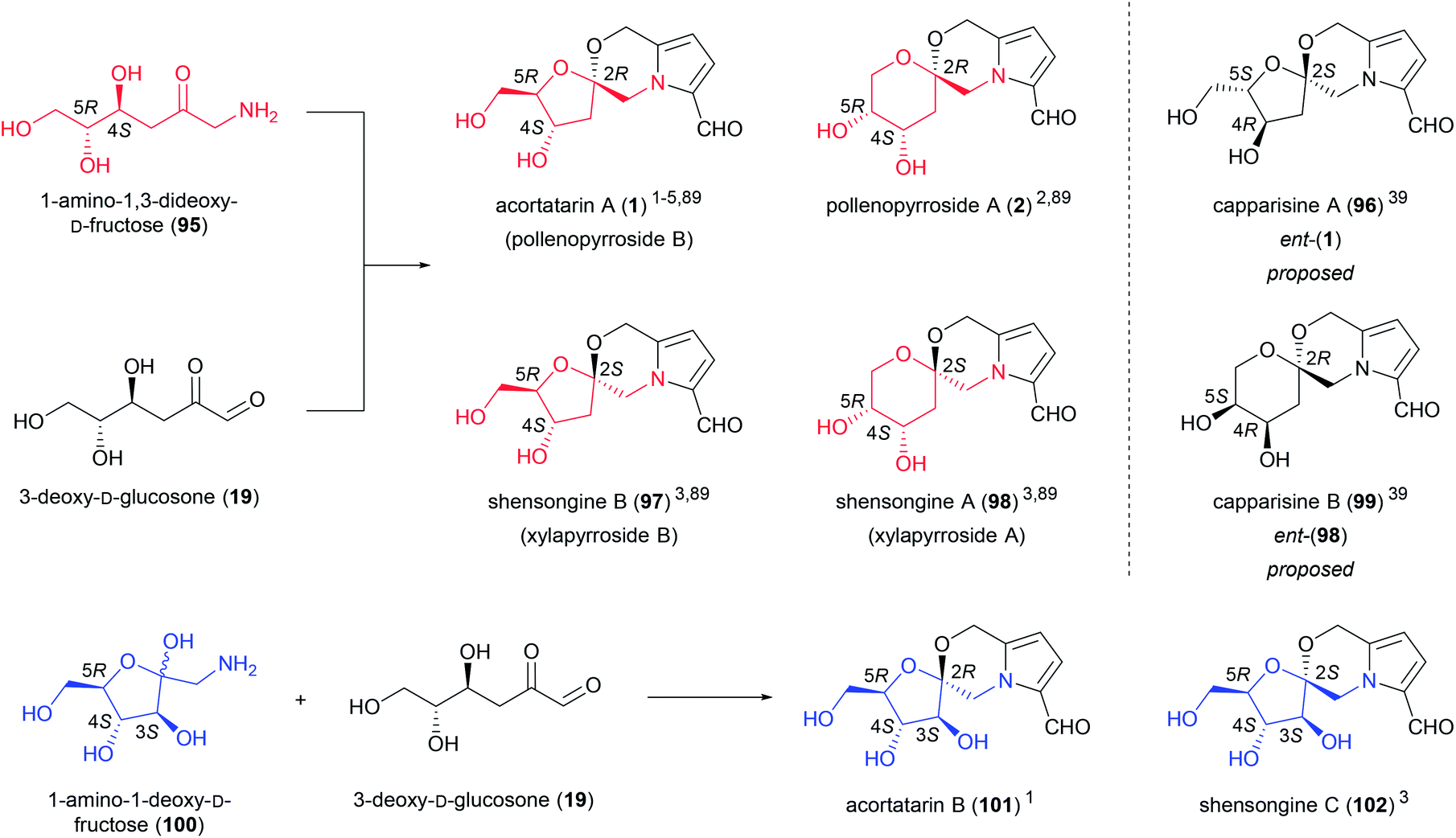 | ||
| Fig. 6 Pyrrolomorpholine spiroketals. | ||
Pyrrolomorpholine spiroketals first appeared in the literature in 2010 when three reports of their isolation emerged independently of each other.1,2,39 Three different trivial naming systems were subsequently proposed in these reports, a problem which would later be compounded by the isolation of additional pyrrolomorpholine spiroketals. In addition to bread crust, these spiroketals have been isolated from the rhizomes of Acorus tatarinowii,1 bee-collected Brassica campestris pollen,2 fruits of Capparis spinosa,39 the traditional Chinese medicine preparation Shensong Yangxin,3 and the fermented mycelia of Xylaria nigripes.89 All of these pyrrolomorpholine spiroketals are derived from D-sugars, with the exception of capparasines A (96) and B (99), the absolute stereochemistry of which was proposed from X-ray crystal structure.39 While this enabled unambiguous assignment of the relative stereochemistry of capparisines A and B, insufficient evidence was provided for the assignment of the absolute stereochemistry of these two compounds, which are likely to be the misassigned structures of acortatarin A (1) and shensongine A (98), respectively.
Pyrrolomorpholine spiroketals inhibit the high glucose-induced production of reactive oxygen species (ROS) in mesangial cells. High glucose-induced ROS production is implicated in diabetic nephropathy, the leading cause of end-stage renal disease in the Western world.90 ROS-inhibition was originally established for acortatarin A (1) and B (101),1 however Verano and Tan later demonstrated that all members of the pyrrolomorpholine spiroketal family inhibit high glucose-induced ROS production.91 Acortatarin A (1) and shensongine C (102) are the most active of the isolated compounds, with IC50 values of 4.6 and 4.8 μM respectively and a maximum inhibition of ROS production of 100%. The synthetic analogues 103 and 104 are the most potent pyrrolomorpholine spiroketals tested to date (Fig. 7), with IC50 values of 0.52 and 0.27 μM respectively, however they can only effect 80% and 60% maximum inhibition of ROS production.
 | ||
| Fig. 7 Synthetic analogues of the pyrrolomorpholine spiroketals with improved inhibition of high glucose-induced ROS production. | ||
A report by Nie and co-workers in 2013 elucidated the biological action of acortatarin A (1).10 Pre-incubation of rat glomerular mesangial cells with acortatarin A (1) attenuates high-glucose phosphorylation of PKC isoforms PKCα and PKCβ1, PLCγ1 and the p85 regulatory subunit of PI3K. Inhibition of the PI3K–PLCγ1–PKC signalling pathway, which is an upstream regulator of NADPH oxidase activation, inhibits high glucose-induced ROS production. In turn, inhibition of high glucose-induced ROS production prevents overproduction of extracellular matrix proteins by mesangial cells, which has been closely correlated with deterioration of renal function.
In addition to their effects on high glucose-induced ROS production, shensongine A (98) and C (102) were found to shorten action potential duration in rat myocardial cells, with noticeable effects at concentrations as low as 1 μM.3 It was speculated that shensongine A (98) and C (102) might either inhibit L-type calcium channels, or facilitate the action of potassium channels.
4. Synthesis
Aside from the isolation of 2-formylpyrroles as minor products in amine–sugar reaction mixtures, efforts have been dedicated towards the selective synthesis of these compounds. Many of these syntheses employ the Paal–Knorr reaction to form pyrrole ring systems around readily available amines, while other approaches involve N-alkylation of a pyrrole substrate (Scheme 4). Both of these strategies often involve late stage modification of the pyrrole ring substituents in order to furnish the 5-hydroxymethylpyrrole-2-carbaldehyde system. More recently, Maillard chemistry-inspired methodologies have been developed to forge the 5-hydroxymethylpyrrole-2-carbaldehyde ring system cleanly in a single step from amines and sugars, or sugar surrogates. This section will focus on the total syntheses of 5-(hydroxymethyl)-1-[(R)-tetrahydro-2′-oxofur-3′-yl)-1H-pyrrole-2-carbaldehyde] (64), funebral (5) and the pyrrolomorpholine spiroketals, which best encompass the different synthetic strategies towards 2-formylpyrroles.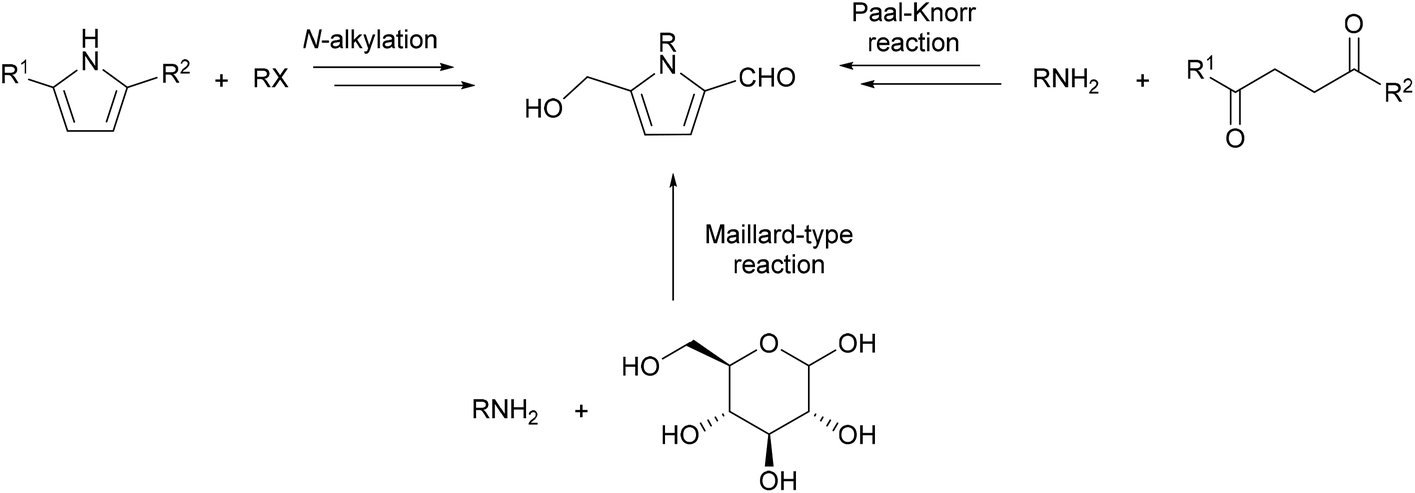 | ||
| Scheme 4 Different synthetic approaches to the 5-hydroxmethylpyrrole-2-carbaldehyde system. | ||
4.1. 5-(Hydroxymethyl)-1-[(R)-tetrahydro-2′-oxofur-3′-yl)-1H-pyrrole-2-carbaldehyde] (64)
The first total synthesis of a 2-formylpyrrole was reported in conjunction with the isolation and biological evaluation of 5-(hydroxymethyl)-1-[(R)-tetrahydro-2′-oxofur-3′-yl)-1H-pyrrole-2-carbaldehyde] (64) by Lynn and co-workers in 1987 (Scheme 5).52 The pyrrole ring system was constructed by the Paal–Knorr reaction of D-homoserine lactone (105) and 2,9-dimethyldeca-2,8-dien-4,7-dione (106), which proceeded with some racemisation of the α-stereocentre. Ozonolysis of bis(isobutenyl)pyrrole 107 and selective reduction of the resultant biscarbaldehyde with diborane afforded 2-formylpyrrole 64. This Paal–Knorr strategy has since been employed in many syntheses of 2-formylpyrroles. | ||
| Scheme 5 Synthesis of 5-(hydroxymethyl)-1-[(R)-tetrahydro-2′-oxofur-3′-yl)-1H-pyrrole-2-carbaldehyde] (64) by Lynn and co-workers (yields omitted where not given). | ||
A racemic synthesis of 2-formylpyrrole 64 was reported by Neier and co-workers in 1993 (Scheme 6).92 This synthesis also employed a Paal–Knorr condensation, using asymmetric dione equivalent 110 to enable a different end-game approach to the 5-hydroxymethylpyrrole-2-carbaldehyde system. Dione equivalent 110 was obtained by oxidation of 2-methylfuran (108) and subsequent hydrogenation of dihydrofuran 109 using RANEY® nickel. The Paal–Knorr condensation of dione equivalent 110 with rac-homoserine lactone (105) proceeded in excellent yield. Vilsmeier–Haack formylation and oxidation of the 5-methyl substituent with lead(IV) acetate afforded acetate 113, hydrolysis of which furnished 2-formylpyrrole 64.
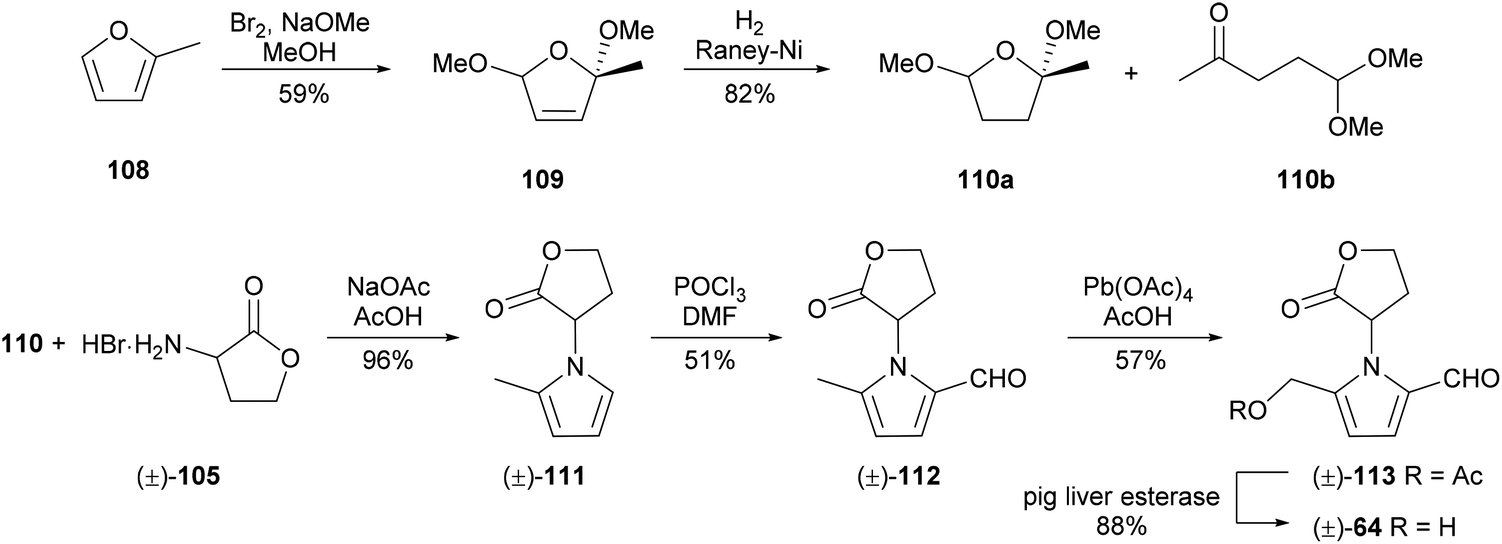 | ||
| Scheme 6 Synthesis of 5-(hydroxymethyl)-1-[(R)-tetrahydro-2′-oxofur-3′-yl)-1H-pyrrole-2-carbaldehyde] (64) by Neier and co-workers. | ||
4.2. Funebral (3)
Funebral (3) has been the subject of four different total syntheses to date. While it shares a high degree of structural similarity to the cellular arrest inhibitor 5-(hydroxymethyl)-1-[(R)-tetrahydro-2′-oxofur-3′-yl)-1H-pyrrole-2-carbaldehyde] (64), funebral (3) has no known bioactivity. Interest in this compound has been driven largely by the synthetic challenge posed by the three contiguous stereocenters of its γ-butyrolactone ring and the sterically crowded pyrrole ring.The first total synthesis of (±)-funebral (3) and (±)-funebrine 63 was reported by Le Quesne and co-workers in 1995,93–95 which utilised 2,9-dimethyldeca-2,8-dien-4,7-dione (106) to construct the pyrrole ring from α-amino-γ-butyrolactone 120 (Scheme 7). Racemic α-amino-γ-butyrolactone 120 was afforded in 14% yield over eight steps, which included a diastereoselective Claisen rearrangement and iodolactonisation. Synthesis of the sterically crowded bis(isobutenyl)pyrrole 121 by Paal–Knorr condensation of α-amino-γ-butyrolactone 120 and dione 106 required titanium(IV) isopropoxide to proceed. (±)-Funebral (3) was afforded from bis(isobutenyl)pyrrole 121 by osmium-catalysed oxidative olefin cleavage and monoreduction. Treatment of (±)-funebral (3) with excess α-amino-γ-butyrolactone 120 provided (±)-funebrine 63.
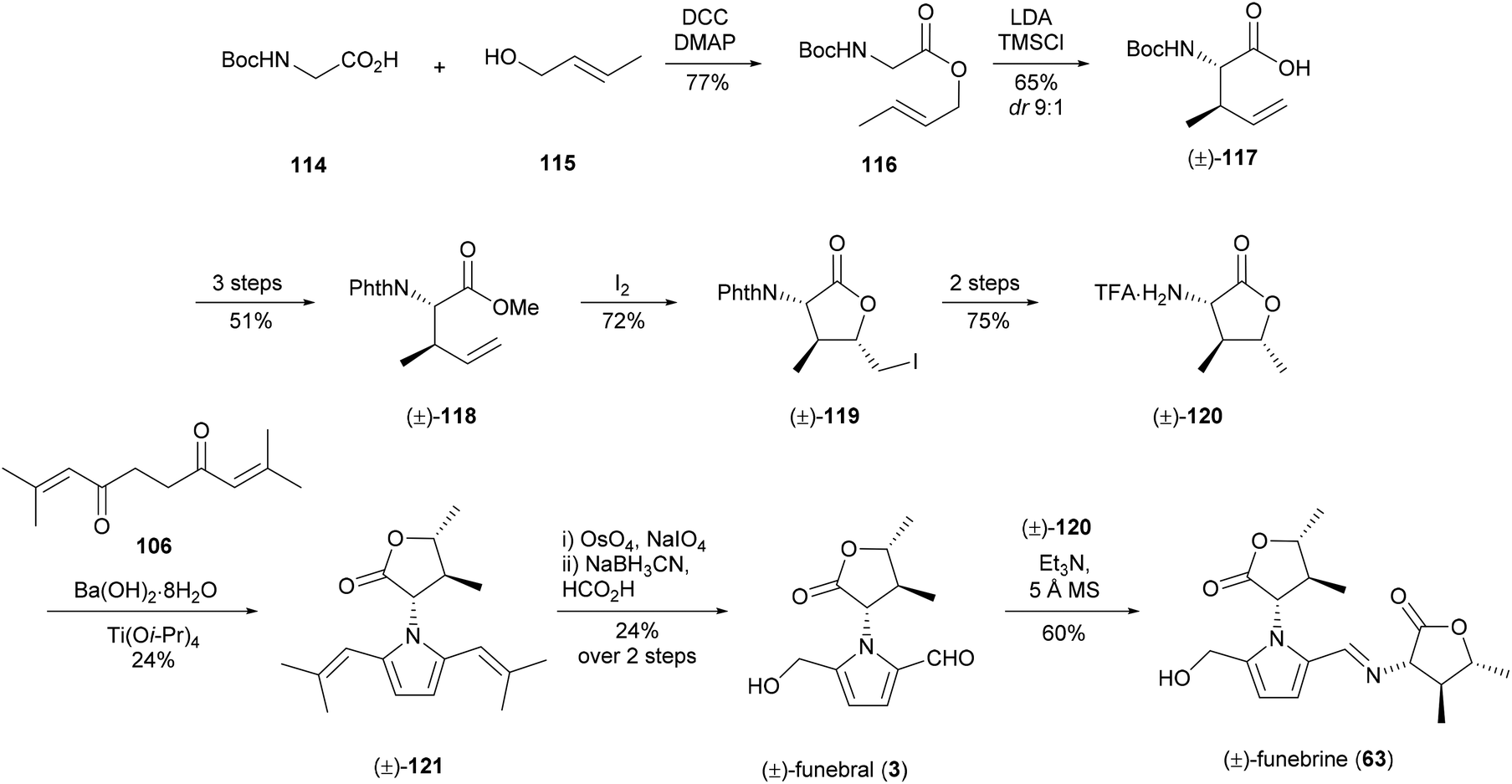 | ||
| Scheme 7 Racemic synthesis of funebral (3) and funebrine (63) by Le Quesne and co-workers. | ||
The first enantioselective synthesis of funebral (3) and funebrine (63) was reported by Ishibashi and co-workers in 2003 (Scheme 8).96 Their preparation of (−)-α-amino-γ-lactone 120 involved a [2,3]-cycloaddition of (E)-crotyl alcohol (115) with nitrone 124, which in turn was derived from methyl glyoxylate (123) and an oxime 122 bearing an L-gulose-based chiral auxiliary. Cleavage of the auxiliary and translactonisation provided lactone 126 in excellent yield, which was elaborated to α-amino-γ-lactone 120. Funebral (3) and funebrine (63) were accessed via the same strategy employed by Le Quesne and co-workers93 with further optimisation of reaction conditions. Interestingly, the Paal–Knorr conditions reported by Le Quesne and co-workers were not reproducible and titanium(IV) ethoxide was instead employed as a Lewis acid catalyst in this key step.
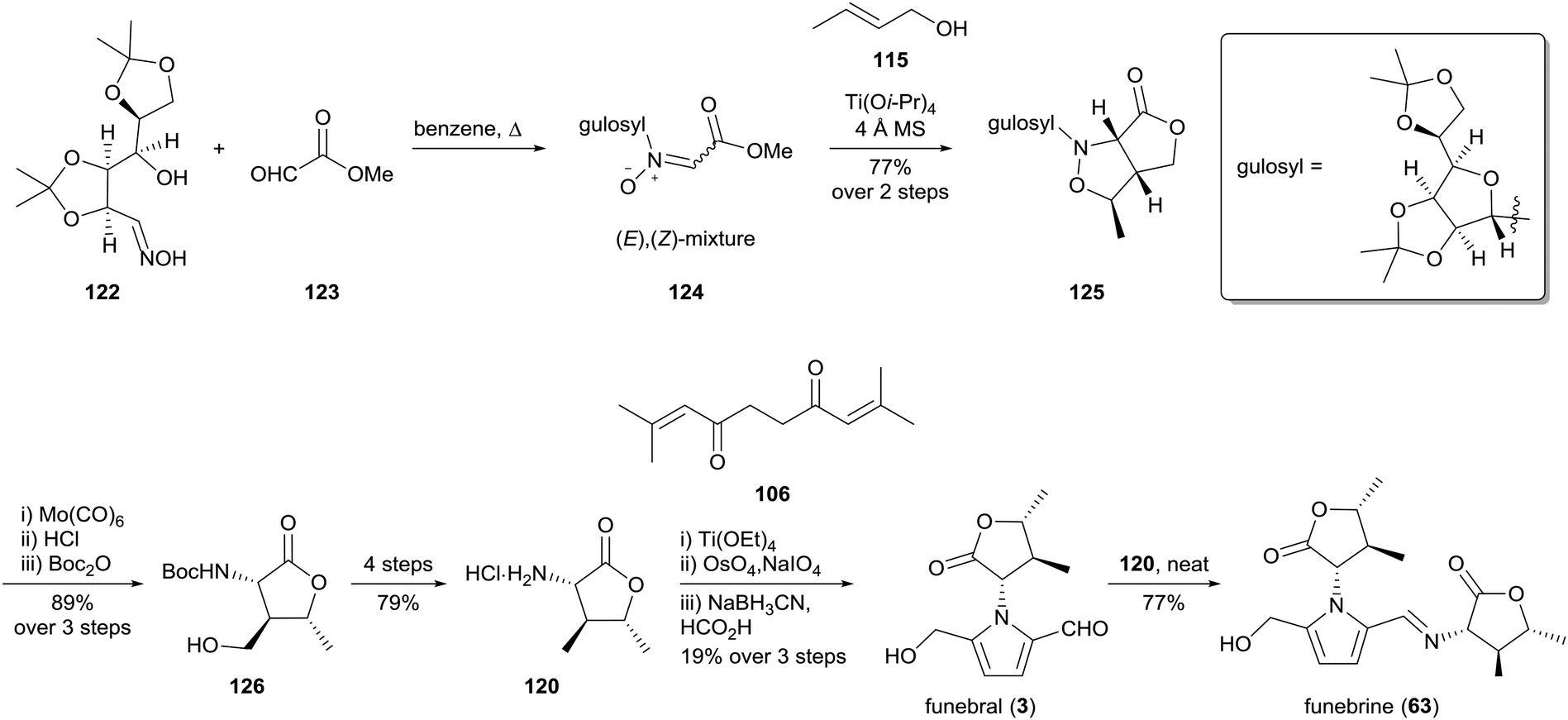 | ||
| Scheme 8 Enantioselective synthesis of funebral (3) and funebrine (63) by Ishibashi and co-workers. | ||
Sakaguchi and co-workers reported a synthesis of funebral (3) and funebrine (63) in 2011, utilising dione 129 for the Paal–Knorr reaction of α-amino-γ-lactone 120 (Scheme 9).97 Dione 129 was prepared over five steps from hydroxymethylfurfural (127), while α-amino-γ-lactone 120 was prepared from 2-butyn-1-ol (130) and Boc-glycine over ten steps which included a diastereoselective Claisen rearrangement of propargyl ester 131 and gold(I)-catalysed cyclisation of the resultant allenylsilane 132. The Paal–Knorr reaction of amine 120 with dione 129 provided pyrrole 134, oxidation of which afforded access to funebral (3) and funebrine (63) in good yield.
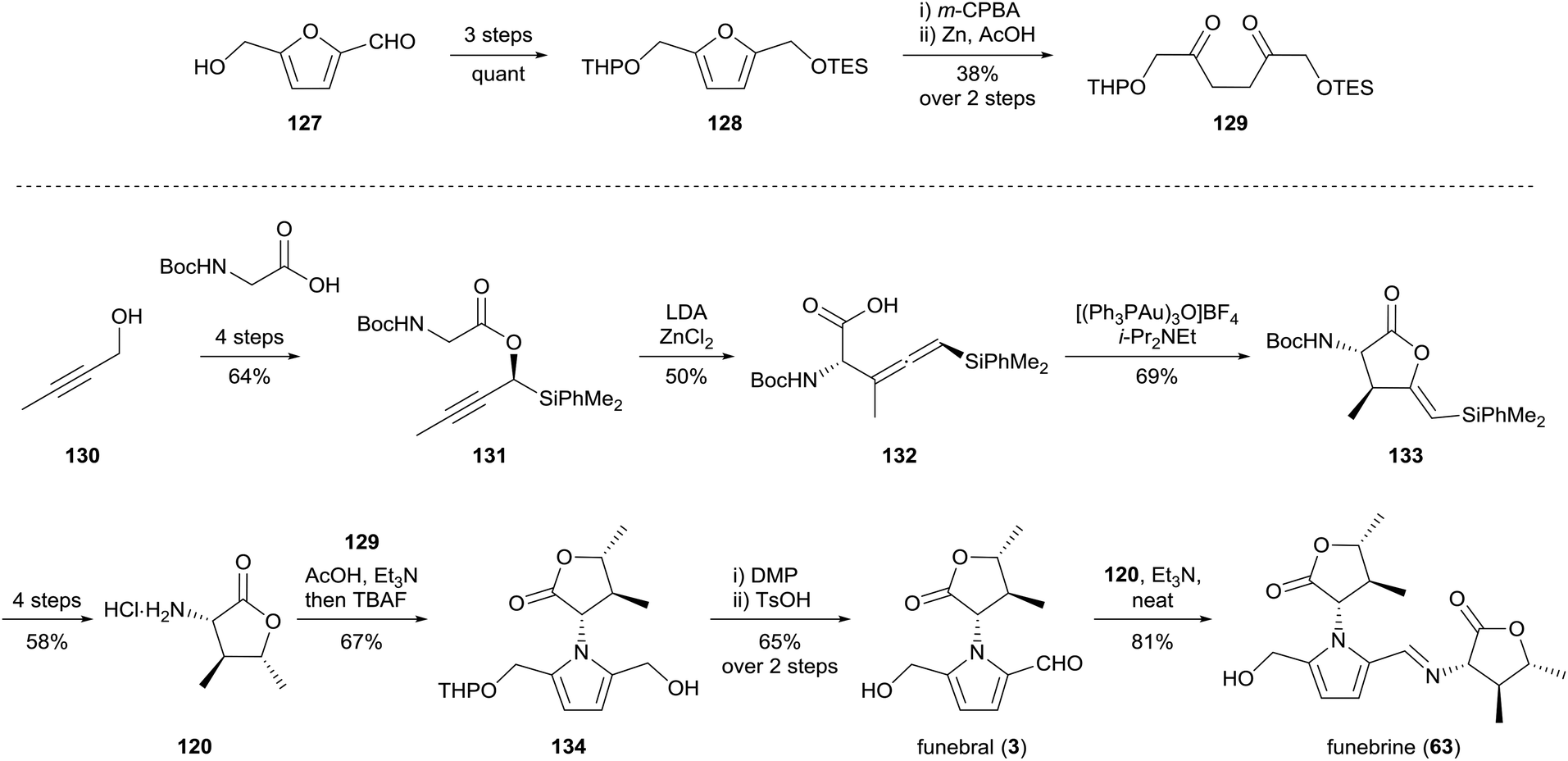 | ||
| Scheme 9 Synthesis of funebral (3) and funebrine (63) by Sakaguchi and co-workers. | ||
In 2014, Brimble and co-workers reported a concise synthesis of funebral (3) by a Maillard-type condensation approach, which forged the pyrrole substituents in the correct oxidation state (Scheme 10).98 α-Amino-γ-butyrolactone 120 was prepared by an L-proline-catalysed Mannich reaction of ethyl glyoxal-derived imine 137 with 2-butanone. α-Amino-γ-butyrolactone 120 was afforded as a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of diastereomers by sodium borohydride reduction. Dihydropyranone 140, which had previously been prepared by the group,99 underwent condensation with four equivalents of amine 120 to provide TBS-protected funebral 141a, along with its diastereomer 141b and funebrine-type adducts 142. Desilylation of TBS-protected adduct 141a afforded funebral (3) in a longest linear sequence of six steps.
:1 mixture of diastereomers by sodium borohydride reduction. Dihydropyranone 140, which had previously been prepared by the group,99 underwent condensation with four equivalents of amine 120 to provide TBS-protected funebral 141a, along with its diastereomer 141b and funebrine-type adducts 142. Desilylation of TBS-protected adduct 141a afforded funebral (3) in a longest linear sequence of six steps.
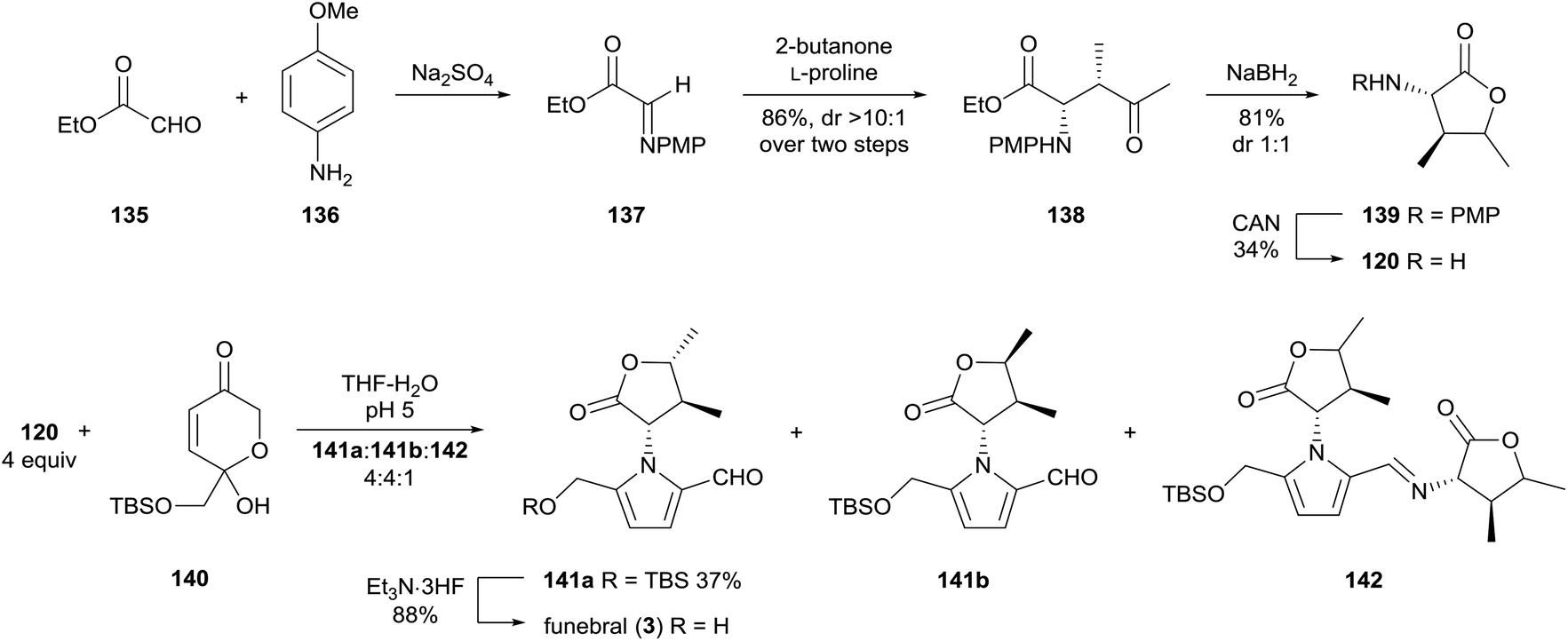 | ||
| Scheme 10 Synthesis of funebral (3) by Brimble and co-workers. | ||
4.3. Pyrrolomorpholine spiroketals
In 2011, Sudhakar and co-workers reported the first total synthesis of acortatarin A (1) and B (101) (Scheme 11).100 Incidentally, the acid-catalysed spiroketalisation conditions used also afforded the anomers of the acortatarins; shensongine B (97) and C (102), preceding their isolation from natural sources by three years. The synthesis strategy employed 2-deoxy-D-ribose and D-arabinose as chiral pool materials. Homologation and epoxidation of these sugars provided electrophiles for N-alkylation of pyrrole 149, which had the requisite aldehyde and hydroxymethyl substituents already installed. Oxidation of secondary alcohols 150 and 151, deprotection and acid-catalysed spirocyclisation afforded α-spiroketal anomers acortatarin A (1) and shensongine C (102) as major products, while β-spiroketal anomers shensongine B (97) acortatarin B (101) were afforded in minor quantities. The efficiency of this approach would inspire later syntheses of pyrrolomorpholine spiroketals by the groups of Kuwahara101 and Hu,89 which both used THP-protected pyrrole carbaldehyde 149.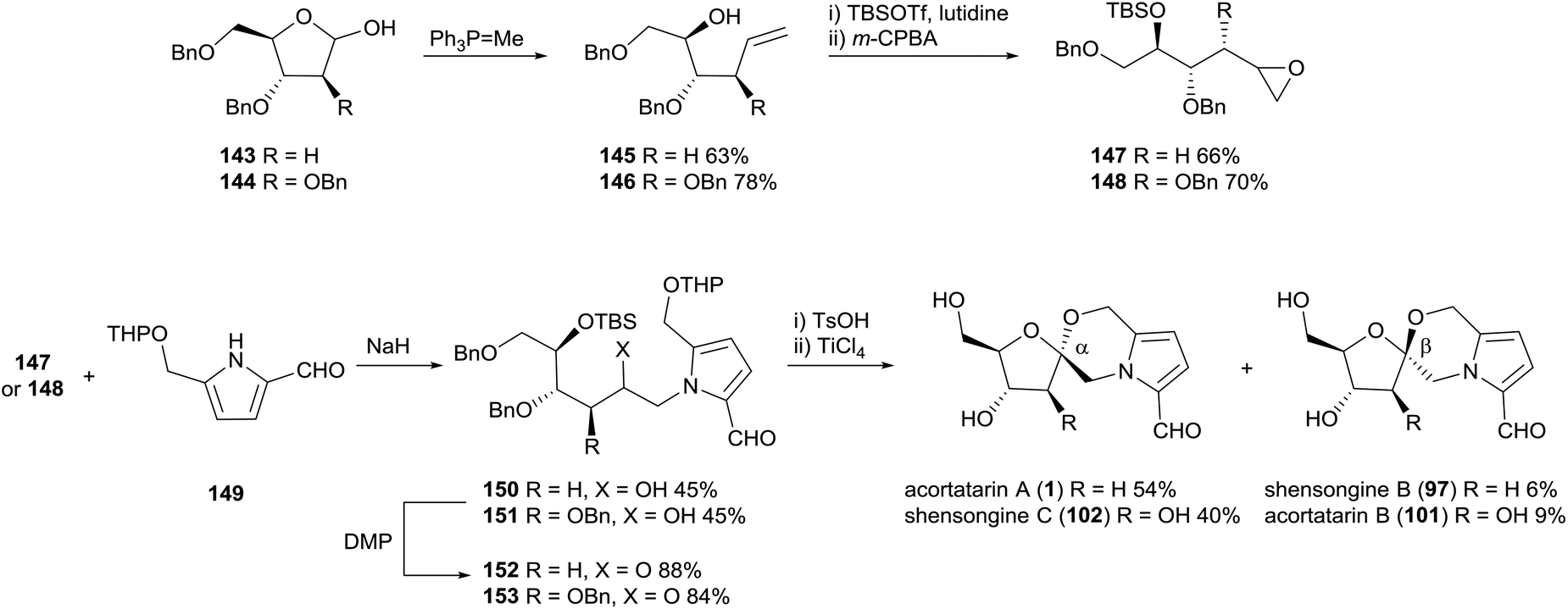 | ||
| Scheme 11 Synthesis of acortatarins A (1) and B (101) and shensongines B (97) and C (102) by Sudhakar and co-workers. | ||
Brimble and co-workers utilised a Maillard-type approach to synthesise acortatarin A (1) in 2012 (Scheme 12).99,102 Dihydropyranone 140, which would be used to construct the pyrrole ring system of acortatarin A (1), was prepared by Achmatowicz rearrangement of furfuryl alcohol 155, which in turn was prepared from furfuryl silyl ether 154. Homoallylic alcohol 157 was obtained by the known allylation of (R)-glyceraldehyde derivative 156. Subsequent protecting group manipulations and functional group interconversions provided aminohydrin 159, which underwent condensation with dihydropyranone 140 to afford TBS-protected adduct 160. Oxidation of alcohol 160, acid-catalysed spirocyclisation and deprotection then furnished acortatarin A (1) as the major product along with β-anomer shensongine B (97) in a diastereomeric ratio of 3:2.
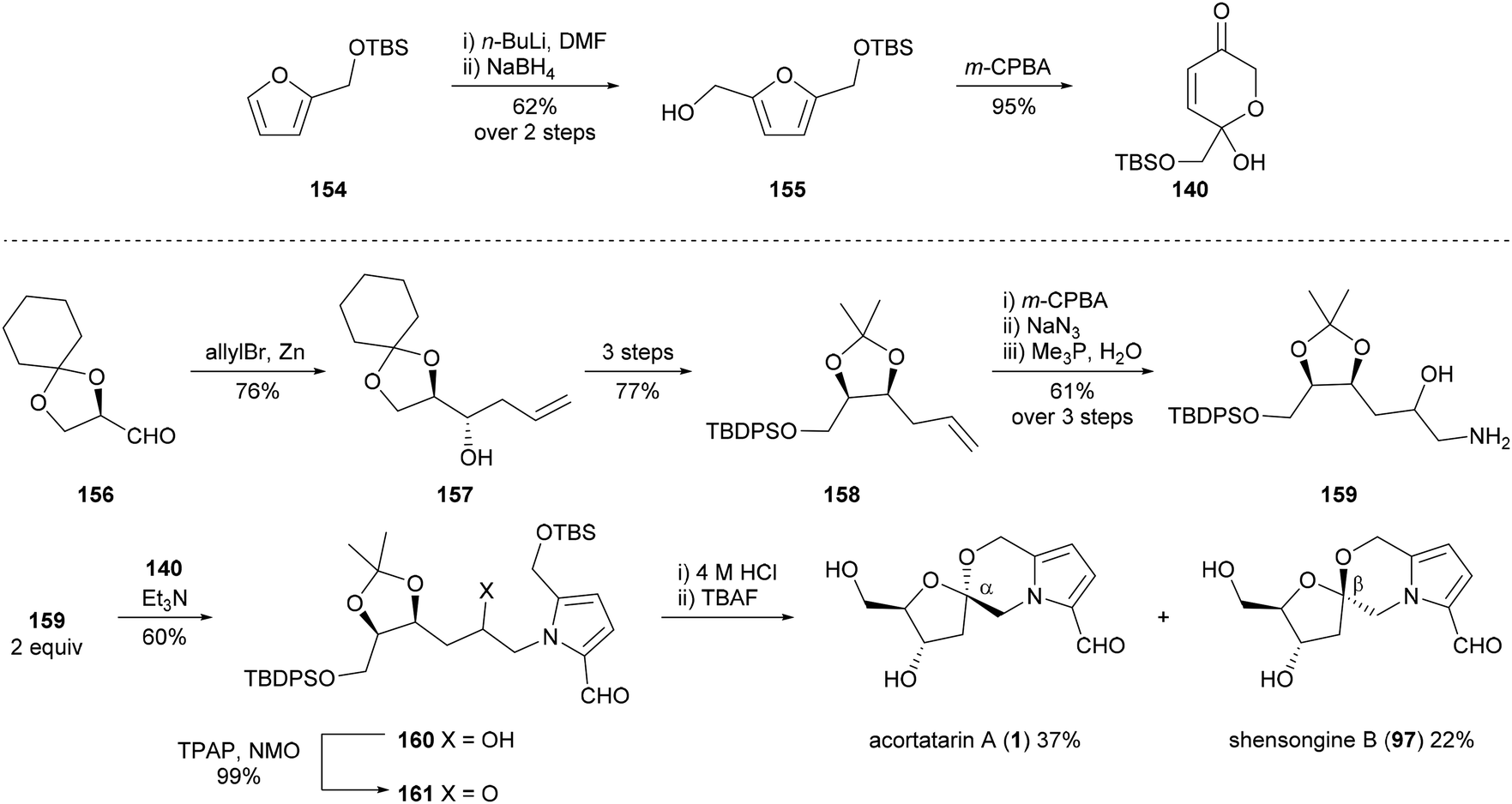 | ||
| Scheme 12 Synthesis of acortatarin A (1) and shensongine B (97) by Brimble and co-workers. | ||
Brimble and co-workers employed the same synthetic strategy to access pollenopyrroside A (2) in 2016 (Scheme 13).103 The Maillard-type condensation of amine 162, which was accessed in six steps from 2-deoxy-D-ribose, proved challenging and required the more reactive acetylated dihydropyranone 163 to proceed. Oxidation, silyl ether deprotection and spiroketalisation provided two [6,6]-spiroketal anomers with little diastereoselectivity. Deprotection of the acetonide protecting group with pyridinium para-toluenesulfonate in methanol resulted in concomitant epimerisation of the spiroketal ring, favouring pollenopyrroside A (2) over shensongine A (98) in a 7:1 ratio.
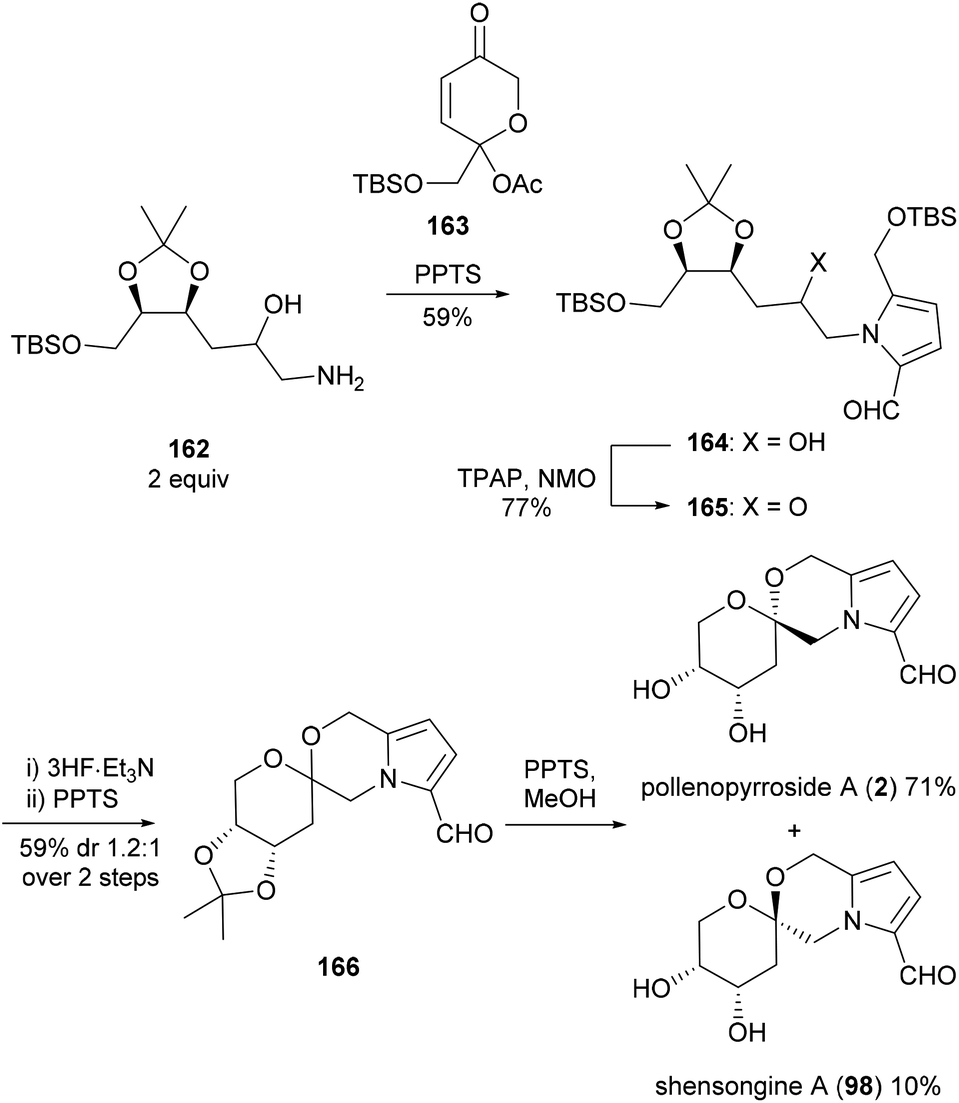 | ||
| Scheme 13 Synthesis of pollenopyrroside A (2) and shensongine A (98) by Brimble and co-workers. | ||
Tan and co-workers reported a synthesis of acortatarins A (1) and B (101), and shensongine B (97) in 2012, using D-thymidine (167) as a chiral pool starting material (Scheme 14).104 The carbon skeleton of the natural products was forged by a high-yielding alkylation of pyrrole-2,5-dicarbaldehyde (170) with iodomethylglycal 169. The [5,6]-spiroketal core of acortatarin A (1) was prepared diastereoselectively by mercury-mediated oxidative cyclisation of the hydroxymethyl pyrrole substituent onto the gycal, followed by borohydride reduction of the mercurial adduct. Deprotection of each anomer gave acortatarin A (1) and shensongine B (97), respectively. From the common glycal intermediate 171, acortatarin B (101) was prepared by diastereoselective β-epoxidation using DMDO, followed by a one pot reduction of the pyrrole-2,5-dicarbaldehyde and spirocyclisation onto the epoxide.
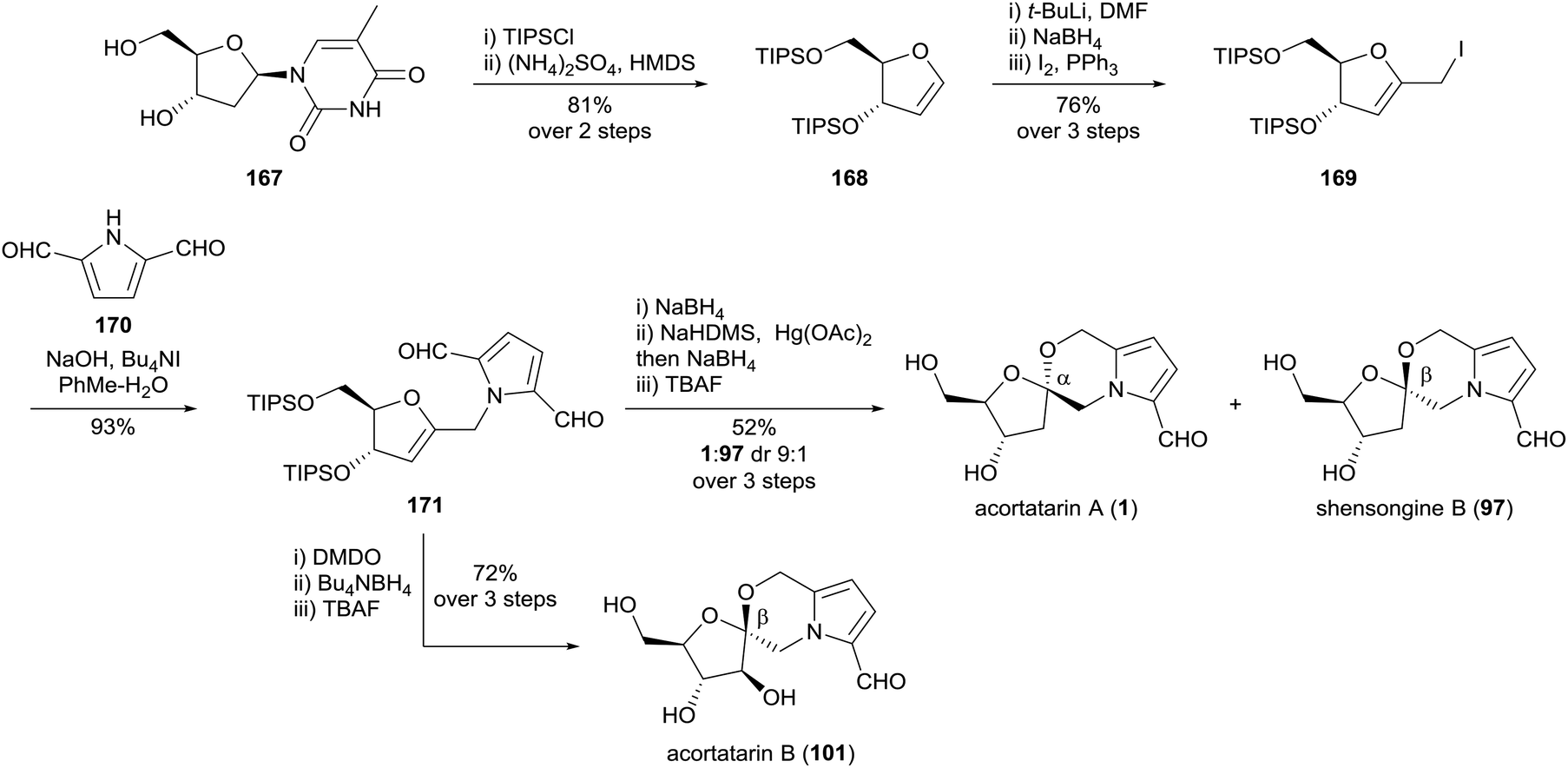 | ||
| Scheme 14 Synthesis of acortatarins A (1) and B (101) and shensongine B (97) by Tan and co-workers. | ||
Verano and Tan reported a follow-up synthesis of the [6,6]-pyrrolomorpholine spiroketals shensongine A (98) and pollenopyrroside A (2) in 2017 using a similar N-alkylation of pyrrole-2,5-dicarbaldehyde (170) (Scheme 15).91 It was noted that biphasic conditions were required for the N-alkylation to prevent dimerization of pyrrole species. Monoreduction of dicarbaldehyde 174, treatment with catalytic Brønsted acid and silyl deprotection afforded shensongine A (98) exclusively. Stereoselective synthesis of pollenopyrroside A (2) was achieved by anti-epoxidation and methanol-catalysed spirocyclisation of glycal epoxide 174, affording the kinetic α-spiroketal anomer 175. Barton–McCombie deoxygenation and desilylation provided pollenopyrroside A (2).
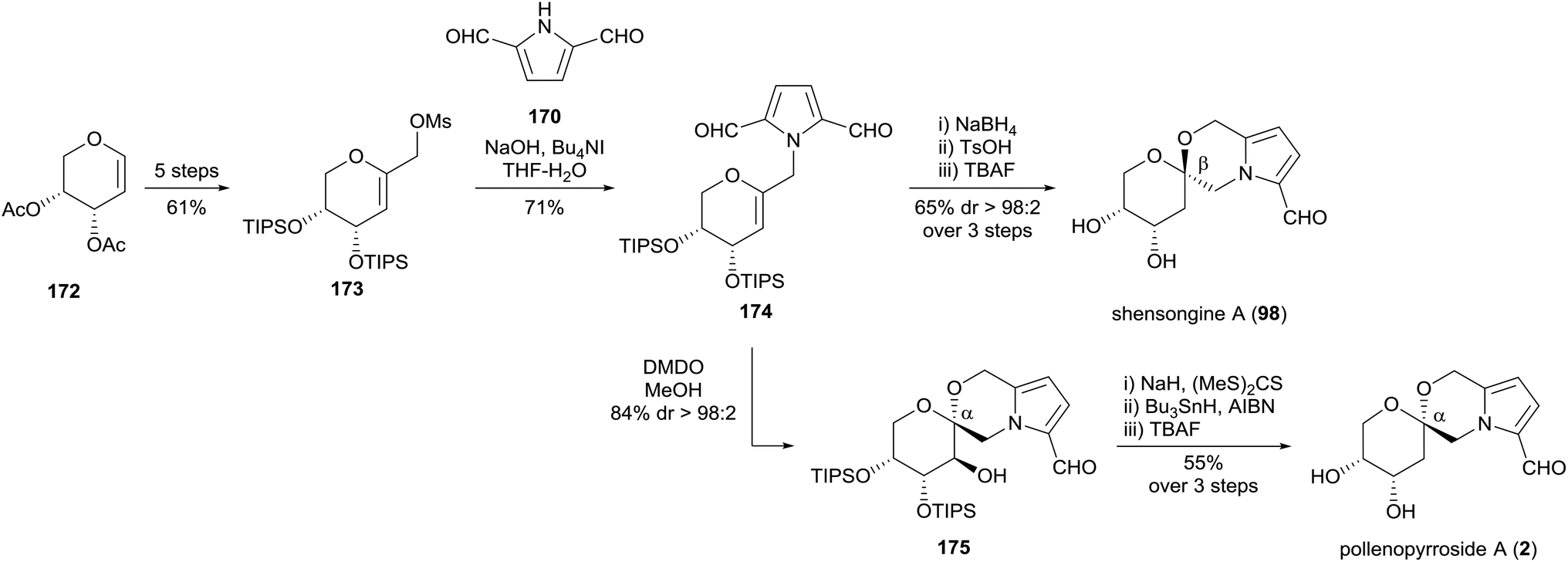 | ||
| Scheme 15 Synthesis of shensongine A (98) and pollenopyrroside A (2) by Verano and Tan. | ||
Borrero and Aponick reported a total synthesis of acortatarin A (1) and shensongine B (97) in 2012, featuring a palladium(II)-catalysed allylic transposition reaction developed within the group (Scheme 16).105 Bromoketone 177 was synthesised over seven steps from known δ-hydroxyester 176, which in turn was derived from L-tartrate. Union of 5-formylpyrrole-2-carboxamide 178 and bromoketone 177 proceeded best with caesium carbonate, which supressed competing elimination of the benzyloxy group. Reduction of pyrrole carbaldehyde 179 and treatment with bis(benzonitrile)palladium(II) chloride resulted in allylic transposition, forging the [5,6]-spiroketal system as a mixture of anomers 180 and 182. The terminal olefins 180 and 182 were advanced separately, both affording acortatarin A (1) as the major product upon titanium(IV) chloride-catalysed benzyl cleavage.
 | ||
| Scheme 16 Synthesis of acortatarin A (1) and shensongine B (97) by Borrero and Aponick. | ||
Zhao and co-workers reported a synthesis of shensongine A (98) and pollenopyrroside A (2) in 2015 (Scheme 17).106 The synthetic strategy involved functionalisation of pyrrole 184, the synthesis of which had been described in an earlier publication by the group from pyrrole and D-fructose.107 A microwave-accelerated protocol was developed to enable efficient bishydroxymethylation of pyrrole 184, with subsequent MnO2 oxidation affording the pyrrole-2,5-dicarbaldehyde 185. Protecting group manipulations, followed by selective reduction of one carbaldehyde group gave 2-formylpyrrole 186, which underwent acid-catalysed cyclisation to provide a 3:1 mixture of β-anomer 187 and α-anomer 188. These two spiroketals were advanced separately to shensongine A (98) and pollenopyrroside A (2) by modified Barton–McCombie deoxygenation and benzyl deprotection.
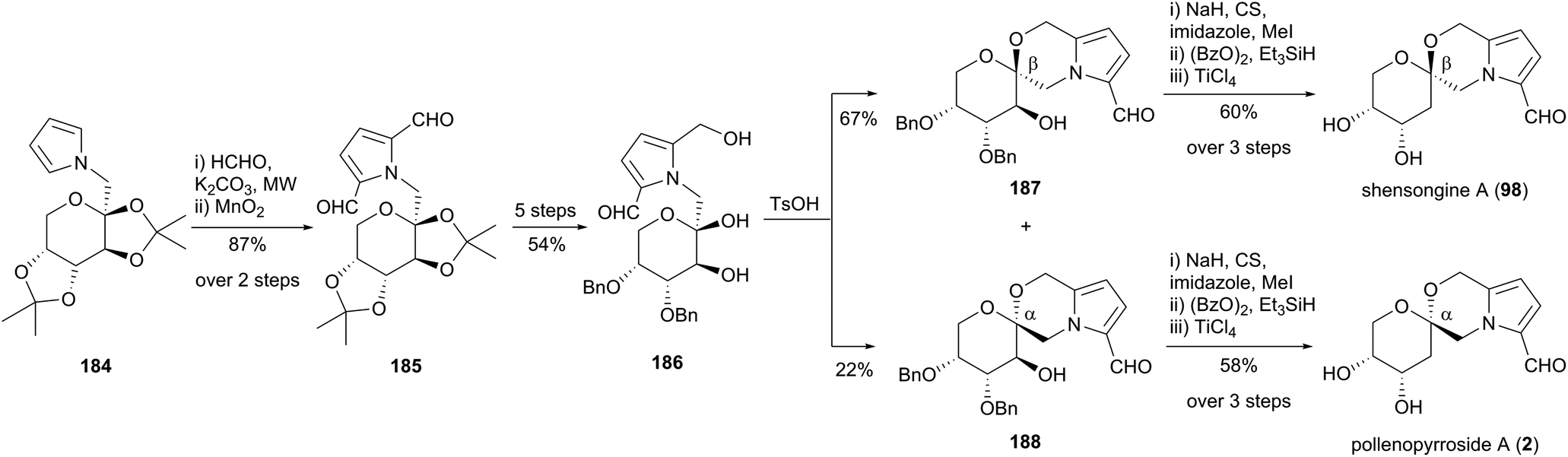 | ||
| Scheme 17 Synthesis of shensongine A (98) and pollenopyrroside A (2) by Zhao and co-workers. | ||
In 2017, Pale and co-workers reported a novel synthesis of acortatarin A (1) and shensongine B (97) which showcased the utility of zeolite-based organic synthesis (Scheme 18).108 Five of the eleven steps in the synthesis were performed using native or metal-doped zeolite catalysts. The acortatarin skeleton was assembled by ynamide coupling of alkynyl bromide 192 and pyrrole 193. This reaction was catalysed by copper(I)-doped zeolite, which performed better than both homogenous copper catalysts, and polystyrene and mesoporous silica-based heterogeneous copper catalysts. Sequential silver-catalysed alkyne hydroxylation and acid-catalysed cyclisation afforded spiroketal 196 as an anomeric mixture. The end-game synthesis-strategy involved redox manipulations to install the pyrrole carbaldehyde substituent and benzyl deprotection, providing acortatarin A (1) and shensongine B (97) as a 3:1 mixture of anomers.
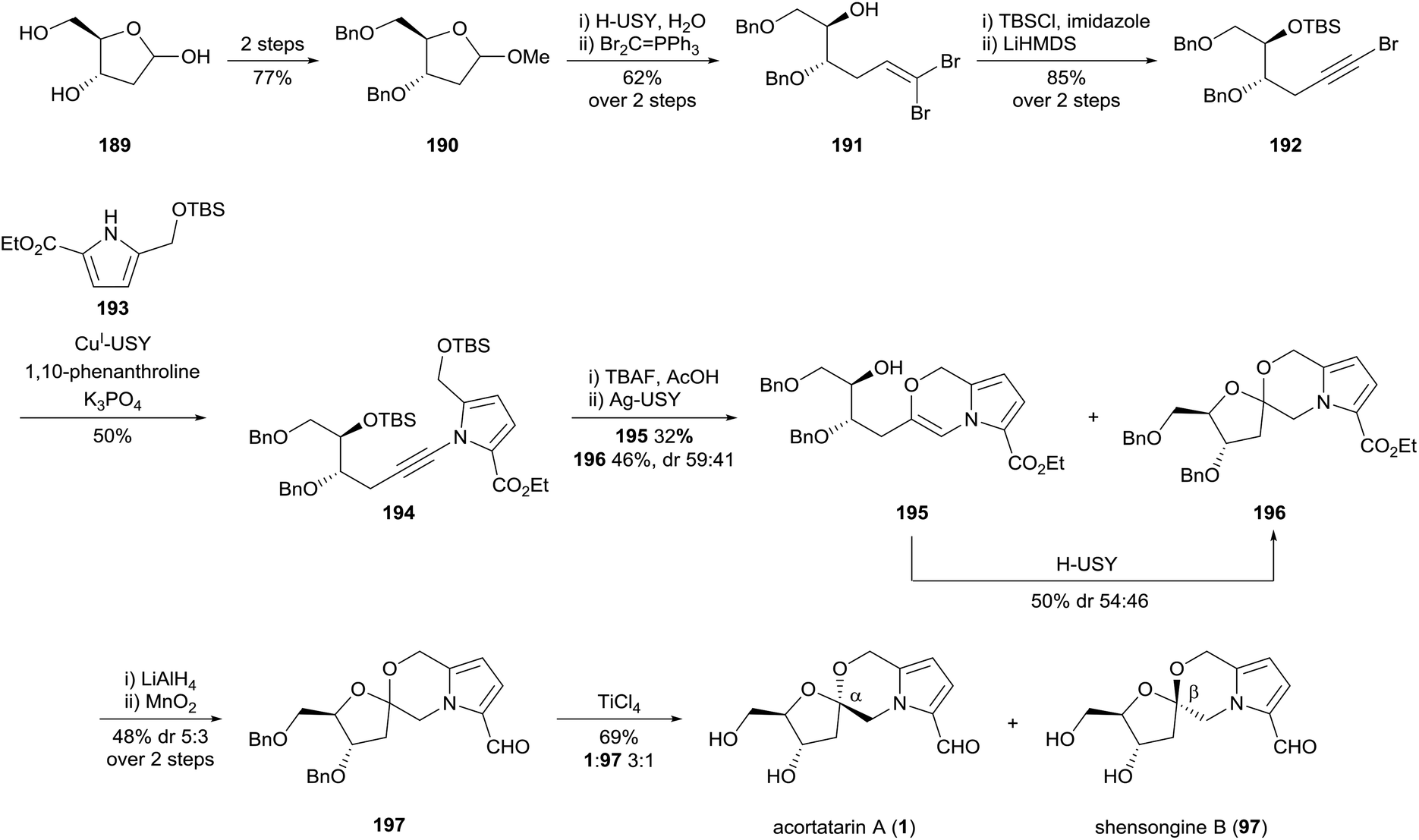 | ||
| Scheme 18 Synthesis of acortatarin A (1) and shensongine B (97) by Pale and co-workers. USY = ultrastable Y-type zeolite. | ||
4.4. Methodologies for 2-formylpyrrole synthesis
In addition to the considerable efforts towards the total synthesis of complex 2-formylpyrroles, two recent methodology studies have explored the reaction of sugars with amines to deliver synthetically useful yields of 2-formylpyrroles. In 2015, Koo and co-workers reported conditions for the synthesis of 2-formylpyrroles directly from various hexose reducing sugars and amines (Scheme 19).109 This Maillard-type reaction was performed in dimethylsulfoxide at 90 °C with one equivalent of oxalic acid to facilitate dehydration of D-glucose (7) in the presence of the basic amine substrates. These conditions were used to form the 2-formylpyrroles of a variety of amines as well as the lactonised pyrrole adducts 38 and 39 of L-phenylalanine methyl ester (198) and L-alanine methyl ester (199), respectively. The Maillard-type reaction of D-glucose (7) and γ-aminobutyric acid methyl ester (200) enabled access to lobechine (69) in three steps. | ||
| Scheme 19 Maillard-type reaction developed by Koo and co-workers. | ||
In 2016, Zhao and co-workers reported conditions for the synthesis of oxazine-fused pyrroles from amino acids and D-fructose (9) (Scheme 20).110 Extensive optimisation led to the solvent mixture of acetic acid and triethylamine (4:3) and use of five equivalents of D-fructose (9) relative to the amino acid. Chiral HPLC analysis of the products indicated that partial racemisation of the α-stereocenter had occurred during the reaction. For substrates D-valine, D-isoleucine, D-phenylalanine and D-tyrosine, this racemisation was minimal, however substrates D-alanine and D-leucine suffered from 25% and 10% racemisation respectively.
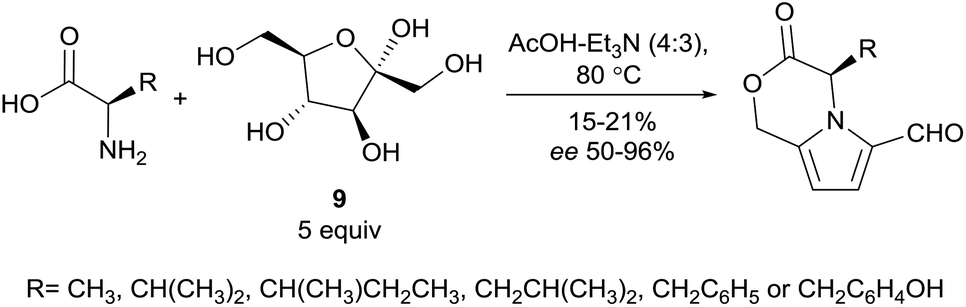 | ||
| Scheme 20 Maillard-type reaction developed by Zhao and co-workers. | ||
5. Conclusions
Initially observed as trace products in amine–sugar reactions mixtures, 2-formylpyrroles have since been isolated from a broad range of natural sources. Derived from amines and reducing sugars by non-enzymatic Maillard pathways, these pyrroles are representative of amine metabolites present in the natural source, yet often possess unique biological activities. The most studied group of pyrrole-2-carbaldehydes — the pyrrolomorpholine spiroketals — may provide a basis for drug discovery directed towards therapeutic intervention of diabetic nephropathy and have therefore enjoyed sustained interest from the synthetic community, succumbing to eleven total syntheses from nine research groups. Inspired by the Maillard pathways responsible for 2-formylpyrroles, other research has realised the preparation of pyrroles from sugars and amines in synthetically useful yields. Such synthetic advances will aid ongoing investigations into the biological activity of 2-formylpyrroles and may even enable the use of this compound family as highly functionalised sustainable platform chemicals.6. Conflicts of interest
There are no conflicts to declare.7. Acknowledgements
We thank the University of Auckland for the award of a Doctoral Scholarship (JMW).8. Notes and references
- X.-G. Tong, L.-L. Zhou, Y.-H. Wang, C. Xia, Y. Wang, M. Liang, F.-F. Hou and Y.-X. Cheng, Org. Lett., 2010, 12(8), 1844–1847 CrossRef CAS.
- J.-L. Guo, Z.-M. Feng, Y.-J. Yang, Z.-W. Zhang and P.-C. Zhang, Chem. Pharm. Bull., 2010, 58(7), 983–985 CrossRef CAS.
- B. Ding, Y. Dai, Y.-L. Hou and X.-S. Yao, J. Asian Nat. Prod. Res., 2015, 17(5), 559–566 CrossRef CAS PubMed.
- Q. Bin, D. Jiang, I. H. Cho and D. G. Peterson, Flavour Fragrance J., 2012, 27(6), 454–458 CrossRef CAS.
- D. Jiang and D. G. Peterson, Food Chem., 2013, 141(2), 1345–1353 CrossRef CAS PubMed.
- Y.-W. Chin, S. W. Lim, S.-H. Kim, D.-Y. Shin, Y.-G. Suh, Y.-B. Kim, Y. C. Kim and J. Kim, Bioorg. Med. Chem. Lett., 2003, 13(1), 79–81 CrossRef CAS.
- S. B. Kim, B. Y. Chang, Y. H. Jo, S. H. Lee, S.-B. Han, B. Y. Hwang, S. Y. Kim and M. K. Lee, J. Ethnopharmacol., 2013, 145(1), 393–396 CrossRef CAS.
- K. H. Kim, E. Moon, K. S. Kang, S. Y. Kim, S. U. Choi, K. R. Lee, K. H. Kim, E. Moon, K. S. Kang and S. Y. Kim, et al. , J. Braz. Chem. Soc., 2015, 26(1), 3–8 CAS.
- M.-J. Don, W.-F. Chiou, C.-C. Shen, H.-J. Yu, C.-H. Chiang, C.-C. Chen and W. Chang, Heterocycles, 2005, 65(5), 1215 CrossRef.
- Z. F. Zhao, L. L. Zhou, X. Chen, Y. X. Cheng, F. F. Hou and J. Nie, Chin. Med. J., 2013, 126(7), 1230–1235 CAS.
- H. Li and S.-J. Yu, J. Sci. Food Agric., 2018, 98, 3225–3233 CrossRef CAS.
- B. B. Butler and A. Aponick, in Strategies and Tactics in Organic Synthesis, ed. M. Harmata, Academic Press, 2015, vol. 11, pp. 1–28 Search PubMed.
- M. Faisal, D. Shahzad, F. A. Larik and P. Dar, Fitoterapia, 2018 DOI:10.1016/j.fitote.2018.03.014.
- T. M. Zenni, J. M. Cassady and R. F. Raffauf, J. Nat. Prod., 1986, 49(4), 695–698 CrossRef.
- T. Matsumoto, S. Nakamura, T. Ohta, K. Fujimoto, M. Yoshikawa, K. Ogawa and H. Matsuda, Org. Lett., 2014, 16(11), 3076–3078 CrossRef CAS.
- H.-J. Yu, C.-C. Chen and B.-J. Shieh, J. Nat. Prod., 1998, 61(8), 1017–1019 CrossRef CAS PubMed.
- T. Nakayama, F. Hayase and H. Kato, Agric. Biol. Chem., 1980, 44(5), 1201–1202 CAS.
- F. Hayase, R. H. Nagaraj, S. Miyata, F. G. Njoroge and V. M. Monnier, J. Biol. Chem., 1989, 264(7), 3758–3764 CAS.
- M. Hellwig and T. Henle, Angew. Chem., Int. Ed., 2014, 53(39), 10316–10329 CrossRef CAS PubMed.
- J. E. Hodge, J. Agric. Food Chem., 1953, 1(15), 928–943 CrossRef CAS.
- H. Kato and M. Fujimaki, Agric. Biol. Chem., 1970, 34(7), 1071–1077 CrossRef CAS.
- G. R. Jurch and J. H. Tatum, Carbohydr. Res., 1970, 15(2), 233–239 CrossRef CAS.
- F. Ledl, in The Maillard Reaction in Food Processing, Human Nutrition and Physiology, ed. P. A. Finot, H. U. Aeschbacher, R. F. Hurrell and R. Liardon, Birkhaeuser, Basel, 1990, pp. 19–42 Search PubMed.
- E. F. L. Anet, Aust. J. Chem., 1960, 13(3), 396–403 CrossRef CAS.
- J. E. Hodge, in Advances in Carbohydrate Chemistry, ed. M. L. Wolfrom, Academic Press, 1955, vol. 10, pp. 169–205 Search PubMed.
- M. O. Lederer and M. Baumann, Bioorg. Med. Chem., 2000, 8(1), 115–121 CrossRef CAS.
- C. T. Walsh, S. Garneau-Tsodikova and A. R. Howard-Jones, Nat. Prod. Rep., 2006, 23(4), 517–531 RSC.
- S. Lautru, L. Song, L. Demange, T. Lombès, H. Galons, G. L. Challis and J.-L. Pernodet, Angew. Chem., Int. Ed., 2012, 51, 7454–7458 CrossRef CAS.
- A. Vingadassalon, F. Lorieux, M. Juguet, G. Le Goff, C. Gerbaud, J.-L. Pernodet and S. Lautru, ACS Chem. Biol., 2015, 10(2), 601–610 CrossRef CAS.
- J. P. Dickerson, D. L. Roberts, C. W. Miller, R. A. Lloyd and C. E. Rix, Tobacco, 1976, 178(9), 71–77 CAS.
- R. A. Lloyd, C. W. Miller, D. L. Roberts, J. A. Giles, J. P. Dickerson, N. H. Nelson, C. E. Rix and P. H. Ayers, Tob. Sci., 1976, 20, 125–133 CAS.
- Y. Guo, X. Li, J. Wang, J. Xu and N. Li, Fitoterapia, 2005, 76(2), 273–275 CrossRef CAS.
- S. B. Kim, B. Y. Chang, B. Y. Hwang, S. Y. Kim and M. K. Lee, Bioorg. Med. Chem. Lett., 2014, 24(24), 5656–5659 CrossRef CAS.
- J. Xiong, Y. Huang, X.-Y. Wu, X.-H. Liu, H. Fan, W. Wang, Y. Zhao, G.-X. Yang, H.-Y. Zhang and J.-F. Hu, Helv. Chim. Acta, 2016, 99(1), 83–89 CrossRef CAS.
- P. Wang, F. Kong, J. Wei, Y. Wang, W. Wang, K. Hong and W. Zhu, Mar. Drugs, 2014, 12(1), 477–490 CrossRef CAS.
- F. O. Chagas, A. M. Caraballo-Rodríguez, P. C. Dorrestein and M. T. Pupo, J. Nat. Prod., 2017, 80(5), 1302–1309 CrossRef CAS PubMed.
- E. Abe, Y. Nakatani, T. Yamanishi and S. Muraki, Proc. Jpn. Acad., Ser. B, 1978, 54(9), 542–547 CrossRef CAS.
- A. Sannai, T. Fujimori and K. Kato, Agric. Biol. Chem., 1982, 46(2), 429–433 CAS.
- T. Yang, C. Wang, G. Chou, T. Wu, X. Cheng and Z. Wang, Food Chem., 2010, 123(3), 705–710 CrossRef CAS.
- R. C. Anderson, A. G. Kelly and J. S. Roberts, J. Agric. Food Chem., 1983, 31(2), 458–459 CrossRef CAS.
- N.-N. Yang, S.-Z. Huang, Q.-Y. Ma, H.-F. Dai, Z.-K. Guo, Z.-F. Yu and Y.-X. Zhao, Chem. Nat. Compd., 2015, 51(4), 730–732 CrossRef CAS.
- W.-Y. Liu, W.-D. Zhang, H.-S. Chen, Z.-B. Gu, T.-Z. Li and Yun-Zhou, J. Asian Nat. Prod. Res., 2003, 5(3), 159–163 CrossRef CAS.
- U. J. Youn, J. Y. Lee, Y.-S. Kil, A.-R. Han, C. H. Chae, S. Y. Ryu and E.-K. Seo, Arch. Pharmacal Res., 2016, 39(3), 321–327 CrossRef CAS.
- U. Joung Youn, Y.-S. Kil, J.-W. Nam, Y. Jin Lee, J. Kim, D. Lee, J.-H. Lee and E.-K. Seo, Helv. Chim. Acta, 2013, 96(8), 1482–1487 CrossRef.
- J.-H. Choi, T. Suzuki, T. Kawaguchi, K. Yamashita, A. Morita, K. Masuda, K. Yazawa, H. Hirai and H. Kawagishi, Tetrahedron Lett., 2014, 55(26), 3596–3599 CrossRef CAS.
- J. Li, L. Pan, C. B. Naman, Y. Deng, H. Chai, W. J. Keller and A. D. Kinghorn, J. Agric. Food Chem., 2014, 62(22), 5054–5060 CrossRef CAS.
- S.-I. Kayano, H. Kikuzaki, T. Ikami, T. Suzuki, T. Mitani and N. Nakatani, Biosci., Biotechnol., Biochem., 2004, 68(4), 942–944 CrossRef CAS.
- S.-I. Kayano, H. Kikuzaki, N. F. Yamada, A. Aoki, K. Kasamatsu, Y. Yamasaki, T. Ikami, T. Suzuki, T. Mitani and N. Nakatani, BioFactors, 2004, 21(1–4), 309–313 CrossRef CAS.
- S. B. Kim, B. Ahn, M. Kim, H.-J. Ji, S.-K. Shin, I. P. Hong, C. Y. Kim, B. Y. Hwang and M. K. Lee, J. Ethnopharmacol., 2014, 151(1), 478–484 CrossRef CAS.
- R. F. Raffauf, T. M. Zennie, K. D. Onan and P. W. Le Quesne, J. Org. Chem., 1984, 49, 2714–2718 CrossRef CAS.
- R. E. Schultes, Bot. Mus. Leafl., Harv. Univ., 1957, 17(9), 247–264 Search PubMed.
- D. G. Lynn, K. Jaffe, M. Cornwall and W. Tramontano, J. Am. Chem. Soc., 1987, 109(19), 5858–5859 CrossRef CAS.
- T. Matsumoto, S. Nakamura, S. Nakashima, T. Ohta, M. Yano, J. Tsujihata, J. Tsukioka, K. Ogawa, M. Fukaya and M. Yoshikawa, et al. , J. Nat. Med., 2016, 70(3), 376–383 CrossRef CAS.
- J. M. Wood, D. P. Furkert and M. A. Brimble, J. Nat. Prod., 2017, 80(6), 1926–1929 CrossRef CAS.
- L. Fowden, Biol. Rev., 1958, 33(4), 393–441 CrossRef CAS.
- Y. Ogawa and T. Konishi, Chem. Pharm. Bull., 2009, 57(10), 1110–1112 CrossRef CAS.
- G. P. Rizzi, Food Rev. Int., 2008, 24(4), 416–435 CrossRef CAS.
- M. Matsui, Y. Sato, H. Bando, M. Murayama, T. Osawa, T. Miura and Y. Oshima, Nat. Med., 1998, 52(3), 232–235 CAS.
- S. Sang, A. Lao, Y. Wang, C.-K. Chin, R. T. Rosen and C.-T. Ho, J. Agric. Food Chem., 2002, 50(22), 6318–6321 CrossRef CAS.
- S. F. Farag, Y. Kimura, H. Ito, J. Takayasu, H. Tokuda and T. Hatano, J. Nat. Med., 2009, 63(1), 91 CrossRef CAS.
- Z. Zhou, J. Luo, K. Pan and L. Kong, Nat. Prod. Res., 2014, 28(14), 1065–1069 CrossRef CAS.
- X.-F. Wang, L. Yu, W.-J. Hao, L. Ma, L. Yin and X.-Y. Fu, Chem. Nat. Compd., 2016, 52(4), 769–770 CrossRef CAS.
- H. J. Jung, H. A. Jung, S. S. Kang, J.-H. Lee, Y. S. Cho, K. H. Moon and J. S. Choi, Arch. Pharmacal Res., 2012, 35(10), 1771–1777 CrossRef CAS.
- W. Chen, X.-A. Shou, Y. Chen, N. Qin, W. Qiao, S.-A. Tang and H.-Q. Duan, Chem. Nat. Compd., 2014, 50(6), 989–993 CrossRef CAS.
- P.-C. Kuo, T.-L. Hwang, Y.-T. Lin, Y.-C. Kuo and Y.-L. Leu, Arch. Pharmacal Res., 2011, 34(5), 715–722 CrossRef CAS.
- Z. Feng, Z. Zhan, Y. Yang, J. Jiang and P. Zhang, Bioorg. Chem., 2017, 74, 10–14 CrossRef CAS.
- Z. Feng, Z. Zhan, Y. Yang, J. Jiang and P. Zhang, Sci. Rep., 2016, 6, 25443 CrossRef CAS.
- L.-Y. Li, Y. Ding, I. Groth, K.-D. Menzel, G. Peschel, K. Voigt, Z.-W. Deng, I. Sattler and W.-H. Lin, J. Asian Nat. Prod. Res., 2008, 10(8), 765–770 CrossRef CAS.
- W.-G. Shan, Y. Wang, L.-F. Ma and Z.-J. Zhan, J. Chem. Res., 2014, 38, 245–246 CrossRef.
- Y.-P. Yang, M.-J. Cheng, C.-M. Teng, Y.-L. Chang, I.-L. Tsai and I.-S. Chen, Phytochemistry, 2002, 61(5), 567–572 CrossRef CAS.
- J.-H. Choi, N. Ozawa, Y. Yamakawa, K. Nagai, H. Hirai and H. Kawagishi, Tetrahedron, 2011, 67(35), 6649–6653 CrossRef CAS.
- G. A. Zou, S. Mansur, S. C. Hu, H. A. Aisa and K. M. Shakhidoyatov, Chem. Nat. Compd., 2012, 48(4), 635–637 CrossRef CAS.
- L. Han, C. Gao, Y. Jiang, P. Guan, J. Liu, L. Li, L. Xu and X. Huang, J. Nat. Prod., 2014, 77(12), 2605–2610 CrossRef CAS.
- J.-J. Xia, Y.-D. Li, X.-M. Liu, Y. Lu, Y.-Q. Wu and Y.-H. Qin, J. Asian Nat. Prod. Res., 2016, 18(8), 779–783 CrossRef CAS.
- G.-H. Xu, Y.-H. Kim, S.-J. Choo, I.-J. Ryoo, J.-K. Yoo, J.-S. Ahn and I.-D. Yoo, Arch. Pharmacal Res., 2009, 32(9), 1215–1220 CrossRef CAS.
- Q. Li, A.-J. Deng, L. Li, L.-Q. Wu, M. Ji, H.-J. Zhang, Z.-H. Li, L. Ma, Z.-H. Zhang and X.-G. Chen, et al. , J. Nat. Prod., 2017, 80(8), 2189–2198 CrossRef CAS.
- X.-F. Yang, X.-H. Cao, Y.-M. Ma and K. Qiao, Nat. Prod. Res., 2018, 32(3), 302–307 CrossRef CAS.
- J. Yu, X. Wang, H. Yan, Y. Geng, X. Wang and H. Zhao, Chn. Pat., 107698510A, February 16, 2018.
- T. Kikuchi, A. Ikedaya, A. Toda, K. Ikushima, T. Yamakawa, R. Okada, T. Yamada and R. Tanaka, Phytochem. Lett., 2015, 12, 94–97 CrossRef CAS.
- M.-D. Wu, M.-J. Cheng, I.-S. Chen, Y.-S. Su, S.-Y. Hsieh, H.-S. Chang, C.-W. Chang and G.-F. Yuan, Chem. Biodiversity, 2013, 10(3), 493–505 CrossRef CAS.
- A. Hiermann, S. Kedwani, H. W. Schramm and C. Seger, Fitoterapia, 2002, 73(1), 22–27 CrossRef CAS.
- L. Xiong, C. Peng, X.-F. Xie, L. Guo, C.-J. He, Z. Geng, F. Wan, O. Dai and Q.-M. Zhou, Molecules, 2012, 17(8), 9939–9946 CrossRef CAS.
- H. Li, J. Xiao, Y.-Q. Gao, J. Tang, A.-L. Zhang and J.-M. Gao, J. Agric. Food Chem., 2014, 62(17), 3734–3741 CrossRef CAS.
- M.-J. Don, C.-C. Shen, Y.-L. Lin, W.-J. Syu, Y.-H. Ding and C.-M. Sun, J. Nat. Prod., 2005, 68(7), 1066–1070 CrossRef CAS.
- L.-L. Liu, J.-L. Yang and Y.-P. Shi, J. Asian Nat. Prod. Res., 2011, 13(10), 920–929 CrossRef CAS.
- N. W. Fan, H. S. Chang, M. J. Cheng, H. Y. Chan, S. Y. Hsieh, T. W. Liu, S. W. Chen, G. F. Yuan and I. S. Chen, Chem. Nat. Compd., 2016, 52(4), 585–590 CrossRef CAS.
- M.-J. Cheng, H.-Y. Chan, Y.-C. Cheng, M.-D. Wu, J.-J. Chen, Y.-L. Chen, S.-Y. Hsieh, G.-F. Yuan and Y.-S. Su, Chem. Nat. Compd., 2015, 51(3), 515–518 CrossRef CAS.
- H. Zhou, R. Zhao and J. Yang, Nat. Prod. Res., 2013, 27(8), 687–690 CrossRef CAS.
- M. Li, J. Xiong, Y. Huang, L.-J. Wang, Y. Tang, G.-X. Yang, X.-H. Liu, B.-G. Wei, H. Fan, Y. Zhao, W.-Z. Zhai and J.-F. Hu, Tetrahedron, 2015, 71(33), 5285–5295 CrossRef CAS.
- S. Giunti, D. Barit and M. E. Cooper, Minerva Med., 2006, 97(3), 241–262 CAS.
- A. L. Verano and D. S. Tan, Chem. Sci., 2017, 8(5), 3687–3693 RSC.
- P. Hayoz, A. Aeby and R. Neier, Chimia, 1993, 47, 230–232 CAS.
- Y. Dong, N. N. Pai, S. L. Ablaza, S.-X. Yu, S. Bolvig, D. A. Forsyth and P. W. Le Quesne, J. Org. Chem., 1999, 64(8), 2657–2666 CrossRef CAS.
- S. L. Ablaza, N. N. Pai and P. W. L. Quesne, Nat. Prod. Lett., 1995, 6(1), 77–80 CrossRef CAS.
- S.-X. Yu and P. W. Le Quesne, Tetrahedron Lett., 1995, 36(35), 6205–6208 CrossRef CAS.
- O. Tamura, N. Iyama and H. Ishibashi, J. Org. Chem., 2004, 69(5), 1475–1480 CrossRef CAS.
- T. Okada, K. Sakaguchi, T. Shinada and Y. Ohfune, Tetrahedron Lett., 2011, 52(44), 5744–5746 CrossRef CAS.
- T.-Y. Yuen, S. E. Eaton, T. M. Woods, D. P. Furkert, K. W. Choi and M. A. Brimble, Eur. J. Org. Chem., 2014, 2014(7), 1431–1437 CrossRef CAS.
- H. M. Geng, J. L.-Y. Chen, D. P. Furkert, S. Jiang and M. A. Brimble, Synlett, 2012, 23(06), 855–858 CrossRef CAS.
- G. Sudhakar, V. D. Kadam, S. Bayya, G. Pranitha and B. Jagadeesh, Org. Lett., 2011, 13(20), 5452–5455 CrossRef CAS.
- T. Teranishi, M. Kageyama and S. Kuwahara, Biosci., Biotechnol., Biochem., 2013, 77(3), 676–678 CrossRef CAS.
- H. M. Geng, L. A. Stubbing, J. Li-yang Chen, D. P. Furkert and M. A. Brimble, Eur. J. Org. Chem., 2014, 2014(28), 6227–6241 CrossRef CAS.
- J. M. Wood, D. P. Furkert and M. A. Brimble, Org. Biomol. Chem., 2016, 14(32), 7659–7664 RSC.
- J. M. Wurst, A. L. Verano and D. S. Tan, Org. Lett., 2012, 14(17), 4442–4445 CrossRef CAS.
- N. V. Borrero and A. Aponick, J. Org. Chem., 2012, 77(19), 8410–8416 CrossRef CAS.
- Z. Cao, Y. Li, S. Wang, X. Guo, L. Wang and W. Zhao, Synlett, 2015, 26(07), 921–926 CrossRef CAS.
- S. Huo, Y. Li, C. Liang, J. Liu and W. Zhao, J. Carbohydr. Chem., 2011, 30(2), 75–84 CrossRef CAS.
- E. Wimmer, S. Borghèse, A. Blanc, V. Bénéteau and P. Pale, Chem.–Eur. J., 2017, 23(7), 1484–1489 CrossRef CAS PubMed.
- N. D. Adhikary, S. Kwon, W.-J. Chung and S. Koo, J. Org. Chem., 2015, 80(15), 7693–7701 CrossRef CAS.
- Z. Cao, Y. Li, S. Wang, B. Tang, X. Guo, L. Wang and W. Zhao, Tetrahedron Lett., 2016, 57(21), 2219–2221 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |