
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNonribosomal antibacterial peptides that target multidrug-resistant bacteria
Yuan
Liu
a,
Shuangyang
Ding
b,
Jianzhong
Shen
abc and
Kui
Zhu
 *ab
*ab
aBeijing Advanced Innovation Center for Food Nutrition and Human Health, College of Veterinary Medicine, China Agricultural University, No. 2 Yuanmingyuan West Road, Beijing 100193, China. E-mail: zhukcau@gmail.com
bNational Center for Veterinary Drug Safety Evaluation, College of Veterinary Medicine, China Agricultural University, China
cBeijing Key Laboratory of Detection Technology for Animal-Derived Food Safety, Beijing Laboratory for Food Quality and Safety, Beijing, China
First published on 16th October 2018
Abstract
Covering: 2000 to 2018, particularly from 2010 to early 2018
The increase in the incidence of antibiotic resistant infections is threatening to overwhelm healthcare practices worldwide. Most antibiotics in clinical use are becoming ineffective, so therefore it is imperative to develop new antibiotics and novel therapeutic strategies. Traditionally, the chemical and mechanistic diversity of nonribosomal antibacterial peptides (NRAPs) as lead compounds have meant that their structures are ideal for antibiotic discovery. Here, we summarize the state of our current knowledge about the mechanisms of antibiotic resistance, which can be used to guide the development of new antibiotics. Furthermore, we provide an overview of NRAPs for treating multi-drug resistant bacteria, including innovative approaches for screening NRAPs from new sources and the underlying mechanisms of antibacterial activity. Finally, we discuss the design of NRAP scaffolds for precise medicine and combinatorial NRAP therapies with existing antibiotics to overcome resistance, which will help to control infections in the post-antibiotic era.
 Yuan Liu | Yuan Liu received his PhD from China Agricultural University in June 2018. His PhD study focused on the discovery of novel nonribosomal antibacterial peptides and antibiotic adjuvants to combat multi-drug resistant bacteria. He has been a University Professor at the Institute of Comparative Medicine and College of Veterinary Medicine at Yangzhou University (Jiangsu, China) since July 2018. |
 Shuangyang Ding | Shuangyang Ding is a Professor at the College of Veterinary Medicine at China Agricultural University. She obtained her PhD in 1999 from China Agricultural University. Currently, she is the deputy director of the National Veterinary Drug Residue Reference Laboratory of China. Her research focuses on the development of methods to analyze veterinary drug residues in animal-derived foods and veterinary drug safety evaluation. |
 Jianzhong Shen | Jianzhong Shen is a Professor and the Dean of the College of Veterinary Medicine at China Agricultural University. He has been an academician of the Chinese Academy of Engineering since 2015. His research focuses on animal-derived food safety, antibiotic resistance and the development of novel antimicrobial agents. |
 Kui Zhu | Kui Zhu is a Professor at the College of Veterinary Medicine at China Agricultural University. He obtained his Doctor of Veterinary Medicine degree (DVM) from Ludwig Maximilian University of Munich (LMU Munich, Germany). After that, he did postdoc training at Duke University in the USA. His research focuses on the development of novel antibiotics and alternative strategies against multi-drug resistant bacteria. |
1 Introduction
The rapid emergence and widespread distribution of antibacterial resistance is now recognized as one of the most serious global threats to human health.1,2 Consequently, the Centers for Disease Control and Prevention (CDC) recently revealed that more than two million people suffer from antibiotic-resistant infections and at least 23![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 people die as a result per year in the United States alone.3 Worryingly, the increase in the incidence of multi-drug resistant (MDR) Gram-negative bacteria, such as plasmid-mediated resistance to carbapenems4 and colistin5–7 in Enterobacteriaceae, is threatening to overwhelm healthcare practices worldwide. A similar situation has also been observed for Gram-positive bacteria, such as the notorious methicillin-resistant Staphylococcus aureus (MRSA)8 and vancomycin-resistant enterococci (VRE).9,10 Collectively, it means that no effective antibiotic is available for combating infections caused by either Gram-positive or Gram-negative superbugs. New antibiotics or alternative therapeutics are urgently required for clinical treatments.
000 people die as a result per year in the United States alone.3 Worryingly, the increase in the incidence of multi-drug resistant (MDR) Gram-negative bacteria, such as plasmid-mediated resistance to carbapenems4 and colistin5–7 in Enterobacteriaceae, is threatening to overwhelm healthcare practices worldwide. A similar situation has also been observed for Gram-positive bacteria, such as the notorious methicillin-resistant Staphylococcus aureus (MRSA)8 and vancomycin-resistant enterococci (VRE).9,10 Collectively, it means that no effective antibiotic is available for combating infections caused by either Gram-positive or Gram-negative superbugs. New antibiotics or alternative therapeutics are urgently required for clinical treatments.
To accelerate the process of antibiotic discovery, we need to develop a mechanistic understanding of the diverse ways in which bacteria survive antibiotic treatments. Such an understanding is critical for developing new antibiotics and designing therapeutic approaches to revitalize existing antibiotics. For example, aspergillomarasmine A, a fungal natural product, selectively removes the zinc ion from metallo-β-lactamases such as NDM-1 and VIM-2, to restore their susceptibility to carbapenems in the treatment of Enterobacteriaceae.11 Additionally, given that antibiotic-producers are always equipped with self-resistance to avoid suicide, inspired by the co-evolution between antibiotic-producers and diverse competitors in natural niches,12–14 resistance-based approaches to mining novel antibiotic candidates will be more efficient.
In the golden era of antibiotics, the chemical and mechanistic diversity of antibacterial natural product lead compounds provided interesting and useful scaffolds for antibiotic discovery.15–17 Heretofore, nonribosomal antibacterial peptides (NRAPs), such as penicillin (the first antibiotic introduced in modern medicine) are well-known as antibiotics in the clinical setting.18–20 Additionally, vancomycin21 and colistin22 are recognized as last resort antibiotics against Gram-positive and Gram-negative pathogens, respectively. NRAPs are a subclass of nonribosomal peptides (NRPs) with antibacterial activities, produced by giant nonribosomal peptide synthetases (NRPSs).23 In particular, NRPSs are composed of multiple modular sections, each of which is responsible for the incorporation of one defined amino acid (not limited to the 20 proteinogenic amino acids) into the final peptide-like products.24,25 The flexible biosynthetic mechanism of NRAPs leads to compounds with structural diversity. Nevertheless, the dissemination of multiple resistant genes, such as β-lactamase associated genes and the van and mcr series of genes,5,26 has ultimately paralyzed the use of such antibiotics in the clinical setting. Fortunately, new leads and NRAP scaffolds have been continually reported in the past decade, with potent antibacterial activity against MDR bacteria.
In this review, we will first provide a brief overview of the molecular mechanism of antibiotic resistance to better guide antibiotic discovery. Then, we will describe the recent progress on NRAPs, including new sources, methodologies, structure–activity relationships and modes of action. Finally, further perspectives on developing effective NRAPs and their derivatives will also be discussed.
2 Molecular mechanisms of antibiotic resistance
Antibiotics, used either for treating infections in human beings, as growth promoters in food-animal production, or as the waste of pharmaceutical plants, serve as a driving force to select antibiotic-resistant bacteria from the persistent coexistence of antibiotic-sensitive and antibiotic-resistant bacterial strains in natural environments. Bacteria have evolved multiple strategies to tolerate antibiotic treatments. Here, we will focus on the antibiotic resistance of individual cells, including the inactivation and modification of antibiotics, prevention of access to antibiotics and structural changes of antibiotic targets (Fig. 1A). Antibiotic resistance mediated by a bacterial population, such as quorum sensing and formation of biofilm, and by hosts (in vivo), such as hiding in the cytosols in the form of a “Trojan horse”, will not be discussed here (Fig. 1B).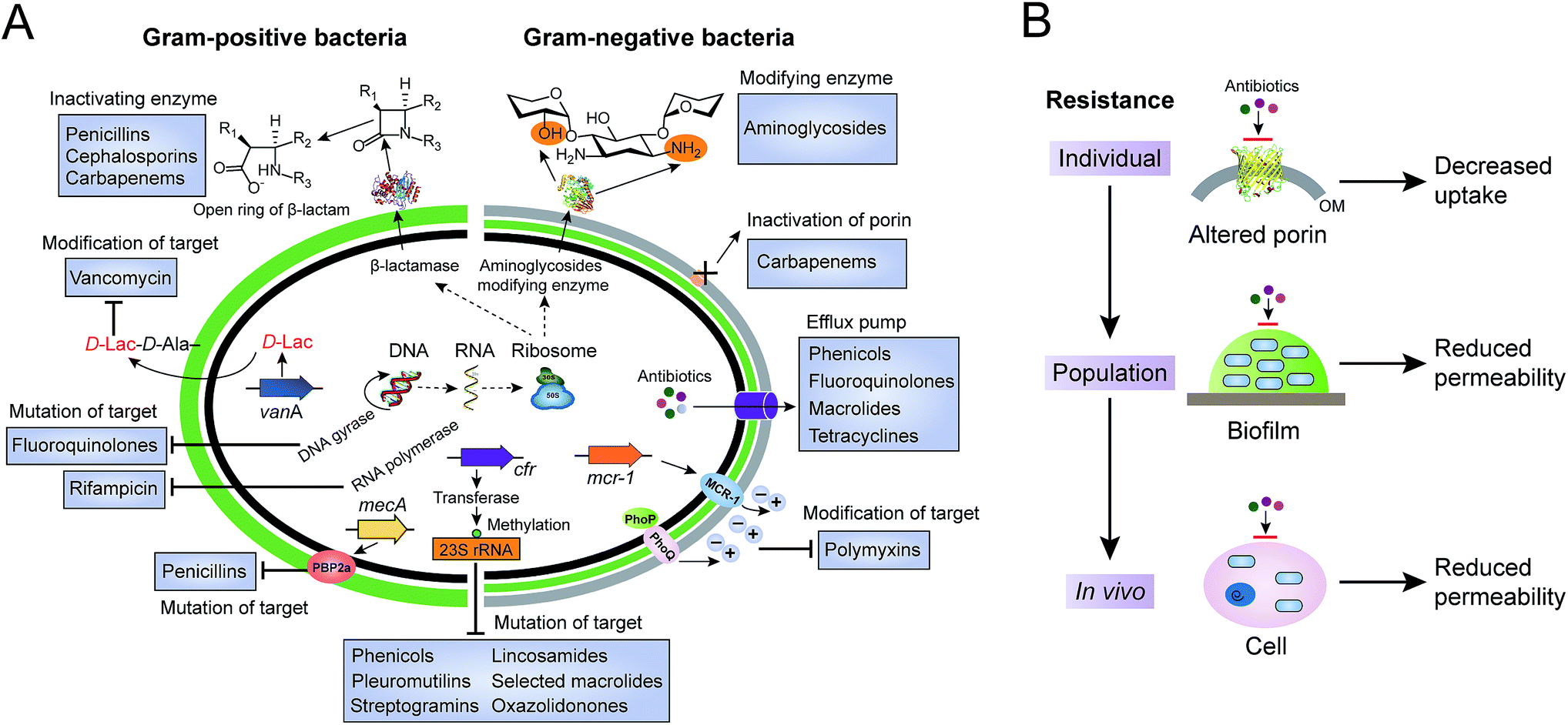 | ||
| Fig. 1 Molecular mechanisms of antibiotic resistance in Gram-positive and Gram-negative bacteria. (A) Antibiotic resistance in single bacterial cells, including inactivation and modification of antibiotics by enzymes, reduced permeability due to inactivation or down-regulated expression of porins, increased efflux pumps, and mutation or modification of antibiotic targets. (B) Antibiotic resistance at the levels of individual cells, the population and in vivo. Bacterial population mediated resistance is known for the formation of biofilm, which serves as a barrier or unfavourable environment against antibiotic treatments. In addition, bacteria can invade and survive in host cells, to circumvent the use of antibiotics. | ||
2.1 Inactivation and modification of antibiotics
Bacteria have evolved various strategies to impair or tolerate antibiotic assaults, of which bacteria can directly break and/or modify the structures of antibiotics to avoid growth inhibition or being killed. Enzymatic degradation and modification are effective means of antibiotic resistance that has brought about resistance to several major classes of existing antibiotics, including β-lactams and aminoglycosides.The process of hydrolysis, carried out by a diverse range of hydrolases, has been identified to inactivate multiple antibiotics. The co-evolution of β-lactam antibiotics and β-lactamases is an excellent example to illustrate the arms race between antibiotics and antibiotic resistance. The β-lactamases serve as work horses to degrade β-lactam antibiotics, such as penicillins, cephalosporins and carbapenems, by breaking the core β-lactam ring open, through either serine nucleophilic attack or the metal-based activation of a water molecule. For example, the first β-lactamase discovered was penicillinase,27 which was first isolated from Escherichia coli K-12 even before the introduction of penicillin in the clinical setting.
Compared to the β-lactams fused to five-membered rings in penicillins, in cephalosporins such as ceftiofur, the β-lactams fused to six-membered rings were developed to resist such β-lactamases. However, cephalosporins were challenged by the subsequent emergence of a new group of enzymes, the extended-spectrum β-lactamases (ESBLs). Fortunately, carbapenems were introduced for clinical use due to their high stability to ESBLs and other β-lactamases. In turn, the increasing numbers of clinical isolates carrying carbapenemases, such as serine Klebsiella pneumoniae carbapenemase (KPC) and New Delhi metallo-β-lactamase (NDM), were observed and became prevalent worldwide. For instance, NDM deactivates the activity of carbapenems by cleaving the β-lactam ring using a Zn2+-activated water molecule. Nowadays, the ndm gene has been found to be widespread in the pathogens of Enterobacteriaceae since its first description in 2009.28 Rapid dissemination of the ndm genes that often located on conjugative plasmids is assisted by the extreme mobility of IS Aba125, an element upstream of such genes.29 Additionally, approximately 20 types of NDM variants have been reported from bacterial isolates of both human and animal origins. Recently, NDM-17 was discovered that has three amino acid substitutions (at the V88L, M154L and E170K positions) and was found to significantly confer enhanced carbapenemase activity to tested β-lactam antibiotics, including penicillin G, ceftazidime, ertapenem, imipenem, and meropenem.30 However, it should be noted that NDMs confer resistance to all β-lactam antibiotics, except for aztreonam, which belongs to the monocyclic β-lactam family of antibiotics.31
Unlike β-lactamases, bacterial enzymes can add different chemical groups to vulnerable sites of antibiotics, by preventing modified antibiotics from binding to the corresponding targets. Compared to other antibiotics, aminoglycoside antibiotics tend to be easily modified due to the abundant amide and hydroxyl groups on the surface of aminocyclitol nuclei linked to amino sugars. Nucleotidyl-transferases, phosphotransferases and acetyltransferases are three main classes of aminoglycoside modifying enzymes that can catalyze the modification at different amide or hydroxyl groups of 2-deoxystreptamine nuclei or sugar moieties.32 Worryingly, a novel genomic island that can encode multiple aminoglycoside-modifying enzymes was found in Campylobacter isolates from a food-producing animal origin that conferred high-level resistance to gentamicin and kanamycin.33 Meanwhile, new aminoglycoside-modifying enzymes are still being discovered, even in nonpathogenic susceptible bacterial species. For example, type VIII and type IX aminoglycoside 3′-O-phosphotransferases were reported in Acinetobacter rudis and A. gerneri in 2017,34 respectively.
2.2 Prevention of access to antibiotic targets
The accumulation of enough antibiotic in bacteria is a prerequisite for antibacterial activity.35 Decreasing the concentrations of intracellular antibiotics to increase resistance can be achieved through reduced permeability or enhanced efflux.Reduced permeability of antibiotics in bacteria can be achieved either in an individual bacterium, by collective behavior or by host cells, as shown in Fig. 1B. Here, we concentrate on the prevention of access to targets in individual bacterial cells. In contrast to Gram-positive bacteria, Gram-negative bacteria are intrinsically resistant to many hydrophilic antibiotics due to the highly impermeable barrier of their outer membrane. Such antibiotics cross the outer membrane by harnessing the porin proteins anchored on the outer membrane.36 Unlike the previous model of drug-binding sites in the channels of porins, reduced permeability of the outer membrane is modulated either by the down-regulation of porins or by the presence of more-selective porins. Therefore, expression of porin variants or reduced expression of porin related genes leads to antibiotic resistance. For instance, decreased expression of the outer membrane porin D (OprD) causes clinically high-levels of resistance to meropenem (a β-lactam antibiotic) in Pseudomonas aeruginosa, in the absence of carbapenemase production.37 Likewise, inactivation of another main porin, CarO, contributes towards increased resistance to carbapenems in A. baumannii.38
Efflux pumps are active transporters that contribute to both intrinsic and acquired resistance to antibiotics,39 particularly in Gram-negative bacteria.40,41 Compared to efflux pumps with narrow substrate specificity (e.g. tetracycline-specific pumps),42 MDR efflux pumps can transport a wide range of structurally diverse antibiotics.43 Resistance-nodulation-cell division (RND) pumps are the most important transporters of mediated resistance.44 A typical RND efflux transporter is located in the inner membrane, which interacts with a periplasmic fusion protein and an outer membrane channel protein to form a tripartite complex, to pump out antibiotics. Upregulation or overexpression of the efflux pump can enhance resistance to various antibiotics.45 Intriguingly, functionally enhanced pumps with single or multiple amino acid substitutions have also been reported. For instance, a single amino acid substitution (G288D) in an AcrB transporter is sufficient to enhance the efficiency of the pump, resulting in clinically relevant resistance in Salmonella.46 Similarly, a resistance-enhancing variant of the predominant efflux pump CmeABC (RE-CmeABC) was characterized in Campylobacter, which confers increased resistance to major classes of existing antibiotics, including chloramphenicol, ciprofloxacin, erythromycin, and tetracycline.47
2.3 Structural changes of antibiotic targets
As well as destroying and modifying antibiotics outside or preventing antibiotic access to intracellular targets, bacteria can form resistance by inducing structural changes of antibiotic targets. Such a strategy is a perfect example of the saying “If you cannot change the world, change yourself”.Bacteria can circumvent antibiotic therapeutics by altering the original targets, resulting in them being able to survive and cause infections. Similar to the direct modification of antibiotics by various modifying enzymes, protection by modification of antibiotic targets is also widely utilized by different bacterial species. Multiple types of antibiotics can target the ribosome to inhibit or block protein synthesis, such as the classes of phenicols, aminoglycosides and tetracyclines. Correspondingly, methylation of the ribosome by methyltransferases has been characterized in diverse bacterial species to resist their action. For example, wide dissemination of plasmid encoded chloramphenicol–florfenicol resistance (cfr) methyltransferase specifically methylates the adenine of position 2503 in the 23S rRNA,48 which has been observed in the isolates of Gram-positive and Gram-negative pathogens from both human and animal origins.49–52 Such an enzyme thereby confers resistance to a wide range of antibiotics, including phenicols, pleuromutilins, streptogramins, lincosamides, selected 16-membered macrolides and also oxazolidonones (such as linezolid).
Modifying enzymes play a crucial role in driving resistance to clinically relevant NRAPs, such as polymyxins and daptomycin. Polymyxins, consisting of polymyxin B and polymyxin E (also known as colistin), are positively charged cyclic NRAPs with hydrophobic fatty acid chains.53 Although colistin has been reported to be responsible for serious toxicity,54 it has become a last-resort antibiotic against MDR Gram-negative pathogens, particularly for carbapenem-resistant Enterobacteriaceae (CRE),55 owing to a barren antibiotic development pipeline. The bactericidal activity of colistin is proposed to occur through the disruption of both the outer membrane and cytoplasmic membrane of the bacteria. Due to the wide use of colistin in the clinical setting and in food-producing animals, there has been a rise in colistin resistance. The first plasmid-mediated colistin resistant gene mcr-1 in Enterobacteriaceae has been reported in China.5 The mcr-1 gene encodes phosphoethanolamine (pEtN) transferase in E. coli, to catalyze the addition of pEtN of lipid A in lipopolysaccharides (LPS). As a result, the affinity between colistin and LPS significantly decreases, because the negatively charged lipid A becomes positively charged. Notably, the global distribution of mcr-1 and a series of variants (mcr-2/3/4/5/6/7/8) has been reported,56–62 which might be due to their high transferability among different bacterial species. Interestingly, a very recent investigation suggested that aquaculture is a potential reservoir of mcr-1.63
To protect critical antibiotic targets for physiological functions, bacteria may evolve different strategies by which to resist antibiotic stresses. Compared to the modification of lipid A with pEtN, recent studies have shown that reduced colistin binding to lipid A can be achieved through the addition of 2-hydroxymyristate, 4-amino-4-deoxy-L-arabinose and palmitate. For example, deletion of mgrB and overexpression of the PhoPQ two-component system increased resistance to polymyxins via different lipid A modifications in K. pneumoniae.64,65 Importantly, these modifications in K. pneumoniae were accompanied by enhanced virulence through lowering the antimicrobial peptide susceptibility and attenuating the activation of early host defense responses. Additionally, despite daptomycin displaying remarkable selectivity against Gram-positive bacteria, the underlying mechanism of daptomycin resistance is not fully understood. The membrane protein multiple peptide resistance factor (MprF) plays a crucial role in the induction of daptomycin resistance, by transferring lysine to modify the membrane lipid phosphatidylglycerol (PG).66 Meanwhile, both LiaF and a GdpD-family protein involved in the cell envelope and cell membrane events, also appear to contribute to daptomycin resistance.67 These results suggest that there is an urgent need to develop new antibiotics to circumvent previously identified targets.
In addition, bacteria can produce alternative elements to mimic the primary targets, offering resistance. For example, MRSA always carries the mecA gene, encoding the penicillin binding protein 2a (PBP2a).68 PBP2a has a low affinity for β-lactam antibiotics, which retains transpeptidase activity. Consistent with this notion, bacteria resist glycopeptide antibiotics such as vancomycin and semisynthetic derivatives by modification of the bacterial cell wall precursor lipid II. Vancomycin specifically binds to lipid II through the formation of a stable complex between the glycopeptide core and the acyl-D-Ala-D-Ala terminus of lipid II, to hinder subsequent building blocks from the penicillin binding proteins (PBPs), thereby inhibiting transglycosylation and transpeptidation.69 As a consequence, bacteria replace D-Ala-D-Ala with D-Ala-D-Lac, D-Ala-D-Ser or other analogs,70 with sharply reduced binding affinity, resulting in a corresponding 1000-fold loss in antimicrobial activity. For example, the genotypes of vancomycin-resistant enterococci (VRE) have been characterized, including the gene clusters of vanA, vanB, vanC, vanD and vanE in clinically relevant isolates.71 Both vanA and vanB are two common phenotypes of acquired vancomycin resistance, and encode multiple enzymes to synthesize alternative dipeptide D-Ala-D-Lac replacing the original D-Ala-D-Ala in peptidoglycan synthesis.72
Collectively, elucidation of the resistant mechanisms and better understanding of the diverse ways by which bacteria resist clinically useful antibiotics, will shed light on the design of alternative therapeutic approaches and guide the development of new antibiotics.
3 Nonribosomal antibacterial peptides (NRAPs)
NRAPs possess versatile chemical scaffolds, suitable antibacterial activity and unique modes of action,73 making them potent leads for antibiotic discovery. In the past decade, a diverse range of NRAPs have been identified, as shown in Table 1, which indicates that there are still untapped sources for discovering NRAPs with as-yet unknown functions. We will introduce these new compounds in terms of their sources, screening methods and structure–activity relationships (Fig. 2).| NRAPs | Years of discovery | Producers | Sources | Activity | Targets | Ref. |
|---|---|---|---|---|---|---|
| a a/b/c, albicidin/tridecaptin A1/telomycin were isolated in 1983/1978/1957 and re-elucidated in 2015/2016/2016, respectively. PBP, penicillin binding protein. LPS, lipopolysaccharide. C55-P, undecaprenyl phosphate. | ||||||
| Penicillin | 1928 | Penicillium | Soil | G+ | PBP | 18 |
| Polymyxins | 1947 | Paenibacillus polymyxa | Soil | G− | LPS | 81 |
| Vancomycin | 1953 | Amycolatopsis orientalis | Soil | G+ | Lipid II | 82 |
| Daptomycin | 1987 | Streptomyces roseosporus | Soil | G+ | Cell membrane | 83,84 |
| A54145 | 1990 | Streptomyces fradiae | Soil | G+ | Cell membrane | 85 |
| Friulimicins | 2000 | Actinoplanes friuliensis | Soil | G+ | C55-P | 86 |
| Bogorol A | 2001 | Bacillus sp. | Marine | MRSA, VRE | Unknown | 87 |
| Tolaasins | 2004 | Pseudomonas tolaasii | Soil | G+ | Unknown | 88 |
| Mannopeptimycins | 2005 | Streptomyces hygroscopicus | Soil | G+ | Lipid II | 89 |
| Bogorols B-E | 2006 | Brevibacillus laterosporus | Marine | MRSA, VRE, E. coli | Unknown | 90 |
| Tauramamide | 2007 | Brevibacillus laterosporus | Marine | Enterococcus sp. | Unknown | 91 |
| Sansanmycin | 2007 | Streptomyces sp. SS | Soil | M. tuberculosis, P. aeruginosa | Translocase I | 92 |
| PAX 3 | 2009 | Xenorhabdus nematophila | Insect | M. luteus | Unknown | 93 |
| Entolysin | 2010 | Pseudomonas entomophila | Soil | S. aureus | Unknown | 94 |
| Pseudofactin | 2010 | Pseudomonas fluorescens | Water | G+ and G− | Unknown | 95 |
| Battacin | 2011 | Paenibacillus tianmuensis | Soil | G− | Cell membrane | 96 |
| Paenibacterin | 2012 | Paenibacillus sp. | Soil | G+ and G− | Unknown | 97 |
| Pekiskomycin | 2013 | Actinomycetes | Soil | G+ | Lipid II | 98 |
| Taromycin A | 2014 | Saccharomonospora sp. | Marine | G+ | Cell membrane | 99 |
| Paenilamicin | 2014 | Paenibacillus larvae | Insect | P. larvae | Unknown | 100 |
| Sevadicin | 2014 | Paenibacillus larvae | Insect | B. megaterium | Unknown | 101 |
| Gageotetrins | 2014 | Bacillus subtilis | Marine | G+ and G− | Unknown | 102 |
| N-Acetylmureidomycins | 2015 | Streptomyces roseosporus | Soil | P. aeruginosa | Translocase I | 103 |
| Lysocin E | 2015 | Lysobacter sp. | Soil | G+ | Menaquinone | 75 |
| Teixobactin | 2015 | Eleftheria terrae | Soil | G+ | Lipid II and lipid III | 76 |
| Albicidin | 2015a | Xanthomonas albilineans | Plant | G+ and G− | DNA gyrase | 104 |
| Cyclohexylgriselimycin | 2015 | Streptomyces | Soil | M. tuberculosis | DnaN | 105 |
| Tridecaptin A1 | 2016b | Paenibacillus terrae | Soil | G− | Lipid II | 106 |
| Humimycins | 2016 | Unidentified | Human | G+ | Lipid II flippase | 107 |
| Telomycin | 2016c | Streptomyces canus | Soil | S. aureus, B. subtilis | Cardiolipin | 108 |
| Lugdunin | 2016 | Staphylococcus lugdunensis | Human | G+ | DNA, RNA, protein and cell wall | 109 |
| Paenipeptins | 2017 | Paenibacillus sp. | Mushroom | G+ and G− | Unknown | 110 |
| Bacaucin | 2017 | Bacillus subtilis | Soil | G+ | Cell membrane | 111 |
| Ilamycins | 2017 | Streptomyces atratus | Marine | M. tuberculosis | Unknown | 112 |
| Ulleungmycins | 2017 | Streptomyces sp. | Soil | G+ | Unknown | 113 |
| Malacidins | 2018 | Unidentified | Soil | G+ | Lipid II | 77 |
| Octapeptin C4 | 2018 | Bacillus circulans | Soil | G− | LPS | 114 |
| Odilorhabdins | 2018 | Xenorhabdus nematophila | Nematode | G+ and G− | Ribosome | 115 |
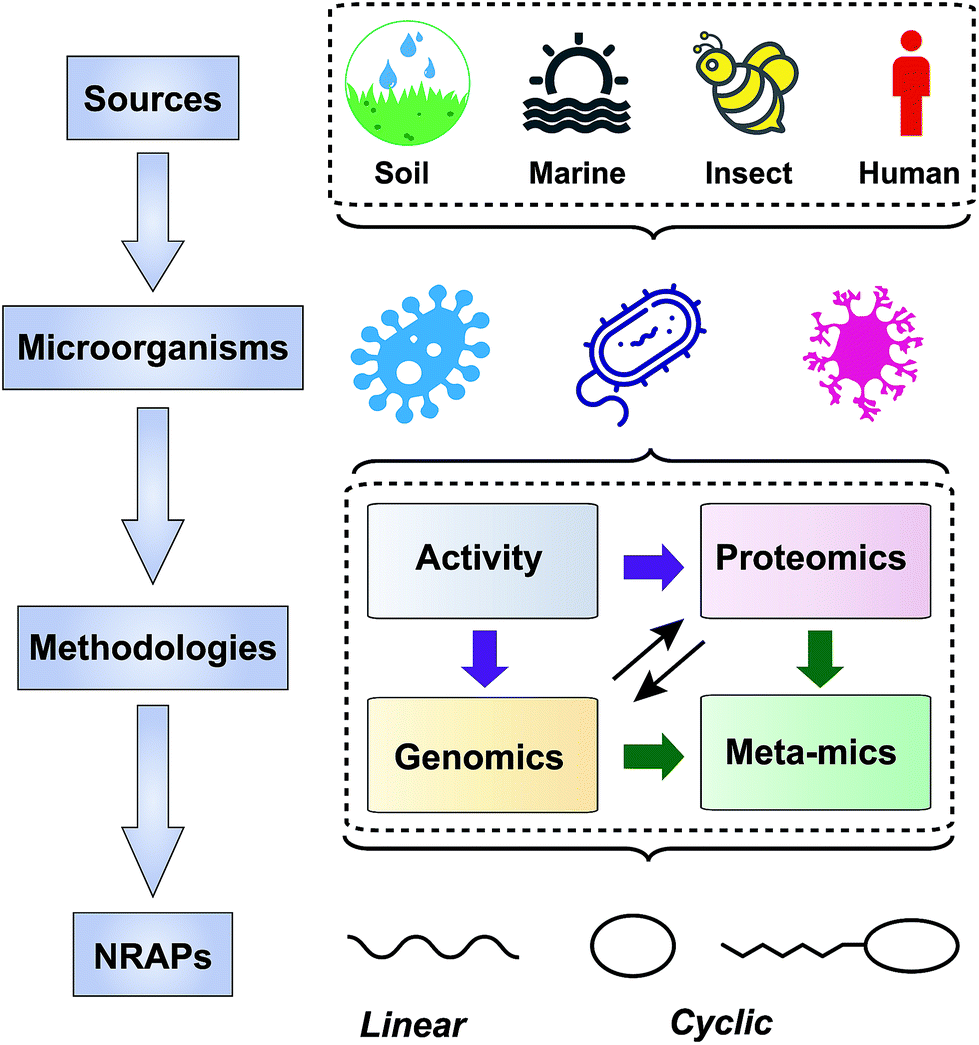 | ||
| Fig. 2 Scheme of approaches taken to screen NRAPs for combating multi-drug resistant pathogens. The producers are mainly from soil, marine, insect and human microorganisms. Screening strategies are based on activity, genomics, proteomics and meta-mics. | ||
3.1 New sources
Most medically important antibiotics are isolated from terrestrial sources, and soil is still an intermittent source of surprise discoveries. Traditionally, the metabolites of many species of microorganisms in artificial media can be extracted with or without further modification to obtain antibiotic candidates.74 Benefitting from the rapid development of biotechnology, more previously unidentified and uncultured bacteria from soil have become new producers for NRAPs, such as lysocin E,75 teixobactin76 (Fig. 3) and malacidins.77 Lysocin E was isolated from Lysobacter sp. RH2180-5 using the silkworm infection model. Meanwhile, teixobactin was characterized from uncultured Eleftheria terrae. This indicates that uncultured bacteria are of importance in potent antibiotic discovery, because uncultured bacteria make up approximately 99% of all species in external environments.78,79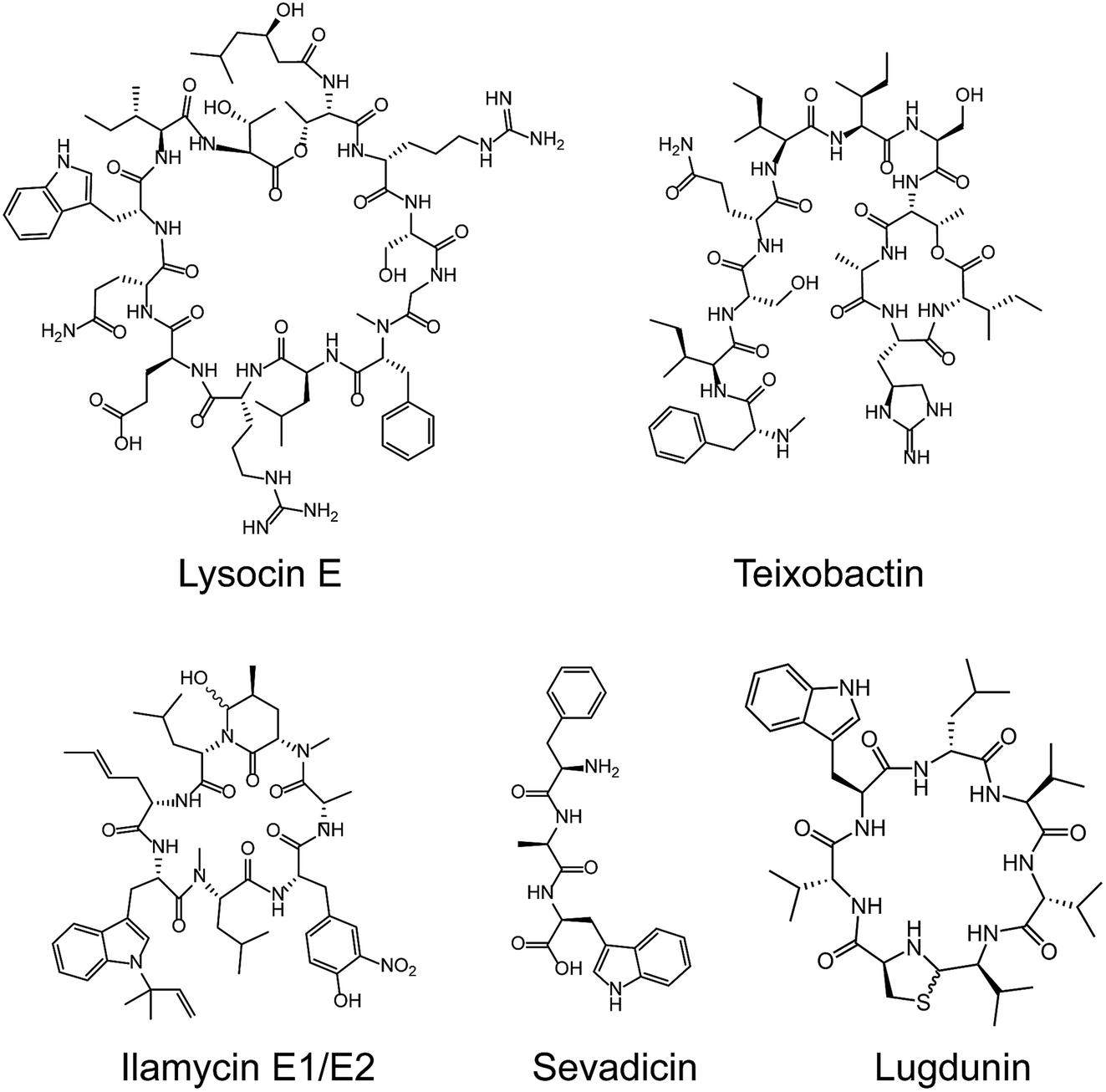 | ||
| Fig. 3 Representative NRAPs from different sources of bacteria discovered since 2010. Lysocin E (culturable soil), teixobactin (unculturable soil), ilamycin (marine), sevadicin (insect) and lugdunin (human). | ||
Given the tremendous biodiversity of organisms,80 the marine environment actually represents another prominent source for antibiotic discovery. Sponges, corals and marine animals contain compounds with interesting scaffolds. For example, ilamycins were isolated and identified from deep sea-derived Streptomyces atratus SCSIO ZH16, of which the ilamycins E1/E2 (Fig. 3) were found to show highly potent anti-tuberculosis activity against Mycobacterium tuberculosis.112 Furthermore, gageotetrins A–C, isolated from marine Bacillus subtilis, are a unique class of linear lipopeptides consisting of repeated Leu and Glu units alongside a fatty acid chain.102 Most unexploited marine microorganisms have been isolated for a long time away from many bacterial pathogens that inhabit the earth, which may provide a solution to combat MDR pathogens.
Evolution between bacterial pathogens and microbial symbioses of insects and other arthropods, provides other sources for discovering new NRAPs. Paenilamicins are encoded by a hybrid NRPS/PKS biosynthetic gene cluster from the bee pathogen Paenibacillus larvae.100 Paenilamicins are employed by producers to fight ecological niche competitors, resulting in American Foulbrood (AFB), the most destructive bacterial disease for honey bees. Similarly, the isolation and structure elucidation of other antibacterial metabolites from P. larvae,116 such as bacillibactin (siderophores), paenilarvins (iturin-like lipopeptides) and sevadicin (nonribosomal tripeptide), have also been reported. Notably, sevadicin (Fig. 3), D-Phe-D-Ala-Trp, is the shortest linear natural NRAP that acts upon bacilli.101
Similarly, very recent studies have revealed that human commensal bacteria produce NRAPs against bacterial pathogens, which has been previously reviewed (see ref. 117). For example, lugdunin (Fig. 3) from the human nasal bacteria Staphylococcus lugdunensis, has been shown to have selective antibacterial activity against MDR Gram-positive bacteria, including MRSA and VRE.109
Although we are facing the dilemma that it's difficult to identify novel compound structures due to increased re-discovery of known antibiotics or their analogues from soil microbes such as Actinomycetes, potent antibacterial compounds from other ecological niches may become valuable sources for new types of NRAPs. Taken together, these findings show how attractive NRAPs from new sources are in combating resistant pathogens and give deep insight into the underlying structure–activity relationships.
3.2 Screening methodologies
Screening methods play a critical role in mining potent bioactive compounds. The simplicity and effectiveness of the Waksman platform led to a golden era in antibiotic discovery, contributing to the discovery of most of the existing antibiotics used in the clinical setting. Due to the high re-discovery rates of previous antibiotics and their derivatives, more robust methods are required to exploit antibacterial leads against antibiotic resistant pathogens from new sources.Importantly, NRAPs with antibacterial activity obtained by in vitro screening may be challenged by their inappropriate properties in vivo, such as poor activity, severe side effects and pharmacological drawbacks. Therefore, different animal infection models are directly utilized to evaluate the therapeutic effects of identified NRAPs. Insect infection models are more suitable for large scale screening due to low cost, fewer ethical concerns and adequate body size for handling.118 For instance, lysocin E, a cyclic NRAP, was successfully obtained based on a silkworm infection model. Briefly, 2794 of 14651 (19%) culture supernatants showed inhibition of S. aureus in vitro, whereas only 23 of the 2794 (0.8%) supernatants showed therapeutic activity in a silkworm infection model, indicating the high efficiency of such a model in excluding candidates without therapeutic potential in vivo.75
The rapid development of DNA sequencing technologies has greatly potentiated the acquisition of genomic data. Candidate BGCs can be identified and analyzed from draft genome sequences using widely used bioinformatics tools such as antiSMASH120 (antibiotics and secondary metabolite analysis shell), PRISM121 (prediction informatics for secondary metabolomes), NRPSPredictor2,122 Minowa123 and Stachelhaus.124 These are open access algorithms for predicting genetically encoded NRPs. For example, a cyclic telomycin from Streptomyces canus was predicted by directly mining biosynthetic scaffolds using PRISM and characterized with a new antibacterial mechanism by being used to target cardiolipin.108 Similarly, humimycin was synthesized by solid phase peptide synthesis (SPPS) based on the bioinformatic analysis of the human microbiome, with a unique antibacterial mechanism of targeting the lipid II flippase in MRSA and other Gram-positive bacteria.107
Another intriguing challenge is that most microorganisms produce far fewer metabolites of interest under artificial growth conditions than genomes suggest. Many specialized metabolite BGCs are poorly expressed, or not expressed at all, under laboratory growth conditions. Therefore, suitable methods for activating such silent BGCs are a prerequisite to enable the perspective of genomics-driven approaches for the discovery of NRAPs. Summarized strategies to induce the expression of silent BGCs to produce structurally diverse specialized metabolites have been reviewed elsewhere,119 which are generally categorized into two groups including pleiotropic methods and pathway-specific methods. For example, two chlorinated cyclic hexapeptides (ulleungmycins A and B) were discovered from Streptomyces sp. KCB13F003 based on the presence of a cryptic gene cluster encoding NRPS and flavin-dependent halogenase and by manipulating the culture conditions.113 Interestingly, subinhibitory concentrations of ribosome-targeting antibiotics (e.g. chloramphenicol) can enhance the production of NRAPs from Streptomyces through global metabolic perturbation.125 Compared to pleiotropic strategies, pathway-specific approaches provide more genetically stable producers for predictable metabolites through sophisticated operations. Taromycin A, similar to clinically approved daptomycin, was obtained by manipulating pathway-specific regulatory genes in marine Saccharomonospora sp. CNQ490.99 The development of gene editing techniques offers alternative methods to awaken silent BGCs, by engineering the transcription and translation machineries, manipulating regulators, replacing natural promoters and heterologous expression. For instance, a one-step robust CRISPR-Cas9 knock-in strategy was established to active multiple silent BGCs in Streptomycetes species.126 Additionally, malacidins, a class of calcium dependent NRAPs, were discovered very recently to integrate the complete malacidin BGC for heterologous expression in Streptomyces albus.77 These results indicate that genomics driven screening may enable the development of a remarkable range of NRAPs and open avenues for the discovery of new antibacterial compounds for applications in medicine and other fields.
Successes in multi-omics technologies mean that meta-omics is a promising approach for new NRAP discovery. Meta-omics combines the advantages of genomics, proteomics, metabolomics, and transcriptomics analysis tools. The construction of a metagenomics library, direct DNA sequencing and single cell technologies make the sequencing data significantly more available, while bioinformatics facilitates the rapid mining of BGCs.130 Similarly, metaproteomics consistently provides insights into the discovery and analysis of NRPSs, providing the groundwork for NRAP development.
Inspired by versatile heterologous expression systems and achievements in synthetic biology,131,132 meta-omic technologies will enable access to a range broad range of NRAPs that have therapeutic applications in medicine.
3.3 Structure–activity relationships
The diverse range of new NRAP scaffolds leads to comprehensive structure–activity relationships (SARs) and sheds light on the design of natural NRAP derivatives with improved antibacterial activity. We focus on the SARs of peptide scaffolds, amino acid types and chemical decorations in terms of antibacterial activity, in either newly identified or previously described NRAPs. | ||
| Fig. 4 Co-evolution of β-lactam antibiotics and β-lactamases. | ||
A wide variety of NRAPs produced by microorganisms, that can be structurally categorized into cyclic and linear scaffolds, have been known for decades and show a wealth of activities. Unlike the ring opening of β-lactam antibiotics, which is ineffective, there has been an increasing number of reports that show that linear NRAPs and their analogs demonstrate comparable or even better antibacterial activity against MDR bacteria. Linear NRAPs represent a new class of antibiotic candidates that are structurally distinct from clinically used cyclic NRAPs, such as daptomycin, polymyxins and vancomycin, and may help to circumvent resistance. Linear NRAPs usually contain short peptide chains and different chemical accessories, in particular modified lipid tails, which have been reported for decades in compounds such as cerxins135 and tridecaptins.136 Recently, lots of linear NRAPs have been discovered from a diverse range of sources using different approaches, including lipopeptides such as humimycins, gageotetrins,102 paenipeptins,110 (Fig. 5) and peptides without any further modifications, such as bacaucin-1 and sevadicin. For example, the peptide skeletons of humimycins107 were bioinformatically predicted from primary sequence data of human-associated bacteria and then chemically synthesized by SPPS, and showed moderate antibacterial activity against MRSA and a synergic effect with β-lactam antibiotics (such as carbenicillin and dicloxacillin). Similarly, antibacterial syn-BNP 1 (an N-acylated 13-mer linear peptide) and antifungal syn-BNP 2 (an N-acylated nonapeptide) can be obtained using the same synthetic-bioinformatic natural product (syn-BNP) approach.137 Interestingly, linear heptapeptide bacaucin-1,111 a ring-opened NRAP of bacaucin without lipid modification, has shown specific antibacterial activity against MRSA in both in vivo and in vitro models. It demonstrates an elegant example of improving antibacterial properties by removing the fatty acid tail and opening the ring structure of the parent compound. Bacaucin-1 consists of all L-type amino acids and the cationic guanidino group under physiological conditions plays a crucial role in its selective activity, suggesting that the design of a linear peptide can be used as an alternative for next-generation precise antibiotics.138 Notably, sevadicin (D-Phe-D-Ala-Trp), the shortest natural linear tripeptide, has been found, which shows activity against bacilli.101 Collectively, the absence of a macrocycle within linear NRAPs makes them more easily accessible as they are easier to synthesize.
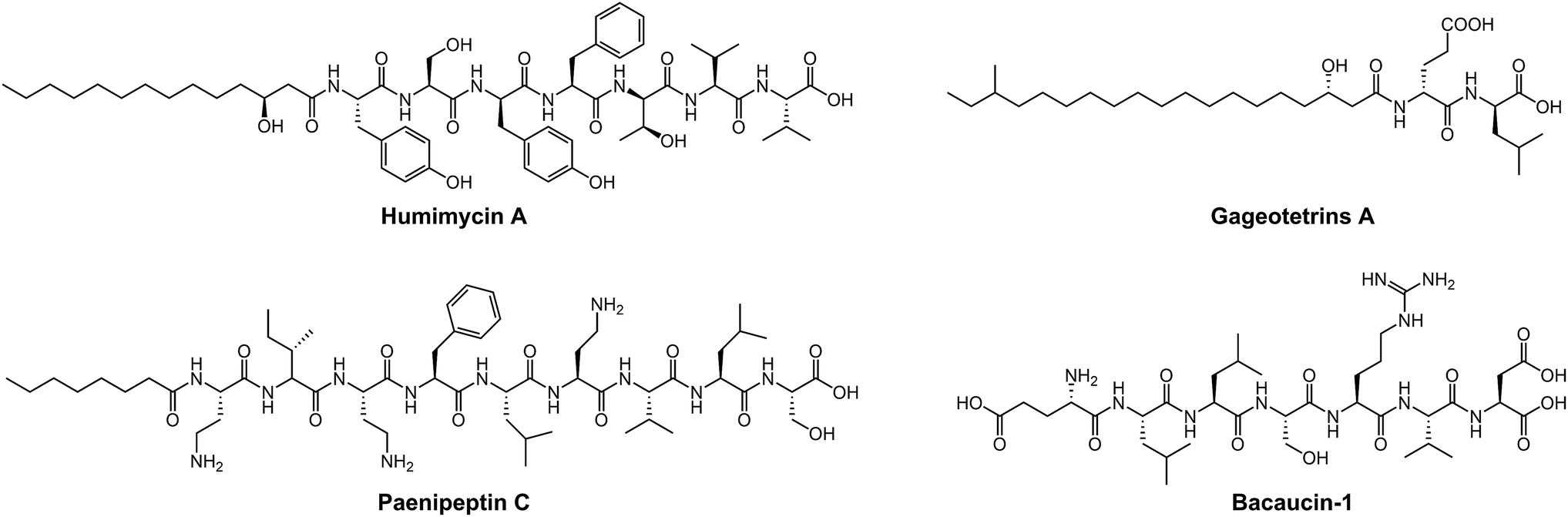 | ||
| Fig. 5 Linear NRAPs and derivatives from a diverse range of sources. Humimycin A (human microbiome), paenipeptin C (mushroom), gageotetrins A (marine) and bacaucin-1 (chemically synthetic derivative of bacaucin) (soil). | ||
In addition, nonproteogenic amino acids can also be incorporated into the peptide skeleton, further expanding the diversity of NRAPs and enhancing their antibacterial activities. For instance, teixobactin is able to kill a series of clinically relevant Gram-positive bacteria, including MRSA, VRE, Streptococcus pneumoniae, Mycobacterium tuberculosis, Clostridium difficile and Bacillus anthracis, with values of minimal inhibitory concentration (MIC) ranging from 0.005 to 0.5 μg mL−1. Such ability is mainly due to the introduction of enduracididine, methyl-phenylalanine and four D-type amino acid residues.76 Interestingly, the introduction of rare L-3-nitrotyrosine and L-2-amino-4-hexenoic acid endows ilamycins E1/E2 with highly selective anti-tuberculosis activity, with an MIC value of 9.8 nM.112 Furthermore, lugdunin, the first example of a new class of macrocyclic thiazolidine NRAPs, has been shown to have potent antimicrobial activity against MRSA and VRE, and a wide range of Gram-positive pathogens with MIC values ranging from 1.5 to 12 μg mL−1.109 However, the detailed SARs of these new identified NRAPs remain largely unclear, and further studies are still required.
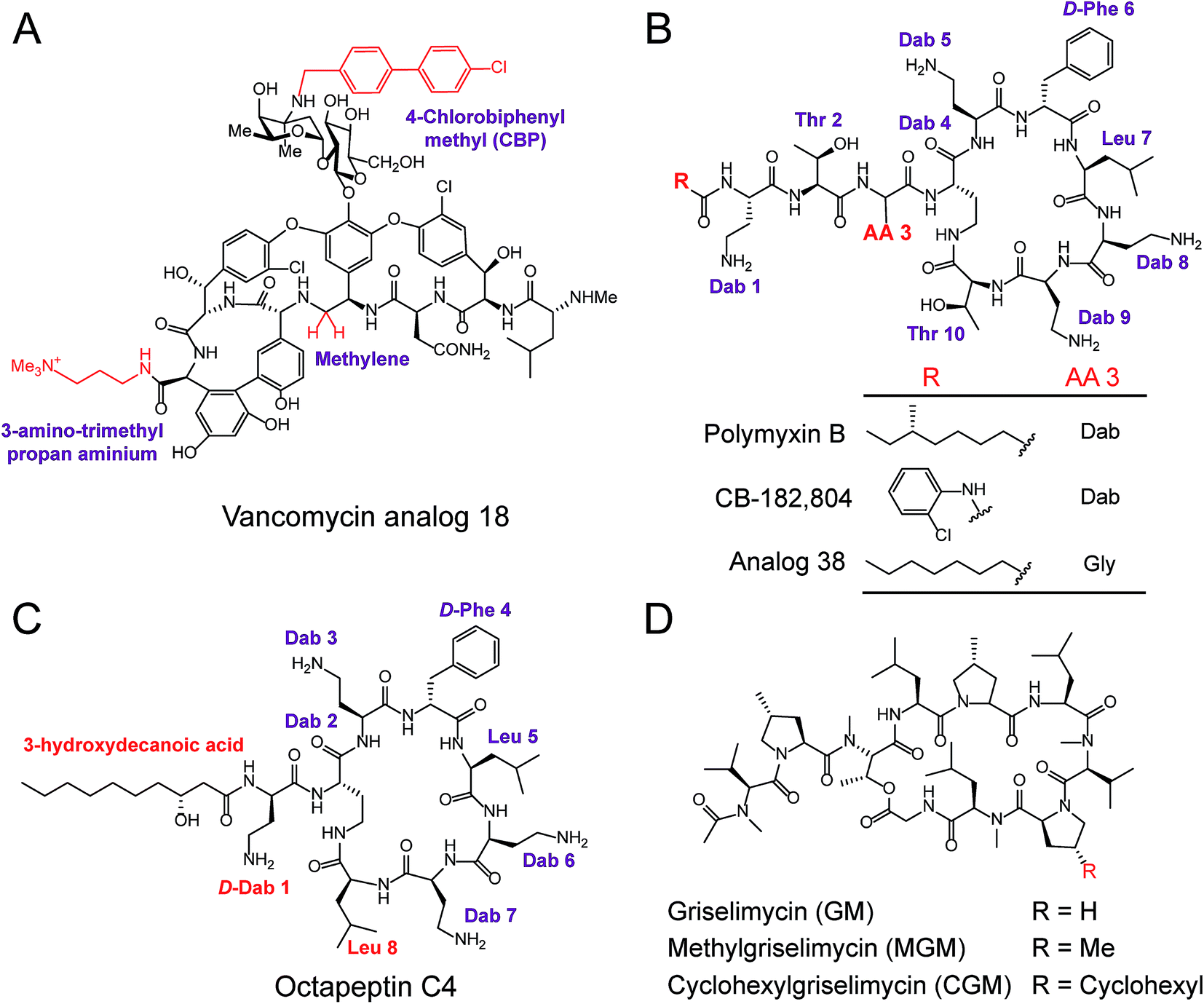 | ||
| Fig. 6 Modification and optimization of NRAP antibiotics. (A) The peripheral modification of vancomycin exhibits enhanced activity against VRE. (B) Structures of polymyxin B and derivatives synthesized through the modifications of N-terminal fatty acyl chains and amino acid substitutions at the third amino acid residue (AA 3). Structures of octapeptin C4 (C) and the derivatives of griselimycin (D). | ||
Polymyxins including polymyxin B and polymyxin E (also known as colistin), are narrow-spectrum antibiotics that are used against MDR Gram-negative pathogens, in particular CRE. Importantly, the side effects of polymyxins involve nephrotoxicity and neurotoxicity in the case of long-term high-dose administration, that accompany their antibacterial activity. To reduce their intrinsic toxicity, systematic SARs have been performed to optimize and design new polymyxin analogs, mainly through the modifications of N-terminal fatty acyl chains and amino acid substitutions. For example, analog CB-182804 (ref. 141) was obtained by deleting the fatty acid chain moiety and adding 2-chlorophenylisocyanate to the N-terminal free amino group in plymyxin B (Fig. 6B). CB-182804 shows comparable antibacterial activity to that of plymyxin B/colistin, whereas decreased cytotoxicity in the kidney proximal tubule cells of rats was observed with a half maximal inhibitory concentration (IC50) of more than 1000 μg mL−1.142 On the other hand, reduced cytotoxicity brought about by the replacement of Dab-3 with Gly and using an octanoic acid at the N-terminal was achieved in Analog 38,143 which retained antibacterial activity with low cytotoxicity to HepG2 cells (IC50 > 300 μM). Detailed SARs of polymyxins have been emphasized and recently reviewed in ref. 144. Despite many efforts being made to obtain better analogs of polymyxin, unfortunately, no analog has been approved for clinical use. Therefore, further studies and alternative approaches are still required, especially in the mining of natural compounds. For example, octapeptin C4, a colistin-like NRAP, is active against MDR bacteria, including polymyxin-resistant bacteria (Fig. 6C),114 and also exhibits reduced nephrotoxicity. Compared to polymyxins, octapeptin C4 and its analogs are N-terminally acylated with longer nonanoyl (C9) or decanoyl (C10) β-hydroxy fatty acyl chains. The change in Thr to Leu at position 8 in the heptapeptide ring of octapeptins is the significant difference between polymyxins and octapeptins.
Owing to the urgent need for novel antibiotics and advanced technologies, rich sources of antibacterial lead compounds have been re-discovered to revitalize previously neglected antibiotics. An exciting example of this is the optimization of the cyclic NRAP griselimycin (GM), which is used for tuberculosis therapy.105 GM from Streptomyces with potential antibacterial and antimycobacterial activities has been known for decades,145 but it still has unfavorable pharmacokinetic properties. Given its high activity, new studies have focused on GM and alkylation of the proline ring at position 8 has been shown to improve its pharmacokinetic properties (Fig. 6D). Attractively, the derivative (CGM) with the addition of a cyclohexyl group on the GM was found to be metabolically stable, and can penetrate the thick mycobacterial cell wall due to increased lipophilicity. CGM exhibited enhanced activity with a MIC value as low as 0.06 μg mL−1 and no cross-resistances with current anti-tuberculosis drugs, suggesting that such derivatives possess an unique mechanism of action differing from other mycobactericidal drugs in use.
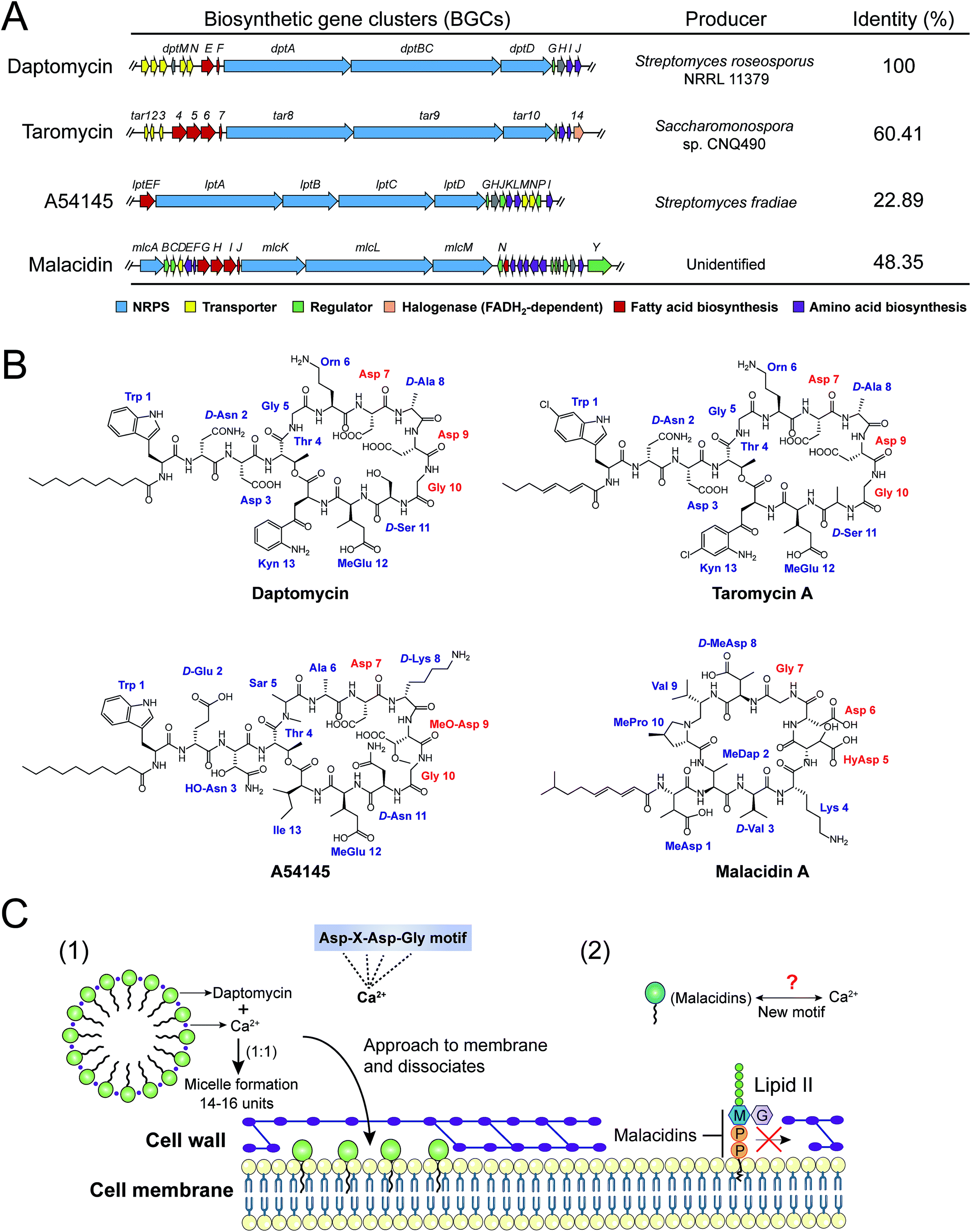 | ||
| Fig. 7 Biosynthetic gene clusters (BGCs) (A), structures (B) and modes of action (C) of four calcium-dependent NRAPs. The whole biosynthetic gene clusters of daptomycin (AY787762), taromycin (KF301601), A54145 (DQ118863) and malacidin (KY654519) were downloaded from GenBank, and the accession numbers are indicated in parentheses. The identities were analyzed against the daptomycin (dpt) gene cluster as a reference, by the Basic Local Alignment Search Tool (BLAST, https://blast.ncbi.nlm.nih.gov/Blast.cgi). | ||
Intriguingly, a new class of calcium-dependent NRAPs, malacidins,77 are challenging this model. The biosynthesis (mlc) gene clusters carry more accessories, suggesting structural differences between daptomycin, A54145 and taromycin A. Malacidins do not contain the DXDG motif incorporated from rare 3-hydroxyl aspartic acid (HyAsp) and also lack the variable spacer residue (Fig. 7B). Additionally, malacidins can specially inhibit lipid II, which indicates that malacidins may harness a new calcium-binding motif (Fig. 7C2). These observations again prove that natural products are, as always, extremely generous in supplying leads to achieve the same therapeutic purpose. Therefore, non-cationic and calcium-dependent NRAPs will be eagerly expected in the future to achieve robust antibacterial activity with reduced side effects.
4 Modes of action
Advanced technologies in chemical biology facilitate the understanding of the mode of action (MOA) of many old NRAPs, such as tridecaptin A1 and GM, and recently identified ones, such as teixobactin, ilamycin and humimycin. We will summarize the validated targets (Table 1) for advancing hits in NRAP discovery programs, while the involved cellular pathways leading to bacterial death or growth inhibition are not discussed here. Meanwhile, Brown and co-workers have rigorously reviewed the strategies for target identification up to 2015.149 Heretofore, many unique antibacterial targets have been identified, such as menaquinone, cardiolipin, flippase and translocase I, which open up new avenues to develop leads for combating resistant bacteria. Generally, inhibition of cell wall synthesis, membrane disruption and blocking critical intracellular processes are the major approaches utilized by NRAPs, as shown in Fig. 8.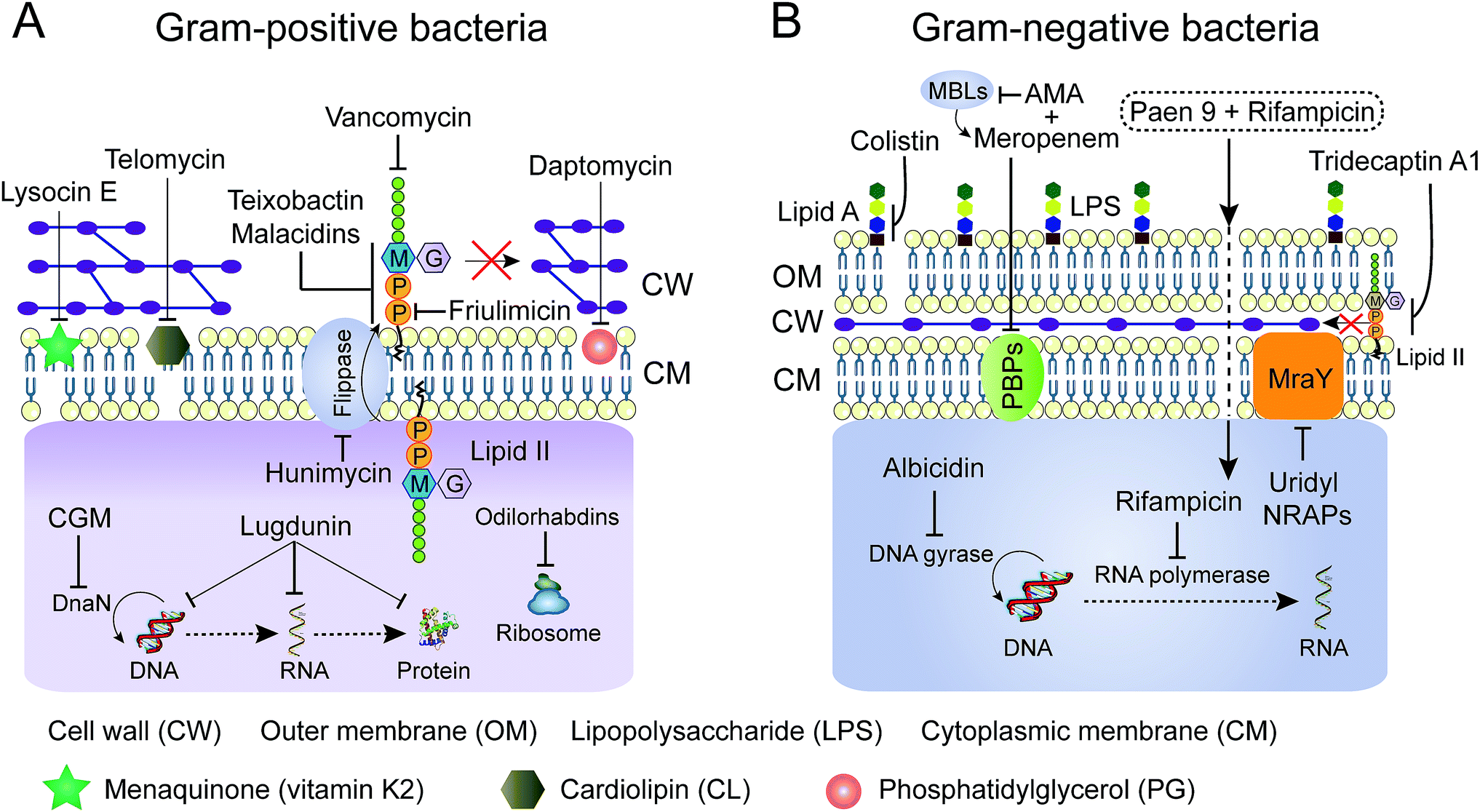 | ||
| Fig. 8 Schematic representation of mechanisms of NRAPs against Gram-positive (A) and Gram-negative bacteria (B). MBLs, metallo-β-lactamases. AMA, aspergillomarasmine A. | ||
4.1 Inhibition of cell wall synthesis
The cell wall provides bacteria with both structural support for performing sophisticated physiological tasks and protection against harsh environmental stresses and invaders, and is readily accessible in both Gram-positive bacteria and Gram-negative bacteria. The bacterial cell wall is composed of peptidoglycan, which is made of polysaccharide chains constructed from cross-linked peptides, as shown in Fig. 8. Lipid II and lipid III are two membrane-anchored precursors used for the biosynthesis of peptidoglycan and teichoic acid (a type of polysaccharide) in Gram-positive bacteria, respectively. Thus, different types of NRAPs predominantly target the bacterial cell wall biosynthesis machinery. For example, teixobactin inhibits cell-wall biosynthesis in S. aureus by binding to a highly conserved motif of lipid II and lipid III.76 Given that lipid II is only present in bacteria, it serves as a specific antibacterial target in the absence of cytotoxicity to mammalian cells.150 Additionally, no resistant S. aureus was obtained after serial passage at the sub-inhibitory concentrations of teixobactin. Similarly, malacidins bind to lipid II in a calcium-dependent manner,77 which is different to the binding of previously characterized calcium-dependent NRAPs such as daptomycin (Fig. 7C).Hydrophilic lipid II is synthesized in bacterial cytoplasm and is transported by lipid carrier molecules, such as undecaprenyl phosphate (C55-P) and flippases, to cross the cytoplasmic membrane. Correspondingly, friulimicins bind to C55-P to perturb cell-wall biosynthesis.86 Humimycins have been demonstrated to specifically inhibit lipid II flippase, in particular for MRSA and other Streptococcus species.107
Although the single layer of peptidoglycan in Gram-negative bacteria is much thinner than that in Gram-positive bacteria, it still a potent target for many NRAPs. The lipopeptide tridecaptin A1 was first isolated in 1978 and was elucidated in 2016. It exerts a bactericidal effect by selectively binding to lipid II in Gram-negative bacteria.106 However, tridecaptin A1 cannot interact with lipid II in Gram-positive bacteria. It remains unclear whether such difference is caused by the change of meso-diaminopimelic acid (DAP) in lipid II in Gram-negative bacteria, to lysine in Gram-positive bacteria. Akin to many other lipopeptides, the fatty acid tail, and D-Dab at position 8 are crucial for disrupting the proton motive force (PMF) and killing bacteria. Furthermore, nucleosidyl NRAP involved sansanmycin92 can selectively target bacterial translocase I (phospho-MurNAc-pentapeptide translocase, also known as MraY), which is essential for the synthesis of lipid I, a key intermediate in mycobacterial peptidoglycan synthesis. And, novel mureidomycin analogues such as N-acetyl-mureidomycin are competitive inhibitors of MraY, exhibiting activity against Pseudomonas aeruginosa.103
4.2 Membrane disruption
Most cationic NRAPs can destroy the bacterial cell membrane by electrostatic interaction. After attachment, the intrinsic fatty acid chains of lipopeptides enable them to efficiently interfere with the most commonly found biological zwitterionic phospholipids in the cell membrane. Impressively, colistin is composed of five positively charged Dab residues and lipophilic moieties. The cationic and hydrophilic region can bind to the outer membrane, in particular lipid A of LPS in Gram-negative bacteria, displacing the Ca2+ and Mg2+ ions and causing destabilization of the LPS layer. Furthermore, a new and ambiguous MOA of daptomycin suggests that moderately amphiphilic daptomycin builds up to 14 to16 units upon the addition of a 1:1 ratio of Ca2+ ions. Subsequently, the complex approaches the cell membrane, dissociates and inserts into the fluid membrane microdomains,84 which might cause oligomerization and lead to the pores depolarizing the membrane, ultimately leading to bacterial death (Fig. 7C1).151 Additionally, consistent with the notion that daptomycin targets phosphatidylglycerol (PG) and triggers membrane blebbing in S. aureus, it is interesting to observe that the release of abundant PG results in the inactivation of its antibacterial activity (Fig. 8A).152 Therefore, other bacterial cell membrane phospholipids153 such as phosphatidylethanolamine (PE) and cardiolipin (CL, also known as diphosphatidylglycerol) are potential candidates for screening new antibiotics. Analogously, telomycin, a predicted cyclic depsipeptide, and its natural analogues, possess a new antibacterial mode of action by inhibiting CL, causing rapid lysis of S. aureus and B. subtilis.108,154 Meanwhile, cinnamycin, a tetracyclic antibiotic, binds to PE.136 Compared to PE and CL, PG is more restricted to bacteria not found in mammalian cells,155 thereby targeting PG would ensure the low cytotoxicity of antibacterial leads. Besides lipids, the bacterial membrane contains a variety of biological molecules. NRAPs can specifically target certain unique membrane components, to trigger inhibitory or killing processes. Unlike any other antibiotics, for the first time, lysocin E has been validated to target menaquinone to achieve potent bactericidal activity.75 Menaquinone (vitamin K2), similar to the electron carrier ubiquinone (coenzyme Q), plays a key role in the electron transport system of respiratory bacteria.156
4.3 Targeting intracellular bacterial components
By bypassing the cell wall and cell membrane barriers, the translocation of certain NRAPs into bacterial cytoplasm can disturb various intracellular machineries, which are responsible for crucial cellular processes such as molecular synthesis and enzymatic activity. For example, cyclohexylgriselimycin (CGM, Fig. 6D), a new derivative of griselimycin (GM), displays high activity against M. tuberculosis by inhibiting the DNA polymerase sliding clamp DnaN.105 Binding of CGM to DnaN inhibits the interaction between DnaN and the α subunit of polymerase III (DnaE1), blocking NDA replication, leading to DNA strand breakages. Interestingly, albicidin, encoded by NRPS/PKS machinery from the sugarcane pathogenic bacterium Xanthomonas albilineans, is a potent DNA gyrase inhibitor for both Gram-positive and Gram-negative bacteria (Fig. 8B).104 Albicidin affects the catalytic DNA cleavage–religation cycle, which is distinct from the widely used fluoroquinolones that act by forming complexes between bacterial DNA and gyrase or topoisomerase IV.157 Furthermore, odilorhabdins, natural linear NRAPs produced by the nematode-symbiotic bacterium Xenorhabdus nematophila, exhibit broad bactericidal activities against Gram-positive and Gram-negative pathogens through binding to the new ribosomal site.115In an attempt to reach intracellular components, many antibiotics cannot penetrate the thick cell wall in Gram-positive bacteria or the low permeability of the outer membrane in Gram-negative bacteria. Intriguingly, several analogues of paenipeptin, such as paen 9 (Fig. 8B), have been shown to increase the activity of rifampicin and clarithromycin against carbapenem resistant and polymyxin resistant pathogens.158 This indicates that many hydrophobic antibiotics such as rifampicin, which targets DNA-dependent RNA polymerase, can be revitalized in combination with NRAPs that disrupt the membrane integrity and destroy the thick cell wall.
In fact, NRAPs are often versatile enough to affect several cellular events. An exciting example is lugdunin,13 a new class of macrocyclic thiazolidine NRAP produced by human nasal S. lugdunensis. Lugdunin simultaneously inhibits the biosynthesis of DNA, RNA, protein or cell-wall precursors and leads to the rapid collapse of bacterial energy resources. Such synchronous inhibition of multiple targets dramatically increases the fitness cost for bacteria to evolve resistance.
5 Future perspectives
5.1 Challenges and solutions
Although the discovery of NRAPs is blooming, the three crucial obstacles in the development of NRAP candidates for clinical trials are high cost, poor protease stability and nonspecific toxicity in both in vivo and in vitro models. Most NRAPs have unique structures with complicated decorations and tend to be very expensive drugs. Therefore, leads with simple and non-cationic scaffolds are promising candidates. As shown in Fig. 5, increased numbers of linear lipopeptides or their derivatives in the absence of accessories achieve potent activity against bacterial pathogens. The development of linear NRAPs or analogues in lieu of cyclic ones will be economically beneficial, because they can significantly accelerate the synthetic process and thus reduce the cost. Most importantly, a large number of such lead compounds can be produced using the standard protocol of solid-phase peptide synthesis (SPPS). Rationally engineered linear derivatives of paenipeptin A/B/C110 and bacaucin-1 (ref. 111) reveal that the peptide cyclization is sometimes not essential for their antibacterial activity. Surprisingly, bacaucin-1, synthesized by all natural amino acids by SPPS without any fatty acid tail, shows even better antibacterial activity against MRSA than its cyclic parent bacaucin. Collectively, NRAP inspired synthetic linear peptides represent a new paradigm in the discovery of better antibiotics.To improve the protease resistance of NRAPs, several approaches have been proposed, including the replacement of natural amino acids with mimics including D-type amino acids, non-natural amino-acid analogues and appropriate formulations, to render them protease resistant. For instance, a recent study showed that D(KLAKLAK)2, a membrane active all-D-enantiomer antimicrobial peptidomimetic, is resistant to proteolytic degradation and is a great prototype drug that targets certain Gram-negative pathogens.159 Proteases or other polypeptidases usually recognize specific peptide sequences or certain side chains of amino acid residues to trigger hydrolysis of peptide bonds. For example, trypsin is the work horse in the digestive system of animals and human beings, cleaving peptide chains mainly at the carboxyl sites of lysine or arginine.160 Thus, rearrangement of the peptide sequence based on the elucidated SAR may provide an alternative way of obtaining candidates.
In practice, the synthesis of most compounds with antibacterial activity is terminated due to their unfavorable pharmacokinetic properties or toxicity. Some antibiotics that are still in use have been continuously criticized for their toxicity, including hemolysis, cytotoxicity, apoptosis and degranulation of mast cells. Long-term and high-dose administration of colistin always results in nephrotoxicity and neurotoxicity.54 To avoid such nonspecific toxicity, the most straightforward way is to reserve the active site by deleting toxic motifs. By removing the fatty acid chain of bacaucin, lead bacaucin-1 is successfully obtained with improved specificity to target MRSA and no detectable toxicity.111 As discussed in Section 4.2, lipophilic fatty acid tails are utilized by many lipopeptides to disrupt the integrity of bacterial membrane. To reduce the side effects, nonspecific toxicity can be addressed by masking these tails based on advanced drug delivery systems or pharmaceutical techniques. For instance, 1-dodecanethiol functionalized gold nanodots with surfactin, a cyclic lipopeptide, can not only alleviate the nonspecific cytotoxic and hemolytic activity of surfactin, but also enhance the ability to treat wounds and skin infections caused by MDR bacteria.161
5.2 Precise antibiotics
Since the introduction of penicillin for treating bacterial infections, broad-spectrum agents have been advocated due to the lack of sensitive and reliable diagnostic methods and time pressures, which has led to the practice of empirical therapy and overprescription.16 Nowadays, more and more studies are demonstrating that broad-spectrum antibiotics are being challenged in infection control and prevention, because they can result in serious side effects, including the triggering of hyper-inflammatory responses and, most notably, the disruption of the beneficial microbiome.162 Therefore, the treatment of infections should be transformed into an era of precise medicine. Precise antibiotics or narrow-spectrum antibiotics, the cornerstone of precise medicine against infections, are urgently required to perform these tasks.NRAPs and their derivatives are promising candidates for the next generation of precise antibiotics that can selectively target the bacterial pathogen of choice, without destroying the beneficial microbes in the hosts. For example, ilamycins,112 heptapeptides from marine-derived Streptomyces atratus, show selective activity against two mycobacteria, including M. smegmatis and M. tuberculosis, while they fail to be effective against another six types of bacteria. This indicates that ilamycins are quite prominent as lead compounds for anti-tuberculosis agents. Similarly, bacaucin-1, a heptapeptide derivative of bacaucin, specifically targets MRSA and some Staphylococcus species.111 These findings suggest that NRAPs hold great promise for generating fewer off-target effects on the gut microbiota and decrease the stress that results in the evolution of resistance.
Advanced achievements in the accuracy of point-of-care tests (POCT) and better understanding of the pathogenesis of bacterial pathogens, in particular resistant pathogens, will accelerate the screening, design and development of precise antibiotics. Most notably, the switch to the era of precise antibiotics is not limited to the targeting of specific bacterial components. Alternative strategies for combating bacterial virulent factors and biofilm are promising approaches to promote personalized therapies that exclusively prevent infections.
5.3 NRAP derived adjuvants
Combinatorial treatment consisting of an existing antibiotic and an adjuvant to potentiate antibacterial activity against MDR pathogens offers a potential approach to minimize the emergence of resistance. Consistent with the elucidation of the mechanism of resistance (Fig. 1A) and MOA of NRAPs (Fig. 8), potential adjuvants can be developed by designing inhibitors of enzymes that inactivate or modify antibiotics, by disruption of permeable barriers and by restoration of antibiotic-target affinity, based on a further understanding of the molecular mechanism of resistance. The use of adjuvants to revitalize antibiotics against resistant bacteria is exemplified by the extensive co-administration of β-lactamase inhibitors, such as clavulanic acid,163 with β-lactam antibiotics, such as amoxicillin. Another exciting example is that of aspergillomarasmine A (AMA), a fungus-derived natural product, and potent inhibitor of metallo-β-lactamases (MBLs) through the chelation of Zn2+ ions from clinically relevant NDM and VIM.11 The synergy between AMA and meropenem fully restores the activity against the enterobacteriaceae, Acinetobacter and Pseudomonas carrying either VIM or NDM-type alleles, in both in vitro and in vivo models. Additionally, hydrophilic antibiotics are intrinsically ineffective against Gram-negative bacteria due to their highly impermeable barrier. To reach intercellular components, adjuvants can be developed to tear or disrupt the outer membrane to facilitate the access of antibiotics to the cytoplasm, and ultimately treat infections. For example, paenipeptin analogues at sub-inhibitory concentrations can significantly enhance the antibacterial activity of rifampicin and clarithromycin against A. baumannii and Klebsiella pneumoniae.158Studies of mechanisms and resistance evolution of new NRAPs and their derivatives are a mandatory requirement in the screening and development of NRAP-based antibiotics and/or adjuvants. Exposure of hidden targets also contributes to the re-sensitization of resistant bacteria to antibiotic therapy. For example, the activity of penicillin can be reversed to kill MRSA in the presence of statins, cholesterol-lowering drugs, through disassembly of penicillin binding protein (PBP2a) oligomerization.164 In addition, some β-lactam antibiotics such as ceftaroline can specially target PBP2a to destroy the cell wall, which augments the activity of daptomycin against resistant strains in the clinical setting.165 This indicates that knowledge about the underlying mechanisms of resistance and synergy will certainly facilitate the development of new therapeutics to revitalize existing antibiotics and minimize the emergence of resistance. Such a strategy is crucial in the near future, as new antibiotics or solutions are not likely to enter the clinical setting immediately.
6 Conclusions
The evolution, dissemination and accumulation of multi-resistant pathogens pose a severe threat to human health, and calls for the development of new antibiotics or novel antibacterial strategies. The achievement and development of antibiotics from a diverse range of sources offers promising alternatives to tackle resistance and to treat resistant bacterial pathogen associated infections. Innovative approaches such as high-throughput screening in genome sequencing and bioinformatics tools accelerate the discovery of new NRAPs. Compared to conventional antibiotics, NRAPs are less prone to causing resistance due to their unique mechanisms of action. Furthermore, advances in the understanding of antibiotic resistance at the molecular level will shed light on how to design new scaffolds of NRAPs as precise antibiotics to generate fewer off-target effects on the host microbiome and to minimize the stress that facilitates resistance. Finally, the combination of NRAP based antibiotic adjuvants with existing antibiotics will enhance antibacterial activity and reverse resistance. Such strategy will be well positioned to fill the gap before new antibiotics are introduced into the clinical setting, in particular for infections caused by Gram-negative pathogens, because many NRAPs can disrupt the impermeable outer membrane and increase the accumulation of intracellular antibiotics. Collectively, NRAPs provide highly potent leads for the production of next-generation antibiotics against bacterial pathogens in the resistance era.7 Conflicts of interest
There are no conflicts to declare.8 Acknowledgements
This work is supported by the National Key Research and Development Program of China (2017YFC1600305), the National Natural Science Foundation of China (31772796) and the Fund of Modern Agriculture Industry System Innovation in Beijing City Team (BAIC06-2017).9 References
- K. Kupferschmidt, Science, 2016, 352, 758–761 CrossRef CAS PubMed.
- J. O'Neill, Antimicrobial resistance: tackling a crisis for the health and wealth of nations, Review on antimicrobial resistance, 2014, http://archive.wphna.org/wp-content/uploads/2015/06/2014-UK-paper-on-superbugs-projected-to-2050.pdf Search PubMed.
- M. McKenna, CDC Threat Report: We will soon be in a post-antibiotic era, 2013, https://www.wired.com/2013/09/cdc-amr-rpt1/ Search PubMed.
- T. R. Walsh, J. Weeks, D. M. Livermore and M. A. Toleman, Lancet Infect. Dis., 2011, 11, 355–362 CrossRef PubMed.
- Y.-Y. Liu, Y. Wang, T. R. Walsh, L.-X. Yi, R. Zhang, J. Spencer, Y. Doi, G. Tian, B. Dong, X. Huang, L. Yu, D. Gu, H. Ren, X. Chen, L. Lv, D. He, H. Zhou, Z. Liang, J. Liu and J. Shen, Lancet Infect. Dis., 2016, 16, 161–168 CrossRef CAS PubMed.
- Y. Wang, G.-B. Tian, R. Zhang, Y. Shen, J. M. Tyrrell, X. Huang, H. Zhou, L. Lei, H.-Y. Li and Y. Doi, Lancet Infect. Dis., 2017, 17, 390–399 CrossRef CAS PubMed.
- Y. Wang, R. Zhang, J. Li, Z. Wu, W. Yin, S. Schwarz, J. M. Tyrrell, Y. Zheng, S. Wang and Z. Shen, Nat. Microbiol., 2017, 2, 16260 CrossRef CAS PubMed.
- H. F. Chambers and F. R. DeLeo, Nat. Rev. Microbiol., 2009, 7, 629–641 CrossRef CAS PubMed.
- R. D. Gonzales, P. C. Schreckenberger, M. B. Graham, S. Kelkar, K. DenBesten and J. P. Quinn, Lancet, 2001, 357, 1179 CrossRef CAS.
- E. Tacconelli and M. A. Cataldo, Int. J. Antimicrob. Agents, 2008, 31, 99–106 CrossRef CAS PubMed.
- A. M. King, S. A. Reid-Yu, W. Wang, D. T. King, G. De Pascale, N. C. Strynadka, T. R. Walsh, B. K. Coombes and G. D. Wright, Nature, 2014, 510, 503–506 CrossRef CAS PubMed.
- V. Tracanna, J. A. De, M. H. Medema and O. P. Kuipers, FEMS Microbiol. Rev., 2017, 41, 417–429 CrossRef CAS.
- N. Ziemert, M. Alanjary and T. Weber, Nat. Prod. Rep., 2016, 33, 988–1005 RSC.
- V. M. D'Costa, C. E. King, L. Kalan, M. Morar, W. W. Sung, C. Schwarz, D. Froese, G. Zazula, F. Calmels and R. Debruyne, Nature, 2011, 477, 457–461 CrossRef.
- A. L. Harvey, R. Edrada-Ebel and R. J. Quinn, Nat. Rev. Drug Discovery, 2015, 14, 111–129 CrossRef CAS PubMed.
- E. D. Brown and G. D. Wright, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- J. Clardy, M. A. Fischbach and C. T. Walsh, Nat. Biotechnol., 2006, 24, 1541–1550 CrossRef CAS.
- A. Fleming, Br. J. Exp. Pathol., 1929, 10, 226–236 CAS.
- G. Banko, A. L. Demain and S. Wolfe, J. Am. Chem. Soc., 1987, 109, 2858–2860 CrossRef CAS.
- A. R. Awan, B. A. Blount, D. J. Bell, W. M. Shaw, H. Jch, R. M. Mckiernan and T. Ellis, Nat. Commun., 2017, 8, 15202 CrossRef.
- Antibiotic resistance threats in the United States, CDC webpage, 2013, https://www.cdc.gov/drugresistance/threat-report-2013/index.html Search PubMed.
- A. Corona and D. Cattaneo, Clin. Infect. Dis., 2017, 65, 870 CrossRef.
- M. Strieker, A. Tanović and M. A. Marahiel, Curr. Opin. Struct. Biol., 2010, 20, 234–240 CrossRef CAS.
- M. Winn, J. Fyans, Y. Zhuo and J. Micklefield, Nat. Prod. Rep., 2016, 33, 317–347 RSC.
- C. Walsh, Nat. Prod. Rep., 2016, 33, 127–135 RSC.
- D. Hughes, Nat. Rev. Genet., 2003, 4, 432–441 CrossRef CAS PubMed.
- B. Jaurin and T. Grundström, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 4897–4901 CrossRef CAS.
- L. Dortet, L. Poirel and P. Nordmann, BioMed Res. Int., 2014, 2014, 249856 Search PubMed.
- P. Nordmann, L. Poirel, T. R. Walsh and D. M. Livermore, Trends Microbiol., 2011, 19, 588–595 CrossRef CAS.
- Z. Liu, Y. Wang, T. R. Walsh, D. Liu, Z. Shen, R. Zhang, W. Yin, H. Yao, J. Li and J. Shen, Antimicrob. Agents Chemother., 2017, 61, e02233-16 CrossRef.
- A. Saxon, A. Hassner, E. A. Swabb, B. Wheeler and N. F. Adkinson Jr, J. Infect. Dis., 1984, 149, 16–22 CrossRef CAS.
- M. Ramirez and M. Tolmasky, Drug Resist. Updates, 2010, 13, 151–171 CrossRef CAS.
- H. Yao, D. Liu, Y. Wang, Q. Zhang and Z. Shen, Antimicrob. Agents Chemother., 2017, 61, e00112–e00117 CrossRef CAS PubMed.
- E.-J. Yoon, C. Grillot-Courvalin and P. Courvalin, J. Antibiot., 2017, 70, 400–403 CrossRef CAS.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Nature, 2017, 545, 299–304 CrossRef CAS.
- S. Cowan, T. Schirmer, G. Rummel, M. Steiert, R. Ghosh, R. Pauptit, J. Jansonius and J. Rosenbusch, Nature, 1992, 358, 727–733 CrossRef CAS.
- H. Chalhoub, Y. Sáenz, H. Rodriguez-Villalobos, O. Denis, B. C. Kahl, P. M. Tulkens and F. Van Bambeke, Int. J. Antimicrob. Agents, 2016, 48, 740–743 CrossRef CAS.
- X. Wu, J. D. Chavez, D. K. Schweppe, C. Zheng, C. R. Weisbrod, J. K. Eng, A. Murali, S. A. Lee, E. Ramage and L. A. Gallagher, Nat. Commun., 2016, 7, 13414 CrossRef CAS.
- L. J. Piddock, Nat. Rev. Microbiol., 2006, 4, 629–636 CrossRef CAS.
- M. Webber and L. Piddock, J. Antimicrob. Chemother., 2003, 51, 9–11 CrossRef CAS.
- H. Nikaido, J. Bacteriol., 1996, 178, 5853–5859 CrossRef CAS.
- M. Linkevicius, L. Sandegren and D. I. Andersson, Antimicrob. Agents Chemother., 2016, 60, 789–796 CrossRef CAS.
- J. M. Blair, G. E. Richmond and L. J. Piddock, Future Microbiol., 2014, 9, 1165–1177 CrossRef CAS.
- E.-J. Yoon, Y. N. Chabane, S. Goussard, E. Snesrud, P. Courvalin, E. Dé and C. Grillot-Courvalin, mBio, 2015, 6, e00309–00315 CrossRef.
- Z. Shen, X.-Y. Pu and Q. Zhang, Appl. Environ. Microbiol., 2011, 77, 7128–7133 CrossRef CAS.
- J. M. Blair, V. N. Bavro, V. Ricci, N. Modi, P. Cacciotto, U. Kleinekathöfer, P. Ruggerone, A. V. Vargiu, A. J. Baylay and H. E. Smith, Proc. Natl. Acad. Sci. U. S. A., 2015, 112, 3511–3516 CrossRef CAS.
- H. Yao, Z. Shen, Y. Wang, F. Deng, D. Liu, G. Naren, L. Dai, C.-C. Su, B. Wang and S. Wang, mBio, 2016, 7, e01543-16 CrossRef.
- J. Shen, Y. Wang and S. Schwarz, J. Antimicrob. Chemother., 2013, 68, 1697–1706 CrossRef CAS.
- G. Morales, J. J. Picazo, E. Baos, F. J. Candel, A. Arribi, B. Peláez, R. Andrade, M.-Á. de la Torre, J. Fereres and M. Sánchez-García, Clin. Infect. Dis., 2010, 50, 821–825 CrossRef CAS.
- L. Diaz, P. Kiratisin, R. E. Mendes, D. Panesso, K. V. Singh and C. A. Arias, Antimicrob. Agents Chemother., 2012, 56, 3917–3922 CrossRef CAS.
- Y. Liu, Y. Wang, C. Wu, Z. Shen, S. Schwarz, X.-D. Du, L. Dai, W. Zhang, Q. Zhang and J. Shen, Antimicrob. Agents Chemother., 2012, 56, 1650–1654 CrossRef CAS.
- Y. Wang, T. He, S. Schwarz, D. Zhou, Z. Shen, C. Wu, Y. Wang, L. Ma, Q. Zhang and J. Shen, J. Antimicrob. Chemother., 2012, 67, 1094–1098 CrossRef CAS.
- T. Velkov, P. E. Thompson, R. L. Nation and J. Li, J. Med. Chem., 2009, 53, 1898–1916 CrossRef.
- T. Velkov, C. Dai, G. D. Ciccotosto, R. Cappai, D. Hoyer and J. Li, Pharmacol. Ther., 2018, 181, 85–90 CrossRef CAS.
- D. Van Duin, K. S. Kaye, E. A. Neuner and R. A. Bonomo, Diagn. Microbiol. Infect. Dis., 2013, 75, 115–120 CrossRef CAS.
- W. Yin, H. Li, Y. Shen, Z. Liu, S. Wang, Z. Shen, R. Zhang, T. R. Walsh, J. Shen and Y. Wang, mBio, 2017, 8, e00543-17 CrossRef.
- A. Carattoli, L. Villa, C. Feudi, L. Curcio, S. Orsini, A. Luppi, G. Pezzotti and C. F. Magistrali, Eurosurveillance, 2017, 22 CrossRef.
- M. Borowiak, J. Fischer, J. A. Hammerl, R. S. Hendriksen, I. Szabo and B. Malorny, J. Antimicrob. Chemother., 2017, 72, 3317–3324 CrossRef CAS.
- B. B. Xavier, C. Lammens, R. Ruhal, S. Kumar-Singh, P. Butaye, H. Goossens and S. Malhotra-Kumar, Eurosurveillance, 2016, 21, 30280 CrossRef.
- M. Abuoun, E. J. Stubberfield, N. A. Duggett, M. Kirchner, L. Dormer, J. Nunezgarcia, L. P. Randall, F. Lemma, D. W. Crook and C. Teale, J. Antimicrob. Chemother., 2017, 72, 2745–2749 CrossRef CAS.
- Y. Q. Yang, Y. X. Li, C. W. Lei, A. Y. Zhang and H. N. Wang, J. Antimicrob. Chemother., 2018, 73, 1791–1795 CrossRef.
- X. Wang, Y. Wang, Y. Zhou, J. Li, W. Yin, S. Wang, S. Zhang, J. Shen, Z. Shen and Y. Wang, Emerging Microbes Infect., 2018, 7, 122 CrossRef.
- Y. Shen, H. Zhou, J. Xu, Y. Wang, Q. Zhang, T. R. Walsh, B. Shao, C. Wu, Y. Hu, L. Yang, Z. Shen, Z. Wu, Q. Sun, Y. Ou, Y. Wang, S. Wang, Y. Wu, C. Cai, J. Li, J. Shen, R. Zhang and Y. Wang, Nat. Microbiol., 2018, 3, 1054–1062 CrossRef CAS.
- T. J. Kidd, G. Mills, J. Sá-Pessoa, A. Dumigan, C. G. Frank, J. L. Insua, R. Ingram, L. Hobley and J. A. Bengoechea, EMBO Mol. Med., 2017, 9, 430–447 CrossRef CAS.
- A. Cannatelli, T. Giani, M. M. D'Andrea, V. Di Pilato, F. Arena, V. Conte, K. Tryfinopoulou, A. Vatopoulos, G. M. Rossolini and C. S. Group, Antimicrob. Agents Chemother., 2014, 58, 5696–5703 CrossRef.
- A. S. Bayer, T. Schneider and H. G. Sahl, Ann. N. Y. Acad. Sci., 2013, 1277, 139–158 CrossRef CAS.
- C. A. Arias, D. Panesso, D. M. Mcgrath, X. Qin, M. F. Mojica, C. Miller, L. Diaz, T. T. Tran, S. Rincon and E. M. Barbu, N. Engl. J. Med., 2011, 365, 892–900 CrossRef CAS.
- D. Lim and N. C. Strynadka, Nat. Struct. Mol. Biol., 2002, 9, 870–876 CAS.
- P. E. Reynolds, Eur. J. Clin. Microbiol. Infect. Dis., 1989, 8, 943–950 CrossRef CAS.
- A. Müller, A. Klöckner and T. Schneider, Nat. Prod. Rep., 2017, 34, 909–932 RSC.
- Y. Cetinkaya, P. Falk and C. G. Mayhall, Clin. Microbiol. Rev., 2000, 13, 686–707 CrossRef CAS.
- P. Courvalin, Clin. Infect. Dis., 2006, 42, 25–34 CrossRef.
- R. D. Suessmuth and A. Mainz, Angew. Chem., Int. Ed., 2017, 56, 3770–3821 CrossRef CAS.
- R. H. Baltz, Curr. Opin. Pharmacol., 2008, 8, 557–563 CrossRef CAS.
- H. Hamamoto, M. Urai, K. Ishii, J. Yasukawa, A. Paudel, M. Murai, T. Kaji, T. Kuranaga, K. Hamase, T. Katsu, J. Su, T. Adachi, R. Uchida, H. Tomoda, M. Yamada, M. Souma, H. Kurihara, M. Inoue and K. Sekimizu, Nat. Chem. Biol., 2015, 11, 127–133 CrossRef CAS.
- L. L. Ling, T. Schneider, A. J. Peoples, A. L. Spoering, I. Engels, B. P. Conlon, A. Mueller, T. F. Schaberle, D. E. Hughes, S. Epstein, M. Jones, L. Lazarides, V. A. Steadman, D. R. Cohen, C. R. Felix, K. A. Fetterman, W. P. Millett, A. G. Nitti, A. M. Zullo, C. Chen and K. Lewis, Nature, 2015, 517, 455–459 CrossRef CAS PubMed.
- B. Hover, S. Kim, M. Katz, Z. Charlop-Powers, J. Owen, M. Ternei, J. Maniko, A. Estrela, H. Molina, S. Park, D. Perlin and S. Brady, Nat. Microbiol., 2018, 3, 415–422 CrossRef CAS.
- M. S. Rappé and S. J. Giovannoni, Annu. Rev. Microbiol., 2003, 57, 369–394 CrossRef.
- K. Lewis, Nat. Rev. Drug Discovery, 2013, 12, 371–387 CrossRef CAS.
- J. W. Blunt, B. R. Copp, W.-P. Hu, M. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2009, 26, 170–244 RSC.
- P. G. Stansly, R. G. Shepherd and H. J. White, Bull. Johns Hopkins Hosp., 1947, 81, 43–54 CAS.
- M. H. Mccormick, J. M. Mcguire, G. E. Pittenger, R. C. Pittenger and W. M. Stark, Antibiot. Annu., 1955, 3, 606–611 Search PubMed.
- F. Ehlert and H. C. Neu, Eur. J. Clin. Microbiol., 1987, 6, 84–90 CAS.
- A. Müller, M. Wenzel, H. Strahl, F. Grein, T. N. Saaki, B. Kohl, T. Siersma, J. E. Bandow, H. G. Sahl and T. Schneider, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 7077–7086 CrossRef.
- L. Boeck, H. Papiska, R. Wetzel, J. Mynderse, D. Fukuda, F. Mertz and D. Berry, J. Antibiot., 1990, 43, 587–593 CrossRef CAS.
- W. Aretz, J. Meiwes, G. Seibert, G. Vobis and J. Wink, J. Antibiot., 2000, 53, 807–815 CrossRef CAS.
- T. Barsby, M. T. Kelly, S. M. Gagné and R. J. Andersen, Org. Lett., 2001, 3, 437–440 CrossRef CAS.
- C. Bassarello, S. Lazzaroni, G. Bifulco, P. Lo Cantore, N. S. Iacobellis, R. Riccio, L. Gomez-Paloma and A. Evidente, J. Nat. Prod., 2004, 67, 811–816 CrossRef CAS.
- H. He, Appl. Microbiol. Biotechnol., 2005, 67, 444–452 CrossRef CAS.
- T. Barsby, K. Warabi, D. Sørensen, W. T. Zimmerman, M. T. Kelly and R. J. Andersen, J. Org. Chem., 2006, 71, 6031–6037 CrossRef CAS.
- K. Desjardine, A. Pereira, H. Wright, T. Matainaho, M. Kelly and R. J. Andersen, J. Nat. Prod., 2007, 70, 1850–1853 CrossRef CAS.
- Y. Xie, R. Chen, S. Si, C. Sun and H. Xu, J. Antibiot., 2007, 38, 158–161 CrossRef.
- M. Gualtieri, A. Aumelas and J. O. Thaler, J. Antibiot., 2009, 62, 295–302 CrossRef CAS.
- V. G. Isabelle, N. Alexey, A. Luis, L. Peter, B. Gérard, P. T. Maria, C. Pierre, K. Christoph, C. Martine and L. Bruno, Appl. Environ. Microbiol., 2010, 76, 910–921 CrossRef.
- T. Janek, M. Łukaszewicz, T. Rezanka and A. Krasowska, Bioresour. Technol., 2010, 101, 6118–6123 CrossRef CAS.
- C.-D. Qian, X.-C. Wu, Y. Teng, W.-P. Zhao, O. Li, S.-G. Fang, Z.-H. Huang and H.-C. Gao, Antimicrob. Agents Chemother., 2012, 56, 1458–1465 CrossRef CAS.
- Y. Guo, E. Huang, C. Yuan, L. Zhang and A. E. Yousef, Appl. Environ. Microbiol., 2012, 78, 3156–3165 CrossRef CAS.
- M. N. Thaker, W. Wang, P. Spanogiannopoulos, N. Waglechner, A. M. King, R. Medina and G. D. Wright, Nat. Biotechnol., 2013, 31, 922–927 CrossRef CAS.
- K. Yamanaka, K. A. Reynolds, R. D. Kersten, K. S. Ryan, D. J. Gonzalez, V. Nizet, P. C. Dorrestein and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 1957–1962 CrossRef CAS.
- S. Müller, E. Garcia-Gonzalez, A. Mainz, G. Hertlein, N. C. Heid, E. Mösker, H. van den Elst, H. S. Overkleeft, E. Genersch and R. D. Süssmuth, Angew. Chem., Int. Ed., 2014, 53, 10821–10825 CrossRef.
- E. Garcia-Gonzalez, S. Muller, P. Ensle, R. D. Sussmuth and E. Genersch, Environ. Microbiol., 2014, 16, 1297–1309 CrossRef CAS.
- F. S. Tareq, M. A. Lee, H. S. Lee, Y. J. Lee, J. S. Lee, C. M. Hasan, M. T. Islam and H. J. Shin, Org. Lett., 2014, 16, 928–931 CrossRef CAS.
- L. Jiang, L. Wang, J. Zhang, H. Liu, B. Hong, H. Tan and G. Niu, Sci. Rep., 2015, 5, 14111 CrossRef CAS.
- S. Cociancich, A. Pesic, D. Petras, S. Uhlmann, J. Kretz, V. Schubert, L. Vieweg, S. Duplan, M. Marguerettaz, J. Noell, I. Pieretti, M. Hugelland, S. Kemper, A. Mainz, P. Rott, M. Royer and R. D. Sussmuth, Nat. Chem. Biol., 2015, 11, 195–197 CrossRef CAS.
- A. Kling, P. Lukat, D. Almeida, A. Bauer, E. Fontaine, S. Sordello, N. Zaburannyi, J. Herrmann, S. Wenzel, C. König, N. Ammerman, M. Barrio, K. Borchers, F. Bordon-Pallier, M. Brönstrup, G. Courtemanche, M. Gerlitz, M. Geslin, P. Hammann, D. Heinz, H. Hoffmann, S. Klieber, M. Kohlmann, M. Kurz, C. Lair, H. Matter, E. Nuermberger, S. Tyagi, L. Fraisse, J. Grosset, S. Lagrange and R. Müller, Science, 2015, 348, 1106–1112 CrossRef CAS PubMed.
- S. A. Cochrane, B. Findlay, A. Bakhtiary, J. Z. Acedo, E. M. Rodriguez-Lopez, P. Mercier and J. C. Vederas, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, 11561–11566 CrossRef CAS.
- J. Chu, X. Vila-Farres, D. Inoyama, M. Ternei, L. J. Cohen, E. A. Gordon, B. V. Reddy, Z. Charlop-Powers, H. A. Zebroski, R. Gallardo-Macias, M. Jaskowski, S. Satish, S. Park, D. S. Perlin, J. S. Freundlich and S. F. Brady, Nat. Chem. Biol., 2016, 12, 1004–1006 CrossRef CAS.
- C. W. Johnston, M. A. Skinnider, C. A. Dejong, P. N. Rees, G. M. Chen, C. G. Walker, S. French, E. D. Brown, J. Berdy, D. Y. Liu and N. A. Magarvey, Nat. Chem. Biol., 2016, 12, 233–239 CrossRef CAS.
- A. Zipperer, M. C. Konnerth, C. Laux, A. Berscheid, D. Janek, C. Weidenmaier, M. Burian, N. A. Schilling, C. Slavetinsky, M. Marschal, M. Willmann, H. Kalbacher, B. Schittek, H. Brotz-Oesterhelt, S. Grond, A. Peschel and B. Krismer, Nature, 2016, 535, 511–516 CrossRef CAS PubMed.
- E. Huang, X. Yang, L. Zhang, S. H. Moon and A. E. Yousef, FEMS Microbiol. Lett., 2017, 364, fnx049 Search PubMed.
- Y. Liu, S. Ding, R. Dietrich, E. Märtlbauer and K. Zhu, Angew. Chem., Int. Ed., 2017, 56, 1486–1490 CrossRef CAS.
- J. Ma, H. Huang, Y. Xie, Z. Liu, J. Zhao, C. Zhang, Y. Jia, Y. Zhang, H. Zhang and T. Zhang, Nat. Commun., 2017, 8, 391 CrossRef PubMed.
- S. Son, Y. S. Hong, M. Jang, K. T. Heo, B. Lee, J. P. Jang, J. W. Kim, I. J. Ryoo, W. G. Kim and S. K. Ko, J. Nat. Prod., 2017, 80, 3025–3031 CrossRef CAS PubMed.
- T. Velkov, A. Gallardo-Godoy, J. Swarbrick, M. Blaskovich, A. Elliott, M. Han, P. Thompson, K. Roberts, J. Huang, B. Becker, M. Butler, L. Lash, S. Henriques, R. Nation, S. Sivanesan, M. Sani, F. Separovic, H. Mertens, D. Bulach, T. Seemann, J. Owen, J. Li and M. Cooper, Cell Chem. Biol., 2018, 25, 380–391 CrossRef CAS.
- L. Pantel, T. Florin, M. Dobosz-Bartoszek, E. Racine, M. Sarciaux, M. Serri, J. Houard, J. M. Campagne, R. M. de Figueiredo, C. Midrier, S. Gaudriault, A. Givaudan, A. Lanois, S. Forst, A. Aumelas, C. Cotteaux-Lautard, J. M. Bolla, C. Vingsbo Lundberg, D. L. Huseby, D. Hughes, P. Villain-Guillot, A. S. Mankin, Y. S. Polikanov and M. Gualtieri, Mol. Cell, 2018, 70, 83–94 CrossRef CAS.
- S. Müller, E. Garcia-Gonzalez, E. Genersch and R. D. Süssmuth, Nat. Prod. Rep., 2015, 32, 765–778 RSC.
- W. K. Mousa, B. Athar, N. J. Merwin and N. A. Magarvey, Nat. Prod. Rep., 2017, 34, 1302–1331 RSC.
- S. Panthee, A. Paudel, H. Hamamoto and K. Sekimizu, Front. Microbiol., 2017, 8, 373 Search PubMed.
- P. J. Rutledge and G. L. Challis, Nat. Rev. Microbiol., 2015, 13, 509–523 CrossRef CAS.
- T. Weber, K. Blin, S. Duddela, D. Krug, H. U. Kim, R. Bruccoleri, S. Y. Lee, M. A. Fischbach, R. Müller and W. Wohlleben, Nucleic Acids Res., 2015, 43, W237–W243 CrossRef CAS.
- M. A. Skinnider, C. A. Dejong, P. N. Rees, C. W. Johnston, H. Li, A. L. Webster, M. A. Wyatt and N. A. Magarvey, Nucleic Acids Res., 2015, 43, 9645–9662 CAS.
- M. Röttig, M. H. Medema, K. Blin, T. Weber, C. Rausch and O. Kohlbacher, Nucleic Acids Res., 2011, 39, W362 CrossRef.
- Y. Minowa, M. Araki and M. Kanehisa, J. Mol. Biol., 2007, 368, 1500–1517 CrossRef CAS.
- T. Stachelhaus, H. D. Mootz and M. A. Marahiel, Chem. Biol., 1999, 6, 493–505 CrossRef CAS.
- Y. Tanaka, M. Izawa, Y. Hiraga, Y. Misaki, T. Watanabe and K. Ochi, Appl. Microbiol. Biotechnol., 2017, 101, 4417–4431 CrossRef CAS.
- M. M. Zhang, F. T. Wong, Y. Wang, S. Luo, Y. H. Lim, E. Heng, L. Y. Wan, R. E. Cobb, B. Enghiad and E. L. Ang, Nat. Chem. Biol., 2017, 13, 607 CrossRef CAS.
- S. B. Bumpus, B. S. Evans, P. M. Thomas, I. Ntai and N. L. Kelleher, Nat. Biotechnol., 2009, 27, 951–956 CrossRef CAS.
- C. Y. Chen, F. R. Chang, Y. C. Shih, T. J. Hsieh, Y. C. Chia, H. Y. Tseng, H. C. Chen, S. J. Chen, M. C. Hsu and Y. C. Wu, J. Nat. Prod., 2000, 63, 1475–1478 CrossRef CAS.
- J. L. Meier, S. Niessen, H. S. Hoover, T. L. Foley, B. F. Cravatt and M. D. Burkart, ACS Chem. Biol., 2009, 4, 948–957 CrossRef CAS.
- M. M. Schofield and D. H. Sherman, Curr. Opin. Biotechnol., 2013, 24, 1151–1158 CrossRef CAS.
- A. T. Tucker, S. P. Leonard, C. D. Dubois, G. A. Knauf, A. L. Cunningham, C. O. Wilke, M. S. Trent and B. W. Davies, Cell, 2018, 172, 618–628 CrossRef CAS.
- A. R. Awan, B. A. Blount, D. J. Bell, W. M. Shaw, J. C. H. Ho, R. M. Mckiernan and T. Ellis, Nat. Commun., 2017, 8, 15202 CrossRef.
- R. A. Oliver, R. Li and C. A. Townsend, Nat. Chem. Biol., 2018, 14, 5–7 CrossRef CAS.
- Z. Liu, J. Li, X. Wang, D. Liu, Y. Ke, Y. Wang and J. Shen, Front. Microbiol., 2018, 9, 248 CrossRef.
- J. Shoji and H. Hinoo, J. Antibiot., 1975, 28, 60–63 CrossRef CAS.
- J. Shoji, H. Hinoo, R. Sakazaki, T. Kato, Y. Wakisaka, M. Mayama, S. Matsuura and H. Miwa, J. Antibiot., 1978, 31, 652–661 CrossRef.
- X. Vilafarres, J. Chu, D. Inoyama, M. A. Ternei, C. Lemetre, L. J. Cohen, W. Cho, B. V. Reddy, H. A. Zebroski and J. S. Freundlich, J. Am. Chem. Soc., 2017, 139, 1404–1407 CrossRef CAS.
- C. de la Fuente-Nunez, M. D. Torres, F. J. Mojica and T. K. Lu, Curr. Opin. Microbiol., 2017, 37, 95–102 CrossRef CAS.
- A. Okano, N. A. Isley and D. L. Boger, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 5052–5061 Search PubMed.
- A. Markham, Drugs, 2014, 74, 1823–1828 CrossRef CAS.
- R. A. Leese, Patent application WO2010075416, 2010.
- J. Quale, N. Shah, P. Kelly, E. Babu, M. Backer, G. Rosas-Garcia, J. Salamera, A. George, S. Bratu and D. Landman, Microb. Drug Resist., 2012, 18, 132–136 CrossRef CAS PubMed.
- A. Gallardogodoy, C. Muldoon, B. Becker, A. G. Elliott, L. H. Lash, J. X. Huang, M. S. Butler, R. Pelingon, A. M. Kavanagh and S. Ramu, J. Med. Chem., 2016, 59, 1068–1077 CrossRef CAS.
- F. Rabanal and Y. Cajal, Nat. Prod. Rep., 2017, 34, 886–908 RSC.
- B. Terlain and J. P. Thomas, Bull. Soc. Chim. Fr., 1971, 6, 2363–2365 CAS.
- J. N. Steenbergen, J. Alder, G. M. Thorne and F. P. Tally, J. Antimicrob. Chemother., 2005, 55, 283–288 CrossRef CAS.
- Y. Chuang, H. Lin, P. Chen, C. Lin, J. Wang, Y. Chen and S. Chang, Clin. Infect. Dis., 2017, 64, 1026–1034 CrossRef CAS.
- M. Strieker and M. A. Marahiel, ChemBioChem, 2009, 10, 607–616 CrossRef CAS.
- M. A. Farha and E. D. Brown, Nat. Prod. Rep., 2016, 33, 668–680 RSC.
- S. F. Oppedijk, N. I. Martin and E. Breukink, Biochim. Biophys. Acta, 2016, 1858, 947–957 CrossRef CAS.
- L. Robbel and M. A. Marahiel, J. Biol. Chem., 2010, 285, 27501–27508 CrossRef CAS.
- V. Pader, S. Hakim, K. L. Painter, S. Wigneshweraraj, T. B. Clarke and A. M. Edwards, Nat. Microbiol., 2016, 2, 16194 CrossRef CAS.
- C. Sohlenkamp and O. Geiger, FEMS Microbiol. Rev., 2016, 40, 133–159 CrossRef CAS.
- C. Fu, L. Keller, A. Bauer, M. Brönstrup, A. Froidbise, P. Hammann, J. Herrmann, G. Mondesert, M. Kurz and M. Schiell, J. Am. Chem. Soc., 2015, 137, 7692–7705 CrossRef CAS PubMed.
- N. Malanovic and K. Lohner, Biochim. Biophys. Acta, 2016, 1858, 936–946 CrossRef CAS PubMed.
- A. Upadhyay, F. Fontes, M. Gonzalezjuarrero, M. R. Mcneil, D. C. Crans, M. Jackson and D. C. Crick, ACS Cent. Sci., 2015, 1, 292–302 CrossRef CAS PubMed.
- D. Jonas, I. Engels, D. Hartung, J. Beyersmann, U. Frank and F. D. Daschner, J. Antimicrob. Chemother., 2003, 51, 275–280 CrossRef CAS PubMed.
- S. H. Moon, X. Zhang, G. Zheng, D. G. Meeker, M. S. Smeltzer and E. Huang, J. Med. Chem., 2017, 60, 9630–9640 CrossRef CAS.
- D. M. McGrath, E. M. Barbu, W. H. Driessen, T. M. Lasco, J. J. Tarrand, P. C. Okhuysen, D. P. Kontoyiannis, R. L. Sidman, R. Pasqualini and W. Arap, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 3477–3482 CrossRef CAS.
- J. V. Olsen, S. E. Ong and M. Mann, Mol. Cell. Proteomics, 2004, 3, 608–614 CrossRef CAS.
- W. Y. Chen, H. Y. Chang, J. K. Lu, Y. C. Huang, S. G. Harroun, Y. T. Tseng, Y. J. Li, C. C. Huang and H. T. Chang, Adv. Funct. Mater., 2015, 25, 7189–7199 CrossRef CAS.
- Y. R. Nobel, L. M. Cox, F. F. Kirigin, N. A. Bokulich, S. Yamanishi, I. Teitler, J. Chung, J. Sohn, C. M. Barber and D. S. Goldfarb, Nat. Commun., 2015, 6, 7486 CrossRef.
- C. Reading and M. Cole, Antimicrob. Agents Chemother., 1977, 11, 852–857 CrossRef CAS.
- E. Garcia-Fernandez, G. Koch, R. M. Wagner, A. Fekete, S. T. Stengel, J. Schneider, B. Mielich-Suss, S. Geibel, S. M. Markert, C. Stigloher and D. Lopez, Cell, 2017, 171, 1354–1367 CrossRef CAS.
- W. E. Rose, L. T. Schulz, D. Andes, R. Striker, A. D. Berti, P. R. Hutson and S. K. Shukla, Antimicrob. Agents Chemother., 2012, 56, 5296–5302 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2019 |