Chemistry of the fumiquinazolines and structurally related alkaloids†
Diana I. S. P.
Resende‡ab,
Papichaya
Boonpothong‡a,
Emília
Sousa
*ab,
Anake
Kijjoa
bc and
Madalena M. M.
Pinto
ab aLaboratory of Organic and Pharmaceutical Chemistry, Faculty of Pharmaceutical Sciences, University of Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal. E-mail: esousa@ff.up.pt bInterdisciplinary Centre of Marine and Environmental Research (CIIMAR), Terminal de Cruzeiros do Porto de Leixões, Avenida General Norton de Matos, S/N, 4050-208, Matosinhos, Portugal cICBAS-Instituto de Ciências Biomédicas Abel Salazar, Universidade do Porto, Rua de Jorge Viterbo Ferreira, 228, 4050-313 Porto, Portugal
Received
2nd May 2018
First published on 9th August 2018
Abstract
Covering: this review covers the literature from 1992 to 2018
To date, approximately 80 naturally-occurring secondary metabolites which are structurally-related to fumiquinazolines have been isolated, mainly from marine sources. These alkaloids can be classified into twelve different groups and exhibit different structure motifs depending on the amino acids from which they are derived. This review is focused on isolation, structure elucidation, biological activities, biosynthetic pathways, and synthetic studies of these natural products.
1 Introduction
Nature has been an important source of medicinal products for millennia, with many useful drugs developed from terrestrial and marine sources.1 Due to the development of new techniques and methodologies (sampling techniques, established culture methods, genome mining, etc.) marine organisms have been heavily sampled in the last two decades,2 and have become an important source of pharmacologically active metabolites.3–10 Moreover, in the last decades a significant number of natural products, used as drugs and/or leads, are produced “in part” (and in some cases totally), by interactions with endophytic microbes, as highlighted by Newman and Cragg1 in their last review. The fumiquinazolines, which contain a pyrazino[2,1-b] quinazoline-3,6-dione core linked to an indole moiety (Chart 1), are secondary metabolites that have emerged in the last two decades, mainly from terrestrial and marine fungi, and have revealed very promising activities, especially in the field of chemotherapics.11 The quinazolinone scaffold can be recognized as a privileged structure with several representatives in therapy such is the case of the synthetic anticancer agent idelalisib. Although there are already some reviews on this class of natural products, most of them are focused on a particular perspective.12–16 Therefore, this review deals with the latest progress on isolation, biological activities, and chemical and biogenetically synthetic studies of these natural products.
Chart 1 Typical core structural motif of fumiquinazolines and related alkaloids.
In each section, major challenges related to isolation and synthesis are highlighted, particularly in structure elucidation, concerns in avoiding epimerization, and the trend in the discovery of novel chemotherapeutics based on this scaffold, with activities highly dependent on stereochemistry. This review aims at providing researchers with a perspective in the exploration of new sources, new synthetic routes, new derivatives and applications of fumiquinazolines establishing structure–activity relationship, thus paving the way to potential drug candidates.
2 Isolation, biosynthesis, and biological activities
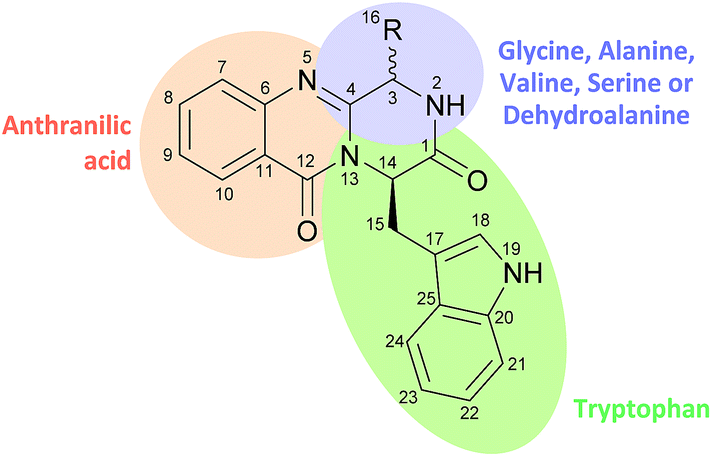
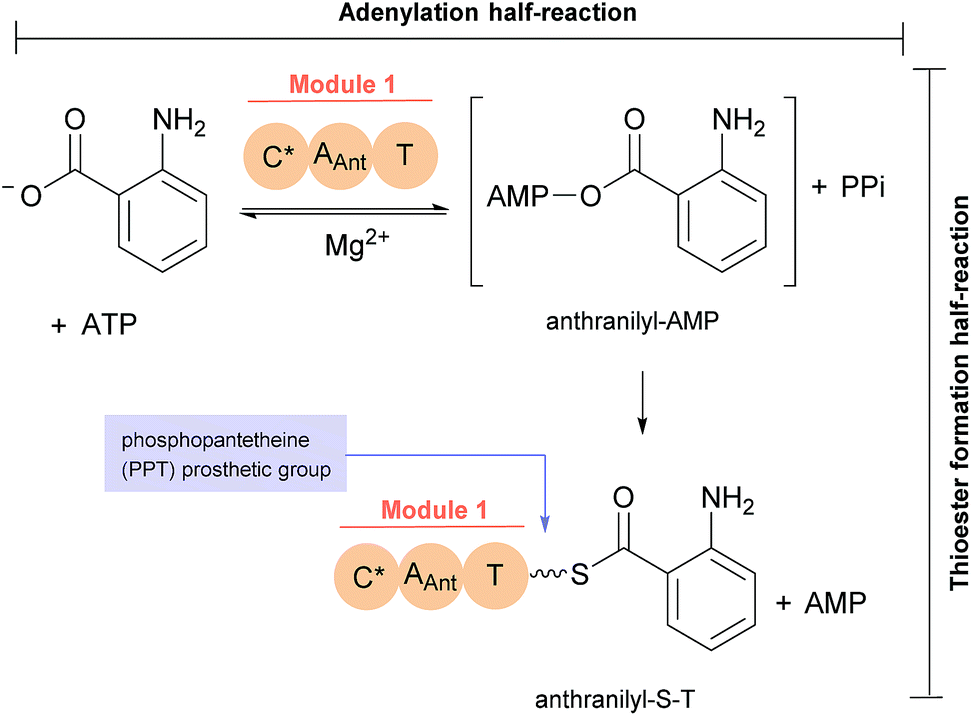
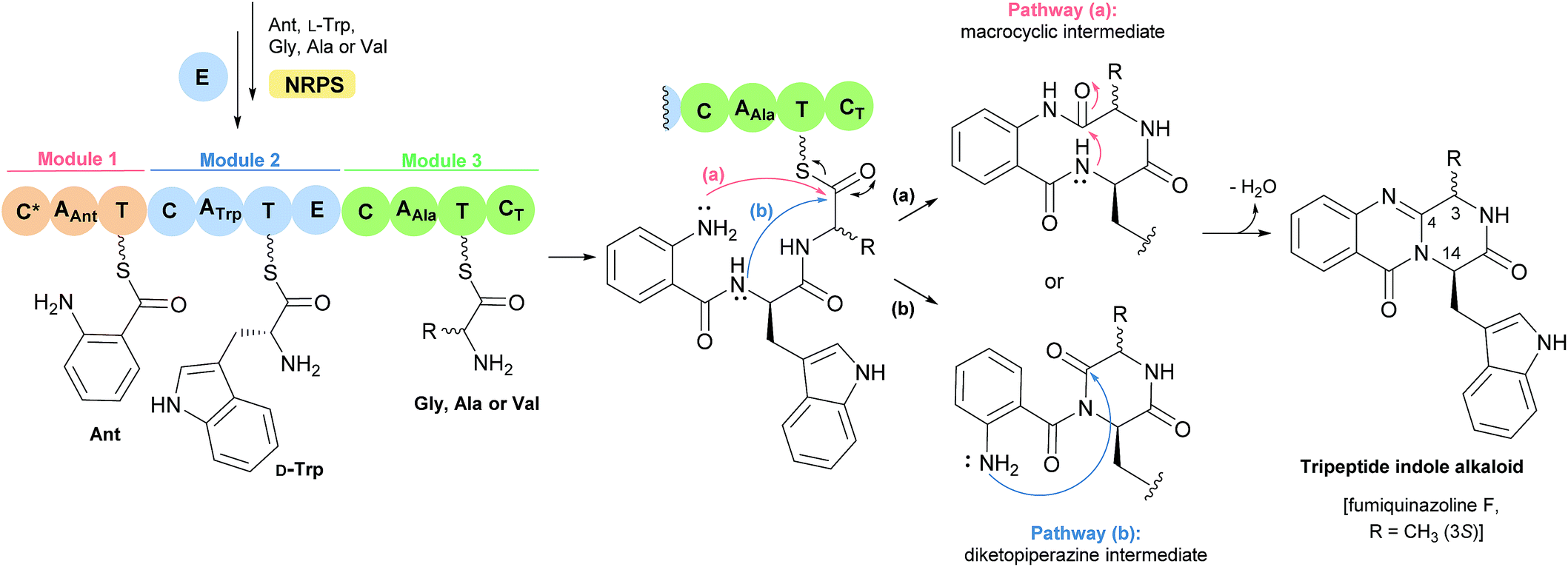
The first reported secondary metabolite with a pyrazino[2,1-b] quinazoline-3,6-dione core linked to an indole moiety, fumiquinazoline A (1, see Chart 2), was characterized in 1992 by spectroscopic and X-ray diffraction analyses.17 Over the next twenty five years, a total of approximately 80 structurally related compounds were isolated mainly from fungi-derived marine sources.3–8 As the number of natural product reports increased, new structural patterns emerged, ranging from compounds containing only a substituted piperazine ring to those with a more complex structure with several fused rings, prenylated indoles, spiro compounds or with complex 3-indolyl groups. The various levels of complexity displayed by these fungal indole alkaloids and their biosynthetic routes are of interest in genetic studies for identification of the metabolite corresponding to each gene cluster.18–30 They are biosynthesized by trimodular non-ribosomal peptide synthetases (NRPSs), through the incorporation of the β-amino acid anthranilate (anthranilic acid, Ant), tryptophan (Trp), and an additional amino acid (Chart 1).22 Glycine (Gly) is incorporated as C-3 and C-4 of the pyazinoquinazolinone ring system, whereas alanine (Ala) or valine (Val) are included as C-3, C-4 and the substituents on C-3. On the other hand, the indole moiety can contain the simple tryptophan structure or a tricyclic imidazoindolone core generated later by the condensation of a fourth amino acid (alanine, valine, isoleucine, 2-aminoisobutyric acid, etc.) with the indole ring of tryptophan. NRPSs are often found as multimodular megasynthases that participate in the biosynthesis of peptidyl scaffolds.20 Minimally, a single NRPS module consists of adenylation (A), peptide carrier protein (PCP)/thiolation (T) and condensation (C) domains arranged in repeating units that assemble non-ribosomal peptides (NRP) by activating and joining amino acid building blocks.20,31 Besides these three domains, some variations of NRPS domains also occur, with the C domain variant epimerization (E), heterocyclization domains and specific domains for N-methylation or formylation being the most common.18,21 The general process of building peptidyl frameworks begins with the A-domain-catalysed activation of the anthranilate as the corresponding anthranilyl-AMP, while ATP is consumed. This domain mediates the attachment to the 4′-phosphopantetheine (ppt) prosthetic arm of the T-domain, forming an acyl-thioester intermediate (Scheme 1).28 Regarding fungal indole alkaloids, the anthranilate is always the first amino acid incorporated by the trimodular NRPS in module one, and usually tryptophan in module two and the third amino acid in module three.21 The tryptophan-activating module has an embedded epimerization domain, presumably to convert the L-Trp-S-T domain thioester into a mixture of the D- and L-isomers, with only the D-Trp-thioester being carried forward.30 The condensation of the three amino acid moieties yields a linear tripeptidyl acyl thioester tethered to the T-domain that is then delivered to the active site of the C-domain (Scheme 2). This domain catalyses the cyclization and the release of the tripeptide either through the formation of a ten-membered macrolactam, that, upon intramolecular cyclization yields the pyrazino[2,1-b]quinazoline-3,6-dione core (pathway a) or through the intramolecular formation of an Ant-diketopiperazine, subsequent cyclization and dehydration to form the tripeptide alkaloid (pathway b).18,20,26
Scheme 1 Anthranilate activation (by adenylation) and anthranilyl transfer (by thiolation) by NRPS module 1.28
Scheme 2 Biosynthesis of tripeptide indole alkaloids.
Taking into consideration the biosynthetic pathways, fumiquinazolines and structurally related alkaloids can be classified into four major groups: alanine-derived alkaloids (fumiquinazolines 1–23, ardeemins 24–28, aniquinazolines 29–32, fumigatosides 33–35), glycine-derived alkaloids (glyantrypines 36–39, cottoquinazolines 40–45, versiquinazolines 46–52), valine-derived alkaloids (fiscalins 53–61, cladoquinazolines 62, 63, quinadolines 64, 65, neosartoryadins 66, 67), and spiroquinazolines 68–77, which have different biosynthetic origins. Biochemical characterizations of fumiquinazolines and derivatives were performed mainly by genetic studies, bioinformatics analysis, and radiolabeling studies, that supported the hypothesized pathways described for the majority of fumiquinazolines groups. More complex structures are formed through further modifications of the simpler structures, such as oxidation and prenylation, with several secondary metabolites sharing the same biosynthetic pathway.32 These studies may serve as an inspiration for biomimetic synthetic procedures using biosynthetic building blocks as starting points and even their combined use with enzymes involved in their biosynthesis.
In the following sections, the fumiquinazolines members and their biological activities are presented detailing their individual biosynthetic tailoring reactions.
2.1 Alanine-derived alkaloids
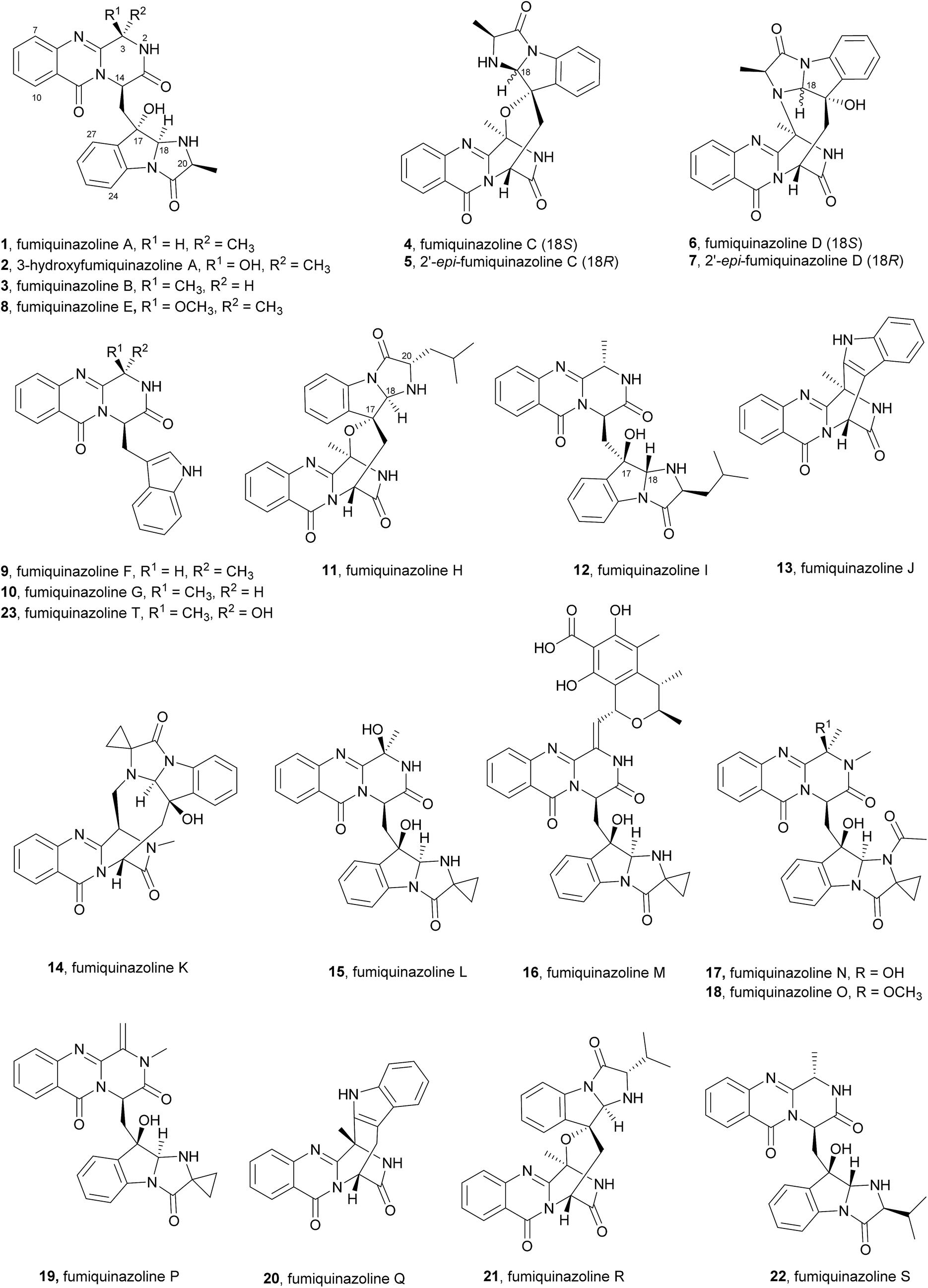
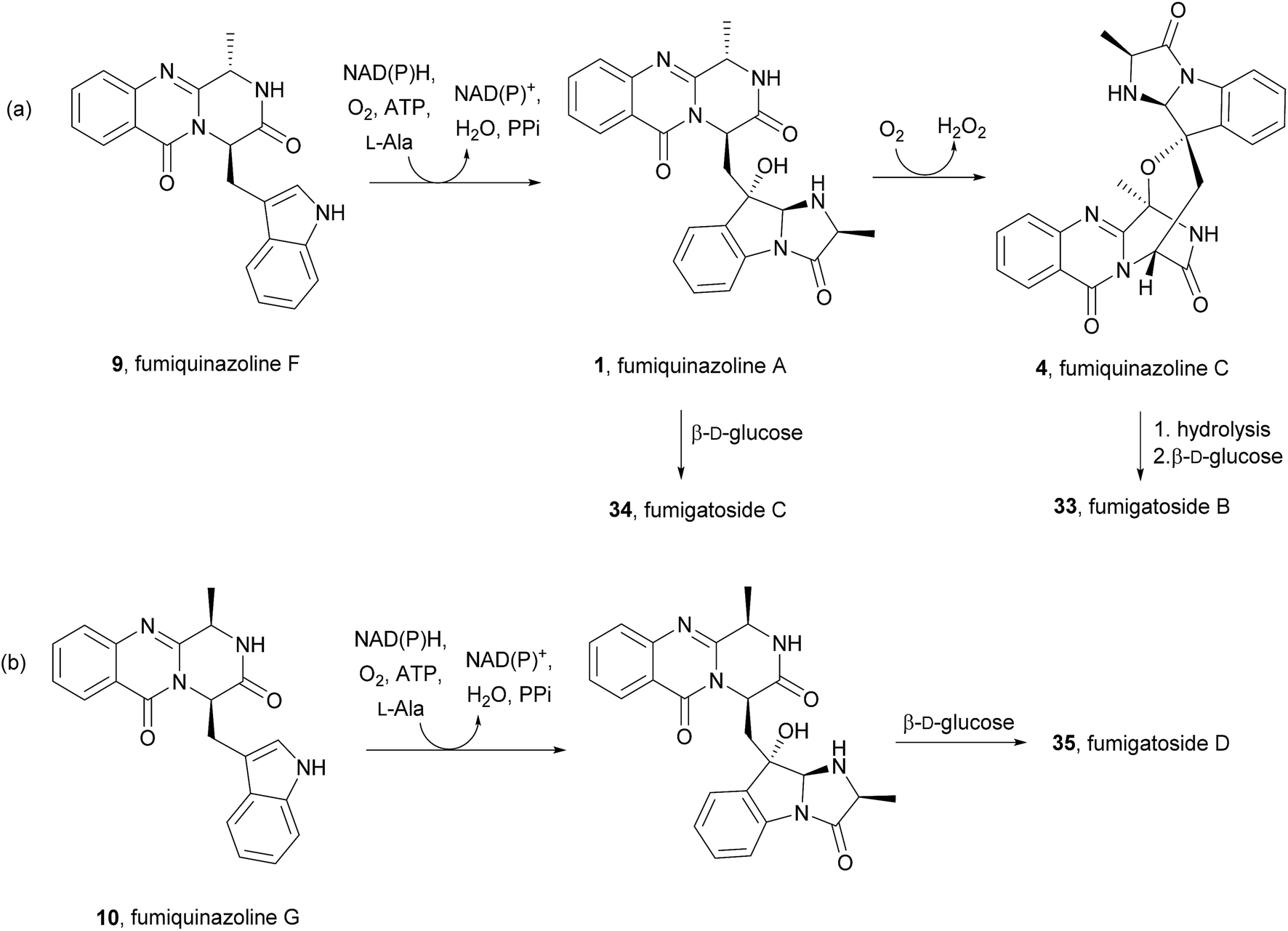
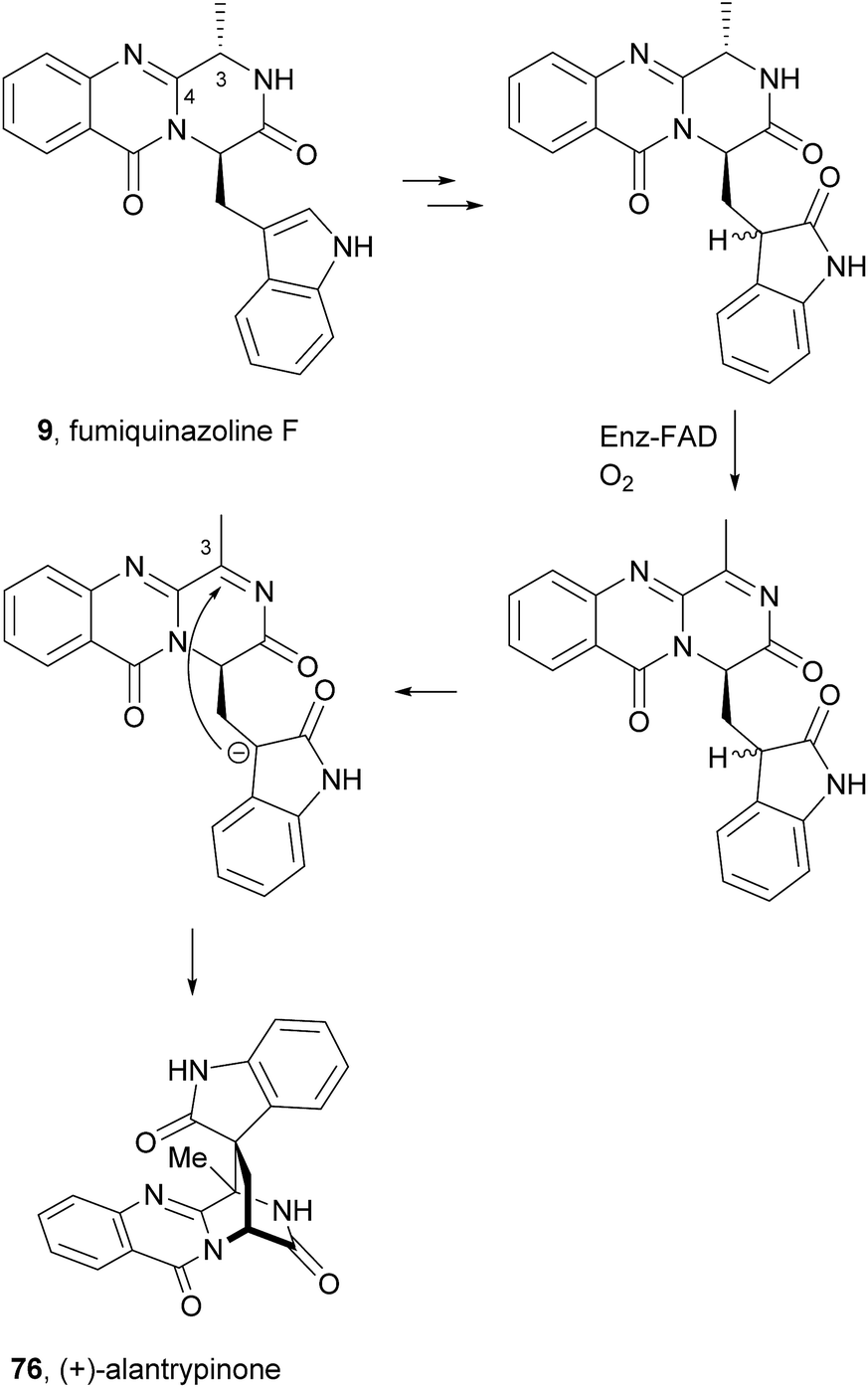
2.1.1 Fumiquinazolines. The structures of fumiquinazolines (1–23, Chart 2) can be presented as quinazolinones fused with a simple piperazine ring system or as more complex spiro moieties. Numata et al.17,33 have first reported the isolation of fumiquinazolines A–G (1, 3, 4, 6, 8–10), cytotoxic fungal metabolites from a strain of Aspergillus fumigatus which was isolated from the marine fish Pseudolabrus japonicas (See ESI Table 1† for detailed biological activities and projections of three-dimensional structures). The stereochemistry and configuration of these compounds were established on the basis of spectral and X-ray analysis and through some chemical transformations (e.g.: hydrogenation, epimerization).17 Since 1992, several other fumiquinazoline derivatives have been isolated from a variety of fungi of the genera Penicillium,34–37Aspergillus,38–46Neosartorya,47Acremonium,48 and Scopulariopsis,49 with fumiquinazoline F (9) being one of the most common metabolites; this secondary metabolite was also one of the most studied concerning its biological effects, exhibiting antimicrobial activity35 and tumour cells proliferation inhibitory activity.39
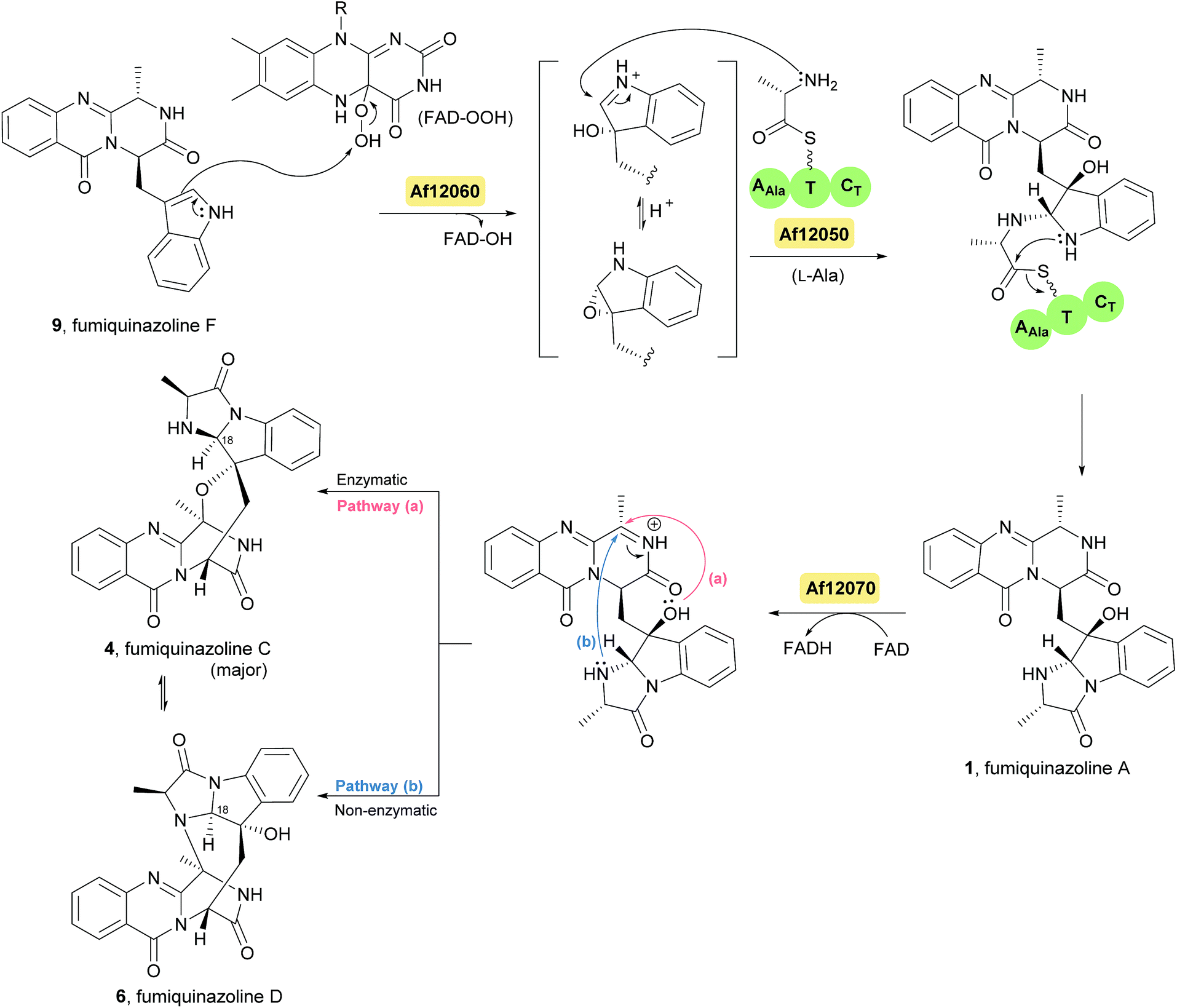
Fumiquinazoline F (9), containing a 6-6-6 tricyclic core was the first biochemically characterized tripeptide indole alkaloid, with its biosynthetic pathway being examined in Aspergillus fumigatus Af293,18,28 a known producer of quinazoline natural products. Biochemical characterization of a trimodular enzyme (NRPS Af12080) in Af293 genome elucidated that fumiquinazoline F (9) is assembled by incorporation of Ant, L-Trp, and L-Ala, following the general process of building tripeptidyl frameworks represented in Schemes 1 and 2. The presence of the epimerization (E) domain in module two of Af12080 is consistent with the epimerization of L-Trp to D-Trp during the assembly to generate the R-stereocenter at C-14 of fumiquinazoline F (9). Tricyclic fumiquinazoline F (9) can be converted into more architecturally elaborated frameworks by three tailoring enzymes encoded in the fumiquinazoline biosynthetic cluster (Scheme 3). Monomodular NRPS Af12050 and a flavoenzyme oxygenase Af12060 are necessary to convert fumiquinazoline F (9) to fumiquinazoline A (1).18 The FAD-enzyme Af12060 acts as an epoxygenase using O2 as cosubstrate and transfers an oxygen atom across the double bond in the pyrrole ring of the indole, generating a product that may be a hybrid between the epoxyindole and the hydroxyiminium. This intermediate is then processed by an alanine-activating NRPS module Af12050 that tethers the L-Ala in thioester linkage to its T-domain with further annulation of the oxidized indole scaffold, creating the 6-5-5 imidazolindolone ring system of fumiquinazoline A (1) (Scheme 3). The fourth enzyme, the monocovalent flavoprotein Af12070, introduces a third level of scaffold complexity. Conversion of fumiquinazoline A (1) to the more complex fumiquinazoline C (4) is catalysed by Af12070, that acts as a flavin adenine dinucleotide (FAD)-dependent amide oxidase, through the formation of an intermediary imine within the pyrazinone ring. Fumiquinazoline C (4) is then converted non-enzymatically to cyclic aminal fumiquinazoline D (6) (Scheme 3).
Scheme 3 Proposed biosynthetic route to fumiquinazolines A (1), C (4) and D (6) in Aspergillus fumigatus Af293. FAD – flavin adenine dinucleotide.
The structure of fumiquinazoline H (11) is closely related to fumiquinazoline C (4), but with the leucine side chain on C-20 instead of a methyl group as well as an opposite configuration at C-17, while fumiquinazoline I (12) is analogous to fumiquinazoline A (1) except that the indole nitrogen has been coupled to a leucine and oxidative cyclization has occurred with the opposite stereochemistry at both C-17 and C-18 on the indole ring (Chart 2).48
Concerning their biological activities two more fumiquinazoline members can be highlighted, fumiquinazoline J (13)39 that inhibited the proliferation of several tumour cells with IC50 values < 20 μM and 2′-epi-fumiquinazoline D (7) that showed interesting anti-insectan activity against the fall armyworm Spodoptera frugiperda.36 Structurally, it is also important to highlight fumiquinazolines K–P (14–19, Chart 2) that are characterized by the presence of a rare 1-aminocyclopropane-1-carboxylic amino acid residue.41 Due to the closeness of their discovery, fumiquinazolines K and L were initially doubly designated; later, these names were revised to fumiquinazolines Q (20) and R (21)50 and the most recent fumiquinazoline derivative was named accordingly, as fumiquinazoline S (22).40
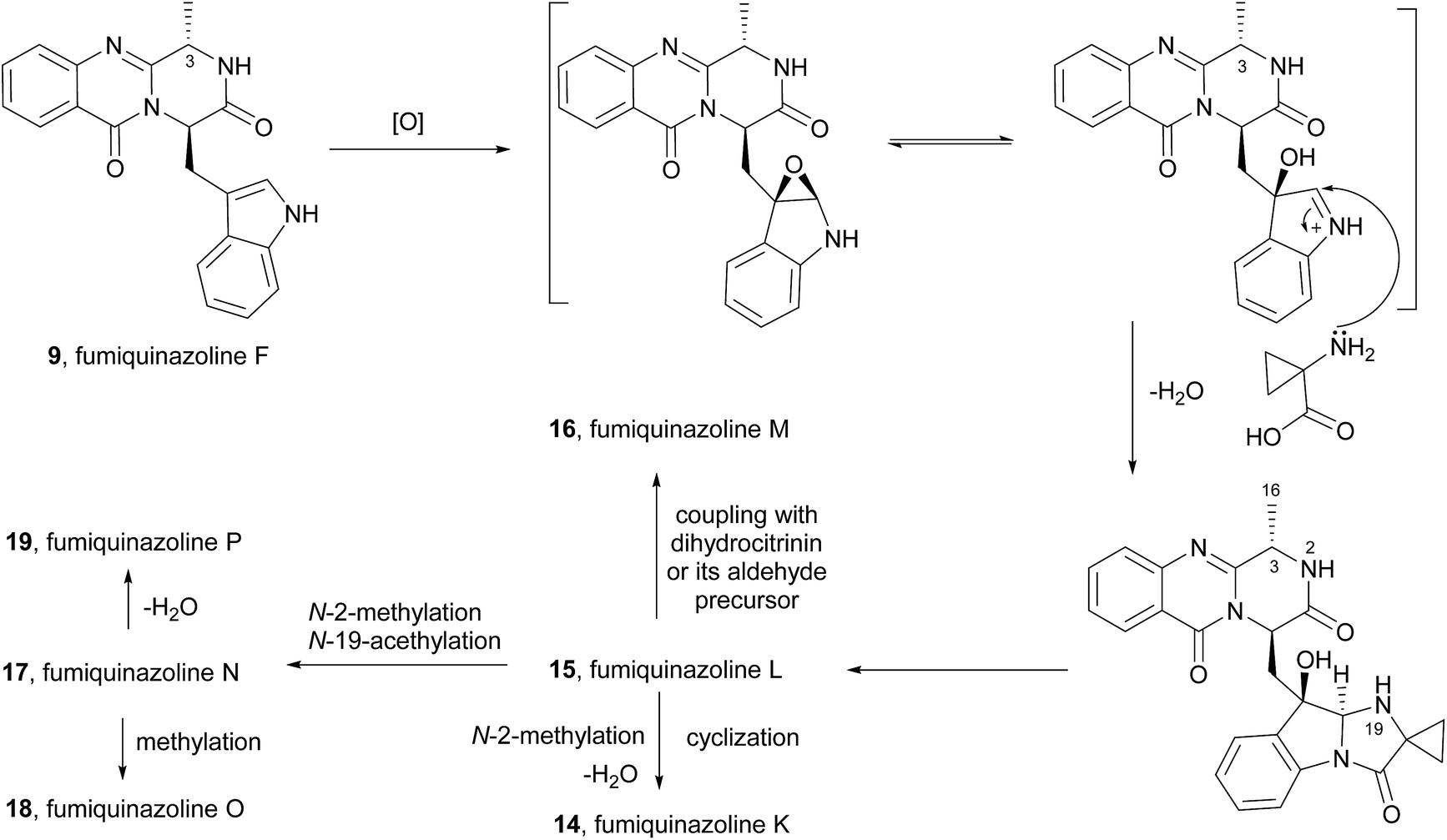
Fumiquinazoline F (9) was also proposed to be the precursor of fumiquinazolines K–P (14–19)41 (Scheme 4). The possible biosynthesis of these compounds would include oxidative coupling of 1-aminocyclopropane-1-carboxylic acid to form the imidazoindolone unit. Oxidation of the resulting intermediate at C-3 would give fumiquinazoline L (15). Methylation and acetylation at N-2 and N-19, respectively, would yield fumiquinazoline N (17) and its methylation would give fumiquinazoline O (18). Dehydration at C-3–C-16 of fumiquinazoline N (17) would result in the formation of fumiquinazoline P (19). The N-19 of the deacetylated intermediate would react with the electrophilic C-16 of the α,β-unsaturated imine moiety in fumiquinazoline P (19) to form fumiquinazoline K (14) by cyclization. Moreover, fumiquinazoline M (16) would be formed by coupling of the dehydrated derivative of fumiquinazoline L (15) with dihydrocitrinin or its aldehyde precursor.
Scheme 4 Proposed biogenetic pathway to fumiquinazolines K–P (14–19).
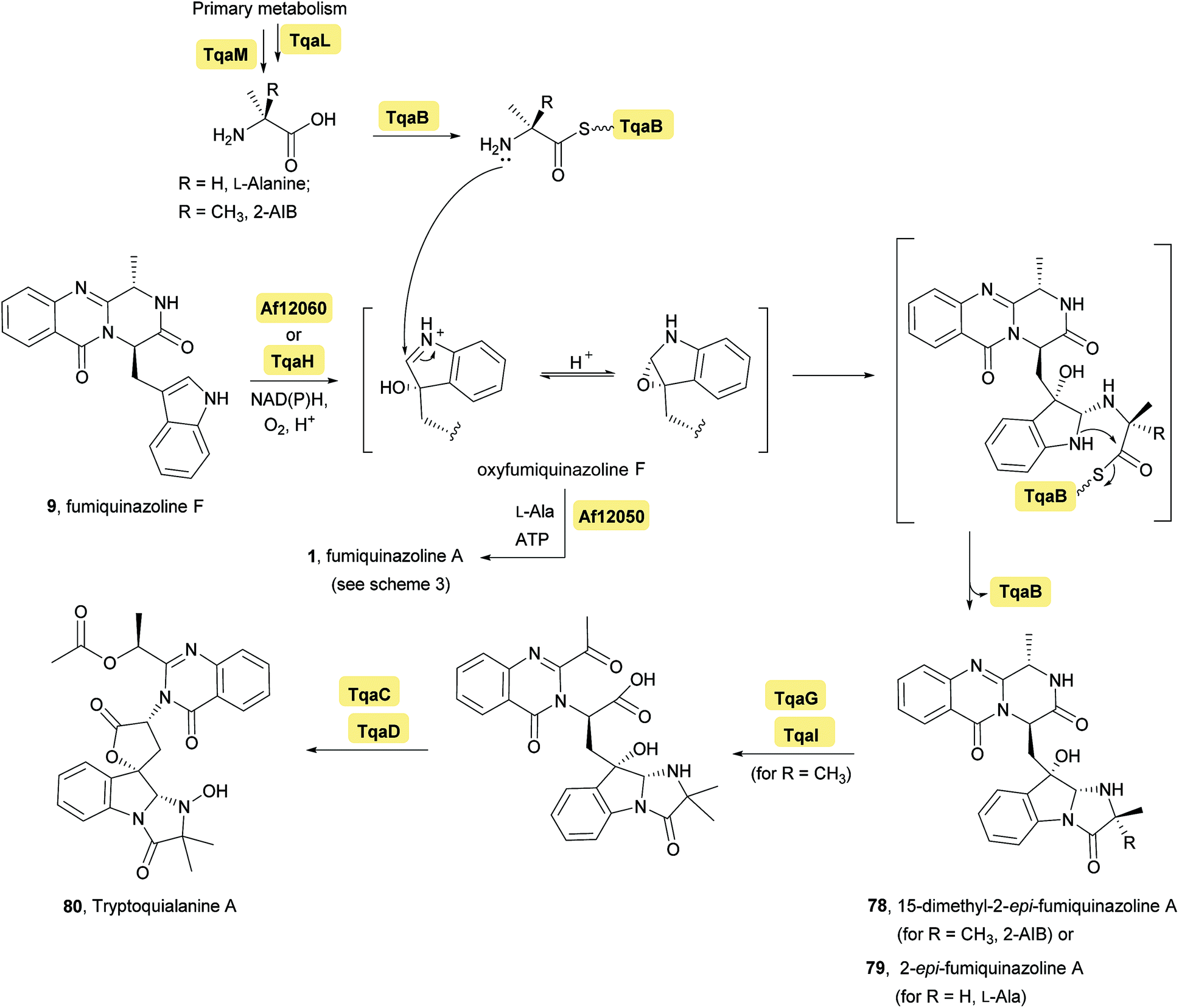
Investigation of the tremogenic mycotoxin tryptoquialanine (80) pathway in Penicilium aethiopicum,27 led to the identification of a gene cluster (tqa) that is involved in the biosynthesis of tryptoquialanine (80), through an intermediate common to the fumiquinazoline pathway. The tqa gene cluster contains 13 genes (named tqaA-tqaM), some of which are similar to NRPS. For example, enzyme TqaA is similar to the aforementioned NRPS Af12080 in Aspergillus fumigatus while TqaB, TqaH, and TqaG are similar to Af12050 (Ala-activating NRPS), Af12060 (epoxidase), and Af12070 (flavin-dependent oxidase), respectively. Because of this investigation, the biosynthesis of tryptoquialanine was hypothesized to proceed first via fumiquinazoline F (9) as intermediate and then, depending on the introduction of 2-aminoisobutyric acid or L-Ala, 15-dimethyl-2-epi-fumiquinazoline A (78) or 2-epi-fumiquinazoline A (79) may be, respectively, biosynthetic precursors of tryptoquialanine (Scheme 5). Concerning the, biosynthesis of 2-epi-fumiquinazoline A (79) by TqaB in Penicilium aethiopicum, the stereochemical control of this reaction was evaluated by Haynes et al.20 The authors mixed and matched the flavoprotein oxygenases Af12060 and TqaH with the A-T-C modular enzymes Af12050 and TqaB to show that the NRPS enzyme control the stereochemical outcome. The terminal 50 kDa condensation domains of Af12050 and TqaB are solely responsible for the stereochemical control as shown by making chimeric forms of these monomodular NRPS enzyme and also by expression, purification, and assay of the excised C-domains. The Af12050 and TqaB condensation domains are thus a paired set of diastereospecific annulation catalysts that act on the fumiquinazoline F (9) scaffold (Scheme 5).
Scheme 5 Enzymes involved in typtoquialanine biosynthesis through an intermediate common to fumiquinazoline pathway and stereochemical divergence of biosynthetic intermediates mediated by the action of a monomodular NRPS, Af12050, or TqaB. ATP – adenosine 5′-triphosphate, NAD(P) – nicotinamide adenine dinucleotide phosphate.
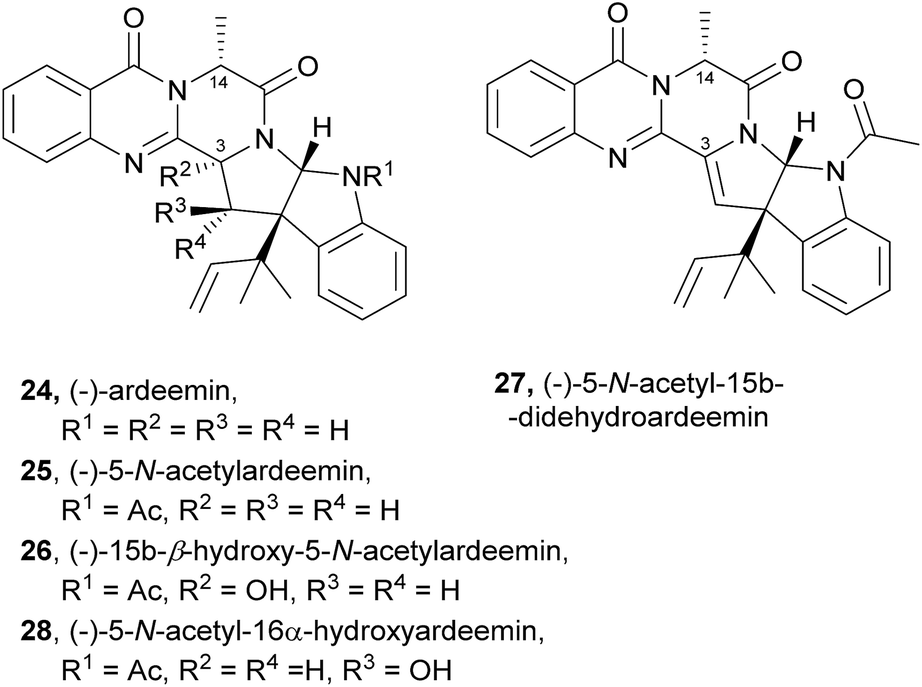
2.1.2 Ardeemins. The ardeemins (24–28, Chart 3) belong to another interesting class of natural products structurally related to fumiquinazolines, containing a hexahydropyrrolo[2,3-b]indole scaffold substituted at the benzylic ring junction with a reverse-prenyl (1,1-dimethylallyl) group. Unlike fumiquinazolines, ardeemins have the indole moiety at C-3, while the methyl group is linked to C-14. Ardeemin (24), 5-N-acetylardeemin (25), and (−)-15b-β-hydroxy-5-N-acetylardeemin (26) were isolated from the fungus Aspergillus fischeri AB 1826M-35 in a screening for compounds which reverse multi-drug resistance (MDR) to antitumour agents.51,52 5-N-acetylardeemin (25) was found to be the most effective of these three compounds in reversing MDR on cancer cells.51 Other ardeemin derivatives, (−)-5-N-acetyl-15b-didehydroardeemin (27)53,54 and (−)-5-N-acetyl-16α-hydroxyardeemin (28)54 were later isolated, along with ardeemins 25 and 26, exhibiting different profiles in reversing, the multidrug-resistant (MDR) phenotype in three cancer cell lines, leukaemia doxorubicin resistant cell K562/DOX, human lung adenocarcinoma cisplatin-resistant cell A549/DDP, and ovarian cancer cisplatin-resistant cell SK-OV-S/DDP. Structure–activity relationship (SAR) studies revealed that the hydroxyl group plays an important role in the MDR reversing effect.54 Bioinformatic analysis on ardeemin (24) showed that three genes are involved in its biosynthesis: a trimodular NRPS, a prenyl transferase, and an acetyltransferase.32,55
Chart 3 Natural occurring ardeemins 24–28. Ac – acetyl.
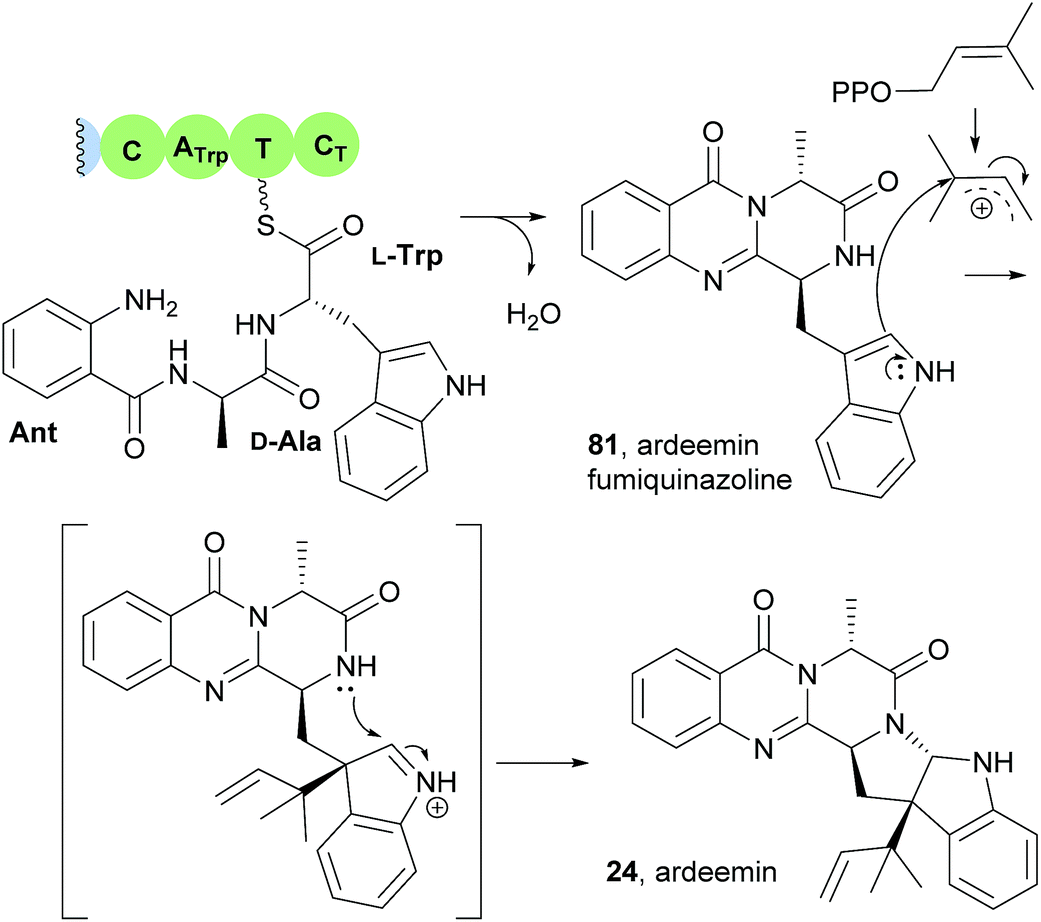
This study also revealed that the product of the NRPS (ArdA) was ardeemin fumiquinazoline (81), a 6-6-6-tricyclic pyrazinoquinazoline with a distinct substitution pattern that probably arises from the Ant-D-Ala-L-Trp tripeptidyl-S-NRPS intermediate instead of Ant-D-Trp-L-Ala-S-NRPS (Scheme 6).21 The mechanism of the double cyclization to form ardeemin fumiquinazoline (81) should be similar to that previously verified for TqaA or Af12080 in fumiquinazoline F (9) biosynthetic pathway (Scheme 2). Further prenylation and cyclization with concomitant generation of the five-member ring leads to ardeemin (24).
Scheme 6 Formation of ardeemin fumiquinazoline (78) and ardeemin (24) via prenyl transfer.
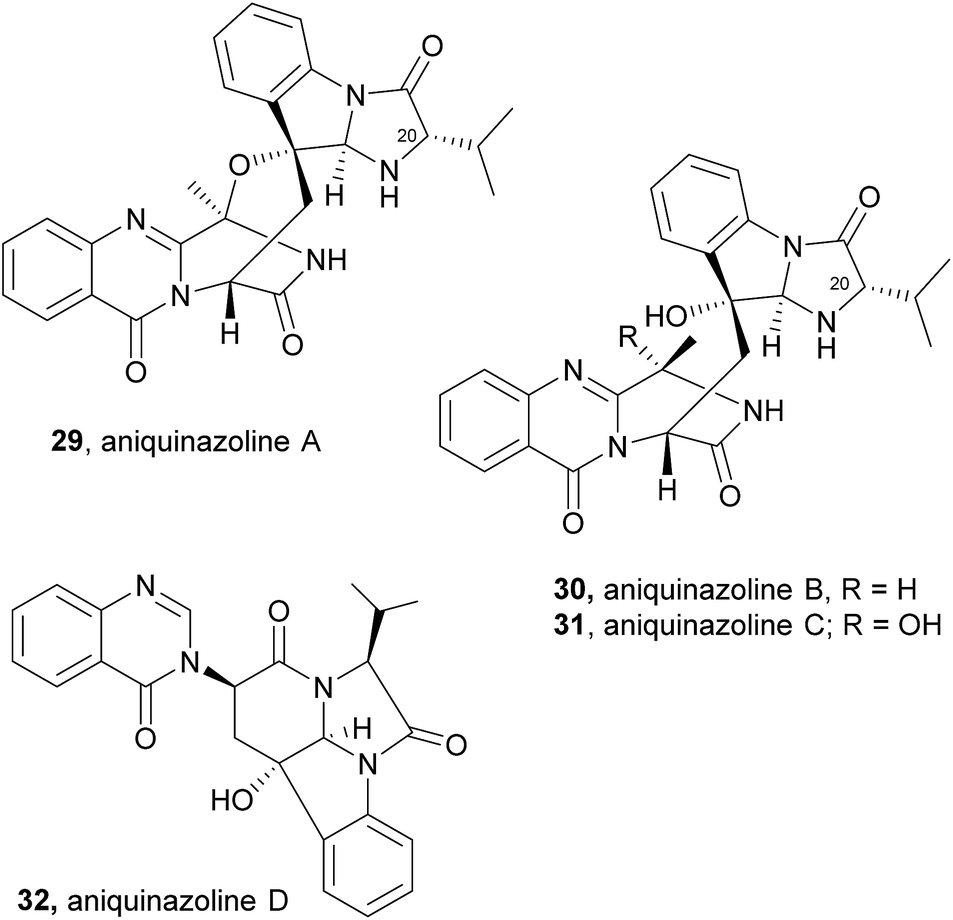
2.1.3 Aniquinazolines. Aniquinazoline A (29, Chart 4) was recently isolated, together with three other derivatives, aniquinazolines B–D (30–32), from the marine-derived endophytic fungus Aspergillus nidulans MA-143.56 The structure of 29 closely resembles that of fumiquinazoline C (4), except for the presence of the isopropyl group on C-20 in the former and the methyl group on the corresponding carbon in the latter. Compounds 30–32 displayed potent toxicity against brine shrimp (Artemia salina) with LD50 values of 1.27, 2.11, 4.95, and 3.42 μM, respectively, which were stronger than that of the positive control colchicine (with LD50 value of 88.4 μM).
Aniquinazolines differ from fumiquinazoline C (4) only in the presence of the isopropyl group instead of a methyl group on C-20, that arises from a Af12050/TqaB variant which activates Val instead of Ala.
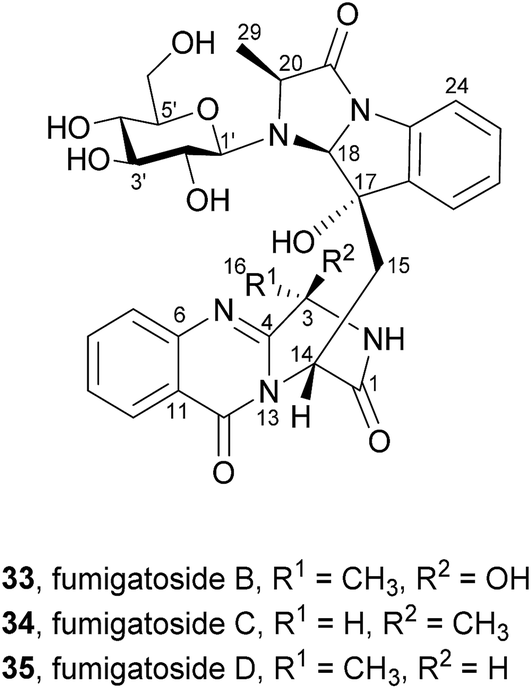
2.1.4 Fumigatosides. Liu et al.57 described the isolation of three new pyrazinoquinazoline indole glucosides, fumigatosides B–D (33–35, Chart 5), from the fungus Aspergillus fumigatus derived from the jellyfish Nemopilema nomurai.
Their structures were elucidated by spectroscopic methods and electronic circular dichroism (ECD) combined with time-dependent density functional theory (TDDFT) calculations. A fourth metabolite was initially designated as fumigatoside A, but due to challenges in its stability the structure could not be unambiguously established.58 Neither antibacterial nor cytotoxic effects were observed when these secondary metabolites were tested against a panel of six different bacterial strains and ten cell lines of solid human tumours.57
Along with their isolation, Liu et al.57 also hypothesized that fumigatosides B (33) and C (34) should arise from an Ant-D-Trp-L-Ala tripeptidyl-S-NRPS with fumiquinazoline F (9) as intermediary (Scheme 7a) and fumigatosides D (35) from an Ant-D-Trp-D-Ala tripeptidyl-S-NRPS with fumiquinazoline G (10) as intermediary (Scheme 7b). However, although fungi produced fumigatosides B (33) and C (34) upon introduction of L-Ala, fumigatoside D (35) was not detected.
Scheme 7 Proposed biogenetic pathways of fumigatosides B–D (33–35). ATP – adenosine 5′-triphosphate, NAD(P) – nicotinamide adenine dinucleotide phosphate, PPi pyrophosphate.
2.2 Glycine-derived alkaloids
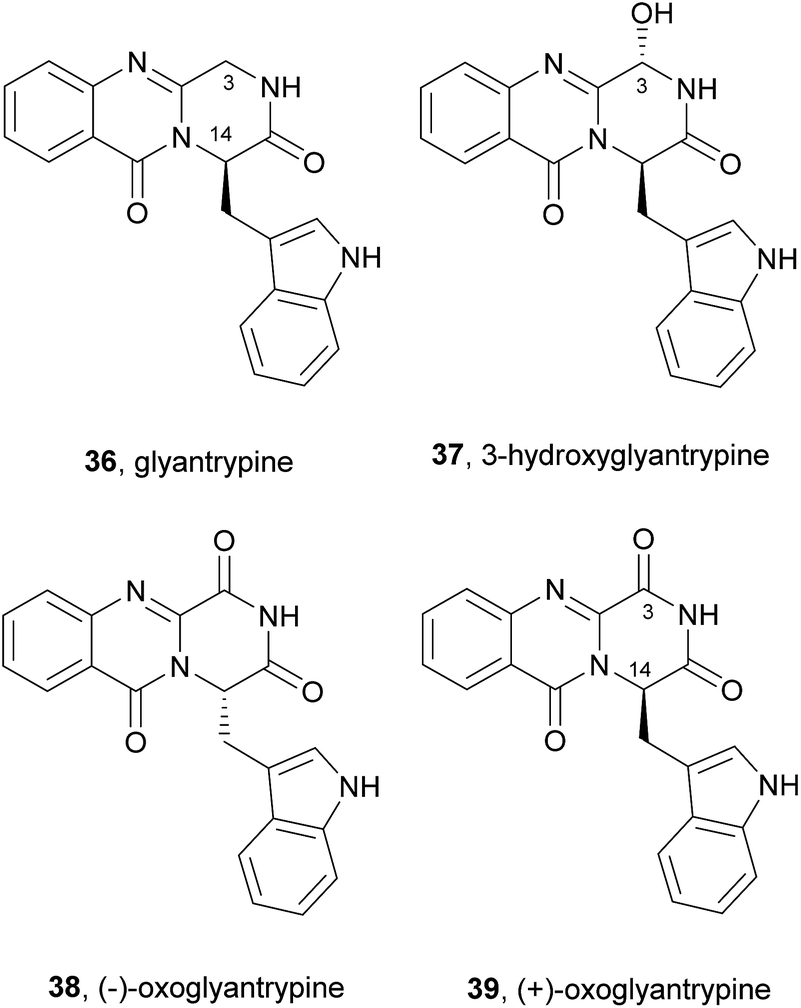
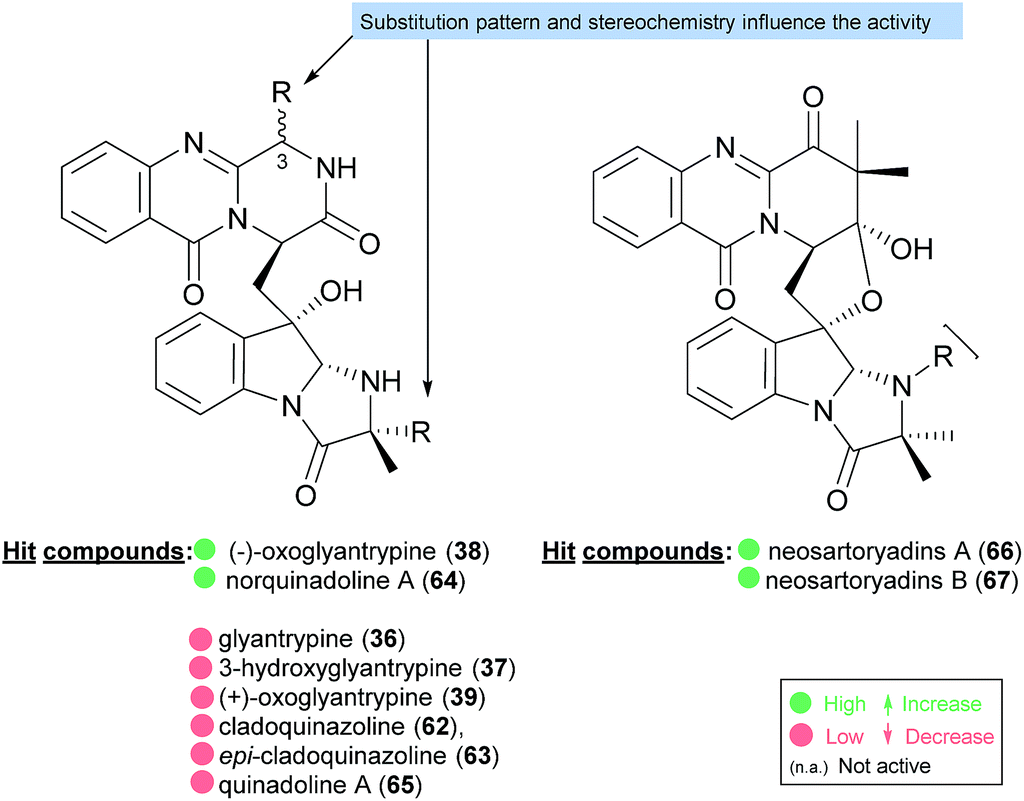
2.2.1 Glyantrypines. The simplest molecular structure belonging to the fumiquinazoline secondary metabolites is glyantrypine (36, Chart 6). This compound was isolated for the first time in 1992 from Aspergillus clavatus,59 and later from other fungal strains.60–62 Other derivatives were also isolated by Peng et al.63 including 3-hydroxyglyantrypine (37), (−)-oxoglyantrypine (38), and (+)-oxoglyantrypine (39). The structures of these derivatives were elucidated primarily by spectroscopic and physical data. The absolute configurations of their stereogenic carbons were established based on comparison of calculated and experimental ECD spectra, nuclear Overhauser enhancement spectroscopy (NOESY) correlations, as well as single-crystal X-ray diffraction analysis. Examination of the antiviral activity of this series of compounds revealed that the hydroxyl group on C-3 of the fumiquinazoline core does not affect the anti-H1N1 activity since 3-hydroxyglyantrypine (37) exhibited weak anti-H1N1 activity with similar IC50 values (100–150 μM) to that of glyantrypine (36). On the other hand, the presence of the carbonyl group on C-3 as well as the stereochemistry of C-14 was found to play a crucial role in the anti-H1N1 activity since (+)-oxoglyantrypine (39), whose C-14 has the same absolute configuration as that of glyantrypine (36) showed a weak anti-H1N1 activity with similar IC50 value (100–150 μM) while 38, with C-14S, exhibited higher anti-H1N1 activity with IC50 values similar to that of the antiviral drug ribavirin.
Chart 6 Naturally occurring glyantrypine (36) and glyantrypine derivatives 37–39.
The biosynthesis of glyantrypine (36) was investigated soon after it was isolated.64 The incorporation of radiolabel from [14C-carboxyl]-anthranilic acid into glyantrypine (36) was able to confirm its role as a direct precursor. NRPS is also involved in glyantrypine (36) biosynthesis in Aspergillus clavatus65 according to the general Scheme 2 for the assembly of tripeptide indole alkaloids via an Ant-Trp-Gly-S-enzyme intermediate that upon double cyclization yields glyantrypine (36) without further tailoring.21 3-Hydroxyglyantrypine (37), (−)-oxoglyantrypine (38), and (+)-oxoglyantrypine (39) are also biosynthesized from the same amino acids as glyantrypine (36) with further oxidation.63
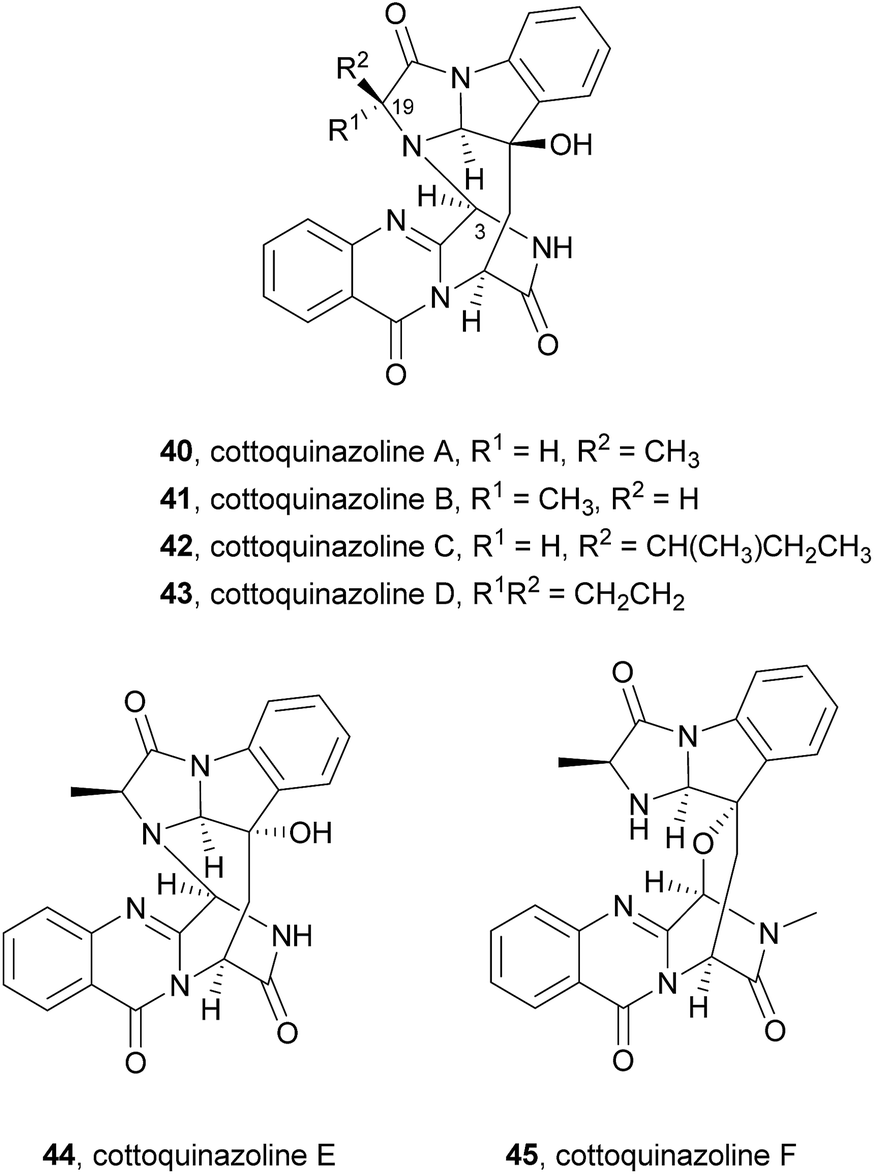
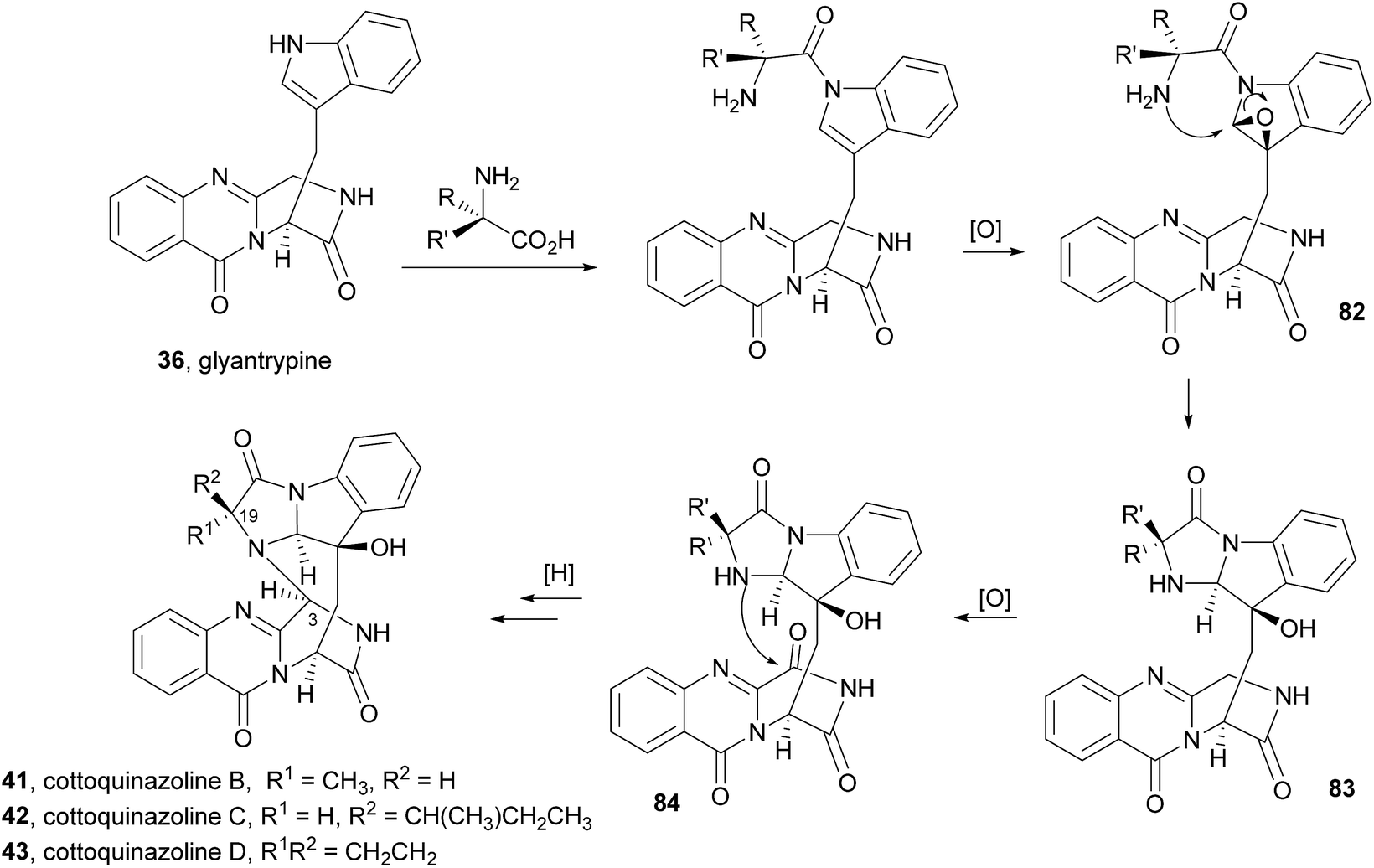
2.2.2 Cottoquinazolines. Cottoquinazoline A (40, Chart 7), a nor analogue of fumiquinazoline D (6), was first isolated in 2009 from an Australian marine-derived isolate of Aspergillus versicolor (MST-MF495).61 The absolute configuration of the stereogenic carbons of 40 was determined by chemical degradation and application of the C-3 Marfey's method for amino acid analysis. This secondary metabolite (40) exhibited potent vasculogenic activity, suggesting that it can be a promising lead compound for cardiovascular diseases. Other members of this group, i.e. cottoquinazolines B–D (41–43) were later isolated from the fungal strain Aspergillus versicolor LCJ-5-4 (ref. 66) and were examined for their in vitro cytotoxicity against tumour cells and for their antimicrobial activity against Escherichia coli, Staphylococcus aureus, Enterobacter aerogenes, Bacillus subtilis, and Candida albicans. Cottoquinazoline D (43), a rare alkaloid with a cyclopropane ring at C-19 (R1R2 = CH2CH2), exhibited an interesting antifungal activity against Candida albicans (MIC = 22.6 μM), while the remaining compounds did not show any relevant antimicrobial activity. Two new norfumiquinazolines, cottoquinazolines E (44) and F (45), were isolated from the fungus Neosartorya fischeri NRRL 181,67 however, their bioactivities were not investigated.
Very recently, the isolation of these compounds from the gorgonian-derived fungus Aspergillus versicolor LZD-14-1 led to the revision of the structures of cottoquinazolines B–D (41–43) by extensive analyses of spectroscopic data combined with ECD data and X-ray analysis which allowed the authors to suggest that the natural compounds are, in fact, enantiomers of the previously described alkaloids.68
Cottoquinazolines B–D (41–43) are also biosynthesized from the same amino acids as glyantrypine (36) involving further condensation with a fourth amino acid and subsequent oxidation that forms the corresponding epoxidation product 82. Intramolecular nucleophilic cyclization of 82 produces intermediate 83 that is oxidized to form 84 (Scheme 8).66 Cottoquinazolines E (44) and F (45) can be formed by a similar process as fumiquinazoline D (6) is generated from fumiquinazoline A (1) (Scheme 3).67
Scheme 8 Proposed biogenetic pathway of cottoquinazolines B–D (41–43).
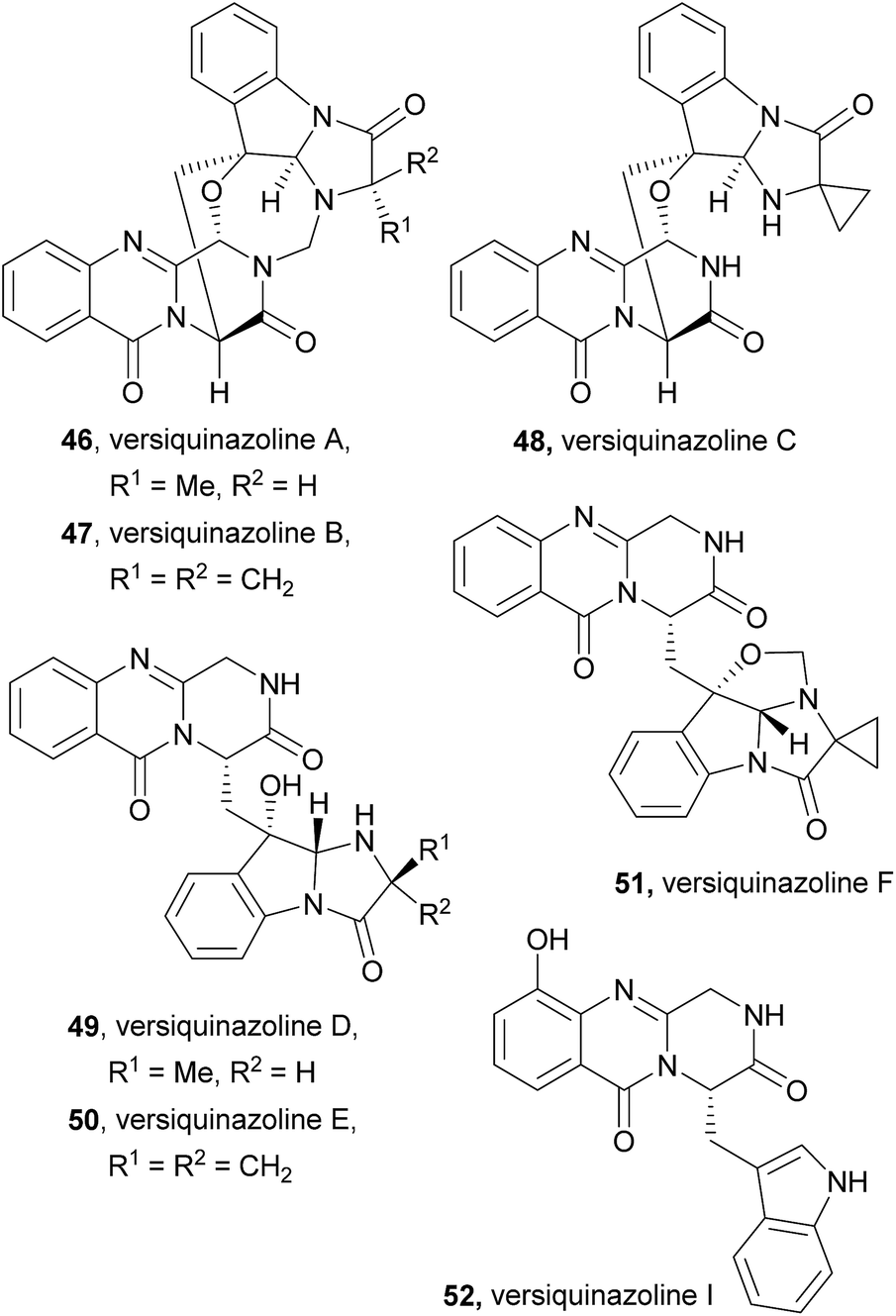
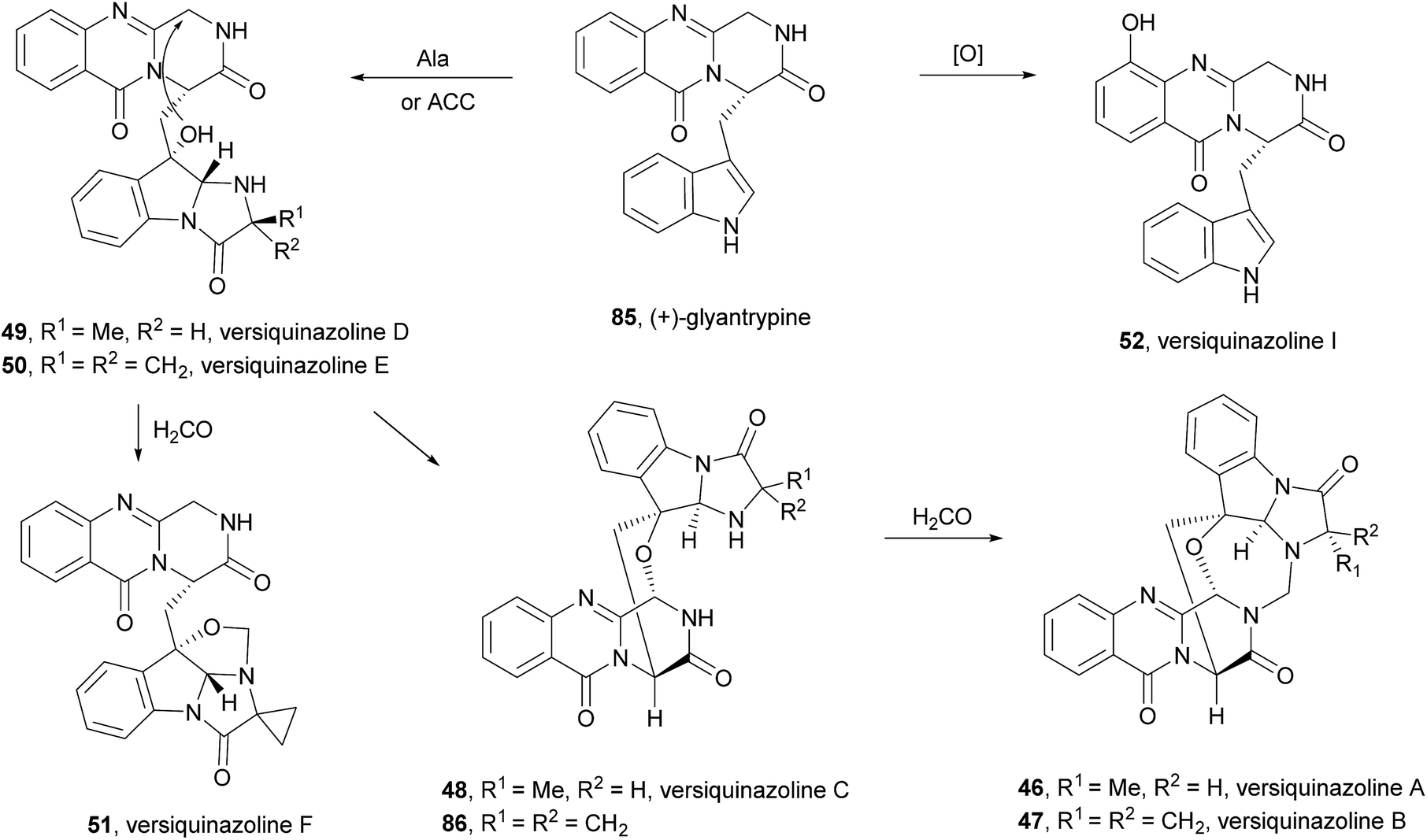
2.2.3 Versiquinazolines. Versiquinazolines (46–52, Chart 8) are another class of fumiquinazoline-type alkaloids which were recently isolated, along with cottoquinazolines B–D (41–43), from the gorgonian-derived fungus Aspergillus versicolor LZD-14-1.68 Among the eleven isolated versiquinazolines, seven of them, i.e. versiquinazolines A–F (46–51) and versiquinazoline I (52), presented the characteristic pyrazino[2,1-b] quinazoline-3,6-dione core linked to the indole moiety, as observed in fumiquinazolines. Other versiquinazolines displayed different structural motifs and will not be considered in this review. Noteworthy, versiquinazolines A (46) and B (47) exhibited significant inhibitory effects against thioredoxin reductase.68 (+)-Glyantrypine (85) was proposed as the biosynthetic precursor of versiquinazolines A–F (46–51) and versiquinazoline I (52) (Scheme 9),68 the latter being formed by a 7-hydroxylation of (+)-glyantrypine (85) while versiquinazolines D (49) and E (50) were proposed to be formed through additional intermolecular condensation with alanine or 1-aminocyclopropanecarboxylic acid (ACC) residue similar to the mechanism shown in Scheme 5. Versiquinazoline C (48) and the related compound 86 can be obtained, through an oxidation-induced formation of an ether bond, from versiquinazoline D (49) and E (50), respectively. Reaction of versiquinazoline C (48), 86, and versiquinazoline E (50) with formaldehyde could generate versiquinazoline A (46), B (47), and F (51), respectively.
Chart 8 Naturally occurring versiquinazolines A–F (46–51) and versiquinazoline I (52).
Scheme 9 Proposed biogenetic pathways of versiquinazolines A–F (46–51) and versiquinazoline I (52). ACC – 1-aminocyclopropanecarboxylic acid.
2.3 Valine-derived alkaloids
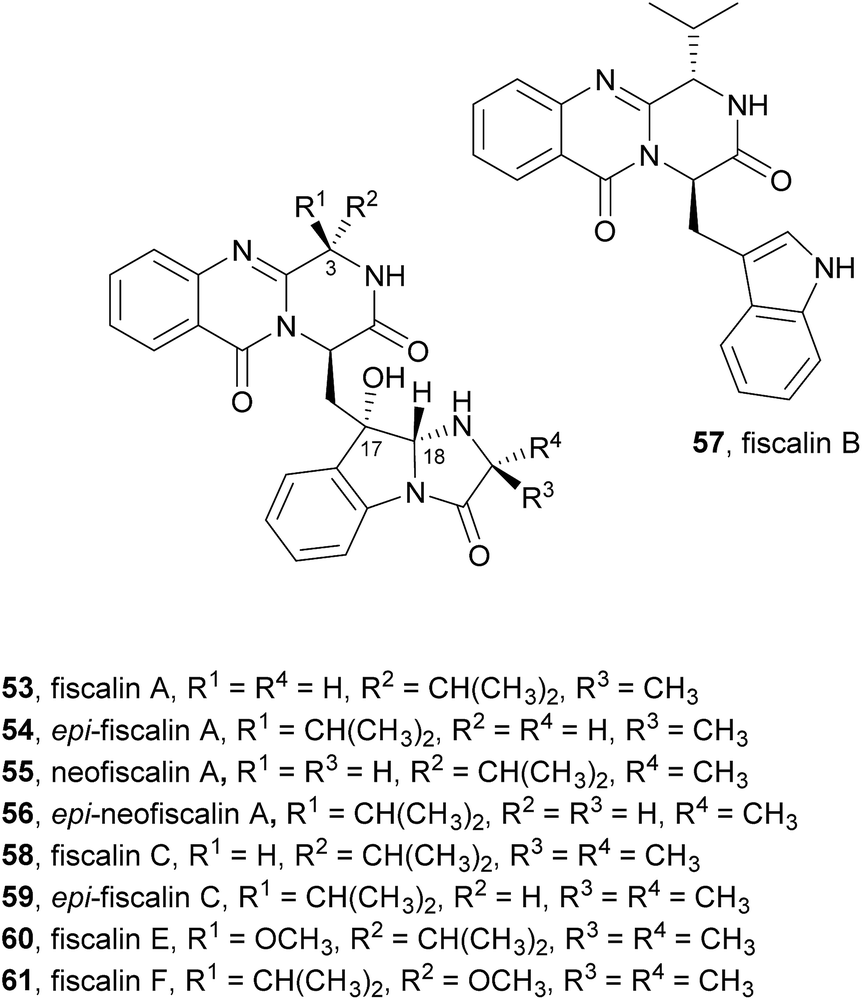
2.3.1 Fiscalins. Another group of alkaloids whose structures are closely related to fumiquinazolines are fiscalins (Chart 9). The only difference between these two groups is the substituent on C-3. While fumiquinazolines have a methyl group at C-3, the fiscalins have an isopropyl group at the corresponding carbon that arises from the incorporation of valine instead of alanine. Moreover, the stereochemistry of C-18 is also different, being 18R in fiscalin A (53) and 18S in fumiquinazoline A (1).69 Fiscalin A (53), B (57), and C (58), first reported in 1993 from Neosartorya fischeri, were found to display an interesting inhibitory activity against substance P.70 Detailed spectroscopic and amino acid analyses revealed that the structures of these compounds consist of an indolyl moiety linked to the anthranilic acid-derived tricyclic system.
Chart 9 Naturally occurring fiscalins 53–61.
Biosynthetically, fiscalin B (57) was found to arise from the Ant-D-Trp-Val-tripeptidyl-NRPS (NFIA_057960) assembly line by the molecular logic presented in Scheme 2.21 Fiscalin A (53) differs from fiscalin C (59) in the involvement of D-Ala instead of 2-aminoisobutyric acid (AIB) in the annulation step.
In our research group a strain of Neosartorya siamensis (KUFC 6349) was isolated from a forest soil at Samaesarn Island, Thailand and among the secondary metabolites isolated from this fungus were fiscalin A (53), epi-fiscalin A (54), neofiscalin A (55), epi-neofiscalin A (56), fiscalin C (58), and epi-fiscalin C (59), in addition to other eight indole alkaloids.71 Further investigation of the in vitro anticancer activity of crude ethyl acetate extracts of the culture of four marine-derived fungi, including Neosartorya siamensis, led one of us to demonstrate for the first time that extracts of Neosartorya paulistensis and Neosartorya siamensis have selective antiproliferative and cell death activities against a panel of three human cancer cell lines: malignant melanoma (A375), hepatocellular carcinoma (HepG2) and colon carcinoma (HCT116).72 These results led to the investigation of the secondary metabolites from the fungi of the marine environment which allowed one of us to isolate fiscalin A (53), epi-fiscalin A (54), neofiscalin A (55), epi-neofiscalin A (56) and epi-fiscalin C (59),73 together with fiscalin C (58) and two other alkaloids previously obtained from Neosartorya siamensis (KUFC 6349), and evaluate them for their antibacterial activity and synergism with currently used antibiotics against multidrug-resistant bacteria. Neofiscalin A (55) showed potent antimicrobial activity (MIC = 8 μg mL−1) against methicillin-resistant Staphylococcus aureus (MRSA) and vancomycin-resistant Enterococcus faecalis (VRE), therefore presenting an excellent candidate for further chemical modifications. This secondary metabolite also exhibited a great potential as antibiofilm agent with no cytotoxicity against a human brain capillary endothelial cell line. Fiscalin C (58) was shown to be a good candidate as an adjuvant in antimicrobial combined therapeutics presenting synergistic activity when combined with oxacillin against MRSA, although by itself it showed no antibacterial effect. The anticancer activity of fiscalin A (53), epi-fiscalin A (54), epi-neofiscalin A (56), and epi-fiscalin C (59) was assessed against HepG2, HCT16, and A375 cells.11 In general, all the tested compounds showed a broad antiproliferative activity at least in one cell line. Epi-fiscalin C (59) was effective in all three cell lines and exhibited the lowest IC50 values in each cell line (from 24 to 86 μM), therefore representing the most effective compound.
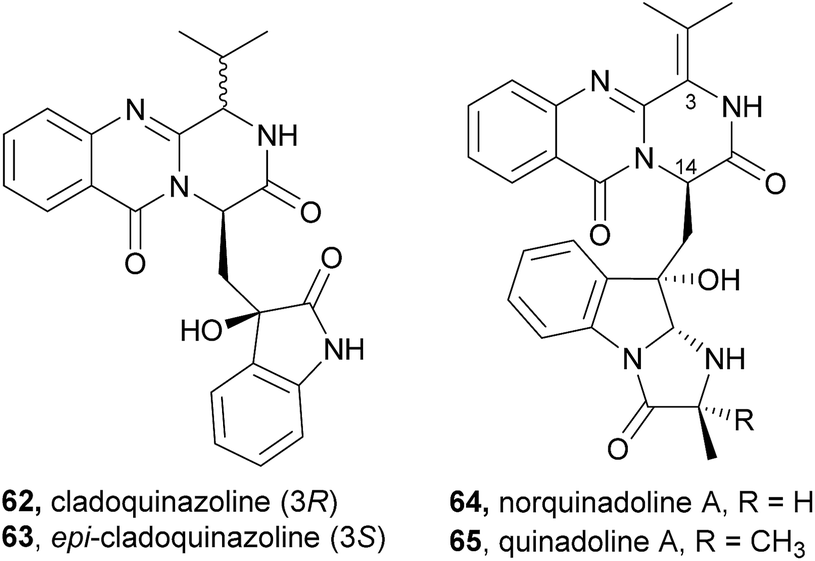
2.3.2 Cladoquinazolines and quinadolines. Cladoquinazoline (62), epi-cladoquinazoline (63), norquinadoline A (64), and quinadoline A (65), were isolated together with six known quinazoline-containing indole alkaloids by Peng et al.63 from the culture of the mangrove-derived fungus Cladosporium sp. PJX-41 (Chart 10). Norquinadoline A (64) whose absolute configuration of C-14 is the same as that of glyantrypine (36), but with the isopropylidene substituent on C-3, exhibited anti-H1N1 activity (IC50 = 82 μM; ribavirin as a positive control, IC50 = 87 μM) while cladoquinazoline (62) and epi-cladoquinazoline (63), with the isopropyl group on C-3, were weakly active (IC50 = 100–150 μM); ribavirin as a positive control, (IC50 = 87 μM), similar to glyantrypine (36).63
Chart 10 Naturally occurring cladoquinazoline (62), epi-cladoquinazoline (63), norquinadoline A (64), and quinadoline A (65).
Like fiscalins, cladoquinazolines and quinadolines should also arise from an Ant-D-Trp-Val-tripeptidyl-NRPS, being D-Val incorporated for cladoquinazoline (62), and L-Val for epi-cladoquinazoline (63).
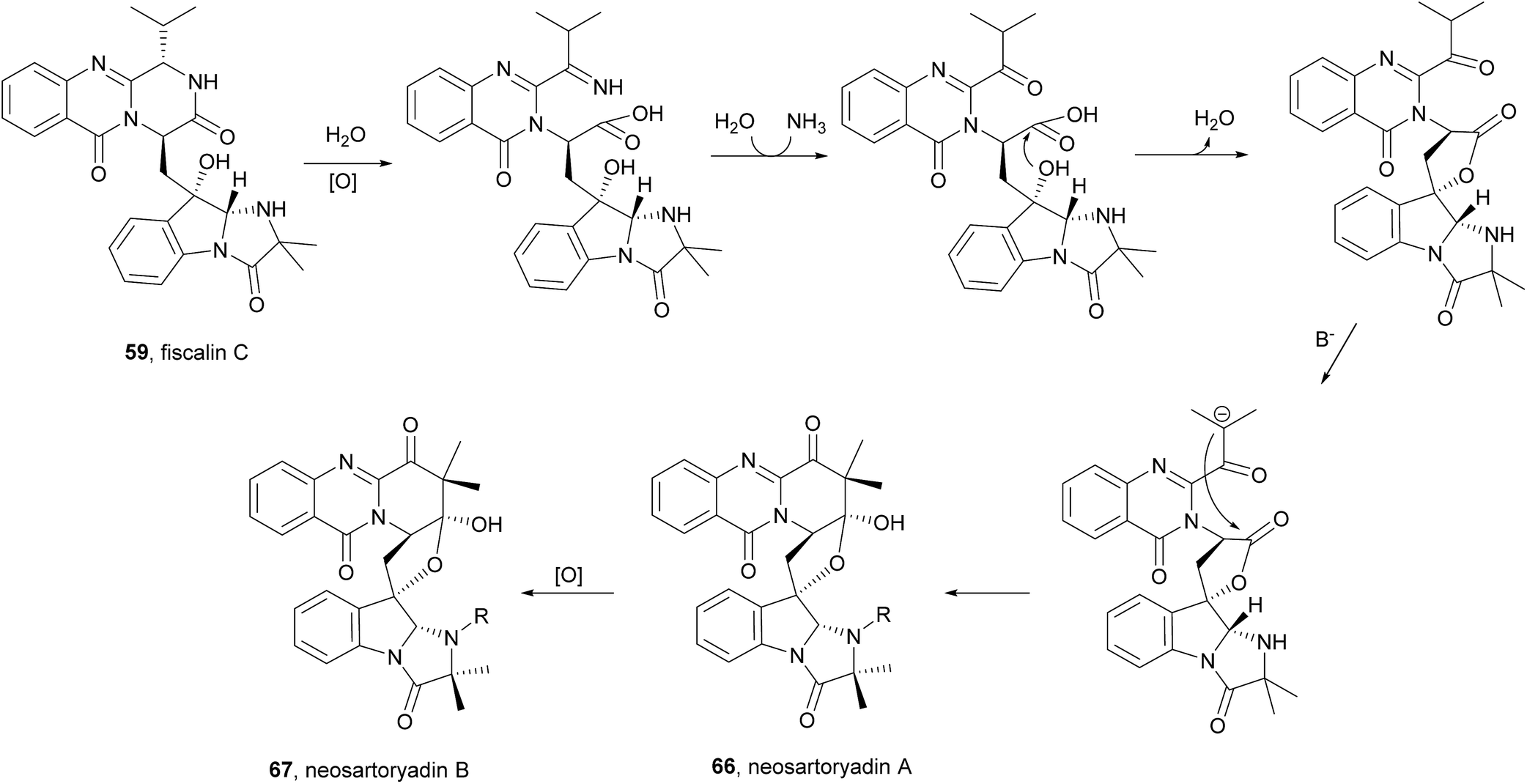
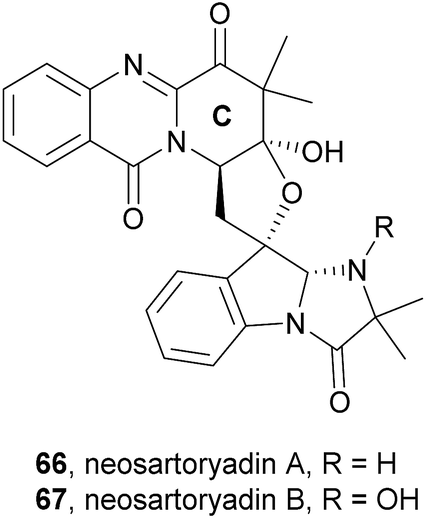
2.3.3 Neosartoryadins. Neosartoryadins contain a pyridinoquinazolinone ring system, i.e. the quinazolinone ring system is fused with a pyridine ring instead of a pyrazino ring as in fumiquinazolines, as well as an additional tetrahydrofuran ring. Since members of this subclass are related to and biosynthetically derived from fiscalin C (58), they were included in this review. These compounds were recently isolated from the endophytic fungus Neosartorya udagawae HDN13-313, isolated from the root of Aricennia marina collected at the mangrove conservation area of Hainan, China.74
Neosartoryadins A (66) and B (67) showed no cytotoxicity against the HL-60 cancer cell line (IC50 > 50 μM) but displayed anti-influenza virus A (H1N1) activity with IC50 values of 66 and 58 μM, respectively.
Along with their isolation, structure elucidation and bioactivity investigations, Yu et al.74 also proposed a plausible biogenetic pathway to neosartoryadins A (66) and B (67), suggesting that these metabolites are also biosynthesized from L-tryptophan, anthranilic acid, L-valine, and AIB (Scheme 10). Neosartoryadins A (66) and B (67) are generated by further modification of fiscalin C (58) by oxidation, hydrolysis, nucleophilic attack by water, dehydration, deprotonation, and subsequent aldol reaction to form the unprecedented C ring (Chart 11).
Scheme 10 Proposed biogenetic pathways of neosartoryadins A (66) and B (67).
Chart 11 Naturally occurring neosartoryadins A (66) and B (67).
2.4 Spiroquinazolines
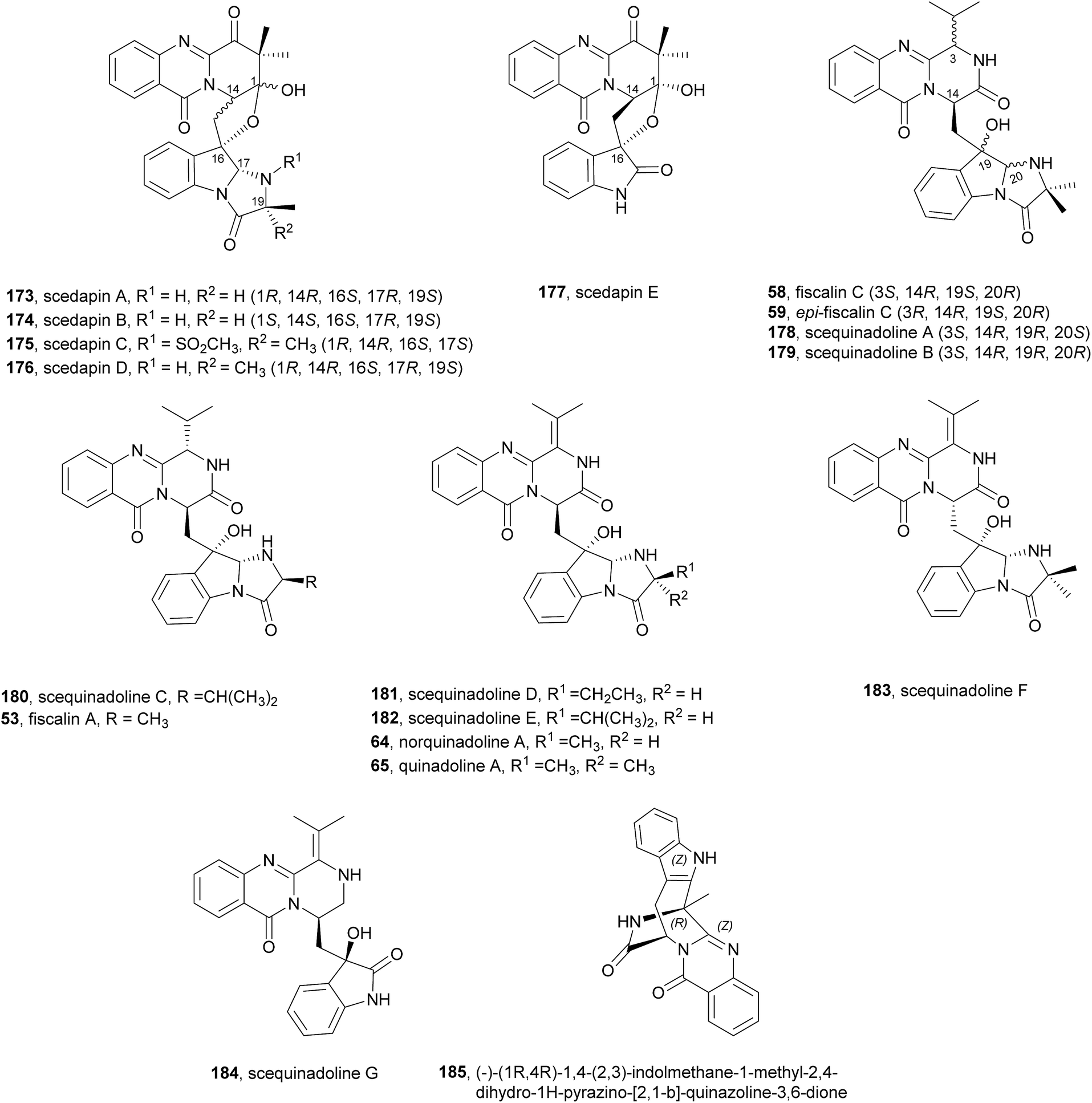
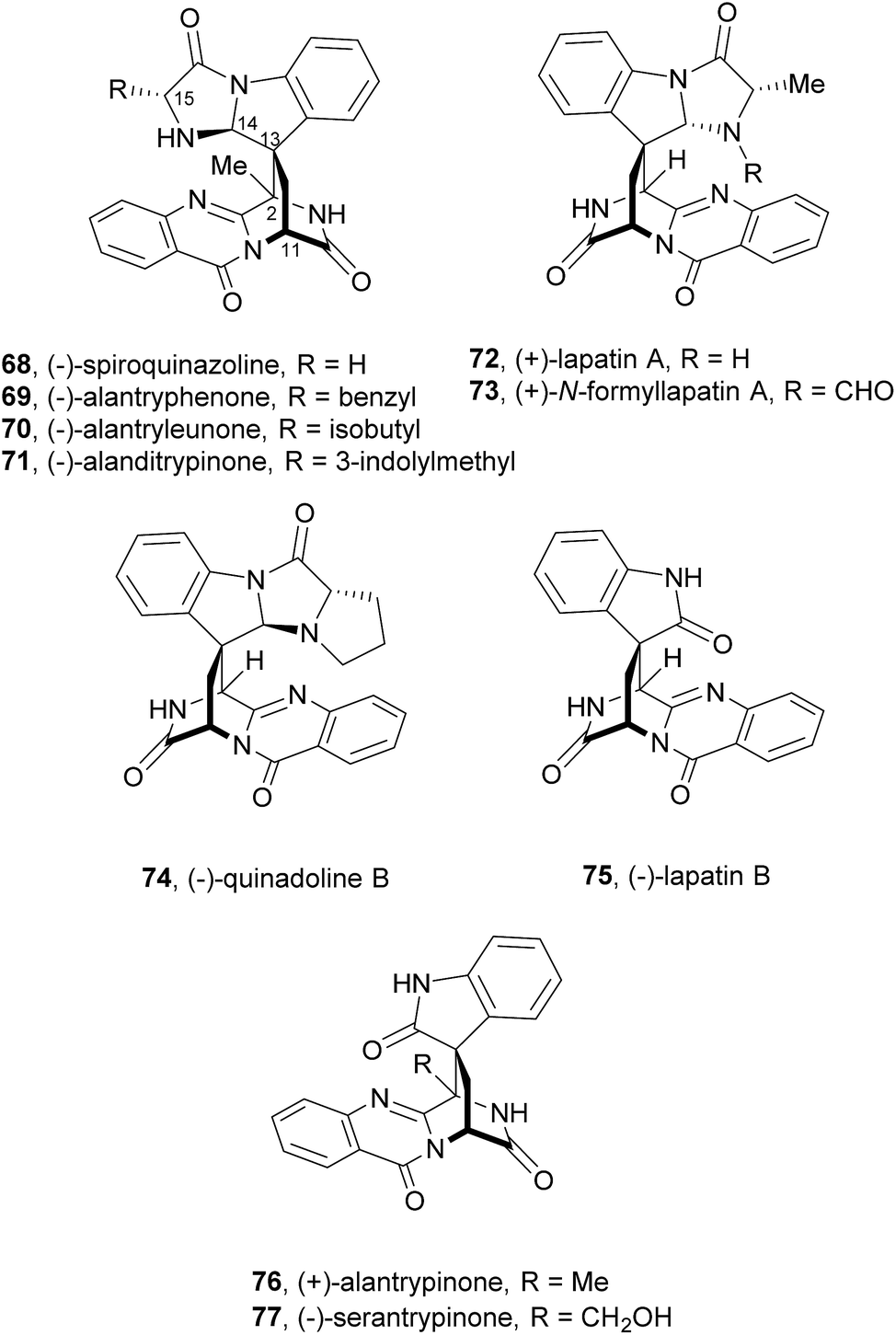
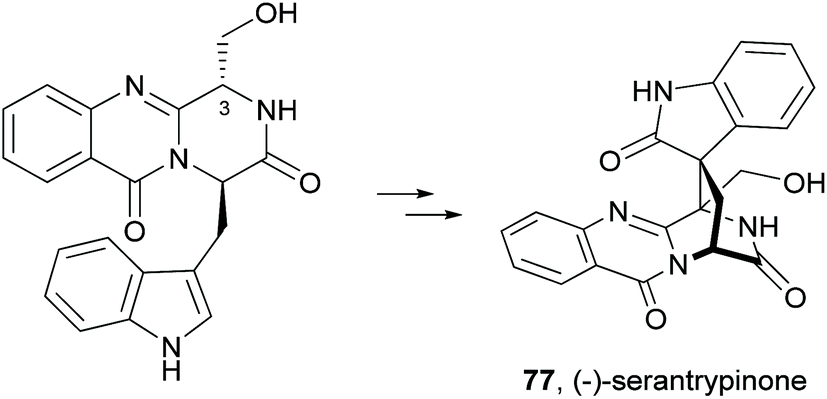
Since the isolation of (−)-spiroquinazoline (68, Chart 12), a substance P inhibitor, from the fungus Aspergillus flavipes in 1994,75 nine other spiroquinazoline alkaloids 69–77 have been isolated from a variety of fungi of the genera Penicillium and Aspergillus.34,60,76–79 All members of the spiroquinazoline alkaloid family have similar structures, containing the pyrimido[2,1-b]quinazoline bridged C-2 and C-11 linked by a methylene group. Structurally, they can be divided in indoline-containing substructures with different C-2 and C-15 substituents [(−)-spiroquinazoline (68),75 (−)-alantryphenone (69),76 (−)-alantryleunone (70),76 (−)-alanditrypinone (71),76 (+)-lapatin A (72),77 (+)-N-formyllapatin A (73),60 and (−)-quinadoline B (74)78] or 3-methyleneoxindole substructures with different C-2 substituents [lapatin B (75),77 alantrypinone (76),34 and serantrypinone (77)79].80N-formyllapatin A (73) displays a unique structural feature since the L-alanine moiety, as a bridge between CH-14 and the indole nitrogen, is N-formylated, representing the first N-formylspiroquinazoline secondary metabolite. The structure elucidation of alantrypinone (76) by Larsen et al.34 was based on the similarity of this new ring system with the skeleton of the spiroquinazoline (68).75 The UV data of serantrypinone (77) were almost identical to those of alantrypinone (76). Although Barrow75 and Larsen34,79 had already used UV-guided isolation of new metabolites, a new algorithm for automated comparison of UV data was only developed a few years later. Larsen reported a new methodology, X-hitting, to track two novel spiroquinazoline metabolites, lapatins A (72) and B (75).77 Since quinazoline derivatives have very similar and characteristic UV spectra due to their anthranilic acid-derived chromophore system, the automated comparison of UV data allowed the authors to track these two spiroquinazoline metabolites in a Penicillium lapatayae extract. The evaluation of the antibacterial activity of the two derivatives, 72 and 75, against four aqua-bacterial Aeromonas hydrophilia, Vibrio alginolyticus, V. anguillarum, and V. harveyi as well as against a human-pathogen, Edwardsiella tarda, revealed that only lapatin B (75) showed potent inhibitory activity against the aqua-bacterial V. harveyi, with MIC value of 16 μg mL−1. It is also important to highlight quinadoline B (74) which moderately inhibited lipid droplet synthesis in mouse macrophages in a dose-dependent manner (50–200 μM) without revealing cytotoxic effects on macrophages even at a concentration of 300 μM.
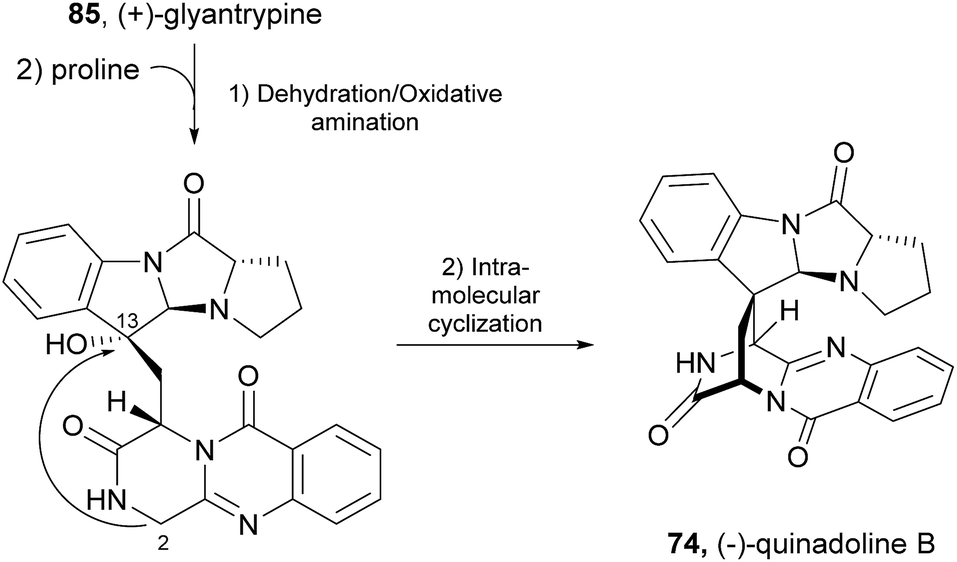
A biosynthetic pathway was hypothesized by Hart who created a model to better understand the origin of this family of alkaloids.81 Regarding alanine-derived spiroquinazoline alkaloids, i.e. compounds 68–71, and 76, anthranilic acid and tryptophan are the precursors and alanine is used to incorporate into C-3, C-4 and the 3-methyl group. NRPS are also involved in the biosynthesis of the two major metabolites of Penicillium thymicola,34 fumiquinazoline F (9) and (+)-alantrypinone (76) (Scheme 11).21 The oxidation of the indole substituent of fumiquinazoline F (9) to 2-indolone and subsequent action of the Af10270-type FAD amine oxidase would yield the equivalent imine in the pyrazinone ring. The new C–C bond in the hexacyclic ring should be formed through the intramolecular capture by the indolone carbanion at C-3. Serantrypinone (77, Scheme 12) is probably formed through a similar Ant-D-Trp-L-Ser-tripeptidyl-NRPS. Spiroquinazolines 72, 74, and 75 are derived from glycine, anthranilic acid and tryptophan. The amino acid bridge that links the tryptophan-derived substructure from C-14 to the indole-derived nitrogen should be formed by alanine in the case of (+)-lapatin A (72) and proline in the case of (−)-quinadoline B (74). Koyama et al.78 suggested a putative biosynthetic pathway for quinadoline B (74) from which the intermediate is generated via intramolecular dehydration of anthranilic acid-tryptophan-glycine cyclotripeptide (74). Subsequent binding of proline to the indoline moiety, via dehydration and oxidative amination, and intramolecular cyclization between C-2 and C-13 leads to a formation of quinadoline B (74) containing a δ-lactam ring (Scheme 13). N-formyllapatin A (73) is likely to be derived from lapatin A (72) which involves several steps such as N-methylation and oxidation, or biosynthesized via intramolecular dehydration from anthranilic acid, tryptophan, glycine, and N-formylalanine.60
Scheme 13 Hypothetical biosynthetic pathway of quinadoline B (74).
2.5 Summary of biological activities and SAR analysis
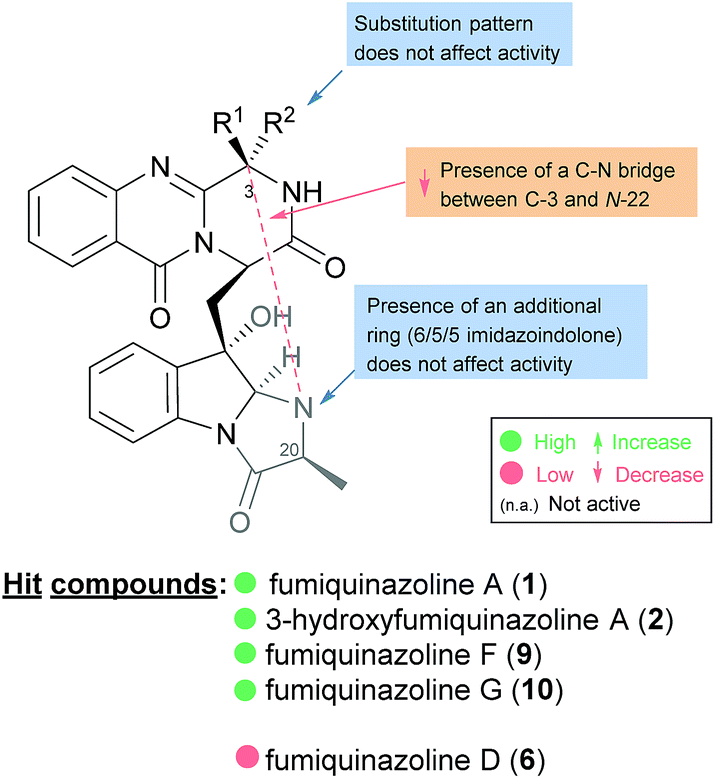
The activities mainly explored for this class of secondary metabolites are in the chemotherapy field, although relevant examples of substance P inhibitors can be found in the fiscalin group [e.g. fiscalins A (53), B (57) and C (58)]. Charts 13–16 represent structure–activity relationship analyses, gathering results of antibacterial, antifungal, antiviral and antitumour activities (ESI Table 1†), respectively, for this class of secondary metabolites. Within antibacterial effects, their broad spectra are not restricted to human pathogens and some members proved their activity also against aqua-pathogenic bacteria such as Vibrio harveyi as reported for glyantrypine (36) and (−)-lapatin B (75). Regarding human pathogens, the linkage of a disubstituted indole core with the quinazoline part through the bridge between C-18 and C-3 adds rigidity to the molecule that might be undesirable for the activity (Chart 13). This fact is noticeable by the complete loss of antibacterial activity in fumiquinazoline Q (20), when compared to fumiquinazoline F (9) [Micrococcus luteus (MIC = 99 μg mL−1) and Staphylococcus aureus (MIC = 137 μg mL−1)]. Regarding fiscalins, the fact that only neofiscalin A (55) was active among all the derivatives that were tested (with low MIC values even against drug-resistant strains), suggests that not only the substituent on C-22 (one methyl group) but also the stereochemistry of C-3 and C-22 (3R, 22R) may be required for this effect. Although the antibacterial activity stands out due to the potencies found, interesting antifungal compounds were also disclosed. SAR suggests that the presence of a C–N bridge between C-3 and N-22 in fumiquinazolines could be detrimental to activity since fumiquinazoline D (6) showed modest inhibitory activity on the growth of phytopathogenic fungi (MICs = 25–50 μg mL−1), whereas fumiquinazolines F (9), G (10), A (1), and 3-hydroxyfumiquinazoline A (2) showed higher (although still modest) antifungal activities (MICs = 12.5–25 μg mL−1) (Chart 14). Both in microbial and cancer models, a key feature of these secondary metabolites is their ability to reverse multidrug resistance which could be related to other cellular mechanisms not yet disclosed. Nonetheless, striking differences can be seen among closely related analogues, with complete loss of activity by changing the stereochemistry of a single chiral centre at C-3 (Chart 15). For instance, (−)-oxoglyantrypine (38) exhibited anti-H1N1 activity while the enantiomer (+)-oxoglyantrypine (39) was only weakly active against the virus. Regarding compounds that have the ability to reverse the multidrug-resistant (MDR) phenotype in cancer cell lines, a OH group at C-15b or C-16 in ardeemins plays an important role in DOX- or DDP-binding transport sites on the P-glycoprotein. Both (−)-5-N-acetyl-15b-didehydroardeemin (27) and (−)-5-N-acetyl-16α-hydroxyardeemin (28) have a OH group at C-15b and C-16, respectively. The former had the highest reversal fold (RF) in SK-OV-S/DDP cell line, and the latter exhibited the strongest reversal activity in K562/DOX and A549/DDP cell lines (Chart 16).
Chart 13 Structure–activity relationship studies for compounds with antibacterial activity.
Chart 14 Structure–activity relationship studies for compounds with antifungal activity.
Chart 15 Structure–activity relationship studies for compounds with anti-influenza virus A (H1N1) activity.
Chart 16 Structure–activity relationship studies for ardeemins with antitumour multi-drug resistance.
Some reports focusing on toxicity studies revealed these natural products to be safe without cytotoxic effects at high concentrations in several in vitro cell models (endothelial cells, macrophages); fumiquinazoline-related compounds could be promising drug candidates but investigations on their physicochemical properties and/or in vivo pharmacokinetic studies are still missing and could help translating these secondary metabolites to useful drug candidates.
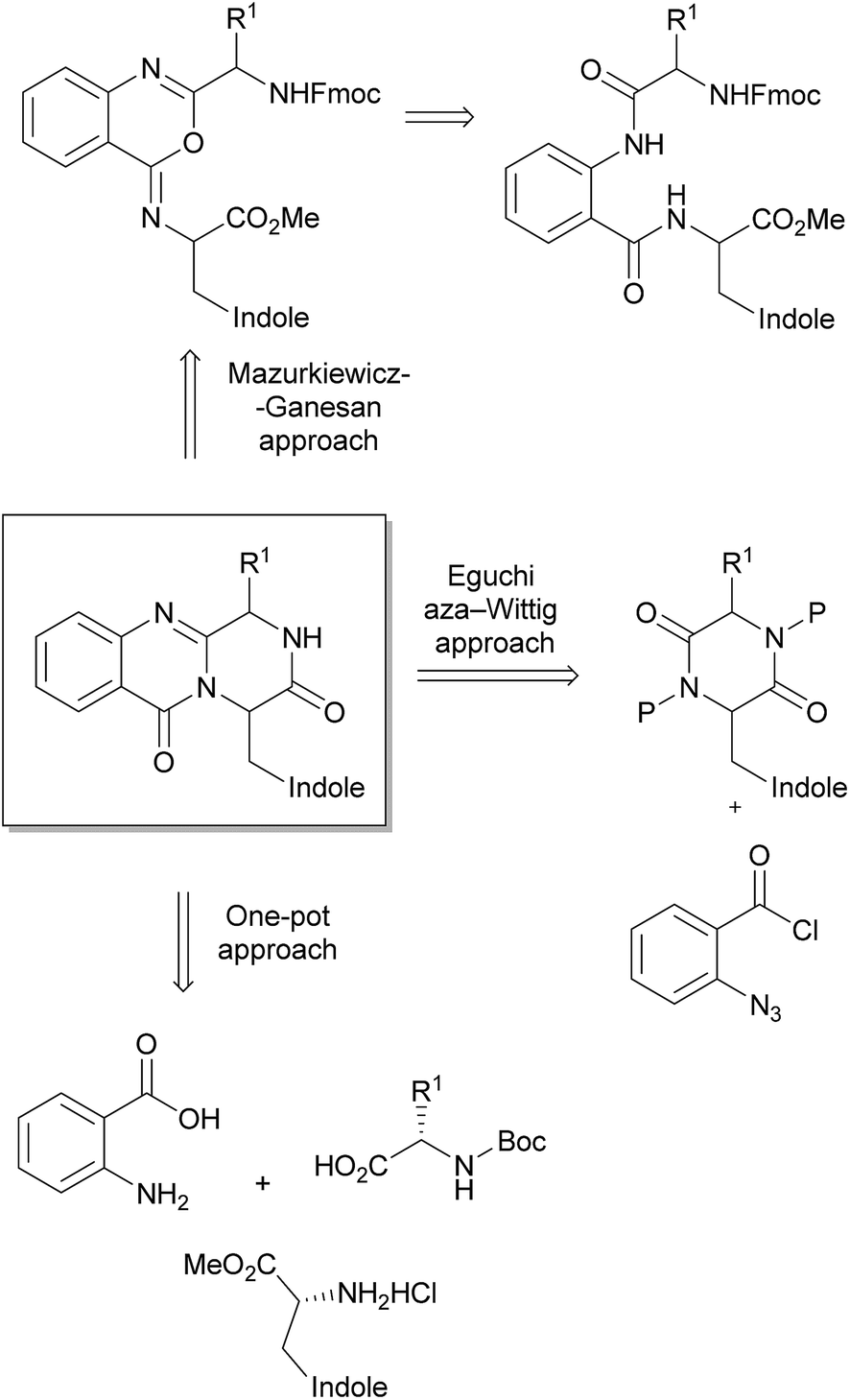
3 Synthesis
The pyrazino[2,1-b]quinazoline-3,6-dione system is the key structural fragment of the fumiquinazoline-derived group of alkaloids herein described. Although there are several approaches for the synthesis of compounds containing this core structure,12–16,82–84 the most common synthetic strategies are based on two main general approaches: (i) the Eguchi-aza Wittig85–87 that consists in a selective acylation of diketopiperazines with o-azido benzoyl chloride, followed by cyclization, and (ii) the Mazurkiewicz approach88 (later modified by Ganesan) which involves a two-step sequence from linear diamides through isomerisation of 4-imino-4H-3,1-benzoxozines to the quinazoline-4-ones (Scheme 14).89,90 However, a third approach, a highly efficient three-component one-pot methodology promoted by microwave irradiation, was more recently reported by Liu et al.91 for the total syntheses of glyantrypine (36), fumiquinazoline F (9), and fiscalin B (57). The following section details these main approaches highlighting the importance of the selection of amino acid protective groups and their removal, the reaction conditions to avoid epimerizations, with the extensive use of 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDAC) and N,N′-dicyclohexylcarbodiimide (DCC) as coupling reagents, and the critical steps of cyclization to the pyrazino[2,1-b]quinazoline-3,6-dione system with organophosphines dominating these dehydrative cyclizations.
Scheme 14 Approaches to the synthesis of the fumiquinazoline-derived alkaloids.
3.1 Eguchi-aza Wittig approach
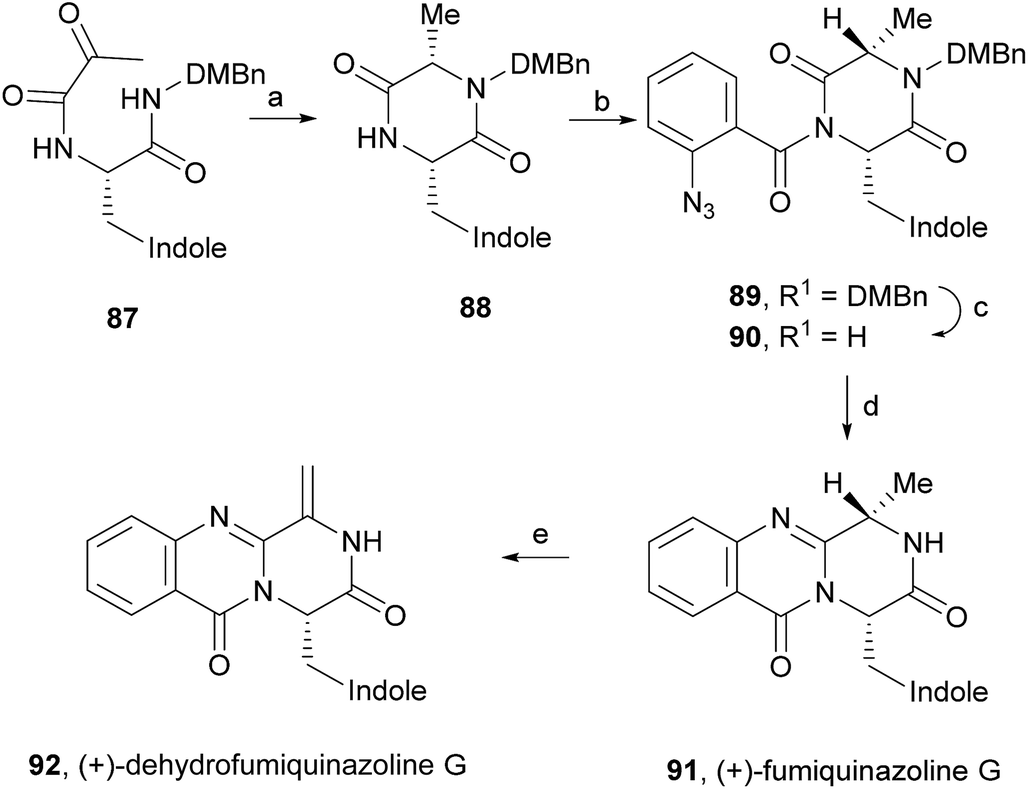
(+)-Fumiquinazoline G (91) or ent-fumiquinazoline G, and (+)-dehydrofumiquinazoline G (92) were first synthesized by He and Snider in 1997,92 using Eguchi's procedure to accomplish the annulation of a quinazoline-4-one onto an amide (Scheme 15). The total synthesis started with the preparation of the pyruvamide 87 from Cbz-L-Trp and dimethoxybenzylamine (not shown). Reduction of the methylene group of the methylenediketopiperazine to give 88 (obtained through dehydration of the intermediary enamide), and acylation of the 4-substituted 2,5-piperazinedione with o-azidobenzoylchloride after formation of the amide anion yielded 89. Deprotection followed by a Staudinger reaction with a phosphine and subsequent cyclization of the corresponding phosphazene, yielded 91 which, after oxidation of the indoline, completed the total synthesis of (+)-fumiquinazoline G (91) in 12 steps from Cbz-L-Trp and dimethoxybenzylamine with 11% overall yield. Oxidation of 91 provided (+)-dehydrofumiquinazoline G (92) in 80% yield.
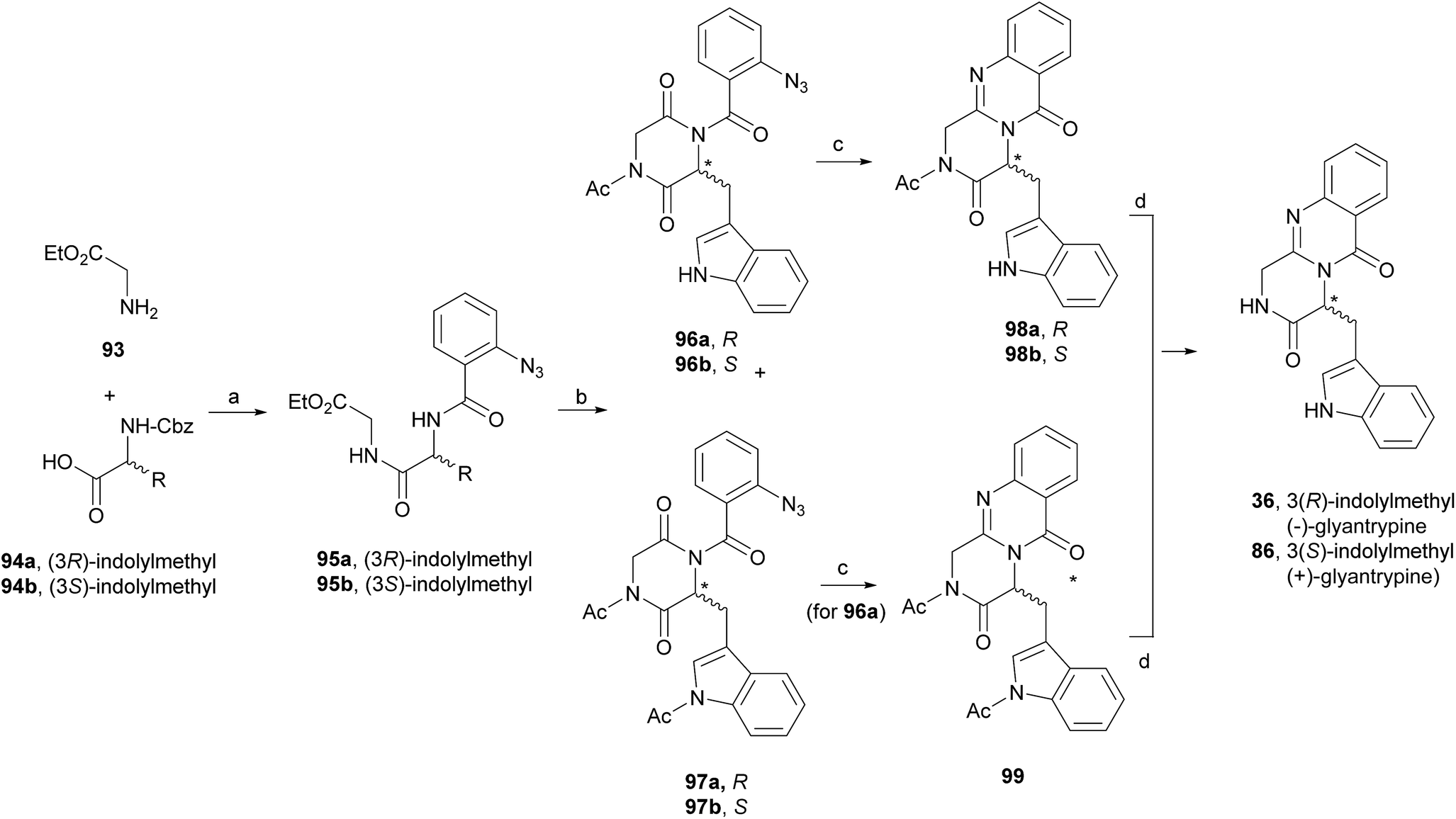
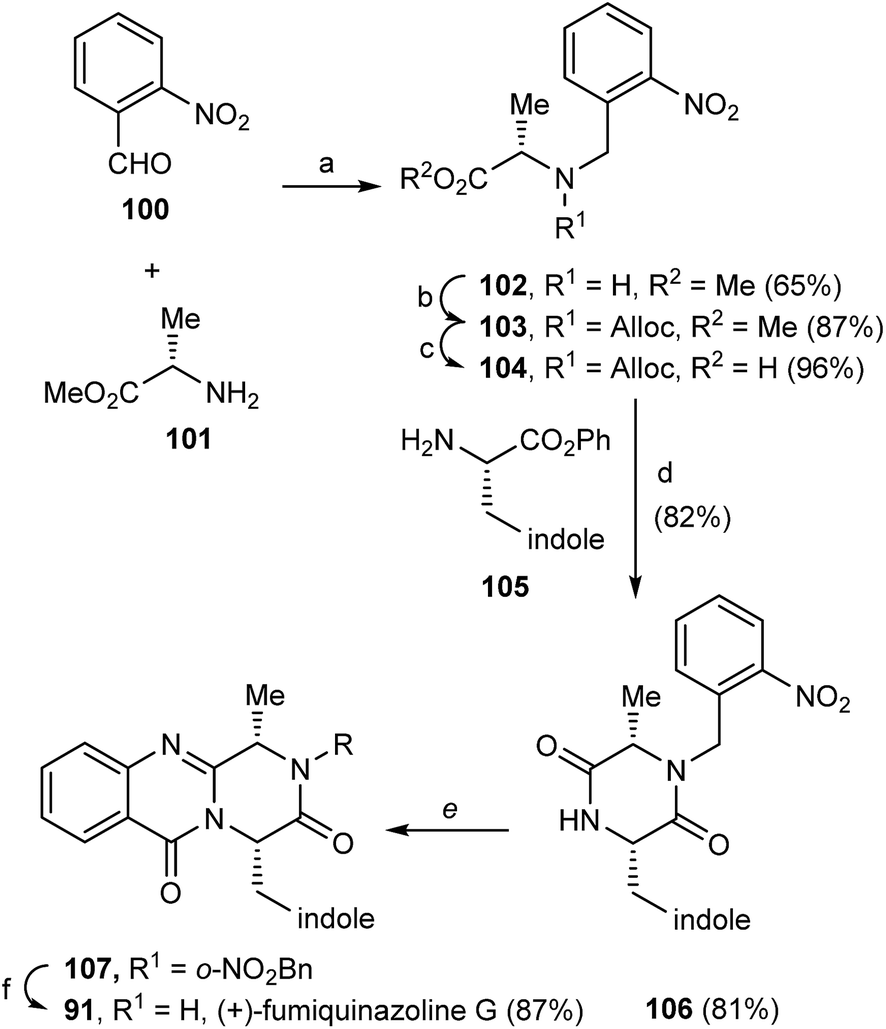
In 2000, Cledera and Avendaño93 published the total synthesis of glyantrypine (36) and ent-glyantrypine (86) based on the double cyclization of open-chain tripeptides. The anthranilic acid unit thereby is the N-terminal residue and bears an azido group as a masked amino function. This synthesis was carried out in three different stages: (a) synthesis of the intermediary tripeptide 95, (b) first cyclization: transformation of 95 into 2,5-piperazinedione derivatives (96 or 97), and finally (c) second cyclization to pyrazino[2,1-b]quinazoline-3,6-dione derivatives through a Staudinger-aza Wittig sequence. Ethyl glycinate (93) and the appropriate N-Cbz-amino acids 94 were used as starting materials for the preparation of tripeptides 95a, b using ethyl chloroformate or 1-ethyl-3-(3-dimethylaminopropyl)-carbodiimide (EDAC) as the coupling agent (Scheme 16). Deprotection and subsequent acylation with o-azidobenzoyl chloride yielded 95a and 95b. The transformation of 95a, b into 2,5-piperazinedione derivatives involved the conversion of these starting materials into the corresponding sodium salts. Subsequent treatment with acetic anhydride yielded a mixture of 96a, b and 97a, b, with complete retention of the stereochemistry. The last step towards the total synthesis of glyantrypine involved a second cyclization via a Staudinger-aza Witting sequence by treatment of 96a, b and 97a, b with tributylphosphine (Bu3P) resulting in 98a, b and 99, respectively. Further deacetylation yielded both stereoisomers of glyantrypine (36 and 85). The synthesis of ent-fumiquinazoline G (91), by Snider and Busuyek,94 demonstrated the utility of their newly developed general procedure for the synthesis of N-(2-nitrobenzyl)diketopiperazines, previously reported for the formation of the benzodiazepinedione of asperlicin (82).17,33,95 The diketopiperazine can be further cleanly deprotected in methanol (MeOH) by irradiation through Pyrex at 254 nm. Also, the use of the 2-nitrobenzyl group as a photochemically labile protecting group for the amide of the diketopiperazines allowed the utilization of the aza-Wittig procedure (Scheme 17). Reductive amination of 2-nitrobenzaldehyde (100) with alanine methyl ester (101) gave 102, followed by coupling with allyl chloroformate to furnish intermediate 103, and subsequent hydrolysis of the methyl ester yielded 104. Condensation of the activated phenyl ester 104 with 105 gave 82% of an intermediary dipeptide that, after deprotection and subsequent cyclization, yielded the desired monoprotected diketopiperazine 106. Then, acylation of 106 afforded quinazolinone 107, followed by photolysis in the last step, to obtain (+)-fumiquinazoline G (91) in 87% yield.
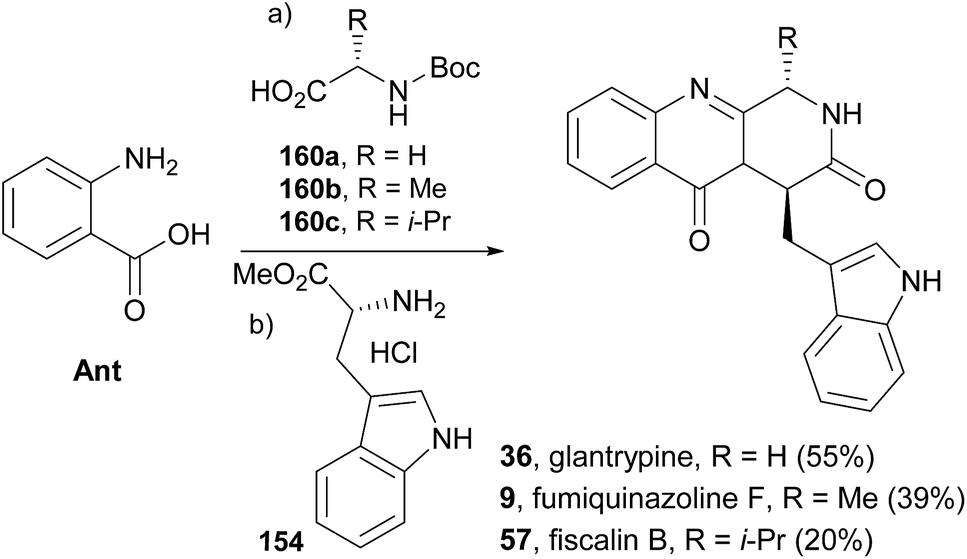
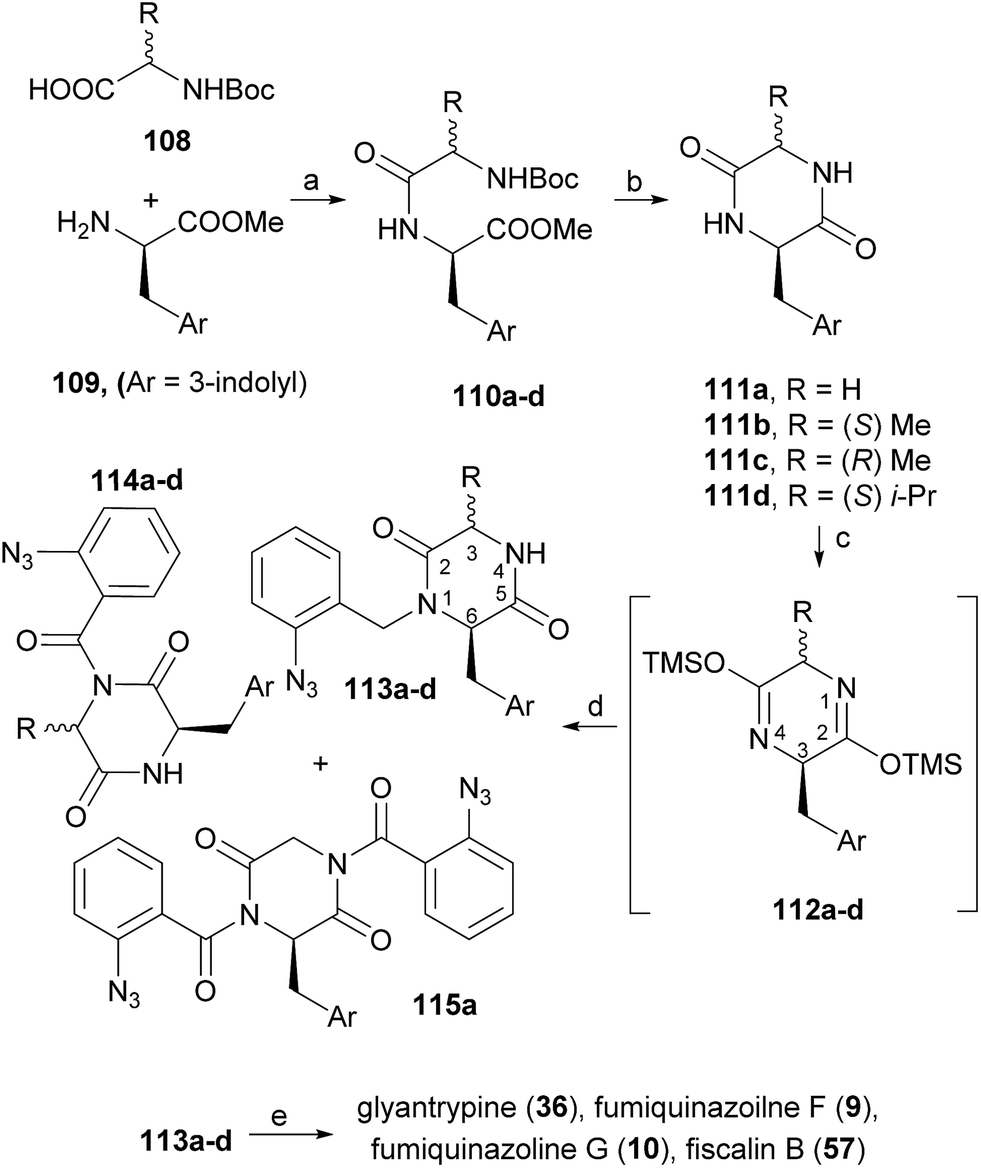
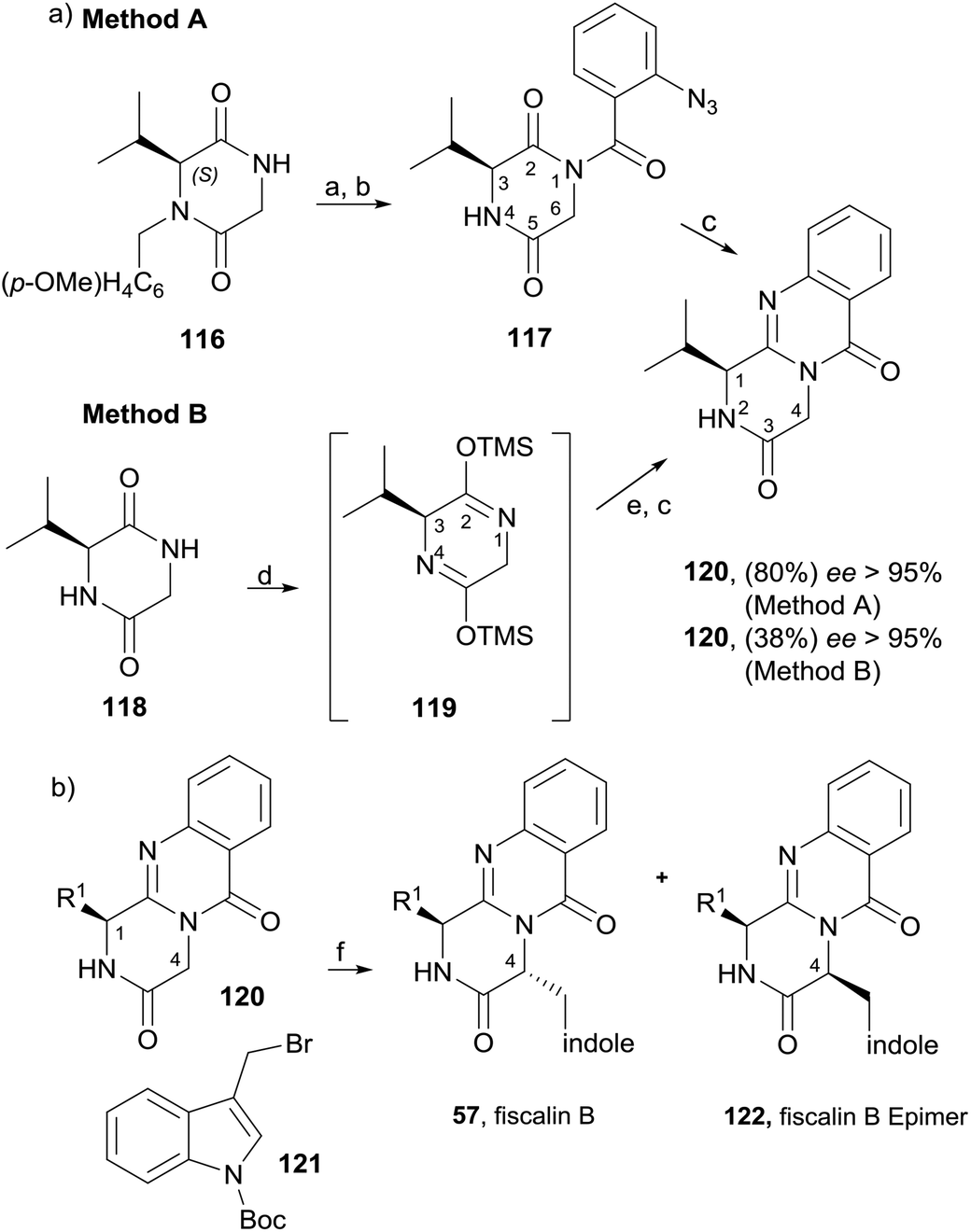
Diketopiperazines (111) are involved in another strategy to construct this type of alkaloids relying on the control of the regioselectivity provided by the folded conformation of the 3-arylmethyl substituent in intermediate 112 (Scheme 18). Hernández and Söllhuber96 reported that 112 adopted a boat-shaped conformation that, in conjunction with the folded conformation of the benzyl group forces the trimethylsilyl group (TMS) on position 2 in such an orientation that it shields the N(1)-position of 112, giving N(1)-acylisomer 113. Based on this premise, the authors developed a four-step total synthesis of glyantrypine (36), fumiquinazoline F (9), fumiquinazoline G (10), and fiscalin B (57). 3-Arylmethyl-diketopiperazines 111a–d were prepared in two steps by previously described methods from the respective N-Boc dipeptides 110a–d, previously obtained from the protected amino acids 108 and 109. Reaction of 111a–d with trimethylsilyl chloride (TMSCl) in the presence of triethylamine (Et3N) gave intermediates 112a–d, and further highly chemoselective acylation with o-azidobenzoyl chloride yielded 113a–d with small amounts of by-products 114a–d and 115a. Intramolecular Staudinger reaction of 113a–d in the presence of Bu3P afforded the desired alkaloids. Regio- and diastereoselective alkylation at C-4 of (1S)-1-isopropyl-2,4-dihydro-1H-pyrazino[2,1-b]-quinazoline-3,6-diones (120, Scheme 19) was used by Hernández et al.97 for the synthesis of fiscalin B (57). The starting materials 120 were prepared by two different methodologies (Scheme 19): method A, which consisted in acylation of 116 with o-azidobenzoyl chloride at the unprotected amine function and subsequent oxidative debenzylation with CAN to give 117, followed by intramolecular Staudinger reaction (Scheme 19); or method B: by acylation of 119 (previously obtained from piperazine-2,5-dione 118) with o-N3C6H4COCl, followed by treatment with tributylphosphine. To accomplish fiscalin B (57), the alkylation reaction of 120 with N-boc-3-indolylmethyl bromide allowed chemo- and diastereoselectivity without the need for N(2)-protecting groups (Scheme 19b).
Scheme 18 Reagents and conditions: (a) EDAC; (b) 200 °C, 4–7 h; (c) TMSCl, Et3N, CH2Cl2; (d) o-N3C6H4COCl, CH2Cl2, rt, 16 h; (e) Bu3P, toluene (dry), rt, 16 h.
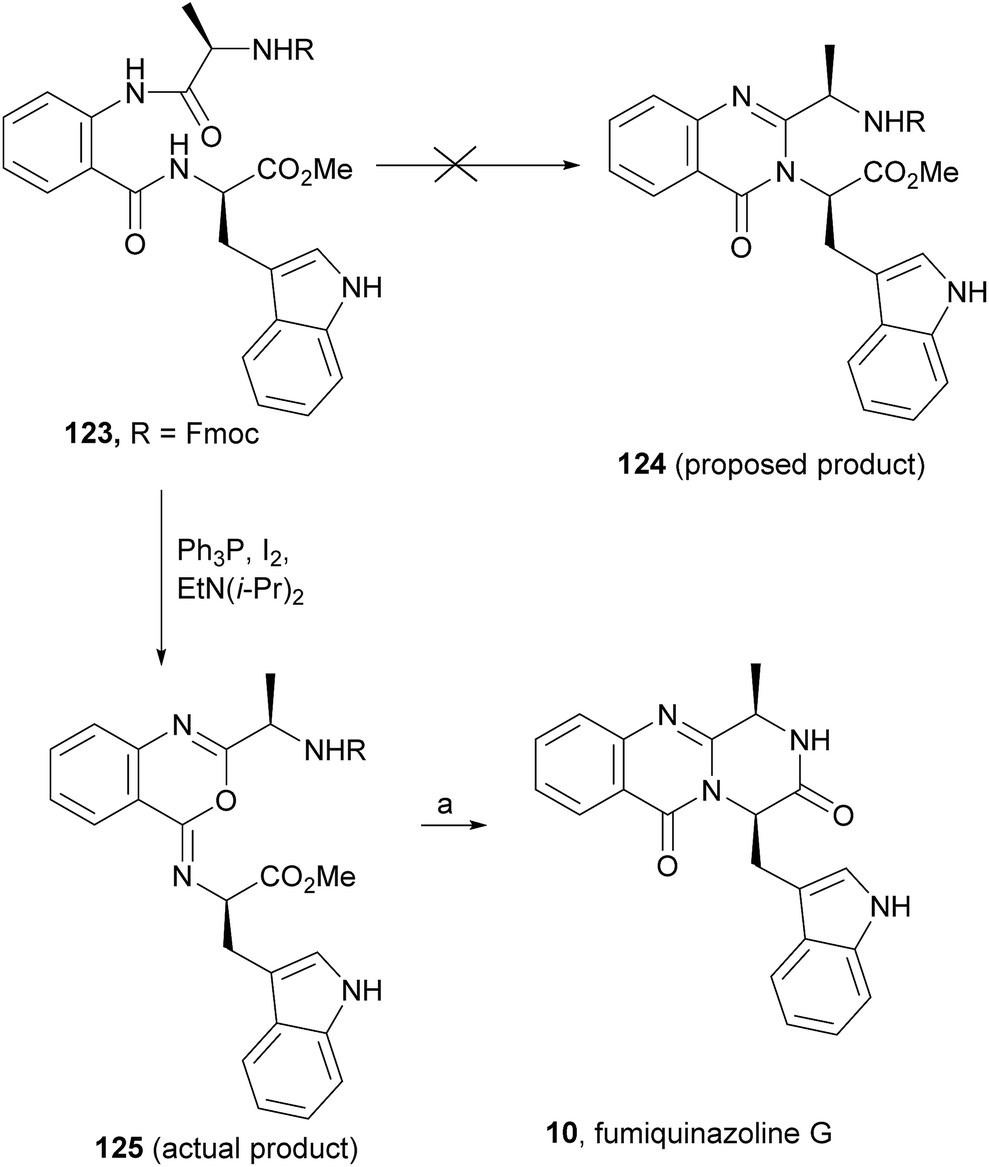
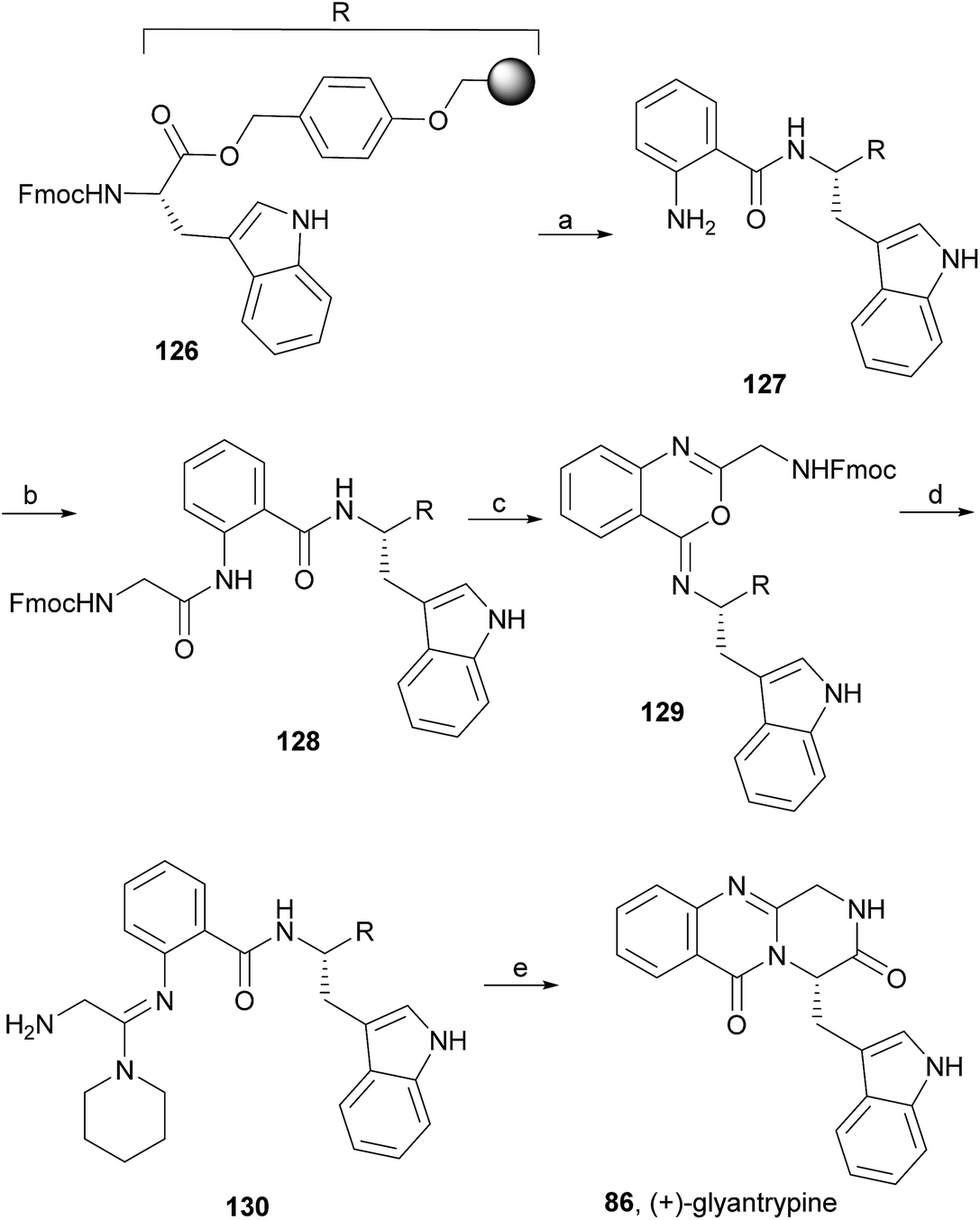
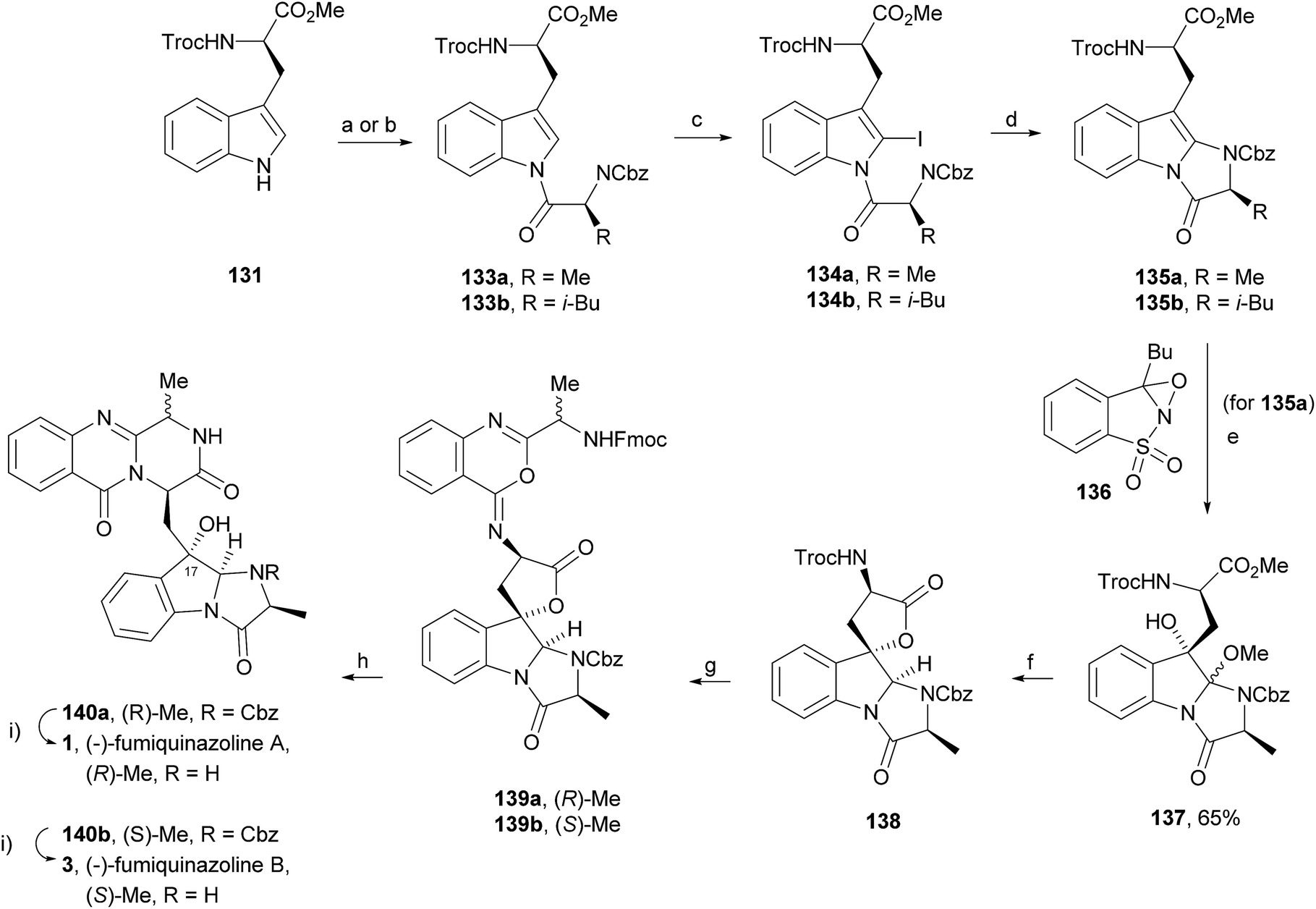
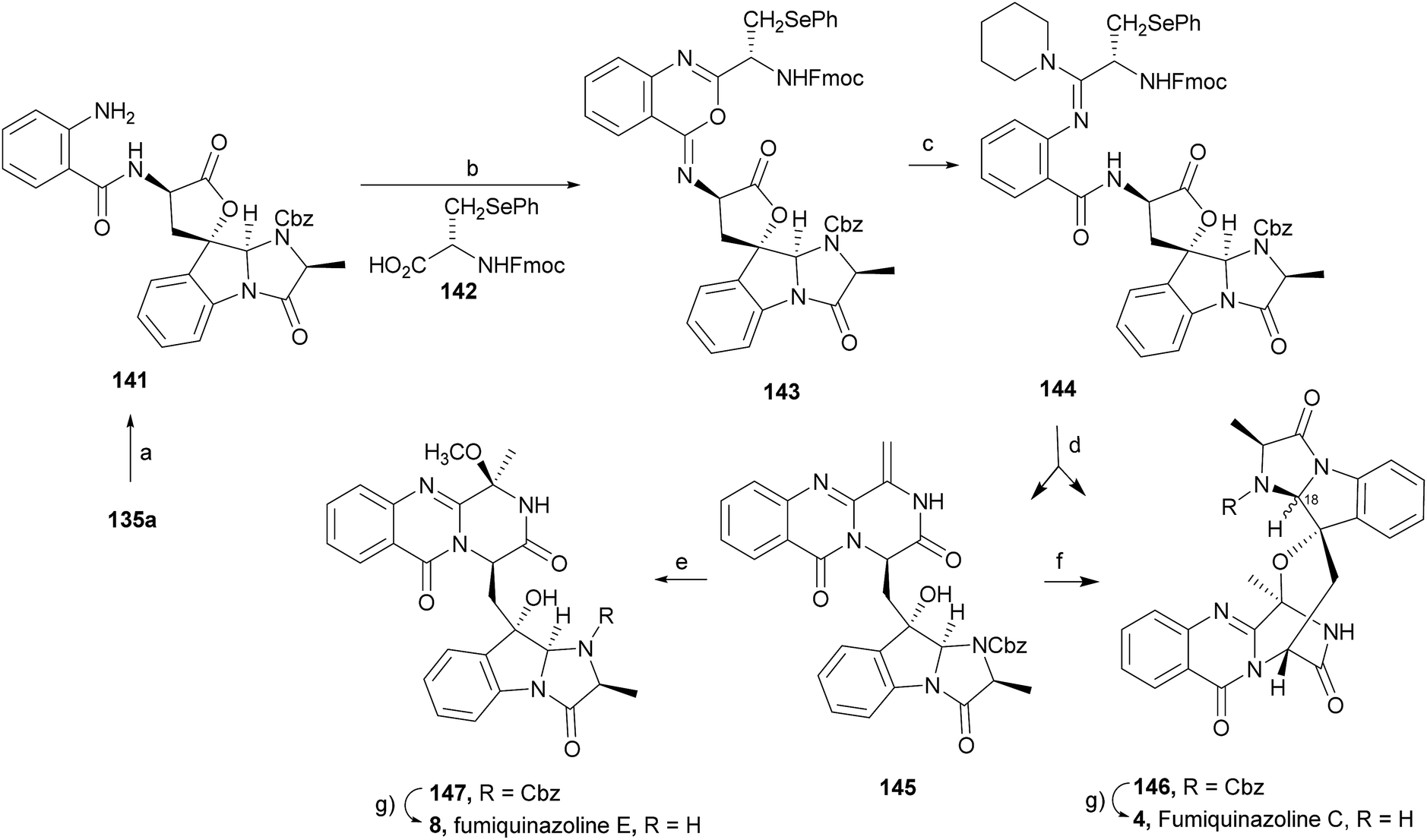
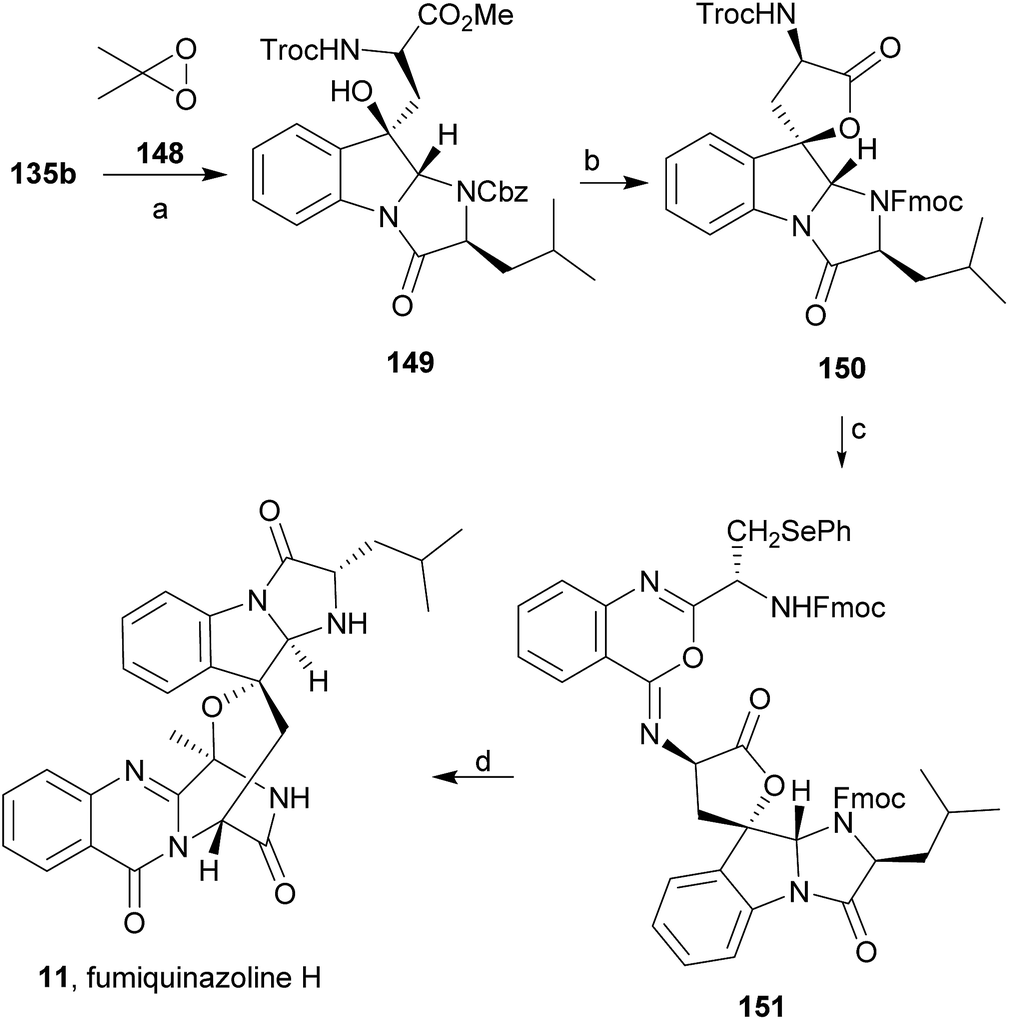
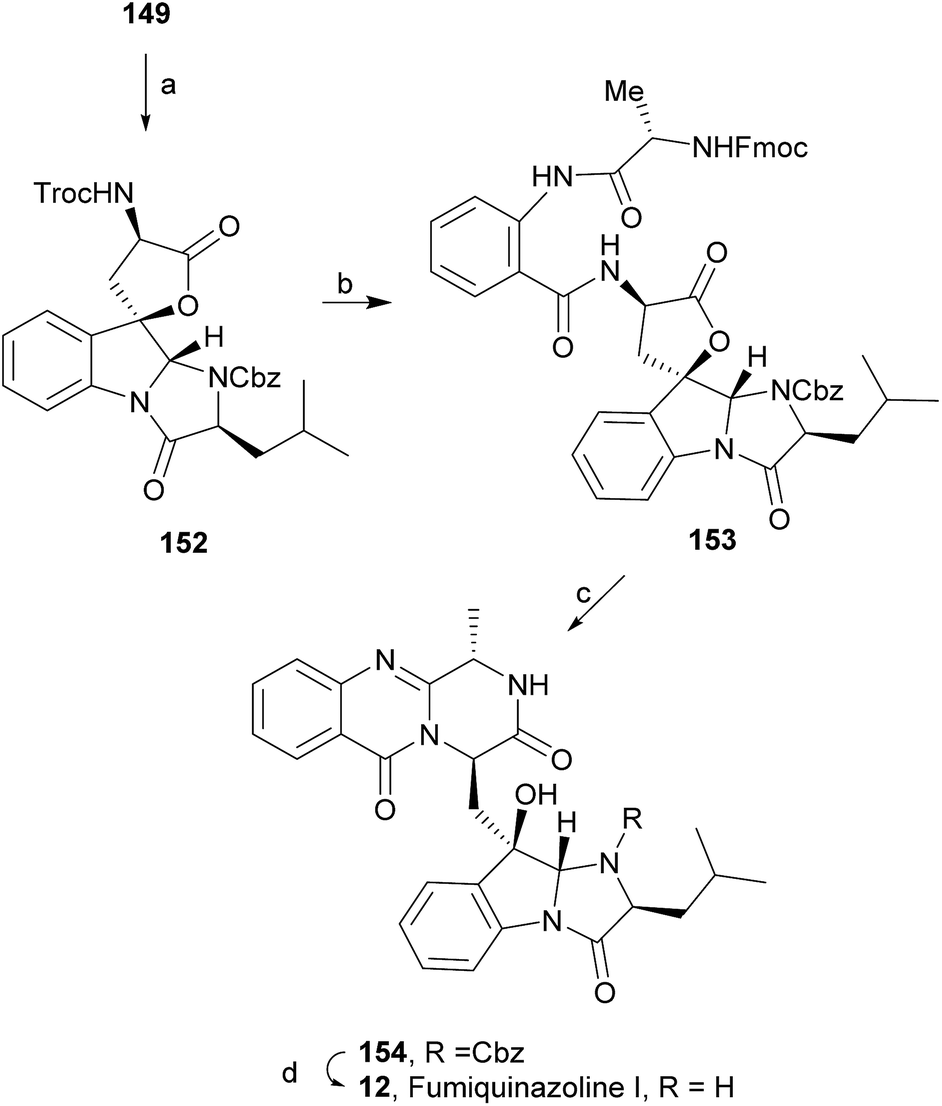
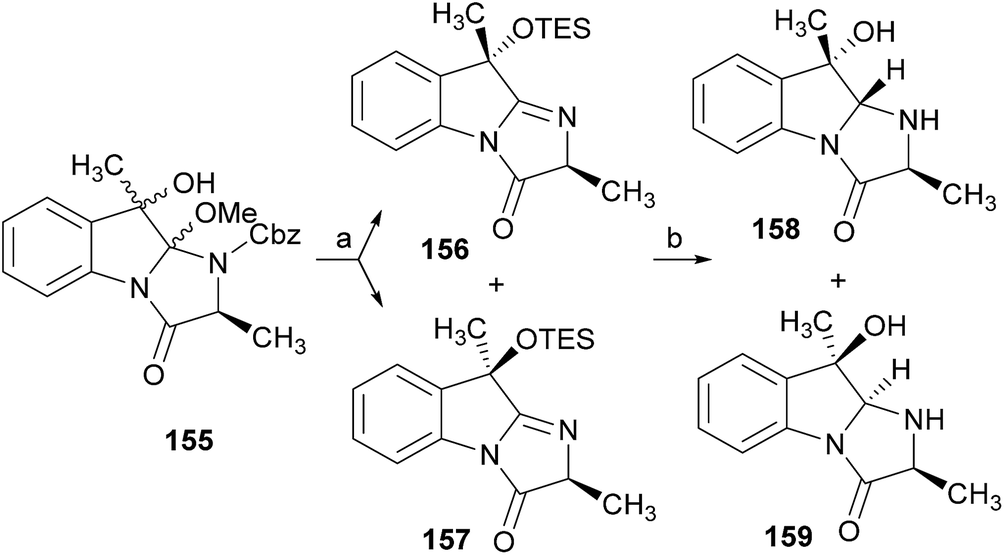
Wang and Ganesan89 reported the total synthesis of fumiquinazoline G (10) and fiscalin B (57), based on a key step involving cyclization of N-acylanthranilamide 123 (Scheme 20). The authors used Wipf's protocol, which involves the application of a Hunig's base, DIEA (the reagent initially developed by Mazurkiewicz88 and later exploited by Wipf),98 along with triphenylphosphine (Ph3P) and iodine to give the cyclization product that they initially described as 4-quinazoline (124). However, soon after the publication, He and Snider99 refuted this hypothesis and proved by NMR experiments that, in fact, the cyclization product was not quinazolinone 124, but 4-imino-4H-3,1-benzoxoazine (125). To accomplish the total synthesis, deprotection of 125 with piperidine afforded an intermediate that was converted to fumiquinazoline G (10) on preparative TLC. This methodology was widely applied to the total syntheses of several natural alkaloids such as alantrypinone (77),100,101 glyantrypine (36),90 anacine,102 verrucines A and B,102 circumdatins C and F,103 fiscalin B (57),89,90 and fumiquinazolines A (1), B (3), C (4), E (8), H (11), and I (12).69,90,104–106 Wang and Ganesan have made use of the same methodology for the synthesis of more complex fumiquinazolines on solid support105 as well as in solution.90 In the solid-phase studies,105 for the total synthesis of (+)-glyantrypine (86), Wang resin loaded with Fmoc-L-Trp (126), a commercially available compound, was deprotected and coupled with anthranilic acid (Scheme 21). Acylation of the amino group of the aniline moiety in 127 with Fmoc-Gly-Cl provided 128; subsequent dehydrative cyclization of this compound yielded 129. Finally, deprotection and rearrangement of the oxacine in 129 gave amidine carboxamide (130). After washing, the resin was refluxed in acetonitrile to induce cyclative cleavage of (+)-glyantrypine (86) with 51% overall yield. Furthermore, the authors explored this synthetic methodology using 4-chloroanthranilic acid instead of anthranilic acid to obtain 9-chloro-glyantrypine in 78% yield. Substitution of Fmoc-Gly-Cl for Fmoc-Phe-Cl and replacement of the starting L-Trp loaded resin with L-Ala, L-Leu, and L-Phe, etc., enabled the preparation by parallel synthesis of a small library of unnatural analogues. In the solution-phase studies, glyantrypine (36), fumiquinazoline F (9), fumiquinazoline G (10), and fiscalin B (57) were synthesized in four steps from tryptophan methyl ester. The key step in the synthesis of these alkaloids is the formation of an anthranilamide residue in a linear tripeptide that is further dehydrated to a benzoxazine that undergo rearrangement to the natural products via an amidine intermediate.90 Snider and Zeng69,104,106 developed several procedures to prepare the bottom half of the more complex members of the fumiquinazoline family of alkaloids and combined it with the Mazurkiewicz–Ganesan cyclization for the top half to obtain these derivatives. The total syntheses of fumiquinazolines A (1), B (3), and I (12) were reported in 2000 (ref. 104) and only two years later, the total syntheses of fumiquinazolines C (4), E (8), and H (11)106 were achieved. The starting point for the synthesis of these alkaloids consists in the modification of Trp indole to introduce the imidazolinone ring and a hydroxyl group (Scheme 22). Thus, the syntheses started with reduction of protected D-Trp (131) to provide the indolines which were acylated with N-Cbz-L-Ala (133a) for (−)-fumiquinazolines A (1), B (3), C (4), and E (8), or with N-Cbz-L-Leu (133b) for (−)-fumiquinazolines H (11) and I (12) (Scheme 22). Mercuration with Hg(OTFA)2 and KI followed by treatment with iodine provided iodoindoles 134 that, upon Buchwald palladium-catalysed condensation, gave the desired key intermediates 135. Another crucial structural aspect is the configuration of the two substituents at C-17 and this selectivity can be easily achieved. Epoxidation of 135a with oxaziridine (136) occurs selectively on the bottom face, leading to the precursors of (−)-fumiquinazolines A (1), B (3), C (4), and E (8). On the other hand, oxidation with dimethyldioxirane proceeds selectively on the top face as needed for (−)-fumiquinazolines H (11) and I (12). The synthesis of (−)-fumiquinazolines A (1) and B (3) starts with the epoxidation of 135a with oxaziridine (136), that yields 137 in 65% as a mixture of diastereomers with the α-hydroxy group and 23% of the corresponding β-hydroxy diastereomers (Scheme 22). Isomer 138 is produced exclusively upon reduction and further lactonization in 66% by stirring with silica gel in dichloromethane for 12 h. Reductive deprotection of 138 affords the amine intermediate which is coupled with anthranilic acid and EDAC in acetonitrile (CH3CN) to yield 85% of the aniline. A second coupling with Fmoc-L-alanine [with the correct configuration for fumiquinazoline A (1)] and with Fmoc-D-alanine [with the correct configuration for fumiquinazoline B (3)] and further dehydrative cyclization yields iminobenzoxazines 139a and 139b. Reaction with piperidine in EtOAc gives the crude amine which, after reflux, affords the protected fumiquinazolines. Hydrogenolysis of the protecting group gives fumiquinazoline A (1) (from 140a) in 90% yield or fumiquinazoline B (3) (from 140b) also in 90% yield. Syntheses of fumiquinazolines C (4) and E (8) present an additional challenge due to a methoxy group at C-3 in fumiquinazoline E (16) and a seven-membered ring formed between C-3 and the oxygen at C-17 in fumiquinazoline C (4). Their syntheses were accomplished through deprotection of fumiquinazoline A (1) intermediate 135a and coupling with anthranilic acid using EDAC to yield 141 (Scheme 23). Reaction of 141 with FmocNHCH(CH2SePh)CO2H (142) and further dehydrative cyclization provided iminobenzoxazine 143. Treatment of 143 with piperidine cleaved the Fmoc group and opened the iminobenzoxazine to give amidine 144. Refluxing 144 in acetonitrile with acetic acid gave Cbz-dehydrofumiquinazoline A (145) and Cbz-fumiquinazoline C (146). In addition, further heating of 145 in 100:1 CH3CN/AcOH at reflux afforded more amount of 146. Treatment of 145 with HCl in methanol provided Cbz-fumiquinazoline E (147) which was hydrogenolyzed to form (−)-fumiquinazoline E (8). In the same way, hydrogenolysis of 146 gave (−)-fumiquinazoline C (4). Epoxidation of 135b with dimethyldioxirane (148) proceeded selectively to furnish 55% of the desired methoxy alcohol with the hydroxyl group cis to the isobutyl group and only 38% of the trans isomer (Scheme 24). Further reduction provided 149, needed for the synthesis of (−)-fumiquinazoline H (11) and I (12). Hydrogenolysis of 149, followed by lactonization through silica gel, gave a tetracyclic lactone which was reacted with FmocCl and i-Pr2NEt to provide 150. Conversion of 150 to 151 proceeded analogously as in 143 to 144. Ring closure of the amidine 151 to the quinazolinone, further formation of the piperazine ring, and elimination of benzeneselenol yields dehydrofumiquinazoline I analogue (not shown). In the absence of the bulky Cbz group, cyclization proceeds spontaneously without epimerization to give (−)-fumiquinazoline H (11) (Scheme 24). Synthesis of (−)-fumiquinazoline I (12) continued from 149 and lactonization yielded 152. Compound 153 was prepared analogously to 143 and 151. Dehydrative cyclization of 153 gave iminobenzoxazine which was deprotected and ring opening with piperidine and then cyclization gave Cbz-fumiquinazoline I (154). Finally, hydrogenolysis of 154 afforded fumiquinazoline I (12) (Scheme 25). Due to the similar structural characteristics showed by both fiscalin A (53) and fumiquinazoline A (1), Snider and Zeng69 also attempted to synthesize fiscalin A (53). The structures of the two compounds differ only in the substituent on the top of the piperazine ring and, more significantly, in the stereochemistry of C-18. Although the authors were not able to accomplish the total synthesis of fiscalin A (53), they successfully prepared the model 158, starting from 155, with the correct stereochemistry (Scheme 26, H and OH anti to each other). Treatment of 155 with TES triflate and subsequent hydrogenation gave a 2.1:1 mixture of imines 156 and 157 that, after hydrogenation and cleavage of the TES ethers gave 45% of 158 with fiscalin A stereochemistry and 25% of 159, which is formed from the minor diastereomer of 155. Unfortunately, when they tried to apply the same methodology used to construct the model for the synthesis of fiscalin A (53), the hydrogenolytic removal of the Cbz group led to a complex mixture of products that did not appear to contain the desired imine.
Scheme 20 Reagents and conditions: (a) (1) 20% piperidine in CH2Cl2, 25 °C, 12 min; (2) preparative TLC.
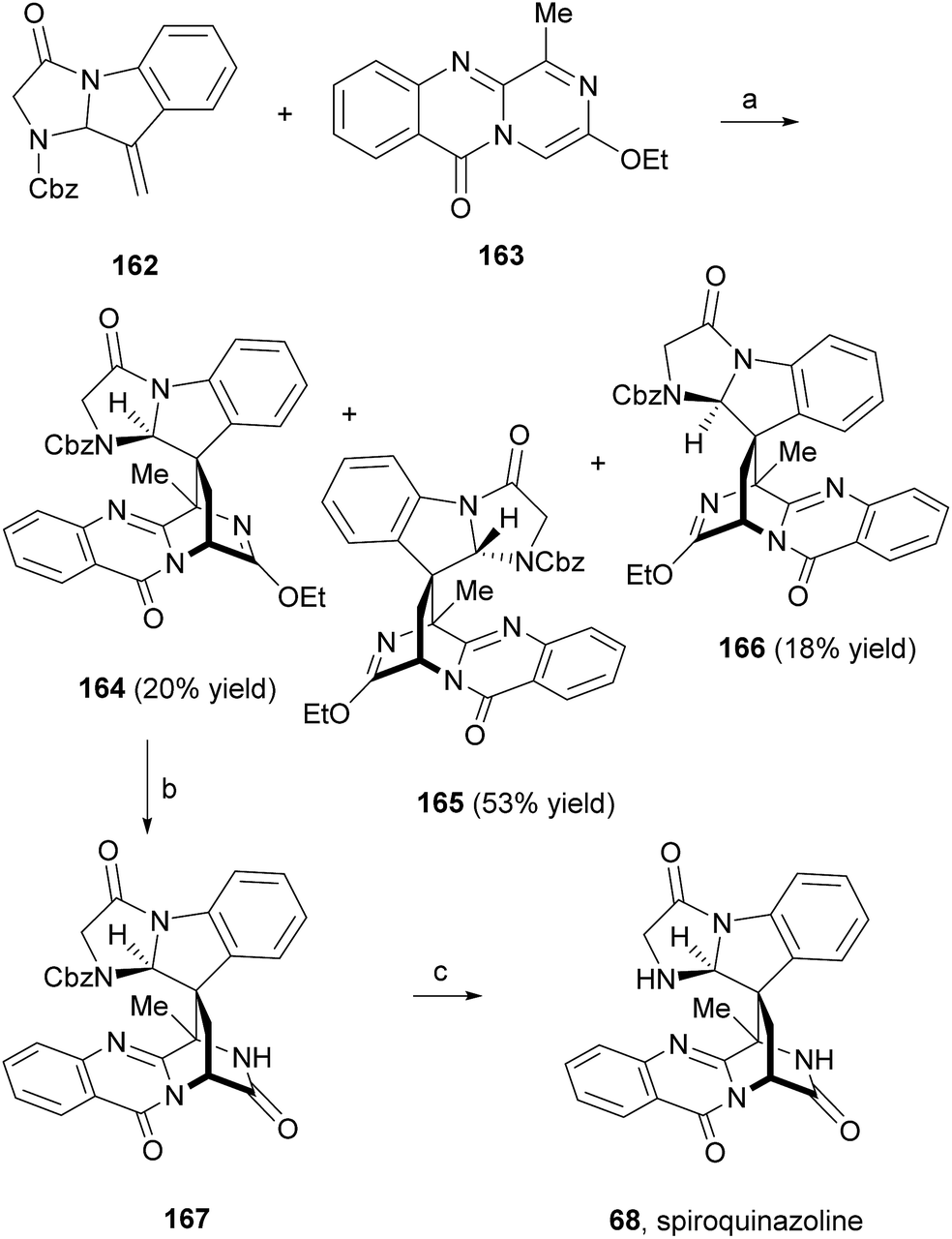
Liu et al.91 reported an easy access to the core pyrazino[2,1-b]quinazoline-3,6-dione scaffold through a novel and highly efficient three-component one-pot reaction sequence promoted by microwave irradiation. The application of this synthetic strategy to the syntheses of glyantrypine (36), fumiquinazoline F (9), and fiscalin B (57) from readily available starting materials (Scheme 27) demonstrated the utility and versatility of this methodology. Until 2010, the total syntheses of lapatin B (75), alantrypinone (76), and serantrypinone (77) were subject to several studies and were already very well documented in the literature.81 Along with those studies, several approaches to synthesize spiroquinazolines were reported but none has succeeded.107,108 The first total synthesis of (±)-spiroquinazoline (68) was described, together with (−)-alantryphenone (69), (+)-lapatin A (72), and (−)-quinadoline B (74) syntheses, in 2013 by Wu and Ma.80 The authors, based on previous studies by Hart81 and Magomedov,109 explored the possibility of using aminal embodied olefins as dienophiles for quick access to indoline-containing spiroquinazoline alkaloids. Olefin 162 was prepared from commercially available 2-nitrophenylacetic acid in eight steps. The aza-Diels–Alder reaction between 162 and azadiene 163 (Scheme 28) furnished the desired adduct 164 (20%), together with isomers 165 (53%) and 166 (16%). Hydrolysis of 162 produced the lactam 167 which, after hydrogenolysis, yielded spiroquinazoline 68. This approach was further applied to the syntheses of other spiroquinazoline alkaloids [(−)-alantryphenone (69), (+)-lapatin A (72), and (−)-quinadoline B (74)] in 11 to 12 steps from commercially available 2-nitrophenylacetic acid.80
Scheme 27 Reagents and conditions: (a) for glyantrypine: (36), 160a and P(OPh)3 py, MW, 150 °C, 10 min; fumiquinazoline F (9) or fiscalin B (57): anthranilic acid, 160b or 159c, and P(OPh)3, py, conventional heating, 55 °C, 16 h; (b) 154, MW, 220 °C, 1.5 min.
Scheme 28 Reagents and conditions: (a) xylene, 130 °C, 3 days; (b) HCl, EtOAc, 95%; (c) Pd/C, H2, EtOAc, 80%.
4 New perspectives
Very recently, Huang et al.110 reported an interesting approach for the in vivo diversification of this type of fungal indole alkaloids. Using an amino acid-directed strategy for inducing the marine-derived fungus Scedosporium apiospermum F41-1 to produce high alkaloid diversity, the authors were able, by feeding a culture of the fungus with diverse amino acids to obtain 22 diverse alkaloids, including 14 new compounds. It is worth highlighting scedapins A-E (173–177, Chart 17) that possess a rare skeleton, only observed in neosartoryadins A (66) and B (67), composed by a pyrazinoquinazolinedione and an imidazoindolone/indolone linked by a tetrahydrofuran ring. Also, scedapin C (175) is the first example in the fumiquinazoline family that contains an aminosulfonyl group.
Chart 17 Compounds obtained by feeding amino acids to the marine fungus Scedosporium apiospermum F41-1.
Some derivatives were tested for anti-hepatitis C virus (HCV) activity: scequinadoline D (181) and scequinadoline A (178) displayed significant anti-HCV activity against J8CC recombinant with the EC50 values of 110 and 129 μM, respectively. This promising approach rendered novel structures to this family of compounds and could be applied to detail SAR studies.
5 Concluding remarks
Fungi-derived naturally occurring fumiquinazolines and related alkaloids with a unique 6/6/6 or 6/6/6/5 quinazoline ring systems connected directly to a 6/5 or 6/5/5 imidazoindolone rings are continuously being discovered (Scheme 29). To date, approximately 80 naturally occurring fumiquinazolines have been isolated from a handful of fungal species collected in different environments around the world, mainly of the genera Penicillium, Aspergillus, and Neosartorya. In this line of research, cocultivation of fungi with bacteria to obtain these secondary metabolites is still underdeveloped and could be a promising approach in revealing new bioactive compounds. Some subclasses of this family of compounds already revealed robust SAR in the field of chemotherapy with the most relevant members revealing potent effects in MDR models.
Scheme 29 Take home message.
Some members of these group of alkaloids have already shown significant potential for further development such as (−)-oxoglyantrypine (38) and norquinadoline A (64) with notable anti-H1N1 activities. In addition, since neofiscalin A (55) showed antimicrobial activity against MRSA and VRE without cytotoxicity to endothelial cells, it can represent a prime candidate for in vivo studies. Fiscalin C (58), presenting synergistic activity when combined with oxacillin against MRSA, also arouses the interest in investigating molecular targets associated with resistance mechanisms.
Although the investigated biological activities are still in preliminary stages, there is no doubt that these are privileged structures with potential as drug candidates. The fused rings, the basic amine and rich stereochemistry are structural ingredients that proved to be important in SAR studies. Nevertheless, mainly due to the limited amounts isolated, most of the studies correspond to phenotypic screening assays and little is known about their molecular targets. Establishing culture methods in order to produce suitable quantities and diversity of compounds is an opportunity in this field of natural products. Over the last years we have been observing an increase of synthetic methods to assemble this scaffold that will allow to provide new derivatives and complete the puzzle in their pharmacodynamic and pharmacokinetic profiles.
The main challenge in synthesis is the intricate stereochemistry found in the more complex structures. Although the three-component one-pot assembly of this scaffold is a very efficient procedure to obtain some of these secondary metabolites, the high temperatures involved resulted in partial epimerization. Also, the pathways concerning cyclization, particularly in the formation of the adjacent ring in the spiro derivatives are quite challenging with reversibility being observed in some reactions, while other approaches failed to obtain complex derivatives such as fiscalin A (53). The total syntheses of fourteen secondary metabolites were already accomplished but these studies did not translate into the intensification of biological activity investigations or SAR definition.
It is hoped that this review can provide medicinal chemists with a comprehensive survey on biosynthetic pathways and existing synthetic strategies merging chemistry and biology of fumiquinazoline derivatives to inspire the use of new approaches in isolation and synthesis for these innovative structures that are emerging in the war against resistance.
6 Conflicts of interest
There are no conflicts of interest to declare.
7 Abbreviations
A
Adenylation
Ac
Acetyl
ACC
1-Aminocyclopropanecarboxylic acid
AIB
2-Aminoisobutyric acid
Ala
Alanine
Ant
Anthranilic acid
ATP
Adenosine 5′-triphosphate
Bn
Benzyl
Bu
Butyl
C
Condensation
CAN
Ceric ammonium nitrate
Cbz
Benzyloxycarbonyl
CD
Circular dichroism
Δ
Reflux
DCC
N,N′-Dicyclohexylcarbodiimide
DDQ
2,3-Dichloro-5,6-dicyano-1,4-benzoquinone
DIEA
N,N-Diisopropylethylamine
DMAP
4-(N,N-Dimethylamino)pyridine
DMB
Dimethoxybenzyl
DMF
Dimethylformamide
DMI
1,3-Dimethyl-2-imidazolidinone
ED50
Half maximal dose effect
Et
Ethyl
EDAC
1-Ethyl-3-(3-dimethylaminopropyl)carbodiimide
Fmoc
Fluorenylmethyloxycarbonyl
FAD
Flavin adenine dinucleotide
Gly
Glycine
IC50
Half maximal inhibitory concentration
KHMDS
Potassium bis(trimethylsilyl)amide
LDA
Lithium diisopropylamide
Me
Methyl
MDR
Multi-drug resistance
MIC
Minimum inhibitory concentration
MRSA
Methicillin-resistant Staphylococcus aureus
NAD
Nicotinamide adenine dinucleotide
NRPS
Non-ribosomal peptide synthetase
Ph
Phenyl
Pr
Propyl
iPr
Isopropyl
py
Pyridine
rt
Room temperature
SAR
Structure–activity relationship
Ser
Serine
T
Thiolation
TES
Triethylsilyl
Tf
Trifluoromethanesulfonyl (triflyl)
TFA
Trifluoroacetic acid
THF
Tetrahydrofuran
TLC
Thin-layer chromatography
TMS
Trimethylsilyl; tetramethylsilane
Tol
Toluene
Trp
Tryptophan
UV
Ultraviolet
VRE
Vancomycin-resistant Enterococcus faecalis
8 Acknowledgements
This work was partially supported through national funds provided by FCT/MCTES – Foundation for Science and Technology from the Minister of Science, Technology and Higher Education (PIDDAC) and European Regional Development Fund (ERDF) through the COMPETE – Programa Operacional Factores de Competitividade (POFC) programme, under the Strategic Funding UID/Multi/04423/2013, the project POCI-01-0145-FEDER-028736 and PTDC/MAR-BIO/4694/2014 (reference POCI-01-0145-FEDER-016790; Project 3599 – Promover a Produção Científica e Desenvolvimento Tecnológico e a Constituição de Redes Temáticas (3599-PPCDT)) in the framework of the programme PT2020 as well as by the project INNOVMAR – Innovation and Sustainability in the Management and Exploitation of Marine Resources (reference NORTE-01-0145-FEDER-000035, within Research Line NOVELMAR), supported by North Portugal Regional Operational Programme (NORTE 2020), under the PORTUGAL 2020 Partnership Agreement, through the European Regional Development Fund (ERDF). Papichaya Boonpothong (ALFABET Scholarship) and Diana I. S. P. Resende (NOVELMAR/BPD_2/2016-019) also acknowledge for their grants.
9 Notes and references
D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRefCAS.
J. A. Beutler, Curr. Protoc. Pharmacol., 2001, 46, 9.11.11–19.11.21 Search PubMed.
J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2012, 29, 144–222 RSC.
J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2013, 30, 237–323 RSC.
J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC.
J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2015, 32, 116–211 RSC.
J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. Munro and M. R. Prinsep, Nat. Prod. Rep., 2016, 33, 382–431 RSC.
J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2017, 34, 235–294 RSC.
M. Saleem, M. S. Ali, S. Hussain, A. Jabbar, M. Ashraf and Y. S. Lee, Nat. Prod. Rep., 2007, 24, 1142–1152 RSC.
T. S. Bugni and C. M. Ireland, Nat. Prod. Rep., 2004, 21, 143–163 RSC.
M. Prata-Sena, A. A. Ramos, S. Buttachon, B. Castro-Carvalho, P. Marques, T. Dethoup, A. Kijjoa and E. Rocha, Phytother Res., 2016, 30, 1862–1871 CrossRefCAS.
U. A. Kshirsagar, Org. Biomol. Chem., 2015, 13, 9336–9352 RSC.
U. A. Kshirsagar and R. S. Rohokale, Synthesis, 2016, 48, 1253–1268 CrossRef.
C. Avendano and J. C. Menéndez, Curr. Org. Chem., 2003, 7, 149–173 CrossRefCAS.
S. Eguchi, Top. Heterocycl. Chem., 2006, 6, 113–156 CAS.
M. Demeunynck and I. Baussanne, Curr. Med. Chem., 2013, 20, 794–814 CAS.
A. Numata, C. Takahashi, T. Matsushita, T. Miyamoto, K. Kawai, Y. Usami, E. Matsumura, M. Inoue, H. Ohishi and T. Shingu, Tetrahedron Lett., 1992, 33, 1621–1624 CrossRefCAS.
B. D. Ames, X. Liu and C. T. Walsh, Biochemistry, 2010, 49, 8564–8576 CrossRefCASPubMed.
B. D. Ames, S. W. Haynes, X. Gao, B. S. Evans, N. L. Kelleher, Y. Tang and C. T. Walsh, Biochemistry, 2011, 50, 8756–8769 CrossRefCAS.
S. W. Haynes, B. D. Ames, X. Gao, Y. Tang and C. T. Walsh, Biochemistry, 2011, 50, 5668–5679 CrossRefCAS.
C. T. Walsh, S. W. Haynes, B. D. Ames, X. Gao and Y. Tang, ACS Chem. Biol., 2013, 8, 1366–1382 CrossRefCAS.
W. Xu, D. J. Gavia and Y. Tang, Nat. Prod. Rep., 2014, 31, 1474–1487 RSC.
F. Y. Lim, B. D. Ames, C. T. Walsh and N. P. Keller, Cell. Microbiol., 2014, 16, 1267–1283 CrossRefCAS.
Y. Tsunematsu, N. Ishikawa, D. Wakana, Y. Goda, H. Noguchi, H. Moriya, K. Hotta and K. Watanabe, Nat. Chem. Biol., 2013, 9, 818–825 CrossRefCAS.
K. A. O'Hanlon, L. Gallagher, M. Schrettl, C. Jöchl, K. Kavanagh, T. O. Larsen and S. Doyle, Appl. Environ. Microbiol., 2012, 78, 3166–3176 CrossRef.
X. Gao, S. W. Haynes, B. D. Ames, P. Wang, L. P. Vien, C. T. Walsh and Y. Tang, Nat. Chem. Biol., 2012, 8, 823–830 CrossRefCAS.
X. Gao, Y.-H. Chooi, B. D. Ames, P. Wang, C. T. Walsh and Y. Tang, J. Am. Chem. Soc., 2011, 133, 2729–2741 CrossRefCAS.
B. D. Ames and C. T. Walsh, Biochemistry, 2010, 49, 3351–3365 CrossRefCAS.
J. F. Sanchez, A. D. Somoza, N. P. Keller and C. C. C. Wang, Nat. Prod. Rep., 2012, 29, 351–371 RSC.
C. T. Walsh, S. W. Haynes and B. D. Ames, Nat. Prod. Rep., 2012, 29, 37–59 RSC.
P. M. Wang, T. Choera, P. Wiemann, T. Pisithkul, D. Amador-Noguez and N. P. Keller, Fungal Genet. Biol., 2016, 89, 102–113 CrossRefCAS.
S. W. Haynes, X. Gao, Y. Tang and C. T. Walsh, ACS Chem. Biol., 2013, 8, 741–748 CrossRefCAS.
C. Takahashi, T. Matsushita, M. Doi, K. Minoura, T. Shingu, Y. Kumeda and A. Numata, J. Chem. Soc. Perkin Trans. 1, 1995, 2345–2353 RSC.
T. O. Larsen, K. Frydenvang, J. C. Frisvad and C. Christophersen, J. Nat. Prod., 1998, 61, 1154–1157 CrossRefCAS.
M. G. Silva, N. A. J. C. Furtado, M. T. Pupo, M. J. V. Fonseca, S. Said, A. A. D. S. Filho and J. K. Bastos, Microbiol. Res., 2004, 159, 317–322 CrossRefCAS.
I. H. Hwang, Y. Che, D. C. Swenson, J. B. Gloer, D. T. Wicklow, S. W. Peterson and P. F. Dowd, J. Antibiot., 2016, 69, 631–636 CrossRefCAS.
M. Lie-Feng, Z. Yang, Q. Hao-Ying, W. Yuan, S. Wei-Guang and Z. Zha-Jun, J. Chem. Res., 2017, 41, 95–97 CrossRef.
F. He, Y.-L. Sun, K.-S. Liu, X.-Y. Zhang, P.-Y. Qian, Y.-F. Wang and S.-H. Qi, J. Antibiot., 2012, 65, 109–111 CrossRefCAS.
X.-X. Han, X.-Y. Xu, C.-B. Cui and Q.-Q. Gu, Chin. J. Org. Chem., 2007, 17, 232–237 CAS.
L. Liao, M. You, B. K. Chung, D.-C. Oh, K.-B. Oh and J. Shin, J. Nat. Prod., 2015, 78, 349–354 CrossRefCAS.
Y. Zhou, A. Debbab, A. Mándi, V. Wray, B. Schulz, W. E. G. Müller, M. Kassack, W. Lin, T. Kurtán, P. Proksch and A. H. Aly, Eur. J. Org. Chem., 2013, 5, 894–906 CrossRef.
S. S. Afiyatullov, O. I. Zhuravleva, A. S. Antonov, A. I. Kalinovsky, M. V. Pivkin, E. S. Menchinskaya and D. L. Aminin, Nat. Prod. Commun., 2012, 7, 497–500 CAS.
H. Tamiya, E. Ochiai, K. Kikuchi, M. Yahiro, T. Toyotome, A. Watanabe, T. Yaguchi and K. Kamei, J. Infect. Chemother., 2015, 21, 385–391 CrossRefCAS.
T. Gauthier, X. Wang, J. Dos Santos, A. Fysikopoulos, S. Tadrist, C. Canlet, M. P. Artigot, N. Loiseau, I. P. Oswald and O. Puel, PLoS One, 2012, 7, e29906 CrossRefCAS.
D. Kang, G. H. Son, H. M. Park, J. Kim, J. N. Choi, H. Y. Kim, S. Lee, S.-B. Hong and C. H. Lee, Fungal Biol., 2013, 117, 211–219 CrossRefCAS.
X.-J. Li, Q. Zhang, A.-L. Zhang and J.-M. Gao, J. Agric. Food Chem., 2012, 60, 3424–3431 CrossRefCAS.
W.-J. Lan, S.-J. Fu, M.-Y. Xu, W.-L. Liang, C.-K. Lam, G.-H. Zhong, J. Xu, D.-P. Yang and H.-J. Li, Mar. Drugs, 2016, 14, 18–30 CrossRef.
G. N. Belofsky, M. Anguera, P. R. Jensen, W. Fenical and M. Köck, Chem.–Eur. J., 2000, 6, 1355–1360 CrossRefCAS.
C.-L. Shao, R.-F. Xu, M.-Y. Wei, Z.-G. She and C.-Y. Wang, J. Nat. Prod., 2013, 76, 779–782 CrossRefCASPubMed.
N. Netz and T. Opatz, Mar. Drugs, 2015, 13, 4814–4914 CrossRefCAS.
J. P. Karwowski, M. Jackson, R. R. Rasmussen, P. E. Humphrey, J. B. Poddig, W. L. Kohl, M. H. Scherr, S. Kadam and J. B. McAlpine, J. Antibiot., 1993, 46, 374–379 CrossRefCAS.
J. E. Hochlowski, M. M. Mullally, S. G. Spanton, D. N. Whittern, P. Hill and J. B. McAlpine, J. Antibiot., 1993, 46, 380–386 CrossRefCASPubMed.
H. W. Zhang, J. Zhang, S. Hu, Z. J. Zhang, C. J. Zhu, S. W. Ng and R. X. Tan, Planta Med., 2010, 76, 1616–1621 CrossRefCASPubMed.
H.-W. Zhang, C. Ying and Y.-F. Tang, Chem. Biodivers., 2014, 11, 85–91 CrossRefCAS.
P. Mai, G. Zocher, L. Ludwig, T. Stehle and S.-M. Li, Adv. Synth. Catal., 2016, 358, 1639–1653 CrossRefCAS.
C.-Y. An, X.-M. Li, C.-S. Li, M.-H. Wang, G.-M. Xu and B.-G. Wang, Mar. Drugs, 2013, 11, 2682–2694 CrossRef.
J. Liu, X. Wei, E. L. Kim, X. Lin, X.-W. Yang, X. Zhou, B. Yang, J. H. Jung and Y. Liu, Tetrahedron, 2015, 71, 271–275 CrossRefCAS.
J. Liu, X. Wei, E. L. Kim, X. Lin, X.-W. Yang, X. Zhou, B. Yang, J. H. Jung and Y. Liu, Org. Lett., 2014, 16, 2574–2577 CrossRefCAS.
J. Penn, P. G. Mantle, J. N. Bilton and R. N. Sheppard, J. Chem. Soc. Perkin Trans. 1, 1992, 1495–1496 RSC.
Y. Liu, X.-M. Li, L.-H. Meng and B.-G. Wang, Phytochem. Lett., 2014, 10, 145–148 CrossRefCAS.
Y.-Q. Fan, P.-H. Li, Y.-X. Chao, H. Chen, N. Du, Q.-X. He and K.-C. Liu, Mar. Drugs, 2015, 13, 6489–6504 CrossRefCASPubMed.
Y. Zhuang, X. Teng, Y. Wang, P. Liu, H. Wang, J. Li, G. Li and W. Zhu, Tetrahedron, 2011, 67, 7085–7089 CrossRefCAS.
J. Peng, T. Lin, W. Wang, Z. Xin, T. Zhu, Q. Gu and D. Li, J. Nat. Prod., 2013, 76, 1133–1140 CrossRefCASPubMed.
J. Penn, M. Purcell and P. G. Mantle, FEMS Microbiol. Lett., 1992, 92, 229–233 CrossRefCAS.
R. Finking and M. A. Marahiel, Annu. Rev. Microbiol., 2004, 58, 453–488 CrossRefCASPubMed.
Y. Zhuang, X. Teng, Y. Wang, P. Liu, G. Li and W. Zhu, Org. Lett., 2011, 13, 1130–1133 CrossRefCAS.

 ab,
Papichaya
Boonpothong‡
a,
Emília
Sousa
ab,
Papichaya
Boonpothong‡
a,
Emília
Sousa


![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :
: