
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Supported heterogeneous catalysts: what controls cobalt nanoparticle dispersion on alumina?†
Matthys J.
Loedolff
,
Bee-Min
Goh
,
George A.
Koutsantonis
 * and
Rebecca O.
Fuller‡
*
* and
Rebecca O.
Fuller‡
*
School of Molecular Sciences M310, The University of Western Australia, Crawley WA 6009, Australia. E-mail: rebecca.fuller@curtin.edu.au
First published on 7th August 2018
Abstract
We investigate how a number of physical parameters control the rate and pattern of nanoparticle assemblage onto a commercially available alumina surface. 8 nm ε-Co nanoparticles supported on polycrystalline alumina are found to have areas of both good dispersion and areas of aggregation. A similar pattern of dispersion was also observed for larger (∼30 nm) polycrystalline ferromagnetic ε-Co nanoparticles. Acid and base treatment of the amphoteric support material prior to the assemblage process is found to have little impact on dispersion of the particles. Using a nonpolar solvent for the assemblage process eliminates the effect of zeta potential and allows for rapid attachment of particles to the support. Performing the assemblage in a polar solvent is found to significantly decrease the rate of the particle attachment to the support. Despite the slower attachment of particles, there is no impact on the nanoparticle distribution pattern. In contrast to the mixed dispersion observed when assembling nanoparticles on an alumina support, ε-Co nanoparticles are found to disperse uniformly across an ordered mesoporous MCM-41 silica support. It seems likely that a specific chemical interaction between the support surface and nanoparticle are dictating the assemblage process.
Introduction
The Fischer–Tropsch (FT) reaction involves the conversion of CO and H2 over a transition metal catalyst into a myriad of valuable hydrocarbons. Although first reported1 in the 1920's, substantial interest in this concept has continued to exist. The FT process provides an alternative to oil for the production of useful end products. On a commercial scale natural gas or coal are utilized to produce the synthesis gas for the FT reaction. In this process, the natural gas (or coal) is firstly converted to a suitable mixture of carbon monoxide and hydrogen, which subsequently involves the hydrogenation of carbon monoxide over an appropriate transition metal catalyst. The range of products that result from the process is affected by a number of factors including the catalyst.2,3 A strong relationship is known to exist between the size of the metal aggregates and the catalytic activity.4 Supported metal clusters do not tend to maintain their form and hence, activity is affected in the reaction stream for industrial processes.5,6 Therefore, metal aggregates supported on inorganic support materials are the industry standard catalytic material.7Metal aggregates can be incorporated onto inorganic supports by a number of methods, these broadly fall into two categories.8 Firstly, metals can be incorporated directly during the synthesis of the support material in co-condensation and ion-exchange processes.9,10 The limitations to the one pot method include low dopant concentration, reduced metal reactivity from the formation of metal species in the walls of porous supports as well migration of the metal to the surface and subsequent agglomeration.10 To address these problems, catalysts can also be produced by the addition of the metal centres after support-synthesis; such methods include wet impregnation and grafting.9,10 Wet impregnation involves metal salts being deposited onto the support and then subsequently reduced into metal aggregates.9 This often results in the formation of aggregates of various sizes and compositions, hence impacting negatively on the selectivity of the catalysts.10,11 Furthermore, there is often a general lack of stability in these materials under the heating required in the reduction of salt or removal of ligand step.12 Traditional heterogeneous FT catalysts have deposited metal catalytic particles with a variable shape, composition and size and which are often mobile on heating.13 The development of new catalysts based on monodisperse nanoparticles is one solution for improving selectivity in the FT process.14
Nanoparticles provide a means for the production of a FT catalyst with finely tuned properties.15 Catalysts based on preformed nanoparticles allow greater control over the active metal species since it is no longer formed in situ but using methodologies that allow the selection of particle size and composition. Nanoparticles composed of FT active metals can be synthesised using hydrothermal techniques which give monodispersed, highly crystalline particles in a number of geometries.16 Particles can then be incorporated directly into the support material allowing greater control over what species are assembled onto the support material. The potential for aggregation of metal clusters in traditional catalysts is minimised. Addition of pre-formed nanoparticles provides an alternative method to wet impregnation and grafting. Despite the ease of preparation of these materials and the demonstrated stability during calcination, there are few reports involving the assemblage of preformed particles onto a support.17–22
Catalytic performance is not only determined by composition, size and shape of the metal species. Dispersion is also a key consideration for the development of catalysts.23 For nanoparticles to find use as heterogeneous catalysts, they must be deposited onto a solid support in a well-controlled way. Such fine-control over the physical structure will lead to improved catalytic processes. Traditional FT catalysts based on metal clusters, have been known to aggregate10 but preformed nanoparticle catalysts have shown both good dispersion in addition to aggregation.21
How particles assemble over a support and the rate at which this occurs is the result of a number of factors including surfactant choice, particle size and morphology, zeta potential, support surface, and temperature.21 These factors influence the competing interactions (van der Waals, steric, magnetic and electrostatic interactions) that occur between the nanoparticles themselves and the nanoparticles and the substrate.24 This competition results in the complex particle dynamics manifesting in deposition patterns on the support material. Theoretically, this area of research is well-established.25 However, experimentally systems are complex and often do not follow expected behaviour; for example, substrate roughness changes the predictions of simple theories.26
While many transition metals display some activity for the FT process, only iron and cobalt are suitable for commercialisation.27 Despite increased costs, cobalt has significantly higher activity, and maintains activity for longer than iron catalysts. Cobalt is the preferred metal for natural gas feedstocks and hence is the favoured catalyst for the petroleum industry.28 As a result, cobalt catalysts are the most developed FT catalysts. Particular focus is on the selectively that results from metal aggregate size.23,29–32 Although a number of catalysts based preformed cobalt particles have been produced,13,20,22,33 dispersion of preformed particles has not been a significant area of focus to date. In this work, we aim to provide an insight into cobalt dispersion across a support. Alumina is the industry standard as an FT catalytic support. We focus on the role of the alumina in the assemblage of particles by changing the surface chemistry of the support through base and acid treatment prior to use. The surface chemistry of the polycrystalline and amphoteric support is complex. Hence we have also investigated the use of a more ordered support material based on silica. The effect of rate of adherence on particle dispersion is explored by altering the solvent used for the particle incorporation into the support.
Experimental section
Co particle synthesis
All synthetic manipulations and work up were carried out using standard Schlenk techniques in an argon atmosphere. Small 8 nm ε-cobalt nanoparticles have been synthesised using the hydrothermal technique.34–37 Larger polycrystalline ε-Co particles (∼30 nm) have also been synthesised using a literature method.38 In both instances, particles were precipitated from the suspension by adding methanol (∼50 mL) to the cooled reaction. The solvent was then removed by vacuum and the product dried under and stored in under argon.Co nanoparticle assemblage onto alumina support
Nanoparticles were assembled onto the alumina support using a modification of our literature procedure.18 In this case, the assemblage is carried out in an inert Ar atmosphere using standard Schlenk procedures. Briefly, 20–30 mL hexane (or chloroform) is added to the cobalt nanoparticles (0.260 g), tridecanol is added to ensure particles are well dispersed. In a separate flask, 20–30 mL hexane (or chloroform) is added to ∼2.75 g of alumina (SASOL PURALOX® SBa200 batch B27013 size <90 micron) calcined in air at 750 °C for 2.5 h (pore diameter ca. 84 Å BET 161 m2 g−1), the slurry is agitated mechanically. The nanoparticle suspension is transferred by cannula to the alumina slurry, significant agitation is maintained for 1–2 hours. The hexane (or chloroform) is subsequently removed by cannula and the light grey powder is then dried under vacuum and stored in under argon.Calcination of Co nanoparticles assembled onto alumina support
The alumina supported nanoparticles were heated at 200 °C for one hour under a flow of nitrogen. The materials were then calcined under a flow of air at the same temperature for a further three hours.Co nanoparticle assemblage onto MCM-41
Our previously developed method of adhering nanoparticles on the support materials is used.18 Nanoparticles are reacted with the well-ordered silicate (MCM-41) that has been prepared via a literature method.39 In this case the assemblage is carried out in an inert Ar atmosphere using standard Schlenk procedures. Briefly, 10–20 mL hexane is added to the nanoparticles (∼8 nm ε-Co nanoparticles). In a separate flask, 10–20 mL hexane is added to calcined MCM-41 (pore diameter 22 Å, BET 1107 m2 g−1) and the slurry is agitated. The nanoparticle suspension is transferred by cannula to the silica suspension and agitated for a further 5 minutes. The solid is recovered from the colourless hexane by filtration and the light grey powder is then dried under vacuum and stored in under argon.Calcination of Co nanoparticles assembled onto MCM-41
The silica supported nanoparticles were heated at either 200 °C or 500 °C for one hour under a flow of nitrogen. The materials were then calcined under a flow of air at the same temperature for a further three hours.Alumina support treatment
The treatment of the PURALOX® alumina is a modification of a previously reported method.40 The alumina (SASOL PURALOX® SBa200 batch B27013 size <90 micron) was calcined at 750 °C in air for 2 hours prior to chemical treatment. Following calcination, the alumina was treated with either a solution of acetic acid at pH 2 or 4 or a solution of NH4OH with pH 8 or 12. The supports were placed in Teflon-lined autoclave for hydrothermal treatment at 220 °C for 2 hours. The supports were collected by filtration and dried in air at 120 °C for 2 hours. The supports were then calcined at 750 °C in air for 2.5 hours. The modified supports were then characterized by BET and XRD.Co nanoparticle assemblages onto treated supports
Nanocrystals of ε-Co (∼8 nm) were assembled onto the four chemically treated supports and subsequently calcined. With the exception of using a chemically treated support, the experiment did not differ to that outlined previously for untreated alumina support.Characterisation
Transmission electron microscopy (TEM) was conducted on a JEOL 3000F operating at 300 kV and equipped with a Gatan Orius SC1000. Compositional analysis in the TEM was performed using energy dispersive X-ray (EDX) spectroscopy (Oxford Instruments, JEOL 3000F) and energy filtered TEM (EFTEM) (Gatan, CM200).Powder X-ray diffraction (XRD) patterns were recorded at room temperature on a Panalytical Empyrean X-ray diffractometer with CuKα 1.54060 Å radiation generated at 40 kV and 40 mA. The amount of Co and Al in the calcined material was determined by X-Ray Fluorescence (XRF) Spectrometry (Ultra Trace Pty Ltd). Samples were cast using a 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :22 flux to form a glass bead that could be analysed by XRF.
:22 flux to form a glass bead that could be analysed by XRF.
N2 absorption and desorption isotherms were measured at 77 K for both the calcined support materials and calcined assembled material catalysts using a TriStar II 3020. Prior to measurement the sample (∼0.1 g) was degassed under vacuum overnight at 130 °C. Specific surface areas were estimated using BET analysis and pore diameters by BJH desorption.
Results and discussion
ε-Co nanoparticle assemblage onto alumina support
Spherical nanocrystals of 8 nm in the ε-Co phase have been synthesised using a previously reported procedure, details of the characterization can be found in the ESI† (Fig. S1). To maintain the integrity of the cobalt particles and prevent surface oxidation prior to assemblage all procedures and storage were carried out in an inert environment. The as-synthesised particles were subsequently reacted with a commercially obtained alumina. Calcination of the alumina was optimised prior to assemblage so that a slight decrease in surface area (BET 185 to 162 m2 g−1) but an increase in the pore volume (BJH 75 to 84 Å) is observed. On addition to the support, the black nanoparticle suspension instantaneously became colourless, suggesting particles adhered to the support quickly in comparison to systems based on iron that typically took a few to several hours.18,19,21 Bright field TEM images of typical uncalcined nanomaterial are shown in Fig. 1. Calcination up to 200 °C did not alter the appearance of the nanomaterial. Supporting the materials should be stable for the reduction steps required prior to use as a catalyst. The calcined material had a surface area (BET ∼ 155 m2 g−1) and pore widths (BJH ∼ 86 Å) with a nanoparticle loading of 3.4%.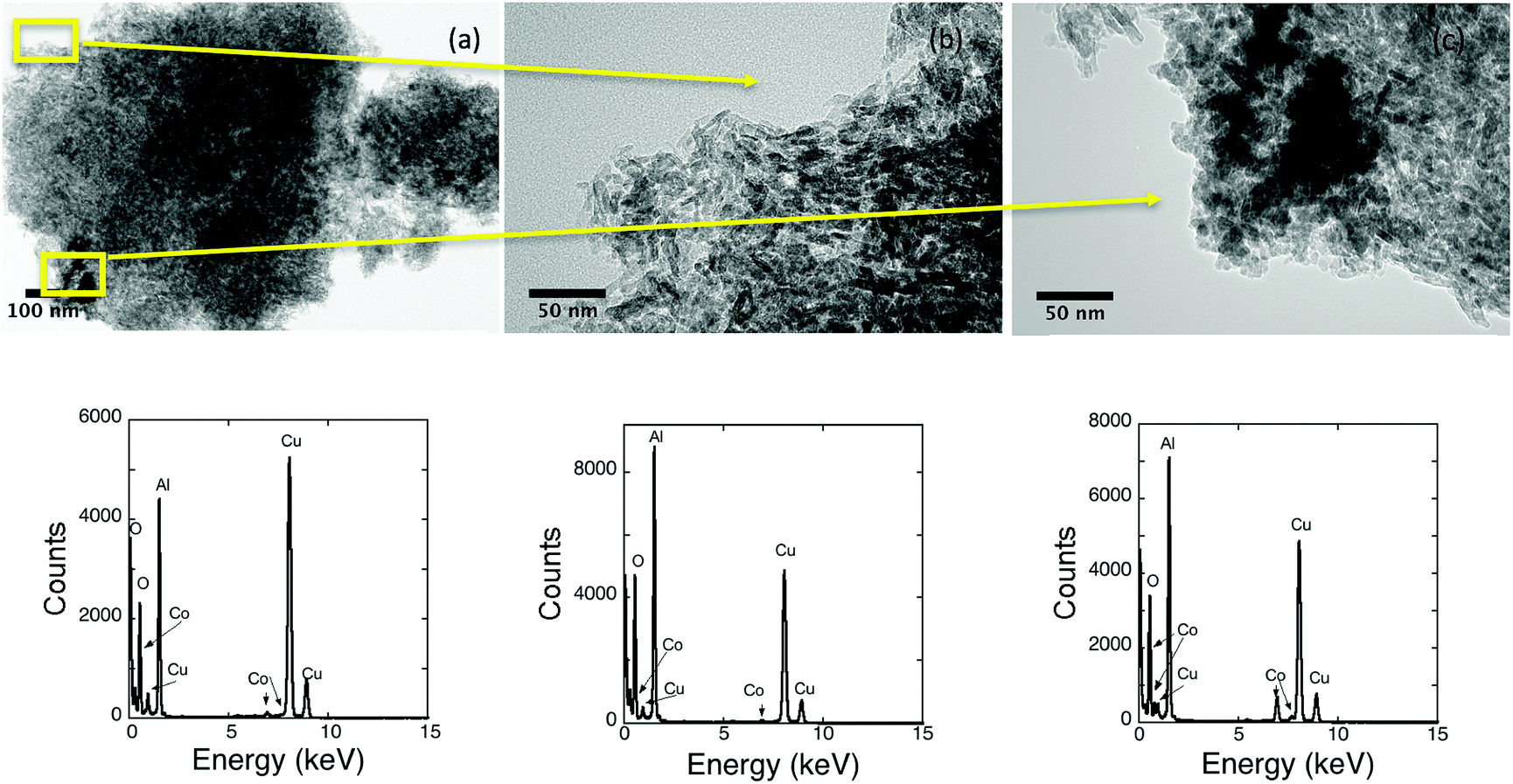 | ||
| Fig. 1 Bright field TEM images of an uncalcined nanomaterial with an EDX spectrum for each shown below. It is difficult to see individual nanoparticles due to the nature of the support. (b and c) are higher resolution images of regions in (a). Although no nanoparticles are seen in (b) cobalt is observed. (c) A darker region on the support is found to have a large cobalt signal. All EDX spectra contain a copper signal due to the grids used to mount the sample. | ||
The polycrystalline nature of the alumina made it difficult to observe the nanoparticle dispersion by TEM. In general, the 8 nm nanoparticles cannot be easily differentiated from the smaller alumina crystals (∼20 nm), which aggregate together to form the support (∼200–1000 nm). The similar size of the pores and the particles make it unlikely that particles are contained inside the pores, it is more likely that they are found exclusively on the surface of the support. Fig. 1 has been used to highlight the difficulties associated with the TEM analysis. In (a) a typical bright field image is shown, the nanoparticles are not evident in the image. Higher magnification has been used to investigate regions of Fig. 1a in conjunction with EDX spectroscopy. In (b), good dispersion is suggested as no observable cobalt particles are found and the EDX spectrum contains both the Kα and Kβ signals of cobalt. The darker region in (c) seems likely to arise from the clustering of cobalt nanoparticles on the surface. The cluster of particles is difficult to characterise for large pieces of support material as the thickness obscures TEM detail. For smaller pieces of alumina, the clustering can be more easily observed. In Fig. 2, a cluster of nanoparticles is clearly evident. The cluster is comprised of individual nanoparticles rather than a single large agglomerate of cobalt.
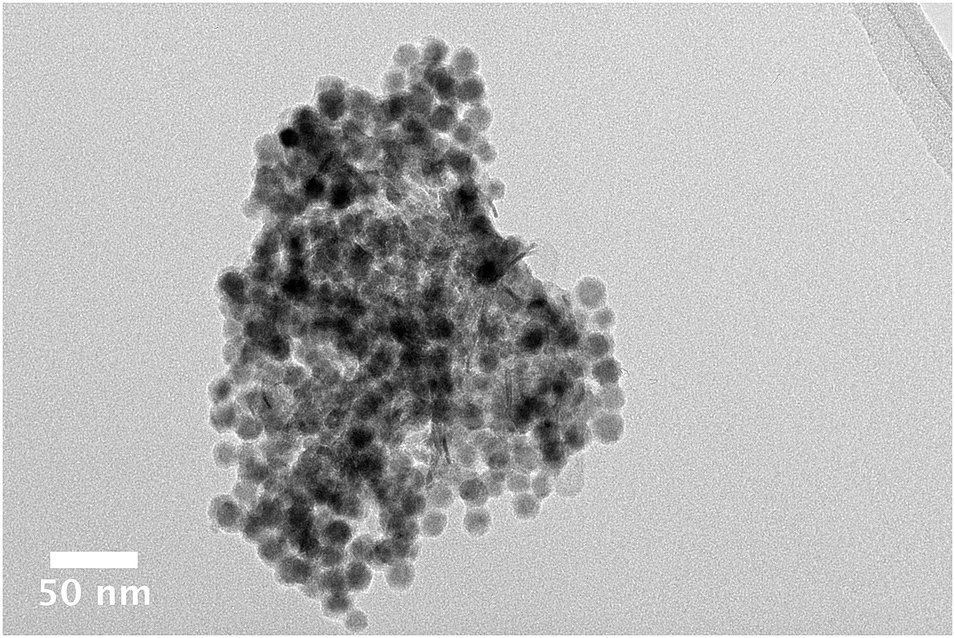 | ||
| Fig. 2 Bright field TEM image of an agglomerate of cobalt nanoparticles on an alumina support. Individual particles are clearly evident. | ||
Although results suggest that the dark regions on a support are the result of the clustering of Co nanoparticles, it cannot be ruled out that it is the result of a thick piece of alumina. The distribution of the particles across the support is clearer with alternative TEM imaging methods. High angle annular dark field scanning TEM (HAADF STEM) is one such method (Fig. 3). Image contrast in this method is based on atomic number so that heavier elements appear brighter in images and can be assigned through the use of EDX spectroscopy.
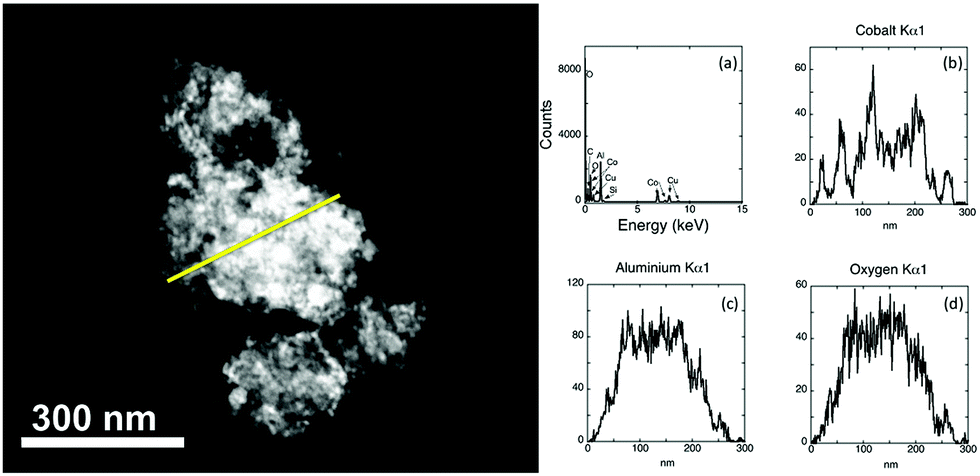 | ||
| Fig. 3 HAADF STEM imaging is used to obtain the electron image (left). A line trace (yellow) has been taken and element analysis through EDX performed. (a) Full EDX spectrum for the material; (b) the cobalt map; (c) the aluminium map and (d) oxygen map. | ||
In Fig. 3 the sample thickness prevents individual cobalt particles being observed directly in the electron image. The presence of cobalt and aluminium is found in the full EDX spectrum. A line trace analysis was performed, allowing the elemental distribution across a region to be analysed. Discrete peaks are noted for cobalt, consistent with the particles having a reasonable dispersion across the support. The intensity of the aluminium and oxygen Kα is relatively uniform, indicating that these elements occur across the length of the sample as a result of support. In addition to HAADF STEM, Energy Filtered TEM (EFTEM) was also used to confirm the location of particles across the thicker pieces of the support. In this method (ESI,† Fig. S2) image maps for each element are acquired. Similar to other characterization methods, EFTEM supports the inference of nanoparticles occurring in areas of both good distribution and as clusters across the support.
The ferromagnetic nature of as-synthesised ε-Co nanoparticles has been shown by others to play a significant role in the assemblage of particles onto a support.17 Particles were observed to form chainlike structures across a silica support material due to constructive magnetic interactions in addition to non-interacting areas with good distribution. However, particle loadings for these systems were significantly more than in the present work i.e. loadings were 6.5 times greater. The significant decrease in the amount of nanoparticle should reduce nanoparticle chain formation in these materials. Indeed, it seems improbable that the ferromagnetism of the particles did not lead to the agglomeration of the cobalt nanoparticles on the support as clustering has also been observed for superparamagnetic iron containing nanoparticles that have also been assembled onto this support material.19 The observation by TEM of nanoparticle clustering on the PURALOX® alumina for both ferromagnetic and superparamagnetic particles suggests that it is unlikely that magnetic interactions are playing a significant role in particle distribution, it is more likely the support itself is affecting how the particles disperse across the surface.
Larger polycrystalline nanocrystals of ε-Co ∼30 nm were also synthesised (ESI,† Fig. S3) and assembled onto an alumina support. Although larger sized particles are not associated with good FT activity, the larger size enables the dispersion across the support to be more easily observed by TEM. In Fig. 4, the TEM image clearly illustrates areas of good particle dispersion and regions of agglomeration that occur for the larger ∼30 nm ε-Co. Despite the larger size, octahedral shape, polycrystalline nature and ferromagnetism of these particles,38 the spatial distribution of the nanoparticles across the alumina support surface at these loadings is analogous to that noted for the smaller cobalt particles. These observations suggest that the role of the support/NP interactions outweigh the inter-nanoparticle interactions in determining the dispersion of NPs across the support.
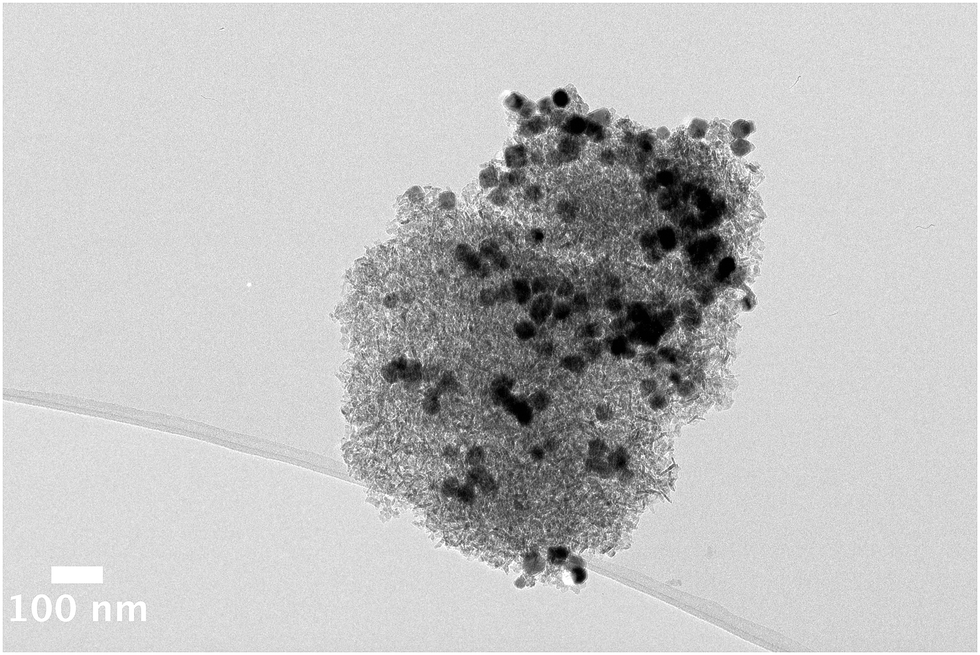 | ||
| Fig. 4 Bright field TEM image of an uncalcined nanomaterial comprised of ∼30 nm ε-Co particles assembled onto an alumina support. The large particles are clearly seen by TEM. | ||
ε-Co nanoparticle assemblage onto MCM-41 support
Preformed cobalt nanoparticles have been previously assembled onto a number of silica supports.17,20 Magnetic interactions were investigated in these materials by incorporating preformed Co nanoparticles at loadings of 22.1% across a silica support SBA-15.17 This composite material contained areas of significant aggregation as well as good distribution. Another study involved the assemblage of monodisperse Co and CoPt particles onto the mesoporous silica, MCF-17 for FT synthesis.20 Unlike the materials made for magnetic studies, the materials designed for FT synthesis have more moderate loadings, hence were less likely to have significant magnetic coupling. The authors of this study did not fully explore the particle dispersion on the support.20 Hence we have assembled cobalt nanoparticles (ESI,† Fig. S4), produced in this work, onto well-ordered and mesoporous silica, MCM-41, (ESI,† Fig. S5) at catalytic loadings, to see whether, as observed for FePt and Fe2O3 NPs, a better dispersion of particles is observed compared to an alumina support.18,21The material produced from the attachment of ε-Co nanoparticles to MCM-41 (Fig. 5) is comprised of highly disperse particles across the support surface. TEM imaging revealed the retention of both the support and nanoparticle structure. The particles are well distributed across the support with no obvious signs of clustering. Heating up to 500 °C did not alter the appearance of the nanomaterial. The calcined material had a surface area (BET ∼ 1122 m2 g−1) and pore widths (BJH ∼ 25 Å) with a nanoparticle loading of 3.6%.
 | ||
| Fig. 5 Bright field TEM images of ε-Co nanoparticles assembled onto MCM-41. The particles are well distributed across the surface with no obvious clustering. | ||
The nature of the support appears to play an important role in how the particles are incorporated as evinced by the results obtained using the different supports cited in this work. The alumina support is comprised of an aggregation of smaller ∼20 nm crystallites, one can envisage a complex support architecture the direct result of the polycrystalline nature leading to a distribution of nucleation sites across the surface. The small irregular stacks of alumina exhibit numerous crystal faces, edges and corners. This surface is not ideal for a specific chemical interaction with the nanoparticles. In comparison, MCM-41 material is uniform with a well-defined hexagonal pore structure. Comprised of silica linkages the acidity is intrinsically low and unlike the alumina support the surface is devoid of charge. The support has high surface concentration of discrete crystal planes i.e. (100), (110) being ideal for specific chemical interactions. Dispersion across the support material under these conditions seems dependent on the exposed crystal facets, thus, the MCM-41 surface provides the ideal conditions for the uniform distribution of metallic nanoparticles.
Treatment of alumina support
Although using an MCM silica seems to improve particle distribution at catalytic loadings they are known to be unstable at elevated temperatures that are required for catalytic reduction cycles (∼500 °C),41 and thus are not suitable for the successive reduction–oxidation–reduction cycles required for the FT process. Alumina is a more suitable support due to its thermal stability. However, the assemblage of a number of different preformed nanoparticles onto the industry standard calcined alumina support, tends to result in partial aggregation of the particles. Altering the surface chemistry of the alumina with a base or acid treatment was anticipated to improve particle dispersion. The commercially available PURALOX® alumina, was treated at a range of pHs (2, 4, 8 and 12). These chemically treated supports were characterised by XRD, TEM, and N2 absorption/desorption isotherms (ESI,† Table S2, Fig. S6 and S7). The crystallinity of the chemically treated supports appears unchanged from the untreated calcined alumina. With the exception of the pH 12 treatment, the TEM images of supports appear similar to the untreated alumina. Some damage is observed to the highest pH treated alumina with the sample tending to be more aggregated. Similarly, with the exception of the pH 12 treated alumina, the surface areas and pore sizes are very similar. For the pH 12 treated sample we see a significant decrease in surface area and a corresponding increase in pore diameter.The support microstructure and macrostructure are known to affect the catalytic activity and selectivity.42 By treating the γ-Al2O3 surface we hope to improve the dispersion of the cobalt nanoparticles across the support. Alumina is amphoteric and as a result the chemistry of the surface is complex. Like others, before assemblage we calcine the γ-Al2O3 at high temperatures (750 °C), to ensure surface hydroxyl groups are removed through dehydration.43,44 In a traditional FT catalyst this leads to an improved interaction between the Co and alumina and, hence, less migration of the catalytically active particles.40 Treatment of the support with ammonia leads to increased pore size of the alumina as well as the deposition of cations (NH4+) across the surface. The change in charge should lead to alteration in the dispersion of the nanoparticles across the support.40
Following calcination, the pH treated supports are subsequently reacted with 8 nm ε-Co particles (ESI,† Table S3, Fig. S8 and S9). The assemblage of the particles on the treated supports is rapid occurring in a few minutes. The nanomaterials thus obtained have been characterised by TEM, XRD and N2 absorption and desorption isotherms (ESI,† Table S9, Fig. S10 and S11). We observed in TEM imaging that the treatment of the support did not improve particle dispersion across the surface. Significant clustering is observed in the acid treated alumina samples. When particles are assembled onto base treated supports, there are regions of both good dispersion and clustering, analogous to the untreated supports. Despite the range of pH treatments used on the alumina support no significant improvements in the particle dispersion across the support were achieved.
Particle assemblage using alternative solvents
Cobalt particles were found to assemble very rapidly, almost instantaneously, onto the alumina supports in hexane i.e. the black suspension rapidly went clear on addition to the support. A non-polar solvent not only provides a means to suspend our preformed nanoparticles coated with a hydrocarbon surfactant coating but also minimises the impact of zeta potential on the system. Solvent selection for the assemblage was also investigated as a way to alter the assemblage rate. High temperature calcination, of γ-Al2O3 is known to lead to the formation of Lewis acid sites at the surface.43,44 These anionic vacancies are a result of dehydration of the surface; where the surface hydroxyl groups combine with neighbouring hydrogen atoms. The Lewis acid sites on the γ-Al2O3 are the active absorption sites. They have been shown to accept non-bonding electrons from a nitrogen atom in N,N′-tetramethylbenzidene45 and indeed can be chlorinated directly by the addition of CCl4 at elevated temperatures.46 The use of chloroform in this work significantly increased the time taken for particles to attach to the alumina support i.e. on addition to the support, the black particle suspension became colourless overnight (>8 hours) cf. seconds to a few minutes in hexane. The absorption of the CHCl3 at the active sites, significantly slowed the rate at which the metallic nanoparticles can interact with the surface. Despite the increase in attachment time, particle distribution for both the ε-Co nanoparticles was similar to materials made by rapid assemblage in hexane; namely, areas of both good dispersion and areas of clustering (ESI,† Fig. S12) are observed. Although the assemblage process occurs over a longer time, chemisorption of the particles onto the support is not altered.Conclusions
The dispersion of nanoparticles across a support surface requires an intense scientific investment to unravel the associated complexities. In this work, we have focused on the assemblage of cobalt nanoparticles onto an alumina support. Particles were observed to have both good areas of dispersion in addition to some aggregation across the support. Since alumina supports are the industry standard for FT catalysts we have attempted to improve dispersion on this support through a number of methods including: (i) altering the surface chemistry of the alumina by base or acid treatment; (ii) changing rate of assemblage by using polar and non-polar solvents; (iii) using nanoparticle with different morphologies and sizes. All of these parameters were found on have little impact on particle dispersion across the support. Improved dispersion was achieved by using a hexagonally ordered mesoporous silica support. It seems likely that the specific chemical interaction between the nanoparticle and support determines the resultant dispersion. In particular, the specific interaction is highly dependent on the exposed crystal facets of the support. Although MCM-41 has the appropriate surface structure, it does not have the thermal stability to be used as a commercial support in FT synthesis. Two paths are evident for future research into understanding nanoparticle dispersion across an alumina support. Firstly, exploration of nanoparticle surfactant may provide another way to improve dispersion in the industrial relevant polycrystalline alumina supports. Surfactant exchange has been used to control assembly in more uniform support materials, and maybe of use in these materials if the right ligand can be employed. Secondly, new systems will be developed based on treated alumina supports that have a more ordered surface comprised of more discrete crystal planes. An ordered thermally stable support is likely to provide the right environment for uniform nanoparticle dispersion while maintaining suitability for multiple reduction cycles of catalysts during the FT process.Conflicts of interest
This work was financially supported by Chevron through the Western Australian Energy Research Alliance, AES 12-P1SR-88.Acknowledgements
The authors would like to acknowledge Chevron, who through the Western Australian Energy Research Alliance (AES 12-P1SR-88) has financially supported the work. The authors acknowledge the facilities, and the scientific and technical assistance of the Australian Microscopy & Microanalysis Research Facility at the Centre for Microscopy, Characterisation & Analysis, The University of Western Australia, a facility funded by the University, State and Commonwealth Governments. ROF would like to thank the University of Western Australia for a UWA ReEntry Fellowship. This work was supported by the Danish National Research Foundation (DNRF93) Center for Materials Crystallography through a stipend to Matthys J. Loedolff.Notes and references
- F. Fischer and H. Tropsch, Brennst.-Chem., 1926, 7, 97–104 Search PubMed.
- P. M. Maitlis, J. Organomet. Chem., 2004, 689, 4366–4374 CrossRef.
- A. Brenner, in Metal Clusters, ed. M. Moskovits, John Wiley, New York, 1986, pp. 264–275 Search PubMed.
- J. Turkevich and G. Kim, Science, 1970, 169, 873–879 CrossRef PubMed.
- D. F. Shriver, H. D. Kaesz and R. D. Adams, The Chemistry of Metal Cluster Complexes, VCH Publishers, Inc., New York, 1990 Search PubMed.
- G. W. Parshall, Homogeneous Catalysis The Applications and Chemistry of Catalysis by Soluble Transition Metal Complexes, Wiley, New York, 1980 Search PubMed.
- F. Schüth, Annu. Rev. Mater. Res., 2005, 35, 209–238 CrossRef.
- P. Munnik, P. E. de Jongh and K. P. de Jong, Chem. Rev., 2015, 115, 6687–6718 CrossRef PubMed.
- K. Moller and T. Bein, Chem. Mater., 1998, 10, 2950–2963 CrossRef.
- J.-l. Shi, Z.-l. Hua and L.-x. Zhang, J. Mater. Chem., 2004, 14, 795–806 RSC.
- S. T. Bromley, G. Sankar, C. R. A. Catlow, T. Maschmeyer, B. F. G. Johnson and J. M. Thomas, Chem. Phys. Lett., 2001, 340, 524–530 CrossRef.
- J. Guzman and B. C. Gates, Dalton Trans., 2003, 3303–3318, 10.1039/b303285j.
- G. L. Bezemer, P. B. Radstake, V. Koot, A. J. van Dillen, J. W. Geus and K. P. de Jong, J. Catal., 2006, 237, 291–302 CrossRef.
- A. Gual, C. Godard, S. Castillón, D. Curulla-Ferré and C. Claver, Catal. Today, 2012, 183, 154–171 CrossRef.
- Y. Zhu, L. P. Stubbs, F. Ho, R. Liu, C. P. Ship, J. Ä. Maguire and N. Ä. Hosmane, ChemCatChem, 2010, 2, 365–374 CrossRef.
- T. Hyeon, Chem. Commun., 2003, 927–934 RSC.
- A. F. Gross, M. R. Diehl, K. C. Beverly, E. K. Richman and S. H. Tolbert, J. Phys. Chem. B, 2003, 107, 5475–5482 CrossRef.
- R. O. Fuller, N. S. Hondow, G. A. Koutsantonis, M. Saunders and R. L. Stamps, J. Phys. Chem. C, 2008, 112, 5271–5274 CrossRef.
- N. S. Hondow, G. A. Koutsantonis, R. O. Fuller, H. Fansuri, M. Saunders, R. L. Stamps and D. Zhang, New J. Chem., 2010, 34, 1286–1294 RSC.
- A. Alayoglu, S. K. Beaumont, F. Zheng, V. V. Pushkarev, H. Zheng, V. Iablokov, Z. Liu, J. Guo, N. Kruse and G. A. Somorjai, Top. Catal., 2011, 54, 778–785 CrossRef.
- N. Hondow and R. O. Fuller, J. Colloid Interface Sci., 2014, 417, 396–401 CrossRef PubMed.
- H. H. Shin, L. Lu, Z. Yang, C. J. Kiely and S. McIntosh, ACS Catal., 2016, 6, 2811–2818 CrossRef.
- A. Barbier, A. Tuel, I. Arcon, A. Kodre and G. A. Martin, J. Catal., 2001, 200, 106–116 CrossRef.
- A. Weddemann, I. Ennen, A. Regtmeier, C. Albon, A. Wolff, K. Eckstädt, N. Mill, M. K. H. Peter, J. Mattay, C. Plattner, N. Sewald and A. Hütten, Beilstein J. Nanotechnol., 2010, 1, 75–93 CrossRef PubMed.
- M. Elimelech, J. Gregory, X. Jia and R. A. Williams, Particle deposition and aggregation: measurement, modelling and simulation, Butterworth-Heinemann, Woburn, 1995 Search PubMed.
- S. A. Bradford and S. Torkzaban, Langmuir, 2013, 29, 3668–3676 CrossRef PubMed.
- W. Chen, I. A. W. Filot, R. Pestman and E. J. M. Hensen, ACS Catal., 2017, 7, 8061–8071 CrossRef PubMed.
- A. Y. Khodakov, W. Chu and P. Fongarland, Chem. Rev., 2007, 107, 1692–1744 CrossRef PubMed.
- R. C. Reuel and C. H. Bartholomew, J. Catal., 1984, 85, 78–88 CrossRef.
- E. Iglesia, S. L. Soled and R. A. Fiato, J. Catal., 1992, 137, 212–224 CrossRef.
- B. G. Johnson, C. H. Bartholomew and D. W. Goodman, J. Catal., 1991, 128, 231–247 CrossRef.
- G. L. Bezemer, J. H. Bitter, H. P. C. E. Kuipers, H. Oosterbeek, J. E. Holewijn, X. Xu, F. Kapteijn, A. J. van Dillen and K. P. de Jong, J. Am. Chem. Soc., 2006, 128, 3956–3964 CrossRef PubMed.
- G. Prieto, A. Martínez, P. Concepción and R. Moreno-Tost, J. Catal., 2009, 266, 129–144 CrossRef.
- V. F. Puntes, K. M. Krishnan and A. P. Alivistos, Science, 2001, 291, 2115–2117 CrossRef PubMed.
- A. C. S. Samia, K. Hyzer, J. A. Schlueter, C.-J. Qin, J. S. Jiang, S. D. Bader and X.-M. Lin, J. Am. Chem. Soc., 2005, 127, 4126–4127 CrossRef PubMed.
- Y. Bao, A. B. Pakhomov and K. M. Krishnan, J. Appl. Phys., 2005, 97, 10J317 CrossRef.
- H. T. Yang, Y. K. Su, C. M. Shen, T. Z. Yang and H. J. Gao, Surf. Interface Anal., 2004, 36, 155–160 CrossRef.
- R. O. Fuller, B.-M. Goh, G. A. Koutsantonis, M. J. Loedolff, M. Saunders and R. C. Woodward, Dalton Trans., 2016, 45, 11983–11989 RSC.
- D. Kumar, K. Schumacher, C. du Fresne von Hohenesche, M. Grün and K. K. Unger, Colloids Surf., A, 2001, 187-188, 109–116 CrossRef.
- J. Zhang, J. Chen, J. Ren and Y. Sun, Appl. Catal., A, 2003, 243, 121–133 CrossRef.
- M. Broyer, S. Valange, J. P. Bellat, O. Bertrand, G. Weber and Z. Gabelica, Langmuir, 2002, 18, 5083–5091 CrossRef.
- M. Trueba and S. P. Trasatti, Eur. J. Inorg. Chem., 2005, 3393–3403 CrossRef.
- J. K. Thomas, Chem. Rev., 1993, 93, 301–320 CrossRef.
- D. Coster, A. L. Biumenfeld and J. J. Fripiat, J. Phys. Chem., 1994, 98, 6201–6211 CrossRef.
- S. Pankasem and J. K. Thomas, Langmuir, 1992, 8, 501–507 CrossRef.
- A. Melchor, E. Garbowski, M.-V. Mathieu and M. Primet, J. Chem. Soc., Faraday Trans. 1, 1986, 82, 1893–1901 RSC.
Footnotes |
| † Electronic supplementary information (ESI) available: Additional characterisation (TEM, EFTEM, SAED, XRD and BET) of nanoparticles and support materials prior to assemblage. See DOI: 10.1039/c8nj03076f |
| ‡ Current address: School of Molecular and Life Sciences, Curtin University, Bentley Western Australia. |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |