
Total synthesis of sesterterpenoids
Jing
Xu  *b
*b
*b
Abstract
Covering: January 2012 to January 2018
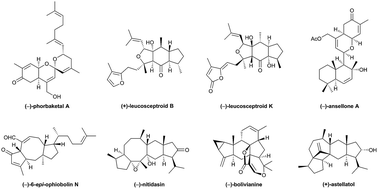
Sesterterpenoids are a small family of terpenes that often possess intriguing biological profiles and complicated chemical structures. Their total syntheses are usually remarkably challenging, requiring methodological and strategic innovation. In this review, we summarize and discuss the total syntheses of sesterterpenoids published during the coverage period, and the key chemical transformations are highlighted.
 Please wait while we load your content...
Please wait while we load your content...

