
Synthesis and antiviral activity of novel spirocyclic nucleosides†

Abstract
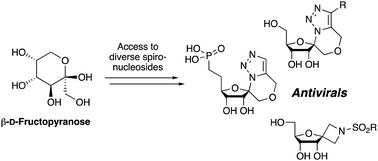
The synthesis of a number of spirocyclic ribonucleosides containing either a triazolic or azetidinic system is described, along with two analogous phosphonate derivatives of the former. These systems were constructed from the same β-D-psicofuranose starting material. The triazole spirocyclic nucleosides were constructed using the 1-azido-1-hydroxymethyl derived sugars, where the primary alcohol was alkylated with a range of propargyl bromides, whereas the azetidine systems orginated from the corresponding 1-cyano-1-hydroxymethyl sugars. Owing to their close similarity with ribavirin, the library of compounds were investigated for their antiviral properties using MHV (Murine Hepatitis Virus) as a model.
- This article is part of the themed collection: Coronavirus articles - free to access collection
 Please wait while we load your content...
Please wait while we load your content...

