
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Halimane diterpenoids: sources, structures, nomenclature and biological activities†
Alejandro M.
Roncero
 ,
Ignacio E.
Tobal
,
Rosalina F.
Moro
,
David
Díez
and
Isidro S.
Marcos
*
,
Ignacio E.
Tobal
,
Rosalina F.
Moro
,
David
Díez
and
Isidro S.
Marcos
*
Departamento de Química Orgánica, Facultad de Ciencias Químicas, Universidad de Salamanca, Plaza de los Caidos 1-5, 37008 Salamanca, Spain. E-mail: ismarcos@usal.es
First published on 27th April 2018
Abstract
Covering: 1970 to 2017
Diterpenes with a halimane skeleton constitute a small group of natural products that can be biogenetically considered as being between labdane and clerodane diterpenoids. Some of these compounds show biological activities, such as antitumour, mosquito repellency, germination inhibition and antimicrobial, as well as being biomarkers for tuberculosis. To the best of our knowledge, there are no reviews on these compounds. In this review, halimane skeleton diterpenoids are classified according to their biogenetic origin, characterization and/or the enzymes involved in their biosynthesis. Herein, a review of their synthesis or synthetic approaches is communicated.
 Alejandro M. Roncero | Alejandro Martín Roncero received his B.Sc. in Chemistry with Honours from the University of Salamanca in 2015. In 2016 he completed an M.Sc. in Evaluation and Development of New Drugs at the same university. Currently, he is a researcher at the University of Salamanca carrying out his PhD under the supervision of Professor I. S. Marcos focusing on the synthesis and biological evaluation of new antitumour lipids. |
 Ignacio E. Tobal | Ignacio E. Tobal obtained his BSc in Chemistry in 2015 at the University of Salamanca and completed his Masters studies in drug discovery and pharmaceutical sciences at the same university. He has worked in antitumour alkylphosphocholines and bioconjugates. Currently, he is a researcher at the University of Salamanca and has initiated his PhD in the area of organic synthesis and natural products under the supervision of Prof. Isidro S. Marcos and Prof. David Diez. |
 Rosalina F. Moro | Rosalina Fernández-Moro obtained her PhD in Organic Chemistry from the Universidad de Salamanca in 1986 under the supervision of Professors J. G. Urones and P. Basabe. She then carried out her postdoctoral research with Professor Steven V. Ley at Imperial College of Science, Technology and Medicine in London (1989–1991), obtaining the D.I.C. in September 1991. Currently she is a Lecturer in Organic Chemistry at the University of Salamanca, where she is researching the synthesis of bioactive compounds, especially indole alkaloids and organocatalysis. |
 David Díez | David Diez received his B.Sc. with Honours in Chemistry from the University of Salamanca in 1982. In 1986 he completed his PhD with Honours at the same University under the supervision of Professors J. G. Urones and I. S. Marcos. Then, he carried out a postdoctoral post (1988–1990, British Council Fellowship) with Professor Steven V. Ley at Imperial College of Science, Technology and Medicine (London), DIC 1991. He became Associate Professor in 1991 at the University of Salamanca and full Professor in 2008 at the same university. He is co-author of more than 180 papers and he acts as referee for many international scientific journals. Currently he is the Dean of the Chemistry Faculty at the University of Salamanca. His current research interests are focused on the transformation of natural products into biologically active compounds, the chemistry of cyclopropanes, sulfones, tetrahydropyrans, and chiral amides, and recently organocatalysis and nanoparticles. |
 Isidro S. Marcos | Isidro Sánchez Marcos received his B.Sc. in Chemistry at the University of Salamanca in 1976 and obtained his PhD from the same University in 1979 under the supervision of Professors J. de Pascual Teresa and J. G. Urones. He was promoted to Lecturer in 1986 and to Full Professor in 2010. The area where he has developed his research is Natural Products. He has isolated and characterized more than 200 compounds and described six new carbon skeletons, in more than 180 publications. Now he is interested in the synthesis of bioactive natural products and the synthesis of structural and functional hybrid compounds of antitumour lipidic ethers and terpenolides. |
1. Introduction and background
Among natural products, terpenes are one of the most numerous and structurally diverse groups. They can be found in nearly all life forms showing a great number of functional activities. Currently, tens of thousands of compounds in this class1,2 (around 55![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000) are known. As soon as new compounds are discovered, from terrestrial or marine origin,3 they are included in reviews on different terpene classes (monoterpenes, sesquiterpenes,4 diterpenes,5,6 sesterterpenes7 and triterpenes8).
000) are known. As soon as new compounds are discovered, from terrestrial or marine origin,3 they are included in reviews on different terpene classes (monoterpenes, sesquiterpenes,4 diterpenes,5,6 sesterterpenes7 and triterpenes8).
Within terpenes, diterpenes are one of the largest families of natural molecules, with more than 18000 compounds derived from GGPP (E,E,E-geranylgeranyl diphosphate). Classification of these compounds is done according to their biogenesis, leading to 126 different carbon skeletons known until now.9
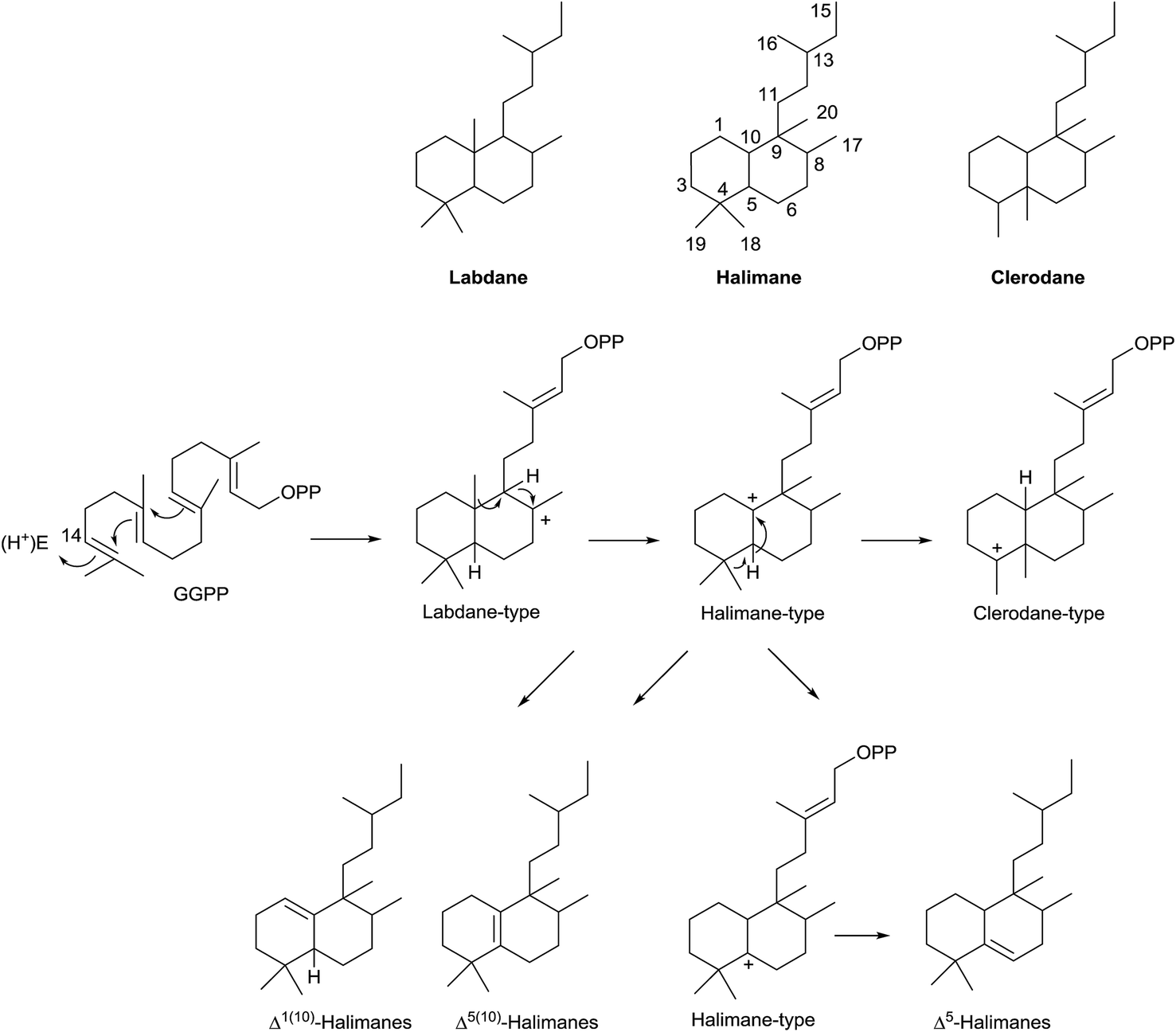
Two big groups can be distinguished among the diterpenes family: those diterpenes in which a pyrophosphate ion is involved in the first biogenetic step and those diterpenes that are generated as a consequence of cyclizations that do not include pyrophosphate ion in the first step.10–13 Labdanes and related diterpenes (like clerodanes14,15) are included in this second group. According to Peters,16 halimanes are found within the labdane and clerodane group, as indicated in the general biogenetic scheme (Scheme 1). The halimane-type carbocation can be in position 10 leading to Δ1(10) or Δ5(10) halimanes or in position 5, due to a hydride 1,2-shift, leading to Δ5 halimanes. Other halimanes can be understood from these intermediates or derivatives, as reported later on.
 | ||
| Scheme 1 | ||
The name ‘halimane’ was introduced in order to provide a simple nomenclature for the isolated ent-halimic acid compound series17 in Halimium viscosum (Fig. 1). Previously in the literature, these compounds were known by generic names, such as rearranged labdanes,18,19 isolabdanes20 or even friedolabdanes.21–25
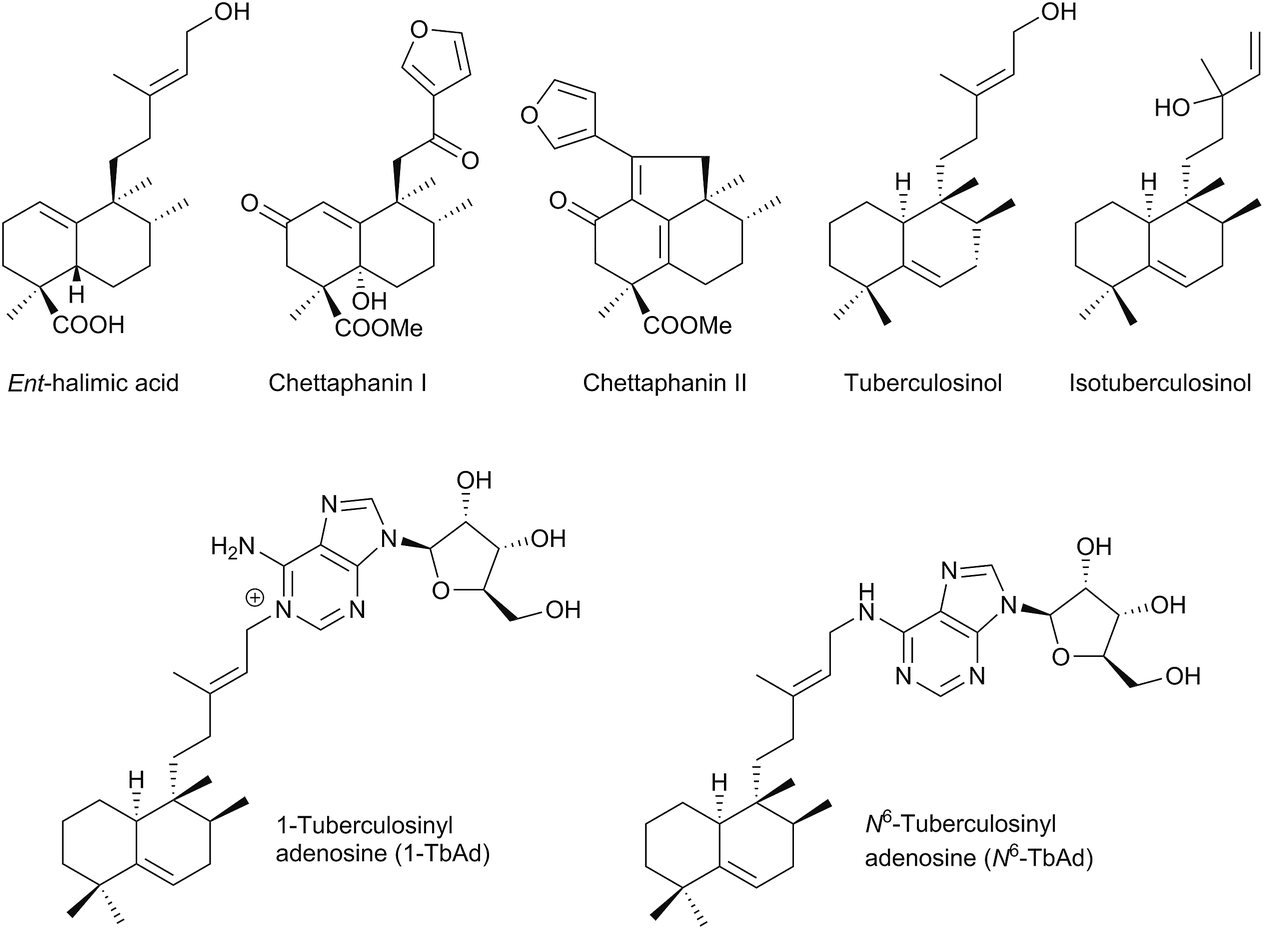 | ||
| Fig. 1 Relevant halimane diterpenoids. | ||
Halimium viscosum is a plant of the Cistaceae family that shows a Hispano–Mauritanian distribution and is abundant in the west of the Iberian Peninsula. Ent-halimic acid and acetoxy ent-halimic acid are found in a 1:1 ratio in considerable amounts in Halimium viscosum extracts (47%), 0.34% with respect of the dry plant weight. Nowadays, a cheap, quick and efficient methodology for the isolation of ent-halimic acid has been achieved. The study of the components of Halimium viscosum and the need for large amounts of ent-halimic acid for the synthesis of biologically active compounds led us to search for Halimium viscosum plants in new locations with better access. In this manner, we found several places for the study of the Halimium viscosum components, such as Villarino de los Aires (Salamanca, Spain),17 La Fregeneda (Salamanca, Spain)26,27 and Valparaiso (Zamora, Spain),28 localities quite close together. To our surprise, in the first location rearranged ent-labdanes (ent-halimanes) with an unsaturated side chain were isolated, in the second ent-halimanes with a saturated side chain were found, and, from the plants of Valparaiso, labdanes with the structural strangeness of having a carboxyl function at C-17 were only isolated. At the time of these studies, to find different chemotypes of the same plant was a novelty. Furthermore, two other chemotypes of Halimium viscosum have been localized in Portugal near the border with Salamanca (Celorico da Beira29 and San João da Pesqueira30).
In Halimium viscosum, in addition to the referred ent-halimanes and labdanes, a series of compounds that show new carbon skeletons has been isolated, characterized and then synthesized. Among them, tricyclic diterpenoids such as valparanes31–35 and valparolanes,36 bicyclic diterpenoids with tormesane skeleton (a new type of sphenolobanes),37,38 tormesolanes39 and new rearranged labdanes with an aromatized ring B, such as fregenedanes40,41 and isofregenedanes,42,43 are found. The biosynthesis of these new carbon skeletons, as valparanes and tormesanes, contrary to the halimane diterpenoids, starts with a cyclization involving the pyrophosphate, so in this plant two different classes of diterpene cyclases should be present.
Owing to the high number of rearranged ent-labdane derivatives isolated in Halimium viscosum and in order to simplify their nomenclature and to facilitate their classification, the name ent-halimane was proposed for that carbon skeleton. Compounds of this class were isolated previously for the first time in the roots of Adenochlaena siamensis, chettaphanin I and chettaphanin II (Fig. 1), with unknown absolute configuration.
To the best of our knowledge, there are no reviews of known halimanes. Owing to the important biological properties that some of them show, and their novelty and structural diversity, we considered it to be very interesting to establish a classification and write a review on these compounds.
Nowadays, the biological activities of these compounds (such as antitumour,44–48 anti-inflammatory,49 antimicrobial,50–52 antifungal,53 germination inhibitors54) are being studied from different points of view. Compounds such as tuberculosinol,55,56 isotuberculosinol57–59 and analogues (Fig. 1) can be considered as virulence factors (VF) in Mycobacterium tuberculosis, a microorganism that causes tuberculosis illness, a major source of morbidity and mortality worldwide.60 These compounds could be targets in a new route to anti-infective therapy.60,61 Recently, tuberculosinyl-adenosine derivatives have been tested as biomarkers for early diagnosis of tuberculosis.62,63
1.1. Sources of halimane diterpenoids
Halimane diterpenoids form a group of secondary metabolites that can be found in different plant species of several families and in other taxonomic group, such as marine organisms and microorganisms (Table 1).| Kingdom | Phylum | Class | Order | Family | Genus | No of species |
|---|---|---|---|---|---|---|
| Plantae | Magnoliophyta | Dicotyledon | Asterales | Asteraceae | Ageratum, Conyza | 2 |
| Compositae | Amphiachyris, Baccharis, Chiliotrichum, Chromolaena, Haplopappus, Koanophyllon, Nardophyllum, Oedera, Ophryosporus, Relhania, Stevia | 15 | ||||
| Caryophyllales | Portulacaceae | Portulaca | 3 | |||
| Fabales | Leguminoseae | Colophospermum, Hymenaea, Tessmania | 4 | |||
| Lamiales | Lamiaceae | Callicarpa, Isodon, Leonurus, Leucas, Marrubium, Plectranthus, Salvia, Teucrium, Vitex | 13 | |||
| Magnoliales | Annonaceae | Polyalthia | 1 | |||
| Malpighiales | Euphorbiaceae | Acalypha, Aparisthmium, Chrozophora, Cladogynos, Croton, Excoecaria, Mallotus | 17 | |||
| Malvales | Cistaceae | Cistus, Halimium | 2 | |||
| Pandanales | Velloziaceae | Vellozia | 3 | |||
| Sapindales | Meliaceae | Aglaia, Dysoxylum | 2 | |||
| Dodonaeaeae | Dodonaea | 1 | ||||
| Marchantiophyta | Jungermanniopsida | Jungermanniales | Geocalycaceae | Heteroscyphus | 1 | |
| Jungermanniaceae | Jungermannia | 3 | ||||
| Plagiochilaceae | Plagiochila | 1 | ||||
| Scapaniaceae | Scapania | 1 | ||||
| Pleuroziales | Pleuroziaceae | Pleurozia | 1 | |||
| Animalia | Chordata | Ascidiacea | Aplousobranchia | Polycitoridae | Cystodytes | 1 |
| Cnidaria | Anthozoa | Alcyonacea | Plexauridae | Echinomuricea | 1 | |
| Mollusca | Gastropoda | Nudibranchia | Aeolidiidae | Spurilla | 1 | |
| Dorididae | Doris | 1 | ||||
| Porifera | Demospongiae | Agelasida | Agelasidae | Agelas | 4 | |
| Axinellida | Raspailiidae | Raspailia | 1 | |||
| Dictyoceratida | Dysideidae | Dysidea | 1 | |||
| Poecilosclerida | Mycalidae | Mycale | 1 | |||
| Bacteria | Actinobacteria | Actinobacteria | Actinomycetales | Micromonosporaceae | Micromonospora | 1 |
| Mycobacteriaceae | Mycobacterium | 1 | ||||
| Streptomycetales | Streptomycetaceae | Kitasatospora | 1 |
The majority of known halimanes have been isolated from Magnoliophyta (Dicotyledon) plants and among them halimanes can be found in nine orders and curiously only in eleven families. It looks like, as occurs in the clerodane diterpenoid family, that the orders and families that contain halimanes are not very numerous. That is, they do not follow the usual taxonomic tendency in which a pyramidal form will be found when going from order to genus.14
Among the families that possess halimane diterpenoids, we can highlight the Compositae, Lamiaceae and Euphorbiaceae families owing to the number of studied species and the number of halimanes found in them. In Cistaceae, it is noteworthy that all the Cistus studied until now, as can be observed in the reviews of diterpenes by Prof. Hanson,6 halimanes have only been found as minor compounds in one of them (Cistus laurifolius). In contrast, the other studied plant of that family (Halimium viscosum) is the species that has provided a major number of halimanes.
The occurrence of halimanes in non-dicotyledon plants (with only seven species studied), in eleven marine organisms (highlighting the sponges), and in three strains of bacteria is much reduced. However, the interest in halimane diterpenoids is definitively much greater because of the presence of halimane purines with important biological activities among them. Besides the mentioned tuberculosinyl adenosine derivatives isolated from Mycobacterium tuberculosis, eleven other halimane purines, mainly isolated from marine sponges, have been characterized. They could constitute a very interesting area for further biological and chemical research owing to their biological activities.
Herein, an up-to-date review of the 246 isolated and characterized natural halimanes is shown.
1.2. Biological activities of halimanes
The halimane purine derivatives present the most relevant biological activities related to tuberculosis biomarkers, antifouling and antimicrobial activities.Currently, two genes (Rv3377c and Rv3378c) found only in virulent species of genus Mycobacterium (such as M. tuberculosis and M. bovis) are known and they are involved in tuberculosinol and isotuberculosinol biosynthesis. Interestingly, these genes could not be found in avirulent species of genus Mycobacterium (such as M. smegmatis and M. avium),59,65 so they may be involved in the infection processes of these bacteria. However, these genes only seem to be functional in M. tuberculosis and not in M. bovis, so that may explain the lower virulence of M. bovis in comparison with M. tuberculosis.66,67
It has been observed that tuberculosinol and isotuberculosinol (Fig. 1), produced in vivo by Mycobacterium tuberculosis68,69 (in a 1:1 ratio), inhibit phagolysosome maturation as well as macrophage phagocytosis, plus a synergistical effect increased by the coexistence of both compounds has been observed. Decrease of the phagocytic capacity could help to explain the pathogenicity of M. tuberculosis.59 Thus, both tuberculosinol/isotuberculosinol biosynthetic proteins (Rv3377c and Rv3378c) are essential for the bacteria's survival inside the macrophage.57,59,70 Because of that, both enzymes are likely to be new potential targets for the development of new drugs.
Recently, two new natural tuberculosinol derivatives (having an adenosine unit attached at diterpene C15) have been isolated from M. tuberculosis, 1-TbAd71 and N6-TbAd62 (Fig. 1). In a comparative lipidomics assay between M. tuberculosis and M. bovis, it has been observed that these two compounds appear in higher amounts in M. tuberculosis and they accumulate to comprise >1% of all M. tuberculosis lipids, so they could serve as an abundant chemical marker of M. tuberculosis.62,71 In addition, in this study it has been proved that the Rv3378c enzyme is responsible for 1-TbAd formation, so this protein appears to be a tuberculosinyl transferase (prenyl transferase).71 In fact, they are being evaluated as biomarkers for tuberculosis.62
Owing to all mentioned above, Rv3377c and Rv3378c are new targets for anti-infective therapies against tuberculosis that block virulence factor (tuberculosinols) formation.60
Nosyberkol (isotuberculosinol) was isolated for the first time from Raspailia sp. and the sponge contains halimane purines too.72 Recently there has been enormous interest in the bioactivities of diterpenyl purines.73
From Agelas sp.,74 nine halimane purines have been isolated that show antibacterial, antifungal, antimalarial, cytotoxic activities, inhibition of adenosine transfer rabbit erythrocytes and Ca2+ channel antagonistic action and α1 adrenergic blockade, among others. Some of these compounds possess antifouling activity against macroalgae. Natural products with this activity are very useful in the fishing industry as an alternative to the antiadherent mixtures that include metals with toxic effects on the marine environment. For this reason, antifouling substances with no or reduced toxicity must be discovered or developed.75
Other halimanes show interesting biological activities, such as antibacterial,50 antiviral,78 antifungal,53 and antimalarial.79 Others exert anti-inflammatory,45 anti-ulcerogenic,80 anti-hyperlipidemic81 or hepatogenic activities. Some halimane diterpenoids act as allelochemicals, regulating the growth of monocotyledon seeds.54 Others behave as allomones acting against insects as repellents or mosquitocidals.82 From the Antarctic nudibranch a series of allomones has been isolated, including diterpene glycerides, which seem to be involved in the defense of those nudibranchs.83
Many of the known halimane diterpenoids have not been biologically evaluated.
2. Biosynthesis
2.1. Biosynthesis overview
Halimane skeleton diterpenoids are formed by cyclization of geranylgeranyl diphosphate (GGPP), catalyzed by class II diterpene cyclases (DTCs).12,13,16 These enzymes are characterized by having an aspartate-rich DXDD motif and are differentiated between class I terpene synthases (TPS) enzymes by not having the characteristic aspartate-rich DDXXD motif that binds divalent metal ions required for catalysis of diphosphate ionization11 (Scheme 2). Several studies have been carried out revealing that the ‘middle’ aspartate in the DXDD motif acts as an acid catalyst.11,84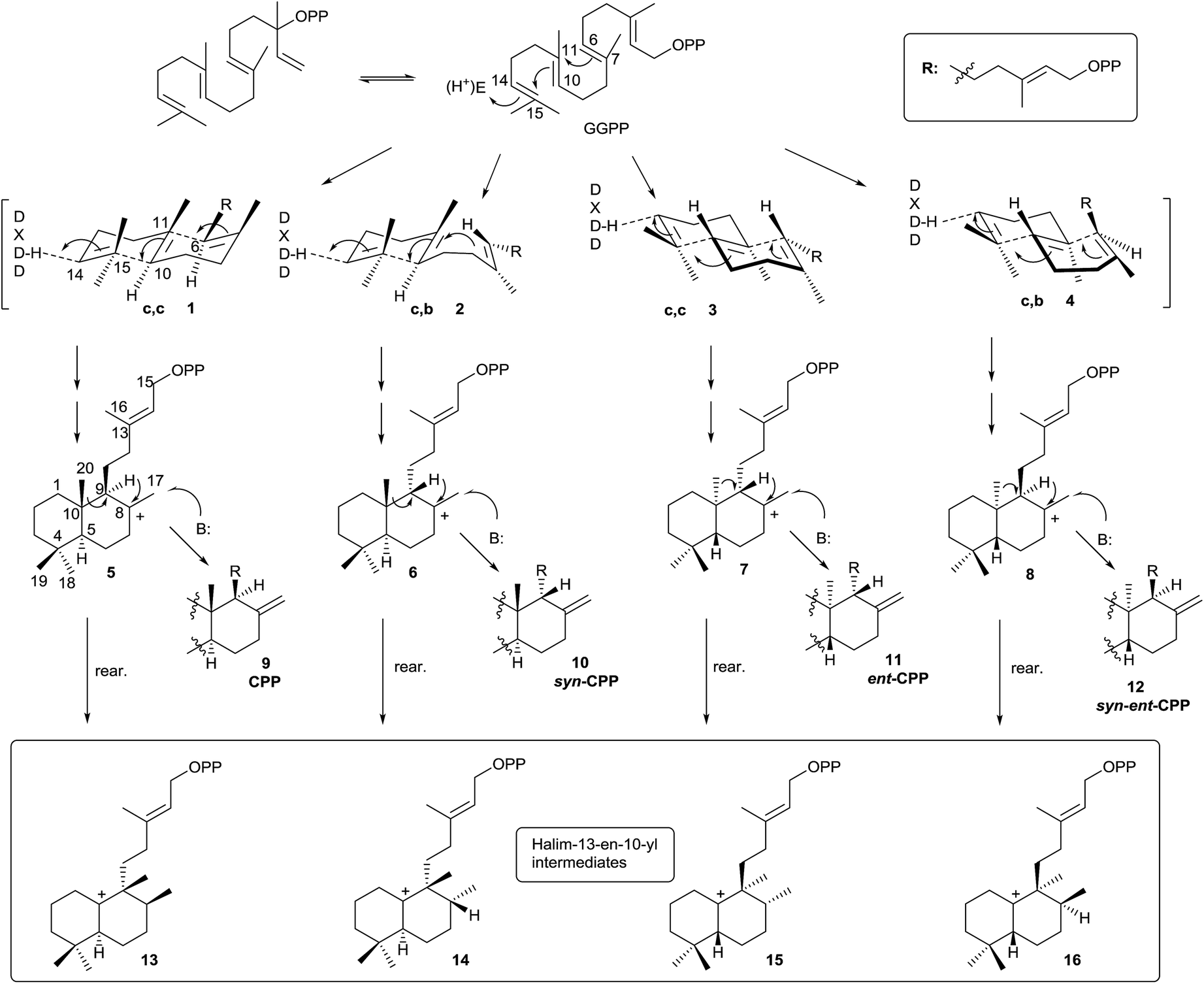 | ||
| Scheme 2 | ||
The DTCs catalyze GGPP bicyclization by a general acid–base mechanism. The process starts by 14,15 double bond protonation of E,E,E-GGPP followed by carbon–carbon double bond anti addition (C10 on C15, then C6 on C11) to give four possible bicyclic products. Effectively, depending on the different prochiral substrate conformer (1, 2, 3 or 4) operating in the cyclization, the corresponding stereoisomer of labda-13-en-8-yl diphosphate intermediate will appear (5, 6, 7 or 8).
In this manner, chair–chair (c,c) conformer 1 leads to 8-carbonium ion 5, which possesses the ‘normal’ anti,anti absolute stereochemistry (between H9–C20–H5). Chair–boat conformer (c,b) 2 leads to carbocation 6 with the ‘normal’ syn,anti absolute stereochemistry (between H9–C20–H5). Antipode conformers of the preceding ones, the chair–chair (c,c) 3 and chair–boat (c,b) 4, lead to the 8-carbonium ions 7 and 8 with anti,anti and syn,anti absolute stereochemistry (between H9–C20–H5), respectively.
Labda-13E-en-8-yl diphosphate intermediates (5, 6, 7 and 8) lead to the different copalyl diphosphate labdane precursors by reaction with a base: copalyl diphosphate (CPP, 9), syn-copalyl diphosphate (syn-CPP, 10), ent-copalyl diphosphate (ent-CPP, 11) and syn-ent-copalyl diphosphate (syn-ent-CPP, 12), respectively.16 Among labdanes, the major ones are ent-CPP 11 (9R,10R) derivatives, followed by the ‘normal’ labdane precursor CPP 9 (9S,10S) derivatives. A lot of class II DTCs that lead to these two precursors are known. However, syn-CPP 10 (9R,10S) derivatives are less common, and only a few syn-ent-CPP 12 (9S,10R) labdane derivatives have been reported.16
However, this is not the only way the intermediate carbocations 5–8 can evolve. A rearrangement by sequential 1,2-hydride shifts (H–C9 to H–C8) and methyl group migration (C10 to C9 methyl migration) can occur instead of producing the stabilization of intermediates 5–8 (Scheme 2) by proton abstraction. In this way, the cationic halim-13E-en-10-yl diphosphate intermediates 13–16 are respectively produced, and they already contain the halimane backbone.
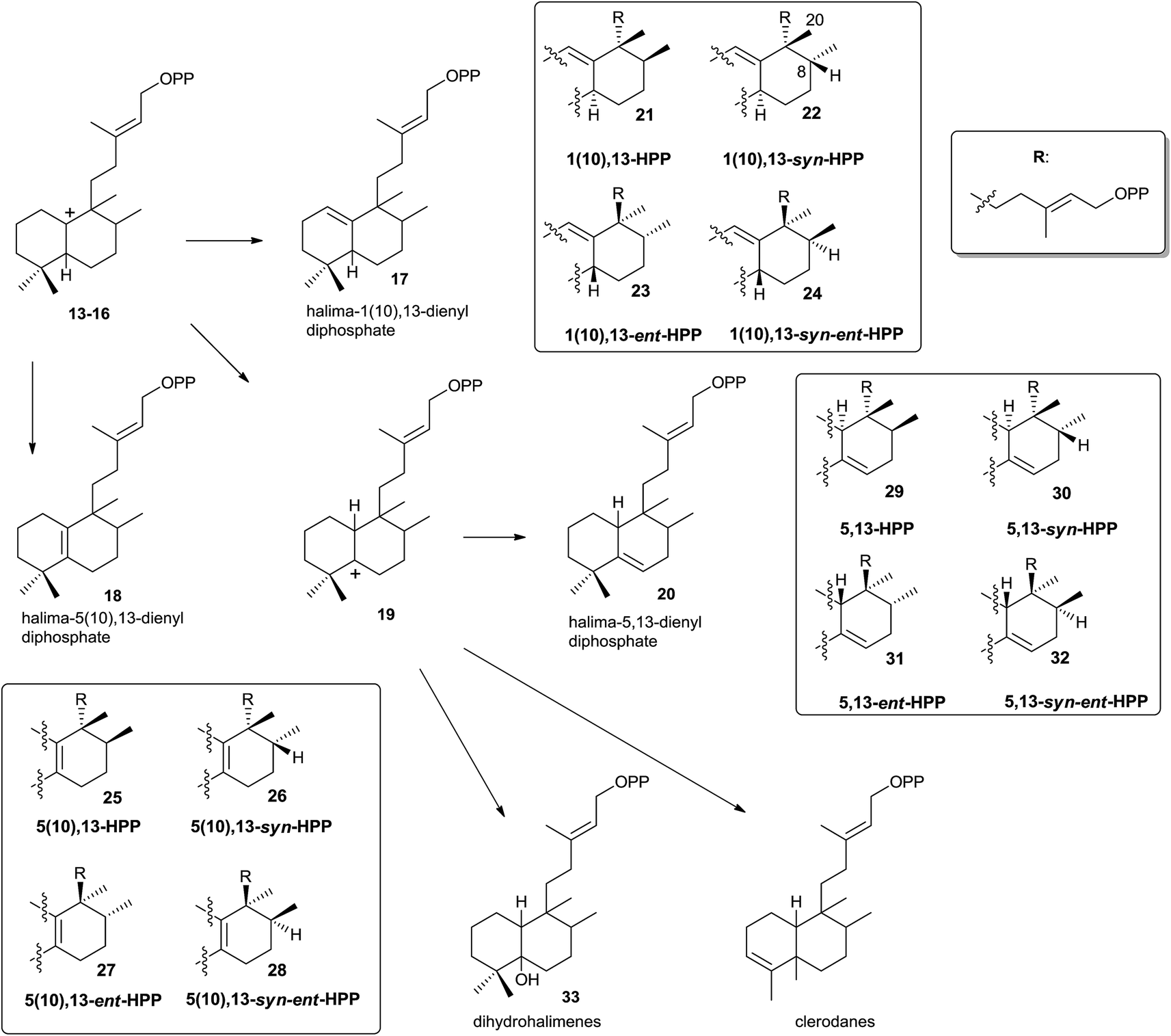
Those halimenyl diphosphate cations 13–16 can evolve by different ways (Scheme 3). Each one can lose a proton providing the halimadienyl diphosphates 17 or 18, or rearrange by hydride shift to form the halimenyl diphosphate cation 19 followed by proton abstraction to give the halimadienyl diphosphate 20. The different diastereoisomer forms (21–24), (25–28) and (29–32) correspond to the halimadienyl diphosphates 17, 18 and 20, respectively (HPP, syn-HPP, ent-HPP, and syn-ent-HPP; where syn prefix refers to cis configuration between H8 and C20). These diphosphates are the precursors of nearly all known halimanes. As has been indicated previously, the final cations 19 usually evolve by deprotonation yielding halimadiene skeleton compounds. However, water addition to the previously cited cations can occur, producing hydroxylated derivatives,85,86 such as the dihydrohalimene 33, thus leading to formal designation of the relevant class II DTCs as hydratases.86 Otherwise, although 1,2-hydride and/or methyl shifts do not appear to be concerted,16 the clerodane skeleton is formed from intermediate 19 by methyl group migration (C4 to C5 methyl migration giving trans and cis clerodanes) and later stabilization, thus generating the more than 1300 compounds that make up the clerodane diterpenoids family.14,15
 | ||
| Scheme 3 | ||
The formation of 3-secohalimenyl derivatives, such as 35 (Scheme 4), can be explained by oxidation of the C3 position of a halimenyl derivative by some specific oxygenase, such as cytochrome P450 monooxygenase.87 It is notable that plants have vastly expanded numbers of cytochrome P450 monooxygenases in their genomes88 providing a ready source of potential downstream acting enzymes. Studies have proved that cytochrome P450 takes part in the biogenesis of a considerable number of tricyclic and tetracyclic diterpenes,16,89 oxidizing these compounds at C3. So, we cannot discard the oxidation of any biogenetic intermediates of 35, achieving a hydroxy derivative like 34. For the formation of intermediates such as 34, in other families of compounds, it has been speculated with the epoxy derivative participation,90 as the GGPP oxide, although in diterpene biosynthesis that intermediate has not been detected until now in nature. Finally, oxidation with rupture of the C3–C4 bond leads to the wide group of 3-secohalimenyl derivatives.
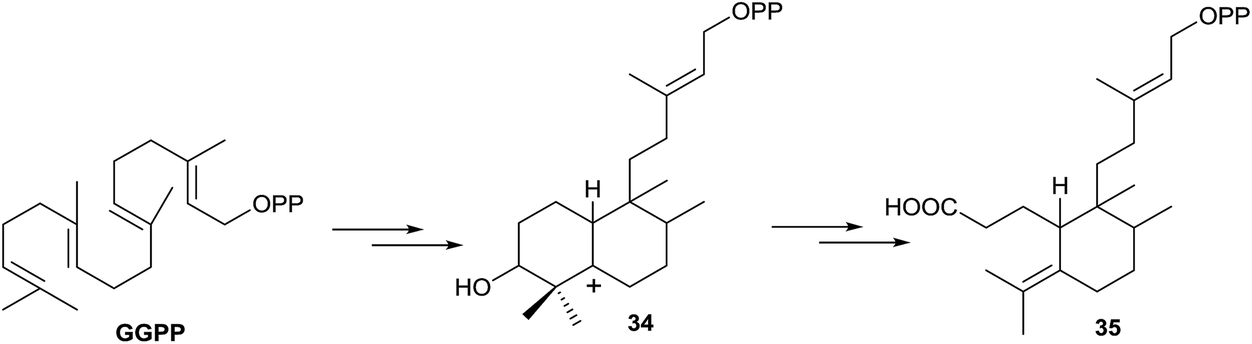 | ||
| Scheme 4 | ||
Rearranged halimanes of different types (I–V) are known (Fig. 2). Each one can be formed by rearrangement of some intermediate cations of other halimanes. In fact, rearranged halimanes have been obtained through biomimetic synthesis starting from natural halimanes.91
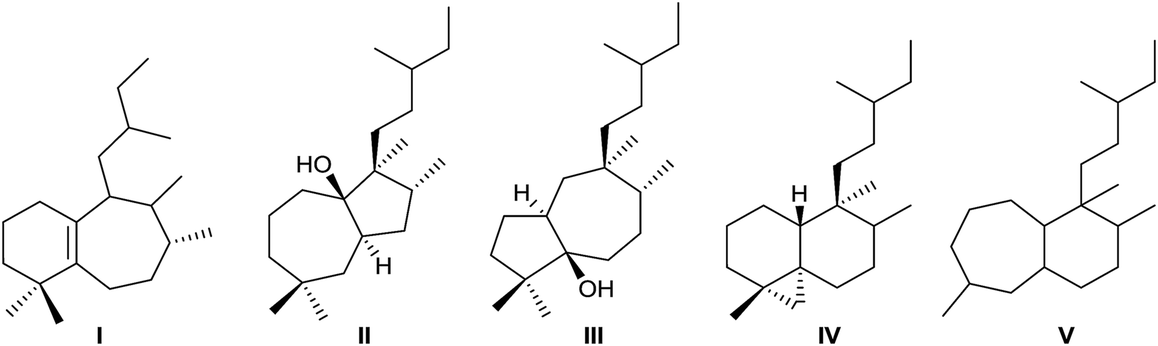 | ||
| Fig. 2 Types of rearranged halimanes. | ||
Type I rearranged compounds should be formed by ring B expansion (Scheme 5). Oxidation of 27 (5(10),13-ent-HPP) at C11 gives 36, the precursor of an intermediate carbocation 36a that facilitates ring B expansion through an intermediate such as 36b, which finally could be stabilized by double bond formation between C9 and C11.
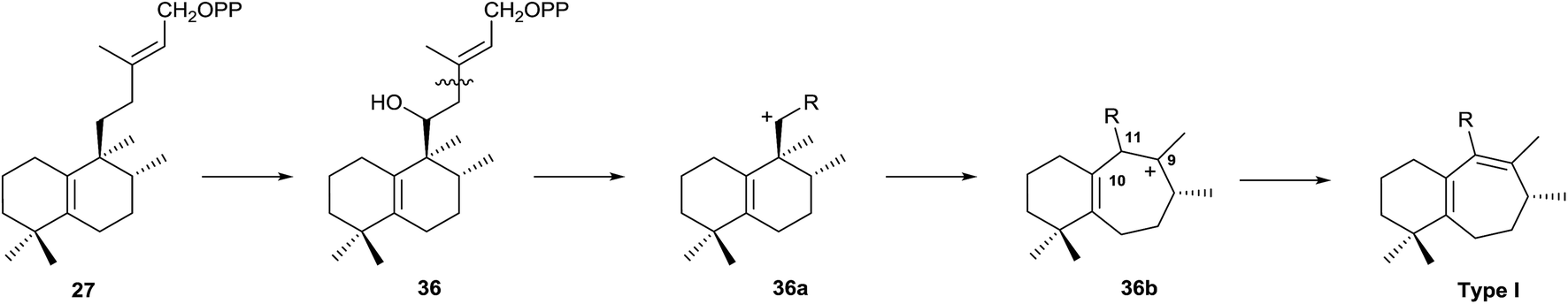 | ||
| Scheme 5 | ||
Type II and III derivatives could be formed by expansion/contraction of the decaline annular system that arises from an intermediate diketone 37a, which will be formed by oxidation of the double bond Δ5(10) through the intermediate 37, as it is shown in Scheme 6.
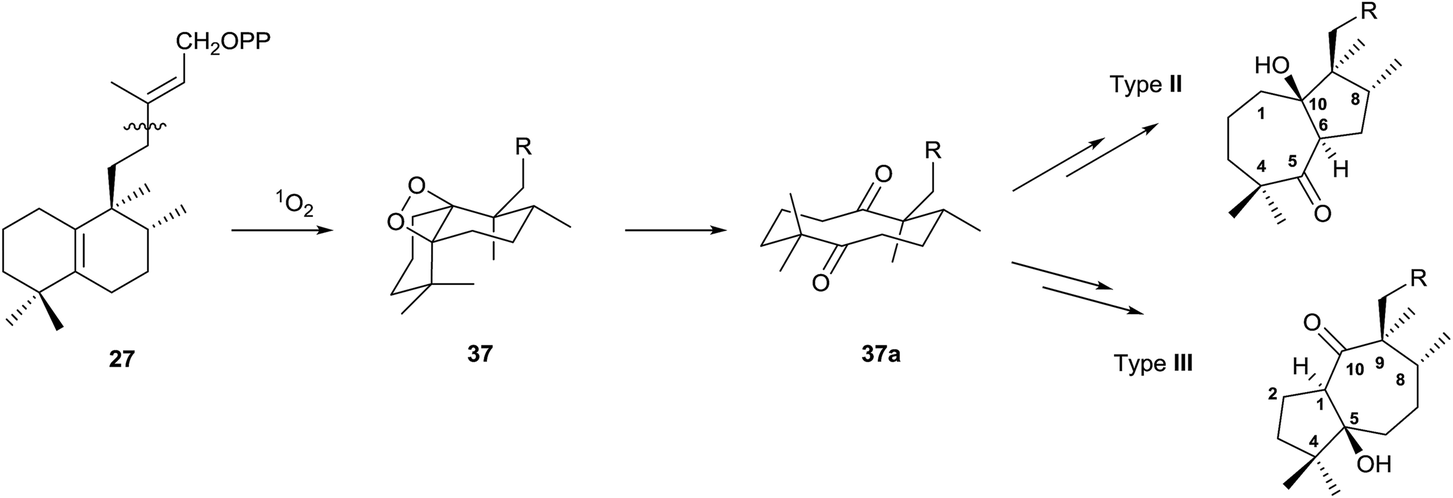 | ||
| Scheme 6 | ||
Type IV derivatives are compounds that can be considered as rearranged halimanes or rearranged clerodanes because C19 is bonded to C4 and C5 forming a cyclopropane ring, and type V derivatives can also be considered rearranged halimanes or clerodanes because C19 has been included in ring A as a consequence of a ring expansion, while other biosynthetic pathways cannot be discarded (Scheme 7). Possibly, the relevant class II diterpene cyclase might promote the cyclopropanation or the ring A expansion as it occurs with the ring contractions in the biosynthesis of permutilin.92
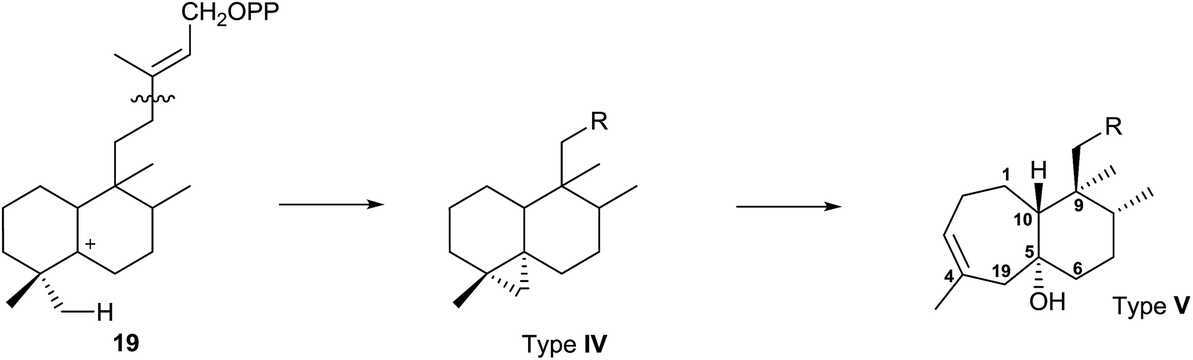 | ||
| Scheme 7 | ||
2.2. Enzymatic and genetic experimental studies
A bacterial class II (B type) DTC that produces halima-5,13-dienyl diphosphate (29) has been identified.16,55 Effectively, Mycobacterium tuberculosis Rv3377c gene has been identified, and the encoded diterpene cyclase has been proved to be responsible for the production of the halimane skeleton (Scheme 8).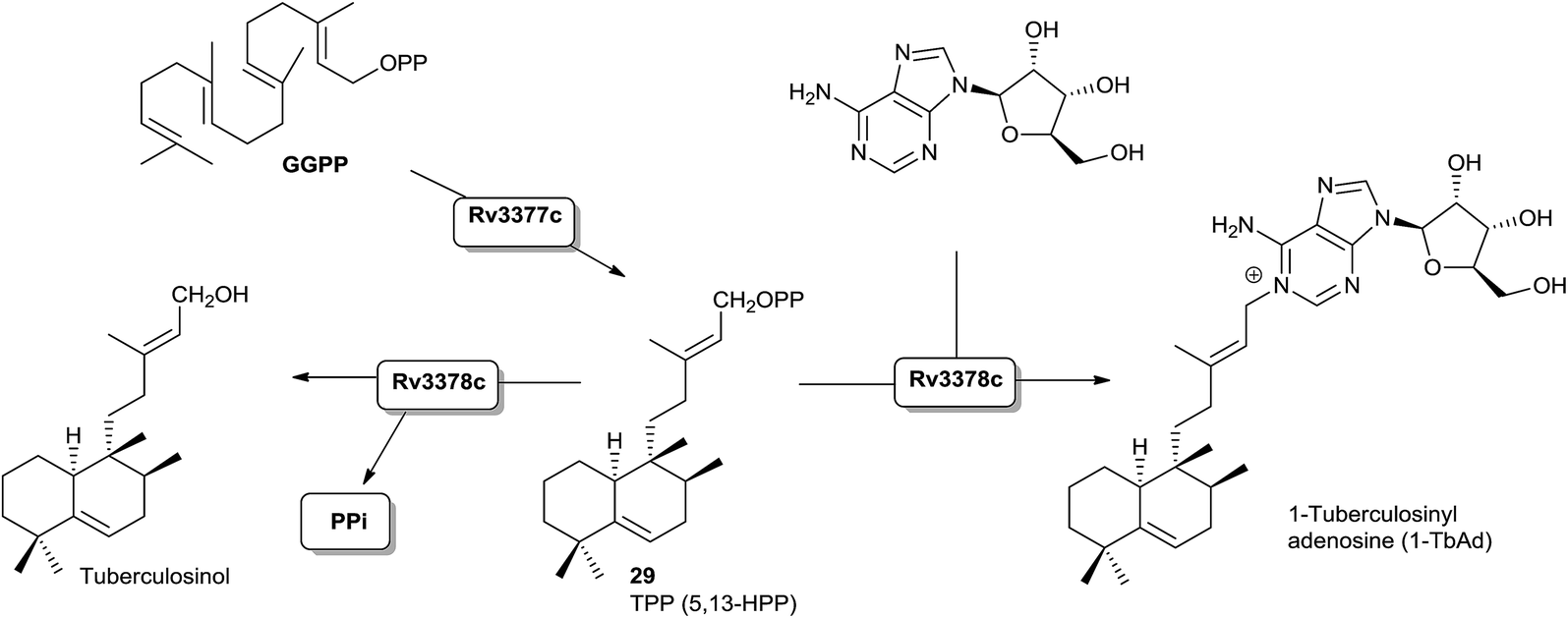 | ||
| Scheme 8 | ||
Recently the structure of the diterpene tuberculosinol/isotuberculosinol synthase (Rv3378c) from Mycobacterium tuberculosis has been reported.60 The biosynthesis of tuberculosinols is catalyzed by two enzymes: Rv3377c, tuberculosinyl (halima-5,13-dien-15-yl) diphosphate synthase, and Rv3378c, tuberculosinol/(R/S)-isotuberculosinol synthase. Both proteins are essential for the bacteria survival inside the macrophage where tuberculosinols inhibit phagolysosome maturation as well as macrophage phagocytosis.57,59,70
Rv3377c is a DTC classified as class II that transforms GGPP into tuberculosinyl diphosphate (TPP) with a halimane core55,93 (Scheme 8), while Rv3378c is a diterpene synthase that converts TPP into tuberculosinol or (R/S)-isotuberculosinols acting as a phosphatase/isomerase.57,59 However, although these two enzymes are sufficient to generate tuberculosinol and isotuberculosinol from GGPP, some studies on the evolution and functional characterization of the biosynthetic operon where these two genes are found have been carried out.94 Recently, a previously unknown type of diterpene-nucleoside, 1-tuberculosinyl adenosine (1-TbAd), was isolated and characterized (Scheme 8). This discovery leads us to consider a reviewed biosynthetic model in which the Rv3378c protein is not a simple phosphatase as currently believed, but that the enzyme acts with combined phosphatase and tuberculosinyl transferase functions by using adenosine as nucleophilic substrate.71
Interestingly, the Rv3377c and Rv3378c genes are found only in virulent Mycobacterium species, and not in avirulent ones.59,66 Another work reported that these genes are only functional in M. tuberculosis, despite being present in other less virulent species, such as M. bovis.67
Recently the incubation of [16,16,16-2H3]GGPP with tuberculosinyl diphosphate synthase (Rv3377c) from M. tuberculosis allowed the stereochemical course of the cyclization reaction to tuberculosinyl diphosphate via chair, chair transition state to be followed, confirming the cyclization pathway.95
New studies with class II DTCs have been carried out where the enzyme has been modified in order to check the fundamental role of the DXDD motif (Scheme 9).11 The high importance of that motif was confirmed and even a single residue modification in that motif can disrupt the normal activity of the enzyme. If the H501 residue is substituted in the rice (Oryza sativa) syn-copalyl diphosphate synthase OsCPS4 forming the mutant OsCPS4-H501D, the rearrangement of the initially formed bicycle is produced, obtaining the novel compound syn-HPP (Scheme 9), whose dephosphorylated derivative structure was characterized and its configuration established by NMR.85
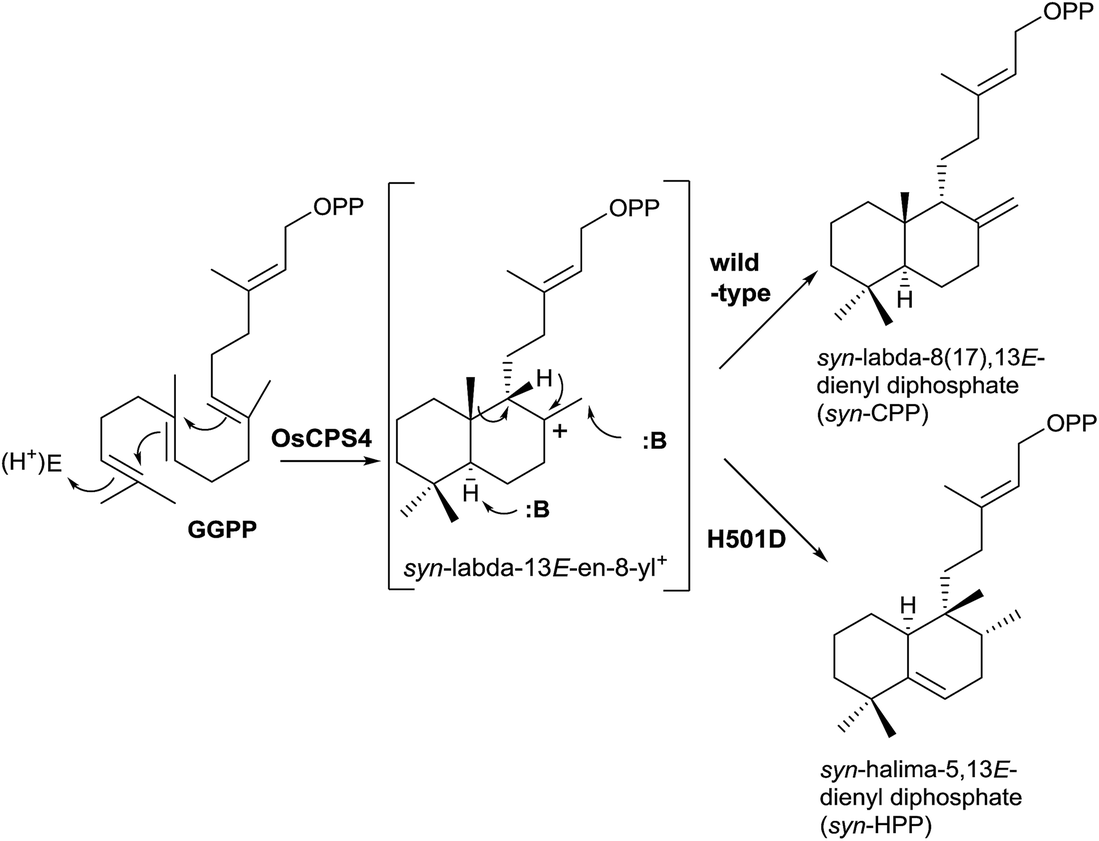 | ||
| Scheme 9 | ||
Recently, Zerbe and co-workers,96 in a work guided to achieve bioactive natural products harnessing the plasticity of these class II diTPS, have realized mutagenic experiments with the horehound (Marrubium vulgare) class II diTPS peregrinol diphosphate synthase (MvCPS1) (Scheme 10). Two double mutants based on the combination of F505 and W323 (W323L:F505Y and W323F:F505Y) produce the rearrangement of the labda-13E-en-8-yl+ intermediate, yielding the halimane skeleton instead of water capture giving the hydroxylabdane derivative.
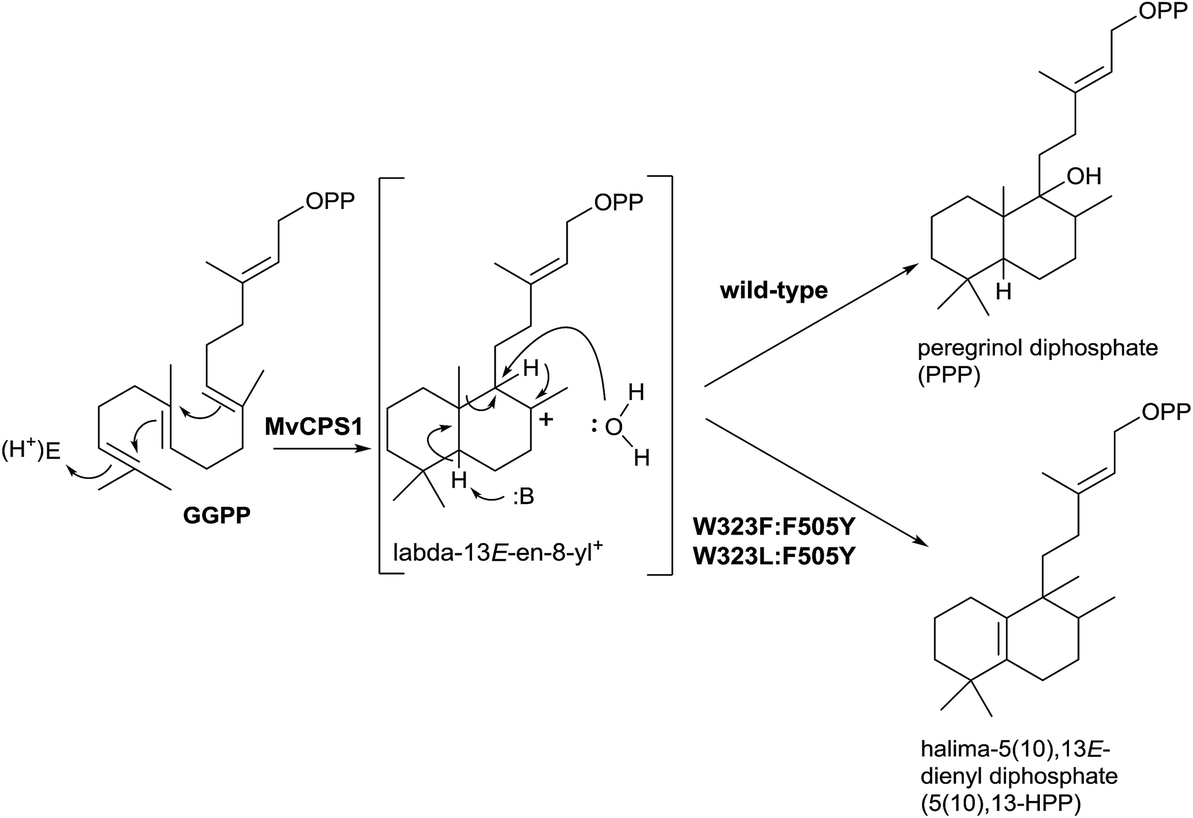 | ||
| Scheme 10 | ||
Biomimetic rearrangements of simplified labdane diterpenoids have been carried out by treatment with a variety of Lewis and protic acids, demonstrating that those rearrangements involve a series of stereospecific 1,2-alkyl and hydride shifts, producing mainly halimanes or a mixture of different dehydration products depending on reaction conditions. However, further rearrangement to clerodane products was not observed, indicating a high degree of enzymatic control for the in vivo formation of these natural products.97 In this work it was shown that the halimane skeleton appears to be inherently more stable than the clerodane structure, as Peters, Tantillo and co-workers demonstrated in their study of quantum chemical calculations.85,98
In many of these plants that contain halimanes, labdanes have also been isolated and in other plants they coexist with clerodanes. For example, in a Cistaceae such as Halimium viscosum ent-halimanes and labdanes coexist,17,26,28,99,100 in Cistus laurifolius labdanes, ent-labdanes, cis-clerodanes and ent-halimanes have been isolated,101–104 and in plants of genus Croton44,105–107 (Euphorbiaceae) labdanes, halimanes and clerodanes coexist. In Haplopappus paucidentatus25 and Nardophyllum lanatum19 (Compositae) labdanes, halimanes and clerodanes coexist. It seems that in Halimium viscosum the biosynthetic route to clerodanes is enzymatically interrupted.
3. Classification
In this review, halimane diterpenoids have been classified according to the endocyclic double bond position and to their corresponding dihydro derivatives. Seco-, nor- and rearranged halimanes have been considered too (Fig. 3).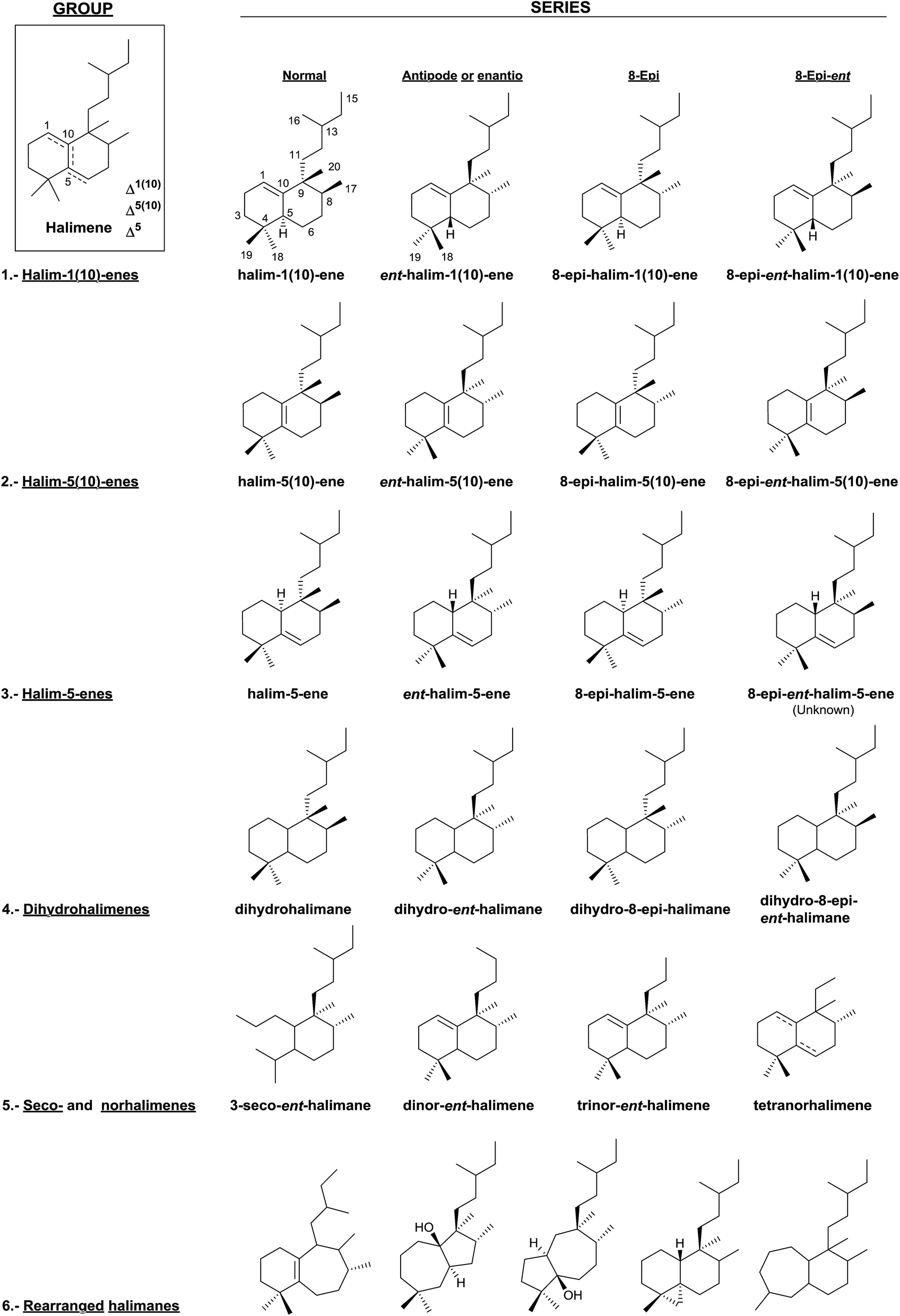 | ||
| Fig. 3 Classification of halimanes. | ||
In this manner, we have divided the halimanes into six different groups: (1) halim-1(10)-enes, (2) halim-5(10)-enes, (3) halim-5-enes, (4) dihydrohalimenes, (5) seco- and norhalimenes and (6) rearranged halimanes. As can be seen in Fig. 2, the first four groups could be divided into four series: ‘normal’, ‘antipode or enantio’, 8-epi and 8-epi-enantio. According to the biogenesis of each group, those derivatives in which methyls Me-17 and Me-20 are cis to each other are denominated halimanes or ent-halimanes, and those in which methyls Me-17 and Me-20 are trans to each other are called 8-epi-halimanes or 8-epi-ent-halimanes (Fig. 3). Derivatives from all these groups of compounds are known, except for 8-epi-ent-halim-5-ene.
To the best of our knowledge, all examples of seco-, nor-, and rearranged halimanes (groups 5 and 6) known nowadays belong to the ‘enantio’ series, except for four tetranorhalimenes and three rearranged halimanes.
4. Natural halimanes
In this section the structures of natural halimanes can be observed; each one is numbered and accompanied by its trivial name. In addition, we summarize the information in tables that appear in the S.I., including the source of isolation, the plant part or organism from which they were isolated, the biological activities and references (Tables S1–S6†).Approximately 70% of the natural halimanes known are of the ‘enantio’ series (ent-halimanes or 8-epi-ent-halimanes) and the remaining 30% are of the ‘normal’ series (halimanes or 8-epi-halimanes).
The halimane structure elucidation has been carried out by extensive spectroscopic techniques, mainly NMR and chemical correlation. X-ray structural analysis has been carried out for several of them, as indicated in the comments of the diverse groups that appear afterwards. The absolute configuration has been established by circular dichroism. However, in some cases, mainly due to the scarcity of natural product, it has not been possible to determine the stereochemistry.
The figures and tables will show the natural products as they have been described and characterized; in most cases the carboxylic acids appear as their respective methyl ester derivatives.
4.1. Halim-1(10)-enes group
The first known halimanes are found in the group of halim-1(10)-enes, which is the most numerous class so we start the classification from them.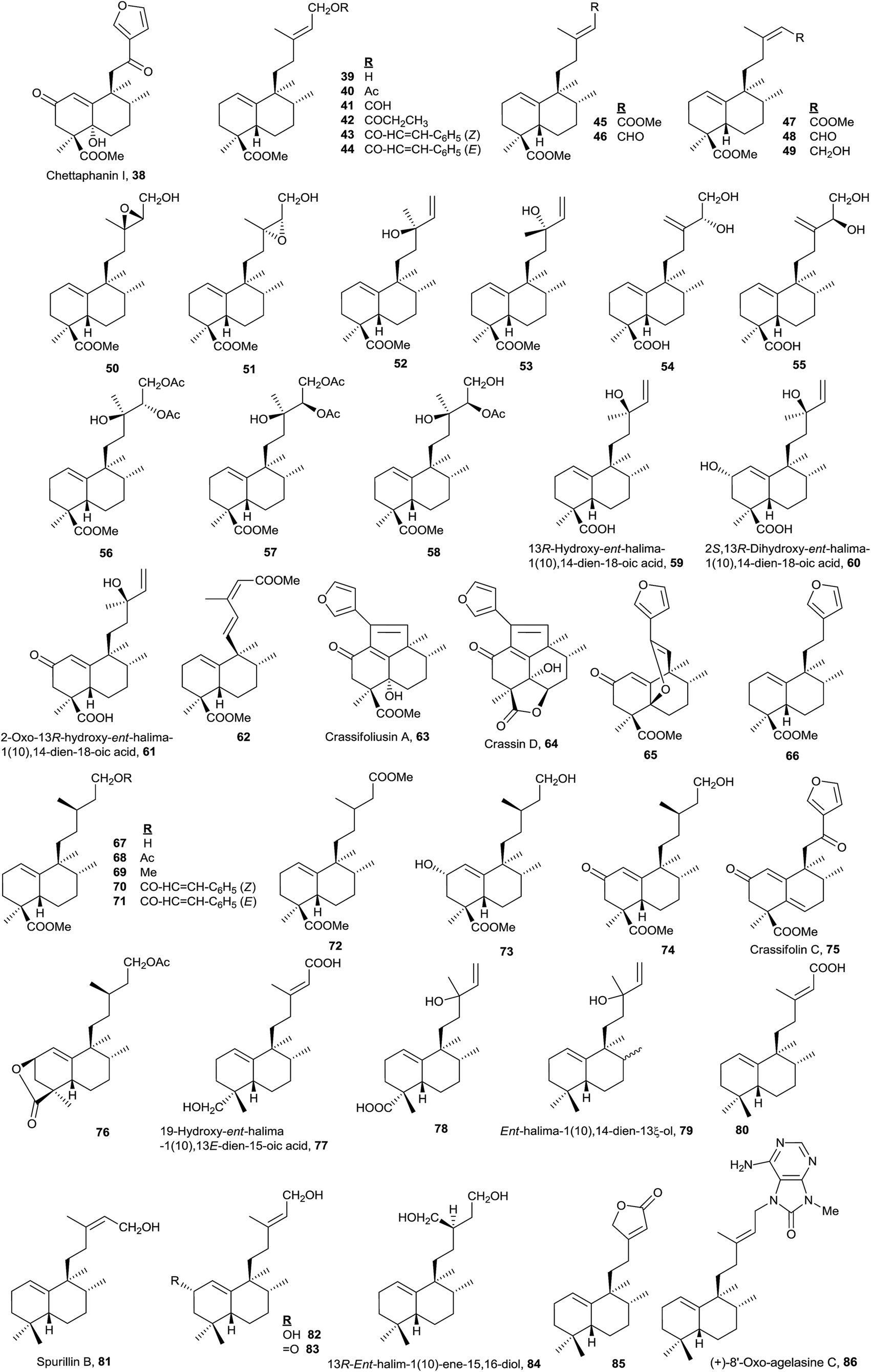 | ||
| Fig. 4 ent-Halim-1(10)-enes. | ||
In literature, the first halimanes were designated as rearranged labdanes, isolabdanes or friedolabdanes, but in order to classify them in a diterpene group, it was decided to name them as halimanes due to the high number of these compounds that appear in plants of genus Halimium.17
The study of genus Halimium plants has made possible to determine the presence of five chemotypes of Halimiun viscosum, known in accordance with the harvesting place: Villarino de los Aires, (Salamanca, Spain),17 La Fregeneda, (Salamanca, Spain),26,27 Valparaiso, (Zamora, Spain),28 Celorico da Beira, (Portugal)29 and San João da Pesqueira, (Portugal).30
In this group, the Euphorbiaceae,78,108,109,111–114 Cistaceae,17,26,27,29,30,110,115–118 Leguminoseae,48,119,120 Compositae,21,24 Jungermanniaceae,121 Velloziaceae52 and Annonaceae122,123 plant families have been studied together with marine organisms of genus Spurilla124 and Agelas.74,125
The most numerous compounds of this group (Fig. 4) are those that show a carboxylic function at C18 and among them the most frequent possess an unsaturated or polyfunctionalized side chain. However, compounds 77 and 78 are the only ones that have an oxygenated function at C19. Compounds with the saturated side chain are abundant too, finding among them those oxidized at C2.
The absolute configuration of 61 was determined by circular dichroism (CD) and the structure of 59 was corroborated by X-ray. These two compounds were isolated from Hymenaea courbaril.48
Only eight compounds 79–86 do not have functionalized C18 or C19. Recently, spurillin B (81), isolated from Spurilla sp (Nudibranchia), has been described, and it is one of the few ent-halimanes with a cis double bond in their side chain.124 8′-Oxo-agelasine C (86) is a novel purine diterpene that was recently isolated from Agelas nakamurai and it is the only halimane purine presenting a carbonyl group at adenine C8.125 Compounds of this type, usually known as agelasines, have been isolated from genus Agelas sponges and are very interesting owing to their antimicrobial and antispasmodic activities and their action as Na,K-ATPase enzyme inhibitors.126
Although this group is quite numerous, only six furohalimane derivatives (38, 63–66 and 75) and a halimanolide (85) are known. These functionalizations are most frequent in the ent-halim-5(10)-enes, as we will explain later. Crassifoliusin A (63)113 and crassin D (64),114 isolated from Croton crassifolius, have a tricyclic system formed by cyclization of C1 with C12 of the halimane side chain.
A lot of these compounds have been isolated from plants used in folk medicine, but the biological activity of many of them has not been determined yet. Only 5948 and 8452 have proved to be active as antitumour drugs.
 | ||
| Fig. 5 Halim-1(10)-enes. | ||
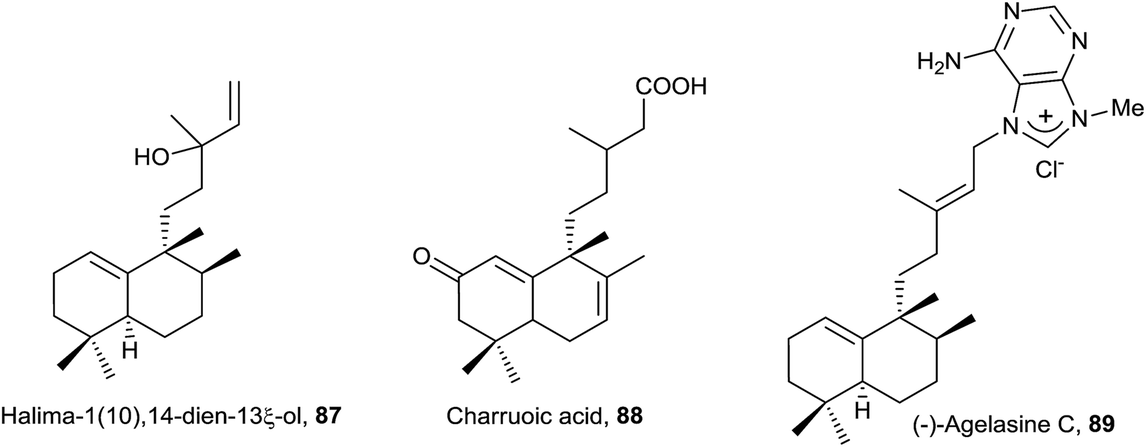
The structure of the natural product agelasine C (89) was established by its enantiomer synthesis128 correcting, in this manner, the original structure proposed by Nakamura and co-workers.126 Agelasine C (89) was isolated from Okinawan sea sponge Agelas sp. and Agelas citrina129 and it exerts antifungal activity.
The structure of 87, halima-1(10),14-dien-13ξ-ol, isolated from Plagiochila barteri, was determined spectroscopically and by comparison with 79.130 In any case, the configuration at C13 has not been determined yet.
 | ||
| Fig. 6 8-epi-Halim-1(10)-enes. | ||
 | ||
| Fig. 7 8-epi-ent-Halim-1(10)-enes. | ||
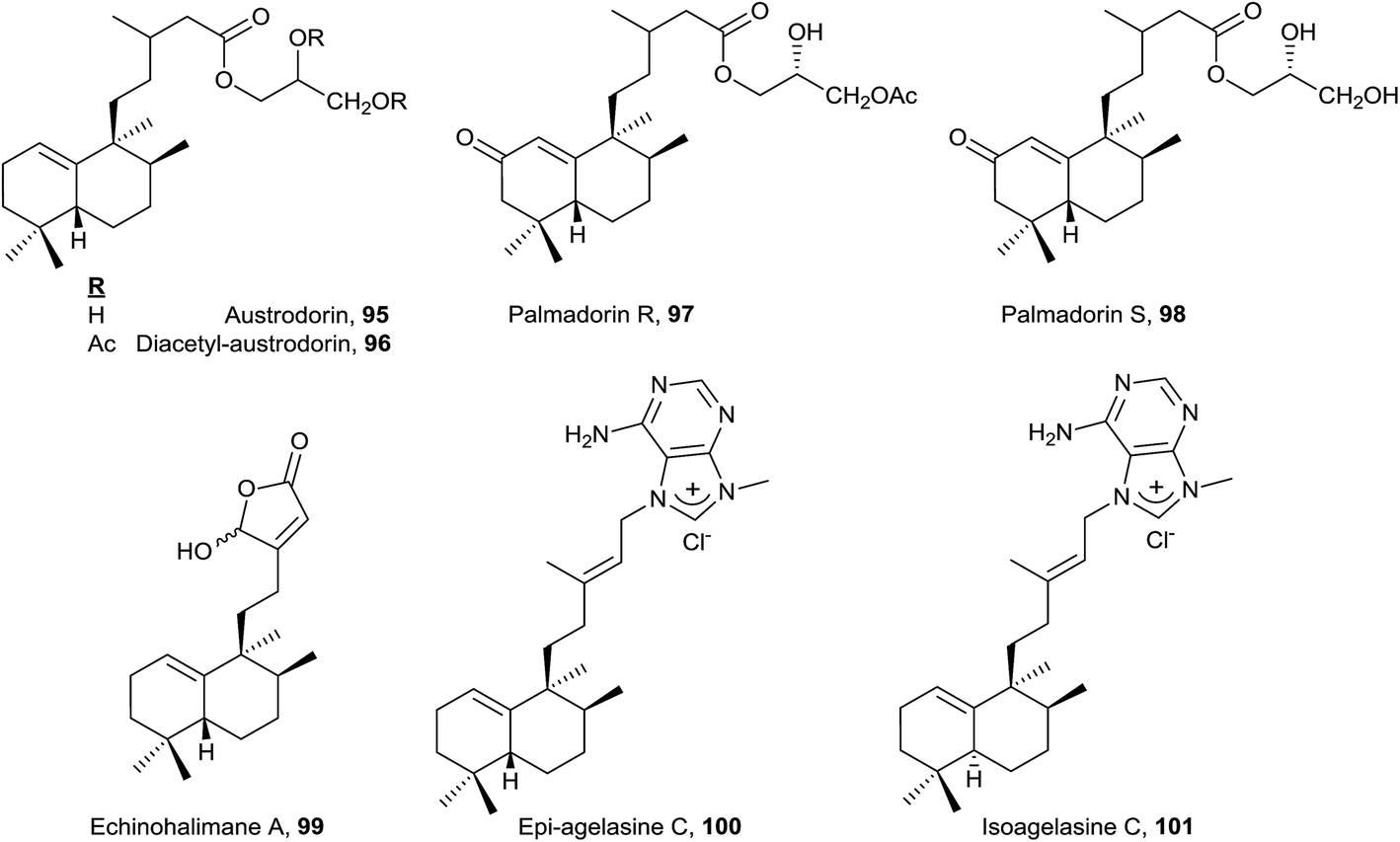
Echinohalimane A (99), which shows a γ-hydroxybutenolide in the side chain, was isolated from a gorgonian identified as Echinomuricea sp.46,47 It is the first halimane isolated from a marine organism belonging to the phylum Cnidaria. This compound was found to exhibit cytotoxicity towards various tumour cell lines and displays an inhibitory effect on the release of elastase by human neutrophils.
The new diterpene alkaloid epi-agelasine C (100) was isolated from the marine sponge Agelas mauritiana as an antifouling substance active against macroalgae.75,128 The original structure proposed for epi-agelasine C, a halim-1(10)-en derivative,75 was corrected to the structure of 8-epi-ent-halim-1(10)-ene derivative for 100128 (Fig. 7). Recently, the structure of isoagelasine C (101), isolated from Agelas nakamurai, has been described and it shows potent antifungal and moderate antibacterial activity.135
4.2. Halim-5(10)-enes group
Together with the halim-1(10)-enes, these compounds form the two largest groups of halimanes.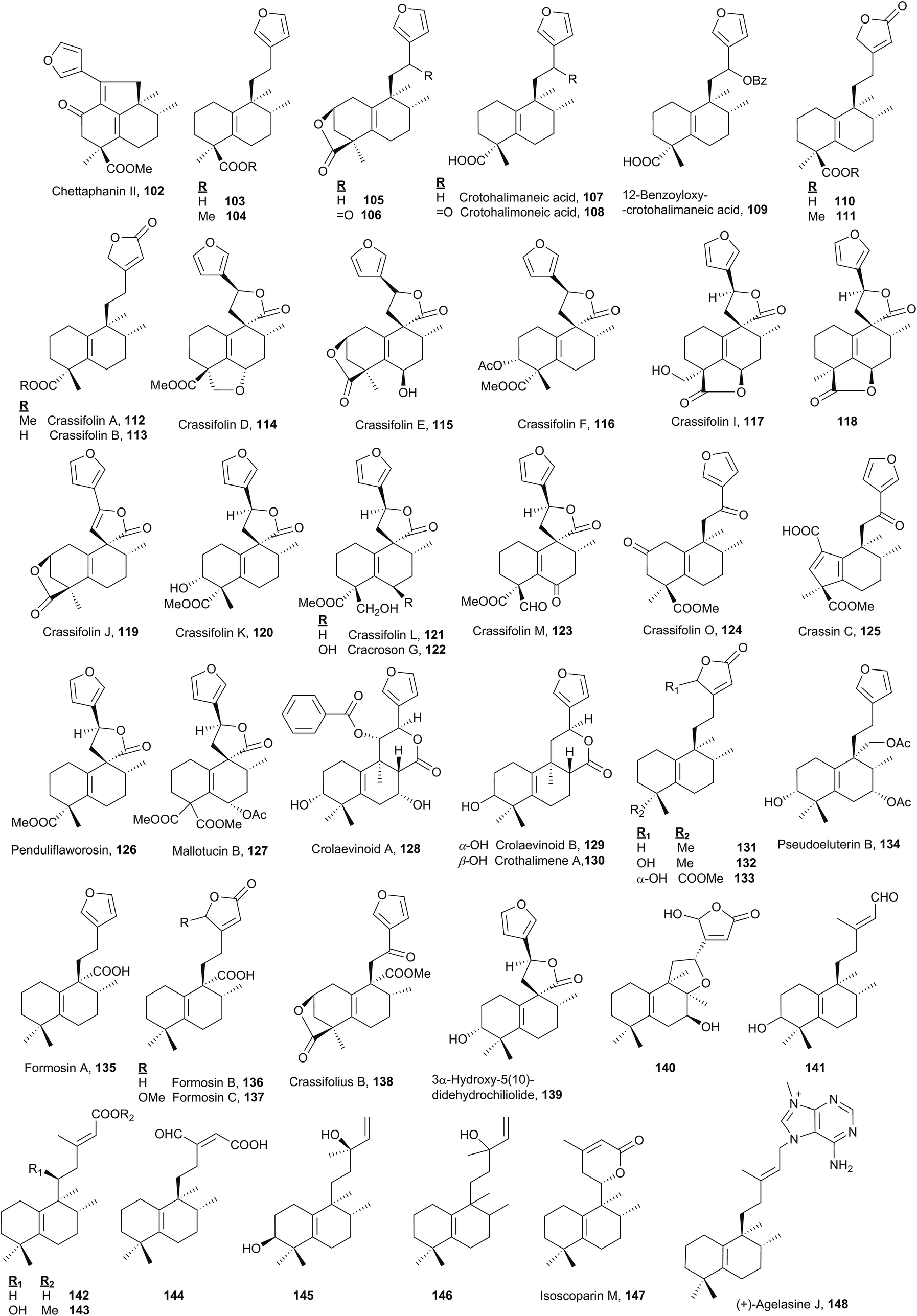 | ||
| Fig. 8 ent-Halim-5(10)-enes. | ||
As chettaphanin II (102), the majority of known ent-halim-5(10)-enes are furo-ent-halimanes. In some of them the furan fragment appears oxidized in the form of 15,16-butenolide. The lactone group can be observed in other positions, for example 20,12-olides. It is usual to find derivatives of this kind with a carboxylic function at C18 or C19. The side chain C12 position usually appears functionalized and ring A can be oxygenated at C2 or C3.
The structure and absolute configuration of lactone 106 were corroborated by synthesis and it shows a moderate activity against HeLa cells.44,78,112,141 Crotohalimaneic acid (107) and crotohalimoneic acid (108) show activity against several human cancer cell lines.140 The structure of crotohalimoneic acid (108) was corroborated by X-ray.
A series of ent-halim-5(10)-enes known as crassifolins 112–117, 119–121, 123–124 and crassin C (125) has been isolated from Croton crassifolius.44,78,144 The absolute configuration of several of them was confirmed by CD. The structure of crassifolin D (114) was corroborated by X-ray; however, its absolute configuration was not determined. As the crassifolins found in Croton crassifolius are included in the ent-halim-5(10)-ene series, the same absolute configuration is proposed for crassifolin D (114) and it was included in this group. Crassin C (125)114 is characterized by its ring A contraction, showing a carboxylic function at C1. This compound could be the result of an oxidation followed by condensation, of any compound that coexists in that plant with functionalization at C2, forming in this manner a [5.6] ring system.
Compounds of this kind functionalized at C6 in ring B are not frequent, only crassifolin D (114), crassifolin E (115), crassifolin I (117),143,144118,138 cracroson G (122),151 crassifolin M (123),44 and mallotucin B (127)136 show this functionalization. Functionalization at C7 is seen in compounds 128, 134 and 140 and at C8 only on compound 140.76
Crassifolin A (112), crassifolin B (113), and penduliflaworosin (126) showed anti-angiogenic activity using a wild-type zebrafish in vivo model,44,78 and crassifolin D (114) shows antiviral and anti-angiogenic activities.44
Crolaevinoids A–B (128–129) and crothalimene A (130) were recently isolated from Croton laevigatus and Croton dichogamus, respectively.105,148 They are the only C17 functionalized derivatives known in this class forming a δ-lactone with carbon C12. The absolute configuration of compounds 128 and 129 was determined by electronic circular dichroism (ECD). These compounds exhibit pronounced inhibition of nitric oxide (NO) production.105
3α-Hydroxy-5(10)-didehydrochiliolide (139) was highly active against a human pancreatic adenocarcinoma cell line at micromolar concentrations45 and 140 shows antitumour activity.76 Formosin A–C (135–137) isolated from Excoecaria formosana were active as antimicrobials,147 and, in addition to crassifolius B (138),150 they are the only compounds that possess a carboxylic acid or methyl ester functionality at C20.
In six of the known ent-halim-5(10)-enes 141–146, no furanic or lactone systems appear in the side chain. Isoscoparin N (143) and isoscoparin M (147),91,155 together with crolaevinoid A (128), are the only ones that show oxygenated functions at C11. The structure of compound 146 has been proposed but its absolute configuration has not been solved.153 Compound 145 shows antitumour activity.154
In this group, the halimane purine agelasine J (148) is included. Agelasine J was isolated from the Solomon Islands marine sponge Agelas cf. mauritiana. It shows antimalarial and antimicrobial activity and MCF7 cell cytotoxicity.79,135
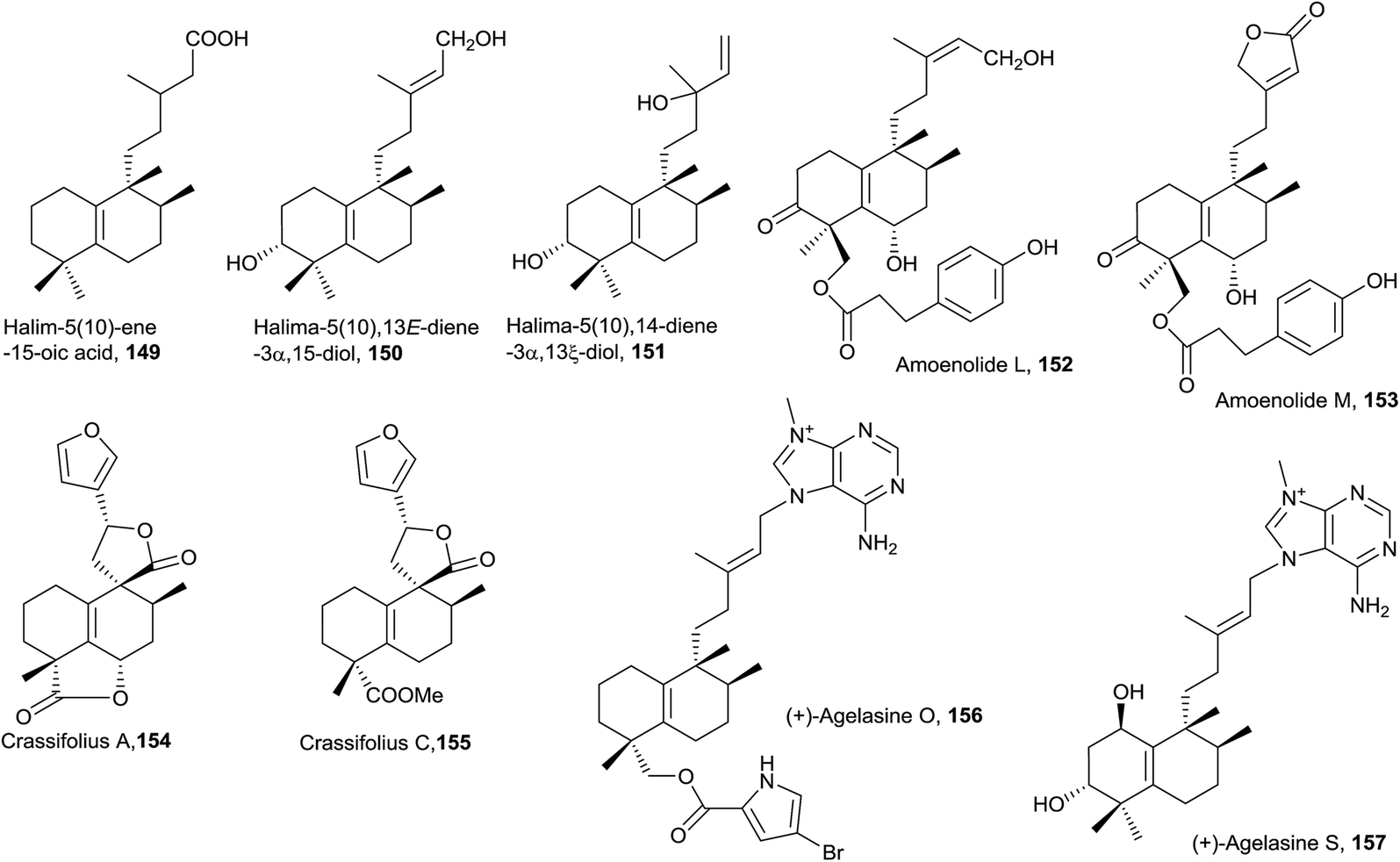 | ||
| Fig. 9 Halim-5(10)-enes. | ||
In this class, two halimane purines isolated from an Okinawan marine sponge Agelas sp. can be found: agelasine O (156) and agelasine S (157).53 Agelasine O (156), in which C18 is esterified with 2-carboxy-4-bromopyrrole, is biologically active as an antibacterial and antifungal.53 Agelasine S (157) exerts antibacterial and antifungal activities and is the only derivative of this type difunctionalized at C1 and C3.
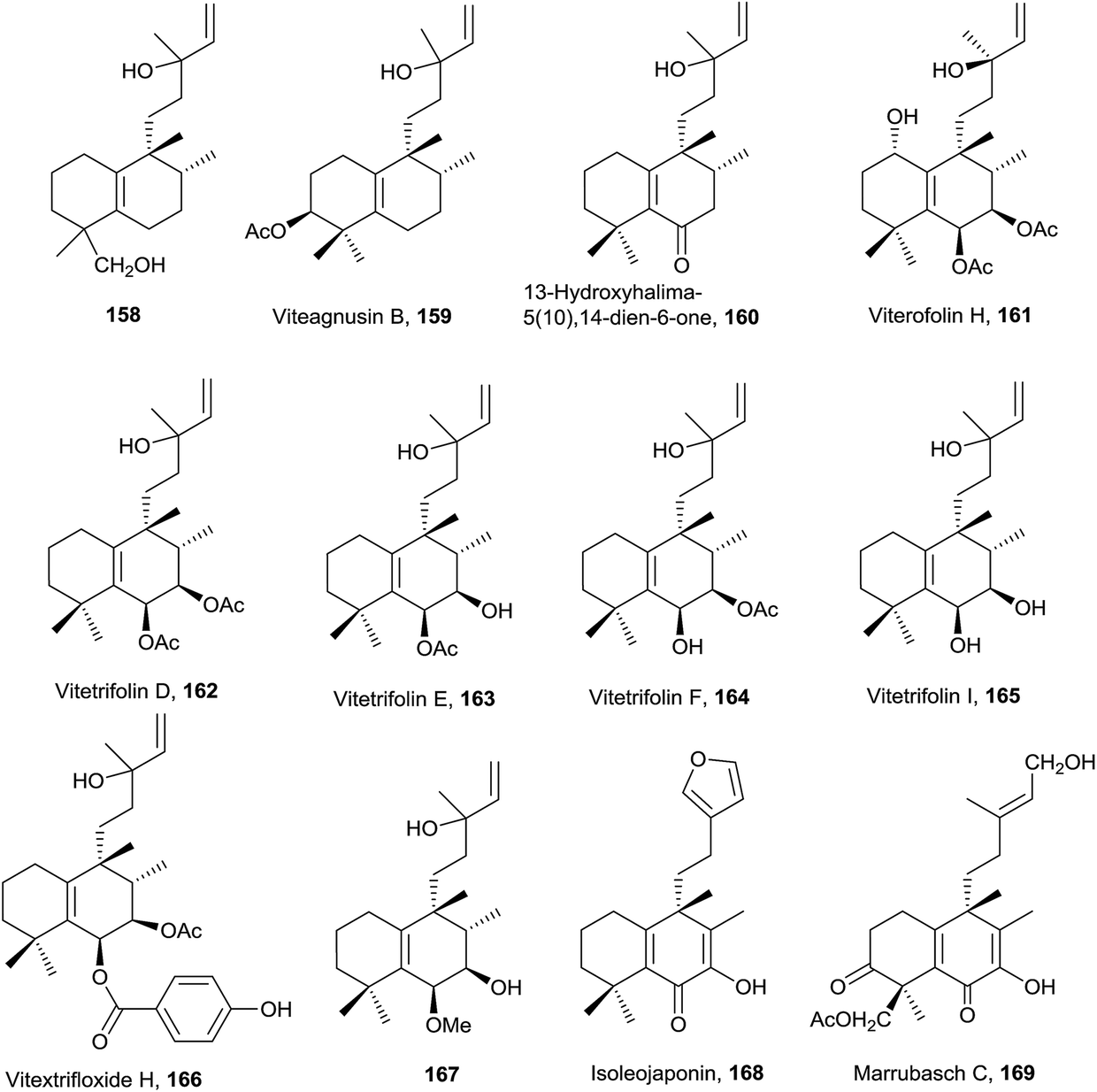 | ||
| Fig. 10 8-epi-Halim-5(10)-enes. | ||
Compound 161 exerts moderate anti-hyperlipidemic activity81 and compounds 162 and 166 are cytotoxic against HCT 116 human colon carcinoma cell line.77,162
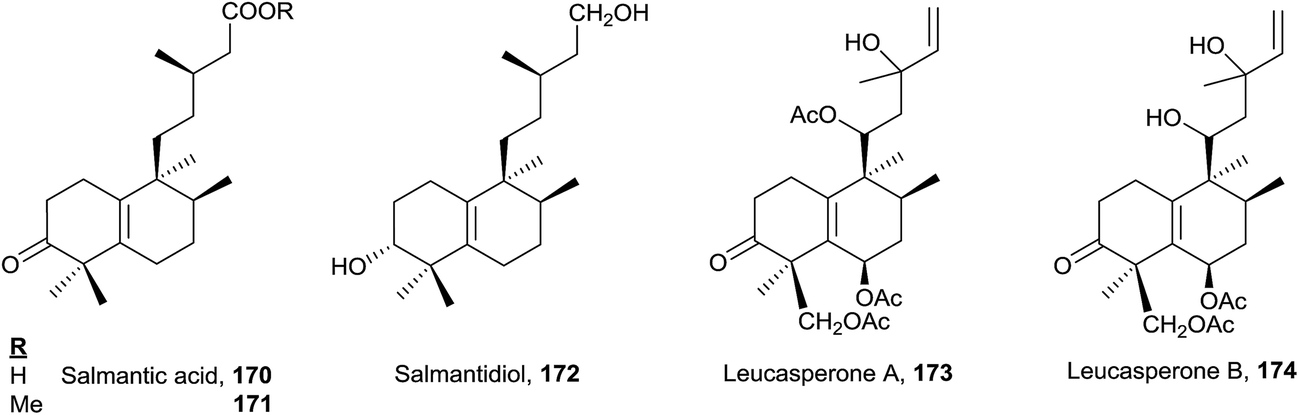 | ||
| Fig. 11 8-epi-ent-Halim-5(10)-enes. | ||
The five compounds of this type are functionalized at C3. Salmantic acid (170), its methyl ester (171) and salmantidiol (172) have the side chain saturated, while leucasperone A (173) and leucasperone B (174), isolated from Leucas aspera,169 show a double bond at C14 and have oxygenated functions at C11 and C13. Leucasperones A and B (173 and 174) possess acetoxyl groups at C6 and C18. Leucasperone A (173) inhibits prostaglandin-induced contractions.169
4.3. Halim-5-enes group
This group of halimanes, having a double bond in ring B, is the most interesting owing to the biological activities that some of them show.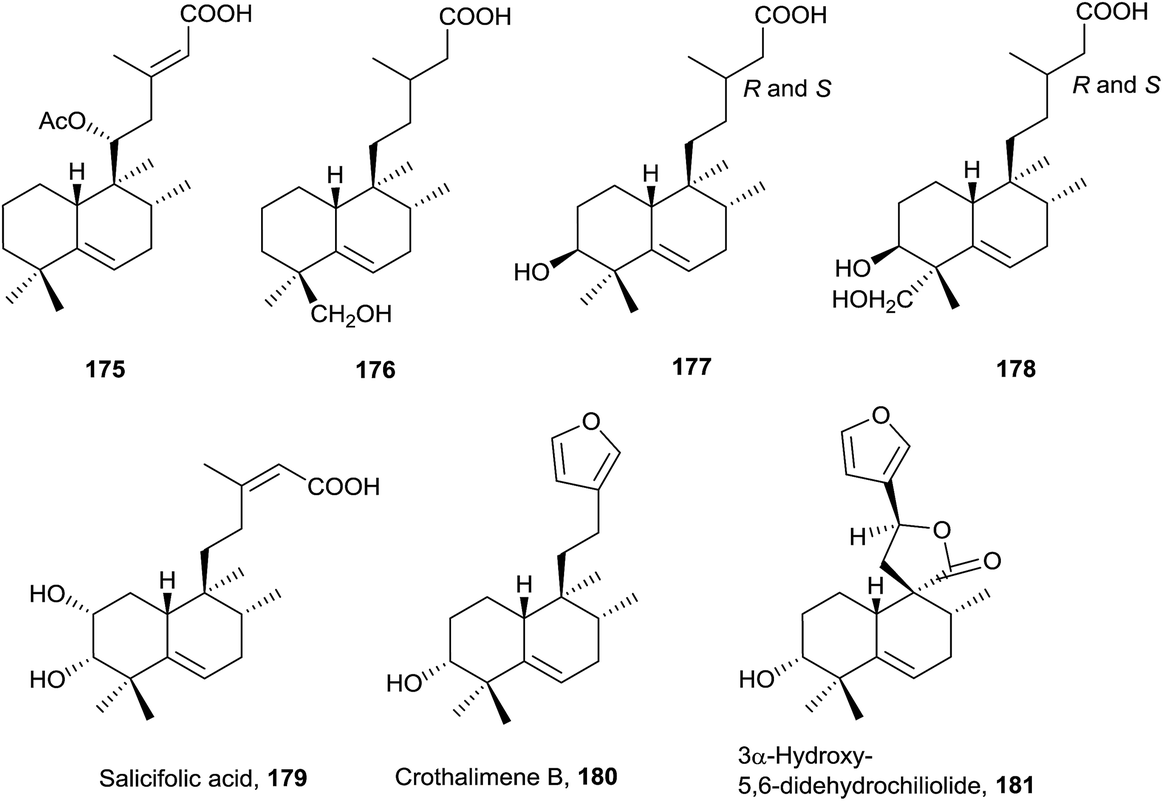 | ||
| Fig. 12 ent-Halim-5-enes. | ||
The side chain can be saturated on 176 (isolated from Haplopappus paucidentatus25), 177 and 178 (Relhania corymbosa and R. squarrosa20), unsaturated on 175 (Plectranthus ornatus171) and 179 (Baccharis salicifolia54) or with furan on 180 (Croton dichogamus148) and 181 (Chiliotrichum rosmarinifolium19 and Nardophyllum bryoides45). Acids 177 and 178 were isolated as their methyl esters and their epimers at C-13 separated, although assignment of their stereochemistry at C-13 was not possible.20
Salicifolic acid (179) regulates the growth of Panicum miliaceum (monocotyledon) seedlings54 and 3α-hydroxy-5,6-didehydrochiliolide (181) was highly active against a human pancreatic adenocarcinoma cell line at micromolar concentrations.45
 | ||
| Fig. 13 Halim-5-enes. | ||
It has been proved that compounds tuberculosinol (182) and isotuberculosinol (183) are produced by Mycobacterium tuberculosis.68,69 None of the studies done detected the presence of 182 or 183 from the cultured cells of 12 nonpathogenic Mycobacterium species.65 It has been observed that tuberculosinol (182) and isotuberculosinol (183) (in a 1:1 ratio, with 183 being a mixture of the diastereomers 13R-isotuberculosinol (183R) and 13S-isotuberculosinol (183S) in a 1:3 ratio) inhibit phagolysosome maturation and macrophage phagocytosis in human-like cells.59 The structures of tuberculosinol (182) and isotuberculosinol (183) have been corroborated and their absolute configuration established by total synthesis.56,58
Recently, two new natural products derived from tuberculosinol have been isolated and characterized: 1-TbAd (200)71 and N6-TbAd (201).62 These two tuberculosinol derivatives possess an adenosine unit bonded at C15 by N1′ or by the nitrogen at C6′ of the adenosine (Fig. 13). Recently it has been observed that compounds 200 and 201 accumulate to comprise >1% of all M. tuberculosis lipids. These diterpene nucleoside compounds are being investigated as biomarkers for tuberculosis.62 The structures of 1-TbAd (200) and N6-TbAd (201) have been corroborated by total synthesis.63
In the halim-5-enes series (Fig. 13), the side chain can be saturated or unsaturated and furans or functionalized butanolides can be found on it.
Tuberculosene (184) has been obtained by enzymatic reaction from a mixture of GGPP with tuberculosinyl diphosphate synthase and CYC2 enzyme from the bacteria Kitasatospora griseola.173,174
In this group, two plants of the Compositae family have been studied. From Koanophyllon conglobatum175 koanophyllic acids 185, 192, 196 and 197, with carboxylic function at C18, were isolated, and from Haplopappus pulchellus23186–191 were isolated, all of them with a saturated side chain. The structure of 195 was spectroscopically determined and its absolute configuration established by ECD51 of their 3-p-bromobenzoate derivatives. This compound possesses antimicrobial activity. From Acalypha macrostachya the 7-oxo derivatives 193 and 194 were isolated.22
Micromonohalimanes A and B (198 and 199, from Micromonospora sp.50), which present antibacterial activity, have been characterized. Micromonohalimane B (199) is the only halimane which includes a chlorine atom in its structure.
 | ||
| Fig. 14 8-epi-Halim-5-enes. | ||
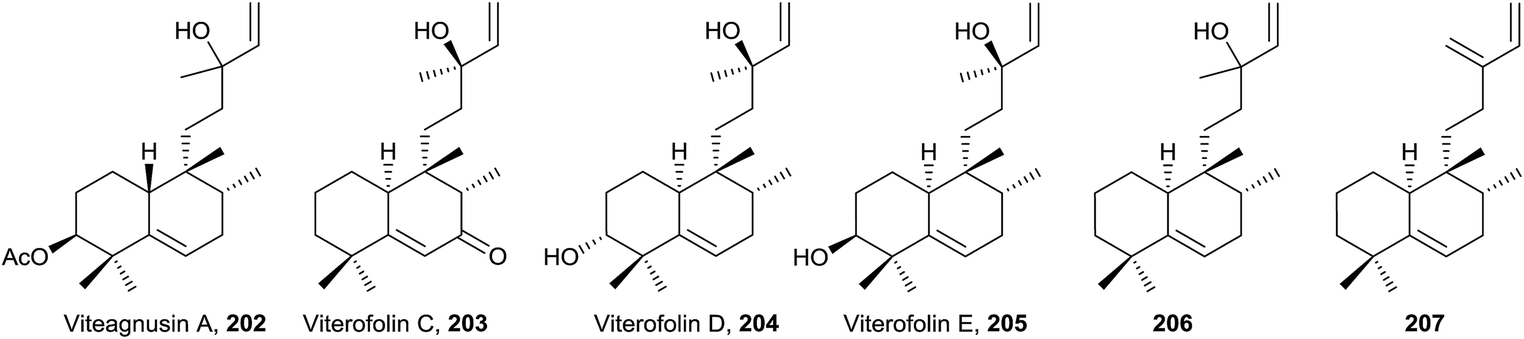
4.4. Dihydrohalimenes group
A structural characteristic of this group of compounds (208–219) is that all of them show oxygenated functions at C5, except diasin (219, from Croton diasii),176 which we include in this group precisely for not having any unsaturation in the decalin system (Fig. 15, Table S4†). Although some members possess acyclic chains (208 and 209, isolated from Pleurozia gigantea18 and Jungermannia truncata,177 and Baccharis salicifolia,54 respectively), the most usual functionalization is the furan (210 and 211 from Dysidea amblia,178212 from Croton crassifolius151 and 213 and 214 from Chiliotrichum rosmarinifolium19) or butanolide fragments (215 and 216 from Heteroscyphus coalitus179 and Polyalthia longifolia,180 respectively). The stereoisomers ambliol B (210) and ambliol C (211) were isolated from Dysidea amblia,178 being the only occasion that halimanes and 8-epi-halimanes coexist in the same organism. Originally, a cis-fused bicyclic ring system was assigned to ambliol B (210), but finally the structure was revised by X-ray analysis.178,181 Compound 209 is a germination inhibitor54 and the structure of compound 214 was corroborated by synthesis.182 | ||
| Fig. 15 Dihydrohalimenes. | ||
Cracroson D (212) shows a new pentacyclic scaffold. It is chemically related to chettaphanin I (38) because 212 is generated from 38 by an intramolecular [2 + 2]-photocycloaddition. The existence of this compound in the extract as a natural product is confirmed by HPLC-MS. Cracroson D (212) exerts moderate cytotoxicity against T24 and A549 cell lines (bladder and lung cancer respectively).
In this group, halimane-purines such as agelasimines A and B (217 and 218, respectively), isolated from Agelas mauritiana,183 are included. In this case, halimane C15 is bonded to purine N7. Both compounds show a wide range of interesting biological activities, such as cytotoxicity, inhibition of adenosine transfer into rabbit erythrocytes, Ca2+ channel antagonistic action and α1 adrenergic blockade.
4.5. Secohalimenes and norhalimenes group
All secohalimenes known 220–232 (Fig. 16, Table S5†) are included in the ent-halimenes series, and are formed by cleavage of the C3–C4 bond. All of them show a furan unit in the side chain, except for 231179 that contains a butenolide in that chain. Frequently C20 is a carboxylic acid that lactonize with a hydroxyl group at C12, 225–230,19,45 or with a hydroxyl group at C5, 231. C3 is always a carboxylic acid (free, lactonized or esterified) except for 229 and 230. C4 is part of a disubstituted olefin (Δ4(18)) 231–232 or tetrasubstituted one (Δ4) 220–230.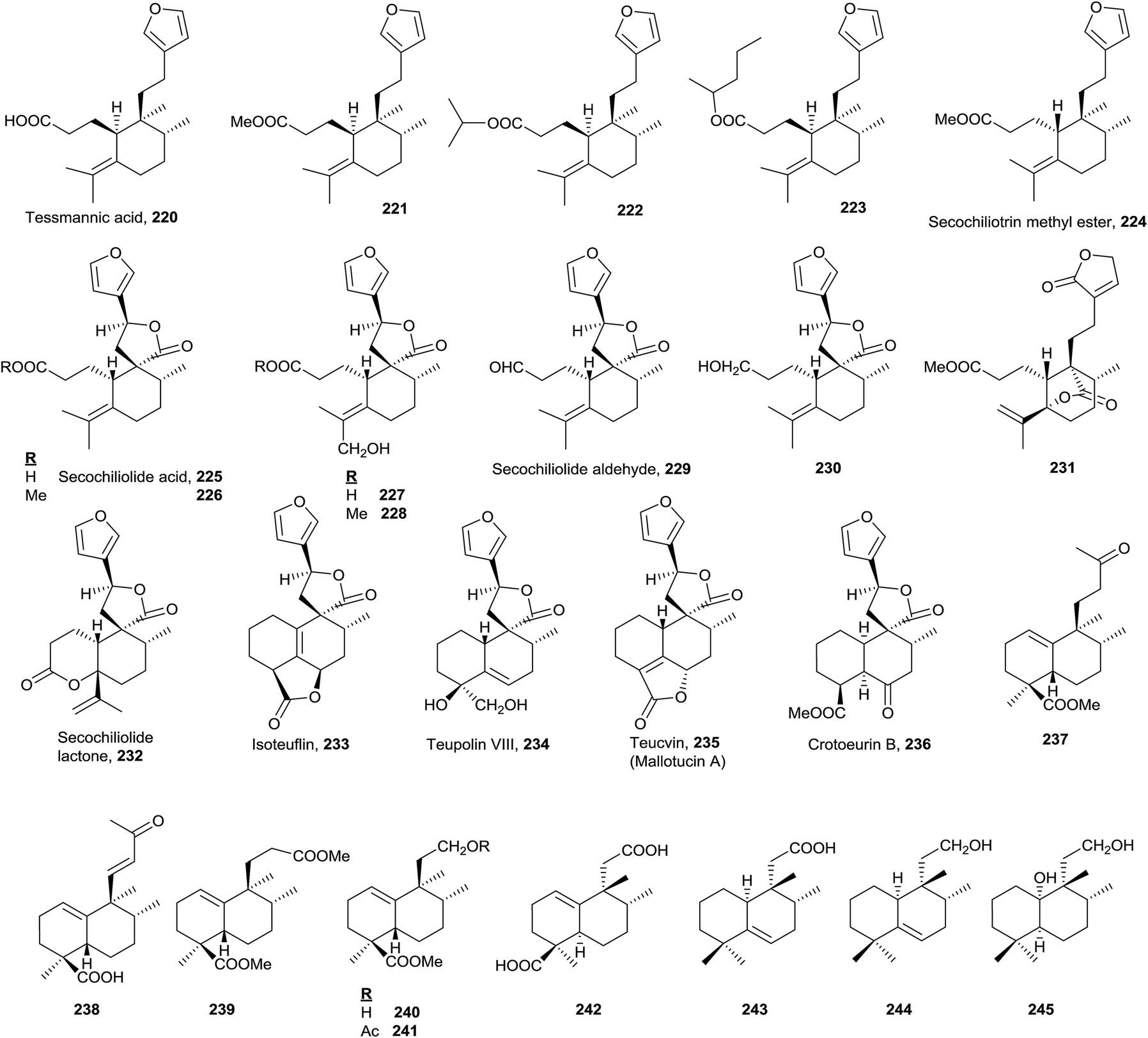 | ||
| Fig. 16 Secohalimenes and norhalimenes. | ||
These compounds are found in the plant families Leguminoseae (from Tessmannia densiflora220–22382), Compositae (Chiliotrichum rosmarinifolium224–226,19Nardophyllum lanatum224–228 and 232,19 and Nardophyllum bryoides225–226 and 23045) and Geocalycaceae (Heteroscyphus coalitus231179).
Tessmannic acid (220) and its methyl ester (221) exhibit antibacterial and antifungal activity.82,184 Butanolides 225, 226 and 230 are biologically active as antitumour agents.45
The known norhalimenes (233–245) can be mono-, di-, tri- and tetra-norhalimene derivatives (Fig. 16).
Compounds such as isoteuflin (233; isolated from Croton crassifolius144 and Teucrium canadense185), teupolin VIII (234; Teucrium polium186), teucvin (mallotucin A; 235; Teucrium viscidum,187,188Mallotus repandus,136Teucrium chamaedrys,189 and Croton jatrophoides190) and crotoeurin B (236; Croton euryphyllus191) belong to a norderivatives group and have always been considered as clerodanes. However, these compounds could be considered norhalimanes as well, since up to now there is no biosynthetic evidence regarding the exact moment at which C19 (or C18) is lost. In any case, a table collecting all norclerodanes (norhalimanes) known to date has been elaborated as a ESI† (Fig. S15–S16, Table S7†). In the table the natural sources of these compounds are presented, as well as their bioactivities and the concerning references.
Among the nine known di-, tri- and tetra-norderivative (237–245), five are ent-halim-1(10)-enes (237–241),17,118,192 all of which are isolated from Halimium viscosum. The other four are 8-epi-halim-1(10)-ene (242; Vellozia stipitata193), 8-epi-halim-5-ene (243 and 244; V. flavicans194) and 8-epi-dihydrohalimene (245; Mycale euplectelloides,195 Porifera). From all of these, the only ones that do not present a carboxylic acid function at C18 are 243, 244 and 245.
4.6. Rearranged halimane group
The known rearranged halimanes are shown in Fig. 17 (Table S6†). They can be classified into four different groups.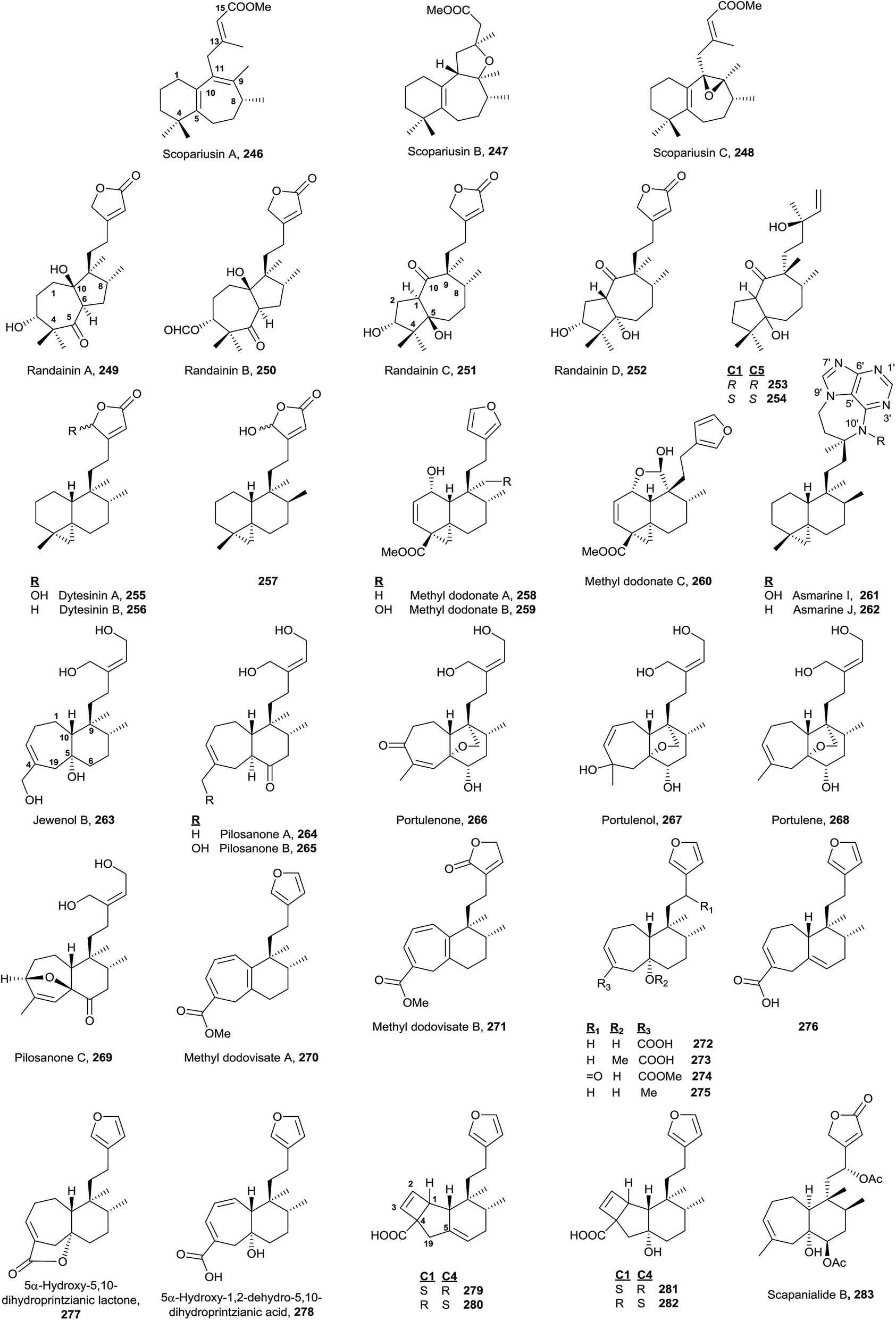 | ||
| Fig. 17 Rearranged halimanes. | ||
• Scopariusins A–C (246–248), isolated from Isodon scoparius,91,155 are characterized for ring B expansion, by incorporation of C11. The structure and absolute configuration of scopariusin A (246) were confirmed by biomimetic synthesis91 from isoscoparin N (143). These compounds were not active as antitumour agents.
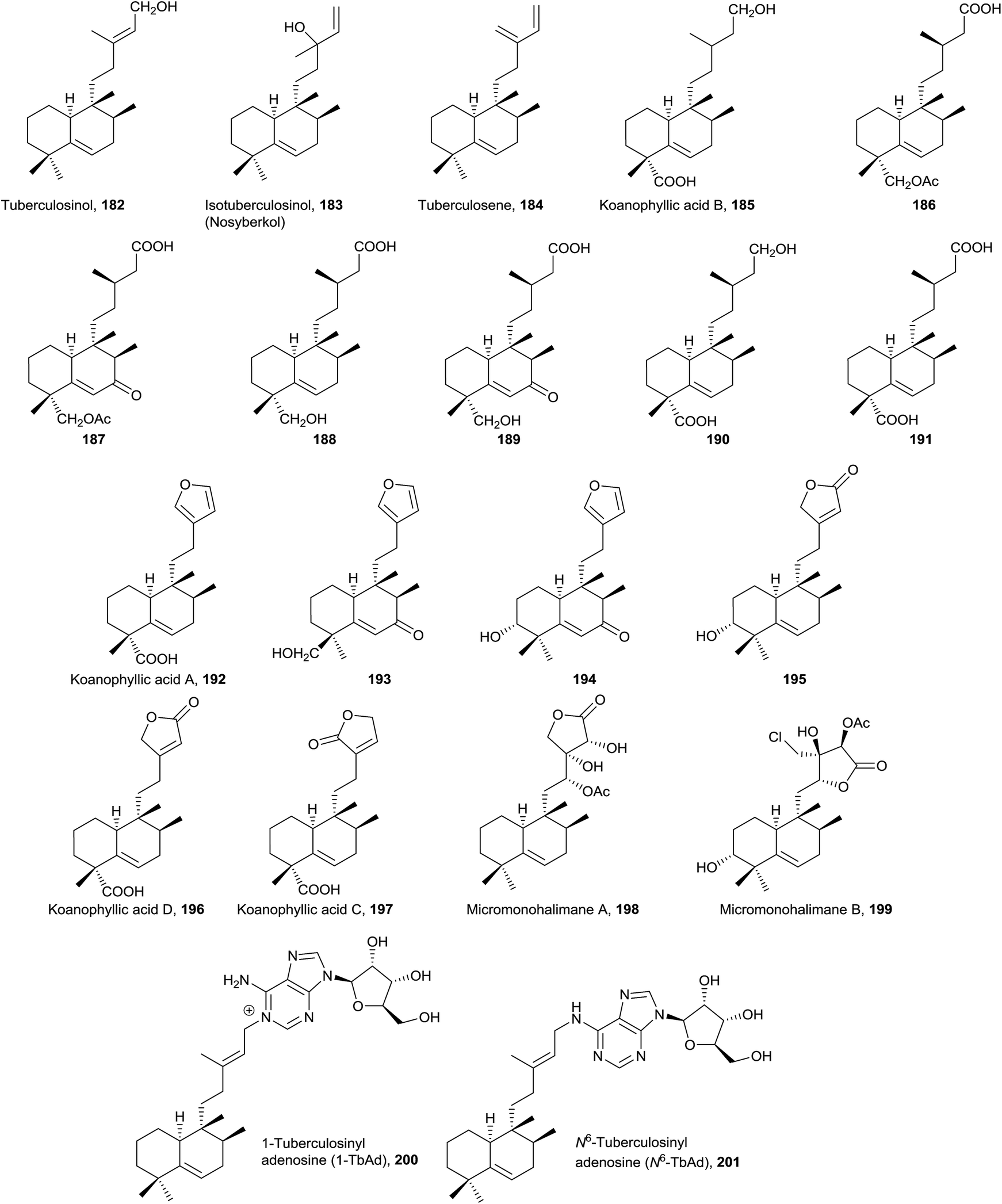
• Randainins A–D (249–252;49 from Callicarpa randaiensis) and viterofolins A–B (253–254;81Vitex rotundifolia) are rearranged compounds having a trans-7/5 ring system or a trans-5/7 ring scaffold in randainins A–B (249 and 250) or randainins C–D (251 and 252) and viterofolins A–B (253–254), respectively, instead of the classic bicyclo[4.4.0]decane system. This is a consequence of contraction-expansion ring processes. Anti-inflammatory activity and cytotoxicity were tested and evaluated for randainins A–D (249–252), and anti-hyperlipidemic activity for viterofolins A–B (253–254). Compound 252 exhibits moderate superoxide anion generation inhibition with an IC50 value of 21.5 ± 2.5 μM.
• Dytesinins A–B (255 and 256, from Cystodytes sp.196), 257 (from Echinomuricea sp.197), methyl dodonates A–C (258–260, from Dodonaea viscosa198), and asmarines I–J (261 and 262, from Raspailia sp.72) are compounds that can be considered rearranged halimanes or rearranged clerodanes because C19 is bonded to C4 and C5 forming a cyclopropane ring. Dytesinins A and B (255 and 256) are ent-halimanes and 257 is an 8-epi-ent-halimane that shows cytotoxic and anti-inflammatory activities. Compounds 255–257 have butenolides or hydroxybutenolides in the side chain. Methyl dodonates A–C (258–260) have C18 functionalized with a methoxycarbonyl group, and they show a furan at the side chain, plus a double bond Δ2. Asmarines I–J (261 and 262) are diterpene-adenine compounds with an 8-epi-ent-halimane skeleton bonded by C15 and C13 to adenine nitrogens N9 and N10. These compounds show cytotoxic activity.
• Compounds 263–283 can also be considered rearranged halimanes or clerodanes because C19 has been included in ring A as consequence of a ring expansion. All of these compounds belong to the ‘enantio’ series except viterofolins A–B (253–254) and scapanialide B (283), which belong to the ‘normal’ series. Compounds 263 (Portulaca cv Jewel199) and 264–269 (isolated from Portulaca pilosa200,201 and Portulaca grandiflora202) have an acyclic side chain, while 270–283 (isolated from Dodonaea viscosa270–271,203Conyza scabrida272–273 and 277–282,204Aparisthmium cordatum274,80Croton cortesianus275205and Scapania parva283206) possess furan or butenolides. All of them show double bonds in different positions of ring A. Compounds 279–282 show a tricyclic skeleton as a consequence of an electrocyclic reaction in ring A.
5. Halimane diterpenoids: synthesis and transformation into bioactive compounds
In the following paragraphs, several natural halimane diterpenoids synthesis and transformations of some of them into bioactive compounds are described.5.1. ent-Halimic acid as precursor of biologically active compounds and other derivatives of interest
ent-Halimic acid, characterized as its methyl ester (39), is very abundant in Halimium viscosum extract (0.34% with respect to the dry plant weight). We have developed a very quick and efficient method to isolate the natural product in multigram quantities by chromatographic separation of the ethyl acetate extract. This compound has an unsaturated side chain, Δ13, and a hydroxyl group at C-15, a carboxyl group at C-18 and a double bond Δ1(10) in the decalin system. All these functionalities make ent-halimic acid a versatile molecule and a very appropriate starting material for the synthesis of natural halimanes, biologically active compounds and other interesting compounds. In Fig. 18 some of the compounds obtained from ent-halimic acid are shown: (1) ent-Halimanolides.123,141,207,208 (2) Chettaphanin I and II.109,209 (3) Bioactive sesterterpenolides.123,210–212 (4) Sesterterpenolides and glycerophospholipids hybrid compounds.213 (5) Rearranged derivatives: ent-labdanes,214 abeopicrasanes215 and propellanes.216 (6) Sesquiterpene-quinone/hydroquinone.217 (7) Sesqui- and diterpene-alkaloids.128,218–222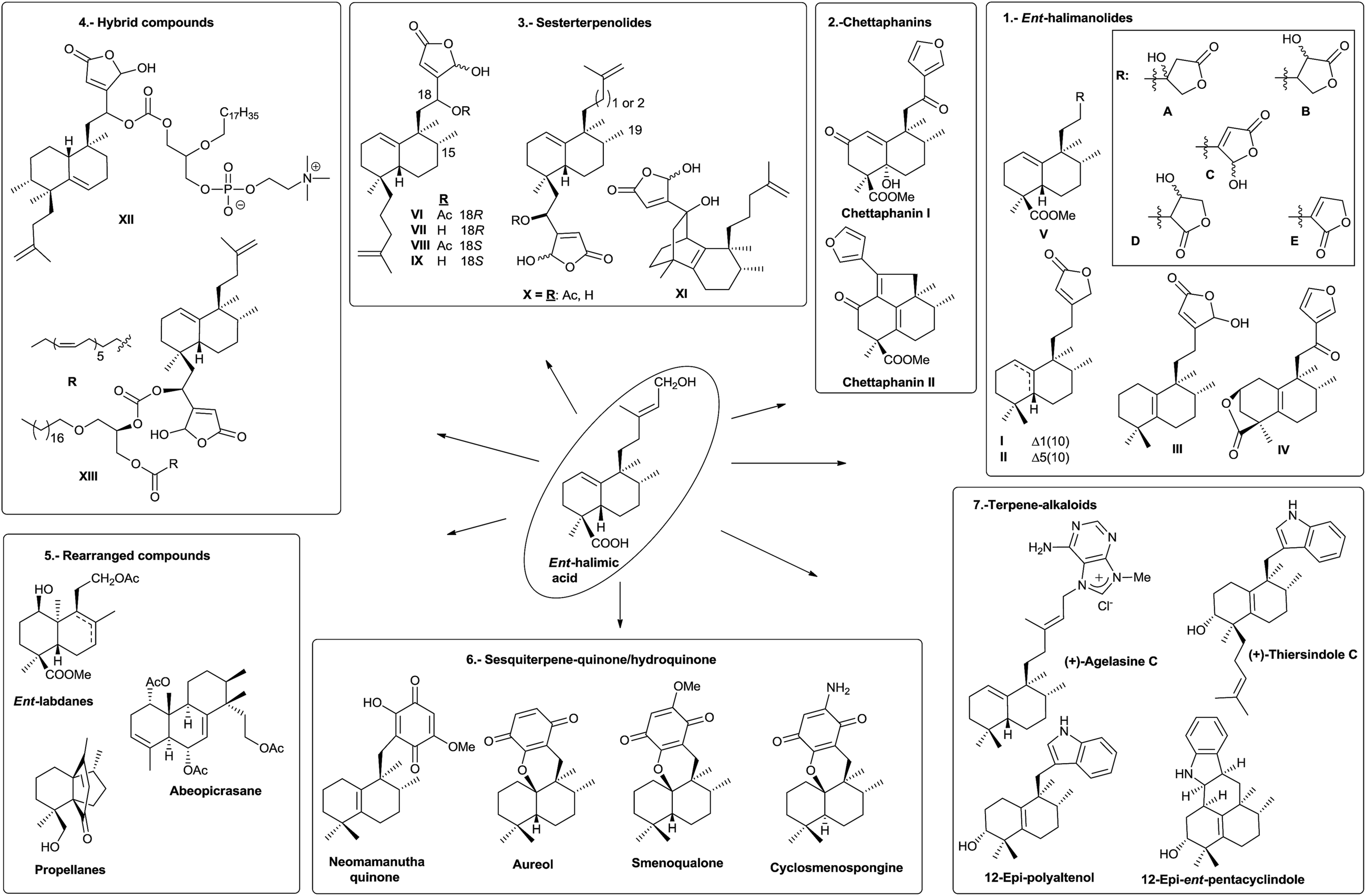 | ||
| Fig. 18 ent-Halimic acid: versatile starting material for the synthesis of bioactive and other interesting compounds. | ||
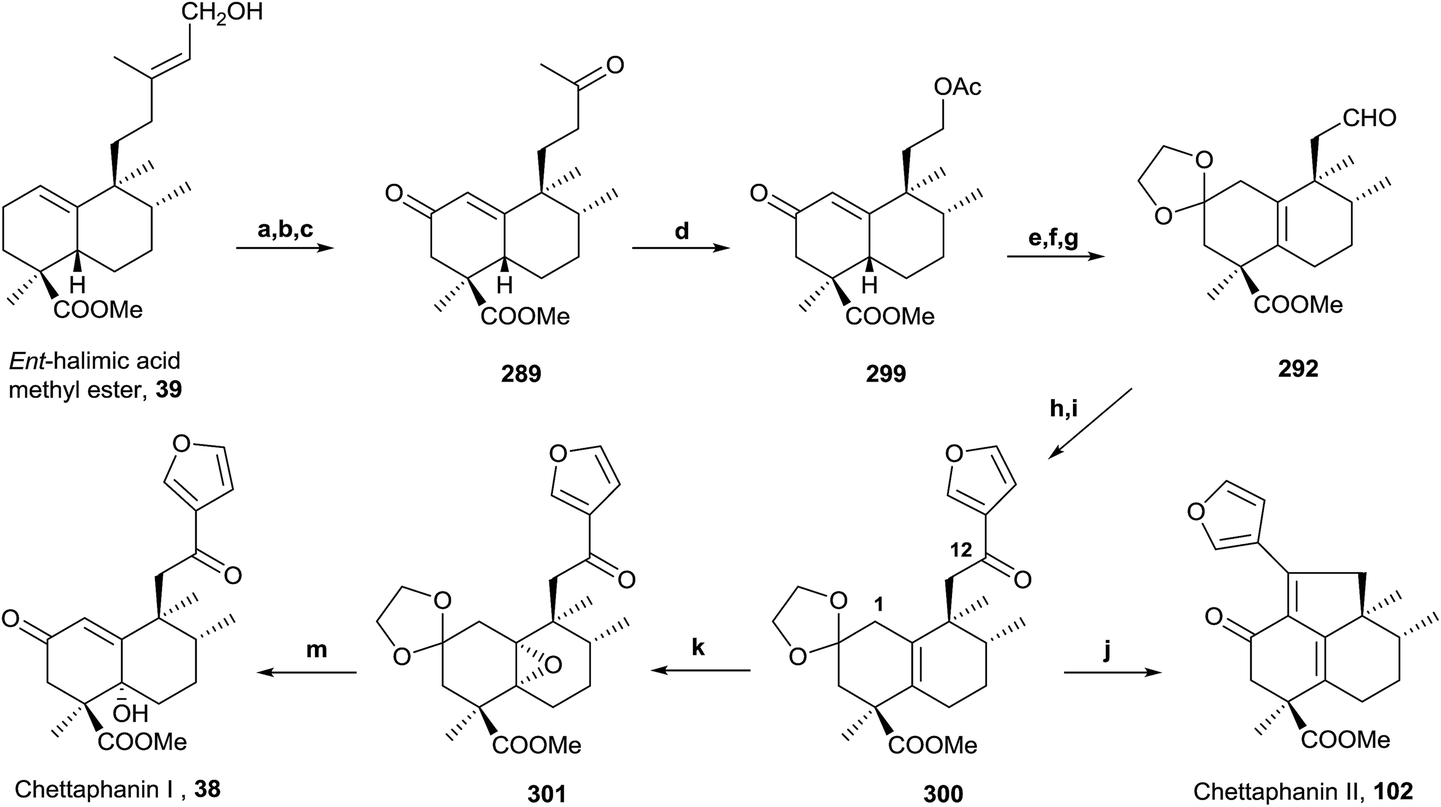
Synthesis from ent-halimic acid methyl ester (39) of several natural halimanes, such as ent-halimanolides, chettaphanin I and II and agelasine C (a diterpene-purine derivative), will be commented on in the following points. These syntheses have made it possible to corroborate their structures.
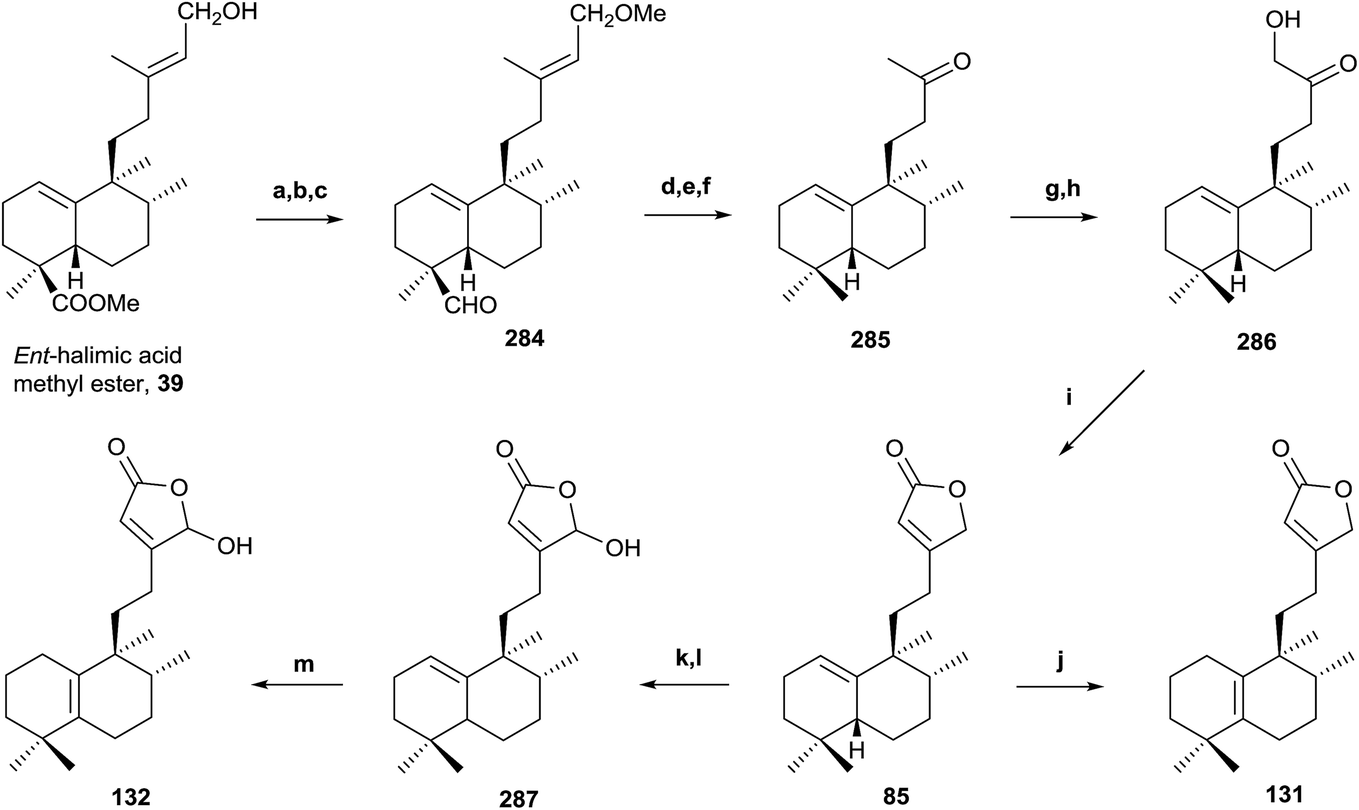 | ||
Scheme 11 (a) NaH, MeI, THF (92%); (b) LAH, Et2O (96%); (c) TPAP, NMO (91%); (d) diethylene glycol, NH2NH2·H2O, KOH, 175–230 °C, 23 h (85%); (e) m-CPBA (92%); (f) H5IO6, THF, H2O (91%); (g) LDA, TMSCl, THF, −78 °C (97%); (h) OsO4, NMO, tBuOH/THF/H2O (7:2:1) (96%); (i) Ph3P![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CCO, C6H6 (91%); (j) I2/C6H6 (10−2 M) (99%); (k) LDA, TBDMSTf, THF, −78 °C (94%); (l) m-CPBA (77%); (m) HI/C6H6 (5 × 10−2 M) (99%). CCO, C6H6 (91%); (j) I2/C6H6 (10−2 M) (99%); (k) LDA, TBDMSTf, THF, −78 °C (94%); (l) m-CPBA (77%); (m) HI/C6H6 (5 × 10−2 M) (99%). | ||
The synthesis of intermediate 285 requires the reduction of the C18 methoxycarbonyl to a methyl and two carbon degradation of the side chain. First of all, the C15 hydroxy group is protected as its methoxy derivative and the resulting compound is treated with LAH followed by TPAP oxidation to obtain 284. Huang-Minlon reduction, followed by chemoselective oxidation and cleavage of the Δ13 double bond with m-CPBA and periodic acid gives the required ketone 285 in very good yield (57% six steps).
Bestmann methodology223 has been used for the synthesis of butenolides in similar systems. In order to apply it, functionalization of C16 as a hydroxy group is necessary. Treatment of 285 with LDA in the presence of TMSCl followed by oxidation of the intermediate silylenolether with OsO4 affords the hydroxyketone 286 in excellent yield. Reaction of the hydroxyketone 286 with Ph3–PCCO gives an intermediate ylide, which cyclizes by intramolecular Wittig reaction with formation of a double bond to give butenolide 85. Isomerization reaction of the double bond Δ1(10) to Δ5(10), with I2 in C6H6, gives butenolide 131 in quantitative yield. The physical properties of the synthesized compounds 85 and 131 are identical to those described for the natural products ent-halima-1(10),13E-dien-15,16-olide and ent-halima-5(10),13E-dien-15,16-olide, respectively.
The synthesis of the γ-hydroxybutenolide 132 has been done by Boukouvalas methodology.224 Treatment of 85 with LDA and TBDMSTf followed by reaction of the intermediate 2-trialkylsilyloxyfuran with m-CPBA afforded 287 in good yield, after column chromatography. Compound 132 was obtained in quantitative yield by acidic isomerization of 287 using HI in benzene at 85 °C. The physical and spectroscopic data of the synthetic product 132 are identical with those reported for the natural product 16-hydroxy-ent-halima-5(10),13-dien-15,16-olide. This synthesis confirms the structures and absolute configurations of the natural products obtained.
Biological assays have been carried out on these compounds and confirmed that compound 85 exhibits cytotoxic and antiviral activity [HeLaM cells (IC50 = 5.0), MDCK (IC50 = 5.1) and influenza virus (IC50 = 6.8)].123
In the same way, an efficient synthesis of ent-halimanolide 106 (15,16-epoxy-12-oxo-ent-halima-5(10),13(16),14-trien-18,2β-olide) has been achieved from ent-halimic acid methyl ester 39, corroborating the structure of the natural compound and establishing its absolute configuration (Scheme 12).141 A new route employing the dinorderivatives 289 and 291 as intermediates allowed the tetranorderivative 292 to be obtained in multigram scale (53% from 39).
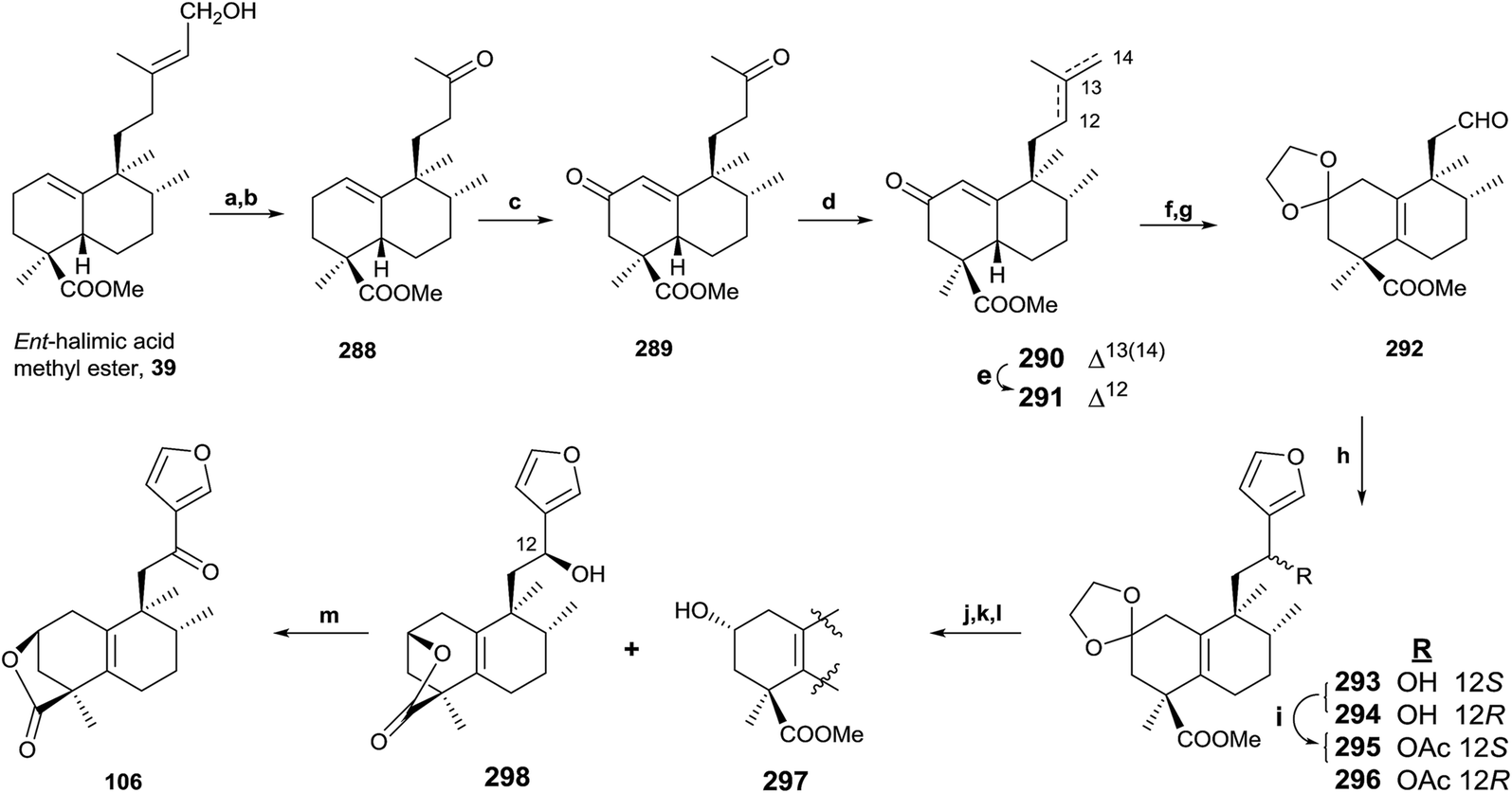 | ||
| Scheme 12 (a) OsO4, NMO, t-BuOH/THF/H2O (7:2:1); (b) Pb(AcO)4, C6H6, 20 min, (94%, two steps); (c) Na2CrO4, Ac2O/AcOH, NaOAc, C6H6, (64%); (d) MePPh3Br, NaHMDS, THF, −78 °C (94%); (e) p-TsOH, C6H6, 60 °C (96%); (f) (CH2OH)2, p-TsOH, C6H6, Dean Stark (97%), (g) (1) OsO4, NMO, t-BuOH, THF, H2O; (2) Pb(AcO)4, C6H6 (96%, two steps); (h) 3-bromofuran, n-BuLi, THF (293: 54%, 294: 39%); (i) Ac2O, pyridine (98%); (j) HCl 2 M, EtOH (96%); (k) Na2CO3, MeOH, 2 h, (96%); (l) NaBH4, EtOH (298: 38%, 297: 43%); (m) TPAP, NMO, DCM, rt, 50 min (92%). | ||
ent-Halimic acid methyl ester (39) oxidation with OsO4 was regioselective. The resulting triol was oxidized with Pb(OAc)4, giving ketone 288 in a 94% global yield for the two steps. Oxidation of 288 with Na2CrO4 gives the α,β-unsaturated ketone 289 (64%). Compound 290 was obtained by chemoselective Wittig reaction of 289. Treatment of 290 in very mild acidic conditions produces double bond isomerization to give 291 in quantitative yield. The synthesis of 292 was achieved in three steps. First, dioxolane carbonyl group protection of 291 gives not only the protection of the carbonyl function, but the double bond isomerization in the bicyclic system to the tetrasubstituted position; and then, OsO4 oxidation and Pb(OAc)4 treatment lead to aldehyde 292. Reaction of 292 with 3-furyllithium gives 293 and 294, which are separated and characterized as their acetylderivatives 295 and 296. The careful hydrolysis of 295 and 296 in acid medium leads to intermediate ketones that produce 297 and lactone 298 by alkalyne hydrolysis and reduction with NaBH4. Oxidation of 298 gives the desired ent-halimanolide 106 in good yield, which is moderately active against HeLa cells (human cervical cancer).
 | ||
| Scheme 13 (a) OsO4, NMO, t-BuOH/THF/H2O (7:2:1); (b) Pb(AcO)4, C6H6 (94%, two steps); (c) Na2CrO4, Ac2O/AcOH, NaOAc, C6H6 (64%); (d) UHP/TFAA (61%); (e) ethylene glycol, p-TsOH, C6H6; (f) 3% K2CO3 in MeOH; (g) PDC, DMF (61%, three steps); (h) 3-bromofuran, nBuLi, (1:1), −78 °C; (i) TPAP/NMO, (99%, two steps); (j) p-TsOH, acetone (72%); (k) m-CPBA, CH2Cl2 (92%); (m) p-TsOH, acetone or HClO4, 30%. | ||
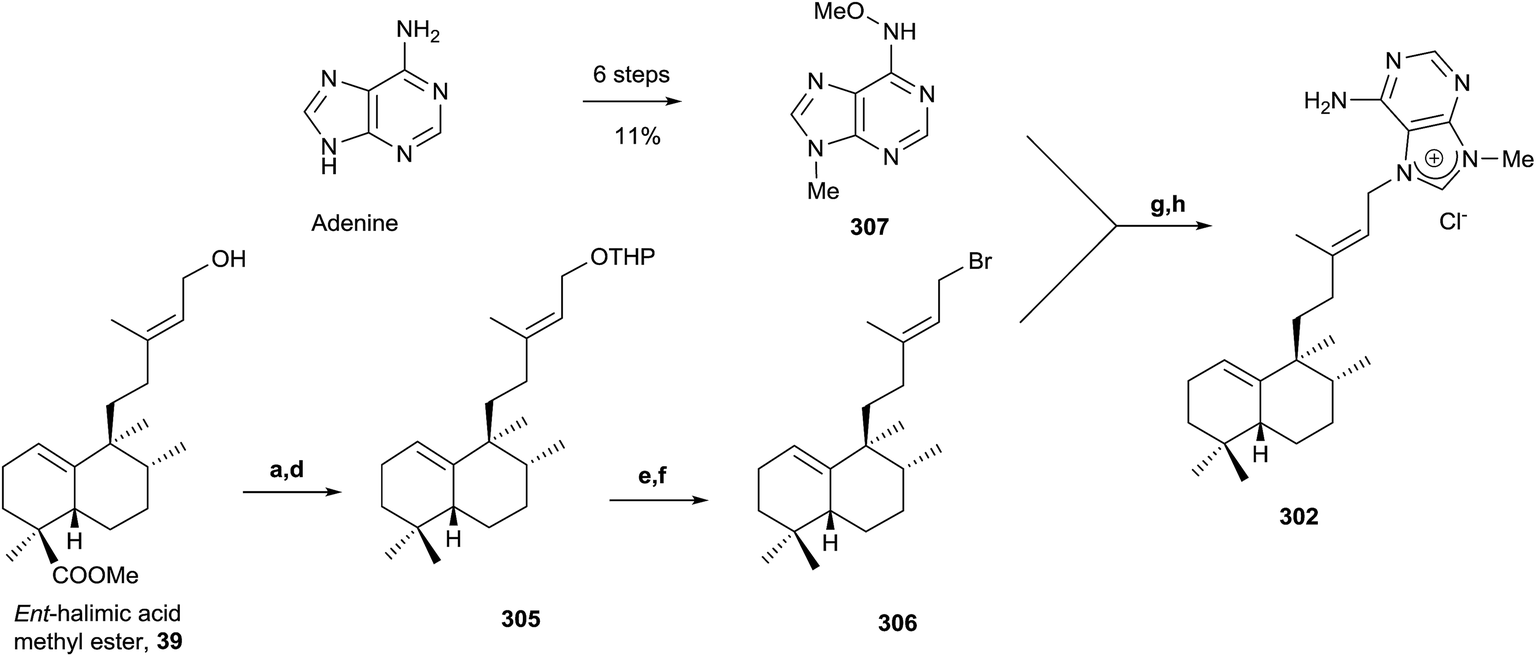 | ||
| Scheme 14 (a) DHP, p-TsOH, C6H6 (98%); (b) LAH, Et2O, 0 °C and then rt (99%); (c) TPAP, NMO (94%); (d) diethylene glycol, NH2NH2·H2O, KOH, 175–230 °C (81%); (e) p-TsOH, MeOH (81%); (f) CBr4, PPh3, DCM (76%); (g) DMA, 50 °C; (h) Zn, MeOH, H2O, AcOH (13%, two steps). | ||
Agelasines are diterpene alkaloid 7,9-dialkylpurine salts, isolated from marine sponges of the genus Agelas.225 Agelasine C is one of the first four agelasines to be isolated by Nakamura and co-workers in 1984126 from the Okinawan sea sponge Agelas sp. (−)-agelasine C showed powerful inhibitory effects on Na,K-ATPase and antimicrobial activities (Fig. 19). In their work, Nakamura and co-workers proposed the structural formula 303 for (−)-agelasine C. epi-Agelasine C (304) was isolated in 1997 by Hattori and co-workers75 from the marine sponge Agelas mauritiana as an antifouling substance active against macroalgae.
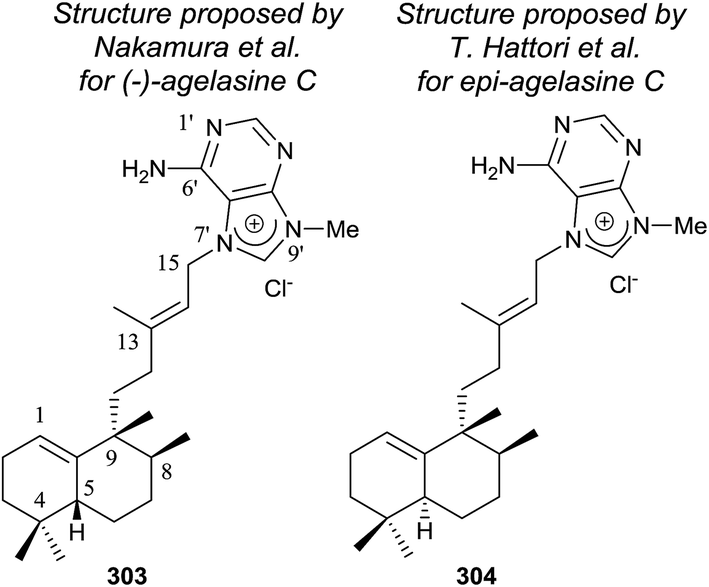 | ||
| Fig. 19 Proposed structures for (−)-agelasine C and epi-agelasine C. | ||
Due to the interest of epi-Agelasine C as an antifouling agent75 and to establish the absolute configuration of this compound, the synthesis of 302 (Scheme 14) was carried out.128 The synthesis of 302 was planned following an analogue design for other agelasines, which consists of coupling the terpenic fragment 306 with a purine derivative such as 307.128
The synthesis of the bromoderivative 306 was achieved in six steps starting from ent-halimic acid methyl ester (39) using the tetrahydropyranyl derivative 305 as an intermediate (Scheme 14) (46%, six steps). Reaction of 39 with DHP in p-TsOH followed by reduction with LAH leads to a hydroxy derivative that was oxidized with TPAP to the carbonyl function and reduced using Huang-Minlon methodology to give 305. Deprotection of the primary hydroxyl group followed by treatment with CBr4 in the presence of PPh3 gave the required bromoderivative 306 (46%, six steps). Alkylation of methoxyadenine 307 with the bromoderivative 306 by heating in dimethylacetamide and subsequent reduction with Zn/AcOH gave compound 302 (13%, two steps).
The physical properties of the synthesized product 302 were very different to those of the natural product epi-agelasine C, thus its proposed structure 304 (Fig. 19) should be revised. On the other hand, when the 1H and 13C NMR spectra of 302 were compared with those for the proposed structure of (−)-agelasine C (303), the two pairs of spectra were identical. However, the optical rotatory power of 302 and natural (−)-agelasine C were similar in absolute value but had a different sign. So, it should be concluded that the structure of the natural product (−)-agelasine C should be corrected to structure 89 (Fig. 20), which is the enantiomer of the synthesized product (+)-agelasine C (302, Scheme 14).
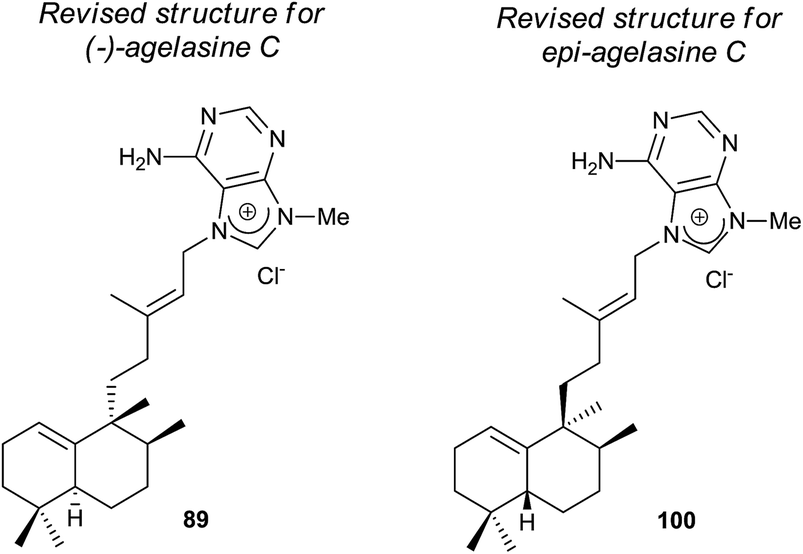 | ||
| Fig. 20 Revised structures for (−)-agelasine C and epi-agelasine C. | ||
Spectroscopic considerations made when comparing the spectra of 302 with those of epi-agelasine C and their specific rotations permitted structure 100 to be suggested for the natural epi-agelasine C, as shown in Fig. 20.
5.2. Synthesis of 3α-hydroxy-5β,10β-epoxychiliolide (214)
The natural product structure of 3α-hydroxy-5β,10β-epoxychiliolide (214) was corroborated by the synthesis of the racemic diterpene (Scheme 15).182 The synthesis starts with the available tetralone 308. Intermediate 309 was obtained in high yield by Mannich reaction, followed by hydrogenation of the resulting exo-methylene bond. In order to introduce the carboxyl group and achieve 310, an epoxidation followed by epoxide isomerization with BF3·Et2O was done. In this manner the corresponding aldehydes, epimers at C9, were obtained, which via Jones oxidation led to 310. Birch reduction of 310 followed by hydrolysis, esterification and protection as 1,3-dioxolane, alkylation with LDA and allyl bromide and deprotection with diluted sulfuric acid on silica gel gave ketone 311 in 72% overall yield. Reduction of 311 with NaBH4 followed by oxidation with osmium tetroxide/N-methyl morpholine-N-oxide and cleavage with sodium periodate gave aldehydes 312a and 312b, which were separated by column chromatography. From ketone 311, the derivative 313 was also obtained by oxidative degradation. Its X-ray analysis allowed the configuration of C8 and C9, quaternary centres, to be established for all compounds. Reaction of epimer 312 with 3-lithiofuran and subsequent lactonization afforded the intermediates 314a–d, and the epoxidation of each led to the isomeric diterpenes 315a–d. Configuration at C12 was established by nOe experiments, but the relative stereochemistry at C3, C5, and C10 still had to be solved. The NMR spectra for 315a was identical to the natural product one. Only one epoxide was obtained with both isomers, so a β-orientation of the epoxy group was more likely. PCC oxidation of 315a gives ketone 316, which gave one isomer by reduction with sodium borohydride, the 3β-hydroxy derivative 315c, so the natural product 214 is the 3α-hydroxy derivative 315a. Through inspection of a model, it was determined that the most favorable entry of the hydride is by the α-face of the molecule, so the relative configuration of all compounds was established in this manner.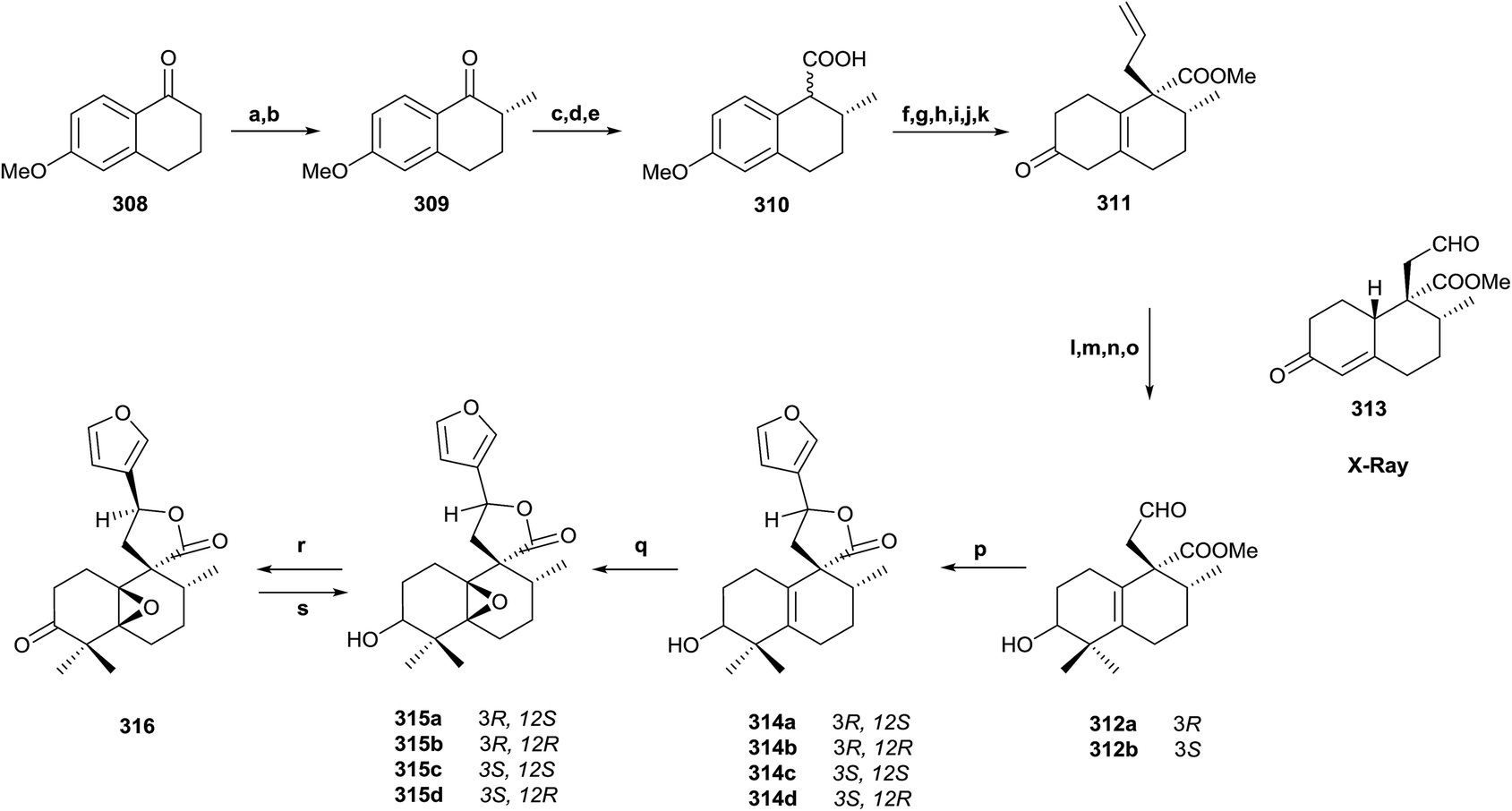 | ||
| Scheme 15 (a) p-Formaldehyde, N-methyl-anilinium-trifluoro acetate; (b) H2, RANEY® nickel; (c) trimethylsulfonium iodide, DMSO, NaH; (d) BF3·Et2O; (e) Jones reagent; (f) Li, NH3; (g) HCl 10%, MeOH, H2O; (h) CH2N2; (i) ethyleneglycol, TMSCl, CH2Cl2; (j) LDA, allyl bromide; (k) SiO2, H2SO4; (l) MeI, t-BuOK, t-BuOH; (m) NaBH4, EtOH; (n) OsO4, NMO; (o) NaIO4, H2O; (p) 3-bromofuran, nBuLi, (1:1), −78 °C; (q) m-CPBA, CH2Cl2; (r) PCC, CH2Cl2; (s) NaBH4, EtOH. | ||
5.3. Synthesis of (+)-agelasimine A (217) and (+)-agelasimine B (218)
Ohba and co-workers226 synthesized the diterpene-adenine derivatives (+)-agelasimine A (217) and (+)-agelasimine B (218) from (+)-trans-dihydrocarvone and, in this manner, they established the absolute configuration of the natural products isolated from the orange sponge Agelas mauritiana (Scheme 16). Previously, Ohba and co-workers followed a similar reaction sequence when they communicated the racemic synthesis of (±)-agelasimine A and (±)-agelasimine B.227,228 In the asymmetric synthesis, the authors used (+)-trans-dihydrocarvone (317) as the starting material. Ozonolysis in methanol of 317 and subsequent treatment with FeSO4-Cu(OAc)2 (ref. 229 and 230) led to (+)-318, which was transformed into (+)-319 by MeLi treatment followed by PCC oxidation.231 Reaction of (+)-319 with vinylmagnesium bromide in the presence of CuBr and Me3SiCl gave stereoselectively a silylenol ether that reacts with formaldehyde to give 320 as a diastereoisomeric mixture. Cyclohexanone 320 was transformed into 321 following the previously described procedure.231 Methylation of 321 and subsequent Huang-Minlon reduction led to 322. Reaction of 322 with 9-BBN followed by Suzuki cross-coupling reaction with E-3-iodo-2-butenoic acid ethyl ester (323) afforded the α,β-unsaturated ester (324). Treatment of 324 with m-CPBA, DIBAL reduction and subsequent treatment with LAH in boiling THF for epoxide reduction led to 325. Reaction of 325 with PBr3 achieved bromination of the primary hydroxy group and subsequent alkylation with 3-methyladenine led to compound 326 after neutralizing the hydrobromide salt. Methylation of (+)-326 with MeI followed by neutralization gave the desired compound (+)-217, which was identical to the natural product (+)-agelasimine A. Reaction of (+)-326 with NaBH4 followed by methylation and neutralization led to (+)-218, which was identical to the natural product agelasimine B. In this manner, the two structures (217 and 218) were corroborated and their absolute configurations established.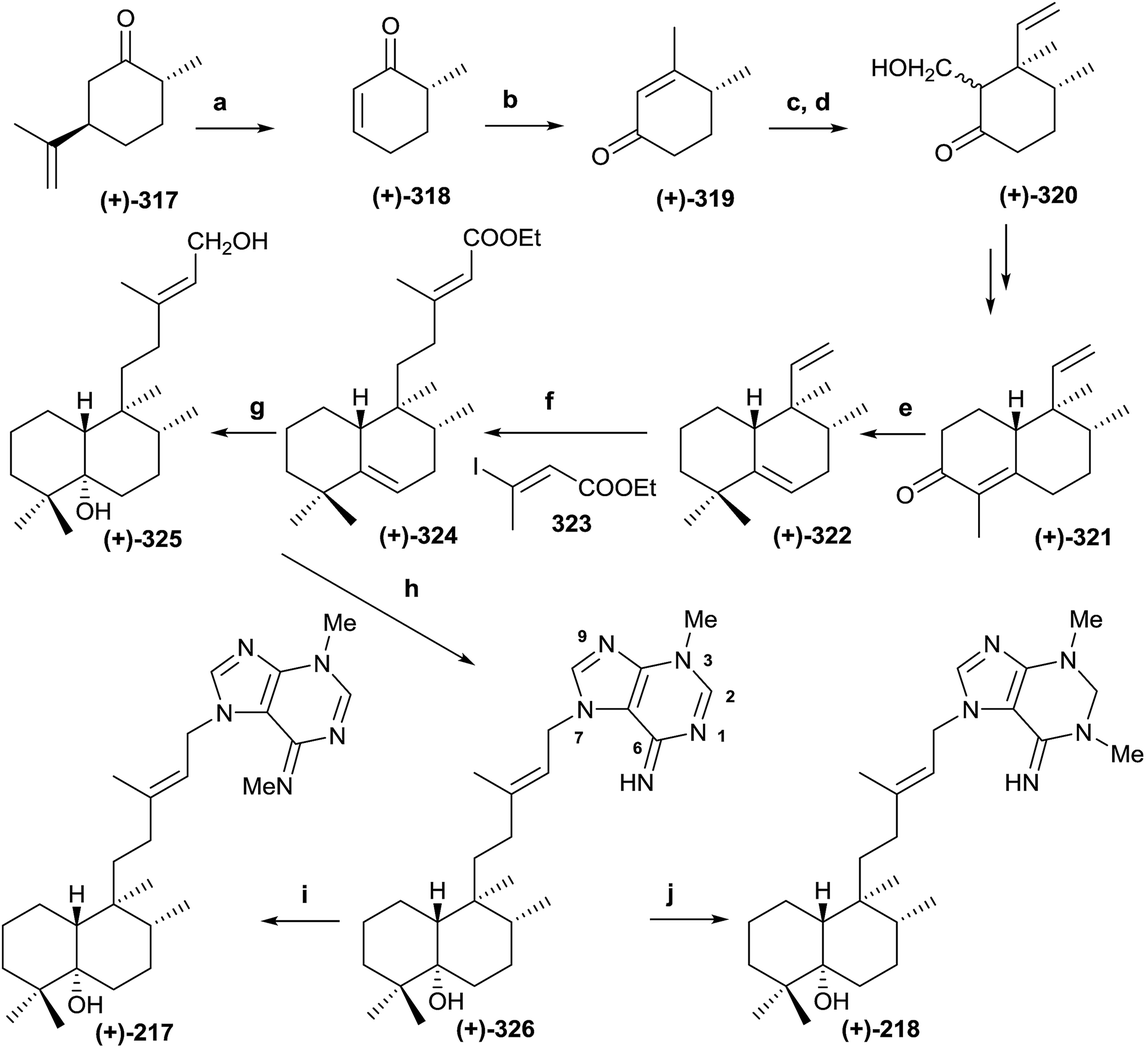 | ||
| Scheme 16 (a) (1) O3, MeOH, (2) FeSO4, Cu(OAc)2 (44%) ; (b) (1) MeLi, (2) PCC (94%); (c) (1) CH2CHMgBr, CuBr·Me2S, Me3SiCl; (d) aq. HCHO, Yb(OTf)3 (75%); (e) (1) MeI, t-BuOK, t-BuOH, (2) NH2NH2·H2O, KOH, diethylene glycol 130 °C 1 h, 190 °C 3 h (72%); (f) (1) 9-BBN, (2) 323, PdCl(dppf), CsCO3, Ph3As (67%); (g) (1) m-CPBA; (2) DIBAL, −78 °C; (3) LAH, THF reflux (51%); (h) (1) PBr3, (2) 3-methyladenine, AcNMe2; (3) aq. NaOH (60%); (i) (1) MeI, AcNMe2; (2) aq. NaOH(61%); (j) (1) NaBH4, (2) MeI, AcNMe2; (3) aq. NaOH (41%). | ||
5.4. Biomimetic synthesis of scopariusin A (246) and isoscoparin N (143)
Scopariusin A (246) was isolated from Isodon scoparius. Its structure was spectroscopically determined and confirmed by biomimetic synthesis from the clerodane isoscoparin O (327) found in the same plant (Scheme 17).91 The structure and absolute configuration of isoscoparin O (327) has been confirmed previously by X-ray analysis and for scopariusin A (246).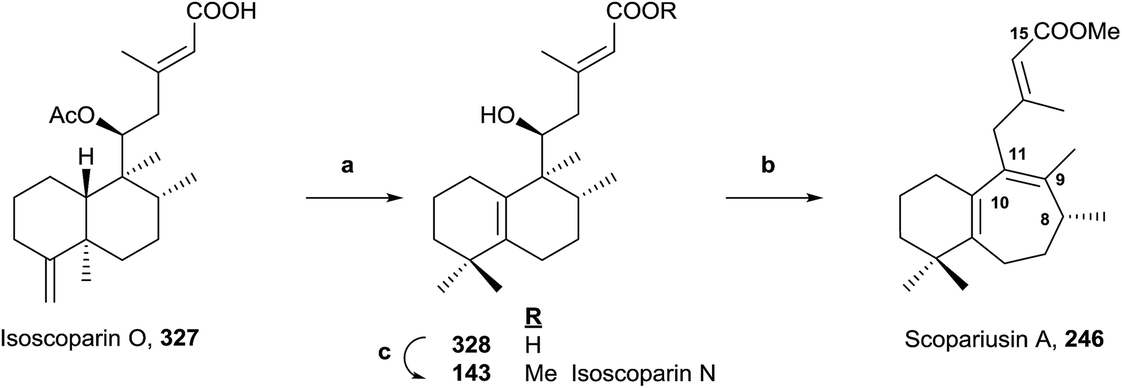 | ||
| Scheme 17 (a) (1) BF3·Et2O, (2) NaOH, MeOH (67%); (b) (1) PTSA, PhCH3, 85 °C, (2) MeI, KOH (64%); (c) MeI, K2CO3 (95%). | ||
Treatment of 327 with BF3·Et2O leads mainly to a halimane derivative, which leads to the hydroxy acid 328 by hydrolysis with NaOH in MeOH. Esterification of 328 with MeI in the presence of potassium carbonate gives 143, whose properties were identical to isoscoparin N isolated from the same extract of Isodon scoparius. Reaction of 328 with p-toluensulfonic acid (PTSA) in toluene and posterior methylation with MeI in KOH-acetone led to 246, whose properties were identical to those of the natural product scopariusin A. In this manner, the structures and absolute configurations of isoscoparin N (143) and rearranged ent-halimane scopariusin A (246) were confirmed.
5.5. Synthesis of tuberculosinol (182) and isotuberculosinol (183)
Tuberculosis, caused by Mycobacterium tuberculosis, is one of the biggest causes of morbidity and mortality worldwide. Developing new drugs effective against those bacteria and new therapies directed to inhibit virulence factor (VF) formation have a great interest.60,63The tuberculosinols tuberculosinol (182) and isotuberculosinol (183; 13R and 13S) are VFs from M. tuberculosis.57,59,93 However, without any doubt the most interesting and promising halimane is tuberculosinol (182). The original proposed structures have been confirmed by synthesis, as can be seen in the following points.56,58
Snider and co-workers' and Sorensen and co-workers' syntheses of tuberculosinol and isotuberculosinol (Schemes 18 and 19, respectively) were published simultaneously and made possible a structural revision of the diterpene obtained from M. tuberculosis, to which the edaxadiene structure was originally assigned, and finally it was revised and reassigned to the same structure of nosyberkol (183; isolated from the Red Sea sponge Raspailia sp.72 extracts), also known as isotuberculosinol.
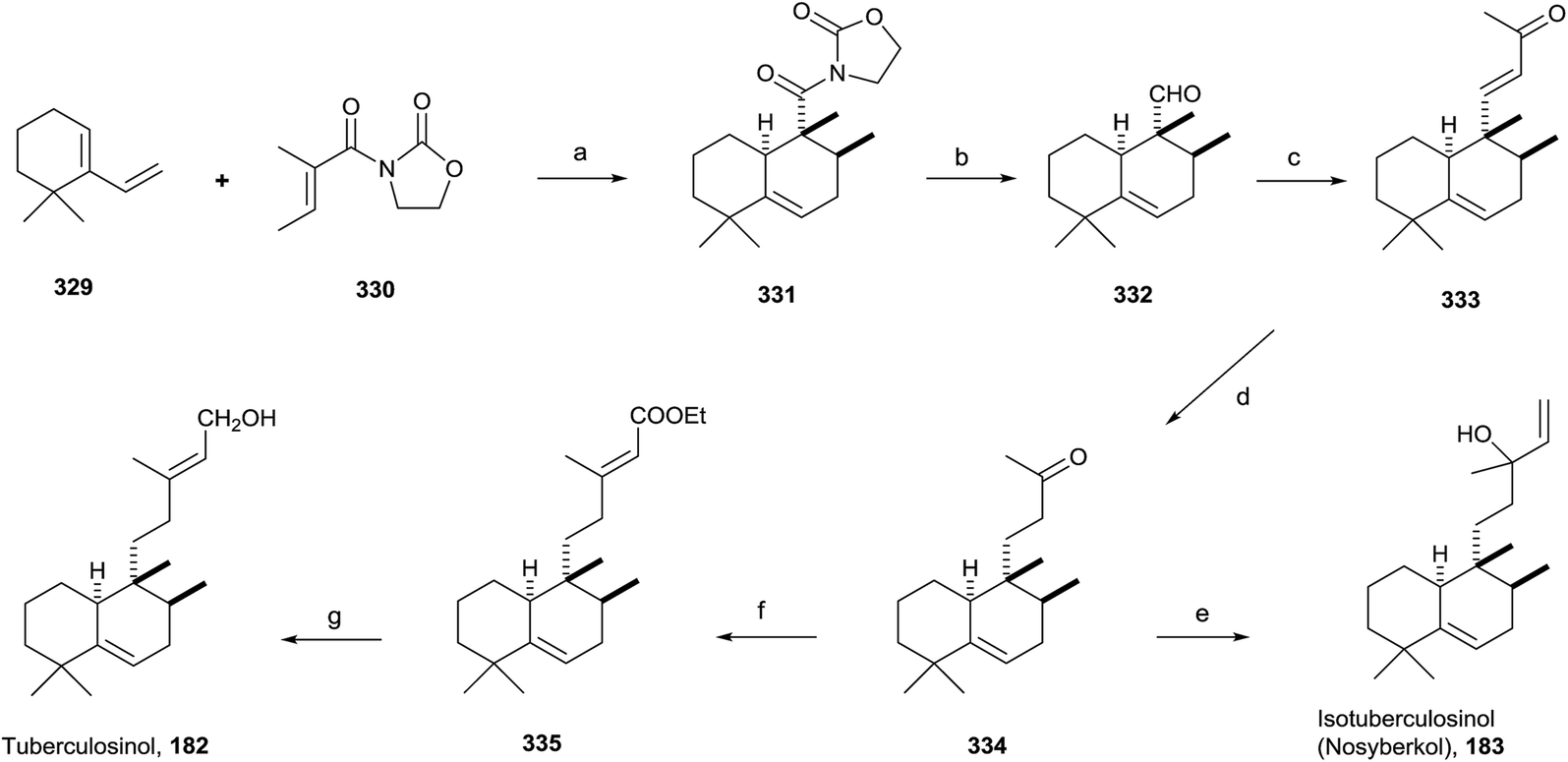 | ||
| Scheme 18 a) Me2AlCl, CH2Cl2 (54%, ∼10:1 exo/endo); (b) (1) LiBH4, THF/H2O; (2) DM periodinane, CH2Cl2 (77%); (c) acetone/MeOH, NaOMe (60%); (d) (1) Li, NH3, EtOH; (2) Jones oxidation (90%); (e) CH2CHMgBr, THF (88%); (f) (EtO)2POCH2COOEt, NaH, THF (84%); (g) DIBAL, CH2Cl2 (92%). | ||
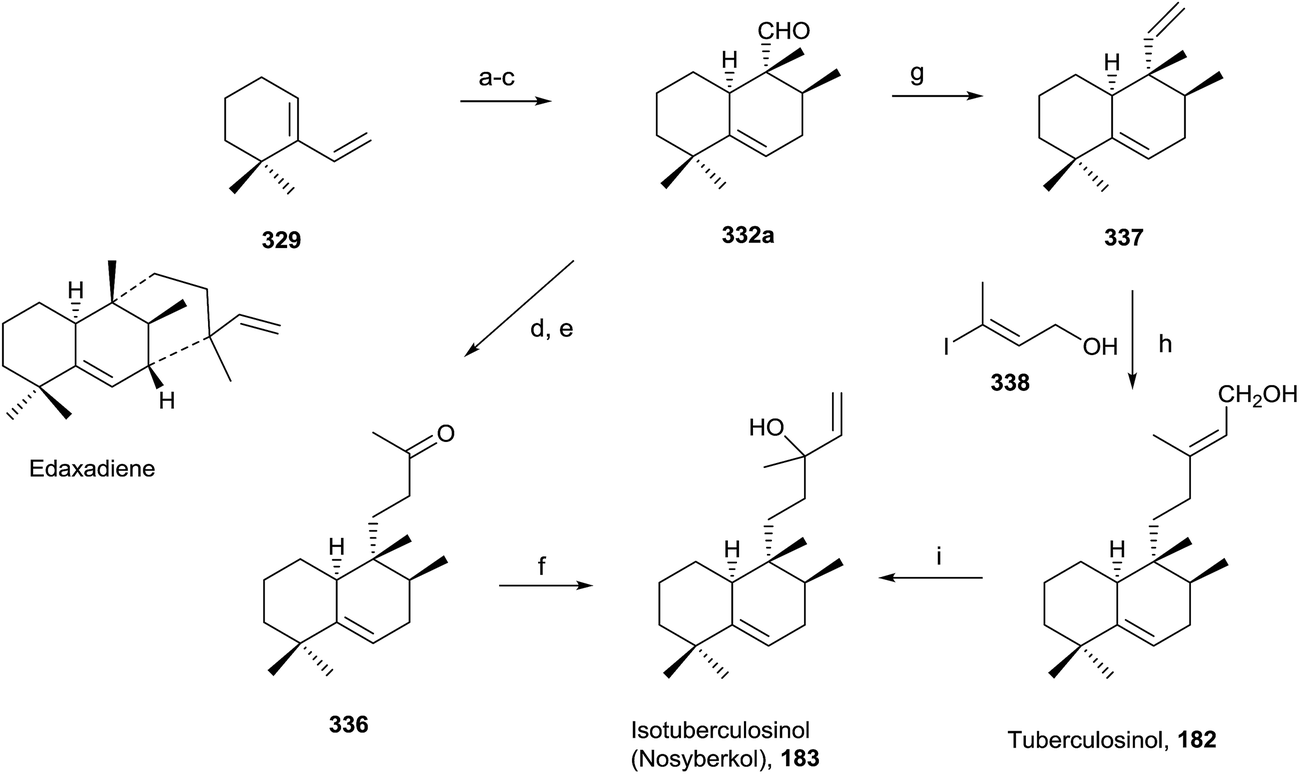 | ||
| Scheme 19 (a) Ethyl tiglate, neat, 160 °C, (71%) (2:1 exo/endo); (b) LAH, THF, (56%); (c) SO3·pyridine, NEt3, CH2Cl2–DMSO (86%); (d) acetone, NaHMDS, THF (87%); (e) 10 mol% Rh(PPh3)3Cl, HSiEt3, CH2Cl2 (83%); (f) vinylmagnesium bromide, THF (93%); (g) Ph3PCH3Br, KHMDS, THF (91%); (h) 9-BBN, THF; then 10 mol% PDCl2(dppf), Ph3As, CsCO3, 338, DMF (73%); (i) 20 mol% CuCl2, acetone (20%). | ||
:1 exo/endo). Reduction of 331 followed by oxidation with Dess–Martin periodinane gives exo aldehyde 332. Reaction of 332 with acetone in the presence of NaOMe leads to 333, which gives ketone 334 by reduction with Li in NH3/THF/EtOH, then by addition of vinylmagnesium bromide provides 183 as a mixture of stereoisomers. The spectroscopic data obtained from the synthetic compound 183 were identical with those reported for both natural nosyberkol and isotuberculosinol.
Reaction of ketone 334 with triethylphosphonoacetate in the presence of NaH leads to 335 in good yield. DIBAL reduction of 335 leads to 182, which is identical to the natural tuberculosinol.55
Tuberculosinol (182) was also synthesized from aldehyde 332a. Methylenation of 332a gives diene 337, which by 9-BBN hydroboration and palladium-mediated cross-coupling with (E)-3-iodobut-2-en-1-ol 338 provides tuberculosinol (182). Isotuberculosinol (nosyberkol; 183) was achieved by treatment of 182 with catalytic copper(II) chloride.
5.6. 1-Tuberculosinyl adenosine (1-TbAd; 200) and N6-tuberculosinyl adenosine (N6-TbAd; 201) syntheses
To date, there is no reliable and fast routine test for the diagnosis of infections caused by M. tuberculosis, in spite of it being one of the more prevalent and fatal causes of infection in the world. Recently, two natural compounds, 1-tuberculosinyl adenosine (1-TbAd; 200) and N6-tuberculosinyl adenosine (N6-TbAd; 201), have been found.62,71 These tuberculosinol derivatives can be used as Mycobacterium tuberculosis (Mtb) specific biomarkers. The N6-TbAd (201) isomer appears that can be generated in vivo by M. tuberculosis, following a Dimroth rearrangement of 1-TbAd (200).62 Recently, 1-TbAd (200) and N6-TbAd (201) syntheses have been carried out (10 and 11 steps, respectively) using as a key step a chiral auxiliary-aided Diels–Alder reaction63 (Scheme 20). This Diels–Alder reaction gives 339 (98% ee, 59%, ∼10:1 exo/endo), which is transformed into tuberculosinol (182) in seven steps. 1-TbAd (200) was obtained in two steps from (182) through the chloride derivative 340 and N6-TbAd (201) was achieved later from 200 by a Dimroth rearrangement.
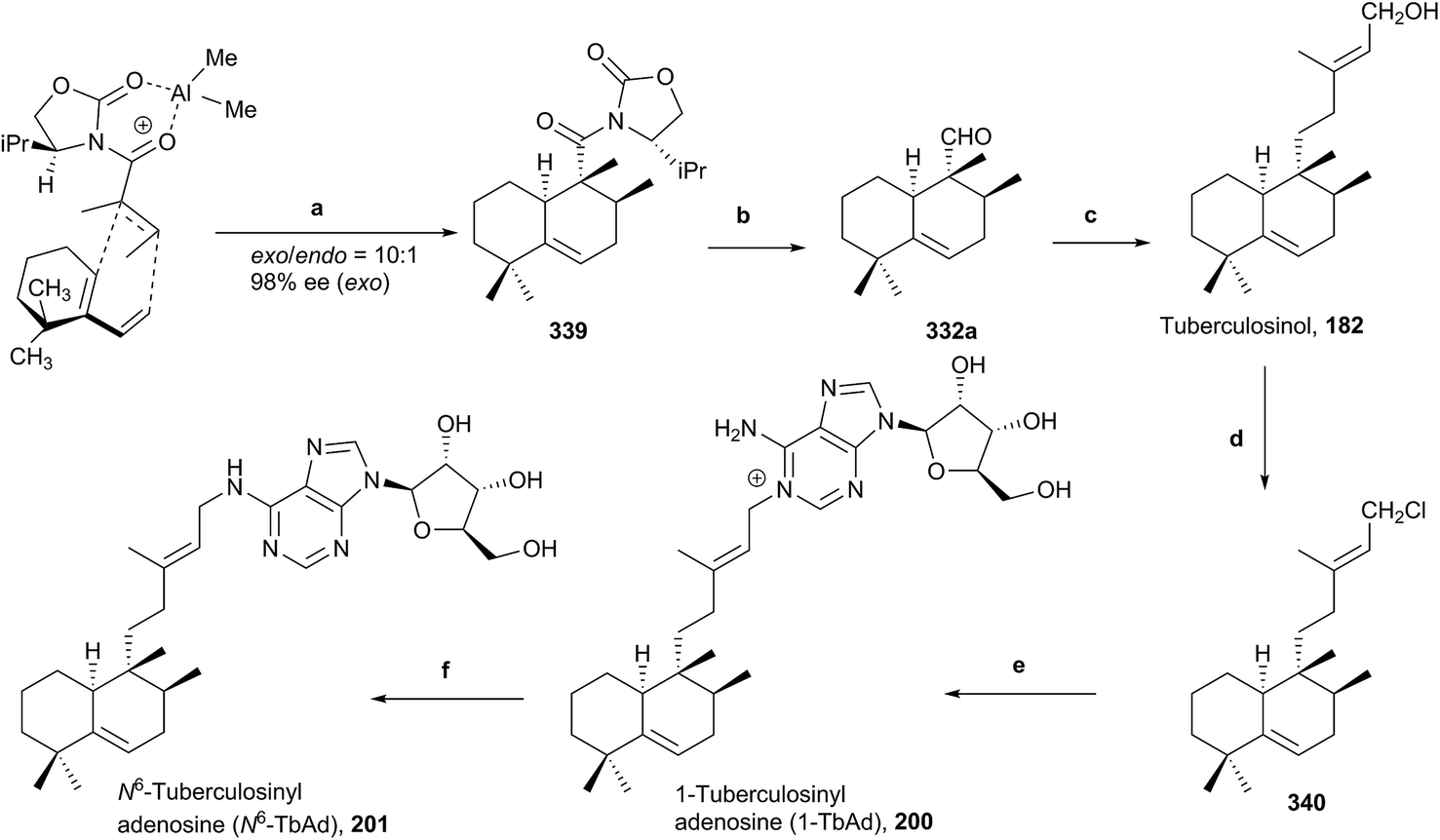 | ||
| Scheme 20 (a) Me2AlCl, CH2Cl2 (59%, ∼10:1 exo/endo); (b) (1) n-BuLi, EtSH, THF; (2) LAH, THF; (3) TPAP, NMO, CH2Cl2 (87%); (c) (1) NaHMDS, acetone, THF; (2) RhCl(PPh3)3, Et3SiH, CH2Cl2; (3) (EtO)2POCH2COOEt, NaH, THF; (4) DIBAL, CH2Cl2 (55%); (d) NCS, Me2S, CH2Cl2 (quantitative); (e) adenosine, NaI, DMF, rt (76%); (f) Me2NH, H2O, rt (quantitative). | ||
6. Conclusion
In this article, the first natural halimane skeleton diterpenoids review is reported. We have classified them into six different groups according to their biogenetic origin. Herein, 246 natural halimanes have been collected, summarizing their structure, natural source and bioactivity.Among the halimane family, the major group corresponds to the ‘antipode or enantio’ series, as also happens in the labdane skeleton diterpenoids. In this manner, ent-HPP and syn-ent-HPP derivatives represent 70% (taking into account nor-, seco-, dihydro-, and rearranged halimanes of these series too) of all known halimanes.
The most interesting reported halimane with the most potential is tuberculosinol (182). The production of isotuberculosinol, tuberculosinol and analogues by M. tuberculosis inhibits the phagocytosis of human macrophage-like cells, thus they can be considered virulence factors (VFs). Recently, two tuberculosinyl adenosines have been isolated. These two natural halimanes, derived from tuberculosinol and isotuberculosinol, are being evaluated as possible biomarkers for early diagnosis of tuberculosis.
Some nor- and rearranged halimanes can be considered as 19-norclerodanes or rearranged clerodanes, respectively, and vice versa. These structures can be classified indistinctly as clerodanes or halimanes without further information on their biosynthetic origin. For this reason, halimanes with a 19-norclerodane structure have been also reviewed and included in the ESI.†
Different halimanes, such as ent-halimic acid (39) and 11R-acetoxy-ent-halima-5,13E-dien-15-oic acid (175), have been used as starting materials for the syntheses of bioactive compounds, for example antibacterial or antitumoral compounds.
7. Conflicts of interest
There are no conflicts to declare.8. Acknowledgements
The authors would like to thank the Spanish Ministry of Economy and Competitiveness, MINECO (SAF2014-59716-R, CTQ2015-68175-R), European Funds for Regional Development, FEDER and Junta de Castilla y León (BIO/SA59/15, UIC21) and Universidad de Salamanca for financial support. AMR and IET are grateful for their fellowships from the FSE (European Social Fund), MINECO and Junta de Castilla y León, respectively. The authors thank Profs. J. G. Urones and P. Basabe for their dedication as teachers, and their guidance and friendship over many years.9. Notes and references
- D. E. Cane and H. Ikeda, Acc. Chem. Res., 2012, 45, 463–472 CrossRef CAS PubMed.
- M. Jia and R. J. Peters, Org. Biomol. Chem., 2017, 15, 3158–3160 CAS.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Nat. Prod. Rep., 2014, 31, 160–258 RSC.
- B. M. Fraga, Nat. Prod. Rep., 2013, 30, 1226–1264 RSC.
- J. R. Hanson, Nat. Prod. Rep., 2016, 33, 1227–1238 RSC.
- J. R. Hanson, Nat. Prod. Rep., 2017, 34, 1233–1243 RSC.
- L. Wang, B. Yang, X.-P. Lin, X.-F. Zhou and Y. Liu, Nat. Prod. Rep., 2013, 30, 455–473 RSC.
- R. A. Hill and J. D. Connolly, Nat. Prod. Rep., 2015, 32, 273–327 RSC.
- J. Buckingham, Dictionary of Natural Products on DVD, CRC, Boca Raton, FL, 2007 Search PubMed.
- D. W. Christianson, Chem. Rev., 2017, 117, 11570–11648 CrossRef CAS PubMed.
- S. Prisic, J. Xu, R. M. Coates and R. J. Peters, ChemBioChem, 2007, 8, 869–874 CrossRef CAS PubMed.
- K. U. Wendt and G. E. Schulz, Structure, 1998, 6, 127–133 CrossRef CAS PubMed.
- J. MacMillan and M. H. Beale, Comprehensive Natural Products Chemistry: Isoprenoids Including Carotenoids and Steroids, Elsevier, 1999, vol. 2 Search PubMed.
- A. T. Merritt and S. V. Ley, Nat. Prod. Rep., 1992, 9, 243–287 RSC.
- R. Li, S. L. Morris-Natschke and K.-H. Lee, Nat. Prod. Rep., 2016, 33, 1166–1226 RSC.
- R. J. Peters, Nat. Prod. Rep., 2010, 27, 1521–1530 RSC.
- J. G. Urones, J. De Pascual Teresa, I. S. Marcos, D. Díez Martín, N. Martín Garrido and R. A. Guerra, Phytochemistry, 1987, 26, 1077–1079 CrossRef CAS.
- Y. Asakawa, X. Lin, M. Tori and K. Kondo, Phytochemistry, 1990, 29, 2597–2603 CrossRef CAS.
- J. Jakupovic, S. Banerjee, F. Bohlmann, R. M. King and H. Robinson, Tetrahedron, 1986, 42, 1305–1313 CrossRef CAS.
- F. Tsichritzis and J. Jakupovic, Phytochemistry, 1990, 29, 3173–3187 CrossRef CAS.
- C. Zdero, F. Bohlmann, R. M. King and H. Robinson, Phytochemistry, 1986, 25, 2841–2855 CrossRef CAS.
- K. Siems, J. Jakupovic, V. Castro and L. Poveda, Phytochemistry, 1996, 41, 851–853 CrossRef CAS.
- C. Zdero, F. Bohlmann and H. M. Niemeyer, Phytochemistry, 1991, 30, 3669–3677 CrossRef CAS.
- J. Jakupovic, E. Ellmauerer, F. Bohlmann, A. Whittemori and D. Gage, Phytochemistry, 1986, 25, 2677–2678 CrossRef CAS.
- J. Jakupovic, R. N. Baruah, C. Zdero, F. Eid, V. P. Pathak, T. V. Chau-thi, F. Bohlmann, R. M. King and H. Robinson, Phytochemistry, 1986, 25, 1873–1881 CrossRef CAS.
- J. De Pascual Teresa, J. G. Urones, P. Basabe, H. Carrillo, M. A. G. Muñoz and I. S. Marcos, Phytochemistry, 1985, 24, 791–794 CrossRef CAS.
- J. De Pascual Teresa, J. G. Urones, H. Carrillo and M. A. G. Muñoz, An. Quim., 1979, 75, 140–143 CAS.
- J. De Pascual Teresa, J. G. Urones, I. S. Marcos, D. D. Martín and V. M. Alvarez, Phytochemistry, 1986, 25, 711–713 CrossRef CAS.
- J. M. L. Rodilla, D. I. M. De Mendonça, J. G. Urones and R. F. Moro, Phytochemistry, 1998, 49, 817–822 CrossRef CAS.
- J. M. L. Rodilla, D. I. D. Mendonça, M. I. G. Ismael, J. A. Figueiredo, M. L. A. Silva and E. Lopes, Nat. Prod. Lett., 2001, 15, 401–409 CrossRef CAS PubMed.
- J. G. Urones, I. S. Marcos, P. Basabe, C. A. Alonso, D. Diez, N. M. Garrido, I. M. Oliva, J. S. Rodilla, A. M. Z. Slawin and D. J. Williams, Tetrahedron Lett., 1990, 31, 4501–4504 CrossRef CAS.
- J. G. Urones, P. Basabe, I. S. Marcos, C. Alonso, I. M. Oliva, N. M. Garrido, D. D. Martín and A. M. Lithgow, Tetrahedron Lett., 1992, 33, 5269–5272 CrossRef CAS.
- J. G. Urones, I. S. Marcos, P. Basabe, C. Alonso, I. M. Oliva, N. M. Garrido, D. D. Martín and A. M. Lithgow, Tetrahedron, 1993, 49, 4051–4062 CrossRef CAS.
- J. G. Urones, I. S. Marcos, P. Basabe, C. Alonso, I. M. Oliva, N. M. Garrido, D. D. Martín and A. M. Lithgow, Phytochemistry, 1993, 34, 747–750 CrossRef CAS.
- I. S. Marcos, R. F. Moro, A. Gil-Mesón and D. Díez, in Studies in Natural Products Chemistry, ed. R. Atta ur, Elsevier, 2016, vol. 48, pp. 137–207 Search PubMed.
- J. G. Urones, I. S. Marcos, P. Basabe, C. A. Alonso, D. Diez-Martin, N. M. Garrido and I. M. Oliva, Tetrahedron Lett., 1990, 31, 5665–5668 CrossRef CAS.
- J. G. Urones, I. S. Marcos, N. M. Garrido, J. de Pascual Teresa and A. S. Feliciano Martín, Phytochemistry, 1989, 28, 183–187 CrossRef CAS.
- J. G. Urones, I. S. Marcos and N. M. Garrido, Phytochemistry, 1990, 29, 3243–3246 CrossRef CAS.
- J. G. Urones, I. S. Marcos and N. M. Garrido, Phytochemistry, 1990, 29, 2585–2589 CrossRef CAS.
- J. G. Urones, I. S. Marcos, N. M. Garrido and R. F. Moro, Phytochemistry, 1990, 29, 3042–3044 CrossRef CAS.
- J. G. Urones, I. S. Marcos, P. Basabe, N. M. Garrido, A. Jorge, R. F. Moro and A. M. Lithgow, Tetrahedron, 1993, 49, 6079–6088 CrossRef CAS.
- J. G. Urones, A. Jorge, I. S. Marcos, P. Basabe, D. Díez, N. M. Garrido, A. M. Lithgow, M. O. C. F. da Fonseca and J. M. L. Rodilla, Tetrahedron Lett., 1996, 37, 1659–1662 CrossRef CAS.
- I. S. Marcos, A. Jorge, D. Diez, P. Basabe, A. M. Lithgow, M. J. Sexmero, N. M. Garrido and J. G. Urones, Phytochemistry, 1996, 41, 1155–1157 CrossRef CAS.
- J.-J. Wang, H. Y. Chung, Y.-B. Zhang, G.-Q. Li, Y.-L. Li, W.-H. Huang and G.-C. Wang, Phytochemistry, 2016, 122, 270–275 CrossRef CAS PubMed.
- M. Sánchez, M. Mazzuca, M. J. Veloso, L. R. Fernández, G. Siless, L. Puricelli and J. A. Palermo, Phytochemistry, 2010, 71, 1395–1399 CrossRef PubMed.
- H.-M. Chung, L.-C. Hu, W.-H. Yen, J.-H. Su, M.-C. Lu, T.-L. Hwang, W.-H. Wang and P.-J. Sung, Mar. Drugs, 2012, 10, 2246 CrossRef CAS PubMed.
- H. Lei, Chem. Biodiversity, 2016, 13, 345–365 CAS.
- M. Abdel-Kader, J. M. Berger, C. Slebodnick, J. Hoch, S. Malone, J. H. Wisse, M. C. M. Werkhoven, S. Mamber and D. G. I. Kingston, J. Nat. Prod., 2002, 65, 11–15 CrossRef CAS.
- H.-H. Cheng, Y.-B. Cheng, T.-L. Hwang, Y.-H. Kuo, C.-H. Chen and Y.-C. Shen, J. Nat. Prod., 2015, 78, 1823–1828 CrossRef CAS PubMed.
- Y. Zhang, N. Adnani, D. R. Braun, G. A. Ellis, K. J. Barns, S. Parker-Nance, I. A. Guzei and T. S. Bugni, J. Nat. Prod., 2016, 79, 2968–2972 CrossRef CAS PubMed.
- K. Du, M. De Mieri, M. Neuburger, P. C. Zietsman, A. Marston, S. F. van Vuuren, D. Ferreira, M. Hamburger and J. H. van der Westhuizen, J. Nat. Prod., 2015, 78, 2494–2504 CrossRef CAS PubMed.
- C. G. Silva, H. M. Santos Júnior, J. P. Barbosa, G. L. Costa, F. A. R. Rodrigues, D. F. Oliveira, L. V. Costa-Lotufo, R. J. V. Alves, E. C. A. Eleutherio and C. M. Rezende, Chem. Biodiversity, 2015, 12, 1891–1901 CAS.
- T. Kubota, T. Iwai, A. Takahashi-Nakaguchi, J. Fromont, T. Gonoi and J. i. Kobayashi, Tetrahedron, 2012, 68, 9738–9744 CrossRef CAS.
- S. del Corral, S. L. Cuffini, S. G. Cardoso, A. J. Bortoluzzid and S. M. Palacios, Phytochem. Lett., 2012, 5, 280–283 CrossRef CAS.
- C. Nakano, T. Okamura, T. Sato, T. Dairi and T. Hoshino, Chem. Commun., 2005, 1016–1018 RSC.
- N. Maugel, F. M. Mann, M. L. Hillwig, R. J. Peters and B. B. Snider, Org. Lett., 2010, 12, 2626–2629 CrossRef CAS PubMed.
- C. Nakano, T. Ootsuka, K. Takayama, T. Mitsui, T. Sato and T. Hoshino, Biosci., Biotechnol., Biochem., 2011, 75, 75–81 CrossRef CAS PubMed.
- J. E. Spangler, C. A. Carson and E. J. Sorensen, Chem. Sci., 2010, 1, 202–205 RSC.
- T. Hoshino, C. Nakano, T. Ootsuka, Y. Shinohara and T. Hara, Org. Biomol. Chem., 2011, 9, 2156–2165 CAS.
- H.-C. Chan, X. Feng, T.-P. Ko, C.-H. Huang, Y. Hu, Y. Zheng, S. Bogue, C. Nakano, T. Hoshino, L. Zhang, P. Lv, W. Liu, D. C. Crick, P.-H. Liang, A. H. J. Wang, E. Oldfield and R.-T. Guo, J. Am. Chem. Soc., 2014, 136, 2892–2896 CrossRef CAS PubMed.
- C.-I. Liu, G. Y. Liu, Y. Song, F. Yin, M. E. Hensler, W.-Y. Jeng, V. Nizet, A. H.-J. Wang and E. Oldfield, Science, 2008, 319, 1391–1394 CrossRef CAS PubMed.
- D. C. Young, E. Layre, S.-J. Pan, A. Tapley, J. Adamson, C. Seshadri, Z. Wu, J. Buter, A. J. Minnaard, M. Coscolla, S. Gagneux, R. Copin, J. D. Ernst, W. R. Bishai, B. B. Snider and D. B. Moody, Chem. Biol., 2015, 22, 516–526 CrossRef CAS PubMed.
- J. Buter, D. Heijnen, I. C. Wan, F. M. Bickelhaupt, D. C. Young, E. Otten, D. B. Moody and A. J. Minnaard, J. Org. Chem., 2016, 81, 6686–6696 CrossRef CAS PubMed.
- A. Koul, E. Arnoult, N. Lounis, J. Guillemont and K. Andries, Nature, 2011, 469, 483–490 CrossRef CAS PubMed.
- T. Sato, A. Kigawa, R. Takagi, T. Adachi and T. Hoshino, Org. Biomol. Chem., 2008, 6, 3788–3794 CAS.
- C. Nakano, M. Oshima, N. Kurashima and T. Hoshino, ChemBioChem, 2015, 16, 772–781 CrossRef CAS PubMed.
- F. M. Mann, S. Prisic, H. Hu, M. Xu, R. M. Coates and R. J. Peters, J. Biol. Chem., 2009, 284, 23574–23579 CrossRef CAS PubMed.
- F. M. Mann, B. C. VanderVen and R. J. Peters, Mol. Microbiol., 2011, 79, 1594–1601 CrossRef CAS PubMed.
- L. Prach, J. Kirby, J. D. Keasling and T. Alber, FEBS J., 2010, 277, 3588–3595 CrossRef CAS PubMed.
- F. M. Mann, M. Xu, X. Chen, D. B. Fulton, D. G. Russell and R. J. Peters, J. Am. Chem. Soc., 2009, 131, 17526–17527 CrossRef CAS PubMed.
- E. Layre, H. J. Lee, D. C. Young, A. Jezek Martinot, J. Buter, A. J. Minnaard, J. W. Annand, S. M. Fortune, B. B. Snider, I. Matsunaga, E. J. Rubin, T. Alber and D. B. Moody, Proc. Natl. Acad. Sci., 2014, 111, 2978–2983 CrossRef CAS PubMed.
- A. Rudi, M. Aknin, E. Gaydou and Y. Kashman, J. Nat. Prod., 2004, 67, 1932–1935 CrossRef CAS PubMed.
- K. K. Wan and R. A. Shenvi, Synlett, 2016, 27, 1145–1164 CrossRef CAS.
- H. Zhang, M. Dong, J. Chen, H. Wang, K. Tenney and P. Crews, Mar. Drugs, 2017, 15, 351 CrossRef PubMed.
- T. Hattori, K. Adachi and Y. Shizuri, J. Nat. Prod., 1997, 60, 411–413 CrossRef CAS.
- E. Scio, A. Ribeiro, T. M. A. Alves, A. J. Romanha, J. D. de Souza Filho, G. A. Cordell and C. L. Zani, Phytochemistry, 2003, 64, 1125–1131 CrossRef CAS PubMed.
- P. Luo, Q. Yu, S.-N. Liu, W.-J. Xia, Y.-Y. Fang, L.-K. An, Q. Gu and J. Xu, Fitoterapia, 2017, 120, 108–116 CrossRef CAS PubMed.
- G.-C. Wang, J.-G. Li, G.-Q. Li, J.-J. Xu, X. Wu, W.-C. Ye and Y.-L. Li, J. Nat. Prod., 2012, 75, 2188–2192 CrossRef CAS PubMed.
- J. Appenzeller, G. Mihci, M.-T. Martin, J.-F. Gallard, J.-L. Menou, N. Boury-Esnault, J. Hooper, S. Petek, S. Chevalley, A. Valentin, A. Zaparucha, A. Al-Mourabit and C. Debitus, J. Nat. Prod., 2008, 71, 1451–1454 CrossRef CAS PubMed.
- C. A. Hiruma-Lima, J. S. Gracioso, W. Toma, A. B. Almeida, A. C. Paula, D. S. B. Brasil, A. H. Muller, A. R. M. Souza Brito and A. R. M. Souza Brito, Phytomedicine, 2001, 8, 94–100 CrossRef CAS PubMed.
- W.-q. Wang, Y.-p. Yin, L. Jun and L.-j. Xuan, Phytochemistry, 2018, 146, 56–62 CrossRef CAS PubMed.
- C. Kihampa, M. H. H. Nkunya, C. C. Joseph, S. M. Magesa, A. Hassanali, M. Heydenreich and E. Kleinpeter, Phytochemistry, 2009, 70, 1233–1238 CrossRef CAS PubMed.
- M. Gavagnin, E. Trivellone, F. Castelluccio, G. Cimino and R. Cattaneo-Vietti, Tetrahedron Lett., 1995, 36, 7319–7322 CrossRef CAS.
- K. U. Wendt, K. Poralla and G. E. Schulz, Science, 1997, 277, 1811–1815 CrossRef CAS PubMed.
- K. C. Potter, M. Jia, Y. J. Hong, D. Tantillo and R. J. Peters, Org. Lett., 2016, 18, 1060–1063 CrossRef CAS PubMed.
- K. Potter, J. Criswell, J. Zi, A. Stubbs and R. J. Peters, Angew. Chem., Int. Ed., 2014, 53, 7198–7202 CrossRef CAS PubMed.
- R. J. Peters, Phytochemistry, 2006, 67, 2307–2317 CrossRef CAS PubMed.
- P. R. Wilderman and R. J. Peters, J. Am. Chem. Soc., 2007, 129, 15736–15737 CrossRef CAS PubMed.
- Q. Wang, M. L. Hillwig, Y. Wu and R. J. Peters, Plant Physiol., 2012, 158, 1418 CrossRef CAS PubMed.
- Y. Matsuda and I. Abe, Nat. Prod. Rep., 2016, 33, 26–53 RSC.
- M. Zhou, H.-C. Geng, H.-B. Zhang, K. Dong, W.-G. Wang, X. Du, X.-N. Li, F. He, H.-B. Qin, Y. Li, J.-X. Pu and H.-D. Sun, Org. Lett., 2013, 15, 314–317 CrossRef CAS PubMed.
- M. Xu, M. Jia, Y. J. Hong, X. Yin, D. J. Tantillo, P. J. Proteau and R. J. Peters, Org. Lett., 2018, 20, 1200–1202 CrossRef CAS PubMed.
- C. Nakano and T. Hoshino, ChemBioChem, 2009, 10, 2060–2071 CrossRef CAS PubMed.
- R. Peters, F. Mann, M. Xu and E. Davenport, Front Microbiol., 2012, 3, 368 Search PubMed.
- C. A. Citron, P. Rabe, L. Barra, C. Nakano, T. Hoshino and J. S. Dickschat, Eur. J. Org. Chem., 2014, 7684–7691 CrossRef CAS.
- S. Mafu, E. Fischer, J. B. Addison, I. Riberio Barbosana and P. Zerbe, ChemBioChem, 2016, 17, 2304–2307 CrossRef CAS PubMed.
- J. H. George, M. McArdle, J. E. Baldwin and R. M. Adlington, Tetrahedron, 2010, 66, 6321–6330 CrossRef CAS.
- K. C. Potter, J. Zi, Y. J. Hong, S. Schulte, B. Malchow, D. J. Tantillo and R. J. Peters, Angew. Chem., Int. Ed., 2016, 55, 634–638 CrossRef CAS PubMed.
- J. G. Urones, I. S. Marcos, P. B. Barcala and N. M. Garrido, Phytochemistry, 1988, 27, 501–504 CrossRef CAS.
- J. G. Urones, I. S. Marcos, D. D. Martin, F. M. S. B. Palma and J. M. Rodilla, Phytochemistry, 1987, 26, 3037–3040 CrossRef CAS.
- J. De Pascual Teresa, J. G. Urones, I. S. Marcos, P. B. Barcala and N. M. Garrido, Phytochemistry, 1986, 25, 1185–1187 CrossRef CAS.
- J. De Pascual Teresa, J. G. Urones, I. S. Marcos, F. Bermejo and P. Basabe, Phytochemistry, 1983, 22, 2783–2785 CrossRef CAS.
- J. De Pascual Teresa, P. Basabe, I. S. Marcos, F. Bermejo and J. G. Urones, An. Quim., 1981, 77C, 184–188 Search PubMed.
- J. De Pascual Teresa, J. G. Urones, P. Basabe, F. Bermejo and I. S. Marcos, An. Quim., 1981, 77C, 290–295 Search PubMed.
- J.-S. Zhang, Y.-Q. Tang, J.-L. Huang, W. Li, Y.-H. Zou, G.-H. Tang, B. Liu and S. Yin, Phytochemistry, 2017, 144, 151–158 CrossRef CAS PubMed.
- A. Aldhaher, M. Langat, B. Ndunda, D. Chirchir, J. O. Midiwo, A. Njue, S. Schwikkard, M. Carew and D. Mulholland, Phytochemistry, 2017, 144, 1–8 CrossRef CAS PubMed.
- A. Salatino, M. L. F. Salatino and G. Negri, J. Braz. Chem. Soc., 2007, 18, 11–33 CrossRef CAS.
- A. Sato, M. Kurabayashi, H. Nagahori, A. Ogiso and H. Mishima, Tetrahedron Lett., 1970, 11, 1095–1098 CrossRef.
- I. S. Marcos, F. A. Hernández, M. J. Sexmero, D. Díez, P. Basabe, A. B. Pedrero, N. García and J. G. Urones, Tetrahedron, 2003, 59, 685–694 CrossRef CAS.
- J. G. Urones, I. S. Marcos, P. Basabe, M. J. Sexmero, H. Carrillo and M. J. Melchor, Phytochemistry, 1994, 37, 1359–1361 CrossRef CAS.
- A. Sato, M. Kurabayashi, A. Ogiso and H. Mishima, Tetrahedron Lett., 1971, 12, 839–842 CrossRef.
- M. Kanlayavattanakul, N. Ruangrungsi, T. Watanabe, M. Kawahata, B. Therrien, K. Yamaguchi and T. Ishikawa, J. Nat. Prod., 2005, 68, 7–10 CrossRef CAS PubMed.
- Z.-X. Zhang, H.-H. Li, F.-M. Qi, H.-Y. Xiong, L.-L. Dong, G.-X. Fan and D.-Q. Fei, Bull. Korean Chem. Soc., 2014, 35, 1556–1558 CrossRef CAS.
- Q.-Q. Yuan, S. Tang, W.-B. Song, W.-Q. Wang, M. Huang and L.-J. Xuan, J. Nat. Prod., 2017, 80, 254–260 CrossRef CAS PubMed.
- J. De Pascual Teresa, J. G. Urones and H. Carrillo, An. Quim., 1978, 74, 488–493 Search PubMed.
- J. G. Urones, I. S. Marcos, M. J. Sexmero, P. Basabe and A. M. Lithgow, Phytochemistry, 1990, 29, 1247–1251 CrossRef CAS.
- D. I. M. D. de Mendonça, J. M. L. Rodilla, A. M. Lithgow and I. S. Marcos, Phytochemistry, 1997, 44, 1301–1307 CrossRef.
- J. G. Urones, M. J. Sexmero, F. A. Hernandez, P. Basabe, A. B. Pedrero, D. Diez and I. S. Marcos, Nat. Prod. Lett., 2001, 15, 387–391 CrossRef CAS PubMed.
- S. F. Khoo, A. C. Oehlschlager and G. Ourisson, Tetrahedron, 1973, 29, 3379–3388 CrossRef CAS.
- A. F. Monteiro, J. M. Batista, M. A. Machado, R. P. Severino, E. W. Blanch, V. S. Bolzani, P. C. Vieira and V. G. P. Severino, J. Nat. Prod., 2015, 78, 1451–1455 CrossRef CAS PubMed.
- F. Nagashima, M. Suzuki and Y. Asakawa, Fitoterapia, 2001, 72, 83–85 CrossRef CAS PubMed.
- N. Hara, H. Asaki, Y. Fujimoto, Y. K. Gupta, A. K. Singh and M. Sahai, Phytochemistry, 1995, 38, 189–194 CrossRef CAS.
- I. S. Marcos, A. B. Pedrero, M. J. Sexmero, D. Diez, P. Basabe, F. A. Hernández and J. G. Urones, Tetrahedron Lett., 2003, 44, 369–372 CrossRef CAS.
- M. L. Ciavatta, S. García-Matucheski, M. Carbone, G. Villani, M. R. Nicotera, C. Muniain and M. Gavagnin, Chem. Biodiversity, 2017, 14, e1700125 Search PubMed.
- L.-L. Hong, J.-B. Sun, F. Yang, M. Liu, J. Tang, F. Sun, W.-H. Jiao, S.-P. Wang, W. Zhang and H.-W. Lin, RSC Adv., 2017, 7, 23970–23976 RSC.
- H. Nakamura, H. Wu, Y. Ohizumi and Y. Hirata, Tetrahedron Lett., 1984, 25, 2989–2992 CrossRef CAS.
- L. S. Favier, M. Nieto, O. S. Giordano and C. E. Tonn, Phytochemistry, 1997, 45, 1469–1474 CrossRef CAS.
- I. S. Marcos, N. García, M. J. Sexmero, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2005, 61, 11672–11678 CrossRef CAS.
- E. P. Stout, L. C. Yu and T. F. Molinski, Eur. J. Org. Chem., 2012, 5131–5135 CrossRef CAS PubMed.
- L. Harinantenaina, S. Kida and Y. Asakawa, ARKIVOC, 2007, 8, 22–29 Search PubMed.
- F. Bohlmann and J. Jakupovic, Phytochemistry, 1979, 18, 631–635 CrossRef CAS.
- M. Ono, Y. Ito and T. Nohara, Chem. Pharm. Bull., 2001, 49, 1220–1222 CrossRef CAS PubMed.
- J. A. Maschek, E. Mevers, T. Diyabalanage, L. Chen, Y. Ren, J. B. McClintock, C. D. Amsler, J. Wu and B. J. Baker, Tetrahedron, 2012, 68, 9095–9104 CrossRef CAS.
- S. Soldatou and B. J. Baker, Nat. Prod. Rep., 2017, 34, 585–626 RSC.
- M.-J. Chu, X.-L. Tang, G.-F. Qin, Y.-T. Sun, L. Li, N. J. de Voogd, P.-L. Li and G.-Q. Li, Chem. Biodiversity, 2017, 14, e1600446 Search PubMed.
- T. Kawashima, T. Nakatsu, Y. Fukazawa and S. Itô, Heterocycles, 1976, 5, 227–232 CrossRef CAS.
- E. K. Adesogan, J. Chem. Soc., Perkin Trans. 1, 1981, 1151–1153 RSC.
- M. C. Kapingu, D. Guillaume, Z. H. Mbwambo, M. J. Moshi, F. C. Uliso and R. L. A. Mahunnah, Phytochemistry, 2000, 54, 767–770 CrossRef CAS PubMed.
- G. Appendino, F. Borrelli, R. Capasso, C. Campagnuolo, E. Fattorusso, F. Petrucci and O. Taglialatela-Scafati, J. Agric. Food Chem., 2003, 51, 6970–6974 CrossRef CAS PubMed.
- S. Roengsumran, S. Pornpakakul, N. Muangsin, P. Sangvanich, T. Nhujak, P. Singtothong, N. Chaichit, S. Puthong and A. Petsom, Planta Med., 2004, 70, 87–89 CrossRef CAS PubMed.
- I. S. Marcos, A. Conde, R. F. Moro, P. Basabe, D. Díez, F. Mollinedo and J. G. Urones, Molecules, 2008, 13, 1120 CrossRef CAS PubMed.
- Y. Hu, L. Zhang, X.-Q. Wen, X.-J. Zeng, W. Rui and Y.-Z. Cen, J. Asian Nat. Prod. Res., 2012, 14, 785–788 CrossRef CAS PubMed.
- W.-H. Huang, G.-Q. Li and J.-G. Li, Heterocycles, 2014, 89, 1585–1593 CrossRef CAS.
- M. Qiu, D. Cao, Y. Gao, S. Li, J. Zhu, B. Yang, L. Zhou, Y. Zhou, J. Jin and Z. Zhao, Fitoterapia, 2016, 108, 81–86 CrossRef CAS PubMed.
- B. Ndunda, M. K. Langat, J. O. Midiwo and L. K. Omosa, Nat. Prod. Commun., 2015, 10, 557–558 Search PubMed.
- J.-T. Song, Y. Han, X.-L. Wang, T. Shen, H.-X. Lou and X.-N. Wang, Fitoterapia, 2015, 107, 54–59 CrossRef CAS PubMed.
- B.-D. Lin, B. Zhou, L. Dong, Y. Wu and J.-M. Yue, Nat. Prod. Bioprospect., 2016, 6, 57–61 CrossRef CAS PubMed.
- A. Aldhaher, M. Langat, B. Ndunda, D. Chirchir, J. O. Midiwo, A. Njue, S. Schwikkard, M. Carew and D. Mulholland, Phytochemistry, 2017, 144, 1–8 CrossRef CAS PubMed.
- Y. Liang, Y. Zhang, G. Wang, Y. Li and W. Huang, Molecules, 2017, 22, 126 CrossRef PubMed.
- J.-L. Tian, G.-D. Yao, Y.-X. Wang, P.-Y. Gao, D. Wang, L.-Z. Li, B. Lin, X.-X. Huang and S.-J. Song, Bioorg. Med. Chem. Lett., 2017, 27, 1237–1242 CrossRef CAS PubMed.
- M. Qiu, J. Jin, L. Zhou, W. Zhou, Y. Liu, Q. Tan, D. Cao and Z. Zhao, Phytochemistry, 2018, 145, 103–110 CrossRef CAS PubMed.
- F. Nagashima, H. Tanaka, Y. Kan, S. Huneck and Y. Asakawa, Phytochemistry, 1995, 40, 209–212 CrossRef CAS.
- F. Nagashima, M. Suzuki, S. Takaoka and Y. Asakawa, J. Nat. Prod., 2001, 64, 1309–1317 CrossRef CAS.
- S.-M. Yang, D.-G. Wu and X.-K. Liu, Z. Naturforsch. C Bio. Sci., 2010, 65, 39–42 CAS.
- M. Liu, W.-G. Wang, H.-D. Sun and J.-X. Pu, Nat. Prod. Rep., 2017, 34, 1090–1140 RSC.
- J. Gu, S.-Y. Qian, G.-G. Cheng, Y. Li, Y.-P. Liu and X.-D. Luo, Nat. Prod. Bioprospect., 2013, 3, 66–69 CrossRef CAS.
- M. J. Pcolinski, D. P. O'Mathúna and R. W. Doskotch, J. Nat. Prod., 1995, 58, 209–216 CrossRef CAS.
- T. Abe, M. Kobayashi, Y. Okawa, T. Inui, J. Yoshida, H. Higashio, H. Shinden, S. Uesugi, H. Koshino and K.-i. Kimura, Fitoterapia, 2016, 113, 188–194 CrossRef CAS PubMed.
- M. Ono, T. Yamasaki, M. Konoshita, T. Ikeda, M. Okawa, J. Kinjo, H. Yoshimitsu and T. Nohara, Chem. Pharm. Bull., 2008, 56, 1621–1624 CrossRef CAS PubMed.
- M. Ono, K. Eguchi, M. Konoshita, C. Furusawa, J. Sakamoto, S. Yasuda, T. Ikeda, M. Okawa, J. Kinjo, H. Yoshimitsu and T. Nohara, Chem. Pharm. Bull., 2011, 59, 392–396 CrossRef CAS PubMed.
- N. Tiwari, J. Thakur, D. Saikia and M. M. Gupta, Phytomedicine, 2013, 20, 605–610 CrossRef CAS PubMed.
- J. Wu, T. Zhou, S.-w. Zhang, X.-h. Zhang and L.-j. Xuan, Planta Med., 2009, 75, 367–370 CrossRef CAS PubMed.
- M. Ono, T. Yanaka, M. Yamamoto, Y. Ito and T. Nohara, J. Nat. Prod., 2002, 65, 537–541 CrossRef CAS.
- C. Lee, J. W. Lee, Q. Jin, H. J. Lee, S.-J. Lee, D. Lee, M. K. Lee, C. K. Lee, J. T. Hong, M. K. Lee and B. Y. Hwang, Bioorg. Med. Chem. Lett., 2013, 23, 6010–6014 CrossRef CAS PubMed.
- H. Wu, S. Wang, Z. Xu, S. Sun, H. Liu, J. Wang, Y. E, Y. Lv, X. Dong, G. Li, L. Zhang and Y. Shi, Molecules, 2015, 20, 839 CrossRef PubMed.
- J.-S. Zhang, Y.-H. Zou, J.-J. Zhao, Y. Chen, J.-M. Bao and G.-H. Tang, Phytochem. Lett., 2016, 16, 241–244 CrossRef CAS.
- T. L. Meragelman, D. S. Pedrosa and R. R. Gil, Biochem. Syst. Ecol., 2004, 32, 45–53 CrossRef CAS.
- J. De Pascual Teresa, J. G. Urones, I. S. Marcos, F. Bermejo, P. Basabe and P. Queimadelos, An. Quim. C, 1983, 79, 451–453 CAS.
- S. K. Sadhu, E. Okuyama, H. Fujimoto and M. Ishibashi, J. Nat. Prod., 2006, 69, 988–994 CrossRef CAS PubMed.
- P. Rijo, C. Gaspar-Marques, M. F. Simões, M. L. Jimeno and B. Rodríguez, Biochem. Syst. Ecol., 2007, 35, 215–221 CrossRef CAS.
- P. Rijo, B. Rodriguez, A. Duarte and M. F. Simoes, Nat. Prod. J., 2011, 1, 57–64 CAS.
- O. Burmistrova, M. F. Simões, P. Rijo, J. Quintana, J. Bermejo and F. Estévez, J. Nat. Prod., 2013, 76, 1413–1423 CrossRef CAS PubMed.
- C. Nakano, T. Hoshino, T. Sato, T. Toyomasu, T. Dairi and T. Sassa, Tetrahedron Lett., 2010, 51, 125–128 CrossRef CAS.
- M. Jia, K. C. Potter and R. J. Peters, Metab. Eng., 2016, 37, 24–34 CrossRef CAS PubMed.
- F. Bohlmann, W.-R. Abraham, R. M. King and H. Robinson, Phytochemistry, 1981, 20, 1903–1906 CrossRef CAS.
- M. A. de Alvarenga, H. E. Gottlieb, O. R. Gottlieb, M. T. Magalhães and V. O. da Silva, Phytochemistry, 1978, 17, 1773–1776 CrossRef CAS.
- M. S. Buchanan, J. D. Connolly, A. A. Kadir and D. S. Rycroft, Phytochemistry, 1996, 42, 1641–1646 CrossRef CAS.
- R. P. Walker, R. M. Rosser, D. J. Faulkner, L. S. Bass, C. H. He and J. Clardy, J. Org. Chem., 1984, 49, 5160–5163 CrossRef CAS.
- M. Toyota, I. Nakamura, S. Takaoka, Y. Kan and Y. Asakawa, Phytochemistry, 1996, 41, 575–580 CrossRef CAS.
- C.-Y. Chen, F.-R. Chang, Y.-C. Shih, T.-J. Hsieh, Y.-C. Chia, H.-Y. Tseng, H.-C. Chen, S.-J. Chen, M.-C. Hsu and Y.-C. Wu, J. Nat. Prod., 2000, 63, 1475–1478 CrossRef CAS.
- R. P. Walker and D. J. Faulkner, J. Org. Chem., 1981, 46, 1098–1102 CrossRef CAS.
- C. Harde and F. Bohlmann, Tetrahedron, 1988, 44, 81–90 CrossRef CAS.
- R. Fathi-Afshar and T. M. Allen, Can. J. Chem., 1988, 66, 45–50 CrossRef CAS.
- S. S. Nyandoro, J. Pharmacogn. Phytochem., 2014, 3, 147–157 Search PubMed.
- M. Bruno, F. Piozzi, G. Savona, M. C. De La Torre and B. Rodríguez, Phytochemistry, 1989, 28, 3539–3541 CrossRef CAS.
- A. Fiorentino, B. D'Abrosca, S. Pacifico, M. Scognamiglio, G. D'Angelo, M. Gallicchio, A. Chambery and P. Monaco, Phytochemistry, 2011, 72, 2037–2044 CrossRef CAS PubMed.
- E. Fujita, I. Uchida, T. Fujita, N. Masaki and K. Osaki, J. Chem. Soc., Chem. Commun., 1973, 793–794 RSC.
- E. Fujita, I. Uchida and T. Fujita, J. Chem. Soc., Perkin Trans. 1, 1974, 1547–1555 RSC.
- F. Fernández-Gadea, C. Pascual, B. Rodríguez and G. Savona, Phytochemistry, 1983, 22, 723–725 CrossRef.
- Z. H. Mbwambo, K. Foubert, M. Chacha, M. C. Kapingu, J. J. Magadula, M. M. Moshi, F. Lemière, K. Goubitz, J. Fraanje, R. Peschar, A. Vlietinck, S. Apers and L. Pieters, Planta Med., 2009, 75, 262–267 CrossRef CAS PubMed.
- Z. Pan, D. Ning, X. Wu, S. Huang, D. Li and S. Lv, Bioorg. Med. Chem. Lett., 2015, 25, 1329–1332 CrossRef CAS PubMed.
- J. G. Urones, I. S. Marcos, M. J. Sexmero, P. Basabe and A. M. Lithgow, Phytochemistry, 1990, 29, 3597–3600 CrossRef CAS.
- Â. C. Pinto, O. A. C. Antunes, M. G. Pizzolatti and V. M. Rumjanek, Phytochemistry, 1996, 42, 771–774 CrossRef.
- A. C. Pinto, O. A. C. Antunes, C. M. Rezende and C. R. D. Correia, Phytochemistry, 1995, 38, 1269–1271 CrossRef CAS.
- M. Salmoun, J. C. Braekman, J. Dewelle, F. Darro, R. Kiss, N. J. De Voogd and R. W. M. Van Soest, Nat. Prod. Res., 2007, 21, 149–155 CrossRef CAS PubMed.
- K. Shimbo, M. Tsuda, E. Fukushi, J. Kawabata and J. Kobayashi, Tetrahedron, 2000, 56, 7923–7926 CrossRef CAS.
- C.-H. Cheng, H.-M. Chung, T.-L. Hwang, M.-C. Lu, Z.-H. Wen, Y.-H. Kuo, W.-H. Wang and P.-J. Sung, Molecules, 2012, 17, 9443 CrossRef CAS PubMed.
- A. Ortega, P. E. García, J. Cárdenas, C. Mancera, S. Marquina, M. L. d. Carmen Garduño and E. Maldonado, Tetrahedron, 2001, 57, 2981–2989 CrossRef CAS.
- A. Ohsaki, N. Ohno, K. Shibata, T. Tokoroyama, T. Kubota, K. Hirotsu and T. Higuchi, Phytochemistry, 1988, 27, 2171–2173 CrossRef CAS.
- A. Ohsaki, K. Shibata, T. Tokoroyama and T. Kubota, J. Chem. Soc., Chem. Commun., 1987, 151–153 RSC.
- A. Ohsaki, Y. Kasetani, Y. Asaka, K. Shibata, T. Tokoroyama and T. Kubota, Phytochemistry, 1995, 40, 205–207 CrossRef CAS.
- O. Ayumi, S. Kozo, T. Takashi, K. Takashi and N. Hideo, Chem. Lett., 1986, 15, 1585–1588 CrossRef.
- H.-M. Niu, D.-Q. Zeng, C.-L. Long, Y.-H. Peng, Y.-H. Wang, J.-F. Luo, H.-S. Wang, Y.-N. Shi, G.-H. Tang and F.-W. Zhao, J. Asian Nat. Prod. Res., 2010, 12, 7–14 CrossRef CAS PubMed.
- F. Bohlmann, M. Grenz, P. Wegner and J. Jakupovic, Liebigs Ann. Chem., 1983, 2008–2020 CrossRef CAS.
- K. Siems, X. A. Dominguez and J. Jakupovic, Phytochemistry, 1992, 31, 4363–4365 CrossRef CAS.
- D.-X. Guo, X.-N. Wang, R.-X. Zhu, N. Liu, J.-C. Zhou, W.-T. Yu and H.-X. Lou, Phytochem. Lett., 2012, 5, 535–540 CrossRef CAS.
- I. S. Marcos, A. B. Pedrero, M. J. Sexmero, D. Diez, N. García, M. A. Escola, P. Basabe, A. Conde, R. F. Moro and J. G. Urones, Synthesis, 2005, 3301–3310 CrossRef CAS.
- I. S. Marcos, J. L. Gonzalez, M. J. Sexmero, D. Díez, P. Basabe, D. J. Williams, M. S. J. Simmonds and J. G. Urones, Tetrahedron Lett., 2000, 41, 2553–2557 CrossRef CAS.
- I. S. Marcos, F. A. Hernández, M. J. Sexmero, D. Díez, P. Basabe, A. B. Pedrero, N. García, F. Sanz and J. G. Urones, Tetrahedron Lett., 2002, 43, 1243–1245 CrossRef CAS.
- I. S. Marcos, A. B. Pedrero, M. J. Sexmero, D. Diez, P. Basabe, F. A. Hernández, H. B. Broughton and J. G. Urones, Synlett, 2002, 105–109 CrossRef CAS.
- I. S. Marcos, M. A. Escola, R. F. Moro, P. Basabe, D. Diez, F. Sanz, F. Mollinedo, J. de la Iglesia-Vicente, B. G. Sierra and J. G. Urones, Bioorg. Med. Chem., 2007, 15, 5719–5737 CrossRef CAS PubMed.
- I. S. Marcos, A. B. Pedrero, M. J. Sexmero, D. Diez, P. Basabe, N. García, R. F. Moro, H. B. Broughton, F. Mollinedo and J. G. Urones, J. Org. Chem., 2003, 68, 7496–7504 CrossRef CAS PubMed.
- A. Gil-Mesón, A. M. Roncero, I. E. Tobal, P. Basabe, D. Díez, F. Mollinedo and I. S. Marcos, Molecules, 2016, 21, 47 CrossRef PubMed.
- I. S. Marcos, M. J. Sexmero, F. Á. Hernández, M. Corrales, P. Basabe, D. Díez and J. G. Urones, Molecules, 2006, 11, 792–807 CrossRef CAS PubMed.
- I. S. Marcos, N. García, M. J. Sexmero, F. A. Hernández, M. A. Escola, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2007, 63, 2335–2350 CrossRef CAS.
- I. S. Marcos, B. Martínez, M. J. Sexmero, D. Diez, P. Basabe and J. G. Urones, Synthesis, 2006, 3865–3873 CrossRef CAS.
- I. S. Marcos, A. Conde, R. F. Moro, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2010, 66, 8280–8290 CrossRef CAS.
- I. S. Marcos, M. A. Escola, R. F. Moro, P. Basabe, D. Diez, F. Mollinedo and J. G. Urones, Synlett, 2007, 2017–2022 CrossRef CAS.
- I. S. Marcos, R. F. Moro, I. Costales, P. Basabe, D. Díez, F. Mollinedo and J. G. Urones, Tetrahedron, 2012, 68, 7932–7940 CrossRef CAS.
- I. S. Marcos, R. F. Moro, I. Costales, P. Basabe, D. Díez, F. Mollinedo and J. G. Urones, Tetrahedron, 2013, 69, 7285–7289 CrossRef CAS.
- I. S. Marcos, R. F. Moro, I. Costales, M. A. Escola, P. Basabe, D. Díez and J. G. Urones, Tetrahedron, 2009, 65, 10235–10242 CrossRef CAS.
- I. S. Marcos, R. F. Moro, I. Costales, P. Basabe, D. Díez, A. Gil, F. Mollinedo, F. Pérez-de la Rosa, E. Pérez-Roth and J. M. Padrón, Eur. J. Med. Chem., 2014, 73, 265–279 CrossRef CAS PubMed.
- H. J. Bestmann, Angew. Chem., Int. Ed., 1977, 16, 349–364 CrossRef.
- J. Boukouvalas and N. Lachance, Synlett, 1998, 31–32 CrossRef CAS.
- H. Rosemeyer, Chem. Biodiversity, 2004, 1, 361–401 CAS.
- M. Ohba, K. Iizuka, H. Ishibashi and T. Fujii, Tetrahedron, 1997, 53, 16977–16986 CrossRef CAS.
- M. Ohba, N. Kawase, T. Fujii, K. Aoe, K. Okamura, R. Fathi-Afshar and T. M. Allen, Tetrahedron Lett., 1995, 36, 6101–6104 CrossRef CAS.
- M. Ohba, N. Kawase and T. Fujii, J. Am. Chem. Soc., 1996, 118, 8250–8257 CrossRef CAS.
- S. L. Schreiber, J. Am. Chem. Soc., 1980, 102, 6163–6165 CrossRef CAS.
- G. Solladie and J. Hutt, J. Org. Chem., 1987, 52, 3560–3566 CrossRef CAS.
- H. Iio, M. Monden, K. Okada and T. Tokoroyama, J. Chem. Soc., Chem. Commun., 1987, 358–359 RSC.
- J. R. Parikh and W. v. E. Doering, J. Am. Chem. Soc., 1967, 89, 5505–5507 CrossRef CAS.
- I. Ojima and T. Kogure, Tetrahedron Lett., 1972, 13, 5035–5038 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8np00016f |
| This journal is © The Royal Society of Chemistry 2018 |