
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
A molecular dynamics study of plasticiser migration in nitrocellulose binders†
Lisa A.
Richards
 *a,
Anthony
Nash
b,
Maximillian Joshua Sebastian
Phipps
c and
Nora H.
de Leeuw
ad
*a,
Anthony
Nash
b,
Maximillian Joshua Sebastian
Phipps
c and
Nora H.
de Leeuw
ad
aDepartment of Chemistry, University College London, 20 Gordon Street, London WC1H 0AJ, UK. E-mail: lisa.richards.14@ucl.ac.uk
bDepartment of Physiology, Anatomy and Genetics, University of Oxford, South Parks Road, Oxford, OX1 3QX, UK
cAtomic Weapons Establishment, Aldermaston, Reading, Berkshire RG7 4PR, UK
dSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, CF10 3AT, UK. E-mail: DeLeeuwN@cardiff.ac.uk
First published on 14th September 2018
Abstract
The migration of the energetic plasticisers 1-nitramino-2,3-dinitroxypropane; 2,4-dinitroethylbenzene; and 2,4,6-trinitroethylbenzene in two nitrocellulose binder mixtures has been investigated by the calculation of diffusion coefficients and activation energies of diffusion from molecular dynamics simulations. The study included parameterisation of force fields for nitrocellulose; 1-nitramino-2,3-dinitroxypropane; the stabilizer ethyl centralite; and the overall nitrocellulose binder mixtures. Simulated densities obtained were in very good agreement with experimental densities. The diffusion coefficients compare favourably with experimental values available for similar systems, when differences such as the proportions of plasticisers are taken into consideration. Examination of the plasticiser diffusion rates suggests that 1-nitramino-2,3-dinitroxypropane migrates more slowly from a nitrocellulose binder than 2,4-dinitroethylbenzene for the nitrocellulose and plasticiser proportions used in this study. Understanding plasticiser migration is essential for the long-term storage of energetic material formulations without significant changes occurring in their properties or compositions.
Introduction
Polymer-bonded explosives (PBXs) are energetic materials containing an explosive component and often other constituents such as binders, stabilisers, plasticisers, oxidisers and fuels.1 The explosive component in a PBX is suspended in a polymeric binder which forms a tough elastomeric rubber when cured in situ, reducing the sensitivity of the explosive. The binder matrix often consists of a cross-linked polymer together with a plasticiser.2 A plasticiser improves the mechanical properties of a binder by lowering the glass transition temperature (Tg), increasing flexibility and improving the overall safety of the energetic formulation. A plasticiser may have other roles such as improving tensile strength or altering explosive performance. One of the earliest binder mixtures was nitroglycerine (NG) thickened with nitrocellulose (NC) to reduce friction and impact sensitivity.2 However, many plasticisers exude from the binder matrix, leading to diminished mechanical properties and reduced shelf-life.3 For example, the exudation of NG from energetic formulations is widely reported.4–6 Ideally, plasticiser migration should be sufficiently low to permit long-term storage of the energetic formulation without significant changes in properties or composition.7As early as 1934, 2,4-dinitroethylbenzene (2,4-DNEB) and 2,4,6-trinitroethylbenzene (2,4,6-TNEB) have been used to plasticise NC. The plasticiser 1-nitramino-2,3-dinitroxypropane (NG-N1) has shown improved properties as a plasticiser of NC compared to the widely used NG plasticiser by an impact sensitivity reduction of 0.2 J for NG to 14 J for NG-N1.8,9 The performance of a mixture of NG-N1 and 2,4-DNEB as an energetic plasticiser of a NC binder for 1,3,5,7-tetranitro-1,3,5,7-tetrazocane (HMX) has been compared to a mixture of 65% 2,4-DNEB and 35% 2,4,6-TNEB, a plasticiser known as K10.10 The NC binder plasticised with NG-N1 and 2,4-DNEB showed increased energetic output compared to the NC binder plasticised with K10,10 but the migration of the plasticiser molecules from the overall NC binder formulations was not investigated. NG migration from an NC binder and K10 migration from polyGLYN binder has been studied using thermogravimetric analysis to determine plasticiser diffusion coefficients to assess plasticiser volatility.3,7 However, before considering time-consuming experimental techniques, such as thermogravimetric analysis, diffusion coefficients can be obtained via molecular dynamics (MD) simulations to initially assess plasticiser migration. The use of MD simulations to determine plasticiser migration rates is particularly attractive for systems that have already been parameterised for simulations to obtain other properties relevant to EM formulations.
In this work, the migration of the energetic plasticisers NG-N1, 2,4-DNEB and 2,4,6-TNEB as part of two composite NC binder systems has been investigated using MD simulations. Thus far, neither in silico nor in vivo studies have assessed the volatility of the concentrations of NG-N1, 2,4-DNEB and 2,4,6-TNEB as part of two NC binder mixtures of 11% NC and 89% plasticiser, where the plasticiser component is made up of either a 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio of 2,4-DNEB and NG-N1, or entirely consists of K10, which itself is a 2:1 ratio of 2,4-DNEB and 35% 2,4,6-TNEB. As such, the first simulated system contains 11% NC, 60% 2,4-DNEB and 29% NG-N1. The second system also contains 11% NC, plus 58% 2,4-DNEB and 31% 2,4,6-TNEB – the latter two components making up the 89% K10 mixture. Both NC binder compositions also contained the stabiliser ethyl centralite (EC) in the standard proportion of ∼1 wt% of the total binder ingredients.7,10 This stabiliser is designed to react with nitrous gases produced during NC degradation, slowing down the degradation process.11,12 Diffusion coefficients were determined for each plasticiser in the two NC binder systems at five temperatures, followed by calculation of activation energies to enable a clear comparison of the migration of NG-N1, 2,4-DNEB and 2,4,6-TNEB. An empirical method based on quantum mechanical calculations, literature values and experimental data was used to parameterise force fields for NC, NG-N1 and EC. Bonding terms and partial charges were derived from the Hessian of quantum mechanical vibrational analysis calculations, whilst non-bonding terms were obtained by refinement of literature values of similar molecules.13,14 The force field used here for 2,4-DNEB and 2,4,6-TNEB had already been derived in previous work.15 The force fields were optimised by adjusting force field parameters until calculated densities of NC, NG-N1, EC and the two NC binder systems matched the available experimental properties.
:1 ratio of 2,4-DNEB and NG-N1, or entirely consists of K10, which itself is a 2:1 ratio of 2,4-DNEB and 35% 2,4,6-TNEB. As such, the first simulated system contains 11% NC, 60% 2,4-DNEB and 29% NG-N1. The second system also contains 11% NC, plus 58% 2,4-DNEB and 31% 2,4,6-TNEB – the latter two components making up the 89% K10 mixture. Both NC binder compositions also contained the stabiliser ethyl centralite (EC) in the standard proportion of ∼1 wt% of the total binder ingredients.7,10 This stabiliser is designed to react with nitrous gases produced during NC degradation, slowing down the degradation process.11,12 Diffusion coefficients were determined for each plasticiser in the two NC binder systems at five temperatures, followed by calculation of activation energies to enable a clear comparison of the migration of NG-N1, 2,4-DNEB and 2,4,6-TNEB. An empirical method based on quantum mechanical calculations, literature values and experimental data was used to parameterise force fields for NC, NG-N1 and EC. Bonding terms and partial charges were derived from the Hessian of quantum mechanical vibrational analysis calculations, whilst non-bonding terms were obtained by refinement of literature values of similar molecules.13,14 The force field used here for 2,4-DNEB and 2,4,6-TNEB had already been derived in previous work.15 The force fields were optimised by adjusting force field parameters until calculated densities of NC, NG-N1, EC and the two NC binder systems matched the available experimental properties.
Theoretical methods
Force field parameterisation
This study utilises the functional form popular across most classical force fields as shown in eqn (1).16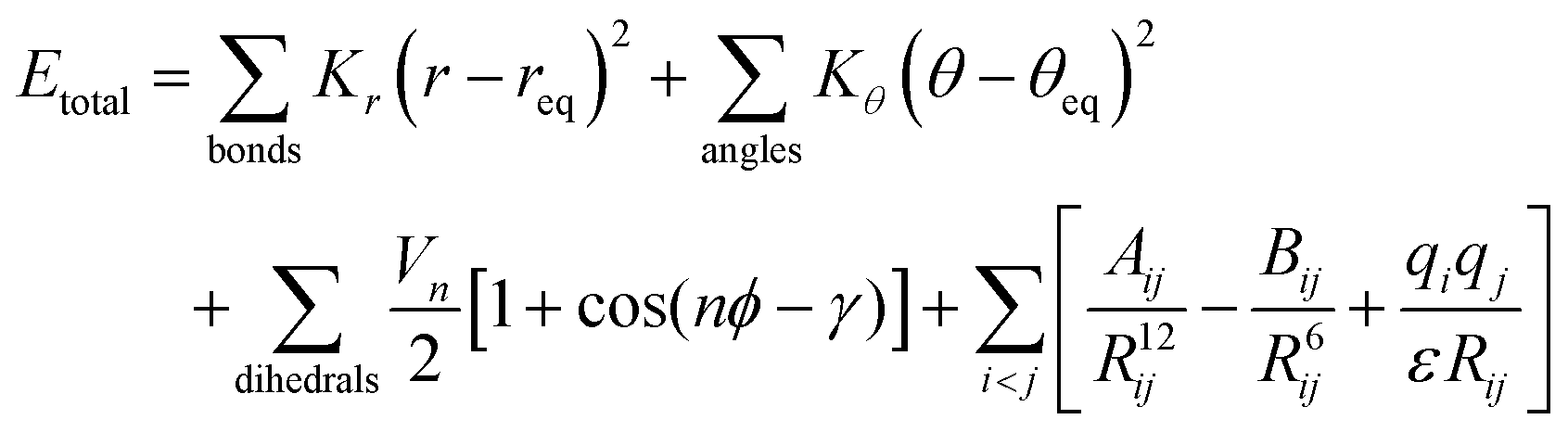 | (1) |
Bond stretching and angle bending are denoted by the first two terms; the equilibrium bond lengths, req, and bond angles, θeq were taken directly from the density functional theory (DFT) geometry-optimised NG-N1 and EC structures and a fully nitrated NC dimer. The DFT geometry-optimised EC and NG-N1 structures and fully nitrated NC dimer are displayed in Fig. 1A, B, C, respectively. The internal force constants for bond stretching, Kr and angle bending, Kθ were obtained from Cartesian second derivatives (Hessian matrices) created in the DFT NG-N1, EC and NC frequency calculations using the Open Access software ForceGen.17,18 NC is produced from cellulose by nitration which results in a mixture of substitution products. Fully nitrated NC was chosen as it contains more than 12.6% nitrogen, the content at which NC has explosive properties making it suitable for an energetic binder. The fully nitrated NC dimer contained the unique bonding environments required to obtain all equilibrium bond lengths, bond angles and corresponding force constants that would be present in a fully nitrated NC polymer chain. Torsional force constants for the dihedrals, displayed in the third term, and the Lennard-Jones (LJ) parameters shown in the fourth term of eqn (1), for NG-N1, EC and NC were taken from the General Amber Force Field (GAFF) and the Optimised Potentials for Liquid Simulations (OPLS) force field.13,14 The latter part of the final term represents the electrostatic potential; qi and qj are the atomic partial charges and ε is the dielectric constant. The RED Server Development package was used to generate the partial charges, using restricted electrostatic potential (RESP) charges from the DFT optimised NG-N1 and EC structures and the fully nitrated NC dimer.19–21 The basis of the parameterisation procedure used in the published biomolecular force fields is that similar chemical groups in different molecules interact in the same way.22 Often parameters are obtained for a number of molecules that contain the functional groups present in all biological macromolecules. An existing biomolecular force field could be used to obtain the bonding parameters as well as the non-bonding parameters for the molecules used in this study, but the terms would be generic. Geometry optimisation of each molecule ensures that the bonding terms derived are unique to the bonding environment of the molecule, whilst refinement of the LJ parameters from the literature establishes a set of parameters fitted specifically to the system under study. The parameters are supplied in the ESI:† NC parameters are shown in Fig. S1–S3 and Tables S1–S4, parameters for NG-N1 are outlined in Fig. S5–S7 and Tables S5–S8 and the parameters for EC are outlined in Fig. S9–S11 and Tables S9–S12.
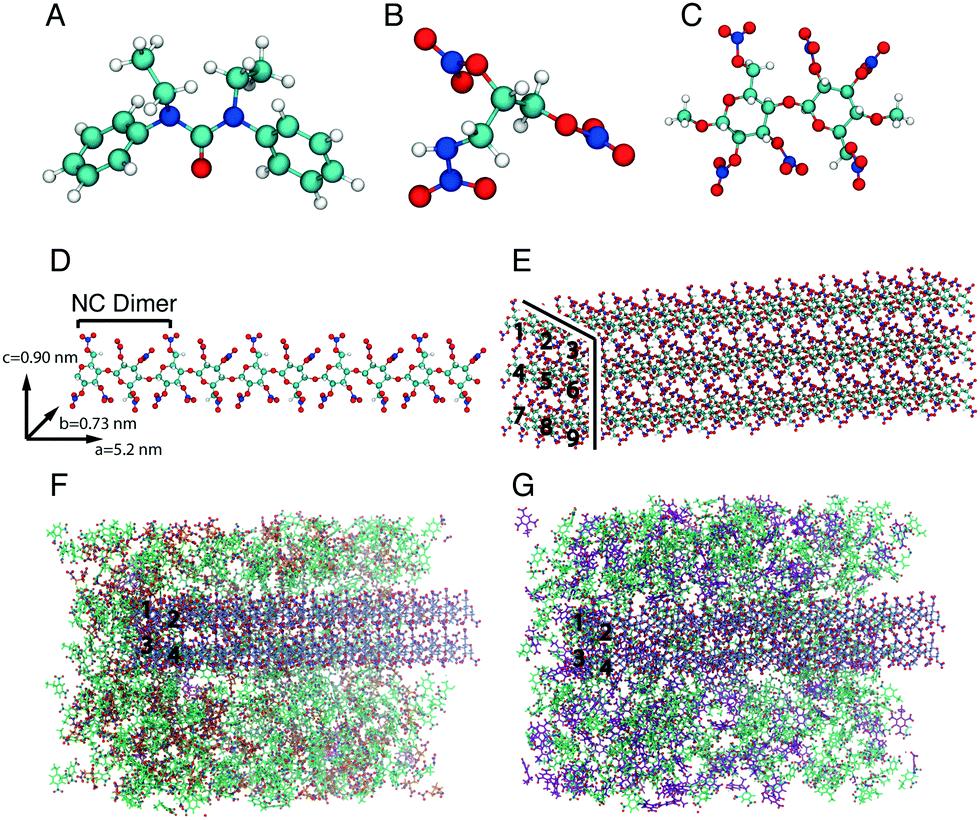 | ||
| Fig. 1 The optimised electronic structure of (A) ethyl centralite (EC), (B) 1-nitramino-2,3-dinitroxypropane (NG-N1), and (C) methyl-capped nitrocellulose (NC) dimer. (D) The unit cell of five NC dimers presented with unit cell periodic boundary dimensions, lattice angles of α = β = γ = 90°, and (E) an extended unit cell of nine NC chains. Both constructs repeat across the unit cell. (F) The pre-equilibration configuration of four extended NC chains within a 2,4-dinitroethylbenzene (2,4-DNEB) and NG-N1 compound with EC, and (G) within a K10 compound with EC. Individual NC chains have been numbered for clarity, running horizontal to the page. Molecules of EC are depicted with pink bonds, 2,4-DNEB with green bonds, NG-N1 with orange bonds and 2,4,6-TNEB with purple bonds. Oxygen and nitrogen have been visualised in red and blue, respectively. | ||
QM calculation details
All DFT calculations were carried out using the Gaussian 09 package.23 Geometry optimisation followed by vibrational frequency calculations were performed using the B3LYP exchange–correlation functional and a 6-311++G** Pople basis-set. Once the average root mean square (RMS) force on all atoms had fallen below 0.01 kcal−1 mol−1 Å−1 the geometry optimisation was deemed complete. To assure that the energy of the geometry-optimised dimer had reached a true energy minimum, frequency calculations were carried out to check the absence of imaginary modes.Validation of parameters
The bond angles and bond lengths obtained from the DFT optimised NG-N1 and EC structures were compared to experimental values obtained by X-ray diffraction.9,24,25 To the best of our knowledge, experimental bond lengths and bond angles are unavailable for NC, and therefore bond lengths and bond angles in the nitro and nitrate ester groups were compared to those in methyl and ethyl nitrate.26,27 Equilibrium constants from QM minimised structures within close agreement to experimental records were used for the parameterisation of force fields. To validate the bonded and non-bonded force field parameters, each molecular component, NG-N1, EC and NC, was simulated separately at 298 K and densities were compared to experimental data. The LJ parameters were altered systematically until the theoretical density was in close agreement with the experimental value.9,25,28 Furthermore, the dipole moment of an NC dimer unit was computed from simulation of the NC at 298 K and compared to available experimental dipole moments (Table 3).29 The densities of the two NC binder mixtures were simulated at 298 K and compared to experimental data. The LJ parameters were adjusted until the simulated densities of the NC, NG-N1 and 2,4-DNEB binder and the NC and K10 binder were all within 2% of the experimental values.10Molecular dynamics simulation details
The Sander module of the Amber 14 package was used for all MD simulations and energy minimisations.16 Prior to the MD simulation, energy minimisation was performed using the steepest descent method, followed by the conjugate gradient method. Once the RMS of the Cartesian element of the gradient was less than 1.0 × 10−4 kcal−1 mol−1 Å−1 the minimisation was stopped. For all simulations, periodic boundary conditions were used, whereas the Particle Mesh Ewald (PME) was used for treatment of long-range electrostatics and a non-bonded cutoff of 8 Å for the pairwise LJ interactions. Bonds to hydrogen atoms were constrained using the SHAKE algorithm.30 A 0.001 ps time step was used and the velocity Verlet algorithm integrated the equations of motion. Equilibration was performed by simulating each system using the NVT ensemble (constant number of particles, volume and temperature) followed by a simulation using the NPT ensemble (constant number of particles, pressure and temperature). Production simulations were performed using the NPT ensemble. All simulations utilised the Anderson temperature-coupling to calculate temperature and an isotropic implementation of the Berendsen barostat was used to maintain constant pressure in the NPT simulations.31,32Molecular systems
The EC and NG-N1 boxes were built using the program Packmol with a tolerance of 2 Å between the molecules.33 A simulation cell (40 Å3 volume) containing 306 molecules of NG-N1 using the unit cell dimensions proposed by Altenburg et al. was constructed,9 and in addition a separate simulation cell of identical dimensions was constructed with 149 EC molecules using EC unit cell dimensions from the literature.24 The dimensions proposed by Meader et al. were used to build a NC polymer.34 Using the UCSF Chimera package a molecular chain of 5 geometry optimised fully nitrated NC dimers was constructed and added to a box with the lattice dimensions a = 5.20 nm, b = 0.73 nm, and c = 0.90 nm and lattice angles of α = β = γ = 90° (see Fig. 1D).34–36An extended unit cell for NC was generated by replicating the single unit cell containing the 5 NC dimer polymer chain: the polymer chain was replicated in the x coordinate and the resulting chain replicated three times in the y and z coordinates (Fig. 1E).35 The NC model used in the binder mixtures was constructed by firstly duplicating the 5 dimers in the x coordinate to create a chain of 10 dimers, the 10 dimers were then replicated in the z coordinate and then the resulting two chains replicated in the y coordinate.35 The compositions of the two NC binder mixtures were the same as those used in experimental studies to compare the energetic output of NG-N1 and 2,4-DNEB as a plasticiser of NC with the plasticiser mixture 2,4-DNEB and 2,4,6-TNEB (K10).10 The binder mixtures contained 11% NC and 89% plasticiser, as previously described.
The NC binder mixture plasticised with NG-N1 and 2,4-DNEB was constructed by adding 640 2,4-DNEB molecules and 286 NG-N1 molecules to a NC simulation cell with dimensions 115.5 Å × 84.4 Å × 68.9 Å, as displayed in Fig. 1F. To construct the NC binder system plasticised with K10, 630 2,4-DNEB molecules and 276 2,4,6-TNEB molecules were added to the NC simulation cell with dimensions 115.5 Å × 89.4 Å × 70.9 Å, as displayed in Fig. 1G. The NC was positioned in both simulation cells so as to cross the x coordinate periodic boundary, thereby emulating a continuous NC polymer chain. The EC added to each mixture was 1% by weight of the total binder ingredients.7,37 Both of the binder mixture configurations were built using the program Packmol. A tolerance of 2.5 Å between the molecules was applied.33 The construction of all simulation cells is outlined in the ESI.†
Molecular dynamics simulations
MD simulations of the pure NC, EC and NG-N1 systems, as well as the binder mixtures, NC plasticised with NG-N1 and 2,4-DNEB and NC plasticised with K10 were performed at 298 K. Equilibration of each system was performed for 30 ps using the NVT ensemble, followed by a 400 ps simulation using the NPT ensemble. A 12 ns production run using the NPT ensemble was performed on the EC and NG-N1 systems and an 18 ns simulation on the NC construct. The NC underwent an additional 2 ns NPT production run at 298 K during which the dipole moment was calculated with respect to the centre of mass of an NC dimer. A 20 ns production run simulation using the NPT ensemble was performed for both binder mixture systems.Calculation of diffusion coefficients
Use of Langevin dynamics or the Anderson thermostat may lead to unreliable diffusion coefficients;38 therefore the NC binder systems were heated and equilibrated using the Anderson thermostat before switching the thermostat to the Berendsen scheme for the production runs.31 As long as the simulation has evolved for a sufficient time-period the centre-of-mass diffusion coefficient, D, can be calculated according to eqn (2):| 2nDt = 〈|ri(t) − ri(0)|2〉 | (2) |
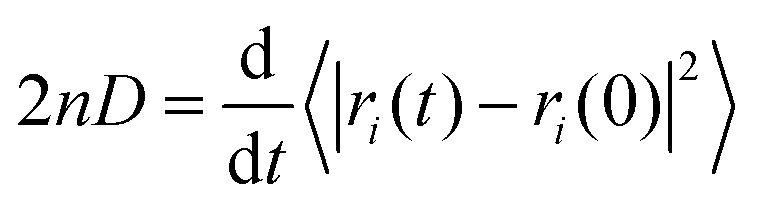 | (3) |
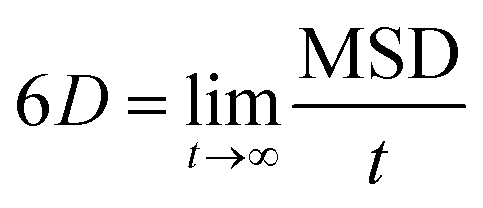 | (4) |
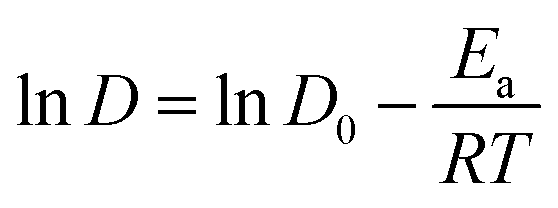 | (5) |
Results and discussion
A comparison of the bond lengths and bond angles between the QM and MD calculations and the experimental values of NG-N1, EC and NC is displayed in Table 1. To assess the credibility of the force field parameter set, simulated densities were compared to the experimental densities for each of the pure systems and for both NC binder systems (Table 2). The root mean squared deviations (RMSD) were averaged over simulation time to give the root mean square fluctuations (RMSF), which are listed in Table 2 for the simulated densities. The dipole moment for an NC monomer calculated from the simulation is displayed in Table 3 alongside experimental values. The diffusion coefficients (D) of 2,4,6-TNEB and 2,4-DNEB in the NC binder plasticised with K10 and the diffusion coefficients for 2,4-DNEB and NG-N1 in the NC binder system plasticised with the 2,4-DNEB and NG-N1 mixture are displayed in Table 4. The standard deviation of the gradient of the MSD versus time was used to calculate the standard errors for the diffusion coefficients given in Table 4. For comparison of the results with experimental values, Table 5 contains diffusion coefficients of NG-N1 simulated at 398 K, together with the experimental value of the diffusion coefficient for NG in a similar system, whereas simulated values of D at 373 K and 398 K for K10 are displayed alongside experimental values for K10 in Table 6. Natural logarithmic plots of diffusion coefficients versus reciprocal temperature for 2,4-DNEB and NG-N1 in the NC, 2,4-DNEB and NG-N1 binder and for 2,4-DNEB and 2,4,6-TNEB in the NC K10 binder, are shown in Fig. 2. The activation energies of diffusion calculated for each plasticiser molecule (in kJ mol−1) are presented in Table 7 with the associated errors calculated from the standard errors of the gradients of the natural logarithm versus 1/T graphs.| % to exp. | Molecular construct | ||
|---|---|---|---|
| NG-N1 | EC | NC | |
| Experimental bond lengths and angles referenced in the Validation of parameterisation section. | |||
| [0–3%] | 63 | 79 | 92 |
| [4–5%] | 23 | 4 | 6 |
| [6–10%] | 4 | 4 | 2 |
| >11% | 10 | 14 | 0 |
| Simulated density (g cm−3) | Experimental density (g cm−3) | |
|---|---|---|
| Experimental densities referenced in the Validation of parameterisation section. | ||
| NG-N1 | 1.801 ± 0.009 | 1.799 |
| EC | 1.159 ± 0.01 | 1.160 |
| NC | 1.619 ± 0.01 | 1.600 |
| NC binder – NG-N1 & 2,4-DNEB | 1.412 ± 0.02 | 1.428 |
| NC binder – 2,4,6-TNEB & 2,4-DNEB (K10) | 1.385 ± 0.02 | 1.400 |
| T (K) | Experimental and simulated dipole moments for an NC monomer (Debye) | |
|---|---|---|
| Experimental | Simulated | |
| Experimental dipole moments referenced in the Validation of parameterisation section. | ||
| 298 | 2.5 | 2.4 |
| 2.3 | ||
| T (K) | NC binder: 2,4-DNEB and 2,4,6-TNEB (K10) | NC binder: 2,4-DNEB and NG-N1 | ||||||
|---|---|---|---|---|---|---|---|---|
| 2,4-DNEB | 2,4,6-TNEB | 2,4-DNEB | NG-N1 | |||||
| D (107 cm2 s−1) | Std. err. (cm2 s−1) | D (107 cm2 s−1) | Std. err. (cm2 s−1) | D (107 cm2 s−1) | Std. err. (cm2 s−1) | D (107 cm2 s−1) | Std. err. (cm2 s−1) | |
| Calculation of diffusion coefficients displayed in Tables S13 and S14 in the ESI. | ||||||||
| 298 | 0.33 | 2.72 × 10−10 | 0.30 | 4.66 × 10−10 | 0.16 | 3.13 × 10−10 | 0.19 | 5.63 × 10−10 |
| 323 | 2.17 | 5.20 × 10−9 | 1.27 | 2.08 × 10−9 | 0.45 | 1.49 × 10−9 | 0.22 | 3.09 × 10−10 |
| 348 | 3.75 | 4.26 × 10−9 | 1.85 | 1.43 × 10−9 | 0.85 | 1.53 × 10−9 | 0.56 | 6.56 × 10−10 |
| 373 | 4.30 | 9.30 × 10−10 | 3.08 | 1.13 × 10−9 | 1.50 | 6.17 × 10−10 | 0.96 | 3.57 × 10−10 |
| 398 | 9.31 | 9.90 × 10−10 | 5.76 | 1.48 × 10−9 | 3.38 | 8.67 × 10−10 | 2.48 | 1.03 × 10−9 |
| T (K) | Diffusion coefficients for NG-N1 and NG (cm2 s−1) | |
|---|---|---|
| NG-N1 | NG | |
| 398 | 2.48 × 10−7 | 1.09 × 10−7 |
| T (K) | Experimental and simulated diffusion coefficients for K10 (cm2 s−1) | |
|---|---|---|
| Experimental | Simulated | |
| 373 | 1.32 × 10−7 | 3.88 × 10−7 |
| 398 | 7.01 × 10−7 | 8.07 × 10−7 |
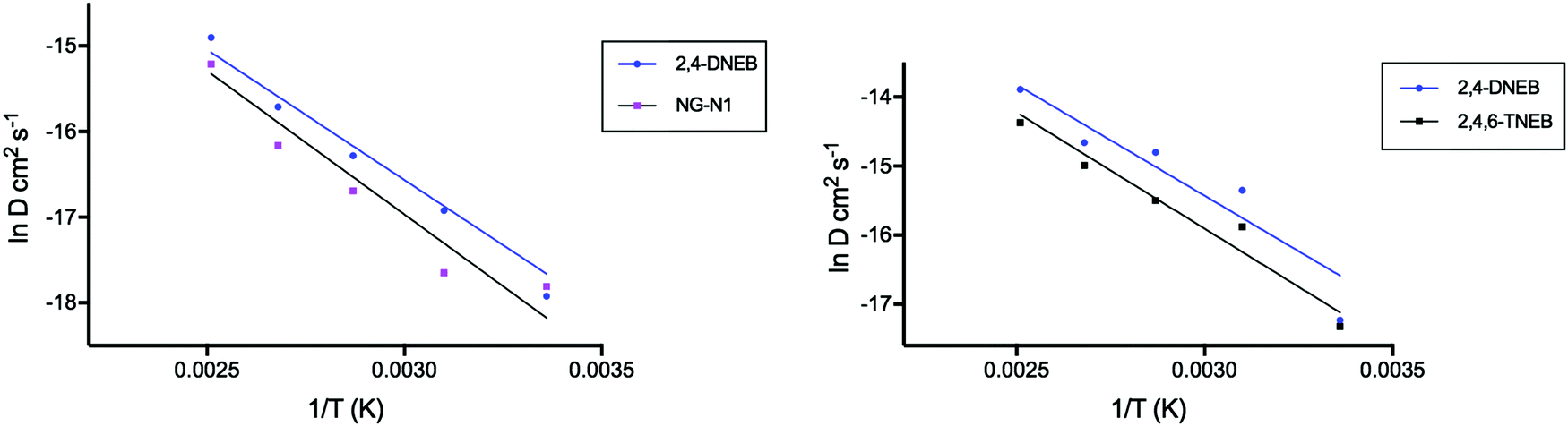 | ||
| Fig. 2 Plot showing the correlation between lnD of 2,4-DNEB and NG-N1, and lnD of 2,4-DNEB and 2,4,6-TNEB (K10), versus the reciprocal of temperature (K). | ||
| NC binder | Plasticiser | Activation energy (kJ mol−1) |
|---|---|---|
| NC, 2,4-DNEB & NG-N1 binder | 2,4-DNEB | 25.3 ± 1.2 |
| NG-N1 | 27.9 ± 2.2 | |
| NC & K10 binder | 2,4-DNEB | 26.7 ± 2.8 |
| 2,4,6-TNEB | 28.0 ± 1.7 |
Parameterisation and validation of NC binder components
The QM-derived bond lengths and bond angles of NC were in excellent agreement with experiment. As much as 92% of the QM bond lengths and bond angles were within 3% of experimental values (Table 1). Of the QM bond lengths and angles for NG-N1 and EC, 86% and 83%, respectively, were within 5% of the experimental values (Table 1). For NG-N1, five of the 20 QM bond lengths, accounting for 10% of the total bonds and angles, were overestimated compared to experiment. For EC, eight of the 21 QM bond lengths, accounting for 14% of the total bonds and angles, were overestimated compared to experiment (Table 1). To evaluate the effect on overall bulk properties, MD simulations of NG-N1 and EC were performed at 298 K using the QM-derived bond lengths and bond angles.The simulated densities of EC and NG-N1 were within 0.1% of the experimental values and the simulated density of NC, using the original GAFF LJ parameters without adjustment, was within 1.2% of experimental value.13 Further simulations of the NC binders were therefore performed with these parameters and without adjustment to the bond lengths.
The simulated densities of the NC binder mixture, either plasticised with NG-N1 and 2,4-DNEB or plasticised with K10, differ from their experimental densities by 1.1% and are within acceptable error, suggesting that comparison between diffusion coefficients for the plasticiser molecules in each NC binder system would be possible.
A dipole moment of 4.753 D from a 2 ns simulation at 298 K of an NC dimer, was halved to enable comparison with the experimental values of a fully nitrated NC monomer. The dipole moment of an NC monomer obtained from simulation was in good agreement with experiment, with the value within 0.1 of the experimental dipole moments.29
The calculation of diffusion coefficients of plasticisers
The diffusion coefficients for 2,4-DNEB and 2,4,6-TNEB in the NC binder plasticised with K10 positively correlate with temperature (Table 4), which is expected due to the increased kinetic energy in the system. Plasticiser migration rates are inversely proportional to the molecular weight of the plasticiser,3 which is evident in the NC binder plasticised with K10: 2,4-DNEB with a lower molecular mass of 196 u has a greater diffusion coefficient at all temperatures compared to 2,4,6-TNEB with a molecular mass of 241 u. The same trends in diffusion coefficients are observed in the NC binder plasticised with a mixture of 2,4-DNEB and NG-N1 (Table 4). The diffusion coefficients for 2,4-DNEB and NG-N1 increase with temperature and are larger for 2,4-DNEB compared to NG-N1 across the 323–398 K temperature range, which is explained by the smaller molecular mass of 2,4-DNEB (196 u) compared to NG-N1 (226 u). Calculated diffusion coefficients are sensitive to small changes in density,41 which is reflected in the NC binder systems, where the diffusion coefficients for 2,4-DNEB and 2,4,6-TNEB in the NC binder plasticised with K10 are greater than those of 2,4-DNEB and NG-N1 at the corresponding temperatures in the NC binder plasticised with the 2,4-DNEB and NG-N1 mixture. The faster diffusion of 2,4-DNEB and 2,4,6-TNEB molecules in the K10 NC binder can be attributed to the lower simulated density of 1.385 g cm−3 compared to the simulated density of 1.412 g cm−3 for the NC binder plasticised with the 2,4-DNEB and NG-N1 mixture.Experimental diffusion coefficients for 2,4-DNEB, 2,4,6-TNEB and NG-N1 in the NC binder systems used in this study are not available. There are, however, experimental diffusion coefficients calculated for similar systems which have been used to assess the reliability of the values. Cartwright conducted thermogravimetric analysis (TGA) at constant temperature to measure the mass loss of various plasticised propellants due to plasticiser vaporisation and used the data obtained to calculate the diffusivities and activation energies of plasticisers.7 One of the experiments calculated diffusion coefficients for nitroglycerin (NG) at three temperatures in a NC propellant containing NC with a 13.25% nitrogen content, a plasticiser content of 40% and 1% EC by weight of the total propellant ingredients.7 The molecular mass of NG is 227 u and NG-N1 has a molecular mass of 226 u, and considering their structural similarity and mass a comparison was therefore made between the diffusion coefficient for NG-N1 at 398 K in the NC, 2,4-DNEB and NG-N1 binder system and the experimental value for NG at 398 K in the experimental NG and NC propellant mixture.7
The diffusion coefficient calculated at 398 K for NG-N1 in the NC, 2,4-DNEB and NG-N1 binder system is 2.48 × 10−7 cm2 s−1, which is the same order of magnitude as the value for the diffusion coefficient calculated from experiment at 398 K for NG in the NC propellant (1.09 × 10−7 cm2 s−1). This value for the diffusion coefficient of 2.48 × 10−7 cm2 s−1 is higher than the experimental value for NG. Although diffusion coefficients are inversely proportional to molecular mass,42 the molecular masses of the molecules are unlikely to be a contributing factor in this case, as NG-N1 has a molecular mass of 226 u and NG has a molecular mass of 227. The reason for the larger diffusion coefficient at 398 K for NG-N1 compared to NG is most likely to be a result of the ratio of NC to plasticiser. A more highly plasticised binder or propellant would exhibit greater diffusivity due to greater mobility within the macromolecular network.7 For example, it has been suggested that the ability of NC to immobilize plasticiser molecules is reduced at higher plasticiser levels.43 The diffusion coefficient of NG-N1 is approximately 2.3 times greater than the experimental diffusion coefficient obtained for NG. It is therefore possible that the increase in the diffusion coefficient is due to the NC binder system plasticised with 2,4-DNEB and NG-N1 containing 89% plasticiser, approximately 2.2 times the 40% plasticiser content of the NC and NG propellant investigated experimentally.7,10
An experimental study by Provatas assessed plasticiser migration rates in a polyGLYN binder containing 15% plasticiser by weight.3 Diffusion coefficients were calculated for the GLYN oligomer and K10 at four different temperatures, of which two temperatures have been utilized in our study. Provatas reported diffusion coefficients for the plasticiser mixture K10, not separate values of the diffusion coefficient for the constituent 2,4-DNEB and 2,4,6-TNEB in the mixture.3 To obtain estimates of the simulated diffusion coefficients of K10 in the NC binder, which could then be compared to Provatas’ experimental values, the diffusion coefficients for 2,4-DNEB and 2,4,6-TNEB were multiplied by their K10 plasticiser proportions (0.65 and 0.35, respectively) and added together. The experimental diffusion coefficients for K10 at 373 K and 398 K in a polyGLYN binder and the estimations of the diffusion coefficient for K10 obtained in this study are displayed in Table 6.
Again, the simulated diffusion constants calculated for K10 are of the same order of magnitude as those obtained for K10 at 373 K and 398 K in the experimental study of the polyGLYN binder,3 although the diffusion coefficients for K10 calculated from the simulations at 373 K and 398 K are both higher than the experimental values. These discrepancies are likely due to the different densities and plasticiser contents of the experimental polyGLYN binder and simulated NC K10 binder. A polyGLYN binder typically has a density of 1.420 g cm−3 compared to 1.385 g cm−3 for the simulated NC and K10 binder, and the polyGLYN binder contained 15% plasticiser by weight compared to 89% for the simulated NC K10 binder.10,44 Higher plasticiser levels result in greater diffusivity of plasticiser molecules and calculated diffusion coefficients are also sensitive to small changes in density.7,41 The lower density of the simulated NC and K10 binder compared to polyGLYN means that plasticiser molecules can move with less restriction. This fact, combined with a much higher plasticiser content, would account for the larger diffusion coefficients for the simulated K10 compared to the experimental study.
The diffusion coefficients calculated for all plasticiser molecules in both NC binder systems increase steadily from 298 K to 373 K, but it is noticeable that there is a large increase in the diffusion coefficient value from 373 K to 398 K (Table 4). The increase in diffusion coefficient at higher temperatures is also apparent in the experimental studies of the NC and NG propellant and the K10 and polyGLYN binder.7 In the simulated NC binder plasticised with 2,4-DNEB and NG-N1, D for NG-N1 increases from 9.59 × 10−8 cm2 s−1 at 373 K to 2.48 × 10−7 cm2 s−1 at 398 K. A large increase is also observed between 381 K and 398 K in the experimental NC and NG propellant, where the diffusion coefficient jumps from 3.54 × 10−8 cm2 s−1 to 1.09 × 10−7 cm2 s−1.7 We suggest that at 398 K the kinetic energy of the molecules has increased compared to 373–380 K, resulting in an increased mobility of molecules and significantly larger diffusion coefficient values. The diffusion coefficient for K10 in the simulated NC K10 binder increases by 4.19 × 10−7 cm2 s−1 between 373 K and 398 K, and in poly(GLYN) the diffusion coefficient of K10 increases by 5.69 × 10−7 cm2 s−1 between 373 K and 398 K.3 A large increase in the diffusion coefficient of K10 is seen in both systems and in both cases this is likely to be due to the increase in kinetic energy of the molecules between 373 K and 398 K. However, the increase in diffusion coefficient for K10 in poly(GLYN) is greater than that for K10 in the NC K10 binder. This is unexpected, as poly(GLYN) has a higher density and greater NC content than the NC K10 binder, which would suggest slower diffusion of molecules. It is possible that the fluidity of the poly(GLYN) binder system did not increase considerably until 398 K, whereas molecule mobility in the NC K10 binder was already greater at lower temperatures due to a higher plasticiser content. Therefore, the increase in diffusion rate of K10 between 373 K and 398 K in the NC K10 binder was less pronounced compared to the increase seen in poly(GLYN).
Plots of the logarithm transformation of the diffusion coefficient versus the reciprocal temperature (1/T) for the plasticisers in both binder systems show linearity (Fig. 2, least-squares fit). The activation energies of diffusion for each plasticiser were calculated using the gradients of the linear fits and their associated errors were calculated from the standard errors of the gradients of the linear fits. The natural logarithmic transformation of each diffusion coefficient is listed in Tables S15 and S16 of the ESI.†
As could be expected from its lower molecular mass, the activation energy of 2,4-DNEB is slightly lower than that of NG-N1 in the NC binder plasticised with a mixture of these two molecules. Experimental studies of low molecular mass migrants in poly(ethylene terephthalate) show the same trend in activation energies of diffusion:45 the lower the mass of a molecule, the higher the diffusion rate and the lower the activation energy of diffusion. The same trend occurs in the NC and K10 binder, where 2,4-DNEB with a lower molecule mass than 2,4,6-TNEB has a slightly lower activation energy of diffusion.
The activation energy of 2,4-DNEB is 25.3 kJ mol−1 in the NC binder plasticised with 2,4-DNEB and NG-N1, compared to 26.7 kJ mol−1 in the NC and K10 binder. This would not be expected, as the lower simulated density of the NC and K10 binder should facilitate diffusion leading to a lower activation energy compared to the value obtained for 2,4-DNEB in the NC, 2,4-DNEB and NG-N1 binder, although the calculated difference in activation energy is only 1.4 kJ mol−1. The activation energy of NG in the experimental study of an NC propellant containing NC with a 13.25% nitrogen content and a plasticiser content of 40% was reported as 89 kJ mol−1, three times the value of NG-N1.7 However, as already discussed, the much greater plasticiser content of the simulated NC binders used in this study is likely to lead to increased mobility of the plasticiser molecules, resulting in lower activation energies of diffusion.
Conclusion
The migration of the energetic plasticisers 1-nitramino-2,3-dinitroxypropane (NG-N1); 2,4-dinitroethylbenzene (2,4-DNEB); and 2,4,6-trinitroethylbenzene (2,4,6-TNEB) in two nitrocellulose (NC) binder mixtures has been investigated via the calculation of diffusion coefficients and activation energies of diffusion from molecular dynamics simulations. Force field parameters for the plasticiser molecules 2,4-DNEB and 2,4,6-TNEB had been derived previously, but parameterization of NC, NG-N1 and ethyl centralite (EC) was required for the construction of two NC binder mixtures. Simulated densities of the new force field parameter sets for NC, NG-N1 and EC were in excellent agreement with published experimental densities. The Lennard-Jones parameters for the two constructed NC binder mixtures were adjusted until the simulated density of each system was within 1.1% of the experimental value.Molecular dynamics simulations of both systems were performed over five temperatures to enable calculation of diffusion coefficients from the mean squared displacements and calculation of activation energies of diffusion using the Arrhenius relationship. Examination of the diffusion coefficients for the plasticisers suggests that NG-N1 will migrate at a slower rate than 2,4-DNEB, when a mixture of these two molecules are used to plasticise a NC binder with a plasticiser-to-NC ratio of 89%:11%. The activation energy of diffusion for NG-N1 is 2.6 kJ mol−1 higher than the activation energy of diffusion for 2,4-DNEB in the NC binder plasticised with a mixture of these two molecules, further confirming a lower migration rate for NG-N1. The diffusion coefficient of NG-N1 (2.48 × 10−7 cm2 s−1) calculated from molecular dynamics simulations of the NC, 2,4-DNEB and NG-N1 binder at 398 K compares favourably to the experimental value for nitroglycerin (1.09 × 10−7 cm2 s−1) in a NC propellant, when the much greater plasticiser content of the simulated NC binder is taken into consideration. A much greater plasticiser content compared to NC results in greater mobility of the molecules, as the ability of NC to immobilise plasticiser molecules is reduced. The rates of diffusion of 2,4-DNEB and 2,4,6-TNEB calculated from simulations of the NC and K10 binder are of the same order of magnitude as experimental values available for diffusion of K10 in binder system containing poly(GLYN) instead of NC. At ∼398 K, the diffusion coefficient for K10 calculated from the simulations was 8.07 × 10−7 cm2 s−1 compared to an experimental value of 7.01 × 10−7 cm2 s−1. The simulated diffusion coefficients of 2,4-DNEB and 2,4,6-TNEB are reasonable, when the different densities and plasticiser contents of the experimental poly(GLYN) binder and simulated NC binder are taken into account. The diffusion coefficients obtained for NG-N1 at all temperatures are lower than those obtained for 2,4-DNEB and 2,4,6-TNEB in the NC and K10 binder at the corresponding temperatures. These results also suggest a favourable migration rate for NG-N1, although the simulated density of 1.412 g cm−3 for the NC, 2,4-DNEB and NG-N1 binder compared to 1.385 g cm−3 for the NC and K10 binder should be weighted, as a higher density will decrease the diffusion rate of all molecules.
In conclusion, the volatility of NG-N1 when combined with 2,4-DNEB in a plasticiser mixture for use in a NC binder of this composition is not significantly higher than that of 2,4-DNEB. Increasing the NC content in the binder could be considered, as even a small increase should reduce the mobility of the molecules and decrease the high diffusion rate of the plasticiser molecules.
Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research was funded by the Engineering and Physical Sciences Research Council (EPSRC) and The Atomic Weapons Establishment (AWE). The authors acknowledge the use of the UCL Legion High Performance Computing Facility (Legion@UCL), and associated support services, in the completion of this work. NHdL acknowledges AWE for a William Penney Fellowship. ©British Crown Owned Copyright 2018/AWE.References
- J. Yang, X. Gong and G. Wang, A Promising Azido Nitrate Ester Plasticiser for Propellant, Comput. Mater. Sci., 2015, 110, 71–76, DOI:10.1016/j.commatsci.2015.07.049.
- A. Provatas, Energetic Polymers and Plasticisers for Explosive Formulations, Melbourne, 2000 Search PubMed.
- A. Provatas, Energetic Plasticiser Migration Studies, J. Energ. Mater., 2003, 21(4), 237–245, DOI:10.1080/713770435.
- D. A. Reese, L. J. Groven and S. F. Son, Formulation and Characterization of a New Nitroglycerin-Free Double Base Propellant, Propellants, Explos., Pyrotech., 2014, 39(2), 205–210, DOI:10.1002/prep.201300105.
- J. P. Agrawal and H. Singh, Qualitative Assessment of Nitroglycerin Migration from Double-base and Composite Modified Double-base Rocket Propellants: Concepts and Methods of Prevention, Propellants, Explos., Pyrotech., 1993, 18(2), 106–110, DOI:10.1002/prep.19930180211.
- W. Wu, Y. Yang, J. Chen, Y. Li and Y. Cheng, Nitroglycerine Migration to Polyurethane Used for Inhibition of Double-Base Propellants through Thermal Analysis Techniques, J. Therm. Anal. Calorim., 2016, 125(2), 881–886, DOI:10.1007/s10973-016-5457-z.
- R. V. Cartwright, Volatility of NENA and Other Energetic Plasticisers Determined by Thermogravimetric Analysis, Propellants, Explos., Pyrotech., 1995, 20(2), 51–57, DOI:10.1002/prep.19950200202.
- G. Hale, Propellant Powder, US Pat., 1963992, 1934 Search PubMed.
- T. Altenburg, T. M. Klapötke and A. Penger, Primary Nitramines Related to Nitroglycerine: 1-Nitramino-2,3-Dinitroxypropane and 1,2,3-Trinitraminopropane, Cent. Eur. J. Energ. Mater., 2009, 6(3–4), 3–4 Search PubMed.
- M. Willcox, J. Padfield and D. McAteer, Demonstration of 1-Nitramino-2,3-Dinitroxypropane as an Energetic Plasticiser Component in an HMX-Based PBX, Shrivenham.
- D. Trache and A. F. Tarchoun, Stabilizers for Nitrate Ester-Based Energetic Materials and Their Mechanism of Action: A State-of-the-Art Review, J. Mater. Sci., 2018, 53(1), 100–123, DOI:10.1007/s10853-017-1474-y.
- P. Krumlinde, S. Ek, E. Tunestål and A. Hafstrand, Synthesis and Characterization of Novel Stabilizers for Nitrocellulose-Based Propellants, Propellants, Explos., Pyrotech., 2017, 42(1), 78–83, DOI:10.1002/prep.201600122.
- J. Wang, R. M. Wolf, J. W. Caldwell, P. A. Kollman and D. A. Case, Development and Testing of a General Amber Force Field, J. Comput. Chem., 2004, 25, 1157–1174 CrossRef CAS PubMed.
- W. L. Jorgensen and J. Tirado-Rives, The OPLS [Optimized Potentials for Liquid Simulations] Potential Functions for Proteins, Energy Minimizations for Crystals of Cyclic Peptides and Crambin, J. Am. Chem. Soc., 1988, 110(6), 1657–1666, DOI:10.1021/ja00214a001.
- L. Chirlian and M. Francl, Atomic Charges Derived from Electrostatic Potentials; A Detailed Study, J. Comput. Chem., 1987, 8(6), 894–905, DOI:10.1002/jcc.540080616.
- W. D. Cornell, P. Cieplak, C. I. Bayly, I. R. Gould, K. M. Merz, D. M. Ferguson, D. C. Spellmeyer, T. Fox, J. W. Caldwell and P. A. Kollman, A Second Generation Force Field for the Simulation of Proteins, Nucleic Acids, and Organic Molecules, J. Am. Chem. Soc., 1995, 117(19), 5179–5197, DOI:10.1021/ja00124a002.
- A. Nash, H. L. Birch, T. Collier and N. De Leeuw, Force Gen: Atomic Covalent Bond Derivation for Gromacs, 2017, https://sourceforge.net/projects/forcegen/.
- J. M. Seminario, Calculation of Intramolecular Force Fields from Second-Derivative Tensors, Int. J. Quantum Chem., Quantum Chem. Symp., 1996, 30, 1271–1277, DOI:10.1002/(SICI)1097-461X(1996)60:7<1271::AID-QUA8>3.0.CO;2-W.
- E. Vanquelef, S. Simon, G. Marquant, E. Garcia, G. Klimerak, J. C. Delepine, P. Cieplak and F. Y. R. E. D. Dupradeau, Server: A Web Service for Deriving RESP and ESP Charges and Building Force Field Libraries for New Molecules and Molecular Fragments, Nucleic Acids Res., 2011, 39(2), 511–517, DOI:10.1093/nar/gkr288.
- F.-Y. Dupradeau, A. Pigache, T. Zaffran, C. Savineau, R. Lelong, N. Grivel, D. Lelong, W. Rosanski and P. Cieplak, The R.E.D. Tools: Advances in RESP and ESP Charge Derivation and Force Field Library Building, Phys. Chem. Chem. Phys., 2010, 12(28), 7821–7839, 10.1039/c0cp00111b.
- P. Cieplak, W. D. Cornell and P. A. Kollman, A Well-Behaved Electrostatic Potential Based Method Using Charge Restraints for Deriving Atomic Charges: The RESP Model, J. Phys. Chem., 1993, 97, 10269–10280 CrossRef.
- L. Monticelli and E. Salonen, in Biomolecular Simulations: Methods and Protocols, ed. L. Monticelli and S. Emppu, Humana Press, New York, 2016 Search PubMed.
- M. J. Frisch and G. W. Trucks, et al., Gaussian’09, Gaussian Inc., Wallingford, CT, 2009 Search PubMed.
- R. Betz, T. Gerber and H. Schalekamp, 1,3-Diethyl-1,3-Diphenylurea, Acta Crystallogr., Sect. E: Struct. Rep. Online, 2011, 67(4), o827, DOI:10.1107/S1600536811008294.
- P. Ganis, G. Avitable, E. Benedetti, C. Pedone and M. Goodman, Crystal and Molecular Structure of N,N′-Diethyl-N,N′-Diphenylurea, Proc. Natl. Acad. Sci. U. S. A., 1970, 67, 426–433 CrossRef CAS.
- P. Cox and S. Waring, Microwave Spectrum, Structure, and Dipole Moment of Methyl Nitrate, Trans. Faraday Soc., 1971, 67, 3441–3450, DOI:10.1021/ja01038a006.
- J. R. Durig and T. G. Sheehan, Raman Spectra, Vibrational Assignment, Structural Parameters and Ab Initio Calculations for Ethyl Nitrate, J. Raman Spectrosc., 1990, 21, 635–644 CrossRef CAS.
- D. M. French, J. Appl. Polym. Sci., 1978, 22, 309–313 CrossRef CAS.
- F. Baker, M. Jones, T. Lewis, G. Privett, D. Crofton and R. Pethrisk, Dielectric Studies of Nitrocellulose- Nitroglycerine Mixtures, Polymer, 1984, 25, 815–820 CrossRef CAS.
- J. P. Ryckaert, G. Ciccotti and H. J. C. Berendsen, Numerical Integration of the Cartesian Equations of Motion of a System with Constraints: Molecular Dynamics of n-Alkanes, J. Comput. Phys., 1977, 23(3), 327–341, DOI:10.1016/0021-9991(77)90098-5.
- H. J. C. Berendsen, J. P. M. Postma, W. F. van Gunsteren, A. DiNola and J. R. Haak, Molecular Dynamics with Coupling to an External Bath, J. Chem. Phys., 1984, 81(8), 3684–3690, DOI:10.1063/1.448118.
- H. C. Andersen, Molecular Dynamics Simulations at Constant Pressure and/or Temperature, J. Chem. Phys., 1980, 72(1980), 2384, DOI:10.1063/1.439486.
- C. Steffen, K. Thomas, U. Huniar, A. Hellweg, O. Rubner and A. Schroer, Packmol: A Package for Building Initial Configurations for Molecular Dynamics Simulations, J. Comput. Chem., 2008, 31(16), 2967–2970, DOI:10.1002/jcc.
- D. Meader, E. D. T. Atkins and F. Happey, Cellulose Trinitrate: Molecular Conformation and Packing Considerations, Polymer, 1978, 19(12), 1371–1374, DOI:10.1016/0032-3861(78)90086-1.
- E. F. Pettersen, T. D. Goddard, C. C. Huang, G. S. Couch, D. M. Greenblatt and E. C. Meng, F. T. UCSF Chimera – a Visualization System for Exploratory Research and Analysis, J. Comput. Chem., 2004, 25(13), 1605–1612 CrossRef CAS PubMed.
- W. Humphrey, A. Dalke and K. Schulten, VMD – Visual Molecular Dynamics, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- B. Zhao, T. Zhang, Z. Wang, S. Sun, Z. Ge and Y. Luo, Kinetics of Bu-NENA Evaporation from Bu-NENA/NC Propellant Determined by Isothermal Thermogravimetry, Propellants, Explos., Pyrotech., 2017, 42(3), 253–259, DOI:10.1002/prep.201600054.
- J. E. Basconi and M. R. Shirts, Effects of Temperature Control Algorithms on Transport Properties and Kinetics in Molecular Dynamics Simulations, J. Chem. Theory Comput., 2013, 9, 2887–2899, DOI:10.1021/ct400109a.
- F. Muller Plathe, Permeation of Polymers a Computational Approach, Acta Polym., 1994, 45(4), 259–293, DOI:10.1002/actp.1994.010450401.
- D. R. Roe and T. E. Cheatham, PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data, J. Chem. Theory Comput., 2013, 9(7), 3084–3095 CrossRef CAS PubMed.
- T. Fox and P. A. Kollman, Application of the RESP Methodology in the Parametrization of Organic Solvents, J. Phys. Chem. B, 1998, 102(41), 8070–8079, DOI:10.1021/Jp9717655.
- A. D. Kirk, The Range of Validity of Graham’ s Law, J. Chem. Educ., 1967, 44(12), 745–750 CrossRef CAS.
- A. J. Phillips, Study of Factors Which Affect the Volatility of Smokeless Powder, New Jersey, USA, 1940 Search PubMed.
- L. T. De Luca, T. Shimada, V. P. Sinditskii and M. Calabro, Chemical Rocket Propulsion: A Comprehensive Survey of Energetic Materials, Springer Berlin Heidelberg, 2016, pp. 863–887 Search PubMed.
- F. Welle and R. Franz, Diffusion Coefficients and Activation Energies of Diffusion of Low Molecular Weight Migrants in Poly(Ethylene Terephthalate) Bottles, Polym. Test., 2012, 31(1), 93–101, DOI:10.1016/j.polymertesting.2011.09.011.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c8nj03464h |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |