
Natural products and ring-closing metathesis: synthesis of sterically congested olefins
C.
Lecourt
,
S.
Dhambri
 ,
L.
Allievi
,
Y.
Sanogo
,
N.
Zeghbib
,
R.
Ben Othman
,
M.-I.
Lannou
*,
G.
Sorin
* and
J.
Ardisson
*
,
L.
Allievi
,
Y.
Sanogo
,
N.
Zeghbib
,
R.
Ben Othman
,
M.-I.
Lannou
*,
G.
Sorin
* and
J.
Ardisson
*
Paris Descartes University, Sorbonne Paris Cité, Faculté des Sciences Pharmaceutiques et Biologiques, Unité CNRS UMR 8638 COMÈTE, 4 Avenue de l'observatoire, 75270 Paris Cedex 06, France. E-mail: marie-isabelle.lannou@parisdescartes.fr; geoffroy.sorin@parisdescartes.fr; janick.ardisson@parisdescartes.fr
First published on 18th January 2018
Abstract
Covering: 2002 to August 2017.
This review highlights recent RCM reactions towards the synthesis of sterically congested natural products. It offers an insight into various synthetic targets and approaches and provides information on the evolution of catalysts as powerful tools enabling the use of increasingly challenging diene precursors.
 C. Lecourt | Camille Lecourt obtained her M.Sc in Chemistry directed towards life sciences from University Paris Descartes in 2013, she then joined the group of Pr Ardisson where she worked on the total synthesis of hemicalide. She obtained her PhD in November 2016 and pursued with a post-doctorate partitioned between the laboratory of Pr Ardisson (Univ. Paris Descartes) and the laboratory of Pr. J. Cossy and DR. C. Meyer (ESPCI Paris Tech). |
 S. Dhambri | Sabrina Dhambri obtained her M.Sc in Chemistry directed towards life sciences from University University Pierre & Marie Curie in 2009 and is currently working as an engineer in the laboratory of Pr. Ardisson. Her research interests range from methodology in organometallic chemistry to total synthesis. |
 L. Allievi | Luca Allievi joined the laboratories of Chimie Paris Tech in 2013 as an Erasmus fellow, and obtained his M.Sc in Chemical Science from the University of Milan in 2014. He has recently defended his Ph.D, on the total synthesis of vibsatins A and B (November 2017). |
 Y. Sanogo | Youssouf Sanogo obtained his M.Sc in Molecular Chemistry from the University Pierre & Marie Curie in 2014. He then joined the group of Pr. Ardisson where he focused on the synthesis of thapsigargin. He has recently defended his Ph.D. (November 2017). |
 N. Zeghbib | Narimane Zeghbib obtained her Ph.D. in 2016 under the supervision of Pr. T. Martens (ICMPE, Thiais). Her Ph.D. project was based on the synthesis and structure–activity relationship of chemical inducers of Parkinson's disease. Since 2016, she is pursuing her career in Paris, as a post-doctoral fellow with Pr. Ardisson (Paris Descartes University) in the field of medicinal chemistry. |
 R. Ben Othman | Raja Ben-Othman obtained her Ph.D. in 2006 in the field of the contribution of ring closing metathesis to access highly functionalized azapolycyclic structures, under the supervision of Pr. Bernard Decroix (URCOM, Le Havre). She got a position of Assistant Professor in Superior School of Science and Technology of Hammam Sousse (ESSTHS, Tunisia). After 3 years of post-doctoral position under the supervision of Prs. Sylvain Routier and Franck Suzenet (ICOA, Orléans), she is pursuing her career at Paris Descartes University as a researcher in Pr. Janick Ardisson group. |
 M.-I. Lannou | Marie-Isabelle Lannou worked as a Ph.D. student under the supervision of Pr J.-L. Namy at University Paris Sud, Orsay in the field of lanthanides chemistry (1999–2002). She then joined the team of Pr Enders in RWTH Aachen as a postdoctoral fellow (2003–2004), where she focuses on the synthesis of crippowellins. Her project as Assistant Professor in Dr A. Pancrazi/Pr J. Ardisson team in Cergy Pontoise, allowed her to develop an approach towards the synthesis of sarcodictyines, then she got a position of Assistant Professor in Pr F. Mongin team in Rennes where she worked on zinc-ate complexes. In 2006, she joined the CNRS as a researcher in Dr A. Pancrazi/Pr J. Ardisson team in Cergy Pontoise and then moved to Paris (Pr J. Ardisson, UMR CNRS 8638, Paris Descartes University). Consequently, her research interests are various, from methodology in organometallic chemistry to total synthesis of natural products and green chemistry. |
 G. Sorin | Geoffroy Sorin obtained his PhD in 2008 in the field of the synthesis and structure–activity relationship of huprine derivatives under the supervision of Pr P.-Y. Renard (IRCOF, Rouen). He pursued his career in Paris, as a post-doctoral fellow with Pr L. Fensterbank (University Pierre and Marie Curie) on the oxidation of alkyl trifluoroborates for tin-free radical chemistry, then with Pr J. Ardisson (Paris Descartes University) on the structural elucidation and synthesis of a new polyketide, hemicalide. In 2011, he got a position of Assistant Professor in Pr J. Ardisson team in Paris (UMR CNRS 8638, Paris Descartes University). Recently, he received CRCT funding for researches in Dixon's group (Oxford, GB) (September 2016-August 2017). His research interests mainly focus on the total synthesis of natural products and new synthetic methods. |
 J. Ardisson | Pr Janick Ardisson received her PhD in 1984 from the University of Orsay (Paris 11) where she worked on the total synthesis of Aspidosperma alkaloids, under the supervision of Pr J. Poisson and Dr N. Kunesch. She joined Pr M. Julia team (ENS, Paris) as a postdoctoral fellow to develop a total synthesis of avermectins (1984–1987) and then returned to the faculty of Pharmacy of Chatenay-Malabry (Paris 11) as Assistant Professor. She then joined the University of Cergy-Pontoise as full Professor and is now full Professor at the Paris Descartes University. Her research interests include total synthesis of bioactive natural products. |
1. Introduction: the RCM reaction
Molecular evolution on Earth has produced an extraordinary library of natural products characterized by a large diversity of chemical structures. Rapid access to the functionalized motifs included in these natural compounds as well as their derivatives represents a great challenge in organic synthesis and numerous efficient methods are required to synthesize them.1In particular, compact and sterically congested natural products, that often possess important biological activities due to their unique three-dimensional structures, have frequently proven difficult to synthesize. The problems associated with forging strained connectivities in complex molecules have led to the development of specific and reliable synthetic strategies.
In this context, the high activity and tolerance to numerous functional groups of the ring-closing metathesis reaction (RCM) could explain its success. RCM has often been efficiently applied to the construction of a wide range of strained molecules of various structures.
This review surveys the RCM reaction of hindered olefins, with emphasis on the recent syntheses of natural products employing this reaction. An overview of the different catalysts developed for this application will be presented. The aim of this article is to highlight delineated examples of relevant strategy related to the achievement of sterically demanding RCM reactions toward the synthesis of natural products.
Throughout the last two decades, RCM reaction has become one of the most important tools in organic chemistry and has therefore been substantially reviewed.2,3 Although earlier reports may partially overlap with this review, the recent development of even more efficient catalysts has pushed away the limits of metathesis of sterically hindered substrates and therefore, has allowed for its widespread application to the synthesis of complex natural products. In particular, there is no review exclusively devoted to the synthesis of congested natural products through RCM reaction, therefore, this is the purpose of this survey.
2. Catalysts and conditions towards RCM of hindered olefins
Formation of carbon–carbon double bonds from simple to highly functionalized alkenes represents a great challenge in natural product synthesis. In this context, olefin metathesis reaction has become one of the most powerful tools for their elaboration. The awarding by the Nobel Prize Committee to Chauvin, Schrock and Grubbs for their work on metathesis reactions, attests to the immense impact of this reaction on the academic and industrial chemical community.2.1 General reactivity of alkenes
In 2003, in the course of cross olefin metathesis reaction (CM), Grubbs provided a useful overview of the general reactivity of alkenes towards metathesis, in relation to their electronic character or substitution.4 More particularly, olefins were classified into four categories (types I to IV), considering their ability to undergo homodimerization and, in parallel, their reactivity [from higher to lower reactivity (for sterically bulky, electron-deficient, etc. olefins)] (Table 1). These empirical considerations could therefore provide precious guidelines to perform challenging RCM reaction.| Olefin type and reactivity | Olefins (examples) |
|---|---|
| Type I (fast homodimerization) | Terminal olefins, 1° allylic alcohols, allyl halides, styrenes (no large ortho substituents, …) |
| Type II (slow homodimerization) | Styrenes (large ortho substituents), acrylates, vinyl ketones, 2° allylic alcohols, … |
| Type III (no homodimerization) | 1,1-Disubstituted olefins, non-bulky trisubstituted olefins, acrylonitrile, 3° allylic alcohols, … |
| Type IV (inert to CM = spectators) | Trisubstituted allylic alcohols, 1,1-disubstituted olefins, disubstituted α,β-unsaturated carbonyls, … |
2.2 Catalyst tuning and associated results
A wide range of catalysts has been shown to initiate metathesis. The most active and widely used catalysts are Mo- or Ru-based complexes. The Mo-based complexes are sometimes more active than their Ru counterparts; however, they suffer from high oxygen and moisture sensitivity. The Ru-based catalysts are much more stable and display a high functional-group tolerance. In particular, the development of ruthenium alkylidene complexes bearing N-heterocyclic carbene ligands (NHC) (i.e. classical Grubbs' II and Hoveyda–Grubbs' II catalysts) has largely broadened the scope and the utility of this reaction. Furthermore, many metathesis catalysts have now become commercially available, thus rendering RCM more widely applicable.2The rate at which precatalysts for olefin metathesis reactions are activated has a strong influence on the rate of substrate conversion. In general terms, fast initiating complexes provide good substrate conversion within a short time or at low temperatures. Slowly initiating precatalysts (latent complexes) lead to slow reactions or require high temperatures. In fact, these precatalysts act as a reservoir, by generating small stationary concentrations of active species and appear to be useful for RCM reactions of challenging hindered substrates. While the initiation step is critical in determining how quickly active species are formed, the behaviour and activity of these species, as well as catalyst decomposition also determine the overall efficiency of the reaction. Based on these concepts, it can easily be understood why the use of different precatalysts results in such different substrate conversions in RCM reactions.2c,5
In 2008, a first general guide was reported by Grela et al., revealing the relative efficacies of different catalysts and providing guidelines for a more rational choice of the reaction conditions.6 By comparing the activity of several classical catalysts (GI 1, GII 2, GHI 3, GHII 4, IndI 5, IndII 6, Gre 7, …), it has been possible to deduce empirical rules (Fig. 1). First-generation catalysts were found to be useful in the metathesis of sterically unhindered substrates. Towards sterically hindered alkenes, second generation catalysts could display good to very high activities under optimised conditions. Noteworthy, if a rise of temperature is often favourable, an increase of the catalyst loading is much less valuable.
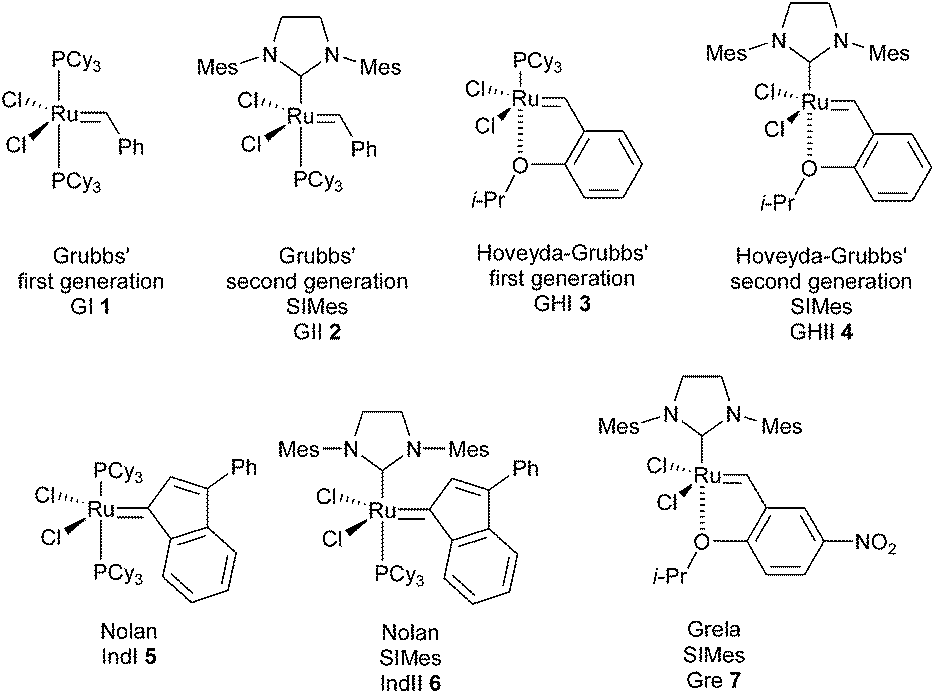 | ||
| Fig. 1 Some of RCM catalysts studied by Grela. | ||
However, despite a significant number of successful applications in the synthesis of compact natural products with GII and GHII catalysts 2 and 4, elaboration of hindered olefins through RCM remained especially challenging and in the last ten years, different catalysts were proposed to improve these reactions. Some examples of catalysts are listed below.
In 2007, Stewart, Grubbs and Schrödi et al. demonstrated that the removal of one ortho substituent of aryl attached to the nitrogen of the NHC ligand as for complexes GIITol 8 and GHIITol 9, was beneficial for the RCM of hindered substrates (Fig. 2).7
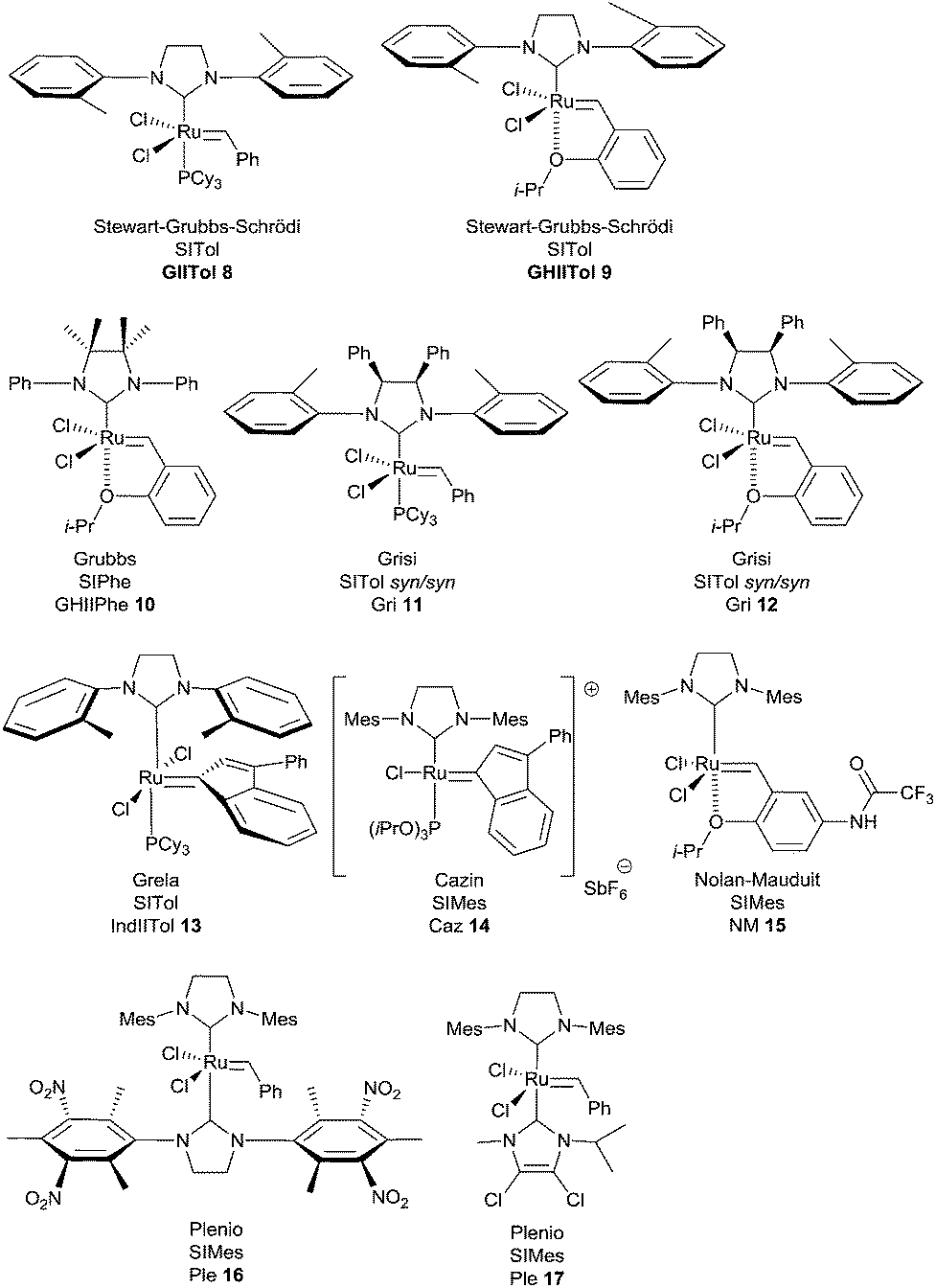 | ||
| Fig. 2 Catalysts for RCM of hindered alkenes. | ||
Based on the same concept, in 2008, Grubbs revealed that N,N′-diphenyl-substituted NHC complex GHIIPhe 10, bearing gem-dimethyl groups on the backbone of the NHC, was resistant to decomposition through C–H activation and proved to be one of the most efficient catalysts in RCM reactions to form tetrasubstituted olefins.8
More recently, Grisi et al. have identified a class of Ru catalysts containing NHC ligands with methyl or phenyl groups on the NHC skeleton.9 Notably, syn/syn complexes with o-tolyl N-substituents (Gri 11 and Gri 12) have been successfully applied in hindered RCM reactions.
In 2007, Nolan et al. described the SIMes-based indenylidene complex IndII 6, more resistant to harsh conditions than its benzylidene counterpart GII 2, owing to its high thermal stability.10 This catalyst therefore led to better results in the ring-closing metathesis of tetrasubstituted dienes.
Taking these results into account, Grela et al. demonstrated that complex IndIITol 13 with an o-tolyl ligand associated the stability of indenylidene complex with the beneficial catalytic effect of sterically less demanding NHC ligands. Specifically, it was highlighted that catalyst 13 was very efficient in several RCM reactions of hindered olefins.11
Interestingly, Cazin et al. have prepared and studied an unprecedented cationic ruthenium indenylidene complex bearing a mixed ligand system NHC/P(OR)3, Caz 14. The high activity of the catalyst and thermal stability of the active species allowed the full conversion of low reactive substrates at high temperatures (140 °C) in xylene, within very short reaction times (only 15 minutes) and using low catalyst loadings (0.1 or 0.2 mol%).12
Complexes of type GHII 4 suffer from somewhat slow activation kinetics. Some activated variants have been developed based on electronic modulation of the coordination environment. In a research program, Grela et al. proved that the nitro-substituted Hoveyda catalyst Gre 7 was dramatically more active than both GII and GHII complexes 2 and 4.13
Following a similar concept, Clavier, Nolan and Mauduit et al. prepared a series of analogues of the Hoveyda complex bearing a trifluoromethylamide group.14 Beneficial activity and stability were observed for the complex NM 15 which was found to be very efficient for sterically hindered olefins.
Finally, Plenio et al. reported a new type of ruthenium complex with mixed NHC ligands: one of the two ligands is electron-rich and the other one is electron-deficient.5b,15,16 Remarkably, the catalytically active species is generated after releasing of the electron-deficient ligand from ruthenium.
Thus, complexes Ple 16 and Ple 17 proved to be highly efficient pre-catalysts for RCM of substituted alkenes [loadings of only 0.25–1 mol% were sufficient when the reaction is carried out in toluene at 80 °C for 3 h (Ple 16) or 20 h (Ple 17)].
When comparing the performances of the catalysts, the ten-times lower catalyst loadings required with Cazin and Plenio catalysts Caz 14, Ple 16 and Ple 17 to achieve successfully RCM of sterically hindered olefins, are noteworthy.
2.3 Reaction conditions and associated results
Several authors demonstrated the usefulness of fluorinated aromatic hydrocarbons (FAH) as solvents and microwave irradiation in promoting difficult RCM reactions.Blechert, Grela, Nolan and Dorta et al. reported that FAH solvents allow substantially higher yields to be obtained, especially in the case of challenging substrates, including natural and biologically active compounds.17 This doping effect might be due to the formation of π–π complexes between N-mesityl groups of the NHC ligand and the FAH solvent molecules. The FAH effect was investigated in the RCM of selected natural or biologically active substrates. Thus, the GII 2 catalysed RCM reaction of diene 18, a derivative of (−)-isopulegol, a monoterpenic alcohol widely employed in the flavour and perfume industry, led to bicyclic compound 19 with an incomplete conversion in 1,2-dichloroethane, while the use of perfluorotoluene allowed the formation of the tetrasubstituted double bond with quantitative conversion (Scheme 1).
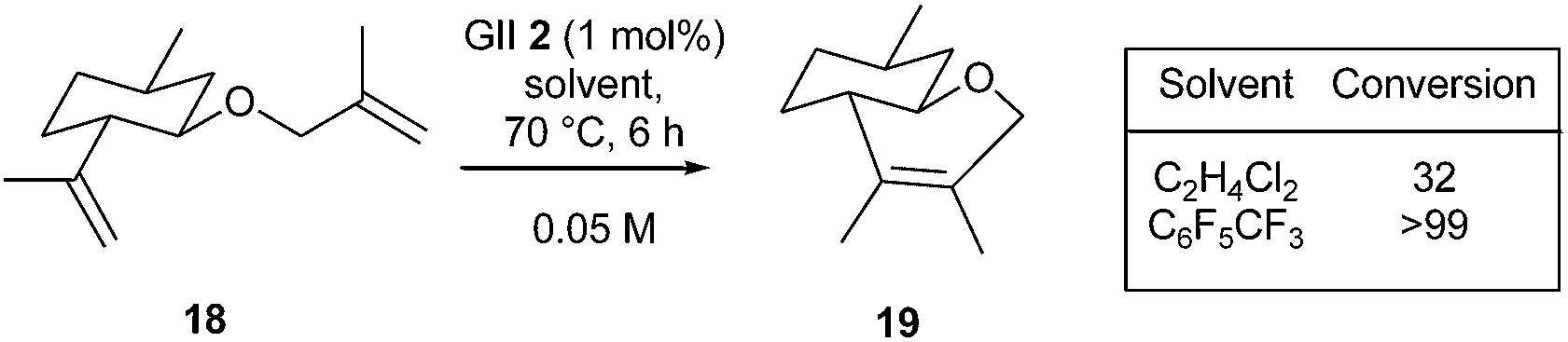 | ||
| Scheme 1 RCM reaction of 18 in FAH. | ||
Additionally, application of microwave irradiation, classical known as a powerful activation method, was also studied.
In this context, Mauduit and Grela et al. have demonstrated that a synergistic effect could be observed in sterically demanding RCM transformations promoted by several commercially available ruthenium complexes and conducted in FAH solvents under microwave irradiation.18 In particular, it was found that, when dienic system 20 was subjected to 2 mol% of commercial IndII catalyst 6 in C6D6 at 100 °C for 24 h, no reaction took place towards the lactone 21 bearing a tetrasubstituted double bond (Scheme 2, entry 1). Interestingly, with the same catalyst loading, using octafluorotoluene, 33% formation of desired lactone 21 was observed at 120 °C (thermal activation) after only 15 min (Scheme 2, entry 2). However, when the same reaction mixture was conducted under microwave irradiation, 57% conversion of 20 into 21 was obtained (Scheme 2, entry 3). Finally, optimized conditions (2 × 2 mol% of 6, microwave irradiation, 2 × 15 min), led to a significantly better conversion of 81% (Scheme 2, entry 4).
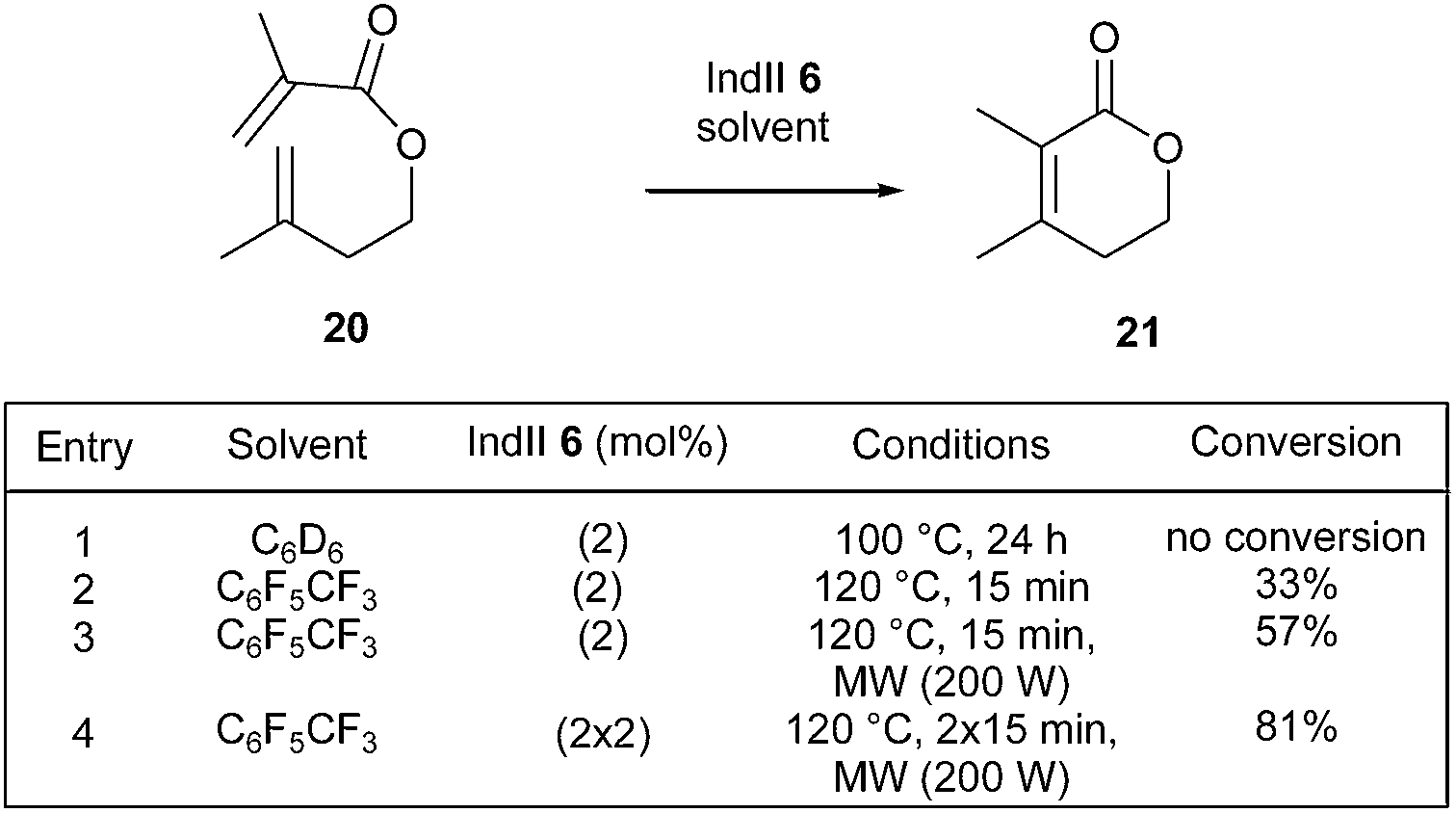 | ||
| Scheme 2 RCM reactions of 20 in FAH and through microwave irradiation. | ||
In summary, in the course of ring-closing metathesis (RCM) reactions involving sterically demanding olefins, recent progresses in ruthenium olefin metathesis catalysts, with the design of more suitable complexes with modified structures of NHC ligands and reactive carbene leaving groups, should now open the way to the synthesis of challenging natural products. However, most of the time, these catalysts are not yet commercially available or readily accessible, which could restrict their use in the synthesis of complex products.
3. Challenging RCM approaches towards di- or tri-substituted alkenes
In this part of the review, we will focus on relevant examples highlighting sterically demanding and/or electronically disfavoured ring-closure metathesis reactions towards challenging natural products including di- or trisubstituted alkenes. This chapter could constitute an interesting starting point for an accurate prediction of the feasibility of a RCM reaction in a multistep reaction sequence. However, selected examples will clearly evidence that many other parameters may influence the reaction among these, hydrogen bonding, spatial orientation of the respective olefins, Thorpe–Ingold effect, neighbouring functions or protecting groups. Consequently, thanks to both the wide variety of reported RCM catalysts and the above-mentioned optimization parameters, synthetic chemists now possess a powerful toolbox for a widespread use of RCM reaction in total synthesis.Many examples were chosen in the area of complex terpenoids, since most of the double bonds present in this class of compounds exist in trisubstituted form owing to the isoprene rule. However, polyketides, carbohydrates and alkaloids also caught our attention.
3.1 RCM of strained motifs
Generally, GII and GHII catalysts 2 and 4 enabled RCM reactions of strained or hindered motifs in fair to excellent yields.γ- and δ-Lactones are common moieties in natural products and RCM represents a strong tool for their elaboration, despite the reduced reactivity of the electron-deficient acrylate type alkene of the precursor, which sometimes requires the use of additives.
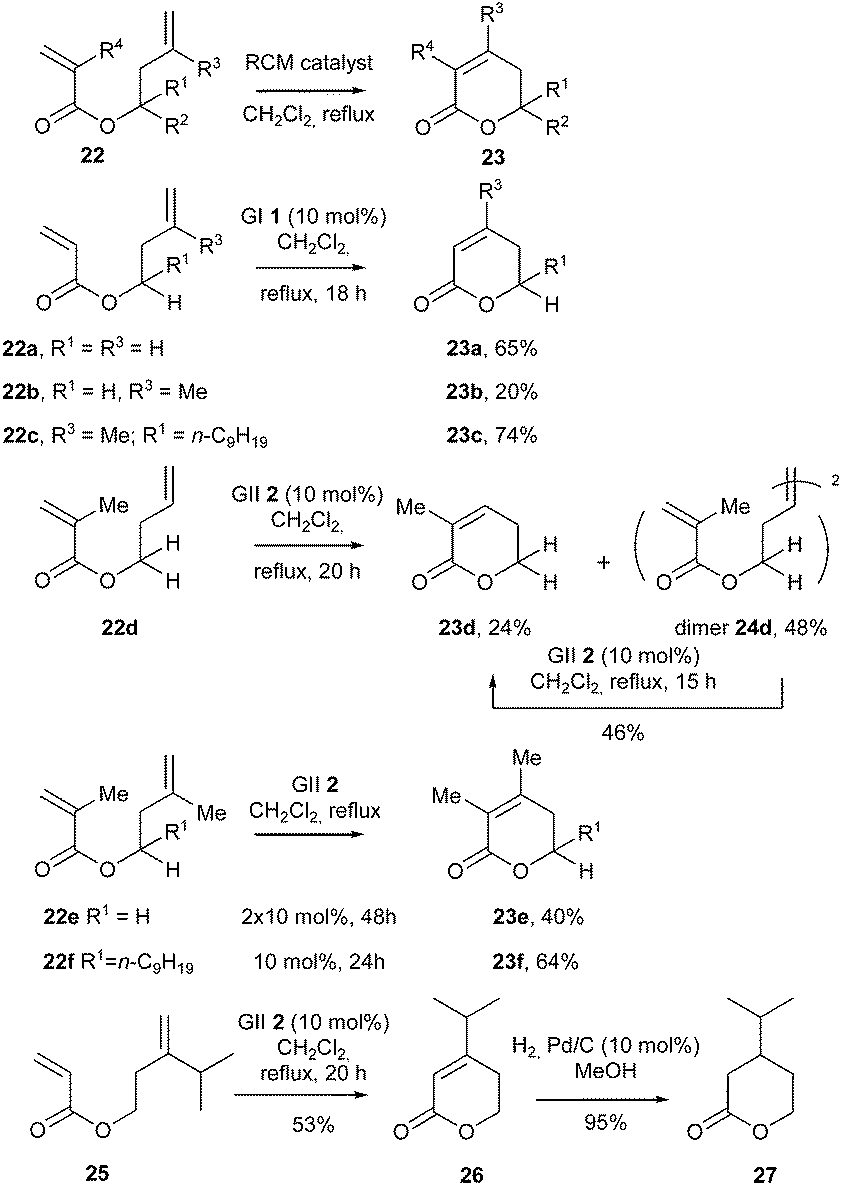
In 2007, an interesting study was reported by D'Annibale et al. about RCM reaction of various homoallylic esters 22 bearing alkyl substituents at different positions, towards the synthesis of the corresponding δ-lactones 23.19 The influence of these structural features on the choice of the appropriate catalyst (GI 1, GII 2, GHI 3, GHII 4) was carefully examined along with reaction parameters, with emphasis on catalyst loading, additives (especially Lewis acids for increasing the reactivity of the electron-poor acrylic double bond by weak coordination of the ester group) and reaction times. Noteworthy, activity of the catalysts was particularly optimized by using high dilution conditions as well as by providing a constant supply of reactive catalyst through portion wise addition or, even better, dropwise addition of solutions of metathesis complex to the reaction mixture.
In particular, it was shown that, under these catalyst dropwise addition conditions, RCM of substrate 22a comporting unhindered double bonds (R3 = R4 = H), was compatible with the use of GI catalyst 1 (formation of the corresponding lactone 23a in 65% yield), while from 22b, with a methyl group on the homoallylic double bond (R3 = Me and R4 = H)], yield of lactone 23b droped to 20%. Nevertheless, the presence of an alkyl group on the homoallylic carbon (22c; R1 = n-C9H19) had a promoting effect in the formation of lactone 23c whose yield rose to 74%, suggesting a significant Thorpe–Ingold effect.
It was also noticed that, from diene 22d [characterized by methyl substitution of the electron-deficient acrylic double bond (R4 = Me and R3 = H)], the homoallylic moiety preferentially underwent a cross-metathesis reaction in the presence of GII catalyst 2, providing the dimeric compound 24d. This dimer could be further converted into the desired α,β-unsaturated δ-lactone 23d in the presence of the same catalyst 2.
The formation of the sterically hindered tetrasubstituted pentenolide 23e from 22e (R3 = R4 = Me) required the addition of a second load of catalyst 2 (2 × 10 mol%) to reach a fair yield of 40%. As previously observed, the presence of an alkyl substituent (R1) on the homoallylic carbon atom (22f, R1 = n-C9H19) increased the yield of the reaction (lactone 23f was formed in 64% yield). Remarkably, in both e and f series, no corresponding dimer was detected.
These results were further applied to the synthesis of two naturally occurring δ-lactones, components of tobacco flavor widely used as aromatizers in the cigarette manufacturing, 26 and 27. In the presence of GII catalyst 2, ester 25 comporting a bulky isopropyl group on the homoallylic double bond, cyclized into δ-lactone 26 in an acceptable 53% yield (Scheme 3).
 | ||
| Scheme 3 D'Annibale's RCM reactions towards synthesis of δ-lactones 23, 26 and 27. | ||
At this stage, it appeared interesting to mention, the recent review published by Bassieta and D'Annibale et al. in 2013, highlighting the RCM-based synthesis of α,β-unsaturated γ- and δ-lactones present in biologically active compounds of natural origin.3c In this account, the authors evidenced RCM reactions of allyl and homoallyl acrylic esters promoted by different ruthenium carbene catalysts toward the formation of five- and six-membered conjugated unsaturated lactones.
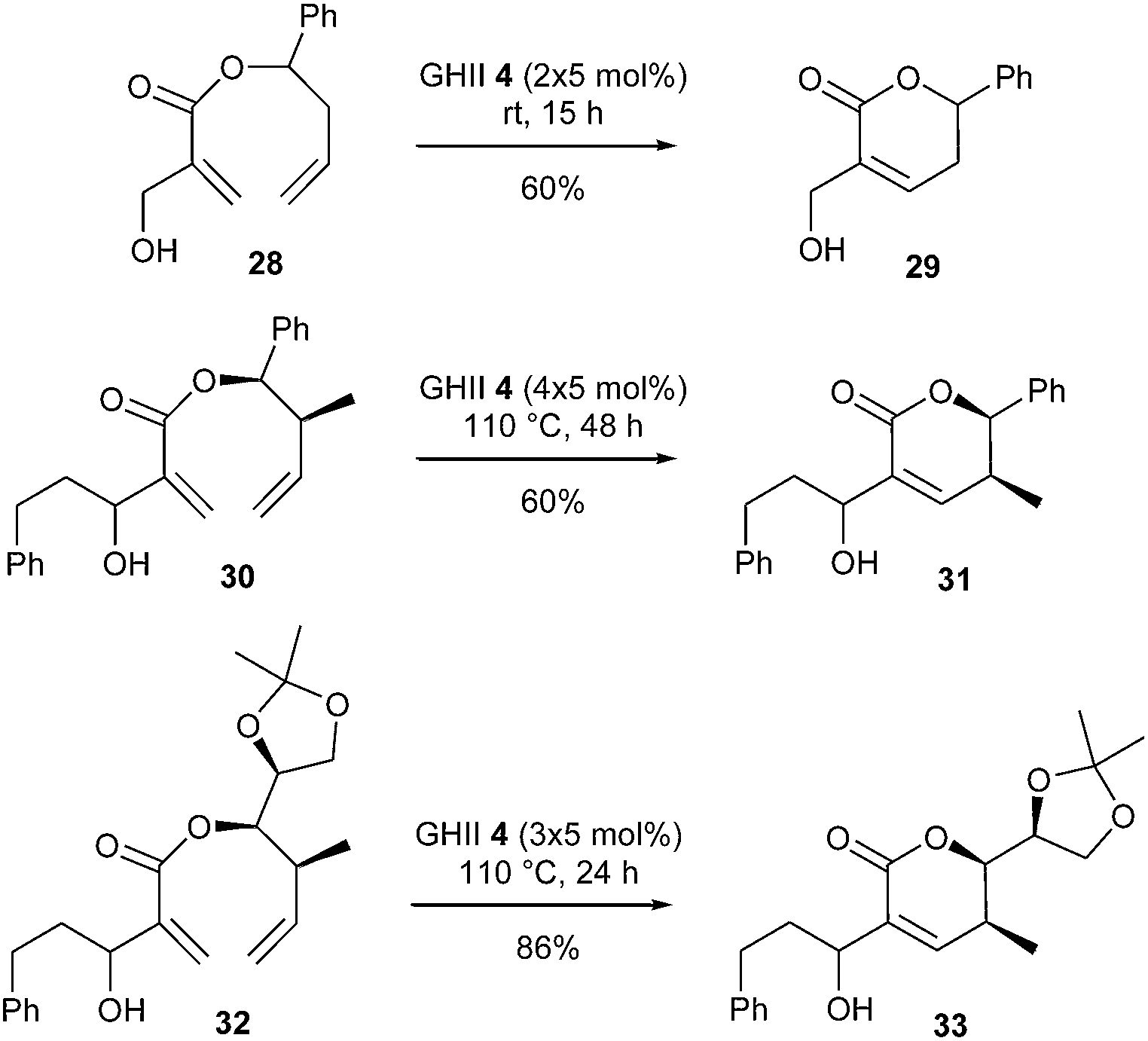
Still in the field of synthesis of natural 5,6-dihydro-α-pyrones, it is possible to describe the recent studies achieved by Shen and Zuo et al., on the challenging RCM reaction of sterically demanding homoallyl 2-(hydroxymethyl)acrylates with an alkyl or aryl substituent on the hydroxymethyl group.20 In a preliminary screening, it was noted that, when the hydroxyl group in the acrylic moiety of substrates 28 and 30 was protected (TBS, Bn, …), the reaction gave mainly self-metathesis products in the presence of GII or GHII catalysts 2 and 4. Interestingly, for substrate 28 with a free primary allylic hydroxyl group, the desired lactone 29 was obtained in 60% yield using GHII catalyst 4 (2 × 5 mol%) at room temperature for 15 h. However, when this hydroxyl group is substituted by a phenylethyl segment, the RCM reaction is negatively affected, and harsher conditions were necessary to reach lactone 31 in 60% yield from diene 30 [GHII catalyst 4 (4 × 5 mol%) at 110 °C for 48 h].
Further exploration surprisingly revealed that diene 32 containing a 1-oxygenated alkyl group at the homoallylic carbon in place of the aromatic group exhibited improved reactivity. The corresponding lactone 33 was isolated in 86% yield using GHII catalyst 4 (3 × 5 mol%) at 110 °C for 24 h. Interestingly, computational studies established that OH⋯Cl hydrogen bonding between the substrate and GHII catalyst promoted the RCM reaction. However, it was also disclosed that this hydrogen bond directed catalytic system to interact with the acrylic double bond instead of the apparently more reactive electron-rich double bond (Scheme 4).
 | ||
| Scheme 4 Shen and Zuo's RCM reactions towards synthesis of δ-lactones 29, 31 and 33. | ||
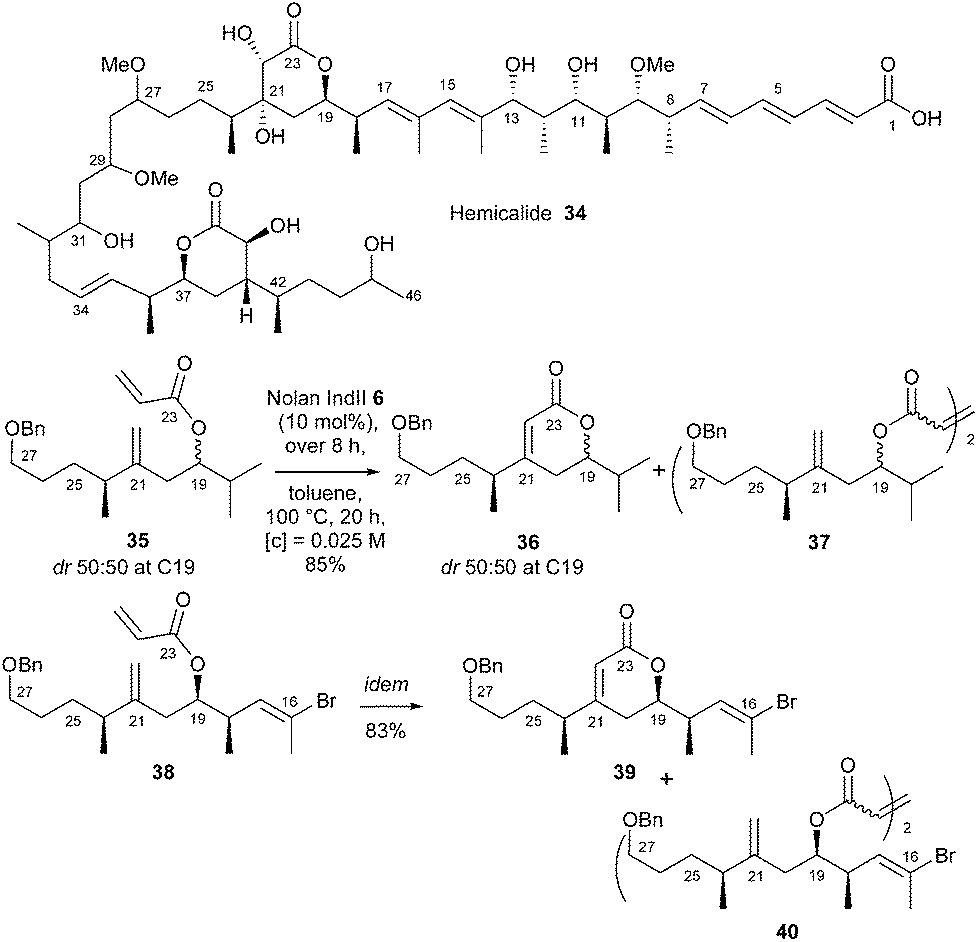
Marine organisms constitute an important source of structurally diverse and biologically active secondary metabolites. Hemicalide 34, a complex polyketide isolated in 2011 from a marine sponge, was reported to display high anti-proliferative potency at subnanomolar concentrations and through a unique mechanism. Our laboratory rose to the challenge of developing a total synthesis of this natural product (or stereoisomers thereof). One of the defining features of the approach was the construction of the central α,β-dihydroxy δ-lactone of hemicalide 34 through RCM reaction of acrylate 38 bearing a hindered chain on the homoallylic double bond.21 An initial survey was performed using easily accessible model acrylate 35, which allowed screening of different catalysts (GI 1, GII 2, GHII 4, GHIITolyl 9, Nolan–Mauduit NM 15, Plenio Ple 16 and Nolan IndII 6) and addition mode. The optimized conditions, i.e. dropwise addition of commercial Nolan catalyst IndII 6 (10 mol%) over 8 h, in toluene at 100 °C for 20 h, authorized the synthesis of pure lactone 36 in an excellent 85% yield along with homodimer 37 as a minor side product (36/37: 93![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :7). It is interesting to note that, under the same conditions, with classical Hoveyda–Grubbs and non-commercial Plenio catalysts GHII 4 and Ple 16, lactone 36 was obtained in fair 67% and 79% yields respectively. These conditions were selected for RCM reaction in the synthesis of the C16–C27 δ-lactone 39 from acrylate 38. Thus, if GHII catalyst 4 already afforded pure lactone 39 in good yield (74%) along with homodimer 40 (39/40: 90:10), an excellent yield (83%) was obtained with Nolan's catalyst IndII 6, (39/40: 93:7) (Scheme 5).
:7). It is interesting to note that, under the same conditions, with classical Hoveyda–Grubbs and non-commercial Plenio catalysts GHII 4 and Ple 16, lactone 36 was obtained in fair 67% and 79% yields respectively. These conditions were selected for RCM reaction in the synthesis of the C16–C27 δ-lactone 39 from acrylate 38. Thus, if GHII catalyst 4 already afforded pure lactone 39 in good yield (74%) along with homodimer 40 (39/40: 90:10), an excellent yield (83%) was obtained with Nolan's catalyst IndII 6, (39/40: 93:7) (Scheme 5).
 | ||
| Scheme 5 Laboratory's RCM reactions towards synthesis of hemicalide 34. | ||
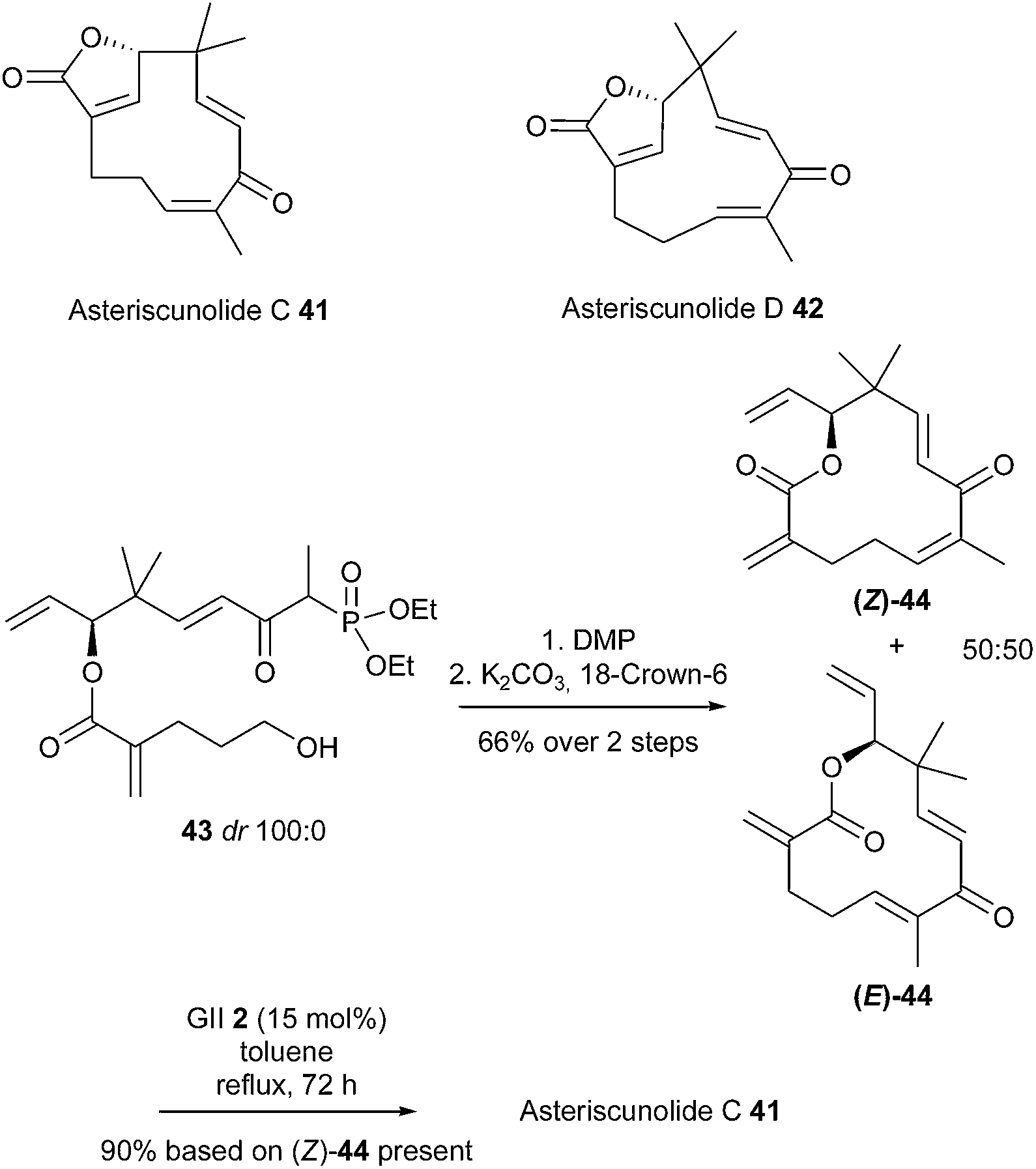
Asteriscunolide C 41 possesses a humulene skeleton with a 11-membered ring and was found to have potent anticancer activity. The first total synthesis of this natural product was accomplished by Fernandes et al. in 2013.22 The key step was the highly ambitious conversion of a 12-membered ring into a strained 11-membered ring with one (E) and two (Z)-double bonds, by means of a late-stage RCM reaction. The sequence involved the formation of the RCM precursor 44 through an intramolecular Horner–Wadsworth–Emmons olefination of phosphonate 43. However, this reaction resulted in a mixture of desired (Z)-44 and (E)-44 in a 50:50 ratio. The final RCM reaction was attempted on the mixture and the best results were obtained using GII catalyst 2 (15 mol%) in refluxing toluene for 72 h. Only asteriscunolide C 41 was isolated in 90% yield, based on the proportion of (Z)-44 present in the substrate. No trace of asteriscunolide D 42 was detected, and the all (E)-isomer (E)-44 could not be recovered. Undoubtedly, the two (E)-double bonds in (E)-44 might be preventing the RCM reaction to generate the third (E) bond in the strained 11-membered macrocycle (Scheme 6).
 | ||
| Scheme 6 Fernandes' RCM reactions for the synthesis of asteriscunolide C 41. | ||
Elaboration of five- and six-membered cycles through RCM reaction will be next regarded.
Apiose is a structurally unusual furanose, whose naturally occurring form has the 3-(R) configuration, found in various biologically active natural products. Thus, highly efficient synthesis of these sugars constitutes an important goal in organic chemistry. Very recently, Rhee et al. reported a unique multicatalytic synthesis of apiose-containing mono- and oligosaccharides.23 The approach comprised three key steps allowing the introduction of all the stereochemical information present in the apiose residue: a first palladium-catalyzed asymmetric intermolecular hydroalkylation of an alkoxyallene (46) leading to the construction of an acyclic chiral O,O-acetal (47), subsequent ruthenium (GI catalyst 1) RCM reaction and osmium-catalyzed dihydroxylation. This sequence, applied to initial reaction between allylic alcohol 48 and alkoxyallene 49 towards formation of the intermediate 50, permitted the challenging convergent synthesis of the apiose-containing trisaccharide moiety 51, which constitutes the key fragment of polygalin G 45 (Scheme 7).
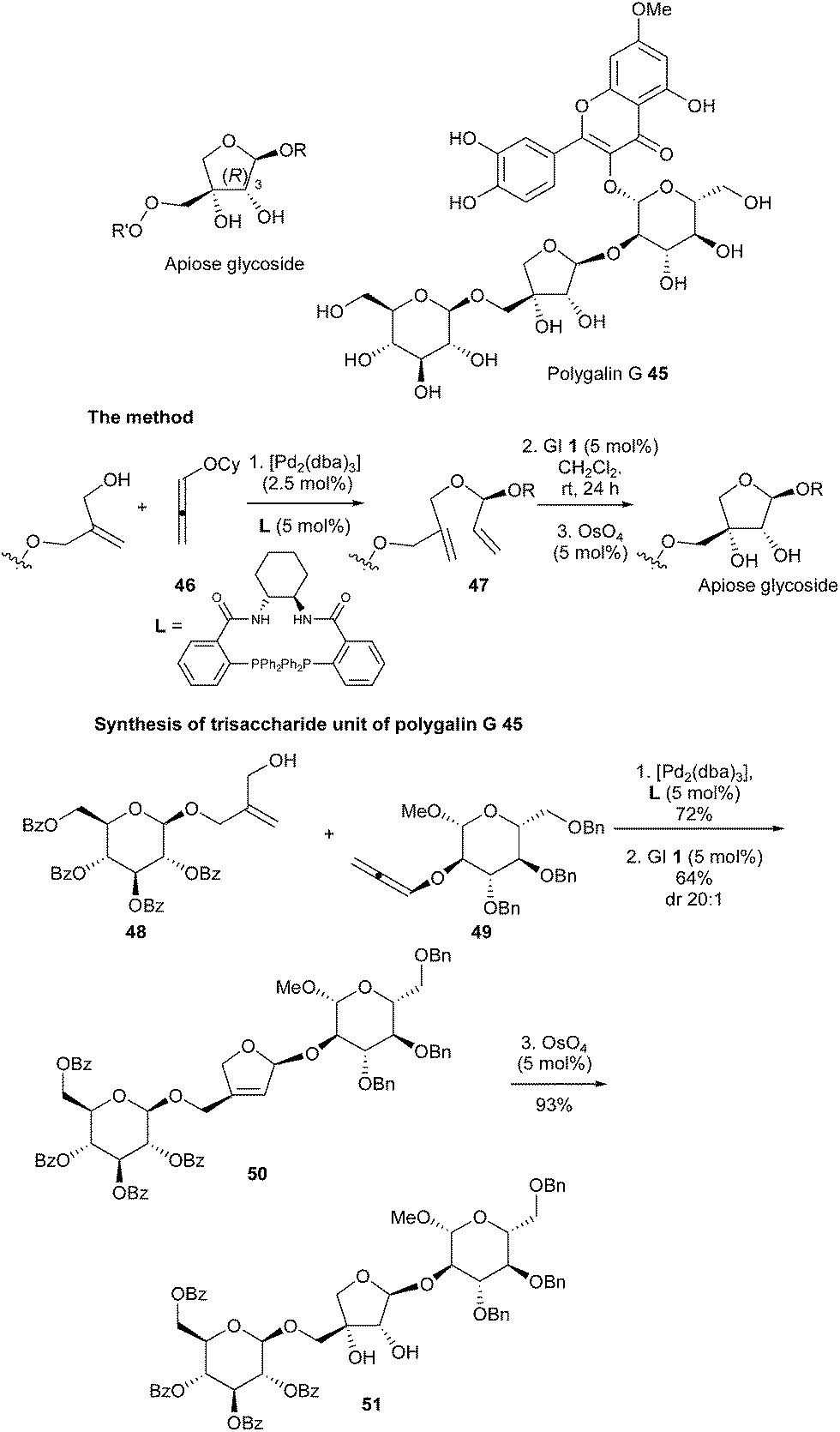 | ||
| Scheme 7 Rhee's RCM reactions for synthesis of the trisaccharide unit 51 of polygalin G 45. | ||
A highly efficient enantioselective desymmetrization of prochiral trienes, catalysed by a stereogenic-at-Mo catalyst 52 (dr 83:17) was developed by Schrock and Hoveyda.24 Malcolmson and Meek et al. demonstrated the efficacy of this catalyst in the enantioselective total synthesis of (+)-quebrachamine 55 featuring a quaternary carbon stereocenter.24 With only 1 mol% catalyst 52 loading, triene 53 was entirely transformed within 1 h into tetracycle 54, in 84% yield and 96% ee. This latter was further converted into the natural product 55 by hydrogenation (Scheme 8). The application of a C1-symmetric chiral ruthenium complex proved to be unsuccessful, leading to racemic diene 54.
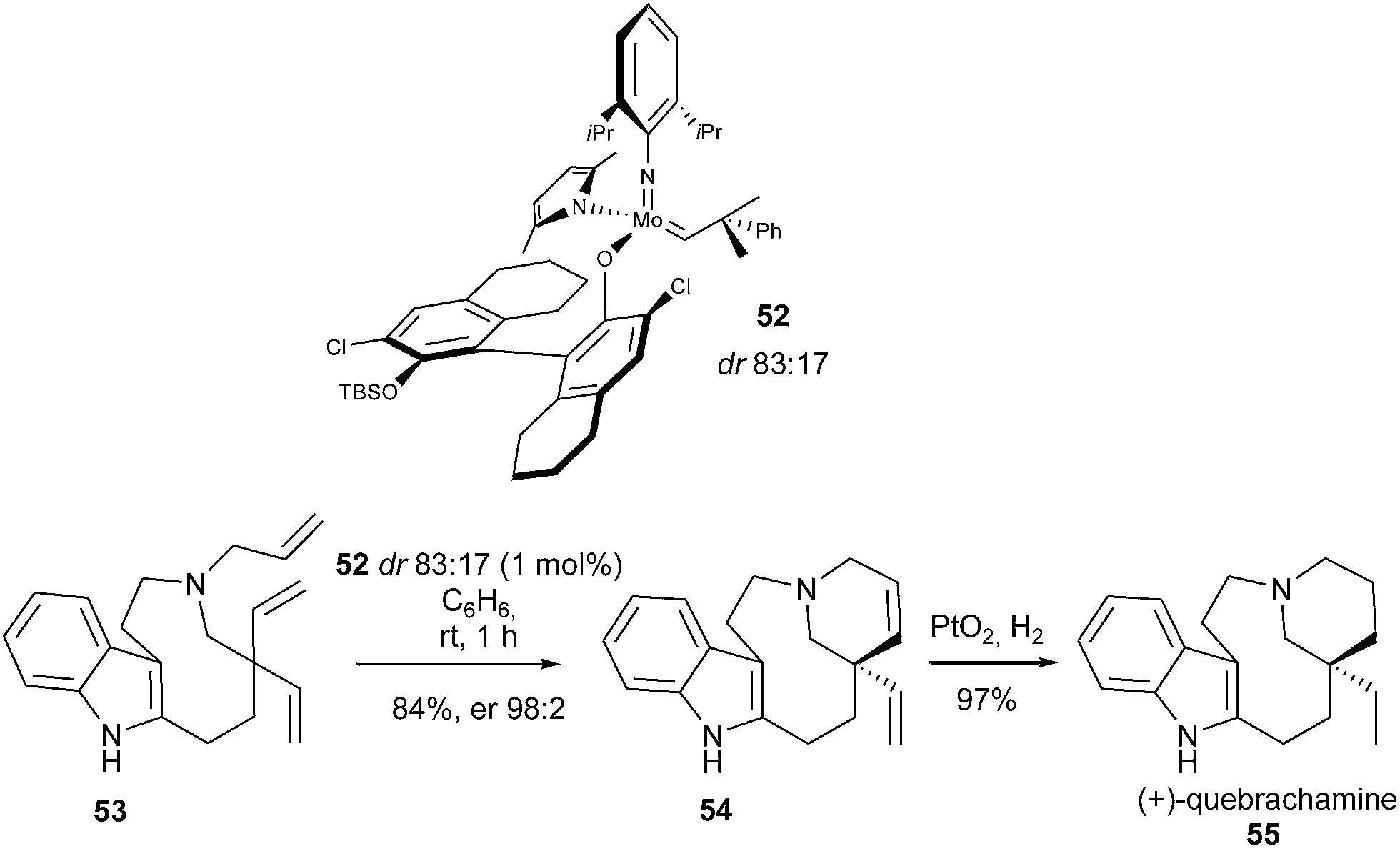 | ||
| Scheme 8 Malcolmson and Meek's desymmetric RCM reactions for synthesis of quebrachamine 55. | ||
Many natural products contain a spirocarbocycle as structural element. For the direct synthesis of these systems, the RCM method offers very mild reaction conditions and flexibility. Ito et al. reported in 2013 a total synthesis of spirocursanone 57, a tricyclic diterpenoid, involving the spiro ring formation by RCM.25 Treatment of tetraene 56 with GII catalyst 2 (2.0 mol%) in dichloromethane at room temperature for 3 h, successfully provided the target tricyclic compound spirocursanone 57 in excellent yield (98%) (Scheme 9).
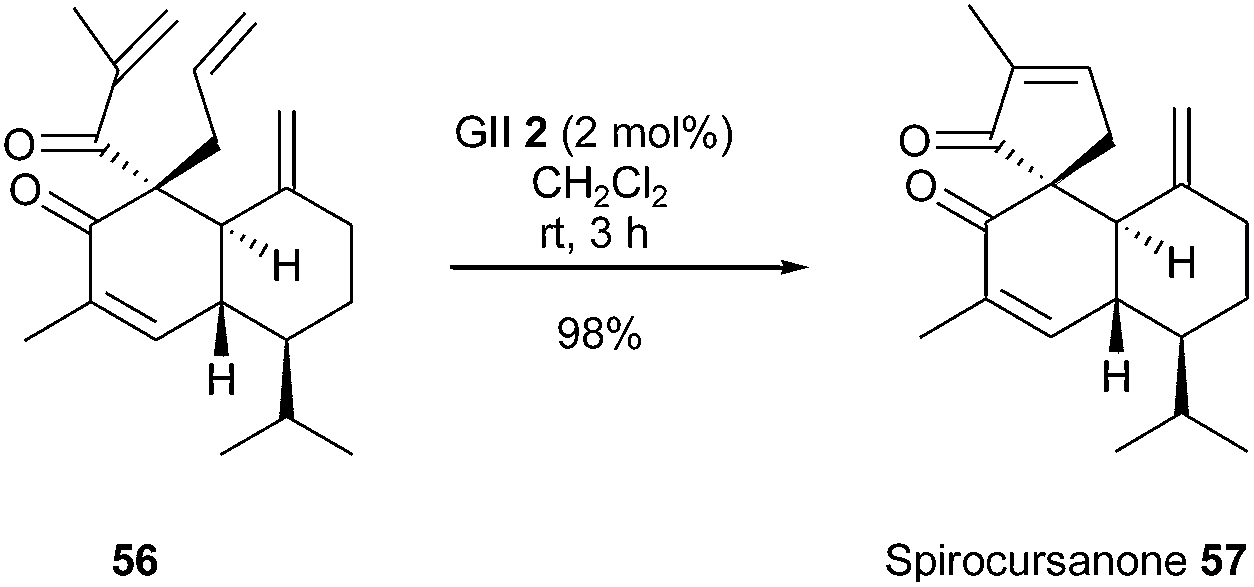 | ||
| Scheme 9 Ito's RCM reaction for the synthesis of spirocursanone 57. | ||
Przewalskin B 58, isolated from a Salvia species, possesses a unique tetracyclic skeleton that contains a spirocyclic enone system and a α-hydroxy-β-ketone moiety. Xie et al. accomplished a concise total synthesis of this compound (±)-58 involving a challenging RCM reaction of a sterically crowded system (i.e. between a 1,1-disubstituted olefin and a sterically hindered terminal alkene) to construct the cyclic enone moiety.26 Treatment of diene (±)-59 with GII 2 or GHII 4 catalysts afforded β-ketoester (±)-60 in low yield (<20%). RCM of corresponding allylic alcohols (±)-61 and (±)-62 in the presence of GII catalyst 2 in toluene at 100 °C, followed by oxidation successfully produced β-ketoester (±)-60 in 65% overall yield (Scheme 10).
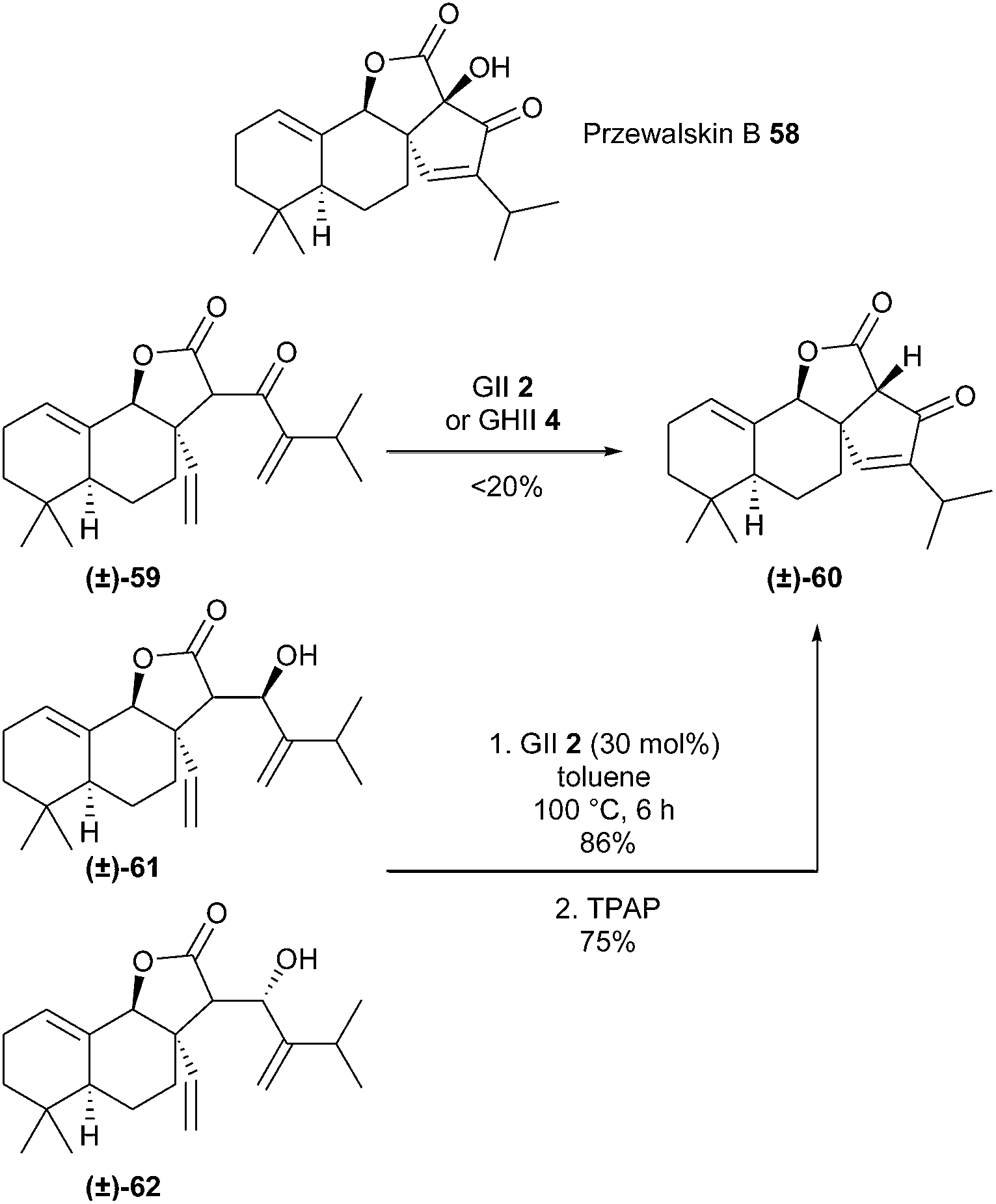 | ||
| Scheme 10 Xie's RCM reactions for the synthesis of przewalskin B 58. | ||
It is well-known that medium and large cycles are often difficult to build owing to unfavourable non-bonding interactions; however, RCM reactions could allow their direct elaboration with satisfactory results, even in case of sterically congested or electronically deactivated dienes.
Chen and Yang et al. reported the total synthesis of schindilactone A 63, which is characterized by 12 stereogenic centers and an unusual steric congestion.27 Two independent synthetic approaches were developed, both of which featured RCM reactions for the construction of the fused/bridged ring system B,C,D,E and/or the δ-lactone H at a late stage of the synthesis. The reaction of diene 64 as a pair of diastereomers at C15 with GII catalyst 2 (10 mol%) and MgBr2 (20 mol%) at 30 °C for 12 h provided the tetracycle 65 in 65% yield as a single isomer. This observed MgBr2-mediated in situ epimerization through dynamic kinetic resolution was in agreement with the results reported by Scholl and Grubbs.28 The construction of lactone H fragment at the end of the total synthesis was quite challenging. Remarkably, when the highly functionalized diene 66 was treated with GII catalyst 2 (50 mol%) in toluene at 100 °C for 1 h, the desired system 67 was obtained in 70% yield (Scheme 11).
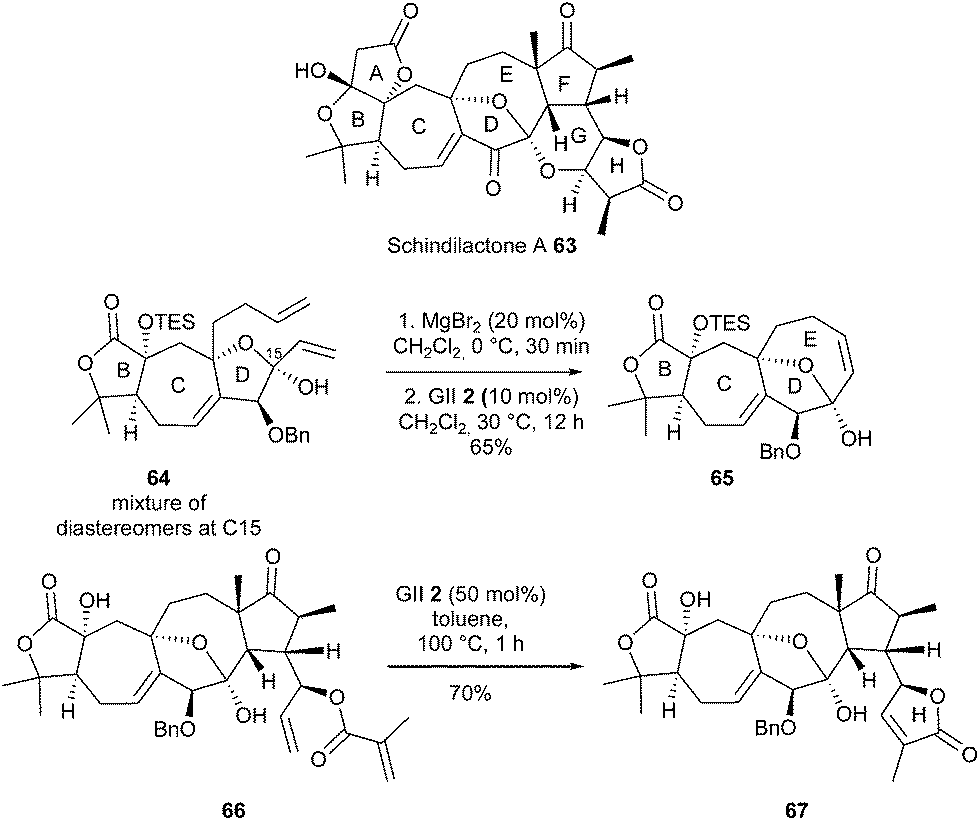 | ||
| Scheme 11 Chen and Yang's RCM reaction for synthesis of schindilactone A 63. | ||
13-Oxyingenol 69, derivative 68 and ingenol 70 are diterpenoids showing strong bioactivities. Kigoshi et al. reported in 2012 the first total synthesis of (−)-13-oxyingenol 69 and its natural derivative 68.29 For the construction of carbocycle B, best results were obtained when α,β-unsaturated ketone 71, which possesses one electro-deficient olefin and a lowered steric hindrance, was used instead of corresponding alcohol (or acetate) at C7 position. Thus, the reaction of 71 with GHII catalyst 4 (30 mol%) in refluxing toluene for 35 h provided the tetracyclic framework 72 in 86% yield. Remarkably, no epimerization of β-diketone moiety in 71 and 72 was observed (Scheme 12).
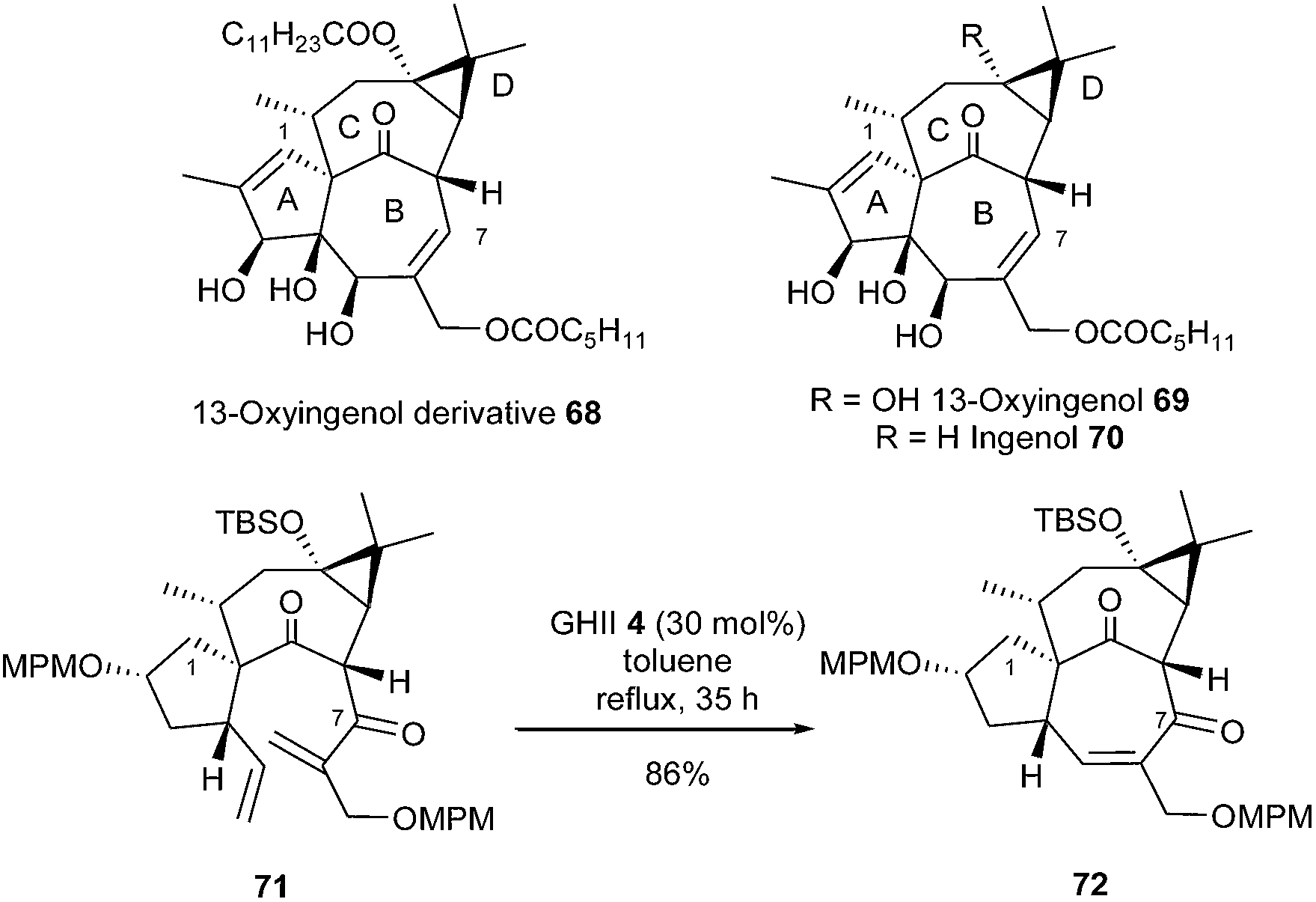 | ||
| Scheme 12 Kigoshi's RCM reactions for the synthesis of oxyingenol 69. | ||
In nature, tropolones (or hydroxycycloheptatrienones) and 3,4-benzotropolones are found in fungi and plants, and several of them have gained patent protection as antimicrobial, antiretroviral and antiobesity agents. 3,4-Benzotropolones could be perceived as thermodynamically favored enol tautomers enol-73 of much less stable 1,2-diketones keto-73. Accordingly, Brückner et al. achieved the synthesis of various 3,4-benzotropolones enol-73 targeting diketones keto-73 as their monoketals (74), following a RCM route.30 In particular, the unsubstituted benzocycloheptadienone monoketal 76 was smoothly synthesized from dienone 75, in the presence of GII catalyst 2 (1 mol%), in dichloromethane at room temperature for 18 h, in nearly quantitative yield. Subsequent hydrolysis and keto–enol tautomerism furnished the desired 3,4-benzotropolone.
On the contrary, 2 and 5 mol% of GII catalyst 2 and heating at 100 °C (for 90 min and 7 h respectively) were required to perform the ring closure on dienones 77 and 79. Bicyclic monoketals 78 and 80 bearing a trisubstituted bond were then obtained in excellent yields. This study established that a nonbenzenoid aromatic such as a tropolone can be synthesized by RCM, although this reaction may not be systematically obvious (Scheme 13).
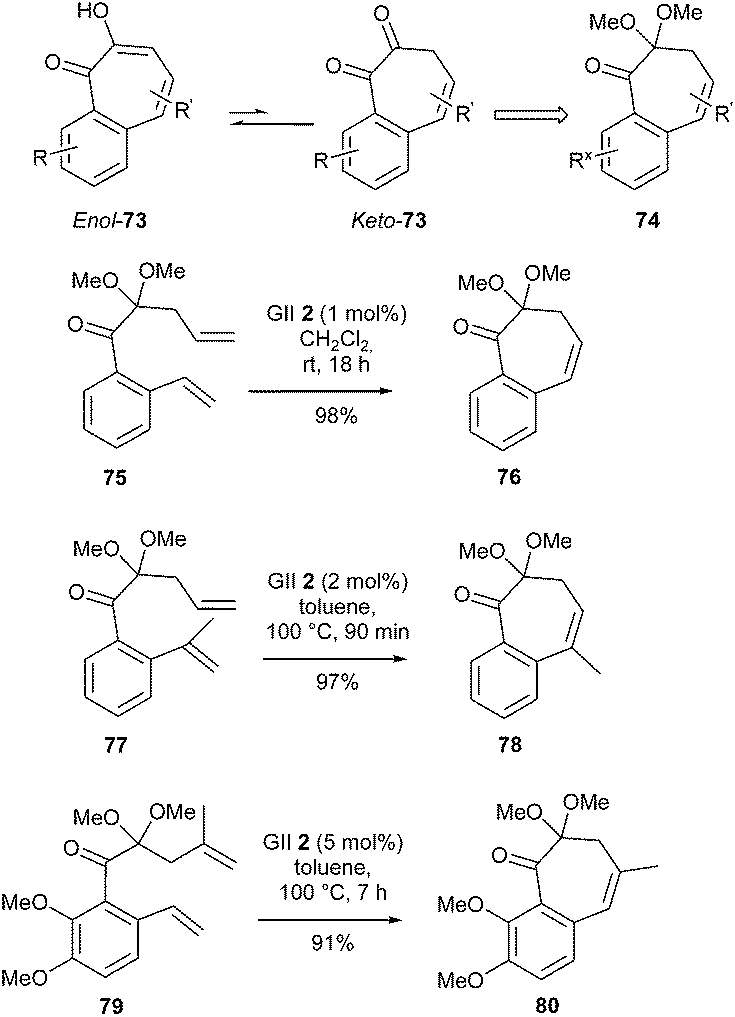 | ||
| Scheme 13 Brückner's RCM reactions for synthesis of 3,4-benzotropolones 73. | ||
Pleuromutilin 81, a diterpene isolated from several species of basidiomycetes, is an antibiotic which inhibits protein synthesis by binding to the 50S ribosomal subunit of bacteria.
Sorensen et al. described a concise synthesis of the tricyclic carbon skeleton of this natural product with the idea of constructing the eight-membered ring by RCM.31 When a solution of diene 82 and GII catalyst 2 (3 × 10 mol%) was heated to reflux for 24 h, the desired eight-membered ring formation took place and afforded cyclized compound 83 in moderate yield (31–47%). With the aim of incorporating functional groups at C14 of pleuromutilin scaffolds, RCM of diastereomeric dienes 84 and 86 was examined. The reaction of diene 84 with GHII catalyst 4 (2 × 11 mol%) in refluxing dichoromethane for one week afforded the tricyclic alkene 85 in 80% yield, however, ester 86 did not undergo an analogous RCM towards 87. The viability of the metathesis process seemed to hinge on the configuration at C14: one explanation may be that a destabilizing, non-bonding interaction between the hydroxyl group at C14 and the methyl group at C12 hinders the formation of the metallocyclobutane (Scheme 14).
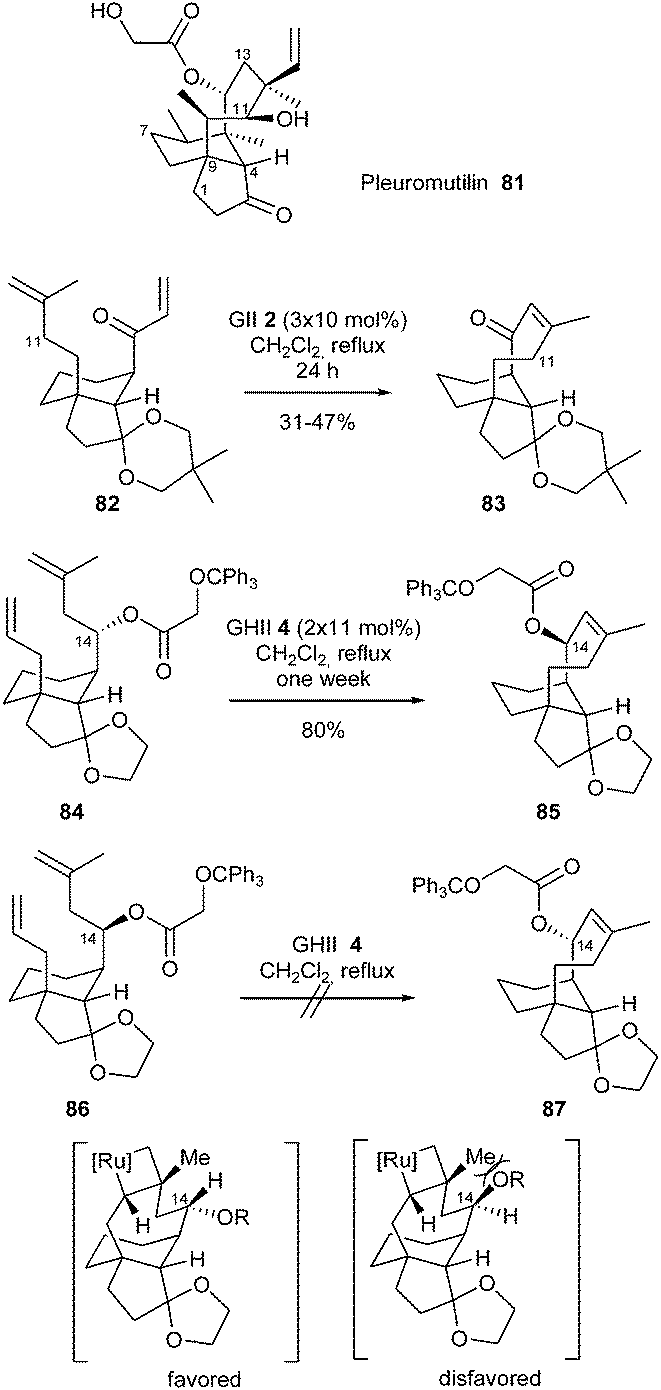 | ||
| Scheme 14 Sorensen's RCM reactions for synthetic approach towards pleuromutilin 81. | ||
In 2013, Nakada et al. reported an enantioselective total synthesis of (+)-ophiobolin A 88, a cytotoxic sesterterpene (biogenetically formed from five isoprene units) characterized by a unique 5-8-5-5 tetracyclic ring system.32 To address the difficulties in forming the medium-sized, eight-membered carbocyclic ring, a RCM was planned at a late stage of the synthesis. At first, RCM of dienes 89 or 91 (bearing an extra alkene to decrease the transannular interactions in the transition state) was attempted using GII 2, GHII 4 or GHIITol 9 catalysts, but no desired products 90 or 92 could be detected and most of the starting material was recovered at the end of the reaction. The authors speculated that the reaction was affected by the bulky TBDPS (t-butyldiphenylsilyl) ether at C5. Thus, the RCM of diene 93 was next examined. In the presence of GII catalyst 2, in toluene at 110 °C, the desired tricyclic system 94 was isolated in 58% yield, whereas with GHII catalyst 4, 94 was only obtained in 39% yield along with the terminal double-bond isomerized diene as major product. Finally, the RCM reaction of 93 with GHII 4 in the presence of 10 equivalents of 1,4-benzoquinone (to avoid isomerization of the C8 double bond) allowed the formation of 94 in an excellent 91% yield. Towards the synthesis of (+)-ophiobolin A 88, the RCM of diene 95 with a benzylic ether at C5 and the hydroxylated form of the isopropenyl group at C6, was examined under these conditions. The reaction successfully afforded the desired product which was used in the next steps without further purification (formation of intermediate 96 in 68% yield over three steps from 95) (Scheme 15).
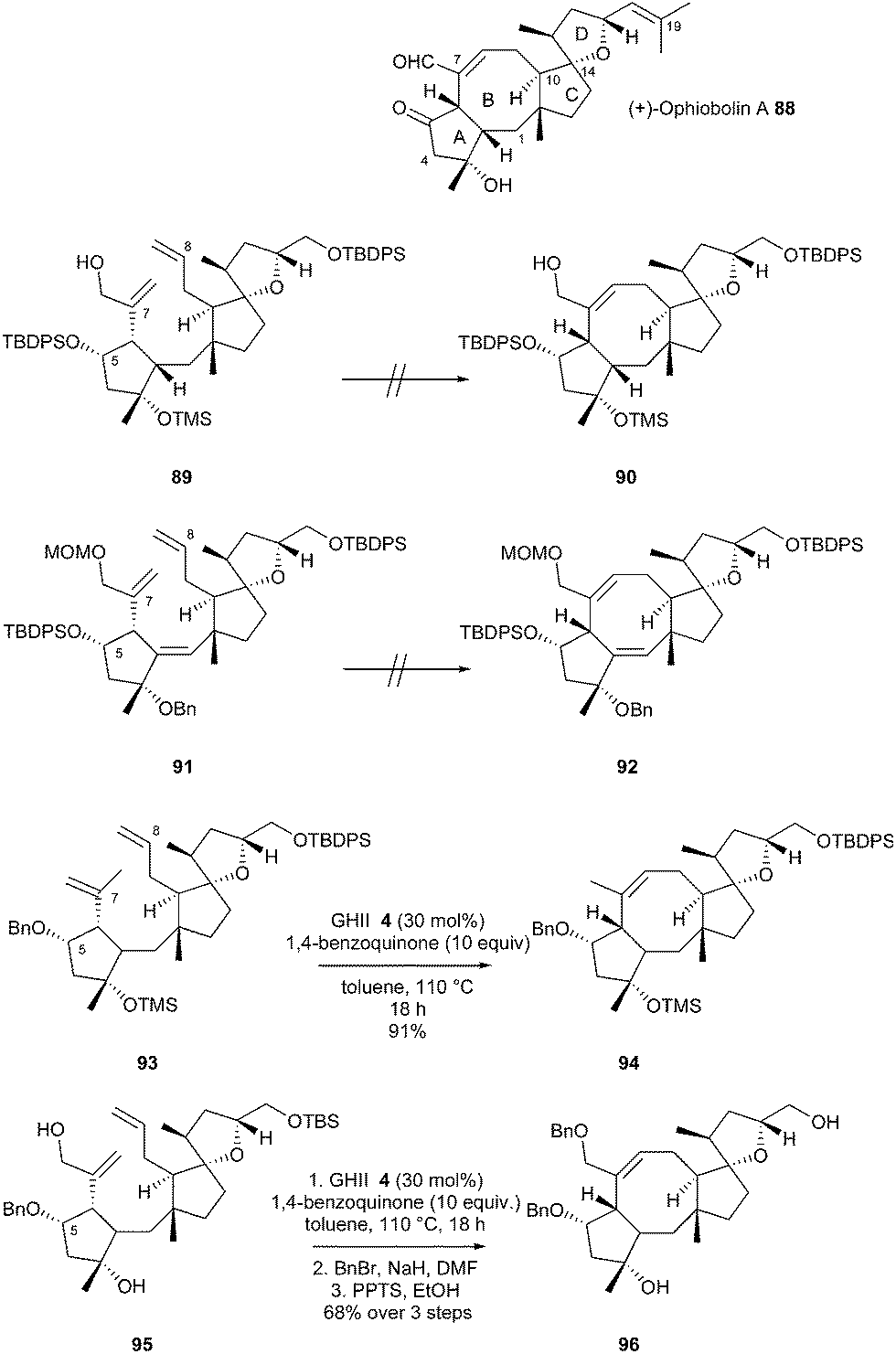 | ||
| Scheme 15 Nakada's RCM reactions for synthesis of ophiobolin A 88. | ||
Steroids constitute an extensive and important class of biologically active polycyclic compounds that are widely used for therapeutic purposes. In this context, steroid-like polycyclic systems containing eight-membered carbocycles represent a large group of compounds of importance. In 2005, Granja et al. reported the construction of steroid-like polycycles 97 with a linear fused 6-8-6 carbocyclic system utilizing RCM to form ring B or a 12-membered precursor, according to the strategy.33 The feasibility of RCM was first explored using models 98a (R = H) and 98b (R = Me). However, with GI catalyst 1, no reaction took place. In the presence of GII 2 (70 mol%), 98a and 98b were respectively converted into 99a and 99b, i.e. six-membered ring formation prevailed over eight-membered ring formation, though this transformation involved reaction with the more substituted olefin. On the other hand, when 100 was subjected to the reaction catalyzed by GI 1 or GII 2 catalysts, product 99a (R = H) was obtained instead of 101. Finally, it was demonstrated that, in the absence of competing double bonds and carbocycle A, formation of the eight-membered ring was possible. Thereby, a one-week RCM reaction of 102 with GI catalyst 1 (70 mol%) in refluxing dichloromethane, provided tricyclic product 103 in 95% yield. The authors proposed that coordination of the OTBS oxygen to the ruthenium could explain both the sluggishness of the kinetics of this reaction and why the Ru methylidene catalyst survived for much longer time than its usual half-life. In the present case, one can wonder about the relevance of the procedure, considering the short half-life of GI 1 catalyst. Subsequent Heck cyclisation, from corresponding homologated (Z)-vinyl iodide at C3-C4, allowed the synthesis of the desired steroid-like systems 97 (Scheme 16).
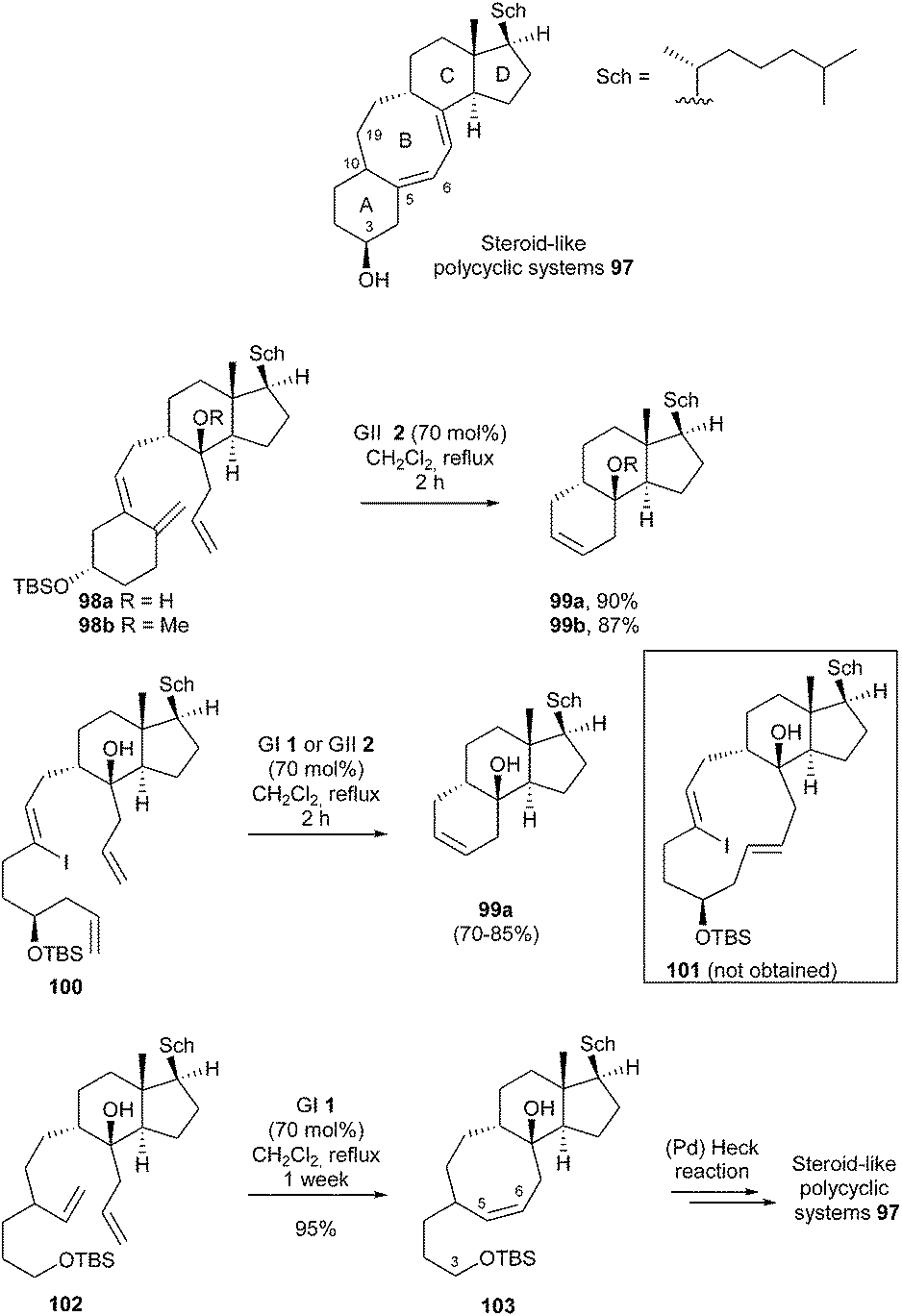 | ||
| Scheme 16 Granja's RCM reactions for synthesis steroid-like polycyclic systems 97. | ||
RCM reactions towards macrolides occasionally remained problematic despite many attempts performed. A modification of the strategy could therefore be required to access the target compound. The following two examples clearly demonstrated that steric hindrance at the allylic position is prohibitive for RCM.
Interesting preliminary studies on the use of RCM for the synthesis of bryostatins were reported very recently (in 2017) by Thomas et al.34 A late-stage RCM approach towards bryostatin 10 104 was envisaged from a fully functionalized precursor 105. However, attempts to carry out this reaction to elaborate macrolide 106, in the presence of GII catalyst 2, were unsuccessful and systematically yielded mixtures of products. Although not fully characterized, main side-products disclosed partial loss of methoxy ketal as well as some isomerization of the double bond of the vinyl tetrahydropyran. The authors hypothesized that the ketone at C19 may coordinate organometallic intermediates formed by metathesis of the proximal double bond at C17, thereby inhibiting the catalytic cycle. Otherwise, steric hindrance generated by the two geminal methyl groups at C18 may also be a critical factor. Thus, RCM of simpler analogues 107 and 109, one without and the other with two geminal allylic methyl groups was investigated. Using GII catalyst 2, a modest 17% yield of the (E)-alkene 108 was obtained from ester 107, but no cyclized product 110 was observed under similar conditions from the reaction with ester 109. These results are consistent with the assumption that geminal methyl substituents at C18 provide additional hindrance. The ester 107 that lacked methyl groups delivered the metathesis product, albeit in very low yield. As a consequence, the authors mentioned that they could not clearly delineate the factors influencing this critical step. Thus, a wider methodological survey was planned to define the scope of this RCM reaction (Scheme 17).
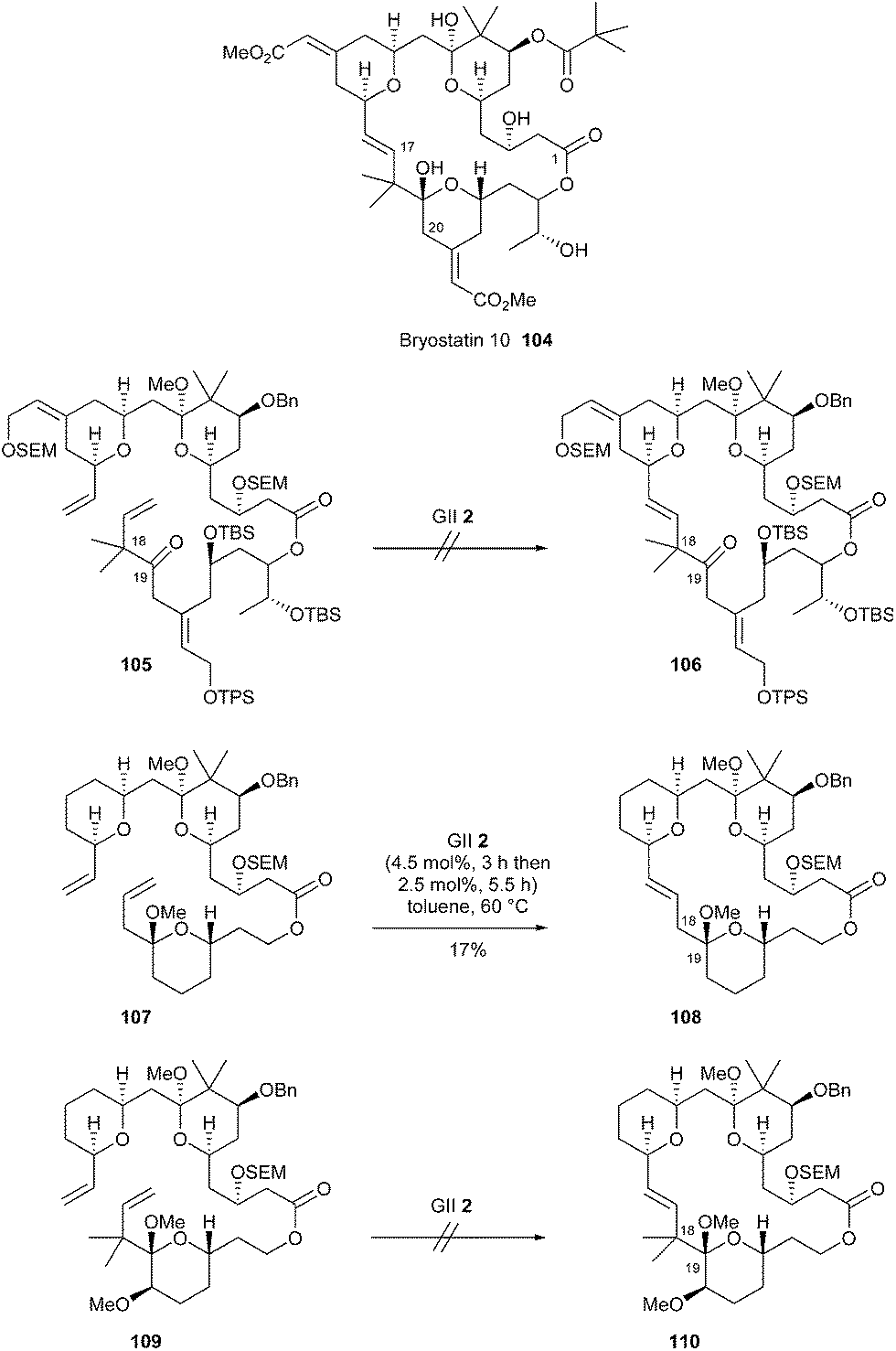 | ||
| Scheme 17 Thomas' RCM reactions for synthesis of bryostatin 10 104. | ||
Leiodolide A 111 is a 19-membered marine macrolide possessing significant cytotoxicity. Towards the synthesis of this molecule, Altmann et al. investigated a RCM based macrocyclization of the two dienes 112 and 114 (R = H or Me) incorporating the complete framework of the macrocyclic core.35 Dienes were subjected to different RCM conditions, employing either GII 2, GHII 4 or Piers 116 catalysts.36 Unfortunately, 112 and 114 revealed unreactive and none of the desired cyclized products 113 or 115 could be detected. Thus, another cyclization approach had to be envisioned and is currently ongoing (Scheme 18).
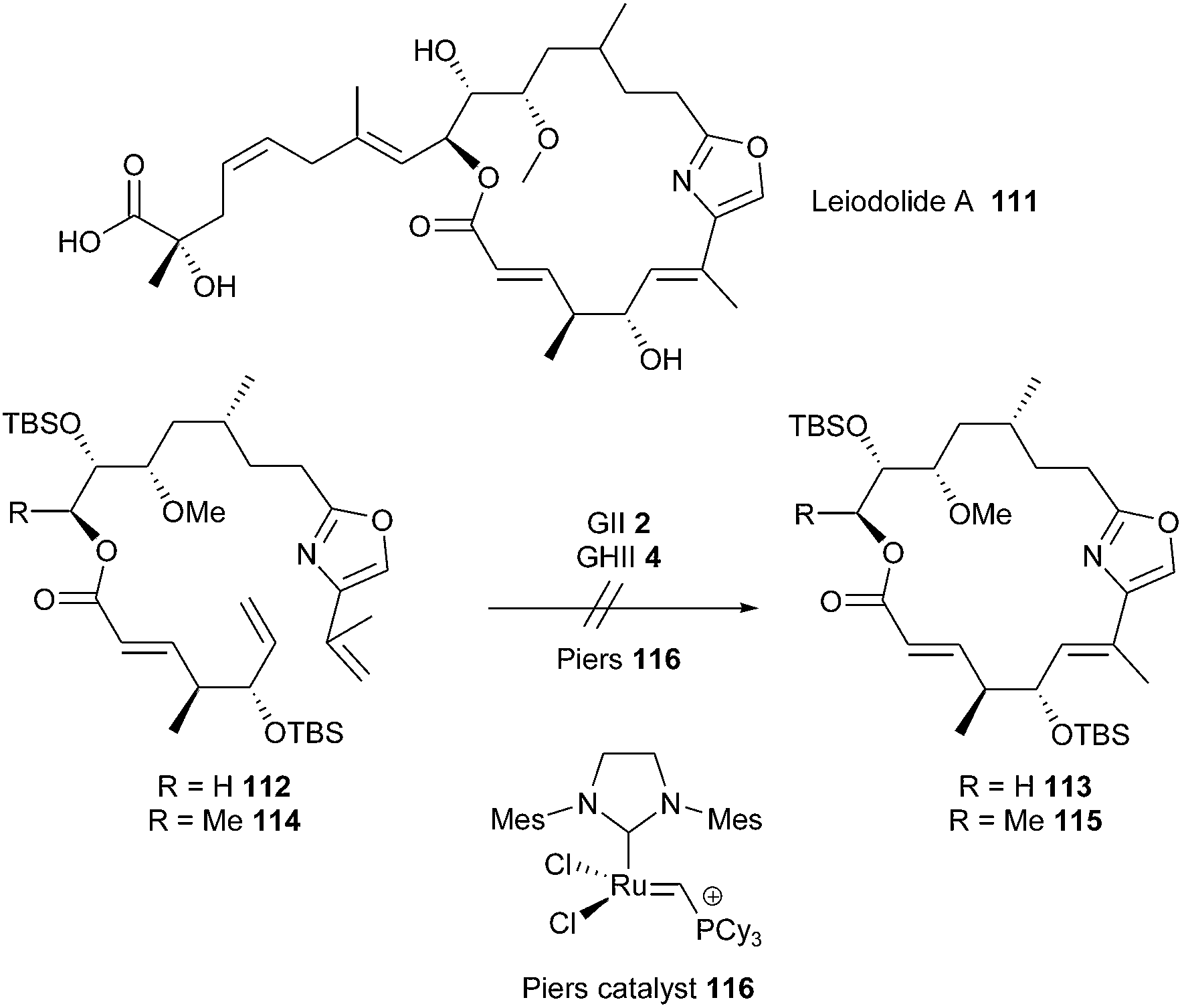 | ||
| Scheme 18 Altmann's RCM reactions for synthesis of leiodolide A 111. | ||
The unique 6-aza-spiro[4.5]decane architecture of halichlorine 117 and its bioactivity has attracted considerable attention. In 2014, Arimoto et al. reported a total synthesis of this molecule and the initially investigated strategy featured a challenging double RCM reaction.37 Unsaturated ester 118 was designed as the potential precursor for halichlorine 117, through six-membered and macrocyclic RCMs, in a one pot process. The six-membered RCM in the presence of C15–C16 terminal olefin was first explored on model 119. The GHII catalyst 4 (10 mol%, 60 °C in toluene) fully converted 119 into the RCM product 120 including a double bond at C3–C15 (the desired product 121 was not detected). All attempts to transform 119 or 120 into 121 failed. This unexpected cyclization into 120 is thought to occur due to the spatial proximity of C3 and C15 centers. Therefore, a stepwise RCM strategy was envisaged. With GHII catalyst 4, compound 122 was smoothly transformed into tricyclic system 123. However, substrate 124 proved to be inert to metathesis conditions (125 was not detected). In this latter case, the authors suggested that concomitant coordination of ruthenium atom by tertiary amine function and C15 olefin could result in catalyst poisoning. To cope with these unexpected results, a modified sequence was performed by carrying out the six-membered RCM at a later stage of the synthesis followed by final macrolactonization. In this context, the six-membered RCM of compound 126 quantitatively afforded the desired tricyclic compound 127 (Scheme 19).
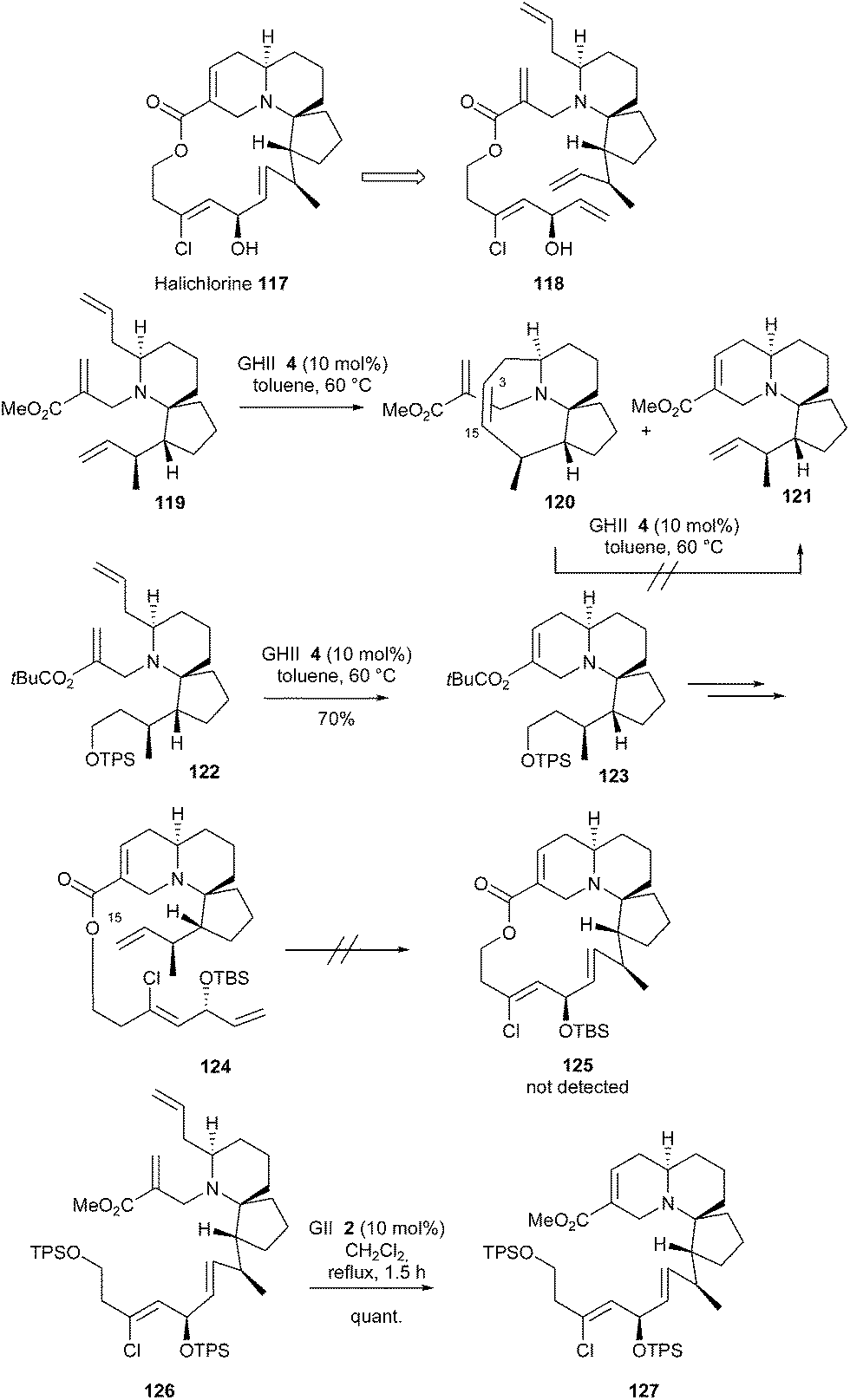 | ||
| Scheme 19 Arimoto's RCM reactions for synthesis of halichlorine 117. | ||
3.2 RRCM as an efficient alternative to classical RCM
For overcoming initial difficulties and improving results, Hoye's relay metathesis (RRCM) strategy has been investigated in the presence of classical GII or GHII catalysts 2 or 4.38 Thus, appropriate modifications could transform poorly reactive RCM substrates into suitable RRCM precursors and thus solve reactivity or selectivity problems.Penostatins are cytotoxic polyketide derivatives isolated from a strain of Penicillium originally separated from a marine alga. The first total synthesis of penostatin B 128 was accomplished by Shishido et al. in 2012, using a relay RCM (RRCM) to install the dihydropyranone ring.39 At first, the RCM of diene 129 has been examined, however, the use of GII catalyst 2 or GHII catalyst 4, combined with Ti(OiPr)4, resulted in no reaction or low yields in desired tricyclic compound 130. Therefore, a RRCM reaction was investigated. When conducted from ester 131 in the presence of GII catalyst 2 (25 mol%) in refluxing dichloromethane for 2 h, the RRCM advantageously provided the dihydropyranone 130 in a good 83% yield. Despite a higher number of steps, this strategy revealed that cyclization could readily occur through the most electron-deficient alkene (Scheme 20).
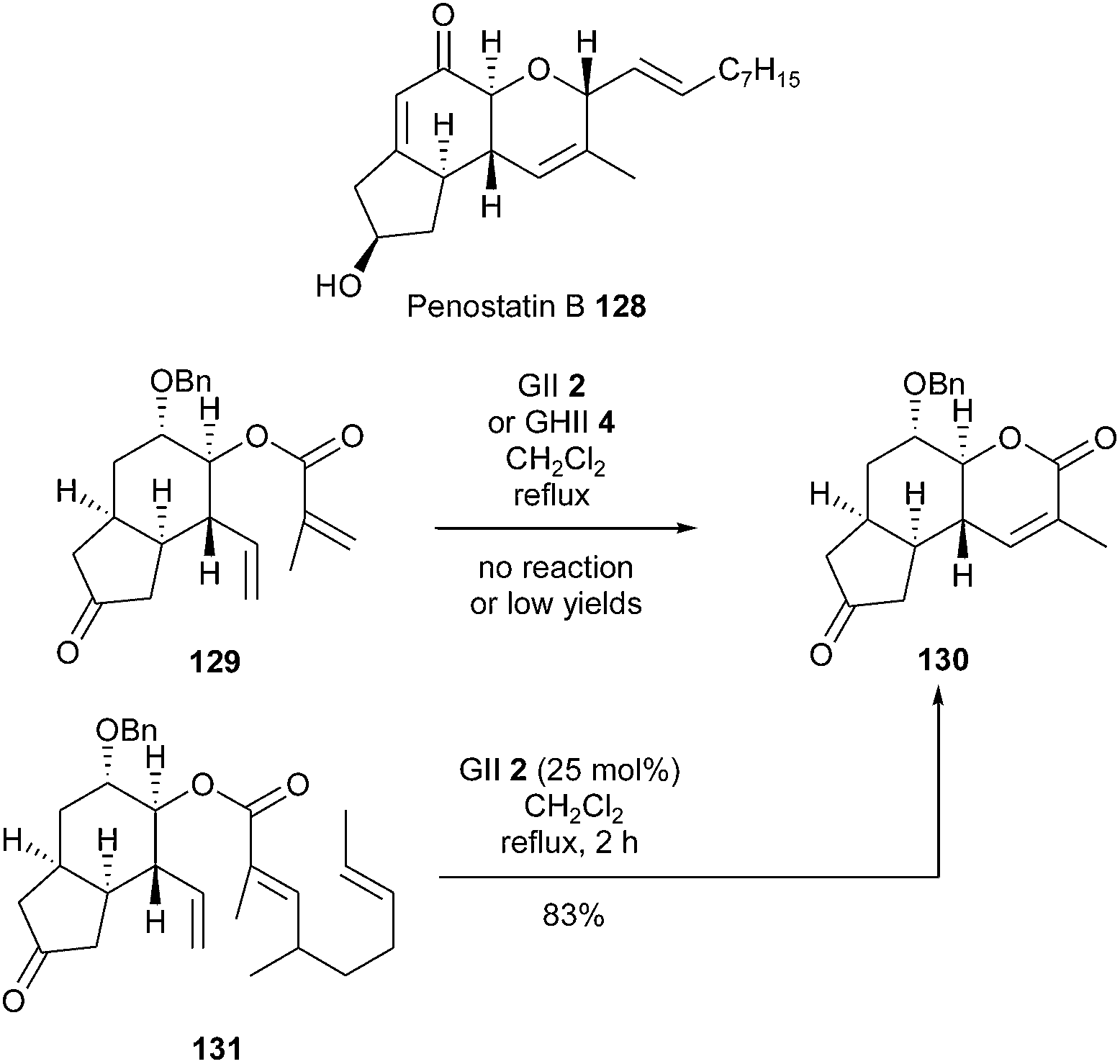 | ||
| Scheme 20 Shishido's RCM reactions for the synthesis of penostatin B 128. | ||
(+)-Dihydro-epi-deoxyarteannuin B 132, a natural product isolated from a species of Artemisia, plays a central role in efforts to develop synthetic routes to semisynthetic artemisinin. Dudley et al. reported a total synthesis of this compound in 2007.40 In a first approach, it was shown that all attempts to accomplish a RCM reaction at the late-stage of the synthesis, i.e. to transform the bicyclic diene 133 into spirolactone 132, were unsuccessful. The authors proposed that, in addition to steric congestion, the alkene 132 may be significantly more strained than the fused bicyclic substrate 133. Interestingly, the conversion of reduced compound 134 into decalin 135 was effective, albeit in low yield (37%). Final improvement was achieved through application of RRCM strategy, the spiro tricyclic system 132 was thus obtained in a good 77% yield from tethered triene 136, in the presence of GII catalyst 2 (Scheme 21).
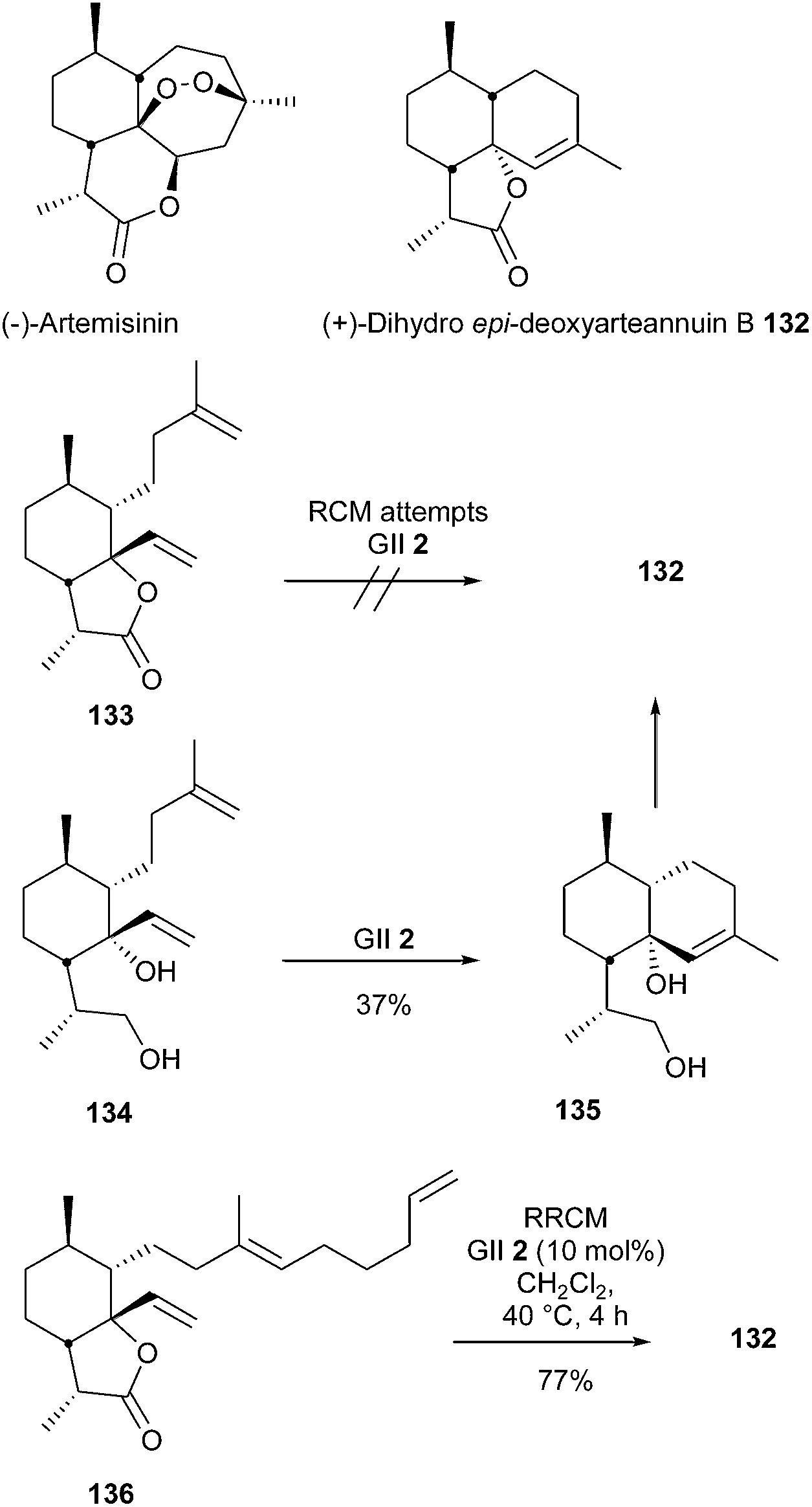 | ||
| Scheme 21 Dudley's RCM reactions for the synthesis of dihydro-epi-deoxyarteannuin B 132. | ||
3.3 Tandem or sequential RCM, RRCM, ROM, CM
The strategies implying tandem or sequential RCM, RRCM, ROM or CM reactions are always very elegant but remain sometimes problematic to carry out.Stemaphylline 137 and stemaphylline N-oxide 138 are two recently disclosed Stemona alkaloids featuring the characteristic central pyrrolo[1,2]azepine core linked to a terminal lactone moiety. In 2013, Lindsley et al. completed the first total synthesis of these natural products through a tandem bis-RCM strategy to construct the azepine ring and γ-lactone.41 Reactivity of tetraene C9a-epi139 was first explored. Acid additives are often added to RCM reaction of substrates bearing an amino function to avoid coordination of the amine to the ruthenium atom and subsequent deactivation of the catalyst. Besides, catalyst Gre 712 has been reported as a Ru complex with improved reactivity, able to perform challenging RCM reactions under mild conditions. Thus, in the presence of CSA and catalyst Gre 7, the desired bis-ring closure product C9a-epi141 was isolated in 52% yield via azepine C9a-epi140 as pyrrolo-azepine intermediate. Actually, Gre 7 and GHII 4 catalysts were found to exhibit comparable activity in this reaction and GHII 4 was finally selected for this transformation in the natural series.
Towards the synthesis of stemaphylline 137, pyrrolidine tetraene 139 was next subjected to these conditions. However, the ring closure proceeded more slowly, and a mixture of bis-ring closure products 141 and C12-epi141 was isolated. The addition of 1,4-benzoquinone, previously reported to suppress undesirable olefin migration, did not modify this result. To elucidate at which stage the epimerization was occurring, the reaction was stopped after closure of the azepine ring. The isolation of mono-ring closure compound 140, as a single product, clearly demonstrated that formation of the γ-lactone was the problematic step.
Molecular modelling revealed that tetraene C9a-epi139 possesses an intramolecular hydrogen bond which could preorganize the substrate for RCM, whereas in contrast, conformation of tetraene 139 is more disordered. Moreover, although the two olefins in azepine intermediate C9a-epi140 are in close proximity, the reacting olefins in 140 are pointed away from one another.
Consequently, the authors investigated a relay ring-closing metathesis (RRCM) strategy to improve both the reactivity and the selectivity. Reaction of RRCM substrate 142, in the presence of CSA and then GHII catalyst 4 (20 mol%), in refluxing toluene for 30 min, provided the desired RCM product 141 in 48% yield with only minor epimerization at C12 (dr 91:9) (Scheme 22).
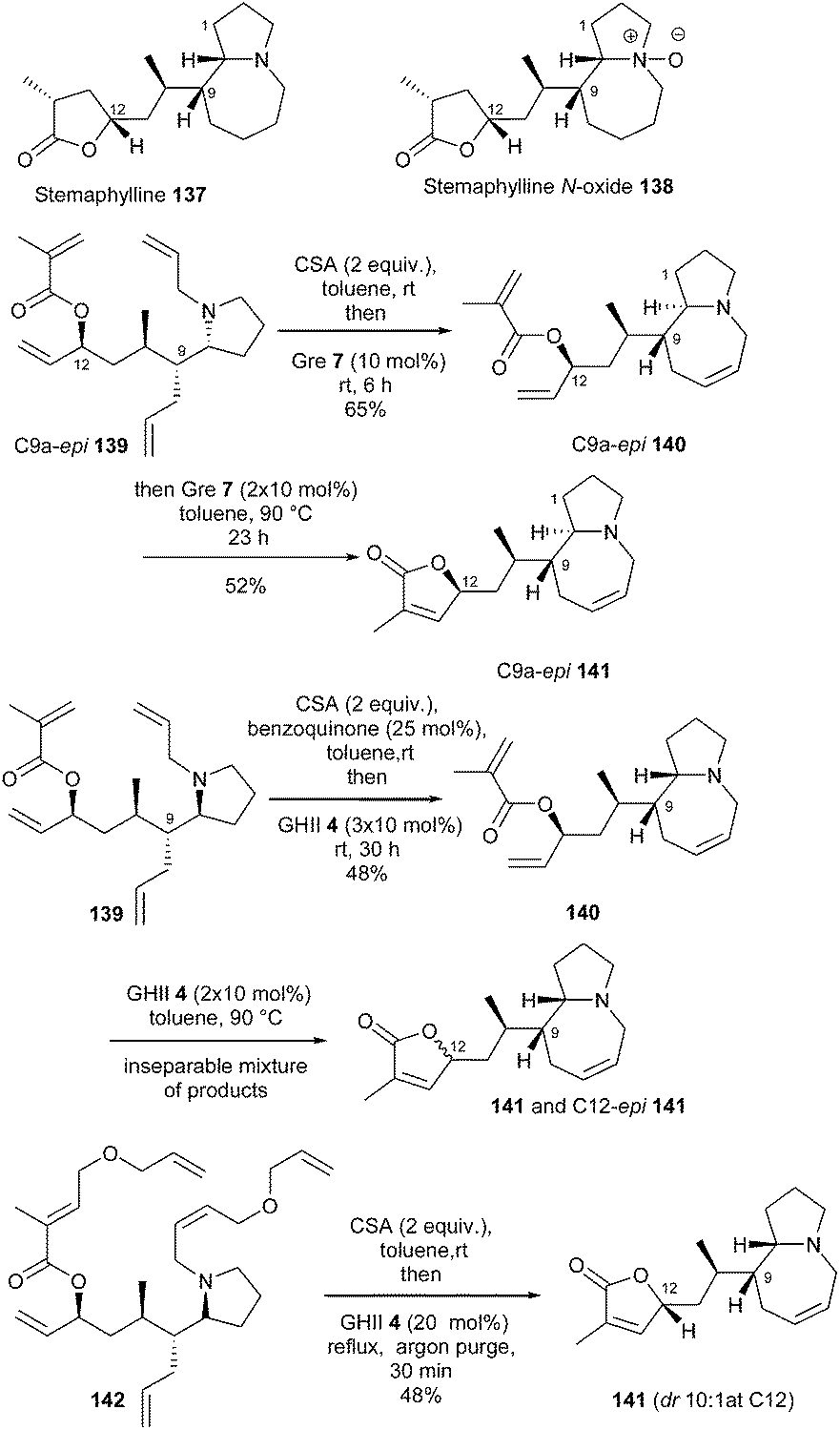 | ||
| Scheme 22 Lindsley's RCM reactions for the synthesis of stemaphylline 137 and stemaphylline N-oxide 138. | ||
The cyanthiwigins, isolated from marine sponges, constitute part of a larger class of diterpene natural products called cyathanes, which display a vast array of biological properties. In 2011, Stoltz et al. described a first generation total synthesis of three cyanthiwigins.42 The synthetic route towards cyanthiwigin B 143 employed an elegant tandem one-pot RCM/cross-metathesis (CM) reaction for the construction of the seven-membered cyathane C-ring. In initial studies, it was proved that the ring-forming process towards 146 was both faster and higher-yielding when GHIITol 9 was employed instead of GHII catalyst 4. Successive addition of GHIITol catalyst 9 (30 min, benzene, 40 °C) and excess vinylboronate 145 (5 equiv., 20 h) to tetraolefin 144, followed by an oxidative work-up, delivered the desired bicyclic aldehyde 147, via146, in 51% yield (Scheme 23).
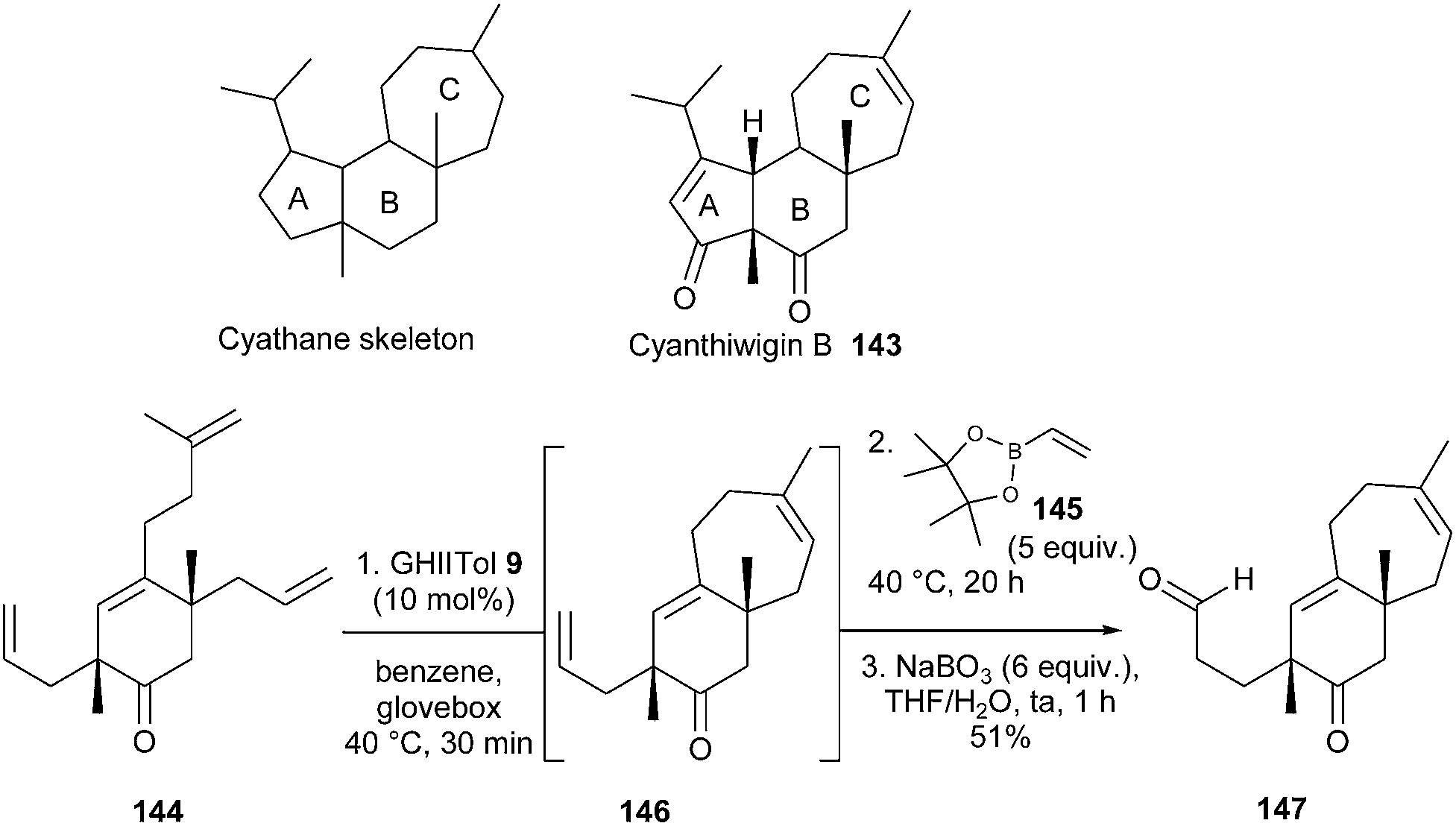 | ||
| Scheme 23 Stoltz's RCM reaction for synthesis of cyanthiwigin B 143. | ||
Ring-rearrangement metathesis (RRM) reactions, involving multiple metathesis processes such as ring-opening (ROM)/RCM, in a one-pot operation, allow the synthesis of complex frameworks difficult to elaborate by conventional methods.3j
In 2005, towards total synthesis of (+)-cyanthiwigin U 148, Phillips reported a two-directional tandem ROM/RCM sequence which could be considered as a cornerstone in RRCM reaction.43 Treatment of bis-enone 150 (elaborated from 149) with GHII 4 catalyst (3 × 7 mol%) in refluxing toluene under an ethylene atmosphere allowed to directly afford the tricycle 151 in 43% yield. Several pathways were proposed: initial ROM (leading to A or B) and subsequent RCM or initial MR between endo enone and C16–C14 olefin (C) followed by ROM (D) and RCM (Scheme 24).
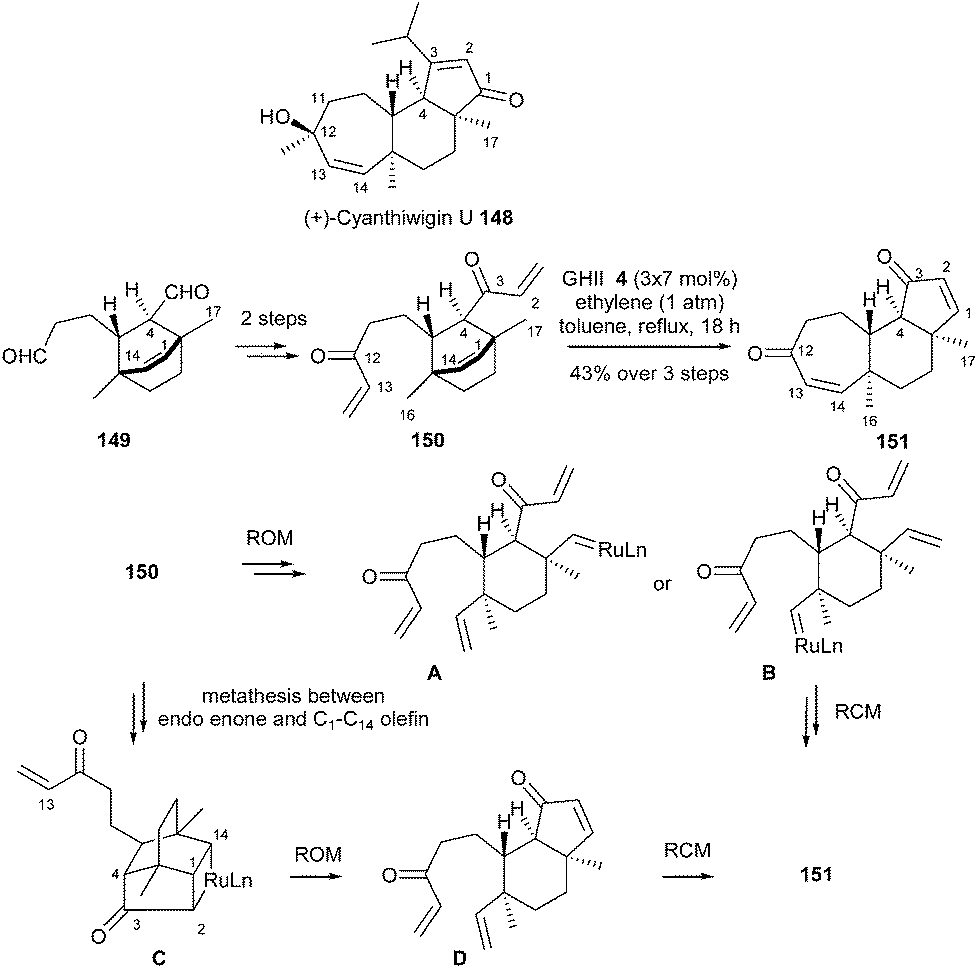 | ||
| Scheme 24 Phillips ROM/RCM reaction for synthesis of (+)-cyanthiwigin U 148. | ||
The humulanolides are a series of sesquiterpene lactones with unique structures. In 2014, Li et al. developed an elegant concise synthesis of several of these natural products, without the need of protecting groups.44 Notably, the 11-membered ring and bridged butenolide moieties in astericunolide D 152 were introduced in one step using a ROM/RCM cascade reaction. Practically, compound 153 readily underwent ROM/RCM cascade in the presence of GHII catalyst 4 (20 mol%) in refluxing toluene to afford astericunolide D 152 in 36% yield along with dimer 154 (7%). The driving force of this reaction was the opening of the highly strained four-membered ring. It was proposed that the ruthenium catalyst preferentially reacted with the monosubstituted terminal olefin of 153 to generate ruthenium carbene complex 155. Subsequent ROM with the strained cyclobutene then afforded butenolide 157via156. Finally, RCM would provide the 11-membered ring of 152. This reaction allowed the challenging formation of two sterically hindered, trisubstituted electron-deficient double bonds, by controlling the chemoselectivity of the transformation from the four olefins in starting material 153. Furthermore, this strategy offered a straightforward access to a library of compounds since astericunolide D 152 was easily converted into a series of other astericunolides (Scheme 25).
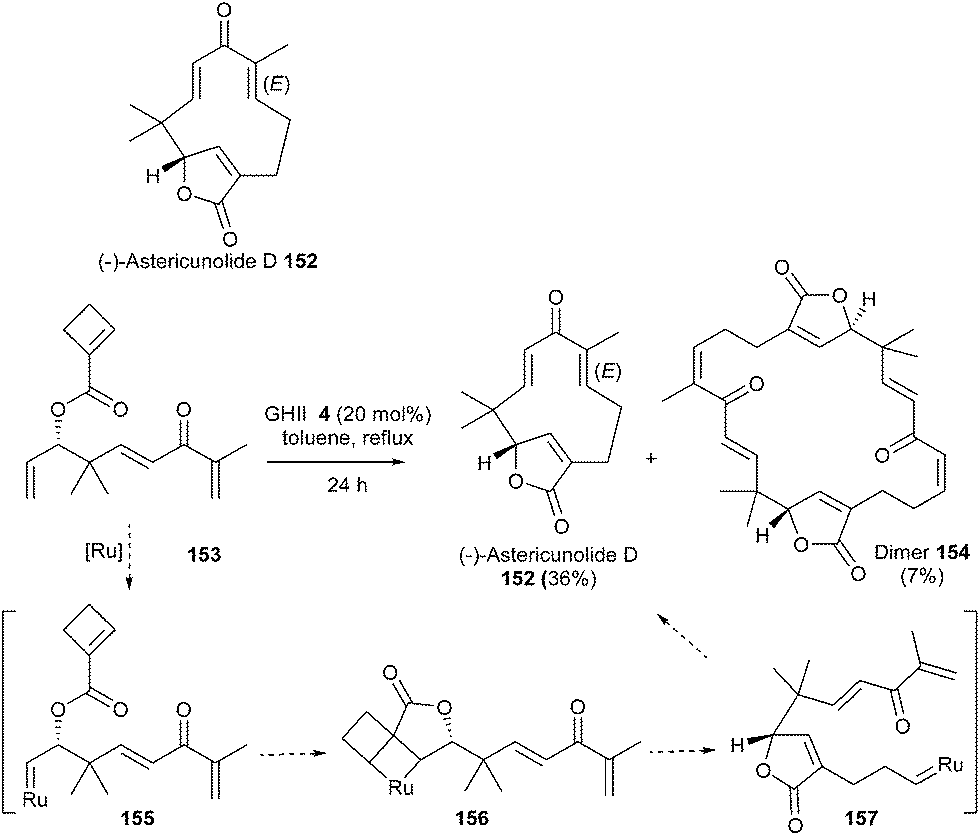 | ||
| Scheme 25 Li's ROM/RCM reaction for synthesis of astericunolide D 152. | ||
4 Challenging RCM approaches towards tetrasubstituted alkenes
RCM reactions leading to tetrasubstituted alkenes are more sterically demanding and often require more specific catalysts such as GHIITol 9.In 2014, Jones et al. accomplished the synthesis of emetine analogue NSC-134754 (±)-159, a potent inhibitor of the Hypoxia-Inductible Factor (HIF) pathway, and concomitantly reassigned its structure.45 Initially, the preparation of a compound possessing the previously reported structure (±)-158 of this analogue46 was achieved. The synthesis included a RCM reaction to build the tetrasubstituted double bond of the key tricyclic intermediate (±)-161. Several catalysts were screened and GHIITol 9 was found to efficiently catalyze this transformation in good yield from diene (±)-160, along with the corresponding aldehyde as a side product (10%). Therefore, this aldehyde was quantitatively reduced to the corresponding alcohol (±)-161, leading to an overall yield of 75%. Nevertheless, both diastereomers of compound (±)-158 were biologically inactive. This led to the reassignment of the structure of NSC-134754 and subsequent development of the new compound (±)-159 using the formerly developed approach involving the RCM transformation of diene (±)-162 into (±)-163. One of the diastereomers of (±)-159 was shown to possess HIF inhibitory properties (Scheme 26).
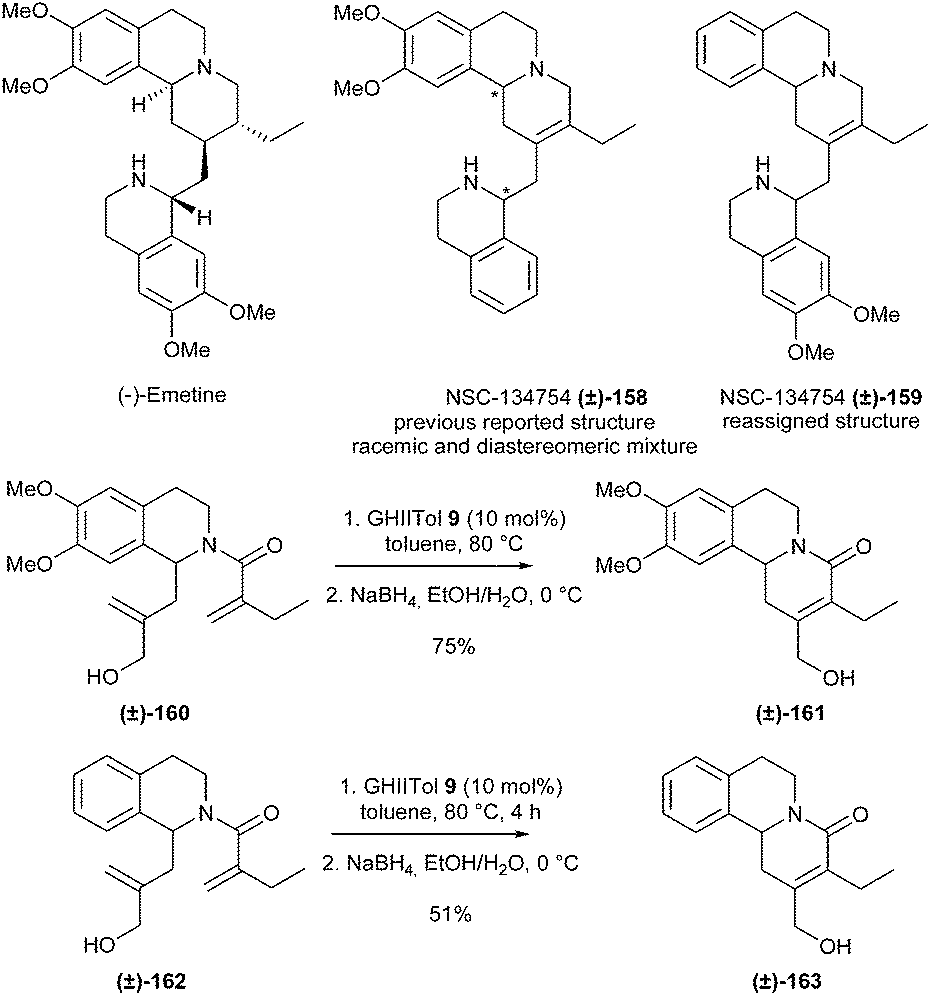 | ||
| Scheme 26 Jones' RCM reactions towards the total synthesis of NSC-134754 159. | ||
The withanolides comprise a family of more than 300 plant-derived steroids possessing diverse bioactivities. Many withanolides contain a trans-hydrindane motif connected to an α,β-unsaturated δ-lactone as a common structural moiety. In general, the bioactivity is encoded in this common part and is further modulated by different substituents.
In this context, the whitanolides sominone 164 and its derivative denosomin 165 could be considered as promising candidates for antidementia therapeutic agents, in light of their high potential as reconstructing agents of neuronal networks. Matsuya et al. achieved the synthesis of these compounds by a strategy relying on a RCM reaction for the construction of the δ-lactone side chain.47 For this purpose, an initial survey was performed using several Ru-based catalysts GII 2, GHII 4, GIITol 8 and GHIITol 9 and it was observed that the cyclization yields were significantly improved with catalyst GHIITol 9 in toluene at 80 °C. From substrate 168, lactone 165, possessing a trisubstituted olefin, was obtained in a satisfying yield of 88% [98% yield based on recovered starting material (brsm)]. Nevertheless, when attempted on compound 166 with the aim of forming a tetrasubstituted olefin, lactone 167 was only synthesized in a low 22% yield after 2 h (92% yield brsm). A recycling process was considered to be better suited for this RCM transformation than longer reaction times (Scheme 27).
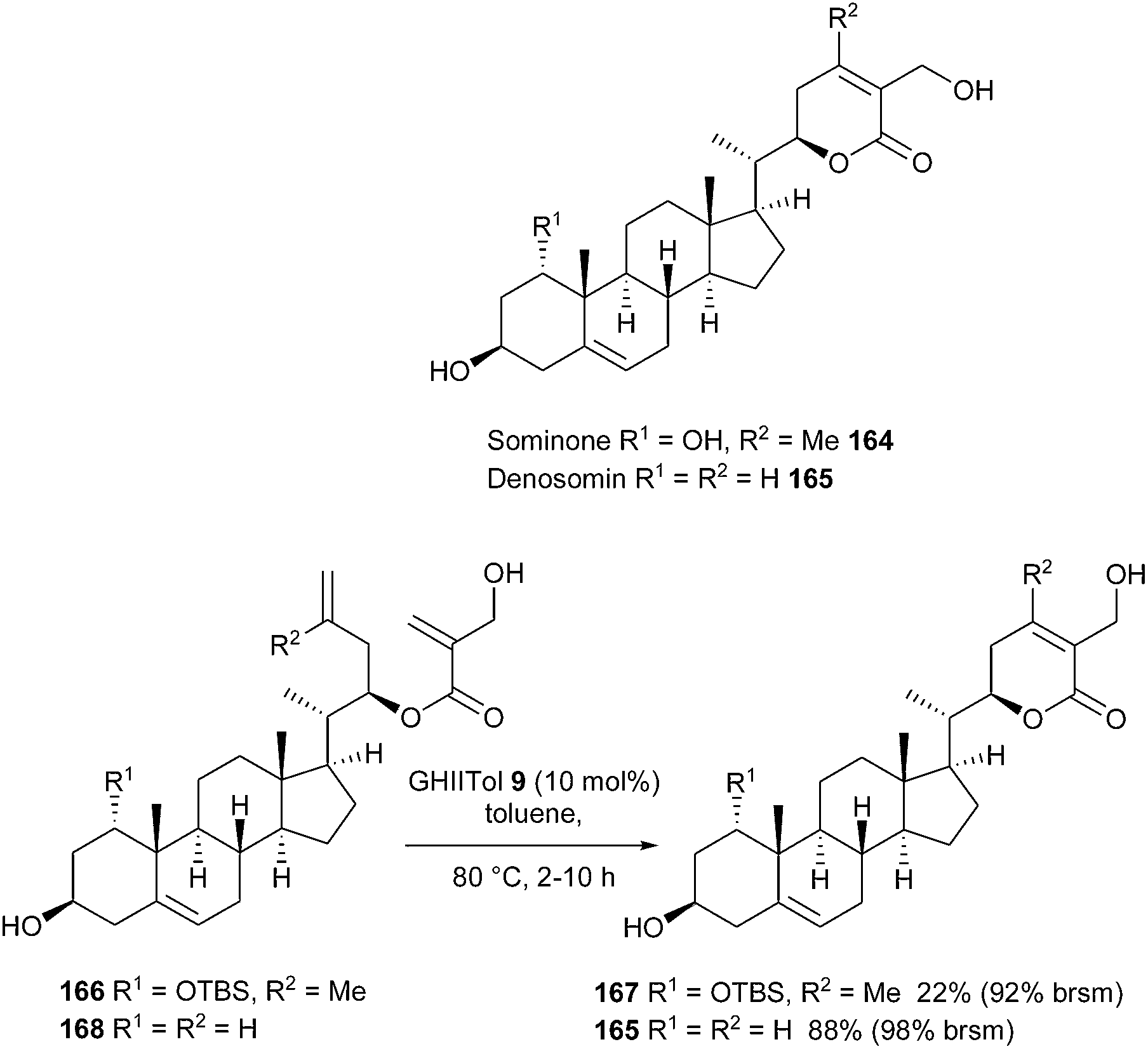 | ||
| Scheme 27 Matsuya's RCM reactions for the synthesis of sominone 164 and denosomin 165. | ||
Similarly, in 2016, Taylor et al. initiated a research program to prepare a wide range of novel steroid analogues derived from the withanolides, using readily available steroids as starting materials.48 In particular, synthesis of the typical α,β-dimethyl- α,β-unsaturated lactone 170 was investigated through RCM reaction from acrylate 169. However, the steric hindrance significantly affected the efficiency of the reaction, since treatment with GHII 4 added in two portions at 24 h interval, in toluene at 120 °C, provided lactone 170 in a disappointing 24% yield (Scheme 28). The authors did not report studies in the presence of other catalysts and particularly, the use of GHIITol 9 could have been relevant in this case.
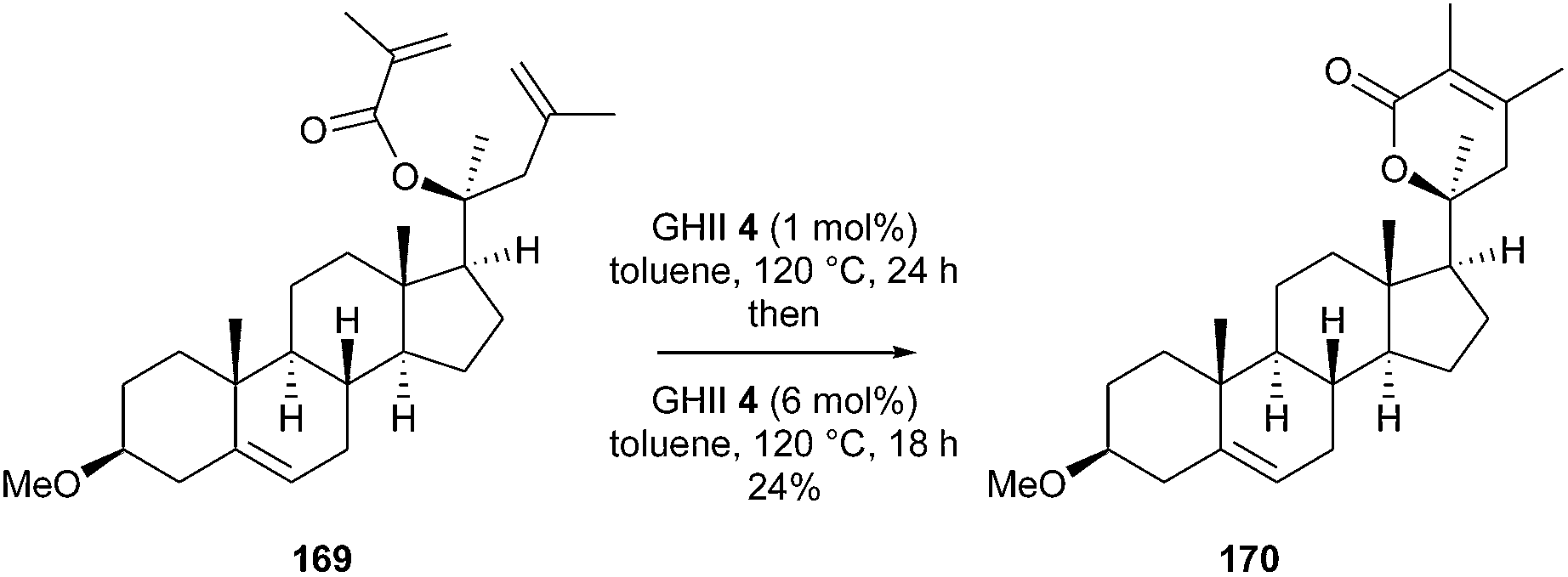 | ||
| Scheme 28 Taylor’ RCM reactions for the synthesis of withanolide-inspired coumpound 170. | ||
Grubbs' investigations on the use of GII catalyst 2 in RCM reactions have led to a general method for the synthesis of coumarins.49 This method allowed for a very convenient access to a variety of such bicyclic systems including either a di- or a trisubstituted olefin (with for instance, conversion of 171 into 172). Nevertheless, in the case of elaboration of the tetrasubstituted coumarin 174 from diene 173, only moderate yield was observed (45%, 99% brsm) although a higher catalyst loading was employed (10 mol% instead of 5 mol%) (Scheme 29).
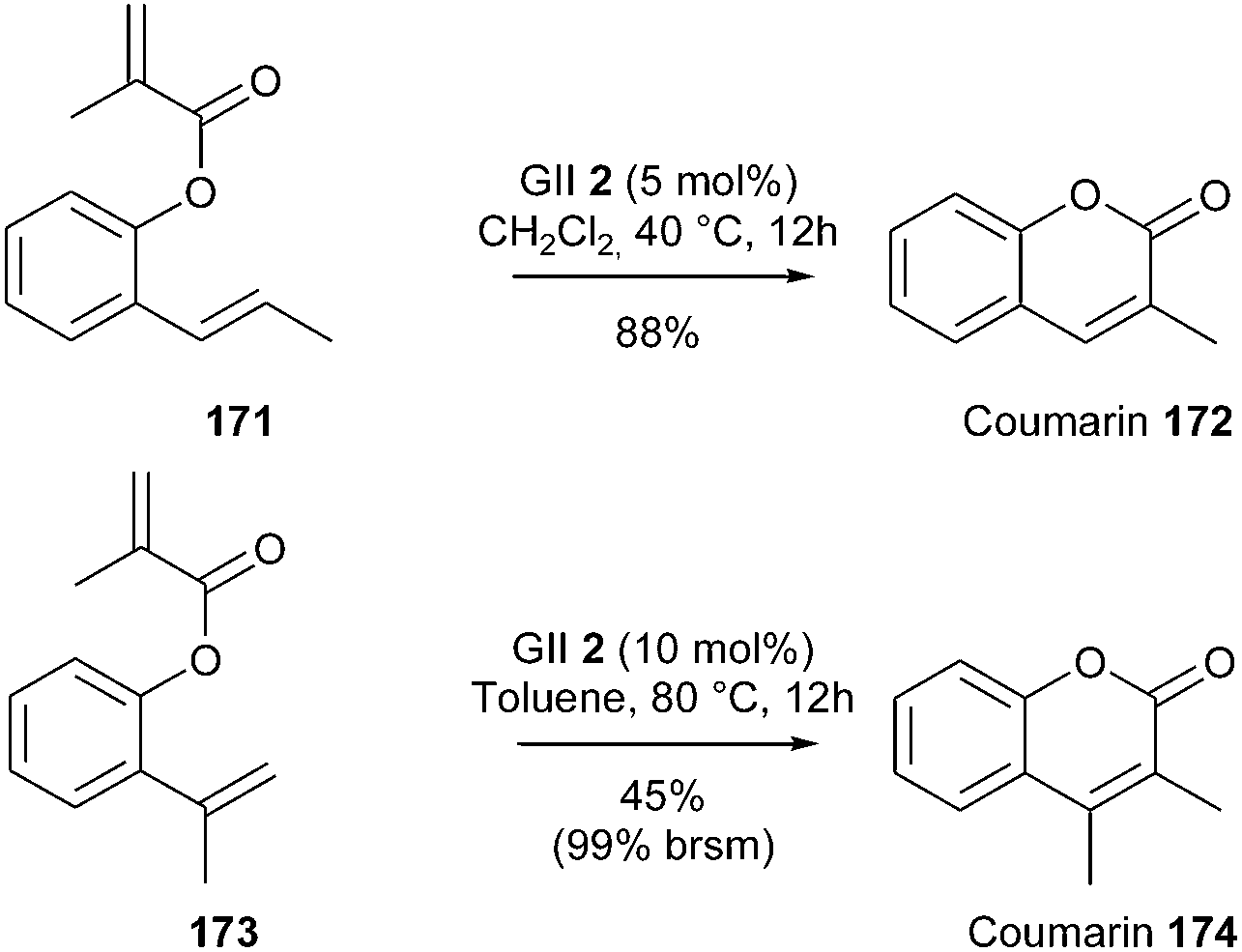 | ||
| Scheme 29 Grubbs' RCM reactions for the synthesis of coumarins. | ||
Maculalactone A 176, isolated from a cyanobacterium, constitutes a promising antifouling agent. A synthesis of this butenolide, based on a late stage RCM reaction, was envisaged by Gademann et al.50 Nevertheless, (±)-maculalactone A 176 was obtained from diene 175 in a low conversion of 11%, in the presence of GII catalyst 2 and Ti(OiPr)4. The potential activity of catalysts GHII 4 or GHIITol 9 was not described. Another strategic approach involving a base-catalysed cyclisation was finally undertaken and provided a successful access to this target (Scheme 30).
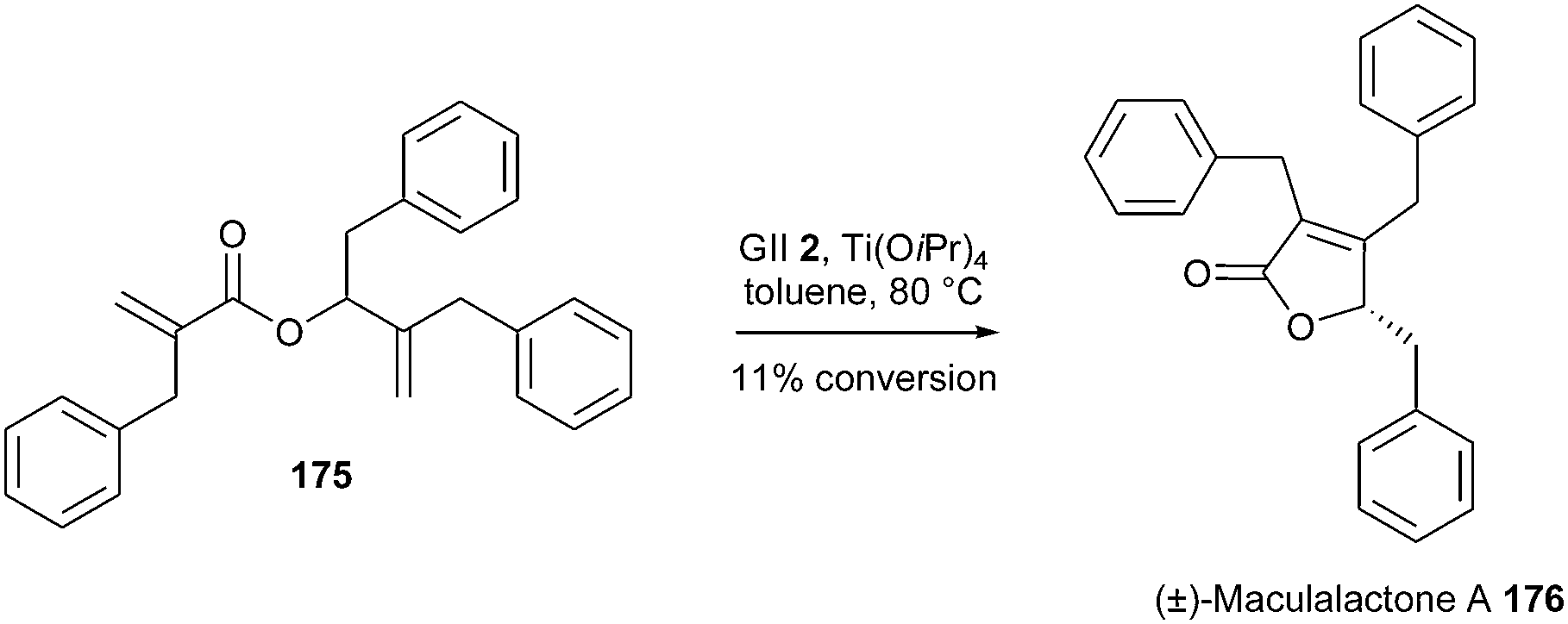 | ||
| Scheme 30 Gademann's RCM reaction for the synthesis of maculalactone A 176. | ||
In 2002, Meyers et al. described a synthetic approach towards the diterpenoids (+)-triptoquinone A 177 and its analogue (+)-triptinin A 178.51 The strategy involved a RCM reaction for the construction of the tricyclic ring systems 180 and 182. Initially, diene 179 containing a chiral Lewis basic oxazoline was treated with GI 1 catalyst (20 mol%), in dichloromethane at reflux, resulting in the formation of the cyclized compound 180 in good yield (90%). Construction of the appropriate tetrasubstituted olefin 182 from 181 was then considered. Noteworthy, in this case, the more reactive molybdenum Schrock catalyst 183 was found to be necessary to yield the desired product 182 in 90% yield.52 It should be noted that, as main drawback, this reaction required careful degassing (Scheme 31).
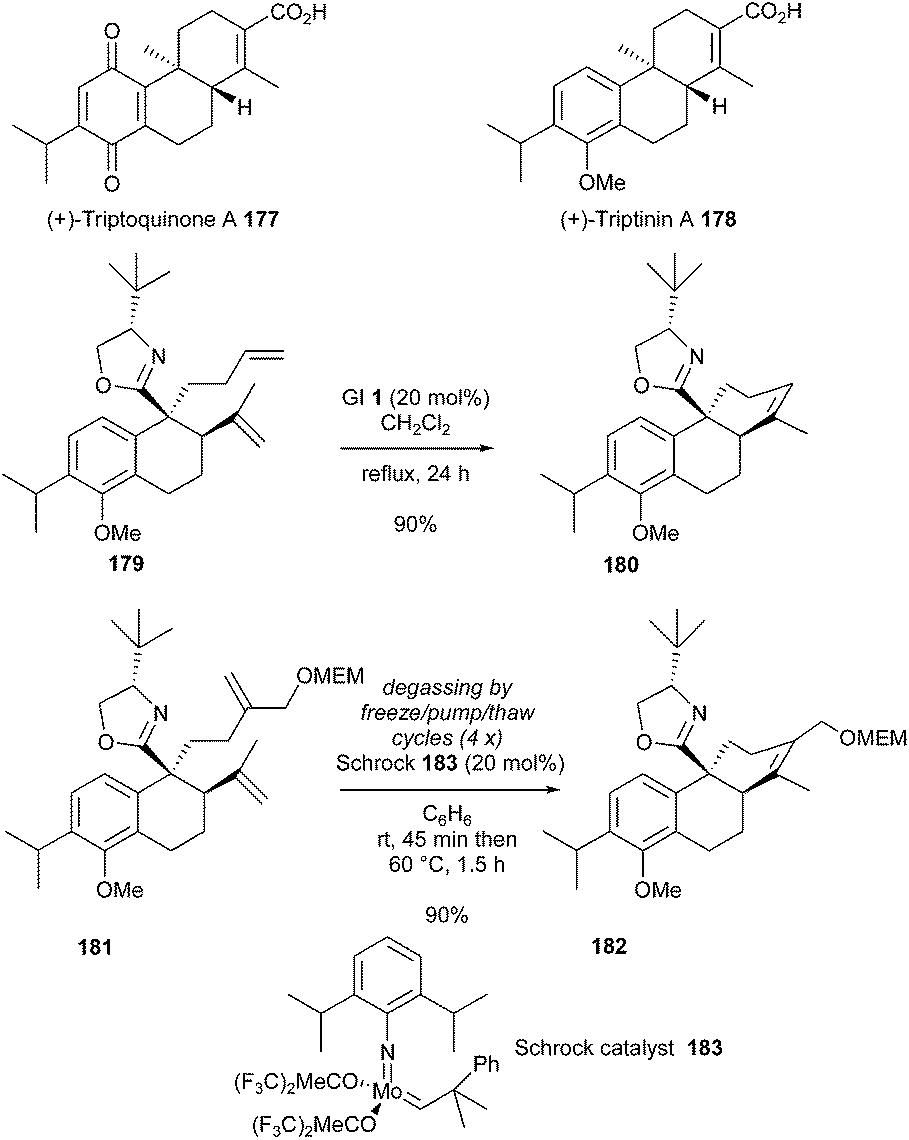 | ||
| Scheme 31 Meyers' RCM reactions for the synthesis of (+)-triptoquinone A 177 and (+)-triptinin A 178. | ||
Chamigrenes, a natural product family of sesquiterpenes with a diverse array of biological activity, are characterized by a spiro[5.5]undecane core. In 2010, Grubbs and Stoltz et al. developed a general synthetic strategy towards the challenging motifs of natural products 184, 185 and 186, based on a RCM reaction, to concomitantly elaborate the B ring tri- or tetra substituted olefin and the spirocyclic system.53 Preparation of elatol 184, one of the most biologically interesting and synthetically demanding chamigrenes, was the ultimate goal of the study. At first, however, they chose to focus on a simpler target, laurencenone B 185. GII and GHII complexes 2 and 4 provided moderate conversion of bis(olefin) (+)-187 to the desired tetrasubstituted olefin product (−)-188. Gratifyingly, catalyst GHIITol 9 afforded rapid and clean conversion delivering spirocycle (−)-188 in 97% yield. However, these RCM conditions proved ineffective when applied towards the synthesis of analogues 190 or 192 (with a contraction or an extension of the homoallyl chain) and 194 (devoid of dimethyl substitution) from respectively dienes (+)-189, (−)-191 and (+)-193 (Scheme 32).
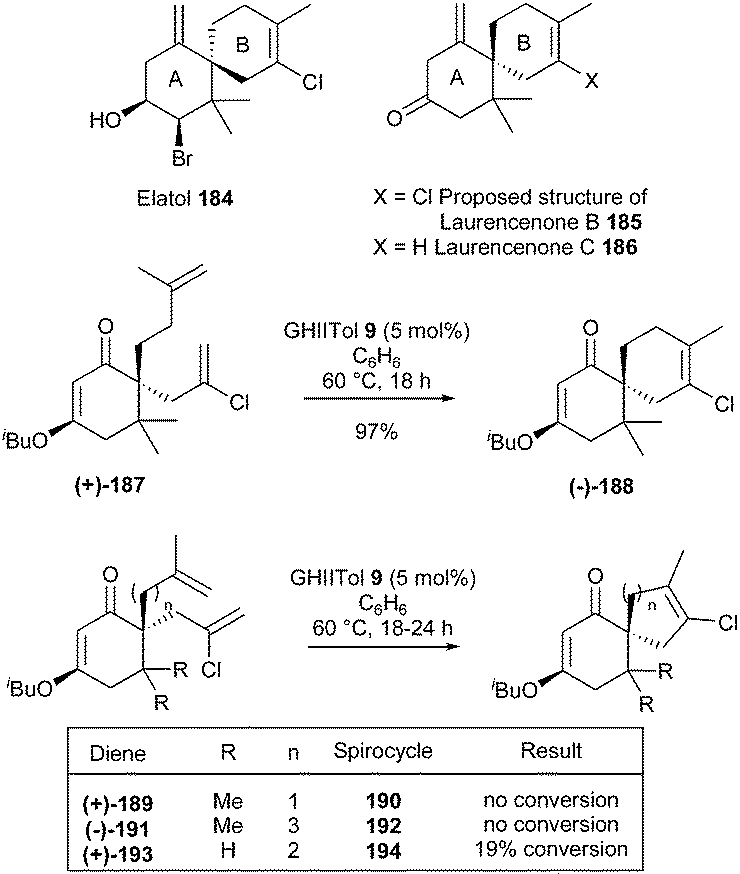 | ||
| Scheme 32 Grubbs and Stoltz' RCM reactions for the synthesis of laurencenone B 185 and elatol 184. | ||
A concise synthesis of (−)-aplysin 195, a sesquiterpene isolated from mollusc species aplysia and red algal laurencia, has been completed by Aggarwall et al.54 A challenging RCM reaction was envisioned to form the key core intermediate 197, comprising a tetrasubstituted alkene adjacent to a quaternary center. Regardless of the conditions used, only a partial conversion of the precursor 196 was observed in the presence of GII catalyst 2. Alternatively, switching to GHII 4 (10 mol% in refluxing toluene for 24 h) afforded olefin 197 in quantitative yield (Scheme 33).
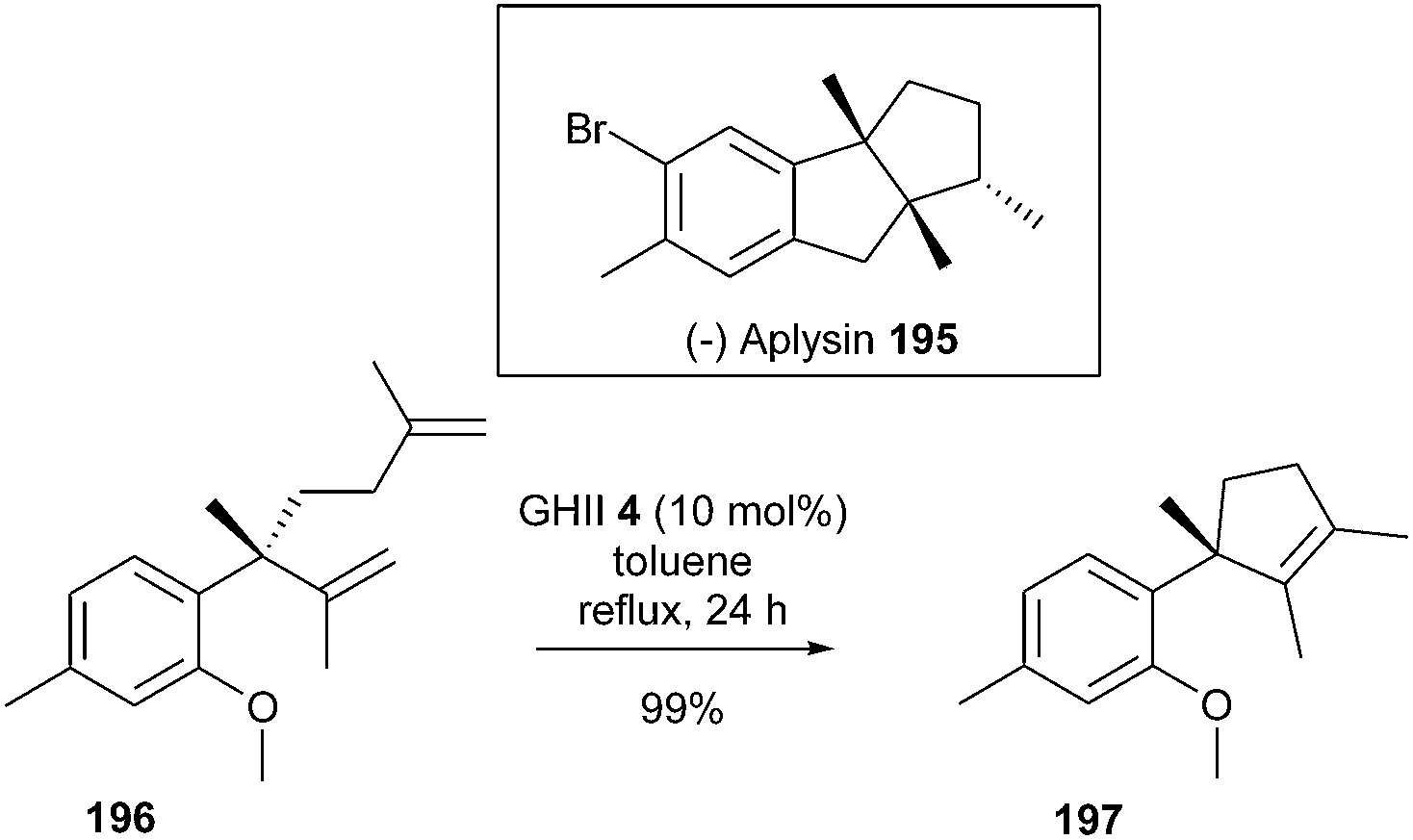 | ||
| Scheme 33 Aggarwall's RCM reaction for the synthesis of (−)-aplysin 195. | ||
One of the key features of the elaboration of spirotenuipesines A and B 198 and 199, reported by Danishefsky et al. in 2008, was the synthesis of tetrasubstituted cyclopentenyl system 201 by RCM.55 When diene 200 was subjected to slow addition of the GII catalyst 2, in benzene at reflux, the desired compound 201 was isolated in 82% yield (Scheme 34).
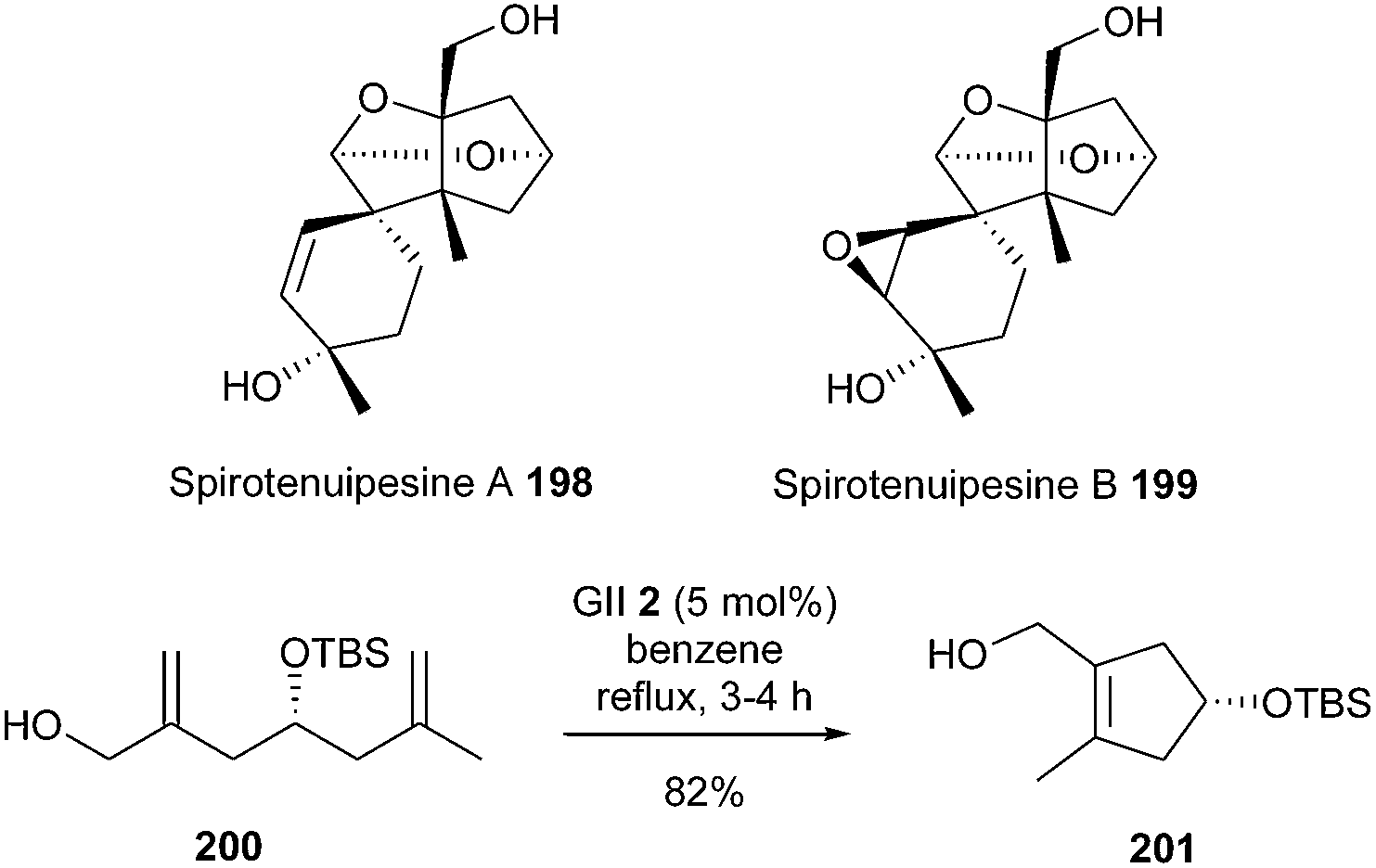 | ||
| Scheme 34 Danishefsky's RCM reaction for the synthesis of spirotenuipesines A and B 198 and 199. | ||
Synthesis of furanic-estrane derivatives, inhibitors of 17β-hydroxysteroid dehydrogenase which is one of the steroidogenic enzymes involved in the biosynthesis of estradiol, was reported by Poirier et al. in 2011.56 The synthetic sequence, involved a GII 2 catalyzed RCM reaction of intermediate 202 and subsequent catalytic hydrogenation of the tetrasubstituted double bond as key steps from a derivative of estradiol 203 (Scheme 35).
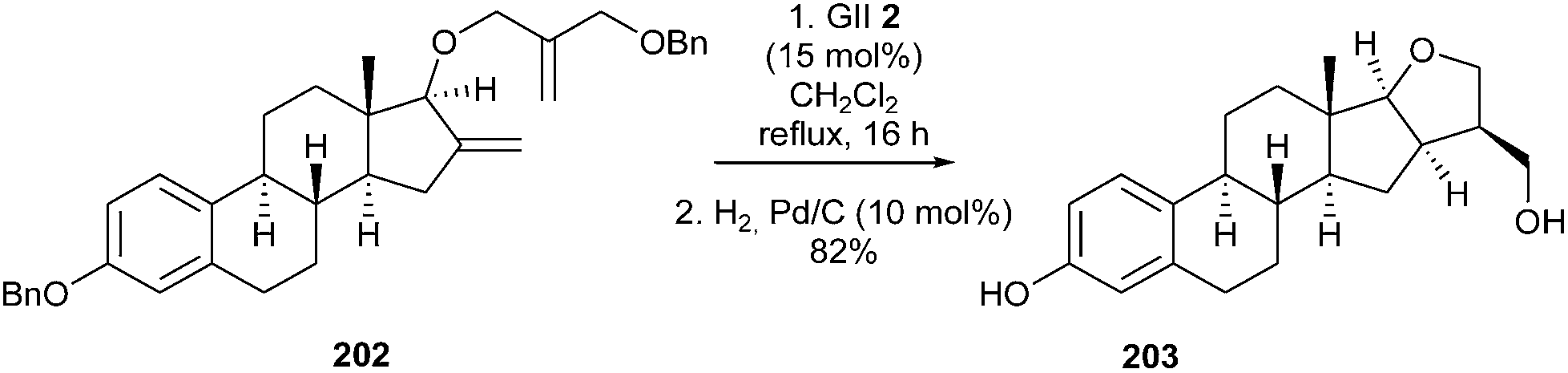 | ||
| Scheme 35 Poirier's RCM reaction for the synthesis of 203. | ||
Arglabin 204 is a guaianolide which activates the RAS proto-oncogene, a process which would play a pivotal role in 20–30% of all human tumors. In 2007, Reiser et al. developed the first enantioselective synthesis of this tricyclic system 204.57 The strategy involved a final challenging RCM reaction, requiring the formation of a tetrasubstituted double bond within a medium-size seven-membered ring. Nevertheless, the combination of GII catalyst 2 (15 mol%, added in three portions) and inert-gas (Ar) sparging at 95 °C for 6 h, allowed the formation of the tricyclic compound 206, precursor of arglabin, in 86% yield from diene 205 (Scheme 35).
Surprisingly, preliminary work reported in 2003, revealed that model diene 207 could not be cyclized into the tricyclic lactone 208 in the presence of GII catalyst 2; in this case, GHII catalyst 4 (2 × 5 mol%) was needed and a moderate yield of 208 was observed (48%).58 In the meantime, Reiser et al. demonstrated that, combining inert gas sparging to purge off ethylene along with microwave irradiation, allowed for a real optimization of this type of RCM reactions.59 Thus, under these conditions, conversion of the TES-protected diene 209 into the tricyclic system 210 was achieved in almost quantitative yield in the presence of GII catalyst 2 (Scheme 36).
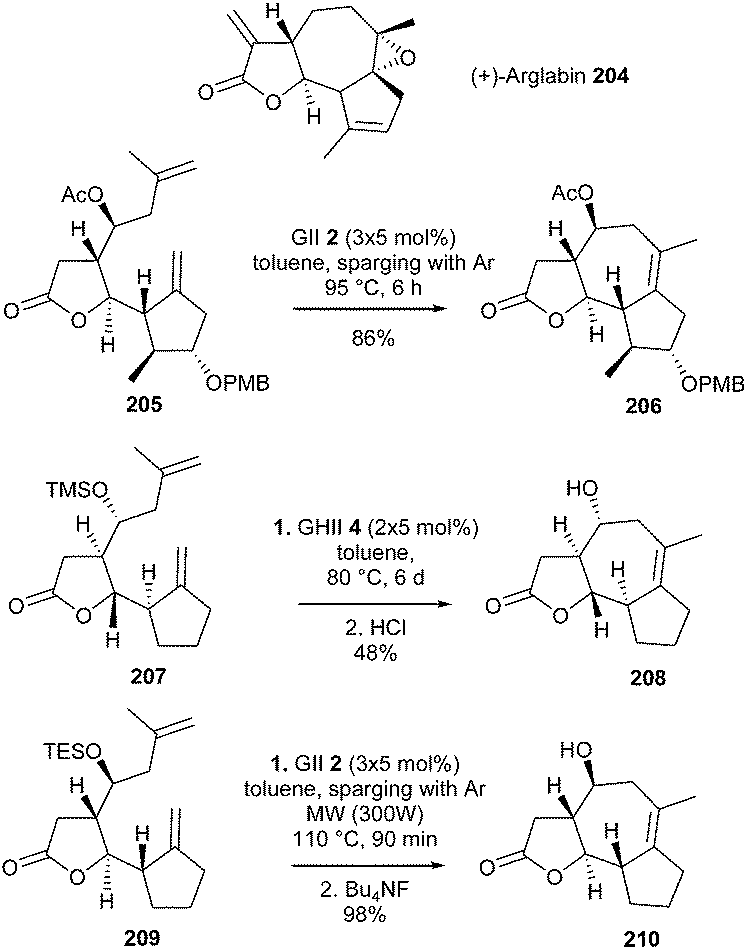 | ||
| Scheme 36 Reiser's RCM reactions for the synthesis of arglabin 204. | ||
Harmata et al., in 2014, demonstrated the possibility of generating functionalized cyclic systems bearing alkenyl boron species through RCM metathesis.60 Thus, diene 212 was transformed into the seven-membered ring 213 in 80% yield, using GHII catalyst 4 (10 mol%) in toluene at 110 °C for 24 h. This compound was readily converted into the polycycle 214 possessing the ABC ring substructure of ingenol 211 in a few steps (Scheme 37).
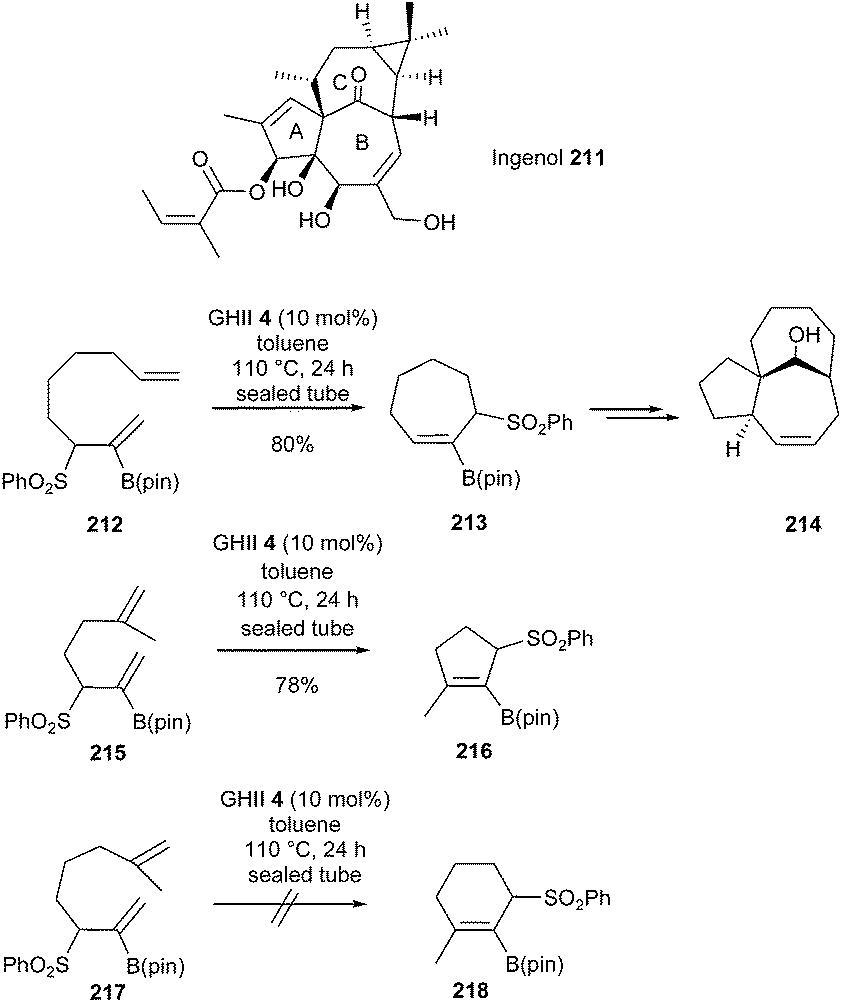 | ||
| Scheme 37 Harmata's RCM reactions towards synthesis of ingenol 211. | ||
Under similar conditions, formation of cyclopentene 216 comprising a tetrasubstituted double bond was possible from diene 215; however, curiously, synthesis of the corresponding cyclohexene 218 from 217 failed.
5. Conclusions
This review reported selected examples of RCM reactions towards the synthesis of sterically hindered olefins included in complex natural products.The RCM reaction has taken an important place in organic synthesis to generate carbo- and heterocycles, therefore, many catalysts have been specifically designed to carry out these reactions.
However, the RCM reaction of sterically hindered olefins still represents a great challenge. Construction of these motifs is often planned at a late stage of the synthetic route on already highly elaborated fragments and an important optimization at this point is challenging. The generally used commercial catalysts, GII 2, GHII 4, GHIITol 9 and occasionally IndII 6, have allowed for a significant number of particularly elegant synthetic achievements in the field of complex and strained natural products synthesis. Moreover, the wide variety of reaction conditions that has been reported constitutes another vast domain of improvement for the RCM of congested systems. Among the most significant parameters that can be either tuned separately or in combination, it is worth mentioning: the dilution, the addition mode of the catalyst (portion wise or slow addition), heating under microwave irradiation and the use of fluorinated aromatic solvents.
Finally, in recent years, RCM reaction of sterically hindered olefins has enabled a powerful access to even more crowded systems. Application to the synthesis of natural products, although widely developed, could still be optimized. Hence, nowadays, the use of non-commercial or difficult to synthesize catalysts, albeit highly suitable for congested systems, remains generally limited to methodological survey.
Conflicts of interest
There are no conflicts to declare.6 Acknowledgements
We thank ANR (AMICAL project, ANR-BLAN-2013/SIMI 7/Agreement: ANR-13-BS07-0011-01) for doctoral fellowship for C. L. We thank Dr C. Meyer for careful reading of the article and constructive comments.7. Notes and references
- P. A. Wender and B. L. Miller, Nature, 2009, 460, 197 CrossRef CAS PubMed.
- For recent general reviews about metathesis reactions, see: (a) A. M. Lozano-Vila, S. Monsaert, A. Bajek and F. Verpoort, Chem. Rev., 2010, 110, 4865 CrossRef CAS PubMed; (b) G. C. Vougioukalakis and R. H. Grubbs, Chem. Rev., 2010, 110, 1746 CrossRef CAS PubMed; (c) D. J. Nelson, S. Manzini, C. A. Urbina-Blanco and S. P. Nolan, Chem. Commun., 2014, 50, 10355 RSC; (d) A. H. Hoveyda, J. Org. Chem., 2014, 79, 4763 CrossRef CAS PubMed; (e) S. Fustero, A. Simón-Fuentes, P. Barrio and G. Haufe, Chem. Rev., 2015, 115, 871 CrossRef CAS PubMed.
- For recent reviews about natural products RCM reactions, see: (a) Total synthesis: A. Fürstner, Chem. Commun., 2011, 47, 6505 RSC; (b) Macrocycles: A. Fürstner and K. Langemann, Synthesis, 1997, 792 CrossRef; (c) Five- and six-membered α,β-unsaturated lactones: M. Bassettia and A. D'Annibale, Curr. Org. Chem., 2013, 17, 2654 CrossRef; (d) Dihydrofuran and -pyran: R. Jacques, R. Pal, N. A. Parker, C. E. Sear, P. W. Smith, A. Ribaucourt and D. M. Hodgson, Org. Biomol. Chem., 2016, 14, 5875 RSC; (e) Butenolides, butyrolactones: B. Mao, M. Fañanás-Mastral and B. L. Feringa, Chem. Rev., 2017, 117, 10502 CrossRef CAS PubMed; (f) Pentenolides and pyranones: M. Špulák, M. Ghavre and M. Pour, Tetrahedron Lett., 2017, 58, 263 CrossRef; (g) Peptides: E. C. Gleeson, W. R. Jackson and A. J. Robinson, Tetrahedron Lett., 2016, 57, 4325 CrossRef CAS; (h) Constraining peptides: A. Glasa and T. N. Grossmann, Synlett, 2015, 26, 1 CrossRef; (i) Guaianolides: A. Santana, J. M. G. Molinillo and F. A. Macías, Eur. J. Org. Chem., 2015, 2093 CrossRef CAS; (j) Ring-rearrangement metathesis: S. Kotha, M. Meshram, P. Khedkar, S. Banerjee and D. Deodhar, Beilstein J. Org. Chem., 2015, 11, 1833 CrossRef CAS PubMed; (k) Conjugated dienes: À. Balla, M. Al-Hashimi, A. Hlil, H. S. Bazzi and R. Tuba, ChemCatChem, 2016, 8, 2865 CrossRef. For a recent book on metathesis reaction, see: (l) Metathesis in Natural Product Synthesis: Synthesis: Strategies, Substrates and Catalysts, ed. J. Cossy, S. Arseniyadis and C. Meyer, Wiley-VCH, Weinheim, Germany, 2010 Search PubMed.
- A. K. Chatterjee, T.-L. Choi, D. P. Sanders and R. H. Grubbs, J. Am. Chem. Soc., 2003, 125, 11360 CrossRef CAS PubMed.
- (a) L. H. Peeck, R. D. Savka and H. Plenio, Chem.–Eur. J., 2012, 18, 12845 CrossRef CAS PubMed; (b) V. Thiel, K.-J. Wannowius, C. Wolff, C. M. Thiele and H. Plenio, Chem.–Eur. J., 2013, 19, 16403 CrossRef CAS PubMed.
- M. Bieniek, A. Michrowska, D. L. Usanov and K. Grela, Chem.–Eur. J., 2008, 14, 806 CrossRef CAS PubMed.
- I. C. Stewart, T. Ung, A. A. Pletnev, J. M. Berlin, R. H. Grubbs and Y. Schrödi, Org. Lett., 2007, 9, 1589 CrossRef CAS PubMed.
- C. K. Chung and R. H. Grubbs, Org. Lett., 2008, 10, 2693 CrossRef CAS PubMed.
- (a) F. Grisi, A. Mariconda, C. Costabile, V. Bertolasi and P. Longo, Organometallics, 2009, 28, 4988 CrossRef CAS; (b) C. Costabile, A. Mariconda, L. Cavallo, P. Longo, V. Bertolasi, F. Ragone and F. Grisi, Chem.–Eur. J., 2011, 17, 8618 CrossRef CAS PubMed; (c) A. Perfetto, C. Costabile, P. Longo and F. Grisi, Organometallics, 2014, 33, 2747 CrossRef CAS.
- H. Clavier and S. P. Nolan, Chem.–Eur. J., 2007, 13, 8029 CrossRef CAS PubMed.
- C. Torborg, G. Szczepaniak, A. Zieliński, M. Malińska, K. Woźniak and K. Grela, Chem. Commun., 2013, 49, 3188 RSC.
- O. Songis, A. M. Z. Slawin and C. S. J. Cazin, Chem. Commun., 2012, 48, 1266 RSC.
- (a) K. Grela, S. Harutyunyan and A. Michrowska, Angew. Chem., Int. Ed., 2002, 41, 4038 CrossRef CAS; (b) A. Michrowska, R. Bujok, S. Harutyunyan, V. Sashuk, G. Dolgonos and K. Grela, J. Am. Chem. Soc., 2004, 126, 9318 CrossRef CAS PubMed.
- (a) D. Rix, F. Caijo, I. Laurent, F. Boeda, H. Clavier, S. P. Nolan and M. Mauduit, J. Org. Chem., 2008, 73, 4225 CrossRef CAS PubMed; (b) H. Clavier, F. Caijo, E. Borré, D. Rix, F. Boeda, S. P. Nolan and M. Mauduit, Eur. J. Org. Chem., 2009, 4254 CrossRef CAS.
- T. Vorfalt, S. Leuthußer and H. Plenio, Angew. Chem., Int. Ed., 2009, 48, 5191 CrossRef CAS PubMed.
- (a) V. Sashuk, L. H. Peeck and H. Plenio, Chem.–Eur. J., 2010, 16, 3983 CrossRef CAS PubMed; (b) L. H. Peeck and H. Plenio, Organometallics, 2010, 29, 2761 CrossRef CAS.
- (a) D. Rost, M. Porta, S. Gessler and S. Blechert, Tetrahedron Lett., 2008, 49, 5968 CrossRef CAS; (b) C. Samojłowicz, M. Bieniek, A. Zarecki, R. Kadyrov and K. Grela, Chem. Commun., 2008, 6282 RSC; (c) L. Vieille-Petit, X. Luan, M. Gatti, S. Blumentritt, A. Linden, H. Clavier, S. P. Nolan and R. Dorta, Chem. Commun., 2009, 3783 RSC.
- C. Samojłowicz, E. Borr, M. Mauduit and K. Grela, Adv. Synth. Catal., 2011, 353, 1993 CrossRef.
- A. D'Annibale and L. Ciaralli, J. Org. Chem., 2007, 72, 6067 CrossRef PubMed.
- L. Feng, Y. Liu, B. Hou, Z. Yuan, F.-C. Yu, T. Yan, Q. Qin, R. Ji, Y.-M. Li, Y. Shen and Z.-L. Zuo, Org. Biomol. Chem., 2016, 14, 10705 CAS.
- C. Lecourt, S. Boinapally, S. Dhambri, G. Boissonnat, C. Meyer, J. Cossy, F. Sautel, G. Massiot, J. Ardisson, G. Sorin and M.-I. Lannou, J. Org. Chem., 2016, 81, 12275 CrossRef CAS PubMed.
- R. A. Fernandes and V. P. Chavan, Chem. Commun., 2013, 49, 3354 RSC.
- M. Kim, S. Kang and Y. H. Rhee, Angew. Chem., Int. Ed., 2016, 55, 9733 CrossRef CAS PubMed.
- S. J. Malcolmson, S. J. Meek, E. S. Sattely, R. R. Schrock and A. H. Hoveyda, Nature, 2008, 456, 933 CrossRef CAS PubMed.
- H. Abe, A. Sato, T. Kobayashi and H. Ito, Org. Lett., 2013, 15, 1298 CrossRef CAS PubMed.
- M. Xiao, L. Wei, L. Li and Z. Xie, J. Org. Chem., 2014, 79, 2746 CrossRef CAS PubMed.
- W.-W. Ren, Z.-X. Chen, Q. Xiao, Y. Li, T.-W. Sun, Z.-Y. Zhang, Q.-D. Ye, F.-K. Meng, L. You, M.-Z. Zhao, L.-M. Xu, Y.-F. Tang, J.-H. Chen and Z. Yang, Chem.–Asian J., 2012, 7, 2341 CrossRef CAS PubMed.
- M. Scholl and R. H. Grubbs, Tetrahedron Lett., 1999, 40, 1425 CrossRef CAS.
- T. Ohyoshi, S. Funakubo, Y. Miyazawa, K. Niida, I. Hayakawa and H. Kigoshi, Angew. Chem., Int. Ed., 2012, 51, 4972 CrossRef CAS PubMed.
- D. Arican and R. Brückner, Org. Lett., 2013, 15, 2582 CrossRef CAS PubMed.
- J. Liu, S. D. Lotesta and E. J. Sorensen, Chem. Commun., 2011, 47, 1500 RSC.
- K. Tsuna, N. Noguchi and M. Nakada, Chem.–Eur. J., 2013, 19, 5476 CrossRef CAS PubMed.
- R. García-Fandiňo, M. J. Aldegunde, E. M. Codesido, L. Castedo and J. R. Granja, J. Org. Chem., 2005, 70, 8281 CrossRef PubMed.
- R. Dumeunier, T. Gregson, S. MacCormick, H. Omori and E. J. Thomas, Org. Biomol. Chem., 2017, 15, 2768 CAS.
- C. W. Wullschleger, J. Li, A. Edenharter and K.-H. Altmann, Synlett, 2016, 27, 2726 CrossRef CAS.
- P. E. Romero, W. E. Piers and R. McDonald, Angew. Chem., Int. Ed., 2004, 43, 6161 CrossRef CAS PubMed.
- S. Xu, D. Unabara, D. Uemura and H. Arimoto, Chem.–Asian J., 2014, 9, 367 CrossRef CAS PubMed.
- T. R. Hoye, C. S. Jeffrey, M. A. Tennakoon, J. Wang and H. Zhao, J. Am. Chem. Soc., 2004, 126, 10210 CrossRef CAS PubMed.
- K. Fujioka, H. Yokoe, M. Yoshida and K. Shishido, Org. Lett., 2012, 14, 244 CrossRef CAS PubMed.
- G. B. Dudley, D. A. Engel, I. Ghiviriga, H. Lam, K. W. C. Poon and J. A. Singletary, Org. Lett., 2007, 9, 2839 CrossRef CAS PubMed.
- M. L. Schulte, M. L. Turlington, S. S. Phatak, J. M. Harp, S. R. Stauffer and C. W. Lindsley, Chem.–Eur. J., 2013, 19, 11847 CrossRef CAS PubMed.
- (a) J. A. Enquist Jr, S. C. Virgil and B. M. Stoltz, Chem.–Eur. J., 2011, 17, 9957 CrossRef PubMed; (b) K. E. Kim and B. M. Stoltz, Org. Lett., 2016, 18, 5720 CrossRef CAS PubMed; (c) J. A. Enquist Jr and B. M. Stoltz, Nature, 2008, 453, 1228 CrossRef PubMed.
- M. W. B. Pfeiffer and A. J. Phillips, J. Am. Chem. Soc., 2005, 127, 5334 CrossRef CAS PubMed.
- J.-c. Han, F. Li and C.-c. Li, J. Am. Chem. Soc., 2014, 136, 13610 CrossRef CAS PubMed.
- J. A. Hickin, A. Ahmed, K. Fucke, M. Ashcroft and K. Jones, Chem. Commun., 2014, 50, 1238 RSC.
- (a) W. R. Jondorf, B. J. Abbott, N. H. Greenber and J. A. R. Mead, Chemotherapy, 1971, 16, 109 CrossRef CAS PubMed; (b) J. D. Donahue, R. K. Johnson and W. R. Jondorf, Br. J. Pharmacol., 1971, 43, P456 Search PubMed.
- (a) Y. Matsuya, Y.-i. Yamakawa, C. Tohda, K. Teshigawara, M. Yamada and H. Nemoto, Org. Lett., 2009, 11, 3970 CrossRef CAS PubMed; (b) K. Sugimoto, H. Yajima, Y. Hayashi, D. Minato, S. Terasaki, C. Tohda and Y. Matsuya, Org. Lett., 2015, 17, 5910 CrossRef CAS PubMed.
- L. Vitellozzi, G. D. McAllister, T. Genskib and R. J. K. Taylor, Synthesis, 2016, 48, 48 CrossRef CAS.
- A. K. Chatterjee, F. Dean Toste, S. D. Goldberg and R. H. Grubbs, Pure Appl. Chem., 2003, 75, 421 CrossRef CAS.
- S. L. Bader, M. U. Luescher and K. Gademann, Org. Biomol. Chem., 2015, 13, 199 CAS.
- L. F. Basil, H. Nakano, R. Frutos, M. Kopach and A. I. Meyers, Synthesis, 2002, 2064 CAS.
- R. R. Schrock, J. S. Murdzek, G. C. Bazan, J. Robbins, M. DiMare and M. O'Regan, J. Am. Chem. Soc., 1990, 112, 3875 CrossRef CAS.
- D. E. White, I. C. Stewart, B. A. Seashore-Ludlow, R. H. Grubbs and B. M. Stoltz, Tetrahedron, 2010, 66, 4668 CrossRef CAS PubMed.
- C. J. Fletcher, D. J. Blair, K. M. P. Wheelhouse and V. K. Aggarwal, Tetrahedron, 2012, 68, 7598 CrossRef CAS.
- M. Dai, I. J. Krauss and S. J. Danishefsky, J. Org. Chem., 2008, 73, 9576 CrossRef CAS PubMed.
- S. Farhane, M.-A. Fournier, R. Maltais and D. Poirier, Tetrahedron, 2011, 67, 2434 CrossRef CAS.
- S. Kalidindi, W. B. Jeong, A. Schall, R. Bandichhor, B. Nosse and O. Reiser, Angew. Chem., Int. Ed., 2007, 46, 6361 CrossRef CAS PubMed.
- B. Nosse, R. B. Chhor, W. B. Jeong, C. Bo1hm and O. Reiser, Org. Lett., 2003, 5, 941 CrossRef CAS PubMed.
- B. Nosse, A. Schall, W. B. Jeong and O. Reiser, Adv. Synth. Catal., 2005, 347, 1869 CrossRef CAS.
- E. Altenhofer and M. Harmata, Org. Lett., 2014, 16, 3 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2018 |