
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis, absolute configuration and in vitro cytotoxicity of deschloroketamine enantiomers: rediscovered and abused dissociative anaesthetic†
Bronislav
Jurásek‡
 ab,
František
Králík‡
c,
Silvie
Rimpelová
d,
Jan
Čejka
e,
Vladimír
Setnička
c,
Tomáš
Ruml
d,
Martin
Kuchař
*ab and
Michal
Kohout
*f
ab,
František
Králík‡
c,
Silvie
Rimpelová
d,
Jan
Čejka
e,
Vladimír
Setnička
c,
Tomáš
Ruml
d,
Martin
Kuchař
*ab and
Michal
Kohout
*f
aForensic Laboratory of Biologically Active Substances, University of Chemistry and Technology Prague, Technická 5, 166 28, Prague 6, Czech Republic. E-mail: martin.kuchar.vscht.cz
bDepartment of Chemistry of Natural Compounds, University of Chemistry and Technology Prague, Technická 5, 166 28, Prague 6, Czech Republic
cDepartment of Analytical Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28, Prague 6, Czech Republic
dDepartment of Biochemistry and Microbiology, University of Chemistry and Technology Prague, Technická 3, 166 28, Prague 6, Czech Republic
eDepartment of Solid State Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28, Prague 6, Czech Republic
fDepartment of Organic Chemistry, University of Chemistry and Technology Prague, Technická 5, 166 28, Prague 6, Czech Republic. E-mail: michal.kohout@vscht.cz
First published on 29th October 2018
Abstract
In this study, we aim to determine differences in cytotoxicity of racemic deschloroketamine and its enantiomers. The synthesized racemate of this recently rediscovered and abused dissociative anaesthetic was resolved by chiral HPLC and the absolute configuration of the enantiomers was assigned using a combination of circular dichroism methods and single-crystal X-ray. Not only the absolute configuration, but also the most preferred conformers present in the crystal were successfully determined by electron and vibrational circular dichroism supported by ab initio calculations, and confirmed by X-ray. The in vitro cytotoxicity of racemic deschloroketamine and its enantiomers was determined for nine different types of cell lines. Generally, (S)-deschloroketamine exhibited higher cytotoxicity in the majority of cases. For human embryonic kidney cells (HEK 293T), the (S)-enantiomer reached the IC50 below 1 mM concentration and, in consequence, proved to be twice as potent as the (R)-enantiomer. On the other hand, live-cell fluorescence microscopy imaging at sub-IC50 concentrations provided evidence for only a minor effect of deschloroketamine racemate and enantiomers on endoplasmic reticulum stress and mitochondria morphology in neuroblastoma cells SH-SY5Y.
Introduction
The prevalence of new psychoactive substances (NPS) that are not controlled under existing legislation is a complex phenomenon affecting the health and safety of citizens on the global level. According to the European Monitoring Centre for Drugs and Drug Addiction (EMCDDA) 2017 update on NPS in Europe, more than 620 NPS are currently being monitored.1 The most important groups of recreational drugs represent cathinones, synthetic cannabinoids, phenethylamines, tryptamines, piperazines and arylcyclohexylamines. Among these substances, there are many pharmaceuticals previously approved for clinical use and subsequently discontinued or even pharmaceuticals that are still being used in human or veterinary medicine.2–4A dissociative anaesthetic ketamine (1) was introduced to the market in 1970 for analgesia and sedation in clinical practice. However, if administered at a sub-narcotic dose, it can cause hallucinations, floating sensations, dissociation without the actual loss of consciousness, therefore, it is often misused as a recreational drug.5–8 Nightmares, changes in perception, flash-backs, tolerance and dependence as well as chronic changes in the urinary tract are frequently reported adverse effects.7,9 These adverse effects have led to a progressive change in the “psychonauts” and “clubbers” communities, which recently started to prefer methoxetamine (2) – a more potent dissociative anaesthetic (marketed as “bladder-friendly” temporarily legal alternative to ketamine).10 Additionally, several novel dissociative anaesthetics with ketamine-like effects, such as deschloroketamine (3), ephenidine (4) and lefetamine (5) (Fig. 1) have recently been introduced and are being used as ketamine replacements.11,12
 | ||
| Fig. 1 Structures of ketamine (1), methoxetamine (2), deschloroketamine (3), ephenidine (4) and lefetamine (5). | ||
Deschloroketamine, an analogue of ketamine, is not a new substance.13,14 Originally, it was proposed as an immunomodulation agent and for the treatment of bacterial, fungal, viral or protozoan infections.15,16 It has, however, never been put in the clinical practice. Recently, it has been rediscovered as a “legal” alternative to ketamine and is now being abused in a similar manner. According to the anecdotal reports of users, it is about 1.5 times more potent than methoxetamine and about three times more potent than ketamine,17 which makes it a real threat to health and safety of the users. This is, in particular, due to the lack of information on deschloroketamine pharmacokinetics, pharmacodynamics, its toxicity as well as the toxicity of its metabolites. In addition, there is no information available on difference in the effect of different stereoisomers of this drug.
Therefore, we have performed a pilot study with this highly potent drug addressing the important features of chirality. We have synthesized racemic deschloroketamine and separated its enantiomers using high-performance liquid chromatography (HPLC) on a chiral polysaccharide column. Furthermore, we have established the absolute configuration of the enantiomers using a powerful combination of vibrational circular dichroism supported by ab initio calculations and X-ray diffraction.18 Subsequently, in vitro cytotoxicity of both enantiomers was studied and related to the toxicity data known for ketamine.19,20 Such information is essential for the potential use of deschloroketamine in human medicine, especially, analogously to ketamine, as a rapid acting antidepressant.
Results
Synthesis and enantioseparation
Deschloroketamine (3) was synthesized in accord with previously published methods (Scheme 1).13,14 First, benzonitrile reacted with cyclopentyl magnesium bromide in diethyl ether for 72 h giving rise to ketone 7. Due to the instability of the intermediates 8 and 9, it is advisable to accomplish the next synthetic steps in a one-pot manner. Regioselective bromination of cyclopentyl phenyl ketone (7) provided bromoketone 8, which was used without further purification in the reaction with methylamine to afford hydroxy imine 9. Again, after evaporating the unreacted species, the crude imine 9 was subjected to a microwave-assisted thermal rearrangement in decalin to provide deschloroketamine (3). The crude product was stabilized in the form of hydrogen chloride salt and purified by multiple crystallizations to provide chemically pure substance, which was subjected to chiral separation.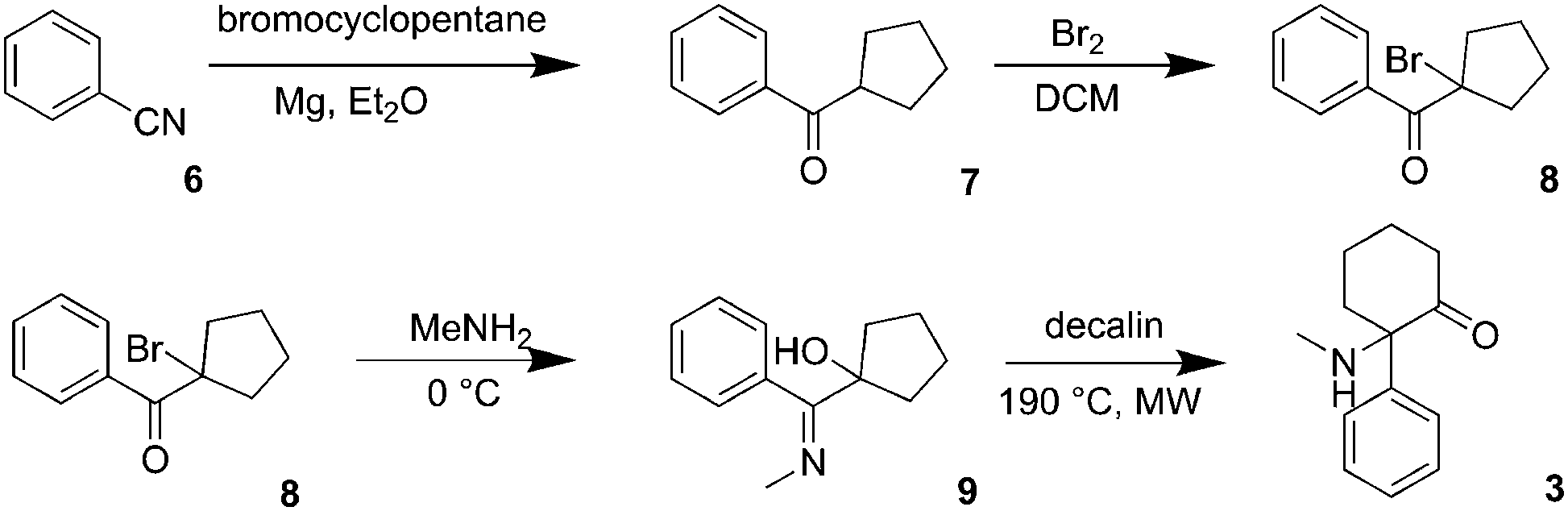 | ||
| Scheme 1 Synthesis of deschloroketamine (3). | ||
Since no data were available on enantioseparation of deschloroketamine, an analytical high-performance liquid chromatographic (HPLC) separation method was developed and subsequently transferred to preparative scale HPLC (for details, see ESI†). The racemic substance was separated into pure enantiomers (Fig. 2) and the purity of collected fractions was verified in the analytical mode (inset of Fig. 2).
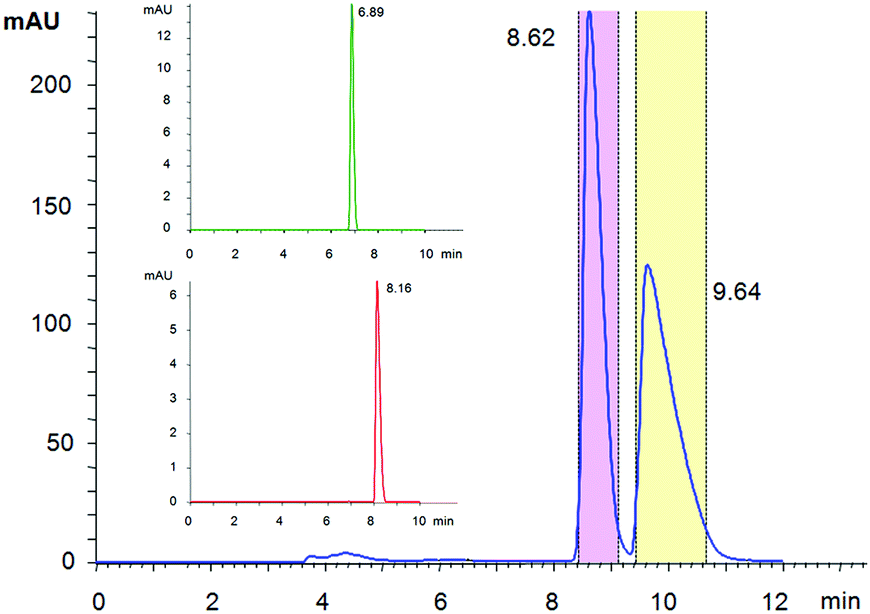 | ||
| Fig. 2 Preparative enantioseparation of deschloroketamine with marked areas of the collected mobile phase. The inset shows verification of the enantiopurity of combined fractions of peak 1 (DCK-1) and peak 2 (DCK-2) in an analytical mode, which was found >99% for both enantiomers. | ||
Determination of the absolute configuration of enantiomers
Conformational analysis. Because most physico-chemical properties are shared by both enantiomers and their geometries are mirror images, it is sufficient to perform geometry optimization only for one of them, in this case the (R)-enantiomer. The key question for successful conformational search is the position of the phenyl and methylammonium (–+NH2CH3) group, which are located at the same carbon atom of the cyclohexane ring. In order to fully cover the conformational possibilities, two basic structural models were created: model A with the phenyl group in the axial position and the –+NH2CH3 group in the equatorial position of the cyclohexane ring, and model B with switched positions of the groups.
For each model, values of dihedral angles defining the mutual orientation of the phenyl and –+NH2CH3 groups were systematically varied in order to produce 12 geometries as starting points for further optimization. The structures were optimized using density functional theory (DFT) approach at CAM-B3LYP/aug-cc-pVDZ and ωB97X-D/TZVP levels of theory using Gaussian 09 program package.21,22
The DFT optimization of the starting structures yielded 3 stable conformers for the model A and 4 stable conformers for the model B (for their relative Gibbs free energies and corresponding Boltzmann weights, see ESI,† Table S1). The calculated energies of all 3 conformers of the model A (i.e. with the phenyl group in the axial position) were significantly lower than those of the model B. This result was slightly unexpected, as it was assumed that the rather bulky phenyl unit should be located in the equatorial position due to lower steric hindrance. However, after closer inspection, the resulting conformers of the model A are stabilized by a hydrogen bond between one hydrogen of the ammonium group and the oxygen atom of the carbonyl group.
According to the Boltzmann weights, only two conformers of the model A seemed to be relevant for further simulations of the spectra, with conformer I being predominantly populated (Fig. 3, left). The major difference between the geometries of the conformations lays in the orientation of the –+NH2CH3 group (see ESI,† Fig. S2 and Table S1).
 | ||
| Fig. 3 Geometries of two lowest-energy conformers of (R)-DCK. | ||
Assignment of absolute configuration of deschloroketamine enantiomers
As expected, the VCD spectra of DCK enantiomers of unknown absolute configuration labelled as DCK-1 and DCK-2 (Fig. 4) were of the opposite sign, confirming that two enantiomers were present. Besides VCD spectra, also IR spectra were recorded (ESI,† Fig. S2) as they may help to correctly assign calculated transitions to experimentally observed bands. Only IR and VCD spectra calculated with ωB97X-D/TZVP are presented as this method provided better results than B3LYP/6-31++G**. The calculated spectra were scaled with the factor of 0.952, which was derived from the CompareVOA algorithm.23 | ||
| Fig. 4 Experimental VCD spectra of DCK-1 and DCK-2 samples (top) compared with the calculated spectra for (R)-DCK (bottom). | ||
A very good agreement was found in a comparison of the calculated VCD spectrum for (R)-DCK with the experimental spectrum of DCK-2, in which all the intense bands were correctly predicted by the calculations. There were two spectral regions, in which it was possible to reliably assign the absolute configuration: first, the region of 1750–1700 cm−1, where an intense band of the valence vibration of carbonyl group is located (negative sign of the band for (R)-DCK) and second, the region of 1350–1280 cm−1, where two positive bands were observed in the spectrum of (R)-DCK. Although the band intensities in the middle part of the spectra (1700–1350) were lower, the spectral profile in this area was also very well predicted by the calculation.
To confirm the results obtained with the VCD spectroscopy, ECD spectroscopy was applied for determining the absolute configuration of DCK enantiomers. Experimental spectra of DCK-1 and DCK-2 (Fig. 5) show again that the previous separation of the enantiomers was successful because of the opposite sign of the spectra. Three intense bands were observed in the experimental spectra: a broad band with the maximum localized approximately at 290 nm and two sharper bands localized at 220 and 194 nm (for major orbital contributions to experimentally observed ECD bands, see ESI†). A very good agreement was found between the calculated ECD spectrum for (R)-DCK and the experimental spectrum of DCK-2, which corresponds with the results obtained from VCD measurements.
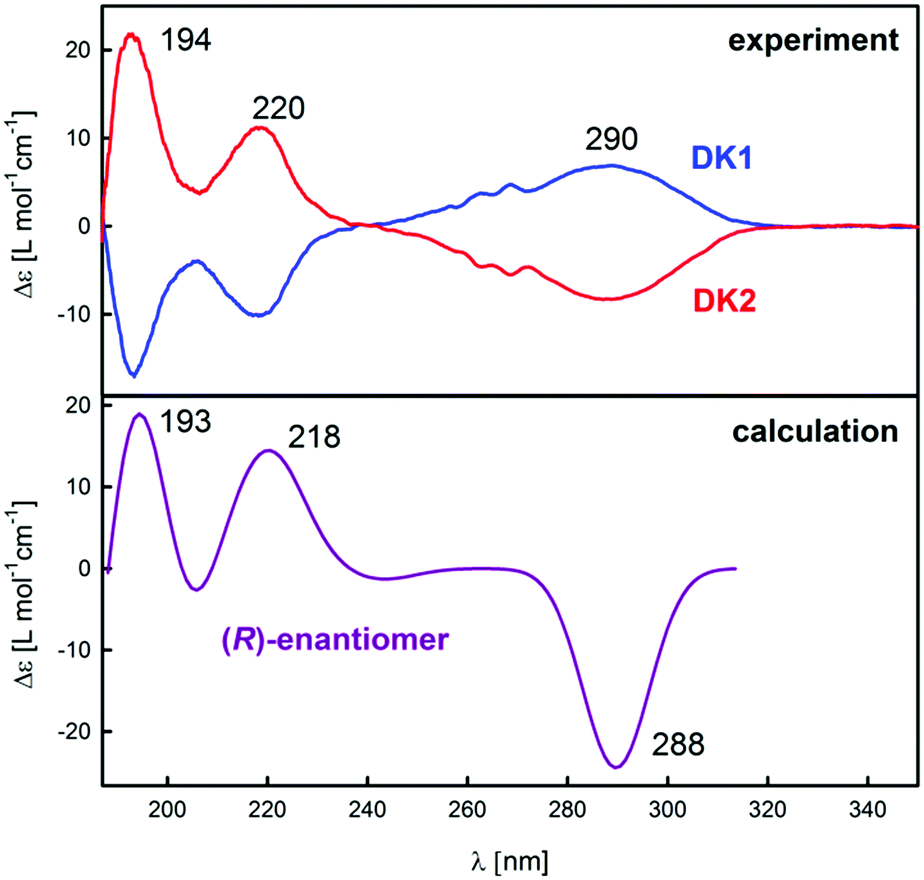 | ||
| Fig. 5 Experimental ECD (top) spectra of DCK-1 and DCK-2 samples compared with the calculated spectra for (R)-DCK (bottom). | ||
X-ray diffraction of hydrochloride salt of enantiomers
In order to confirm the results obtained from spectroscopic methods, X-ray diffraction of single crystals of (S)-DCK, (R)-DCK and racemic forms was performed (for detailed information on crystal system, see ESI†). To this date, powder diffraction pattern of (S)-DCK is the only available data in the literature.24 Therefore, single crystal X-ray analysis was performed and the obtained structures of (R)- and (S)-DCK were compared with the structures predicted by the spectroscopic methods and ab initio calculations. As expected, the resulting crystal structures of the enantiomers were mirror images of each other. Surprisingly, two symmetrically independent molecules were found in the asymmetric unit. Both the spectroscopic methods and X-ray provided identical results. It is interesting to note that the ab initio methods successfully predicted the most favourable conformers of the respective enantiomers, since both these conformers were also detected in the crystal structures (Fig. 6).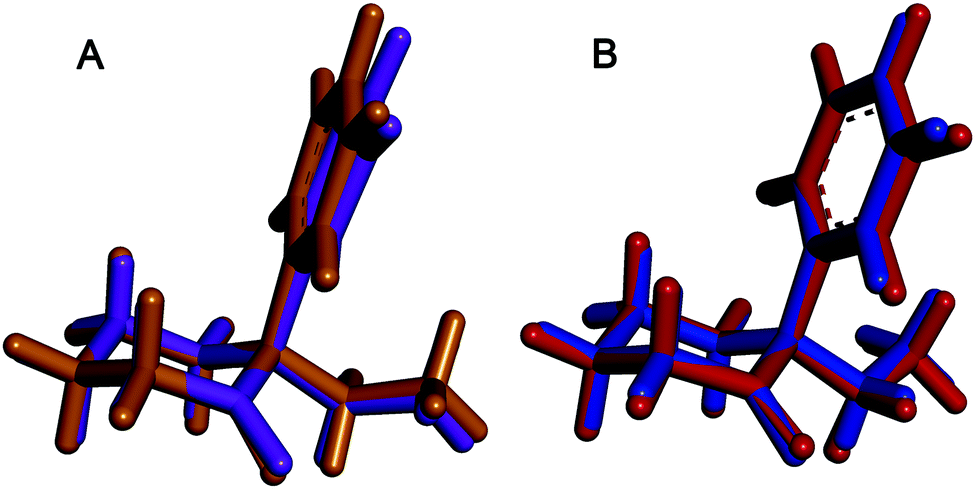 | ||
| Fig. 6 Fitted structures of the most abundant conformers of R-enantiomer (DCK-2) found in the crystal using X-ray analysis (purple and blue) and the most stable conformers predicted by ab initio calculations (ochre and red). | ||
In vitro cytotoxicity of deschloroketamine and its enantiomers
The in vitro cytotoxic profile of deschlorketamine and its two enantiomers was assessed using cell metabolic activity assay WST-1 after 72 h of the compound's treatment. The following cell lines were used to evaluate hepatic, renal, neuronal, cardiac, and urinary toxicity: seven human model cell lines Hep G2, HEK 293T, 5637, HT-29, HMC-3, SH-SY5Y, and HaCaT (derived from liver, kidney, urinary bladder, colon, microglia, neuroblastoma, and epidermis, respectively) as well as one rat H9C2 and one mouse cell line L929 (derived from heart and fibroblasts, respectively). The IC50 values of all tested cell lines after 72 h treatment by (rac)-DCK and its enantiomers are listed in Table 1.| Compound | Cell line | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| Hep G2 | HEK 293T | SH-SY5Y | HMC-3 | HT-29 | 5637 | H9C2 | HaCaT | L929 | |
| IC50 [mM] | |||||||||
| Rac-DCK | 2.10 | 1.43 | 2.48 | 1.54 | 1.58 | 1.52 | 3.00 | 1.82 | 1.86 |
| (S)-DCK | 2.24 | 0.99 | 2.43 | 1.63 | 1.55 | 1.43 | 3.92 | 1.71 | 2.74 |
| (R)-DCK | 2.41 | 1.82 | 3.86 | 1.70 | 1.62 | 1.75 | 4.23 | 2.00 | 2.12 |
The in vitro toxicity of deschlorketamine was concentration-dependent and the IC50 values were in millimolar concentrations. Interestingly, we detected significant differences in cytotoxicities not only among individual compounds but also across the individual cell lines. The highest cytotoxicity was determined for human embryonic kidney cells (HEK 293T), in which (S)-DCK enantiomer reached the IC50 below 1 mM concentration. Moreover, the (S)-DCK was almost two times more potent than (R)-DCK enantiomer. Small differences in the cytotoxicities of individual enantiomers were observed also in an in vitro cardiac model of H9C2 cells and SH-SY5Y cells from neuroblastoma. The latter express opioid, muscarinic and nerve growth factor receptors as well as exhibit neuronal marker enzyme activity.25 Since the differences among the racemate and individual enantiomers were in this cell line rather marginal, we assume that similarly to ketamine,20 the toxicity is not induced via a stereoselective receptor.
Surprisingly, the IC50 of (rac)-DCK and the enantiomers in Hep G2 liver cells, which are a model for hepatotoxicity studies,26 were not significantly decreased compared to other cell lines. This might be caused by very low expression of CYP enzymes in Hep G2 cells. Therefore, the drug metabolites, which are usually the most toxic, cannot be probably properly formed. Nevertheless, at high concentration, the drug may also induce cell death, but without the relation to the CYP activity normally present in primary hepatocytes.27 Even though Hep G2 cells are among the most widely used for this purpose and have many liver-specific functions, some authors have described that they really lack functional expression of most human liver enzyme P450s,28 or that it is ca. 2–3 times lower than in primary hepatocytes.29
Overall, the in vitro cytotoxicity of DCK was higher than what is described for ketamine by, e.g., Braun et al. (2010),20Bai et al. (2011),30 Shen et al. (2015),31 and Kovacova et al. (2016);32 although, needless to say, the IC50 values of ketamine were determined only at 24 and/or 48 h. In clinics, ketamine is used at 1.5 mM concentration33 or even at 93 mM in patients with chronic pain,34 which is concentration much higher than concentrations already inducing toxicity in in vitro evaluations.20 The lower IC50 values of DCK in comparison to ketamine are in agreement with Frison et al. (2016), who reported that DCK is considered to be more potent and longer lasting than ketamine.35
Further cytotoxicity measurement using, e.g., HepaRG cells, by their characteristics resembling primary human hepatocytes, and detailed mechanistic studies will be needed to unravel the toxicity of this NPS.
Fluorescence microscopy
Based on the observations made on ketamine by Braun et al. (2010),20 who described its significant impact on mitochondrial function of iPSc cells already at 20 μM concentration after 24 h treatment (no decrease in cell viability at this concentration) and the fact that, at millimolar concentrations, ketamine induces apoptosis via the mitochondrial pathway independent of death receptor signalling,20 and that 1 mM ketamine induces prolonged elevation of calcium ions in cytosol triggering mitochondrial, and possibly also endoplasmic reticular (ER), stress,36 we have decided to evaluate morphology of mitochondria and endoplasmic reticulum in neuroblastoma cells SH-SY5Y at two sub-IC50 concentrations (250 and 500 μM) of (rac)-DCK and its enantiomers after 72 h treatment by using live-cell fluorescence microscopy imaging, see Fig. 7. | ||
| Fig. 7 Live-cell fluorescence microscopy of SH-SY5Y cells treated with deschlorketamine (rac-DCK) and its enantiomers ((S)-DCK and (R)-DCK) at 500 μM concentration for 72 h. (A, E, I, M) bright field images; (B, F, J, N) endoplasmic reticulum labelled by ER-Tracker Blue-White DPX; (C, G, K, O) mitochondria stained with a mitochondria-specific dye based on a pentamethinium salt; (D, H, L, P) merge. | ||
At 250 μM concentration (72 h), neither (rac)-DCK nor its enantiomers had impact on the morphology of mitochondria and endoplasmic reticulum (data not shown). However, at 500 μM concentration, as it is apparent from Fig. 7, rac-DCK as well as (S)-DCK and (R)-DCK treatment resulted in ER stress pronounced by aggregation of the ER network when compared to control untreated cells (Fig. 7) and cells treated only by the vehicle. Impact of DCK treatment on morphology of mitochondrial network was not apparent at sub-IC50 concentration of 500 μM, nevertheless, we have observed weakened mitochondrial staining in DCK ((S)-DCK, (R)-DCK) treated cells compared to the control. Similar observation was made for ketamine-treated neuron-like cells stained by MitoTracker Red, the fluorescence intensity of which was decreased.20
Discussion
In general, each enantiomer can undergo a different metabolic pathway in the living system, thus, providing different biological response, including differences in adverse effects.37 For ketamine, which we use here as a reference drug, it was reported that the (S)-enantiomer is more potent than the (R)-enantiomer,3 which is in agreement with its higher potency for the inhibition of NMDAR (N-methyl-D-aspartate receptor),38 but also its toxicity.19 The NMDAR inhibitory pathway of ketamine is also responsible for its rapid antidepressant effect, which is vividly studied.39–42 Recently it has been claimed that a major metabolite of ketamine found in human plasma, (2R, 6R)-hydroxynorketamine, acts as a rapid antidepressant in mice through an unspecific activation of AMPAR (α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor).43 This proposed mechanism is not completely accepted yet, since several previous as well as very recent studies point at a significant contribution of NMDAR inhibition in the antidepressant action of ketamine and its metabolites. These studies involve the racemic form, but lately pure (R)- and (S)-ketamine and their enantiomerically pure metabolites, in particular (2R, 6R)-hydroxynorketamine and (2S, 6S)-hydroxynorketamine, seem to play the main role.43–47 Currently, it seems that the long-lasting antidepressant function of ketamine is induced by the hydroxynorketamine metabolites. However, since a long-term treatment, even with very low doses of ketamine, may result in the development of the above-mentioned adverse effects,19,20 alternative drugs with higher potency are required to diminish this risk for patients.Deschloroketamine seems to be an appealing alternative to ketamine, since, according to the anecdotal reports of clubbers and psychonauts, it is 2–3 times more potent than ketamine. This is highly favourable, as deschloroketamine may be most probably administered in much lower doses than ketamine. In addition, it seems to harm the urinary track to a lower extent than ketamine, which may also be beneficial for potential medical application, despite slightly higher toxicity we report here. Considering the enantiomerically pure compounds, the R-enantiomer can be more suitable for potential use in medicine, since it is less toxic than the S-enantiomer. Assuming similarity in the pharmacological profile of deschloroketamine and ketamine, the most active deschloroketamine metabolite i.e. (2R, 6R)-hydroxynordeschloroketamine may also exert rapid and sustained antidepressant activity as well as possess no ketamine-related side effects.43,47
Conclusions
We have synthesized and characterized rac-deschloroketamine and developed a chiral HPLC method to harvest the pure enantiomers, (R)- and (S)-deschloroketamine. The absolute configuration of the single enantiomers has been determined by a combination of ab initio calculated VCD and ECD spectra and their comparison with experimental results. The results have been confirmed by single-crystal X-ray structure analyses of hydrochlorides of both enantiomers. The most stable conformers predicted by calculations have also been found in single crystal structures, which documents high reliability of the chosen combination of spectroscopy with ab initio calculation methods. Similarly to ketamine, (S)-deschloroketamine exhibits higher cytotoxicity than the (R)-enantiomer. However, the observed toxicity is most probably not induced via a stereosensitive receptor. In addition, pathogenic changes on the morphology of mitochondria and endoplasmic reticulum have not been observed at 250 μM concentrations.In conclusion, based on our results, the toxicity of deschloroketamine can be considered similar to ketamine. These results are highly promising, since deschloroketamine is more potent and its effects are longer lasting than those of ketamine. Such properties may be highly beneficial for the potential medical use of deschloroketamine, therefore, its antidepressant effect should be thoroughly investigated.
Experimental section
Chemicals used for the synthesis were obtained from commercial sources (Sigma Aldrich, TCI Europe, Acros Organics). Diethyl ether solution of hydrochloride was prepared from commercial diethyl ether, commercial concentrated hydrochloric acid and calcium chloride as a drying agent. Solvents were acquired from commercial sources and were used after distillation. Solvents denoted as dry, were dried before use by molecular sieves. Other commercial reagents were used without further purification. The reactions were monitored with the aid of thin-layer chromatography (TLC Silica gel 60 F254) and pre-coated reverse phase gel plates (TLC RP-18 F254). All operations involving air-sensitive reagents were performed under an inert atmosphere of dry argon and dried solvents. Thermal rearrangement was carried out in a microwave reactor (Biotage Initiator Classic). Melting points were determined using a melting point apparatus (PGH Rundfunk–Fernsehen), and they are uncorrected. Solvents used for analytical and preparative enantioseparation were of standard HPLC grade and were purchased from VWR International. NMR spectroscopy was performed on an Agilent 400-MR DDR2 spectrometer operating at 400.13 MHz for 1H and 100.62 MHz for 13C. Chemical shifts were referenced internally to the signal of the residual solvent. 2D NMR spectra were obtained using standard sequences as supplied by the manufacturer.Synthetic procedures
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 137.1 (Ar-Cq), 132.8 (Ar-CH), 128.63 (Ar-CH), 128.58 (Ar-CH), 46.5 (CH), 30.1 (2 × CH2), 26.4 (2 × CH2).
O), 134.9 (Ar-Cq), 132.9 (Ar-CH), 130.4 (Ar-CH), 128.3 (Ar-CH), 71.3 (Cq), 41.1 (2 × CH2), 23.6 (2 × CH2).
O), 131.9 (Ar-CH), 131.3 (Ar-CH), 131.0 (Ar-Cq), 129.4 (Ar-CH), 73.0 (Cq), 40.1 (CH2), 32.8 (CH2), 28.6 (CH2), 27.2 (CH3), 22.8 (CH2).
O), 137.1 (Ar-Cq), 132.8 (Ar-CH), 128.63 (Ar-CH), 128.58 (Ar-CH), 46.5 (CH), 30.1 (2 × CH2), 26.4 (2 × CH2).
O), 134.9 (Ar-Cq), 132.9 (Ar-CH), 130.4 (Ar-CH), 128.3 (Ar-CH), 71.3 (Cq), 41.1 (2 × CH2), 23.6 (2 × CH2).
O), 131.9 (Ar-CH), 131.3 (Ar-CH), 131.0 (Ar-Cq), 129.4 (Ar-CH), 73.0 (Cq), 40.1 (CH2), 32.8 (CH2), 28.6 (CH2), 27.2 (CH3), 22.8 (CH2).
Enantioseparation procedure
Analytical enantioseparation of deschloroketamine was performed using Agilent 1100 series system equipped with a diode array detector, binary pump, column thermostat, degasser and an autosampler. Sample concentration was 1 mg mL−1, the flow rate was set to 1 mL min−1 and the temperature was kept at 23 °C (temperature was set to match the room temperature at which the preparative separation was performed). Measurements were carried out on a chiral polysaccharide column ChiralArt Amylose SA.Preparative scale enantioseparation was carried out using AutoPurification System (Waters) equipped with a binary pump, column manager, diode array detector and fraction collector. The enantioseparation was performed on a chiral column Chiralpak IA 250 × 21 mm ID, 5 μm, which contained the same type of a chiral selector as the analytical column, namely tris[3,5-dimethylphenylcarbamoyl]amylose immobilized on the 5 μm silica support. The sample concentration was 10 mg mL−1, the flow rate was set to 15 mL min−1.
Spectroscopic measurements
For all electronic circular dichroism (ECD) measurements, solutions of deschloroketamine hydrochloride were used in demineralised water with a concentration of 7 mg L−1. The ECD spectra were recorded by J-815 spectrometer (Jasco, Japan) in a quartz cuvette (Hellma, Germany) with the optical path length of 1 cm at room temperature. The measurement conditions were set as follows: a spectral region of 185–350 nm, scanning speed of 20 nm min−1, response time of 8 s, resolution of 0.5 nm, band width of 1 nm, and sensitivity of 100 mdeg. The final spectra were obtained as an average of 3 accumulations, which were corrected for the baseline by subtracting the spectra of demineralised water under the same experimental conditions.For all infrared spectroscopy (IR) and vibrational circular dichroism (VCD) measurements, solutions of deschloroketamine hydrochloride in D2O (ISOSAR deuterierte Produkte GmbH, Germany, at least 99.8% of D) with a concentration of 100 g L−1 were used at room temperature. The IR and VCD spectra were recorded in a spectral region of 1750–1275 cm−1 using FT-IR IFS 66/S spectrometer equipped with PMA 37 VCD/IRRAS module (Bruker, Germany). The samples were measured in BioCell (Biotools, USA) with CaF2 windows and the optical path length of 23 μm. The VCD spectra were recorded with the resolution of 4 cm−1 and they are presented as an average of 7–9 blocks, each consisting of 2260 scans.
Computational methods
For all computations, the solute–solvent interactions were considered via the conductor-like continuum model implemented within the polarisable continuum model48 with a default dielectric constant for water of ε = 78.3553. Boltzmann populations, needed for computing the weighted average of spectra of the lowest-energy conformers, were derived from relative Gibbs free energies at a temperature of 298 K.IR and VCD spectra were calculated at the ωB97X-D/TZVP level. The IR and VCD spectra were visualized using Lorentzian band profile with a bandwidth at half-maximum of 6 cm−1. Transition frequencies were scaled in order to improve the similarity of theoretical and experimental spectra23 and to ensure the correct assigning of corresponding bands.
ECD spectra were calculated at the CAM-B3LYP/aug-cc-pVDZ levels. For visualization of the calculated spectra, Gaussian band profile was used. Bandwidths were optimized for individual bands in order to increase similarity between calculated and experimental spectra.
Crystallographic study
X-ray structure analysis was carried out on a Bruker D8 VENTURE system equipped with a multilayer monochromator, a Cu Kα Incoatec microfocus sealed tube (λ = 1.54178 Å) using combined ϕ and ω scans at 180 K. Data collection: APEX3 v2015;49 unit cell refinement: APEX3 v2015; data reduction: SAINT.49 The structure was solved by direct methods in SIR92;50 full matrix least squares refinement on F2 was provided by CRYSTALS.51The X-ray CIF files for the structures have been deposited at the Cambridge Crystallographic Data Center with the deposition numbers CCDC 1838630–1838632.†
All H atoms were located in a difference map, but repositioned geometrically, then, they were initially refined with soft restraints on the bond lengths and angles to regularize their geometry (C–H in the range of 0.93–0.98 Å, N–H set to 0.86 Å, and O–H to 0.82 Å) and Uiso(H) (in the range 1.2–1.5 times Ueq of the parent atom), after which the positions of carbon bound hydrogen atoms were refined with riding constraints. The same set of soft restraints was used during the final stages of the refinement, when the positional parameters of the hetero bound hydrogen atoms were refined. Absolute configuration was determined by the refinement of Flack's x parameter.
Evaluation of in vitro cytotoxicity
Cytotoxicity of deschlorketamine and its R- and S-enantiomer was evaluated in vitro by a WST-1 viability assay (Sigma, USA), based on reduction of tetrazolium salt (WST-1 substrate) into soluble formazan by mitochondrial oxidoreductases in metabolically active cells. The following human cell lines were used for this assay: cells from hepatocellular carcinoma (Hep G2), cells from colorectal adenocarcinoma (HT-29), keratinocytes (HaCaT), embryonic kidney cells (HEK 293T), cells from urinary bladder grade II carcinoma (5637), microglial cells (HMC3), cells from neuroblastoma (SH-SY5Y); one rat cell line derived from heart (H9C2), and one mouse cell fibroblast cell line (L929). The cells were seeded into individual wells of 96-well plates (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 cells per well) in 100 μL of cell culture medium recommended by ATCC (American Tissue Culture Collection) supplemented with 10% fetal bovine serum (FBS). After 16 h of incubation at standard cell cultivation conditions (37 °C, 5% CO2, 95% humidity), the cells were influenced with the tested compounds at final concentrations of 0–5 mM in 100 μL of fresh cell cultivation media with 10% FBS. After 72 h, the cell culture medium was removed and fresh 100 μL of cell culture medium without phenol red with 10 μL of WST-1 was added. Then, after 2 h incubation, the absorbance of formed formazan was measured spectrophotometrically at 450 nm (the reference wavelength of 630 nm) using UV-vis spectrometer (BioRad). The absorbance is directly proportional to the oxidoreductase activity, and thus to the number of metabolically active cells. All samples were tested in quadruplicates. Cells treated only with cell culture medium and cells treated with a vehicle (dimethyl sulfoxide and methanol) served as controls. The IC50 values were determined as the concentration necessary to kill 50% of cells.
000 cells per well) in 100 μL of cell culture medium recommended by ATCC (American Tissue Culture Collection) supplemented with 10% fetal bovine serum (FBS). After 16 h of incubation at standard cell cultivation conditions (37 °C, 5% CO2, 95% humidity), the cells were influenced with the tested compounds at final concentrations of 0–5 mM in 100 μL of fresh cell cultivation media with 10% FBS. After 72 h, the cell culture medium was removed and fresh 100 μL of cell culture medium without phenol red with 10 μL of WST-1 was added. Then, after 2 h incubation, the absorbance of formed formazan was measured spectrophotometrically at 450 nm (the reference wavelength of 630 nm) using UV-vis spectrometer (BioRad). The absorbance is directly proportional to the oxidoreductase activity, and thus to the number of metabolically active cells. All samples were tested in quadruplicates. Cells treated only with cell culture medium and cells treated with a vehicle (dimethyl sulfoxide and methanol) served as controls. The IC50 values were determined as the concentration necessary to kill 50% of cells.
Fluorescence microscopy
SH-SY5Y cells (1 × 105 per well) were seeded into 35 mm glass bottom dishes for live-cell imaging (Ibidi plates, Germany) and left to adhere overnight (ca. 16 h). The attached cells were washed with pre-warmed phosphate buffered saline (PBS, pH 7.4, 37 °C) and incubated with deschloroketamine or its enantiomers (0.1, 0.25 and 0.5 mM concentration) dissolved in complete cell culture medium without phenol red (FluoroBrite DMEM, Thermo Fisher Scientific) for 72 h. Stock solutions of the tested compounds were prepared in methanol, which was always used as a control. After the incubation period, the cells were washed with PBS. Then, cell organelles, such as mitochondria and endoplasmic reticulum were stained with a mitochondria-specific dye (50 nM, 10 min),52 and a commercial marker ER-Tracker Blue-White DPX (Thermo Fisher Scientific; 120 nM, 30 min). After the incubation period, the cells were washed with PBS and fed with fresh phenol red-free medium. The impact of deschloroketamine on cell morphology and physiology of cell organelles was studied by real-time live-cell fluorescence microscopy at 37 °C and in 5% CO2 atmosphere. The images were acquired by an inverse fluorescence microscope Olympus IX-81 equipped with xCellence system (high-stability 150 W xenon arc burner, EM-CCD camera C9100-02, 60× oil immersion objective with NA 1.4) and by OMX Blaze microscope (Softworx, sCMOS camera, 60× oil immersion objective).Abbreviations
| 5637 | Urinary bladder cells |
| AMPAR | α-Amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptor |
| B3LYP | Becke, three-parameter, Lee–Yang–Parr exchange correlation functional |
| CAM | Coulomb attenuated method |
| CYP | Cytochrome P450 enzymes |
| DCK | Deschloroketamine |
| DFT | Density functional theory |
| ECD | Electron circular dichroism |
| EMCDDA | European monitoring centre for drugs and drug addiction |
| ESI | Electronic supplementary information |
| H9C2 | Mouse cell line derived from heart cells |
| HaCaT | Epidermis cells |
| HEK 293T | Embryonic kidney cells |
| Hep G2 | Liver cells |
| HMC3 | Microganglia cells |
| HPLC | High-performance liquid chromatography |
| HT-29 | Colon cells |
| IC50 | Half maximal inhibitory concentration |
| L929 | Mouse fibroblast cells |
| ωB97X | Dual-corrected exchange–correlation functional |
| NMDAR | N-Methyl-D-aspartate receptor |
| NPS | New psychoactive substances |
| SH-SY5Y | Neuroblastoma cells |
| VCD | Vibrational circular dichroism. |
Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
This research was supported by Czech Science Foundation (project No. 16-17689Y), Ministry of Interior of the Czech Republic (MV0/VI20172020056), JIGA 2017 UCT grant and Specific University Research (project 20-SVV/2017 and 21-SVV/2018).Notes and references
- European Monitoring Centre for Drugs and Drug Addiction and Europol, European Drug Report 2017: Trends and Developments, Luxembourg, 2017 Search PubMed.
- J. D. Tobias, L. D. Martin and R. C. Wetzel, Crit. Care Med., 1990, 18, 819–821 CrossRef CAS PubMed.
- Novel psychoactive substances: classification, pharmacology and toxicology, ed. D. M. Wood and P. I. Dargan, Elsevier, London, 2014 Search PubMed.
- J. B. Cole, J. C. Moore, P. C. Nystrom, B. S. Orozco, S. J. Stellpflug, R. L. Kornas, B. J. Fryza, L. W. Steinberg, A. O’Brien-Lambert, P. Bache-Wiig, K. M. Engebretsen and J. D. Ho, Clin. Toxicol., 2016, 54, 556–562 CrossRef CAS PubMed.
- K. L. R. Jansen, J. Psychoact. Drugs, 2000, 32, 419–433 CrossRef CAS PubMed.
- C. J. A. Morgan, L. Muetzelfeldt and H. V. Curran, Addiction, 2010, 105, 121–133 CrossRef PubMed.
- Q. Li, W. M. Chan, J. A. Rudd, C. M. Wang, P. Y. H. Lam, M. S. M. Wai, D. M. Wood, P. I. Dargan and D. T. Yew, in Novel Psychoactive Substances: Classification, Pharmacology and Toxicology, ed. D. M. Wood and P. I. Dargan, Elsevier, London, 2014, ch. 12, pp. 285–307 Search PubMed.
- S. Sassano-Higgins, D. Baron, G. Juarez, N. Esmaili and M. Gold, Depression Anxiety, 2016, 33, 718–727 CrossRef CAS PubMed.
- B. Short, J. Fong, V. Galvez, W. Shelker and C. K. Loo, Lancet Psychiatry, 2018, 5, 65–78 CrossRef PubMed.
- L. Hondebrink, E. E. J. Kasteel, A. M. Tukker, F. M. J. Wijnolts, A. H. A. Verboven and R. H. S. Westerink, Neuropharmacology, 2017, 123, 1–9 CrossRef CAS PubMed.
- S. Beharry and S. Gibbons, Forensic Sci. Int., 2016, 267, 25–34 CrossRef CAS PubMed.
- H. Kang, P. Park, Z. A. Bortolotto, S. D. Brandt, T. Colestock, J. Wallach, G. L. Collingridge and D. Lodge, Neuropharmacology, 2017, 112, 144–149 CrossRef CAS PubMed.
- C. Stevens and D. Parke, US Pat., US3254124, 1966 Search PubMed.
- C. L. Stevens, H. T. Hanson and K. G. Taylor, J. Am. Chem. Soc., 1966, 88, 2769–2774 CrossRef CAS.
- D. Preiss and A. Tatar, US Pat., US6083992 (A), 2000 Search PubMed.
- D. Preiss and A. Tatar, German Pat., DE4409671C1, 1995 Search PubMed.
- PsychonautWiki Contributors, Methoxetamine, https://psychonautwiki.org/w/index.php?title=Methoxetamine&oldid=125222, accessed 22 May 2018.
- M. Kohout, J. Vandenbussche, A. Roller, J. Tůma, J. Bogaerts, P. Bultinck, W. Herrebout and W. Lindner, RSC Adv., 2016, 6, 81461–81465 RSC.
- S. T. Lee, T. T. Wu, P. Y. Yu and R. M. Chen, Br. J. Anaesth., 2009, 102, 80–89 CrossRef CAS PubMed.
- S. Braun, N. Gaza, R. Werdehausen, H. Hermanns, I. Bauer, M. E. Durieux, M. W. Hollmann and M. F. Stevens, Br. J. Anaesth., 2010, 105, 347–354 CrossRef CAS PubMed.
- J.-D. Chai and M. Head-Gordon, Phys. Chem. Chem. Phys., 2008, 10, 6615–6620 RSC.
- M. J. Frisch, et al., Gaussian 09, Wallingford, CT, 2009 Search PubMed.
- E. Debie, E. De Gussem, R. K. Dukor, W. Herrebout, L. A. Nafie and P. Bultinck, ChemPhysChem, 2011, 12, 1542–1549 CrossRef CAS PubMed.
- J. Maixner, B. Jurásek, M. Kohout, M. Kuchař and P. Kačer, Powder Diffr., 2017, 32, 193–195 CrossRef CAS.
- D. J. Barbosa, J. P. Capela, R. Silva, V. Vilas-Boas, L. M. Ferreira, P. S. Branco, E. Fernandes, M. de, L. Bastos and F. Carvalho, Arch. Toxicol., 2014, 88, 455–473 CrossRef CAS PubMed.
- S. Knasmüller, V. Mersch-Sundermann, S. Kevekordes, F. Darroudi, W. Huber, C. Hoelzl, J. Bichler and B. Majer, Toxicology, 2004, 198, 315–328 CrossRef PubMed.
- L. Jia and X. Liu, Curr. Drug Metab., 2007, 8, 822–829 CrossRef CAS PubMed.
- M. Gomez-Lechon, M. Donato, A. Lahoz and J. Castell, Curr. Drug Metab., 2008, 9, 1–11 CrossRef.
- C. Rodríguez-Antona, M. T. Donato, A. Boobis, R. J. Edwards, P. S. Watts, J. V. Castell and M.-J. Gómez-Lechón, Xenobiotica, 2002, 32, 505–520 CrossRef PubMed.
- X. Bai, Y. Yan, S. Canfield and Z. Bosnjak, FASEB J., 2011, 25, 1043.11 Search PubMed.
- C. H. Shen, S.-T. Wang, Y.-R. Lee, S.-Y. Liu, Y.-Z. Li, J.-D. Wu, Y.-J. Chen and Y.-W. Liu, Mol. Med. Rep., 2015, 11, 887–895 CrossRef CAS PubMed.
- V. Kovacova, E. E. E. Abdelsalam, H. Bandouchova, J. Brichta, B. Havelkova, V. Piacek, F. Vitula and J. Pikula, Neuroendocrinol. Lett., 2016, 37, 78–83 Search PubMed.
- Y. Chia, Y. Liu, N. A. Wong, K. Liu and H. Chang, Anesth. Analg., 1998, 86, 265S CrossRef.
- J. H. Vranken, D. Troost, J. T. Wegener, M. R. Kruis and M. H. van der Vegt, Pain, 2005, 117, 231–235 CrossRef CAS PubMed.
- G. Frison, L. Zamengo, F. Zancanaro, F. Tisato and P. Traldi, Rapid Commun. Mass Spectrom., 2016, 30, 151–160 CrossRef CAS PubMed.
- S. C. Baker, S. Shabir, N. T. Georgopoulos and J. Southgate, Am. J. Pathol., 2016, 186, 1267–1277 CrossRef CAS PubMed.
- G.-Q. Lin, J.-G. Zhang and J.-F. Cheng, Chiral Drugs, John Wiley & Sons, Inc., 2011, pp. 3–28 Search PubMed.
- R. Moaddel, G. Abdrakhmanova, J. Kozak, K. Jozwiak, L. Toll, L. Jimenez, A. Rosenberg, T. Tran, Y. Xiao, C. A. Zarate and I. W. Wainer, Eur. J. Pharmacol., 2013, 698, 228–234 CrossRef CAS PubMed.
- R. M. Berman, A. Cappiello, A. Anand, D. A. Oren, G. R. Heninger, D. S. Charney and J. H. Krystal, Biol. Psychiatry, 2000, 47, 351–354 CrossRef CAS PubMed.
- E. Dolgin, Nat. Med., 2013, 19, 8 CrossRef PubMed.
- D. Feifel, B. Malcolm, D. Boggie and K. Lee, J. Affective Disord., 2017, 221, 283–288 CrossRef CAS PubMed.
- I. Singh, C. Morgan, V. Curran, D. Nutt, A. Schlag and R. McShane, Lancet Psychiatry, 2017, 4, 419–426 CrossRef PubMed.
- P. Zanos, R. Moaddel, P. J. Morris, P. Georgiou, J. Fischell, G. I. Elmer, M. Alkondon, P. Yuan, H. J. Pribut, N. S. Singh, K. S. S. Dossou, Y. Fang, X.-P. Huang, C. L. Mayo, I. W. Wainer, E. X. Albuquerque, S. M. Thompson, C. J. Thomas, C. A. Zarate Jr and T. D. Gould, Nature, 2016, 533, 481–486 CrossRef CAS PubMed.
- A. E. Autry, M. Adachi, E. Nosyreva, E. S. Na, M. F. Los, P. Cheng, E. T. Kavalali and L. M. Monteggia, Nature, 2011, 475, 91–95 CrossRef CAS PubMed.
- R. Malinow, Nature, 2016, 533, 477–478 CrossRef CAS PubMed.
- K. Suzuki, E. Nosyreva, K. W. Hunt, E. T. Kavalali and L. M. Monteggia, Nature, 2017, 546, E1–E3 CrossRef CAS PubMed.
- P. Zanos, R. Moaddel, P. J. Morris, P. Georgiou, J. Fischell, G. I. Elmer, M. Alkondon, P. Yuan, H. J. Pribut, N. S. Singh, K. S. S. Dossou, Y. Fang, X.-P. Huang, C. L. Mayo, E. X. Albuquerque, S. M. Thompson, C. J. Thomas, C. A. Zarate and T. D. Gould, Nature, 2017, 546, E4 CrossRef CAS PubMed.
- G. Scalmani and M. J. Frisch, J. Chem. Phys., 2010, 132, 114110 CrossRef PubMed.
- APEX3, S.B. APEX3, SAINT. BRUKER AXS. Inc. Madison, Wisconsin, USA, 2015.
- A. Altomare, G. Cascarano, C. Giacovazzo, A. Guagliardi, M. C. Burla, G. Polidori and M. Camalli, J. Appl. Crystallogr., 1994, 27, 435–436 Search PubMed.
- P. W. Betteridge, J. R. Carruthers, R. I. Cooper, K. Prout and D. J. Watkin, J. Appl. Crystallogr., 2003, 36, 1487 CrossRef CAS.
- S. Rimpelová, T. Bříza, J. Králová, K. Záruba, Z. Kejík, I. Císařová, P. Martásek, T. Ruml and V. Král, Bioconjugate Chem., 2013, 24, 1445–1454 CrossRef PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Details on synthesis and characterization of racemic deschloroketamine, preparative enantiomer separation and single-crystal X-ray parameters is available. CCDC 1838630–1838632. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c8nj03107j |
| ‡ Contributed equally. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |