
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
The many faces and important roles of protein–protein interactions during non-ribosomal peptide synthesis
Thierry
Izoré
 ab and
Max J.
Cryle
*abc
ab and
Max J.
Cryle
*abc
aThe Monash Biomedicine Discovery Institute, Department of Biochemistry and Molecular Biology and ARC Centre of Excellence in Advanced Molecular Imaging, Monash University, Clayton, Victoria 3800, Australia. E-mail: max.cryle@monash.edu
bEMBL Australia, Monash University, Clayton, Victoria 3800, Australia
cDepartment of Biomolecular Mechanisms, Max-Planck Institute for Medical Research, 69120 Heidelberg, Germany. E-mail: max.cryle@mpimf-heidelberg.mpg.de
First published on 12th September 2018
Abstract
Covering: up to July 2018
Non-ribosomal peptide synthetase (NRPS) machineries are complex, multi-domain proteins that are responsible for the biosynthesis of many important, peptide-derived compounds. By decoupling peptide synthesis from the ribosome, NRPS assembly lines are able to access a significant pool of amino acid monomers for peptide synthesis. This is combined with a modular protein architecture that allows for great variation in stereochemistry, peptide length, cyclisation state and further modifications. The architecture of NRPS assembly lines relies upon a repetitive set of catalytic domains, which are organised into modules responsible for amino acid incorporation. Central to NRPS-mediated biosynthesis is the carrier protein (CP) domain, to which all intermediates following initial monomer activation are bound during peptide synthesis up until the final handover to the thioesterase domain that cleaves the mature peptide from the NRPS. This mechanism makes understanding the protein–protein interactions that occur between different NRPS domains during peptide biosynthesis of crucial importance to understanding overall NRPS function. This endeavour is also highly challenging due to the inherent flexibility and dynamics of NRPS systems. In this review, we present the current state of understanding of the protein–protein interactions that govern NRPS-mediated biosynthesis, with a focus on insights gained from structural studies relating to CP domain interactions within these impressive peptide assembly lines.
 Thierry Izoré | TI completed his Ph.D. at the University of Grenoble where he focussed on structural studies of bacterial virulence factors (mainly Gram+ pilus and type III secretion systems) under the supervision of Andréa Dessen. He then joined the group of Jan Löwe at the Laboratory of Molecular Biology in Cambridge to study bacterial and archaeal cytoskeletons using Cryo-Electron microscopy. In November 2016 he moved to Monash University in Australia to work with Max Cryle on the structural biology of NRPS assembly lines. His research efforts concentrate on the complex structures formed by these medically relevant mega-enzymes. |
 Max J. Cryle | MJC obtained his Ph.D. in chemistry from the University of Queensland in 2006. He then moved to the Max Planck Institute for Medical Research in Heidelberg Germany as an HFSP Cross-Disciplinary Fellow and later as an Emmy Noether group leader funded by the DFG. Since 2016 he is an EMBL Australia group leader based within the Biomedicine Discovery Institute at Monash University, where his team focusses on understanding antibiotic biosynthesis (with particular interest in non-ribosomal peptide synthesis) as well as developing new antibiotics. Since 2018 he is funded as a Career Development Fellow by the National Health and Medical Research Council. |
1. Introduction
Non-ribosomal peptide synthetases (NRPSs) are ribosomally independent macromolecular machineries involved in the production of many different classes of peptide-derived natural products.1 The utility of NRPS machineries for producing bioactive peptides stems from two main sources – firstly, the ability to select a wide range of monomers for use in peptide synthesis, and secondly the significant modifications able to be performed to the peptide by a range of catalytic domains found within a specific NRPS. Unlike ribosomes that strictly use a defined set of amino acids to produce peptides and proteins, the NRPS system has evolved to become extremely versatile regarding the substrate monomers that it accepts. Indeed, it has been reported that more than 500 different substrates are accepted by NRPS assembly lines.2 In addition to the 20 well-known L-α-amino acids incorporated in proteins, these building blocks include a broad range of non-proteinogenic α- and β-amino acids3 (including examples such as phenylglycines, cyclic guanidines, alkene-, alkyne-, halo-, hydroxyl- or cyclopropyl-containing amino acids)2 and extend even to homologated amino acids or monomers derived from aminobenzoic acid residues:4 this variety of potential substrates leads to a vast number of conceivable final peptide products. Together with the related polyketide synthase (PKS) systems, megasynthase machineries represent one of the best sources of biologically active compounds for exploitation in medicine. Along with the classic example of peptide production by ACV synthase during penicillin biosynthesis,5 other NRPS-assembled peptides have been found to play diverse roles, with examples including those that act as siderophores, antibiotics (including clinically relevant examples like the glycopeptide antibiotics (GPAs) and daptomycin), cytostatic agents or as regulators in bacterial quorum sensing.6,7The structure of an NRPS plays a vital role in its function. A typical linear NRPS machinery is composed of multiple modules (∼100–200 kDa, depending on domain composition), with each module in the assembly line responsible for the incorporation of one amino acid monomer into the final peptide (Fig. 1). Differing architectures to linear systems include iterative and non-linear NRPS systems: iterative systems possess a limited number of modules with the final peptide being the result of several copies of a shorter peptide fragment, whilst non-linear systems are more complex and less easily defined in terms of traditional modules.1 Whilst both iterative and non-linear NRPS machineries are intriguing from a mechanistic standpoint, the vast majority of medicinally important NRPS-products are produced by linear NRPS machineries. Each module within a linear NRPS can be further described as a series of at least three different domains: the adenylation domain (or A-domain) is responsible for the selection and the activation of amino acids; the peptidyl carrier protein domain (PCP, also known as a thiolation domain) whose role is to shuttle substrates between different domains; and the condensation domain (or C-domain) that catalyses peptide bond formation between PCP bound amino acid monomers and peptides (or amino acids for the initial NRPS module). In addition to these three essential domains, a module can harbour additional domains: examples include epimerisation domains (E-domains), S-adenosylmethionine (SAM)-dependent methyltransferase domains or formylation domains,8 adding yet further potential diversity to the structure of the final NRPS peptide product. The final module of most NRPS machineries contains a terminal thioesterase domain (or TE-domain) that exerts its catalytic activity in releasing the final PCP-bound peptide: this process can also serve to introduce yet further diversity into the final peptide structure, with one common example the cyclisation of a linear peptide. NRPS assembly lines range from a single module as in the Pseudomonas pyreudione synthetase9 up to 18 modules in length and can be encoded by one or more genes. The two longest NRPS machineries encoded in a single gene are kolossin A synthase from Photorhabdus luminescens, an impressive 15 module-long NRPS enzyme,10 and the peptaibol synthetase from Trichoderma virens with 18 modules.11
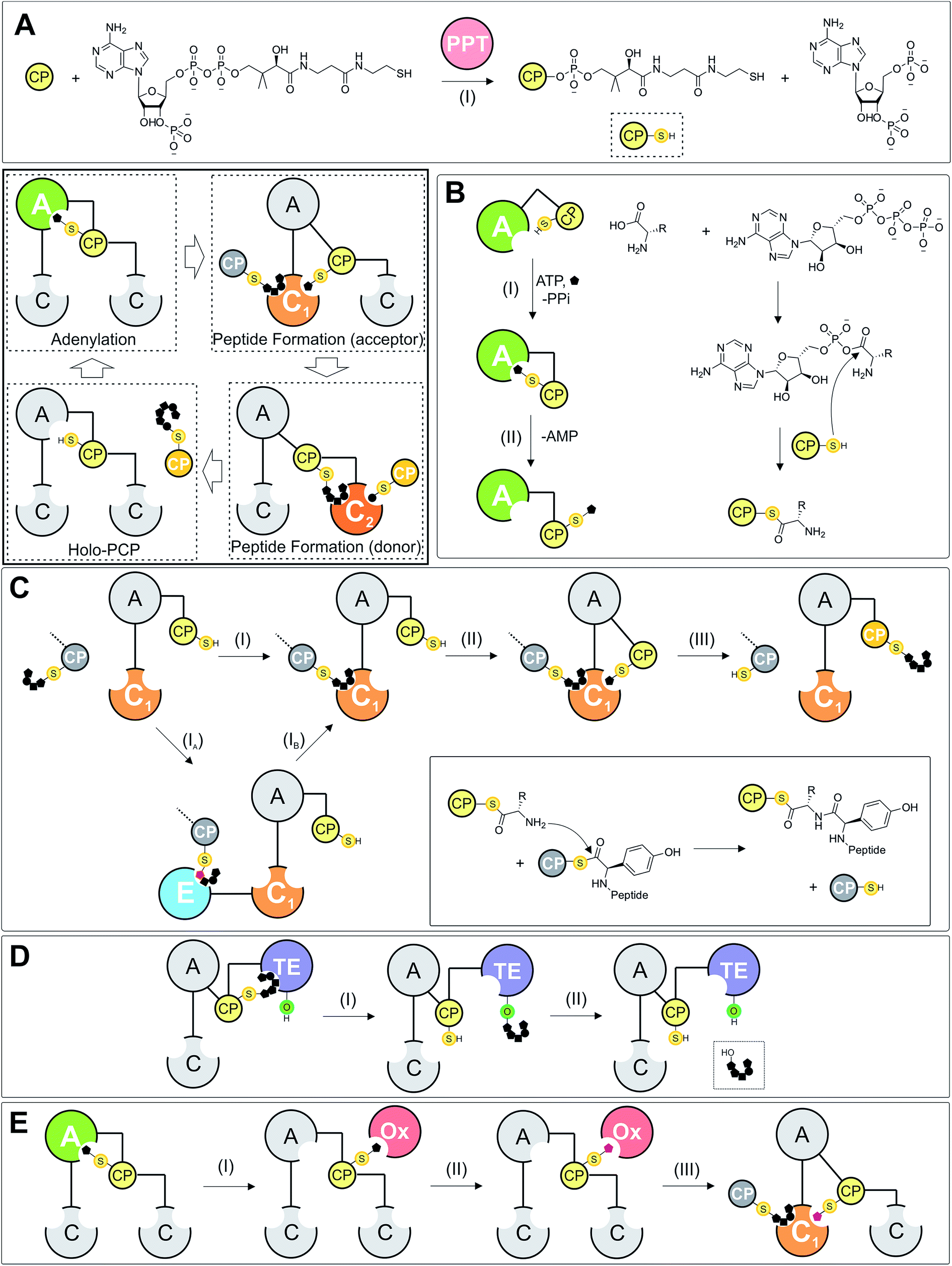 | ||
| Fig. 1 Schematic representation of NRPS-catalysis. (A) Phosphopantetheinyl-transferases (PPTs) perform the initial modification of the PCP (CP) into its holo-form by transferring a PPant-moiety onto the conserved serine of the PCP (CP). (B) Adenylation of amino acid monomers are catalysed by A-domains: following selection and adenylation using ATP, the activated monomer is then loaded onto the PPant moiety of the CP via a thiolation reaction. (C) Condensation domains catalyse peptide bond formation during NRPS mediated peptide synthesis and possess two CP binding sites, known as the donor and acceptor sites. When both sites are occupied by their cognate loaded CP, C-domains are then able to catalyse peptide bond formation between the two CP-bound substrates. Epimerisation (E)-domains are often found in NRPS assembly lines and catalyse the epimerisation of the C-terminal residue of CP-bound peptides for subsequent incorporation within the growing peptide chain by a neighbouring C-domain. (D) The majority of NRPS assembly lines end with a thioesterase (TE) domain that performs the release of the fully-grown peptide; many possible routes to peptide cleavage have been reported in addition to the hydrolytic mechanism shown here. (E) CP-bound amino acid monomers can also be modified during NRPS-mediated peptide assembly in trans by additional biosynthetic enzymes (for example P450 monooxygenases [Ox], halogenases or other enzymes) in a process that must occur prior to the modified, CP-bound monomer being accepted as an acceptor substrate by the neighbouring C-domain. Peptide positioning shown in this schematic figure is indicative only of binding, not specific positioning within the catalytic domains of the NRPS. | ||
Although NRPSs are mostly found in bacteria and fungi and where the product peptides provide a fitness advantage to the producing host, some rare examples of NRPS-like enzymes do exist in higher eukaryotes. However, those that have been identified display altered architectures when compared to standard NRPS machineries, since such machineries are typically composed of an A-domain, PCP-domain and a dedicated C-terminal catalytic domain that is specific to each NRPS.12 Examples of those eukaryotic NRPS are the 2-aminoadipic 6-semialdehyde dehydrogenases (AASDH) involved in lysine metabolism13 and the carcinine and β-alanyl-dopamine synthetase Ebony involved in neurotransmitter recycling and cuticle sclerotisation in insects.14
Given that some of our most critical medicines such as last-resort antibiotics (e.g. vancomycin, daptomycin), anti-tumour drugs (bleomycin, cryptophycin) or immune modulatory compounds (cyclosporine) are NRPS-produced and remain tied to biosynthesis for their production, great interest exists in understanding their biosynthesis.15 Furthermore, the potential to produce new derivatives of such compounds in the future requires effective assembly line reengineering. Because of this, significant efforts have been made in order to understand how NRPS machineries function and the overall structure of these peptide assembly lines.16 Structural data are available for examples of each catalytic domain from standard NRPS machineries – these have been obtained either by NMR, X-ray crystallography or a combination of these techniques. Despite the wealth of information derived from structures of isolated domains, understanding NRPS synthesis as a whole requires imaging domain–domain interactions, module–module interactions and even module–module interactions between modules separated across different proteins. To facilitate the latter, it has been proposed that multi-chain NRPS machineries interact through “communication/docking domains”:17–20 although limited data is available concerning these domains, there is evidence that such interactions can be mediated through a hand/helix interaction.21 Of all domains present in NRPS assembly lines, PCPs are the ones involved in the most numerous interactions. As a domain involved in substrate shuttling, PCPs need to efficiently interact with catalytic domains from both upstream and downstream modules to ensure the effective transfer of amino acids and peptides along the NRPS machinery. Given the essential role of PCPs in all NRPS-processes, this review will summarise the structural data available for NRPS systems with a focus on PCPs and their interaction with other domains of the NRPS assembly line.
2. PCP – the central domain in NRPS mediated synthesis
Carrier protein domains are essential in many processes found in both primary and secondary metabolism.22 In NRPS systems, PCPs play a central role in shuttling amino acids and peptides between different catalytic domains and can also serve as a platform to present the amino acid or peptide chain to in-trans modification enzymes, such as halogenases, transferases or monooxygenase enzymes (Fig. 1E). Typical NRPS assembly lines contain one PCP per module, meaning there are the same number of PCP domains as monomers in the final peptide product of the assembly line. Before they can function in this role, PCPs first require activation by post-translational modification: the inactive apo-form of the PCP is the substrate of a phosphopantetheinyl transferase (PPTase) that catalyses the transfer of a phosphopantetheine moiety (PPant) onto an invariant serine residue located at the start of PCP helix-2 (Fig. 1A). This PPant moiety is derived from coenzyme A (CoASH) and terminates with a reactive thiol group: peptides and amino acids can then be temporarily loaded onto the PCP via this arm in the form of a reactive thioester, which also acts as a flexible arm to allow these tethered substrates to reach the catalytic sites within NRPS domains.Whilst the exact timing of phosphopantetheine modification is not yet known, the highly similar structures of the unmodified apo-form and phosphopantetheine modified holo-form (vide infra) suggest that this modification does not need to be performed co-translationally in order for successful NRPS protein synthesis by the ribosome. PCPs are related to the acyl carrier protein (ACP) domains found in megaenzyme synthases such as fatty acyl synthases and the polyketide synthases, which also adopt a similar fold and require post-translational addition of a phosphopantetheine moiety. One important difference between ACPs and most PCPs is the sequestration of acyl chains within the core of ACPs, which is not observed for PCP-bound substrates (some examples of bound substrate interaction with the PCP have been identified, although these appear to be more on the surface of the PCP).23,24 Substrate sequestration can also lead to minor alterations in ACP structure due to the perturbation of the helical core of the ACP by the substrate.25 A further consequence of ACP sequestration of substrates is that this requires interacting domains to invoke a chain flipping mechanism in order to allow the bound substrates to engage with these interacting domains,26 which is not required for PCPs during NRPS biosynthesis.27
Due to their small size (typically smaller than 100 amino acids, ∼10 kDa) the fold of isolated PCPs has mainly been studied by solution-state NMR,24,28–30 although four structures are also available from X-ray diffraction experiments (ref. 31–33 and unpublished structure 4HKG). These studies have revealed that the PCP folds as a four helix-bundle, with the two N-terminal helices the longest and the remaining helices typically shorter in length (see Fig. 2). All four helices are mostly amphipathic and the hydrophobic sides serve as the interaction surface holding the bundle together. Helices 1 and 2 mostly run parallel to one another and form the back of the domain whilst the shorter helices 3 and 4 pack against the two N-terminal helices. It is also apparent that if helix 4 aligns in a parallel fashion to helices 1 and 2, helix 3 then adopts a perpendicular orientation in regards to the other helices. A long loop connects helices 1 and 2, with the post-translationally modified serine located at the start of helix 2.34 Although not visible on the NMR structures, all crystal structures of PCPs obtained so far (mainly as multidomain structures, vide infra) contain a small additional helix (labelled 1′ in Fig. 2) between helices 1 and 2. In early studies, Koglin et al.30 reported that PCPs adopted different conformations depending on the assembly line catalytic state. As indicated previously, PCPs first need to be “activated” by attachment of a PPant cofactor; it was postulated therefore that in order to reach the different domains with which it needs to interact (i.e. adenylation-, condensation-, thioesterase-domains), the PCP would change conformation depending on its loaded state. This would mean that the PCP would itself “drive” the synthesis machinery by actively shuttling substrates from one catalytic domain to the other. However, this model was first challenged with the crystal structure of BlmI PCP33 and in the light of many more recent studies it is now clear that the PCP can in fact be thought of as a rather rigid domain, with only slight differences between the different catalytic states of the NRPS machinery. This can be seen from a superposition of all available PCP structures deposited into the Protein Data Bank (PDB) in which the PCP has been studied in isolation: here, it is clear that – irrespective of its modification state (apo or holo) – the fold of the PCP remains consistent. To explore whether the PCP undergoes changes in structure during NRPS synthesis (especially considering new multi-domain and full module NRPS structures), a structural alignment of the PCPs from 18 structures was performed. This pool of structures (2VSQ,352FQ1,342JGP,362ROQ,373RG2,384IZ6,394ZXH,404ZXI,405ISW,415ISX,415JA1,425JA2,425T3D,405U89,434DG9,444PWV,455ES8,465EJD,47Fig. 2D and E) represents the conformation of PCPs while interacting with different domains – specifically isochorismate lyase, condensation, thioesterase, adenylation and epimerisation domains. PCPs included in the structural alignment readily superimposed and even the two most dissimilar domains (4XZ1 and 2VSQ) still aligned with a relatively low RMSD value (3.1 Å; calculated using the structure comparison server DALI48). Even though these two PCPs belong to different organisms and are involved in different interactions (4XZ1: thioester-forming state and 2VSQ: condensation state), the structural differences observed in these two cases (centred on helices 1 and 4) are most probably the result of the upstream/downstream linker regions (Fig. 2E). Thus, all available structural evidence to date indicates that PCPs do not undergoing major structural changes during interactions with either their cognate PPant transferase or catalytic NRPS domains. Rather, the motion of PCPs during NRPS-mediated synthesis appears linked to the different states adopted by the adenylation domains (vide infra), which in turn alters the positioning of PCPs due to their close attachment to the mobile subdomain of the A-domain (Asub).
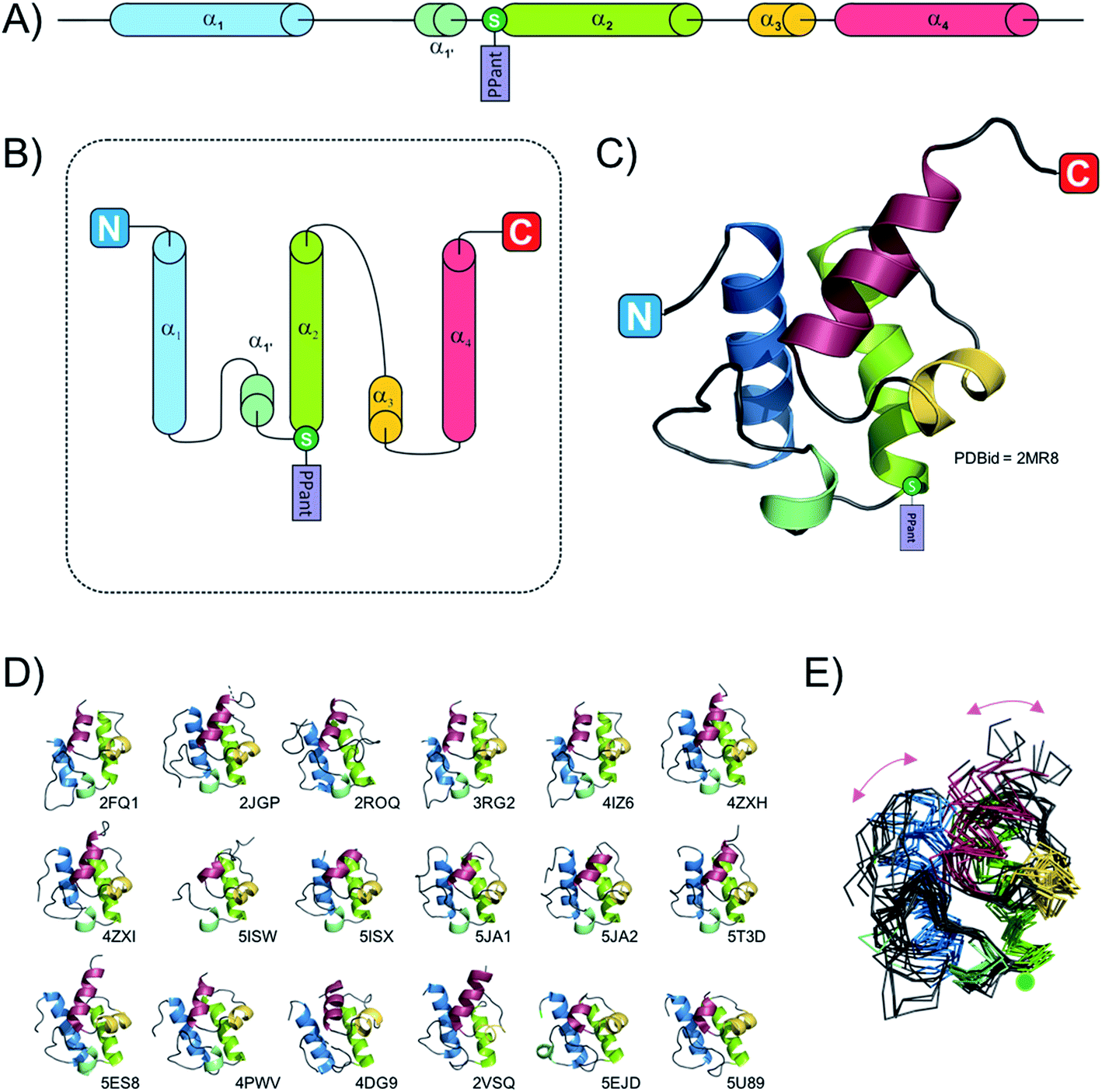 | ||
| Fig. 2 Schematic representation of the PCP domain. (A) linear representation of typical PCP secondary structure. (B) Topology diagram of a canonical PCP. (C) Crystal structure of a PCP from module seven of the teicoplanin-producing NRPS machinery, coloured according to panels (A) and (B). The circled “S” represents the position of the conserved serine residue that bears the phosphopantetheine group (PPant) added after protein synthesis. (D) Gallery of all 18 PCPs utilised in the structural alignment performed within this review. (E) Structural alignment (superposition) of the 18 PCPs shown in panel (D). | ||
Because PCPs play the role of substrate shuttles during peptide synthesis, they need to interact with catalytic domains from both upstream and downstream modules. Owing to this requirement, it could be anticipated that PCPs would not be very specific when it comes to binding other NRPS domains. However, studies from the Ackerley group have cast doubt on such assertions, as their results indicate that PCPs normally found to interact with a condensation domain are unable to interact effectively with a thioesterase (TE) domain in an reengineered NRPS machinery.49 Thus, in order to understand the grounds for the specificity of PCPs, we will now discuss the interaction network between PCPs and other interacting domains based on the structural evidence currently available. To assist the reader, a table of all structures mentioned in this review is included in the text (Table 1).
| PDB code | Protein name | UniProt ID Number | References |
|---|---|---|---|
| 2N5H | PltL-holo | Q4KCZ1 | 24 |
| 2N5L | PltL-pyrrolyl | Q4KCZ1 | 24 |
| 5U3H | PCP1 yersiniabactin | Q7CI41 | 28 |
| 2MR7 | PCP7 teicoplanin | Q70AZ6 | 29 |
| 2MR8 | PCP7 teicoplanin | Q70AZ6 | 29 |
| 2GDW | PCP TycC3 A/H state | O30409 | 30 |
| 2GDX | PCP TycC3 H state | O30409 | 30 |
| 2GDY | PCP TycC3 A state | O30409 | 30 |
| 1DV5 | PCP DltC | P55153 | 31 |
| 4BPH | PCP DltC | P39579 | 32 |
| 4HKG | PCP PksN | V5VHR7 | Non published |
| 2MY6 | PCP KstB | A0A023GUP0 | Non published |
| 4NEO | PCP BlmI | Q9XC48 | 33 |
| 2FQ1 | EntB | P0ADI4 | 34 |
| 2VSQ | SrfA-C | Q08787 | 35 |
| 2JGP | PCP-C TycC5-6 | O30409 | 36 |
| 2ROQ | PCP-TE EntF | P11454 | 37 |
| 3RG2 | A-ACP EntE-EntB | P10378/P0ADI4 | 38 |
| 4IZ6 | A-ACP EntE-EntB | P10378/P0ADI4 | 39 |
| 4ZXH/4ZXI | AB3404 | A0A0X1KH98 | 40 |
| 5ISW/5ISX | Apo/holo PCP-E (GrsA) | P0C062 | 41 |
| 5JA1 | EntF + YbdZ | P11454 + P18393 | 42 |
| 5JA2 | EntF + PA2412 | P11454 + Q9I169 | 42 |
| 5T3D | Holo EntF | P11454 | 40 |
| 5U89 | DhbF | A0A0F6BHX2 | 43 |
| 4DG9 | Holo PA1221 | Q9I4B7 | 44 |
| 4PWV | PCP + P450 skyllamycin | F2YRY5 + F2YRY7 | 45 |
| 5ES8 | LgrA (thiolation state) | Q70LM7 | 46 |
| 5ES5 | LgrA (open and closed states) | Q70LM7 | 46 |
| 5ES9 | LgrA (formylation state) | Q70LM7 | 46 |
| 5EJD | PCP-CT TqaA | F1CWE4 | 47 |
| 1QR0 | Sfp | P39135 | 51 |
| 4MRT | Sfp + PCP (TycC) | O30409 | 52 |
| 4D4I | ApnA A1 | G0WVH3 | 57 |
| 1AMU | PheA | P0C061 | 58 |
| 5WMM | TioS | Q333U7 | 67 |
| 2PST | PA2412 | Q9I169 | 69 |
| 1LC1 | Firefly luciferase | P08659 | 72 |
| 3E7W | DltA | P39581 | 73 |
| 3CW8 | 4-Chlorobenzoate: coA ligase | Q8GN86 | 74 |
| 3FCC | DltA | Q81G39 | 76 |
| 3DLP | 4-Chlorobenzoate: coA ligase | Q8GN86 | 77 |
| 1L5A | VibH | Q9KTV9 | 83 |
| 3CLA | Chloramphenicol acetyltransferase | P00484 | 84 |
| 4JN3 | C1 CDA synthase | Q9Z4X6 | 86 |
| 5DU9 | C1 CDA synthase | Q9Z4X6 | 95 |
| 2XHG | E-Domain TycA | G1K3P2 | 97 |
| 1JMK | TE surfactin | Q08787 | 104 |
| 2CB9 | TE fengycin | Q45563 | 105 |
| 2RON | TE surfactin | Q08788 | 106 |
| 3QMV | TE prodiginine | O54157 | 107 |
| 1KEZ | TE DEBS | Q03133 | 109 |
| 3TEJ | PCP – TE surfactin | P11454 | 110 |
| 3MGX | OxyD vancomycin | Q939Y1 | 113 |
| 3EJB | P450 + ACP biotin | P53554 + P0A6A8 | 116 |
| 4TX3 | X + OxyB teicoplanin | Q70AY8 + Q6ZZJ3 | 121 |
| 4TX2 | X-domain teicoplanin | Q70AY8 | 121 |
3. Activation of the PCP-domain through interaction with a PPTase
The PPTase superfamily was discovered in 199650 and Group II PPTases are involved in the activation of NRPS PCPs (Fig. 3). In contrast to type I and III PPTases, group II PPTases do not oligomerise, but rather are expressed as fused pseudo-dimers (Fig. 3C).51 The first structure of a PPTase in complex with a PCP was solved with the PPTase Sfp, which is required for the activation of the PCP-domains of the surfactin synthetase in B. subtilis.52 In this structure, solved initially by NMR and then later confirmed by X-ray crystallography, Sfp adopts – as expected – a pseudo-dimeric fold where each “monomer” is comprised of a 3-stranded beta-sheet core flanked by three alpha helices. A small additional 2-stranded beta-sheet then covers each “monomer”. Due to the promiscuity of Sfp which needs to interact and activate many different PCPs in the surfactin NRPS, the binding interface between the two proteins does not involve many residues. However, two clear interaction sites have been localised, including one hydrogen bond between PCP backbone carbonyl oxygen of Gln40 and the amide hydrogen from Sfp Tyr36 (Fig. 3E); the rest of the interaction network relies on hydrophobic residues. Sfp displays a small hydrophobic cavity made of residues from the first helix and the preceding loop as well as the third helix and the subsequent loop. In the complex structures, the side chains of Leu46 and Met49 from helix-2 of the PCP occupy this cavity (Fig. 3D). Of interest, multiple sequence alignment of both type-II PPTases and NRPS PCP-domains shows that these hydrophobic positions are widely conserved.53 For these reasons, it is hypothesized that this recognition pattern is widely spread in all NRPS systems, which also aids in explaining the general utility of Sfp for the modification of NRPS PCPs from multiple systems.53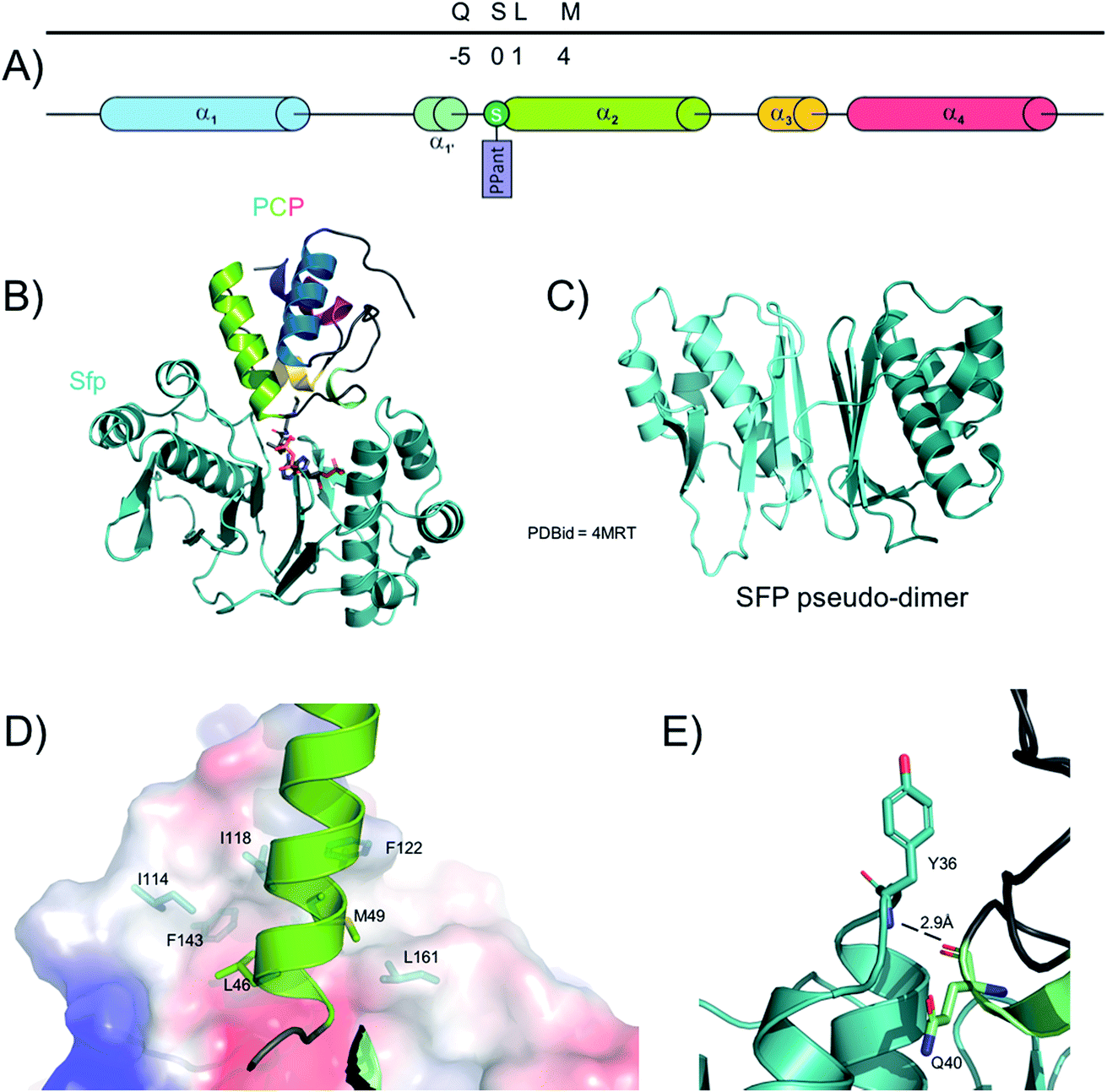 | ||
| Fig. 3 Interaction between the phosphopantetheinyl-transferase Sfp and a PCP from the tyrocidine synthase NRPS. (A) linear localisation of PCP residues involved in interactions with Sfp. (B) Crystal structure of the PCP-Sfp complex. (C) The crystal structure of Sfp showing the pseudo-dimeric fold of this protein. (D) Structural representation of the hydrophobic interactions identified in the Sfp/PCP complex (PCP represented as cartoon, Sfp shown as a protein surface, residues shown as sticks). (E) The single hydrogen bond identified in the Sfp/PCP complex. | ||
4. A-domains – amino acid selection and activation
4.1 Function of adenylation domains
NRPS adenylation domains (or A-domains) belong to a large family of adenylate-forming enzymes (ANL superfamily, class 1a; ∼500 residues, ∼55 kDa) and are key domains within an NRPS assembly line as they are essential for the selection and activation of monomer units for incorporation into the growing peptide.54,55 In a multi-step process, the A-domain first selects the amino acid to be added to the growing peptide chain, then activates this residue via adenylation (ATP + amino acid → aminoacyl adenylate + PPi, Fig. 1B) to make it a competent substrate for the last step – the transfer of the amino acid to the thiol group of the PPant prosthetic group of the PCP. The first step includes the specific recognition of the cognate amino acid by the catalytic pocket of the A-domain. Although typically specific for one substrate, some A-domains have been reported to select several amino acids: examples include the first A-domain of the nostopeptolide A hybrid PKS-NRPS machinery that can bind and activate three branched hydrophobic residues: isoleucine, leucine and valine.56 Another example is the first A-domain of the anabaenopeptin synthetase from Planktothrix agardhii (ApnA A1) that has been shown to activate structurally different amino acids: arginine and tyrosine.57 In this case, X-ray crystallographic studies have revealed that the source of the dual activity is the fact that the arginine residue adopts a conformation that mimics that of a tyrosine within the ApnA A1 catalytic pocket. The specificity of these A-domains for several amino acids could offer great advantages to the source organism since different peptides are produced from only one assembly line, although in both cases the use of proteinogenic amino acids would limit the ability of the organism to select for one amino acid over another.The amazing diversity of non-ribosomal peptides is to a large extent due to the significant diversity of molecules recognised by A-domains. In 1997 the structure of the phenylalanine-activating A-domain PheA from gramicidin synthetase 1 was solved,58 which led to the identification of a specificity code allowing the prediction of the monomer an A-domain would accept as a substrate.59 The refinement of this code and its utility led to the emergence of valuable web-based programs capable of predicting the substrate that an A-domain will accept (see websites NRPSpredictor2 (ref. 60) or NRPS/PKS substrate predictor61). Additionally, the interactions between A-domains and other NRPS domains, such as condensation (C) domains, have recently been shown to alter the selectivity of certain A-domains – this not only indicates the importance of protein–protein interactions in NRPS machineries but also implies that NRPS activity should be assessed in complete modules where possible.62,63
In addition to the great diversity of possible substrates, some A-domains – referred to as “interrupted A-domains” – harbour insertions of modification domains (called auxiliary domains) involved in the alteration of the selected substrate. Auxiliary domains typically exhibit methyltransferase, ketoreductase, oxidase or monooxygenase activities.† Significant research has been performed on auxiliary domains and it has been shown that it is possible to generate a bi-functional A-domain by inserting the sequence of an auxiliary domain in a standard A-domain.65 Similarly, it is possible to generate an uninterrupted A-domain from an interrupted one by deleting the sequence coding for the auxiliary domain.65,66 The potential for engineering specific gain/loss of function within A-domains through the use of auxiliary domains is of great value for reengineering these domains, helping pave the way to generating novel compounds through simple modifications to NRPS machineries. The recent structure of TioS, a natural A-domain interrupted by a methyltransferase auxiliary domain, is the first example of such architecture. In this structure, the adenylation catalytic site is located 60 Å away from the methylation site, which again highlights the crucial role of PCP-bound peptide shuttling in NRPS systems.67
A-domains are often found to form an essential complex with small proteins (c.a. 70 residues), named MbtH-like proteins (MLPs) due to their discovery in M. tuberculosis. MLPs have a consensus sequence identified as NXEXQXSXWPX5PXGWX13LX7WTDXRP68 and share a conserved, relatively flat fold consisting of a core central beta-sheet with three strands covered by a single alpha-helix; some MLPs also possess an extra C-terminal alpha-helix as seen in the first crystal structure of PA2412.69 The role of MLPs is still not fully understood since they have proven to be essential for the activity or solubility of some A-domains69,70 and totally dispensable for others.71
4.2 Structure of adenylation domains
A number of crystal structures of A-domains have been deposited in recent years, which indicate that A-domains share a consistent fold similar to that of firefly luciferase that also catalyses a similar adenylation reaction.58,72 The A-domain fold comprises two distinct domains: a larger N-terminal domain (∼400 residues), also known as Acore and a smaller C-terminal domain (∼100 residues), known as Asub (Fig. 4B) linked together through a small “hinge” loop.40,73 Although the large N-terminal core domain is relatively well constrained, the smaller C-terminal domain has been shown to rotate substantially relative to the Acore. Crystal structures of A-domains in different catalytic states have revealed what is commonly referred to as the “A-domain cycle”, or “domain alternation”. Adenylation domains have been shown to exist in at least two different catalytically relevant states,40,46,73,74 with in solution studies confirming this general mechanism whilst also indicating the potential for different catalytic states to exist in mixed conformations as the reaction progresses.75 One of the clearest examples of domain alternation in an A-domain has been demonstrated for LgrA (linear gramicidin synthetase A) as reported by Reimer et al. in 2016.46 Although structures of A-domains in different catalytic states were available before, Reimer et al. provided snapshots of these catalytic states from the same machinery – LgrA (Fig. 4B) In addition to providing data on the A-domain catalytic cycle, these structures also show how the newly activated amino acid is subsequently passed to a tailoring domain (formylation/F-domain) that adds a formyl group to the amino acid N-terminus via the movement of the PCP. Of further interest, the Acore- and F-domain adopt a very similar conformation in all four structures reported for LgrA. These two domains possess an interaction surface of around 830 Å2 (similar in area to the A/C domain interaction (see below)), which even if not very extensive appears to be sufficient to maintain a constant relative orientation throughout the catalytic cycle of the A-domain. The interaction between the two domains is mostly hydrophobic in nature: specific interactions of note include Phe172 from the formylation domain docking within a hydrophobic cleft in the A-domain formed by Leu516 and Leu520, with other important interactions involving Leu522-Leu187 (residues are from A-domain to F-domain respectively) and Leu184 docking into a hydrophobic cavity onto the F-domain formed by Phe87 and Trp88 (Fig. 4B and D). Although the Acore–F interaction stays consistent throughout the catalytic cycle, the position of Asub, is highly variable. The Asub domain can be seen as a lid above the Acore, controlling access of the substrates and downstream PCP-domain to the catalytic site. At the beginning of the A-domain catalytic cycle, Asub is located above Acore leading to an “open” conformation (Fig. 4B). In this conformation, the active site is accessible to the substrates (amino acid, ATP, Mg2+). Upon substrate binding, the Asub plays a role as a lid and closes access to the catalytic site, hence forming the closed-state (or adenylate-forming state) by rotating by approximately 30 degrees. In doing so, the final loop of Asub moves deeper into the Acore and positions a conserved, essential lysine residue (Lys672 in LgrA) in close proximity to the catalytic site. This lysine residue not only serves to stabilise both the amino acid substrate and ATP but it also stabilises the highly negatively charged reaction intermediate, making it a key component of the active site.76 Completion of the adenylation reaction then triggers the rotation of the Asub domain by around 140 degrees. This considerable rotation allows the release of PPi and drives the PCP-domain to dock onto Acore, thus forming the thiolation state (Fig. 4B and E) (the interaction between the PCP-domain and the A-domain will be detailed in the next section). In the thiolation state, the prosthetic PPant group attached to the PCP-domain is then loaded with the amino acid via a thiolation reaction. After this reaction has been completed, the PCP needs to shuttle its cargo to the formylation domain first, covering a distance of roughly 60 Å and a further rotation of 75 degrees. This is made possible due to the motion of the Asub domain and the effect this has on the neighbouring, linked PCP. With this step, the cycle can reset at step one with the Asub domain in the open conformation (ready for substrates to bind). Of further significance is the structure of AB3404,40 in which a termination module is found in the condensation state (holo-PCP bound to the C-domain) whilst the A-domain is present in the adenylate-forming state. This shows how NRPS machineries have evolved to be an efficient, coupled system in which two catalytic domains are active at the same time within the same module. The residues composing the hinge between Acore and Asub are therefore of extreme importance for the proper continuing of the catalytic cycle: for example, mutation of a hinge residue (invariantly an aspartic acid or a lysine in the linker sequence between the Acore and the Asub) into a proline residue was sufficient to constrain the A-domain in the adenylation-forming state and halt the catalytic cycle in such a mutant.77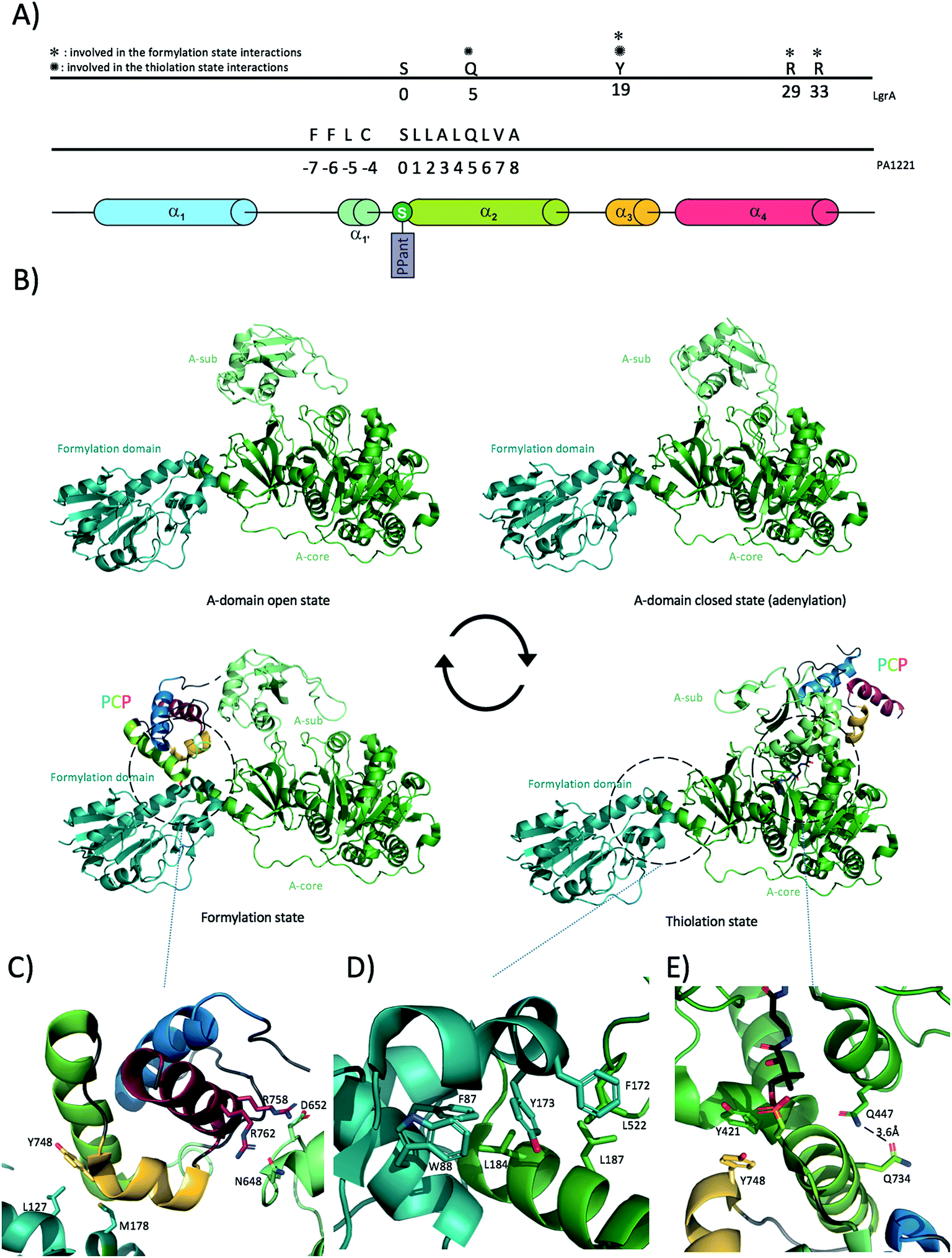 | ||
| Fig. 4 Interaction between PCP and A-domains. (A) Linear localisation of PCP residues involved in interactions with A-domains. (B) Alternation cycle of the LgrA A-domain results in different localisation of the PCP, showing LgrA in the adenylation, thiolation and formylation states. (C) Close-up view of the interactions between the LgrA PCP and A-domains in the formylation state. (D) Close-up view of the interactions between LgrA A- and F-domains. (E) Close-up view of the interactions between PCP and A-domain of LgrA in the thiolation state (PPant arm coloured in black). | ||
4.3 Adenylation domain interactions with PCPs
The transfer of the adenylated substrate onto the PCP needs to be efficiently controlled as the accidental release of such highly reactive intermediates could lead to non-specific protein modification. To avoid such an event, the A-domain needs to interact with the PCP-domain in a configuration that allows the thiolation reaction to occur. Many structures have been solved with an A-domain in a thiolation state with a bound PCP. Isolation and characterisation of such complexes has been enabled by the use of chemical probes that trap the PPant arm of the PCP in the A-domain active site, thus delivering A/PCP complexes for structural characterisation.78The general binding mode in A/PCP complexes involves PCP helix 2, which interacts in a parallel fashion with helix 11 of the Acorevia hydrophobic as well as ionic interactions ((Ar-CP) EntB/EntE (4IZ6, 3RG2),38,39 A-PCP in PA1221 (4DG9),44 EntF A-PCP interaction (5JA1 and 5T3D)).40 In addition “loop 1” between helices 1 and 2 of the PCP forms a network of charged interactions with the last structural motif (loop + strand) of the Asub domain. Mutation analyses confirmed the importance of these regions for the activity of EntB/EntE and assembly lines involved in the formation of pyoluteorin/prodiginine. Indeed, NRPS activity is reduced when point mutations are introduced in the loop 1 motif.40,79 Whilst the PA1221 PCP forms many interactions with its cognate A domain (Fig. 4A), the number of interactions reported in the case of LgrA in thiolation state is much lower. Specifically, Gln734 from PCP helix 2 forms a hydrogen bond with A-domain Gln447 and PCP helix 3 Tyr748 engages into hydrophobic interactions with Tyr421 (Fig. 4E). The analysis of the structure of LgrA in formylation state also reveals that the PCP uses the same Tyr748 from helix 3 to bind a small hydrophobic region on the F-domain composed of Leu127 and Met178 (Fig. 4C). In addition, the structure shows that the Asub participates in positioning the PCP for peptide formylation by creating an electrostatic surface composed of Asn648 and Asp652 allowing interaction with PCP helix 4 Arg residues 758 and 762 (Fig. 4C). It is important to mention here that in contrast to interactions described with any other domain, PCP helix 2 is not involved in any direct binding with the F-domain. Whilst many PCP-A structures show a conserved domain conformation, a recent structure of DhbF with a PCP-A di-domain arranged in the thiolation state varies substantially from the other available structures:43 in this structure, both PCP and Asub domains have moved away considerably from their canonical locations. Indeed, in this structure the Asub domain displays an “open” conformation and the PCP has rotated ∼86 degrees around the PPant arm attachment site. Whether this conformation is biologically relevant or imposed by the crystal packing remains to be assessed.
Interactions between A-domains and their cognate PCPs also include the linker connecting them. It has been demonstrated that over 70% of linkers between A-domains and PCPs have a conserved motif, which follows the essential conserved catalytic lysine of the Asub and displays a LPxP consensus. A mutation of the leucine residue (L958D in EntF) severely hindered the production of enterobactin (reduction of 1000-fold).80 The analysis of this linker in crystal structures of A-PCP di-domain reveals that the leucine residue docks in a conserved hydrophobic pocket created by residues from the beta-sheet in the C-subdomain, thus being important for the positioning of the Asub and the PCP in a conformation competent for the adenylation reaction.
5. Condensation and epimerisation domains
Condensation (C)-domains were first identified as catalytic domains involved in peptide bond formation in NRPS assembly lines in the late 1990's. The presence of these domains (∼450 residues, ∼50 kDa) containing a conserved HHxxxDG motif had been identified to occur the same number of times as the number of condensation and epimerisation events occurred in the peptide synthesis process.81 To evaluate the importance of this conserved motif and assess the catalytic activity of domains bearing it, mutational studies revealed that mutation of the second histidine of the motif into valine was sufficient to disrupt the formation of a linear dipeptide (D-Phe-L-Pro) by a hybrid NRPS assembly line (GrsA A-PCP-E phenylalanine activating module together with TycB C-A-PCP proline activating module).82 These data established the C-domain as the peptide-bond forming domain in NRPS-biosynthesis. Four years after this discovery the first crystal structure of an NRPS C-domain was published.83 This structure revealed the architecture of VibH, a standalone C-domain from the siderophore vibriobactin assembly line (a non-linear NRPS). This structure – which has been shown to be well conserved across all C-domains – is reminiscent of a pseudo-dimer of chloramphenicol acetyl transferase (CAT)84 with additional loss/gain of secondary structure elements. Each “half” of the protein is referred to as the N-terminal lobe or the C-terminal lobe. Both lobes are made up of a central beta-sheet flanked by large alpha-helices. More precisely, the N-terminal lobe possesses a 5-stranded beta-sheet, with one strand originating from a sequence from the C-terminal lobe that is known as “the latch”, a peripheral small 2-stranded beta-sheet, five large alpha-helices and a smaller helix found in the “floor-loop” motif – this also originates from the C-terminal half of the C-domain. In the other half of the C-domain, the C-terminal lobe harbours two central beta-sheets (one with 2- and one with 4 beta-strands) protected on one side by eight alpha helices. The overall fold of the C-domain can be seen as an upright V shape where each half forms one branch of the letter.36 The catalytic site motif HHxxxDG forms part of a loop between the beta strand 6 and the alpha helix 4 in the N-lobe connecting the central strand of the largest beta-sheet with one of the flanking helix. This motif is exposed in the centre of the tunnel formed by the domain two halves.Located at around 15 Å distance from the surface at each side of the end of the tunnel, the catalytic site is placed at the perfect distance from both the donor and acceptor PCP binding sites. No structural intermediate of a C-domain with both donor and acceptor substrates has been obtained so far, however a model of this tri-domain structure is reviewed in ref. 85. Given the pseudo-dimeric nature of the C-domain and the low number of interactions between each sub-domain half (floor loop and latch), it has been reported that C-domains are rather flexible and can be found in different conformations: these range from conformations seen as more “open” to those best described as being more “closed”.86 The relevance of such conformations is not fully understood, although interactions with PCP-bound substrates could be expected to provoke changes in C-domain state. In this way, controlling the specific order of PCP binding would help to maintain efficient NRPS synthesis, with the directionality of NRPS synthesis maintained through the asymmetry of the condensation reaction. Such ordered substrate binding would also provide a hypothesis to explain the increase in hydrolysis of peptides sometimes observed from NRPS assembly lines immediately preceding engineered A-domains:87 in these systems, modification of A-domain specificity can lead to a reduced rate of A-domain activity, which in turn would lead to water being able to competing effectively with the acceptor aminoacyl-PCP for attack of the thioester of the donor peptide substrate due to the slow rate of generation of this intermediate. C-domains can also be seen as crucial gatekeepers in ensuring not only the stereochemistry of the donor peptide (through the presumed dynamic competition for peptidyl-PCP substrate with a neighbouring E-domain) but also in ensuring the correct modification state of the aminoacyl-PCP acceptor substrate through allowing sufficient time for the PCP-bound amino acid to interact with the essential modifying domains, either in cis (such as a methyltransferase domains)67 or in trans (such as a hydroxylase or halogenase enzymes).88,89 Given these important roles that C-domains must play within NRPS catalysis (Fig. 1C), it is clear that many important insights remain to be gained from structural and biochemical investigation of these domains and their PCP-bound complexes.
Analysis of the structures from termination modules C-A-PCP-TE revealed that condensation domains share an extensive interaction surface with neighbouring adenylation domains (total of ∼1100 Å2).35,40 It has been hypothesised that these two domains could act as a catalytic platform possibly arranged in a helical fashion.35,90 However, the relevance of this interaction has been challenged with the structure of the EntF terminal module: the A/C interface in EntF is much less extensive than previously observed (∼780 Å2). The reason behind this discrepancy lies in the fact that in the first two structures the A-domains are seen in the “open-state” where Asub is packed against the C-domain – an interaction that is not present in the EntF structure as the A-domain is in “closed-state”, with Asub folded over the A-core and hence not interacting directly with the C-domain. Additionally, it would appear that whilst the A- and C-domains from the same module interact together, this cannot necessarily be extended to catalytic domains from different modules, since no direct interaction could be seen from a crystal structure of a cross-module NRPS (albeit the only example known to date).43 With a total of only four structures available to date providing insights into A/C interactions, it is difficult to provide a definitive picture of the interaction network between these two important domains and more structural and biochemical data are clearly needed to address this in the future.
C-domains are essential for the process of peptide chain elongation. As described above, both donor and acceptor PCPs bind at a dedicated side of the catalytic tunnel and present the peptides to the catalytic site (Fig. 1C). There, the catalytic histidine (H126 in VibH) (HHxxxDG) has been postulated to act as a general base, enhancing the nucleophilicity of the acceptor aminoacyl PCP and allowing the nucleophilic attack on the carbonyl of the thioester bound amino acid. This then results in the extension of the peptide chain by one residue, which is transferred from the upstream PCP-domain upon peptide bond formation (a mechanism conserved in both CAT and dihydrolipoamide acetyltransferase (E2p)).91 However, this mechanism has been questioned in the light of mutational analysis of several C-domains. Although in the CAT system the equivalent mutant (H195A) is six orders of magnitude less active than the wild type,92 the H126A VibH mutant shows only a minor decrease in catalytic activity (less than two fold).83 In a study by Bergendahl et al.,93 the mutation of the second histidine residue (H146A) in the C-domain of the NRPS TycB was shown to render the enzyme insoluble, suggesting an important structural role. In the same study, the mutation of the aspartate residue of the catalytic motif (D151N) was also reported to yield an inactive enzyme, which has been verified for the equivalent mutations in the other NRPS C-domains VibH (D130A) and EntF (D142A).94 There is evidence that the histidine residue can interact directly with the amino group of the acceptor substrate (gained from the structure of a C-domain from CDA biosynthesis (H157) that was engineered to covalently bind a mimic of the acceptor substrate),95 which could also indicate a role for this residue in positioning the amino group of the acceptor for attack of the donor thioester. Thus, despite being highly conserved, the HHxxxDG motif now appears to play varied roles in different C-domains and thus the specifics of the peptide bond reaction catalysed within C-domains could well vary depending on the specific domain involved.
Delivery of substrates to the catalytic site of the C-domain involves the correct docking of both donor and acceptor PCPs at the surface of the condensation domain. Although no structure of a donor PCP has been solved in complex with a standard elongation C-domain in a productive conformation, the structure of the fungal TqaA PCP-CT complex supports the original hypothesis that the binding location and the binding mode should resemble that of a PCP bound to an epimerisation domain for which structural data are also available (see below).41 Structures of the acceptor PCP-domain bound to the C-domain have, however, been determined: both structures of the terminal modules from SrfA-C and AB3403 are seen in the condensation state, with the acceptor PCP bound to the C-domain (Fig. 5B and C).35,40 When these structures are compared, C-domains superimpose relatively well (RMSD ∼4 Å; calculated using the Matchalign routine in Pymol (368 Cα aligned from a total of 443 Cα)). However, the PCPs are rotated around the PPant attachment site by more than 30° relatively to each other (Fig. 5B). In the case of the structure of AB3403, most of the interactions originate from PCP helix 2 (that carries the PPant attachment site, Ser1006 in this case) as well as the preceding and subsequent loops. In particular, Leu1007 and Val1010 (N-terminal portion of PCP helix 2) are engaged in hydrophobic interactions with Leu22 and Ile80 of the C-domain. Additionally, Val1026, Ala1027 and Ala1030 residues (beginning of PCP helix 3) form hydrophobic interactions with Tyr26 and Leu30 from the C-domain (Fig. 5E). There are limited hydrophilic interactions, with those noted involving the side chain of Lys1011 of the PCP and the main chain carbonyl of Gln78 from the C-domain together with Arg344 from the C-domain interacting with the phosphate of the PPant moiety. In the SrfA-C structure, it is noticeable that PCP helix 2 runs parallel to C-domain helix 1, making possible a number of hydrophobic interactions. Most of the interactions again involve PCP helix 2 and neighbouring loops in the same manner as seen for the AB3403 structure (Fig. 5A). Specific PCP residues include Met1007 and Phe1027 that form hydrophobic interactions with C-domain helix 1 Phe24/Leu28 and helix 10 Tyr337; the importance of these interactions has been probed for the EntB system, which showed the corresponding residues were essential for productive PCP-C interactions.96
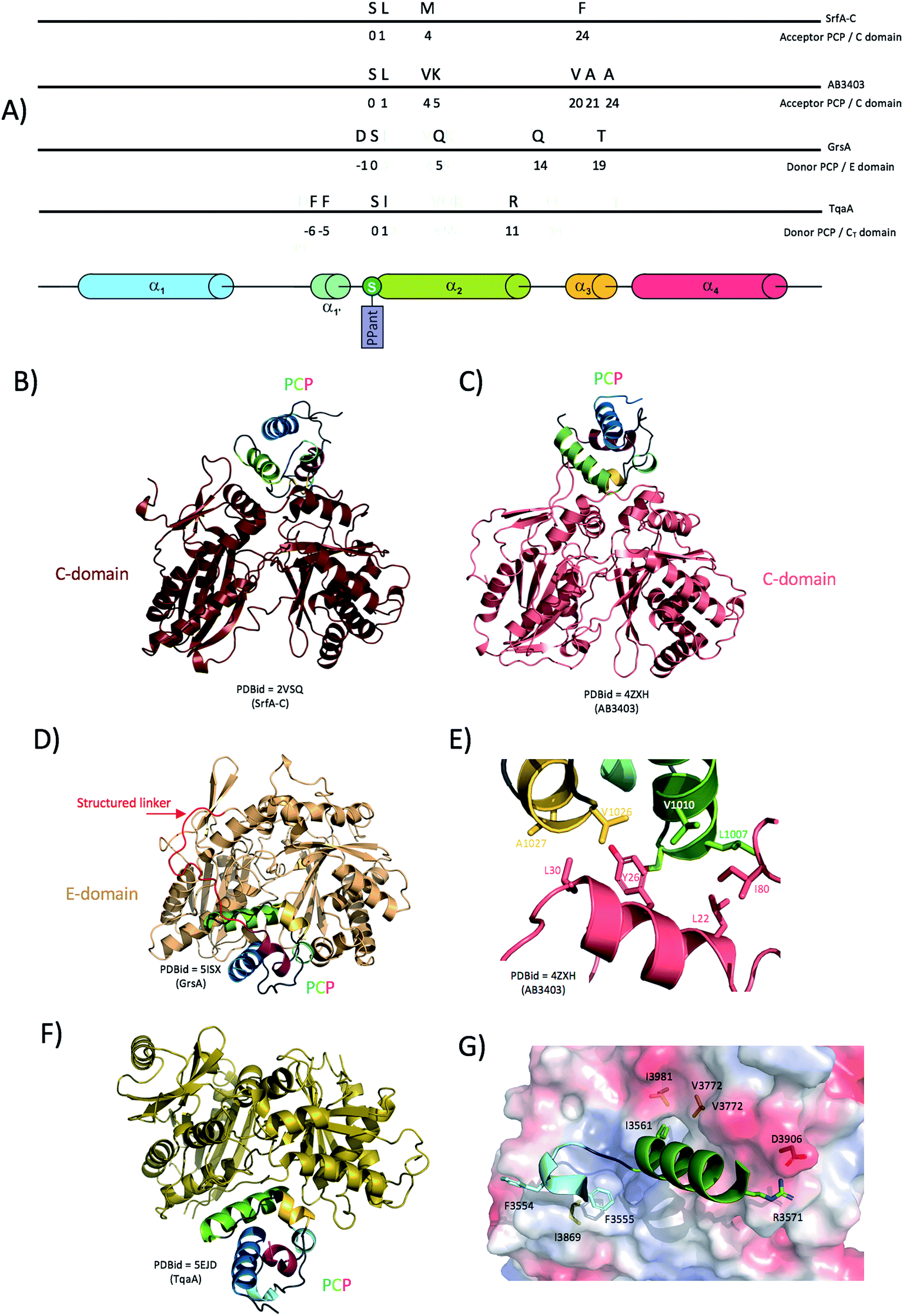 | ||
| Fig. 5 Interaction between condensation/epimerisation domains and PCPs. (A) linear localisation of PCP residues involved in interactions with the either C-domains or E-domains. (B) and (C) Crystal structures of complexes formed between PCPs and C-domains showing the flexible positioning of the PCP (PCPs rotated by 30 degrees between the structures 2VSQ and 4ZXH). (D) Crystal structure of a PCP-domain in complex with an epimerisation domain (5ISX). (E) Close-up of the interactions between the AB3404 PCP and C-domains (coloured as in panel (C)). (F) Crystal structure of a PCP bound to the donor site of a CT domain. (G) Close-up of the interactions identified in the complex of the TqaA PCP (cartoon representation, residues shown as sticks) and CT domains (protein surface representation, residues shown as sticks) coloured as in panel (F). | ||
Epimerisation (E)-domains are non-canonical V-shaped domains that are structurally highly reminiscent of C-domains (Fig. 5D) despite a rather low sequence homology (<20%).83,97,98 E-domains play a vital role in modifying the stereochemistry of amino acids incorporated in the growing peptide chain (i.e. altering the configuration of the C-terminal residue of the PCP-bound peptide from L to D). The first structure of an E domain was of an isolated domain excised from TycA (the first module of the tyrocidine synthetase). From the point of view of the overall structure, E and C-domains initially appear very similar: however, two E-domain specific features have been implicated as playing important roles in their catalytic function. The first feature is found in the so-called “floor loop”, which is extended by at least five residues in E-domains and is postulated to be involved in interactions with the neighbouring PCP-domain. The second important difference is located within the bridge region at the top of the V shaped structure that corresponds to the C-domain binding site for the acceptor PCP. In TycA, this region is blocked by an insertion of eleven residues, which serves to obstruct the catalytic site access from this side of the catalytic tunnel.83,97
The structure of the gramicidin synthetase GrsA PCP-E di-domain41 shows the interaction network required for a functional complex formation and can also be seen as a mimic of an acceptor PCP bound C-domain. One of the most noticeable features of the PCP-E domain interaction is that the linker between the two domains appears to play a prominent role in recognition and binding (Fig. 5D). Indeed, in contrast to usually flexible loops linking C-domains and PCPs, the linker region in this case forms extensive ordered interactions along the surface of the E-domain that are mainly charged/polar in nature. Notably, the residue pairs Arg613/Asp788 and Arg614/Glu785 act as anchor-like electrostatic “hooks” of importance for the localisation of the linker region on the E-domain and the correct positioning of the PCP relative to the E-domain active site tunnel.41 To confirm the significance of the linker interaction with the surface of the E-domain, mutation analyses were carried out revealing that a E785/D788R double mutation was enough to disturb the linker interaction network with the E-domain, which resulted in 20% by-pass of the epimerisation reaction. This result emphasises the importance of the linker in PCP-E domain interactions. This structure also resolved the direct interactions between the PCP and the E-domain, which are largely formed by residues from PCP helices 2 and 3. Four hydrogen bonds stabilise the interface between the domains: PCP/E Gln578 (helix 2)/Asp983, Asp572/Gln979, Gln 587/Glu785 and finally, PCP Thr592 (helix 3) forms a hydrogen bond with Glu898 from the extended floor-loop of the E-domain. It has been suggested that this floor-loop participates in the correct positioning of the PCP-helix 2 (and hence the PPant moiety) to allow catalysis: the recent structure of the unusual CT structure from TqaA, a fungi NRPS C-domain-like involved in macrolactamisation and release of the final product, also demonstrates such a positioning of the PCP in the donor site of the catalytic channel (Fig. 5F).47 Although the first structure of a PCP-C di-domain was obtained earlier for a part of the tyrocidine synthase Tyc6,36 TqaA represents the only structure to date of a donor PCP bound to a C-domain in a catalytic competent state. Upon analysis of the interaction surface between the donor PCP and the C-domain the following multiple interactions have been reported: PCP Arg3571 and CT Asp3906 form the only salt bridge, PCP Phe residues 3554 and 3555 are engaged in hydrophobic interactions with Gly3868 and Ile3869 from the CT domain as well as PCP Ile3561 that docks into a hydrophobic pocket contributed by residues CT Val3772 and Ile3981 (Fig. 5G). Such structural comparisons show that the similar structures of C/E domains are matched by comparable donor PCP-bound states, although the importance of the linker region in E-domains appears to be a crucial difference to C-domains. The example of TqaA also shows that condensation reactions can lead to peptide chain release from an NRPS, although this function is usually the result of a separate thioesterase domain (or less commonly, via reductive cleavage).
6. Thioesterase domains
Thioesterase (TE)-domains play an essential role in catalysing the release of the complete peptide chain at the end of the NRPS-mediated assembly process (Fig. 1D), ensuring the machinery does not stall and is able to perform multiple cycles of catalysis.99 In NRPS assembly lines, two types of thioesterase domains can be found. Type I TE domains are typically the final domain of the last NRPS module whereas type II TE domains are standalone enzymes and are involved in the recognition of incorrectly loaded PCPs. Such misprimed PCPs would lead to the inactivation of the NRPS assembly line and could occur due to modifications blocking the reactive thiol group at the extremity of the PPant moiety (i.e. the incorrect amino acid or an acetyl group from the PPT-catalysed loading of acetyl-CoA). In such cases a trans-acting Type-II TE will exert its enzymatic action to hydrolyse and release the improperly loaded cargo from the PPant moiety of the PCP, ensuring that the machinery is maintained in a productive state.100,101TE domains are relatively small (∼250 residues, ∼30 kDa) and belong to the superfamily of α/β hydrolases that includes a number of lipases and acetylcholinesterases with a catalytic triad typically composed of serine, aspartic acid and histidine residues. TE domains catalyse the release of the substrate from the bound PCP through a two-step reaction: firstly, the TE-domain mediates the transfer of the peptidyl group from the donor PCP onto the activated serine residue in the TE-domain active site, thus forming an O-acyl-enzyme intermediate (the only non-PCP bound intermediate after A-domain activation of amino acid residues). In the second step, a nucleophilic attack on the enzyme tethered ester can take one of several different – and typically highly specific – routes. One common example is hydrolysis, which is triggered when the nucleophile is a water molecule and leads to the release of the linear peptide from the NRPS. Another very important example is macrocyclisation, which occurs when the nucleophile is a functional group from within the linear peptide (i.e. the N-terminal amino group or a nucleophilic side chain) and leads to cyclisation of the peptide with concomitant release from the NRPS. For a comprehensive review on PKS and NRPS release mechanisms, see Du and Lou 2010 (ref. 102) and Horsman et al. 2016.103
Structural approaches have provided insights into both classes of TE domains. The structure of the terminal TE domain from the surfactin assembly line SrfA-C has been excised and structurally characterised as an exemplar of the type I TE-domain fold.104 It exhibits the conserved superfamily fold of a 7-stranded central beta-sheet surrounded by eight α-helices. Of particular interest are the three α-helices known as the “lid”: these cover the active site composed of the conserved serine (Ser80 in SrfA-C) within the signature motif GxSxG together with residues His207 and Asp107. The SrfA-C TE domain was crystallised with two monomers in the unit cell, with the lid regions of each monomer adopting different conformations referred to as the “open” and “closed” forms. The overall structure of the SrfA-C TE domain is reminiscent of a bowl, with a groove under the lid to accommodate the large final peptide substrate.104 The overall architecture of the TE domain was further confirmed by the structure of the excised fengycin NRPS TE-domain (FenTE).105 Aside from a different “lid” conformation, SrfA-C TE and FenTE are closely related structures, with a RMSD of around 1.1 Å when the domain cores are compared (and excluding the lid regions). Structural data concerning type II thioesterases were obtained several years later from the external thioesterase of the surfactin synthetase.106 As has been seen for type I TE domains, the core domain of SrfTEII (surfactin thioesterase type II) also superimposes well onto the core structure of type I TE domains, albeit with some important differences. When compared to SrfA-C TE, SrfTEII possesses an extra helix between the active site residues Asp189 and His216 and also shows a repositioning of the “lid” region. The consequences of those modifications are that the catalytic triad is only partially covered by a short loop, which in turn makes the catalytic site much more accessible than it is in the other (type I TE) structures. In addition, the catalytic pocket in SrfTEII is smaller than the one in SrfA-C TE, which matches well with a role in hydrolysing small groups from the PPant arm as opposed to large peptides. This also ensures that the type II thioesterases do not cleave off the growing peptide chain in a “normal” NRPS process, which would be highly deleterious to their efficiency.
The crystal structure of another type II TE, RedJ, confirmed the shared fold with type I TE and the importance of both the catalytic site pocket and the “lid” to maintain a high degree of selectivity regarding thioesterases' substrates.107 Although TE domains share an overall common fold as described above, examples from the NRPS machineries of the glycopeptide antibiotics and the related GPA-like peptide complestatin possess a longer than usual N-terminal linker to this TE domain. Enzymatic assays carried on the teicoplanin synthesis machinery have recently shown that this linker is important for the activity of the TE domain.108 Of particular interest is the fact that secondary structure predictions show the linker is mostly alpha-helical in nature, which in turn suggests that it could play a structural role. This is supported by the crystal structure of the macrocycle forming TE domain from the clinically relevant erythromycin antibiotic synthase displaying an extended N-terminal linker folded as two additional helices covering the “lid region”.109 Further structural studies of this unusual linker are needed in order to provide new insights into the activity and selectivity of terminal NRPS thioesterase domains in GPA systems.
One of the critical steps in the function of the NRPS assembly line is the recognition of the donor PCP by the TE domain. Structural information has been provided through NMR studies performed on the PCP-TE di-domain structures of the apo EntF37 (type I TE with PCP) and the type II surfactin thioesterase with its cognate PCP.106 The EntF structure reveals that the PCP lies in a small cradle formed by the lid region (residues 226–266, helices 4 and 5) and the core of EntF-TE (Fig. 6B); this lid region covers both PCP and TE domain catalytic sites. The PCP and TE domains mainly interact together through a network of hydrophobic interactions burying a surface of ∼1300 Å2. As in other complexes involving a PCP, interactions are predominantly found to involve PCP helix 2 and the loop between helices 1 and 2 (residues 41 to 55) (Fig. 6A). Within this region, the PCP interacts both with the core of the TE domain as well as with the tip of the lid. It was proposed that Phe41 is structurally important to maintain the 4-helix bundle fold of the PCP through hydrophobic interactions, whereas Phe42 directly interacts with the first beta-strand of the TE core. Specifically, PCP residues Phe41, Phe42 and Met72 act as a hydrophobic clamp on the TE Trp121 (Fig. 6C). These two Phe residues are highly important, since mutations of either result in the loss of interaction between the domains.37 Also, PCP residues Leu49 and His47 dock into a pocket formed at the surface of the TE domain (Fig. 6C). NOE couplings indicate additional interacting residues from the TE lid (Leu240, Ala241, Ala 242 and Gln244) and the TE core (Phe119, Gln122, Leu100 and Leu102). PCP residues involved in the interaction include helix 1/2 loop residue Gly46, helix 2 residues Leu50, and the distant residue Val73. Despite this large interaction network, the PCP-TE di-domain structure shows large movement around the contact region indicative of a continuous breathing/opening motion of the lid. The authors of this study emphasise the fact that this motion is essential for providing the conformational plasticity to the TE in order to accommodate the PCP PPant moiety and allowing the PPant to traverse the catalytic cradle.
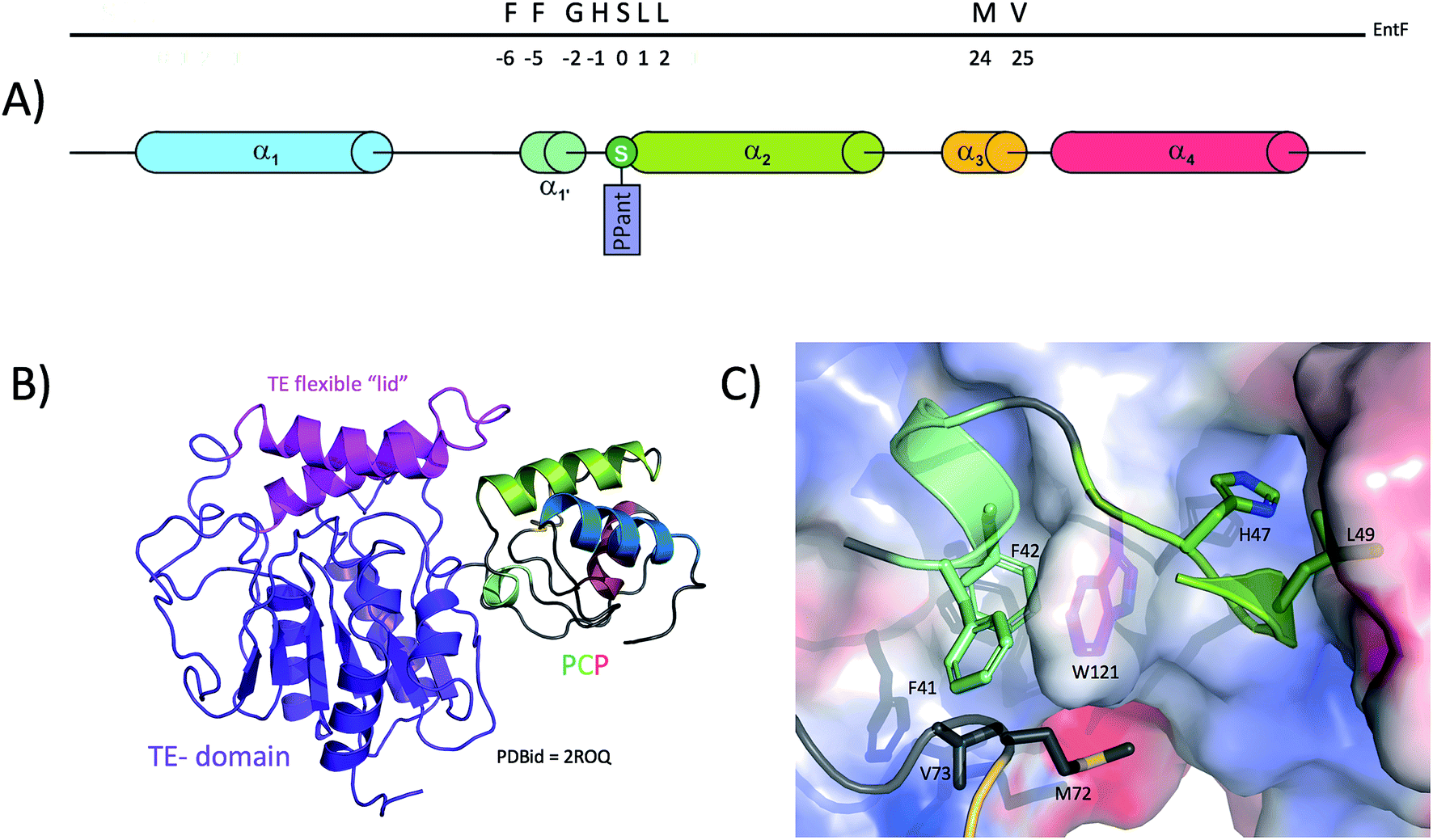 | ||
| Fig. 6 Interaction between thioesterase (TE) domains and PCPs. (A) Linear localisation of PCP residues involved in interactions with TE-domains. (B) Solution structure of a thioesterase domain with a PCP from enterobactin biosynthesis, showing the importance of the lid region in “grasping” the PCP. (C) Close-up view of the interactions between the PCP (cartoon representation, residues shown as sticks) and the TE-domain (protein surface representation, residues shown as sticks) in the complex structure showing the “phenylalanine clamp”. | ||
Although class I and class II TE enzymes are involved in slightly different catalytic activities, NMR interaction studies of SrfTEII with TycC3 PCP indicates a very similar mode of interaction between PCPs and the two classes of thioesterase.106 The interface of the TycC3 PCP is mainly comprised of PCP helix 2 and some additional residues within PCP helix 1 and the PCP C-terminal region. As reported for type I thioesterases, the “lid” region of SrfTEII plays a great role in recognition of PCP helix 2, mostly in the region of the catalytic Ser45 residue. The additional crystal structure of a class I TE (ClbQ) in complex with a donor PCP, confirmed the role of the flexible lid region in substrate binding and specificity.110
7. Other NRPS interactions
Whilst the majority of interactions within NRPS-mediated biosynthesis occurs within the main NRPS machinery (i.e. in cis), there are a number of examples of important modifications that occur in trans during peptide biosynthesis. This requires trans-modifying enzymes to specifically interact with the NRPS-machinery at the desired carrier protein/s, which in turn requires a mechanism to ensure selective interaction of the modifying enzymes solely with the correct carrier protein domains. Limited structural evidence has been gathered considering the range of probable trans-modifying enzymes within NRPS mediated biosynthesis, however two such examples – both related to cytochrome P450 monooxygenase enzymes (P450s) – have been structurally characterised from the biosynthetic machineries producing glycopeptide antibiotics (GPAs) as well as the cyclic depsipeptide skyllamycin (for reviews on the role of such P450s in NRPS-mediated biosynthesis, see ref. 111 and 112).In both these systems, P450 enzymes have been identified as a source of the β-hydroxyl groups found within the final peptide structures of these compounds. In the case of GPA biosynthesis, machineries encoding a homologue of the P450 OxyD have been shown to incorporate β-hydroxytyrosine (Bht) residues directly into the NRPS peptide113,114 (a further subgroup utilises a non-heme iron oxygenase that is believed to act directly against NRPS-bound amino acids during peptide synthesis, although this has yet to be investigated in detail).115 The production of Bht by OxyD also relies on two further proteins – a minimal NRPS module comprising A- and PCP-domains (balhimycin homologue BpsD), and a separate thioesterase (balhimycin homologue Bhp). Mechanistically, OxyD utilises amino acids bound to the PCP-domain of the BpsD protein, which following hydroxylation are then cleaved by the thioesterase for subsequent incorporation into the heptapeptide producing NRPS. The structure of OxyD reveals a well ordered and highly exposed active site, which is unusual for a structure of a substrate-free P450 enzyme.113
Subsequent analysis of the active site residues responsible for orchestrating the open and rigid conformation of the P450 active site revealed that these are highly conserved amongst P450s responsible for the β-hydroxylation of PCP-bound amino acids, suggesting that these P450s recognise and specifically bind to the carrier protein portion of the substrate.88,113 Structural data relating to a P450/PCP complex were obtained in 2014 with the co-crystal structure of a PCP (PCP7) and an P450 from the skyllamycin NRPS machinery.45 In this structure, the PCP adopts the classical 4-helix bundle with helices 2 and 3 arranged as a X-shaped cradle to accommodate residues from helix G of the P450 (Fig. 7B), with the majority of the interactions found within these regions of the two proteins (Fig. 7A). Specifically, a large hydrophobic cavity is formed by PCP residues Phe35, Phe36, Ala45, Phe65, Phe66 and Leu62 to accommodate Trp193 and Leu194 from helix G of the P450 (Fig. 7C). From the P450, residues Ala90, Met94, Leu200 and Leu239 interact with Leu43 from the PCP (the +1 residue from the catalytic Ser42) as well as the two methyl groups of the PPant moiety. It is important to note that except from Ala45, none of those residues belong to PCP helix-2 and mostly belong the PCP-helix 3 and the loop between helices 1 and 2. However, PCP helix-2 still plays an important role in the interaction with the P450, but mainly contributes residues involved in hydrogen bonding. Indeed, Thr46 and Lys47 from PCP helix-2 interact with E235 and Asn197 from the P450 G-helix, whilst PCP helix-3 residues Leu62 and Arg63 interact with Asp191 and Glu198 from the P450 G-helix, respectively. In addition to these protein–protein interactions, a network of hydrogen bonds within the P450 is involved in stabilising the PPant arm of the PCP. The true nature of these interactions is likely perturbed by the PCP cargo present in this structure, which was a small molecule inhibitor mimic of an amino acid that was necessary in order to improve the affinity of the P450/PCP complex for structural analysis.45 At this stage of our analysis of PCPs interaction with other domains, and when comparing TE-PCP and P450-PCP complexes (Fig. 6 and 7), it appears obvious that PCPs interact with these 2 domains in a very similar way, using a group of hydrophobic residues (phenylalanine clamp) to secure a solid anchor to their partner domain.
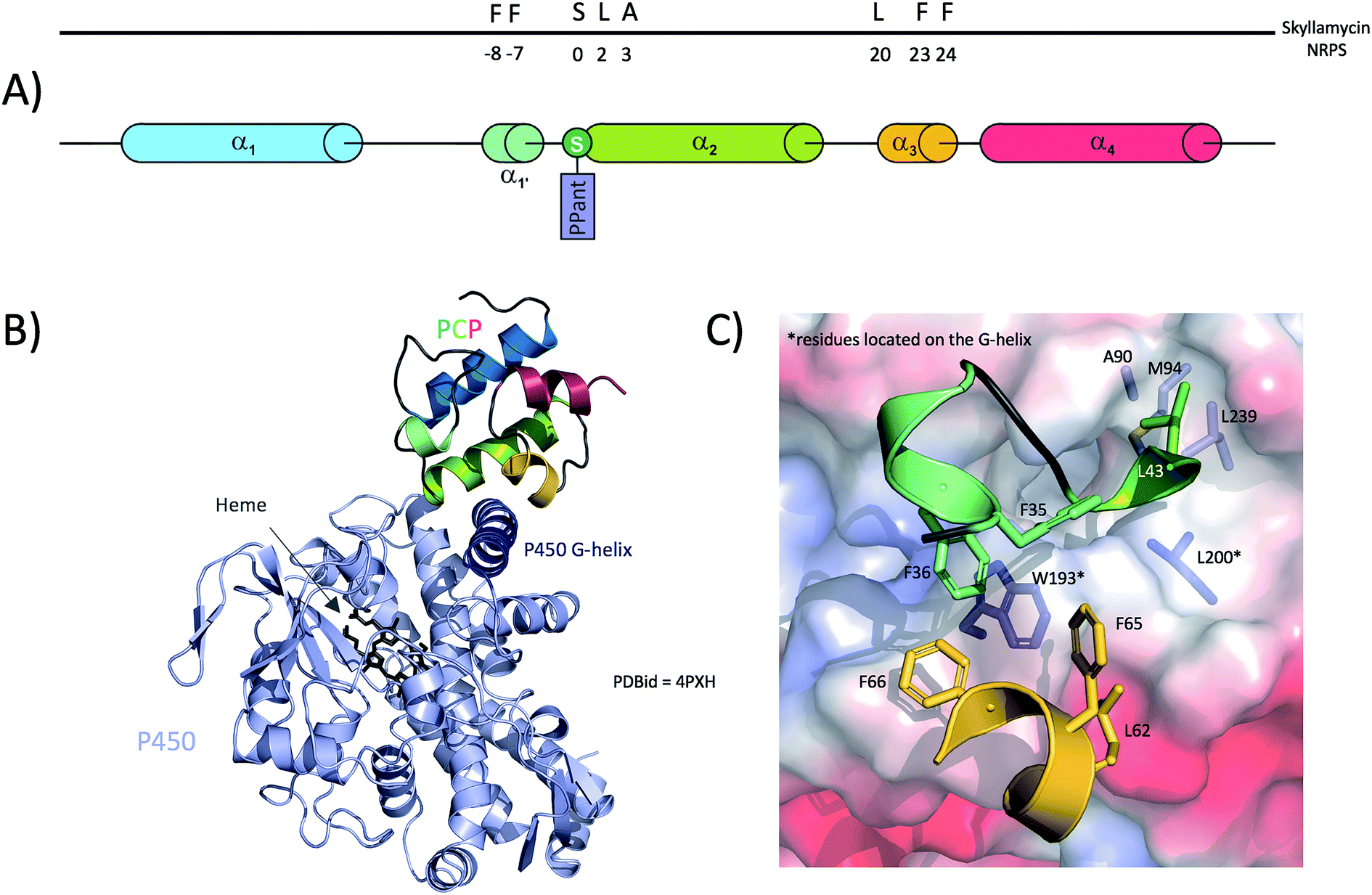 | ||
| Fig. 7 Interaction between a trans-modifying enzyme (P450 monooxygenase) and the PCP from module seven of the skyllamycin biosynthetic NRPS. (A) Linear localisation of PCP residues involved in interactions with the P450 enzyme. (B) Crystal structure of the P450 enzyme in complex with the PCP, showing the importance of the P450 G-helix bound within a hydrophobic groove of the PCP. (C) Detailed interactions between PCP (cartoon representation, residues shown as sticks) and P450 (protein surface representation, residues shown as sticks). | ||
In addition to providing valuable data about P450/PCP complex formation interface, Haslinger et al.45 also discussed the role of the PCP three-dimensional structure in selectively recognising their cognate P450s. Given that PCPs are small and share a high level of sequence conservation, it is unlikely that the amino acid sequence of a PCP would dictate its selectivity. However, it has been shown that subtle changes in tertiary structure can be important for PCP specificity.45 It is important to note that in a comparable P450/CP structure solved from the biotin operon from B. subtilis in which the acyl carrier protein (ACP) is bound to a P450 (P450BioI),116 the ACP protein is located in a very different location on the P450. The differences in the substrate (amino acid vs. fatty acid), carrier protein (amphiphilic PCP vs. acidic ACP) and reaction performed (hydroxylation vs. carbon bond cleavage) likely guide the different binding modes observed in these two complexes, although in both cases these structures reveal how P450 enzymes can use carrier protein binding partners in order to bind and oxidise their desired substrates.
A further, highly complex in trans modification of NRPS-bound substrates has been identified from GPA biosynthesis, in which P450 enzymes perform sequential oxidative cyclisation reactions to generate rigid, biologically active aglycones from the original linear heptapeptide product of the NRPS machinery.117,118 These P450 enzymes (also known as Oxy enzymes) each insert one ring into the final GPA structure: the three enzymes from vancomycin-type GPAs catalyse insertion of the essential C–O–D, D–O–E and AB rings (catalysed in that order by OxyB, OxyA and OxyC, respectively), whilst the non-essential F–O–G ring from teicoplanin-type GPAs is inserted by the enzyme OxyE immediately after the activity of OxyB.119,120 Due to the complexity of the cyclisation cascade, a separate NRPS domain – known as the X-domain – has been implicated in the recruitment of these P450s to the PCP-bound heptapeptide substrate.121 This to date is the only example of a separate recruitment domain for trans-modifying enzymes, with comparable single step modifications (for example the aryl crosslinking observed in arylomycin biosynthesis)122 not requiring such a domain. The essential role of the X-domain in GPA crosslinking has been implied in a number of in vivo118–120 and more recently proven by in vitro experiments, where the use of X-domain containing constructs have allowed the characterisation of both OxyE and OxyA enzymes for the first time.108,121,123–125 Definitive evidence that the X-domain was indeed a binding platform for the Oxy enzymes came with the structure of the complex between the X-domain and OxyB from the teicoplanin NRPS assembly line.121 In this structure, as anticipated, the fold of the X-domain resembled that of a C/E-domain, albeit with insertions that blocked the tunnel usually occupied by the acceptor PCP substrate. In addition, the canonical C-domain catalytic motif was modified in the X-domain making it inactive for peptide bond formation or epimerisation. When compared, structures of the X-domain in the presence or absence of OxyB are extremely similar.121 This observation, also found to be true for OxyB, shows that the formation of this complex does not trigger domain rearrangement and depends solely on a rigid-body type of interaction. Unusually for an NRPS, the interaction forces are mostly driven by hydrogen bonds and salt bridges, with few hydrophobic residues involved. The novel position of the Oxy enzyme within the complex retains space for the simultaneous binding of a PCP bound peptide substrate. This structure together with extensive biochemical evidence supports the notion that the catalytically inactive X-domain acts as a platform onto which the Oxy enzymes can bind in order to affect the complex process of peptide cyclisation during GPA biosynthesis.123 In this case, the subsequent TE-domain also plays a role in proof reading the crosslinked state of the PCP-bound peptide, which only becomes active against fully crosslinked – and thus mature – peptide aglycones.108
Thus, it appears as though the ability to target PCP-domains is sufficient for enzyme targeting in relatively straightforward trans-modification steps of either aminoacyl- or peptidyl-PCP substrates, whilst the highly complex process of GPA crosslinking requires a separate recruitment domain in order to avoid this step from stalling the NRPS machinery.
8. Conclusions
Over recent years, our structural insight into NRPS-mediated peptide synthesis has been rapidly advancing. Examples of this can be found in understanding the importance of PCP-motion coupled to substrate activation via adenylation during A-domain activity, the structural rigidity of PCP-domains during substrate shuttling and mechanistic insights provided by characterising how NRPS domains are assembled into modules. The rapid expansion of our understanding of the potential scope of C-domain catalysis beyond peptide bond formation also adds significantly to our understanding of the catalytic potential of NRPS assembly lines and must also be seen as a major area of future research, in particular the need to determine the structural determinants of C-domain mediated selectivity, novel reactivity and the relevance of coupling A-domain selectivity and rate to effective peptide bond formation in neighbouring C-domains. Furthermore, it is clear that significant work remains in order to fully understand the nature of the interactions between NRPS modules, the process of assembly of NRPS-machineries encoded across multiple proteins and – most intriguingly of all – the higher order structure of NRPS assembly lines. Several models have been postulated for higher order NRPS assemblies, spanning all the way from highly ordered helical-type arrangements through to flexible assemblies with no appreciable ordered structure:40,43,90 here again, our limited access to the structures of larger NRPS assemblies (in this case complete NRPS modules and di-modules) makes it difficult to understand the relevance and accuracy of such models. One technique that will clearly be of great use in this area is cryo-electron microscopy, which has already delivered impressive insights into related, complex assembly lines.126–128 A recent and highly important example of the use of this technique to investigate polyketide synthesis was carried out by the Maier and Townsend groups, who could identify and characterise a functionally relevant asymmetric conformation of the protein that was not apparent from crystallographic studies of the same protein.129 Considering the monomeric nature of NRPS machineries and high degree of variation in module architecture even within one assembly line there is little doubt that cryo-electron microscopy (particularly when coupled with the use of chemical probes to trap the machinery in specific, defined catalytic states) will deliver important contributions to our understanding of NRPS machineries over the years to come. Given the diversity of NRPS systems and their resultant products, it is also conceivable that different NRPS systems will adopt different higher order structures due to the constraints placed on the enzymatic catalysis required to be performed by each individual system. In order to address this, future research into NRPS biosynthesis should prioritize the structural and functional characterization of complete NRPS assembly lines, as it is only with an understanding of complete machineries that we will be able to understand their impressive catalytic and bioengineering potential.9. Conflicts of interest
The authors declare no competing financial interest.10. Acknowledgements
The authors would like to acknowledge the support of Monash University & EMBL Australia; the Australian Research Council (DP170102220, M. J. C.); and the National Health and Medical Research Council (APP1140619, M. J. C.). The authors would also like to acknowledge Professor Mohamed A. Marahiel for the pioneering role he and his research group have played in understanding the fascinating complexity of non-ribosomal peptide synthesis.11. References
- R. D. Süssmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef PubMed.
- C. T. Walsh, R. V. O'Brien and C. Khosla, Angew. Chem., Int. Ed. Engl., 2013, 52, 7098–7124 CrossRef PubMed.
- A. Miyanaga, J. Cieslak, Y. Shinohara, F. Kudo and T. Eguchi, J. Biol. Chem., 2014, 289, 31448–31457 CrossRef PubMed.
- S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noell, I. Pieretti, M. Hugelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Sussmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef PubMed.
- J. F. Martin, The Journal of Antibiotics, 2000, 53, 1008–1021 CrossRef PubMed.
- O. Gonzalez, R. Ortiz-Castro, C. Diaz-Perez, A. L. Diaz-Perez, V. Magana-Duenas, J. Lopez-Bucio and J. Campos-Garcia, Microb. Ecol., 2017, 73, 616–629 CrossRef PubMed.
- D. Schwarzer, R. Finking and M. A. Marahiel, Nat. Prod. Rep., 2003, 20, 275–287 RSC.
- G. Schoenafinger, N. Schracke, U. Linne and M. A. Marahiel, J. Am. Chem. Soc., 2006, 128, 7406–7407 CrossRef PubMed.
- M. Klapper, D. Braga, G. Lackner, R. Herbst and P. Stallforth, Cell Chem. Biol., 2018, 25(6), 659–665 CrossRef PubMed.
- H. B. Bode, A. O. Brachmann, K. B. Jadhav, L. Seyfarth, C. Dauth, S. W. Fuchs, M. Kaiser, N. R. Waterfield, H. Sack, S. H. Heinemann and H. D. Arndt, Angew. Chem., Int. Ed. Engl., 2015, 54, 10352–10355 CrossRef PubMed.
- A. Wiest, D. Grzegorski, B. W. Xu, C. Goulard, S. Rebuffat, D. J. Ebbole, B. Bodo and C. Kenerley, J. Biol. Chem., 2002, 277, 20862–20868 CrossRef PubMed.
- F. Tiburzi, P. Visca and F. Imperi, IUBMB Life, 2007, 59, 730–733 CrossRef PubMed.
- T. Kasahara and T. Kato, Nature, 2003, 422, 832 CrossRef PubMed.
- S. Hartwig, C. Dovengerds, C. Herrmann and B. T. Hovemann, FEBS J., 2014, 281, 5147–5158 CrossRef PubMed.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2016, 79, 629–661 CrossRef PubMed.
- J. M. Reimer, A. S. Haque, M. J. Tarry and T. M. Schmeing, Curr. Opin. Struct. Biol., 2018, 49, 104–113 CrossRef PubMed.
- E. Dehling, G. Volkmann, J. C. Matern, W. Dorner, J. Alfermann, J. Diecker and H. D. Mootz, J. Mol. Biol., 2016, 428, 4345–4360 CrossRef PubMed.
- M. Hahn and T. Stachelhaus, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15585–15590 CrossRef PubMed.
- D. P. Dowling, Y. Kung, A. K. Croft, K. Taghizadeh, W. L. Kelly, C. T. Walsh and C. L. Drennan, Proc. Natl. Acad. Sci., 2016, 113, 12432–12437 CrossRef PubMed.
- C. D. Richter, D. Nietlispach, R. W. Broadhurst and K. J. Weissman, Nat. Chem. Biol., 2008, 4, 75–81 CrossRef PubMed.
- E. Dehling, G. Volkmann, J. C. J. Matern, W. Dörner, J. Alfermann, J. Diecker and H. D. Mootz, J. Mol. Biol., 2016, 428, 4345–4360 CrossRef PubMed.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC.
- A. C. Goodrich, B. J. Harden and D. P. Frueh, J. Am. Chem. Soc., 2015, 137, 12100–12109 CrossRef PubMed.
- M. J. Jaremko, D. J. Lee, S. J. Opella and M. D. Burkart, J. Am. Chem. Soc., 2015, 137, 11546–11549 CrossRef PubMed.
- J. Crosby and M. P. Crump, Nat. Prod. Rep., 2012, 29, 1111–1137 RSC.
- J. E. Cronan, Biochem. J., 2014, 460, 157–163 CrossRef PubMed.
- T. Kittilä, A. Mollo, L. K. Charkoudian and M. J. Cryle, Angew. Chem., Int. Ed., 2016, 55, 9834–9840 CrossRef PubMed.
- B. J. Harden and D. P. Frueh, ChemBioChem, 2017, 18, 629–632 CrossRef PubMed.
- K. Haslinger, C. Redfield and M. J. Cryle, Proteins: Struct., Funct., Bioinf., 2015, 83, 711–721 CrossRef PubMed.
- A. Koglin, M. R. Mofid, F. Lohr, B. Schafer, V. V. Rogov, M. M. Blum, T. Mittag, M. A. Marahiel, F. Bernhard and V. Dotsch, Science, 2006, 312, 273–276 CrossRef PubMed.
- B. F. Volkman, Q. Zhang, D. V. Debabov, E. Rivera, G. C. Kresheck and F. C. Neuhaus, Biochemistry, 2001, 40, 7964–7972 CrossRef PubMed.
- S. Zimmermann, S. Pfennig, P. Neumann, H. Yonus, U. Weininger, M. Kovermann, J. Balbach and M. T. Stubbs, FEBS Lett., 2015, 589, 2283–2289 CrossRef PubMed.
- J. R. Lohman, M. Ma, M. E. Cuff, L. Bigelow, J. Bearden, G. Babnigg, A. Joachimiak, G. N. Phillips and B. Shen, Proteins: Struct., Funct., Bioinf., 2014, 82, 1210–1218 CrossRef.
- E. J. Drake, D. A. Nicolai and A. M. Gulick, Chem. Biol., 2006, 13, 409–419 CrossRef PubMed.
- A. Tanovic, S. A. Samel, L. O. Essen and M. A. Marahiel, Science, 2008, 321, 659–663 CrossRef PubMed.
- S. A. Samel, G. Schoenafinger, T. A. Knappe, M. A. Marahiel and L. O. Essen, Structure, 2007, 15, 781–792 CrossRef PubMed.
- D. P. Frueh, H. Arthanari, A. Koglin, D. A. Vosburg, A. E. Bennett, C. T. Walsh and G. Wagner, Nature, 2008, 454, 903–906 CrossRef PubMed.
- J. A. Sundlov, C. Shi, D. J. Wilson, C. C. Aldrich and A. M. Gulick, Chem. Biol., 2012, 19, 188–198 CrossRef PubMed.
- J. A. Sundlov and A. M. Gulick, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2013, 69, 1482–1492 CrossRef PubMed.
- E. J. Drake, B. R. Miller, C. Shi, J. T. Tarrasch, J. A. Sundlov, C. L. Allen, G. Skiniotis, C. C. Aldrich and A. M. Gulick, Nature, 2016, 529, 235–238 CrossRef PubMed.
- W. H. Chen, K. Li, N. S. Guntaka and S. D. Bruner, ACS Chem. Biol., 2016, 11, 2293–2303 CrossRef PubMed.
- B. R. Miller, E. J. Drake, C. Shi, C. C. Aldrich and A. M. Gulick, J. Biol. Chem., 2016, 291, 22559–22571 CrossRef PubMed.
- M. J. Tarry, A. S. Haque, K. H. Bui and T. M. Schmeing, Structure, 2017, 25, 783–793 e784 CrossRef PubMed.
- C. A. Mitchell, C. Shi, C. C. Aldrich and A. M. Gulick, Biochemistry, 2012, 51, 3252–3263 CrossRef PubMed.
- K. Haslinger, C. Brieke, S. Uhlmann, L. Sieverling, R. D. Sussmuth and M. J. Cryle, Angew. Chem., Int. Ed. Engl., 2014, 53, 8518–8522 CrossRef PubMed.
- J. M. Reimer, M. N. Aloise, P. M. Harrison and T. M. Schmeing, Nature, 2016, 529, 239–242 CrossRef PubMed.
- J. Zhang, N. Liu, R. A. Cacho, Z. Gong, Z. Liu, W. Qin, C. Tang, Y. Tang and J. Zhou, Nat. Chem. Biol., 2016, 12, 1001–1003 CrossRef PubMed.
- L. Holm and L. M. Laakso, Nucleic Acids Res., 2016, 44, W351–W355 CrossRef PubMed.
- J. G. Owen, M. J. Calcott, K. J. Robins and D. F. Ackerley, Cell Chem. Biol., 2016, 23, 1395–1406 CrossRef PubMed.
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol., 1996, 3, 923–936 CrossRef PubMed.
- K. Reuter, M. R. Mofid, M. A. Marahiel and R. Ficner, EMBO J., 1999, 18, 6823–6831 CrossRef PubMed.
- P. Tufar, S. Rahighi, F. I. Kraas, D. K. Kirchner, F. Lohr, E. Henrich, J. Kopke, I. Dikic, P. Guntert, M. A. Marahiel and V. Dotsch, Chem. Biol., 2014, 21, 552–562 CrossRef PubMed.
- J. Beld, E. C. Sonnenschein, C. R. Vickery, J. P. Noel and M. D. Burkart, Nat. Prod. Rep., 2014, 31, 61–108 RSC.
- A. M. Gulick, ACS Chem. Biol., 2009, 4, 811–827 CrossRef PubMed.
- S. Schmelz and J. H. Naismith, Curr. Opin. Struct. Biol., 2009, 19, 666–671 CrossRef PubMed.
- D. Hoffmann, J. M. Hevel, R. E. Moore and B. S. Moore, Gene, 2003, 311, 171–180 CrossRef PubMed.
- H. Kaljunen, S. H. Schiefelbein, D. Stummer, S. Kozak, R. Meijers, G. Christiansen and A. Rentmeister, Angew. Chem., Int. Ed. Engl., 2015, 54, 8833–8836 CrossRef PubMed.
- E. Conti, T. Stachelhaus, M. A. Marahiel and P. Brick, EMBO J., 1997, 16, 4174–4183 CrossRef PubMed.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef PubMed.
- M. Rottig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362–W367 CrossRef PubMed.
- B. I. Khayatt, L. Overmars, R. J. Siezen and C. Francke, PLoS One, 2013, 8, e62136 CrossRef PubMed.
- R. Li, R. A. Oliver and C. A. Townsend, Cell Chem. Biol., 2017, 24, 24–34 CrossRef PubMed.
- S. Meyer, J. C. Kehr, A. Mainz, D. Dehm, D. Petras, R. D. Sussmuth and E. Dittmann, Cell Chem. Biol., 2016, 23, 462–471 CrossRef PubMed.
- K. J. Labby, S. G. Watsula and S. Garneau-Tsodikova, Nat. Prod. Rep., 2015, 32, 641–653 RSC.
- T. A. Lundy, S. Mori and S. Garneau-Tsodikova, ACS Synth. Biol., 2018, 7, 399–404 CrossRef PubMed.
- S. K. Shrestha and S. Garneau-Tsodikova, ChemBioChem, 2016, 17, 1328–1332 CrossRef PubMed.
- S. Mori, A. H. Pang, T. A. Lundy, A. Garzan, O. V. Tsodikov and S. Garneau-Tsodikova, Nat. Chem. Biol., 2018, 14, 428–430 CrossRef PubMed.
- R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2011, 38, 1747–1760 CrossRef PubMed.
- E. J. Drake, J. Cao, J. Qu, M. B. Shah, R. M. Straubinger and A. M. Gulick, J. Biol. Chem., 2007, 282, 20425–20434 CrossRef PubMed.
- S. Lautru, D. Oves-Costales, J. L. Pernodet and G. L. Challis, Microbiology, 2007, 153, 1405–1412 CrossRef PubMed.
- A. M. Gehring, I. Mori and C. T. Walsh, Biochemistry, 1998, 37, 2648–2659 CrossRef PubMed.
- E. Conti, N. P. Franks and P. Brick, Structure, 1996, 4, 287–298 CrossRef PubMed.
- H. Yonus, P. Neumann, S. Zimmermann, J. J. May, M. A. Marahiel and M. T. Stubbs, J. Biol. Chem., 2008, 283, 32484–32491 CrossRef PubMed.
- A. S. Reger, R. Wu, D. Dunaway-Mariano and A. M. Gulick, Biochemistry, 2008, 47, 8016–8025 CrossRef PubMed.
- J. Alfermann, X. Sun, F. Mayerthaler, T. E. Morrell, E. Dehling, G. Volkmann, T. Komatsuzaki, H. Yang and H. D. Mootz, Nat. Chem. Biol., 2017, 13, 1009 CrossRef PubMed.
- K. T. Osman, L. Du, Y. He and Y. Luo, J. Mol. Biol., 2009, 388, 345–355 CrossRef PubMed.
- R. Wu, A. S. Reger, X. Lu, A. M. Gulick and D. Dunaway-Mariano, Biochemistry, 2009, 48, 4115–4125 CrossRef PubMed.
- A. M. Gulick and C. C. Aldrich, Nat. Prod. Rep., 2018 10.1039/c8np00044a.
- M. J. Jaremko, D. J. Lee, A. Patel, V. Winslow, S. J. Opella, J. A. McCammon and M. D. Burkart, Biochemistry, 2017, 56, 5269–5273 CrossRef PubMed.
- B. R. Miller, J. A. Sundlov, E. J. Drake, T. A. Makin and A. M. Gulick, Proteins: Struct., Funct., Bioinf., 2014, 82, 2691–2702 CrossRef PubMed.
- V. De Crecy-Lagard, P. Marliere and W. Saurin, C. R. Acad. Sci., Ser. III, 1995, 318, 927–936 Search PubMed.
- T. Stachelhaus, H. D. Mootz, V. Bergendahl and M. A. Marahiel, J. Biol. Chem., 1998, 273, 22773–22781 CrossRef PubMed.
- T. A. Keating, C. G. Marshall, C. T. Walsh and A. E. Keating, Nat. Struct. Biol., 2002, 9, 522–526 Search PubMed.
- A. G. W. Leslie, J. Mol. Biol., 1990, 213, 167–186 CrossRef PubMed.
- K. Bloudoff and T. M. Schmeing, Biochim. Biophys. Acta, 2017, 1865, 1587–1604 CrossRef PubMed.
- K. Bloudoff, D. Rodionov and T. M. Schmeing, J. Mol. Biol., 2013, 425, 3137–3150 CrossRef PubMed.
- G. C. Uguru, C. Milne, M. Borg, F. Flett, C. P. Smith and J. Micklefield, J. Am. Chem. Soc., 2004, 126, 5032–5033 CrossRef PubMed.
- S. Uhlmann, R. D. Süssmuth and M. J. Cryle, ACS Chem. Biol., 2013, 8, 2586–2596 CrossRef PubMed.
- T. Kittila, C. Kittel, J. Tailhades, D. Butz, M. Schoppet, A. Buttner, R. J. A. Goode, R. B. Schittenhelm, K.-H. van Pee, R. D. Sussmuth, W. Wohlleben, M. J. Cryle and E. Stegmann, Chem. Sci., 2017, 8, 5992–6004 RSC.
- M. A. Marahiel, Nat. Prod. Rep., 2016, 33, 136–140 RSC.
- A. Mattevi, G. Obmolova, E. Schulze, K. Kalk, A. Westphal, A. de Kok and W. Hol, Science, 1992, 255, 1544–1550 CrossRef PubMed.
- A. Lewendon, I. A. Murray, W. V. Shaw, M. R. Gibbs and A. G. W. Leslie, Biochemistry, 2002, 33, 1944–1950 CrossRef.
- V. Bergendahl, U. Linne and M. A. Marahiel, Eur. J. Biochem., 2002, 269, 620–629 CrossRef PubMed.
- E. D. Roche and C. T. Walsh, Biochemistry, 2003, 42, 1334–1344 CrossRef PubMed.
- K. Bloudoff, D. A. Alonzo and T. M. Schmeing, Cell Chem. Biol., 2016, 23, 331–339 CrossRef PubMed.
- J. R. Lai, M. A. Fischbach, D. R. Liu and C. T. Walsh, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 5314–5319 CrossRef PubMed.
- S. A. Samel, P. Czodrowski and L. O. Essen, Acta Crystallogr., Sect. D: Biol. Crystallogr., 2014, 70, 1442–1452 CrossRef PubMed.
- C. Rausch, I. Hoof, T. Weber, W. Wohlleben and D. Huson, BMC Evol. Biol., 2007, 7, 78 CrossRef PubMed.
- A. Schneider and M. A. Marahiel, Arch. Microbiol., 1998, 169, 404–410 CrossRef PubMed.
- D. Schwarzer, H. D. Mootz, U. Linne and M. A. Marahiel, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 14083–14088 CrossRef PubMed.
- E. Yeh, R. M. Kohli, S. D. Bruner and C. T. Walsh, ChemBioChem, 2004, 5, 1290–1293 CrossRef PubMed.
- L. Du and L. Lou, Nat. Prod. Rep., 2010, 27, 255–278 RSC.
- M. E. Horsman, T. P. A. Hari and C. N. Boddy, Nat. Prod. Rep., 2016, 33, 183–202 RSC.
- S. D. Bruner, T. Weber, R. M. Kohli, D. Schwarzer, M. A. Marahiel, C. T. Walsh and M. T. Stubbs, Structure, 2002, 10, 301–310 CrossRef PubMed.
- S. A. Samel, B. Wagner, M. A. Marahiel and L. O. Essen, J. Mol. Biol., 2006, 359, 876–889 CrossRef PubMed.
- A. Koglin, F. Lohr, F. Bernhard, V. V. Rogov, D. P. Frueh, E. R. Strieter, M. R. Mofid, P. Guntert, G. Wagner, C. T. Walsh, M. A. Marahiel and V. Dotsch, Nature, 2008, 454, 907–911 CrossRef PubMed.
- J. R. Whicher, G. Florova, P. K. Sydor, R. Singh, M. Alhamadsheh, G. L. Challis, K. A. Reynolds and J. L. Smith, J. Biol. Chem., 2011, 286, 22558–22569 CrossRef PubMed.
- J. Tailhades, M. Schoppet, A. Greule, M. Peschke, C. Brieke and M. J. Cryle, Chem. Commun., 2018, 54, 2146–2149 RSC.
- S.-C. Tsai, L. J. W. Miercke, J. Krucinski, R. Gokhale, J. C.-H. Chen, P. G. Foster, D. E. Cane, C. Khosla and R. M. Stroud, Proc. Natl. Acad. Sci., 2001, 98, 14808–14813 CrossRef PubMed.
- Y. Liu, T. Zheng and S. D. Bruner, Chem. Biol., 2011, 18, 1482–1488 CrossRef PubMed.
- A. Greule, J. E. Stok, J. J. De Voss and M. J. Cryle, Nat. Prod. Rep., 2018, 35, 757–791 RSC.
- M. J. Cryle, Metallomics, 2011, 3, 323–326 RSC.
- M. J. Cryle, A. Meinhart and I. Schlichting, J. Biol. Chem., 2010, 285, 24562–24574 CrossRef PubMed.
- O. Puk, D. Bischoff, C. Kittel, S. Pelzer, S. Weist, E. Stegmann, R. D. Süssmuth and W. Wohlleben, J. Bacteriol., 2004, 186, 6093–6100 CrossRef PubMed.
- S. Stinchi, L. Carrano, A. Lazzarini, M. Feroggio, A. Grigoletto, M. Sosio and S. Donadio, FEMS Microbiol. Lett., 2006, 256, 229–235 CrossRef PubMed.
- M. J. Cryle and I. Schlichting, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 15696–15701 CrossRef PubMed.
- M. Peschke, M. Gonsior, R. D. Süssmuth and M. J. Cryle, Curr. Opin. Struct. Biol., 2016, 41, 46–53 CrossRef PubMed.
- E. Stegmann, H. J. Frasch and W. Wohlleben, Curr. Opin. Microbiol., 2010, 13, 595–602 CrossRef PubMed.
- B. Hadatsch, D. Butz, T. Schmiederer, J. Steudle, W. Wohlleben, R. Suessmuth and E. Stegmann, Chem. Biol., 2007, 14, 1078–1089 CrossRef PubMed.
- E. Stegmann, S. Pelzer, D. Bischoff, O. Puk, S. Stockert, D. Butz, K. Zerbe, J. Robinson, R. D. Suessmuth and W. Wohlleben, J. Biotechnol., 2006, 124, 640–653 CrossRef PubMed.
- K. Haslinger, M. Peschke, C. Brieke, E. Maximowitsch and M. J. Cryle, Nature, 2015, 521, 105–109 CrossRef PubMed.
- W.-T. Liu, R. D. Kersten, Y.-L. Yang, B. S. Moore and P. C. Dorrestein, J. Am. Chem. Soc., 2011, 133, 18010–18013 CrossRef PubMed.
- M. Peschke, K. Haslinger, C. Brieke, J. Reinstein and M. Cryle, J. Am. Chem. Soc., 2016, 138, 6746–6753 CrossRef PubMed.
- M. Peschke, C. Brieke and M. Cryle, Sci. Rep., 2016, 6, 35584 CrossRef PubMed.
- C. Brieke, M. Peschke, K. Haslinger and M. J. Cryle, Angew. Chem., Int. Ed., 2015, 54, 15715–15719 CrossRef PubMed.
- S. Jenni, M. Leibundgut, T. Maier and N. Ban, Science, 2006, 311, 1263–1267 CrossRef PubMed.
- J. R. Whicher, S. Dutta, D. A. Hansen, W. A. Hale, J. A. Chemler, A. M. Dosey, A. R. H. Narayan, K. Hakansson, D. H. Sherman, J. L. Smith and G. Skiniotis, Nature, 2014, 510, 560–564 CrossRef PubMed.
- T. Maier, S. Jenni and N. Ban, Science, 2006, 311, 1258–1262 CrossRef PubMed.
- D. A. Herbst, C. R. Huitt-Roehl, R. P. Jakob, J. M. Kravetz, P. A. Storm, J. R. Alley, C. A. Townsend and T. Maier, Nat. Chem. Biol., 2018, 14, 474–479 CrossRef PubMed.
Footnote |
| † For a comprehensive review on auxiliary domains, refer to Labby et al. 2015.64 K. J. Labby, S. G. Watsula and S. Garneau-Tsodikova, Nat Prod Rep, 2015, 32, 641–653. |
| This journal is © The Royal Society of Chemistry 2018 |