
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Comparative effects of trifluoromethyl- and methyl-group substitutions in proline†
Vladimir
Kubyshkin
 *a,
Stanislav
Pridma
b and
Nediljko
Budisa
*a
*a,
Stanislav
Pridma
b and
Nediljko
Budisa
*a
aBiocatalysis Group, Institute of Chemistry, Technical University of Berlin, Müller-Breslau-Str. 10, Berlin 10623, Germany. E-mail: kubyshkin@win.tu-berlin.de; nediljko.budisa@tu-berlin.de
bEnamine Ltd, Chervonotkatska 78, Kyiv 02094, Ukraine
First published on 9th July 2018
Abstract
Proline is one of a kind. This amino acid exhibits a variety of unique functions in biological contexts, which continue to be discovered and developed. In addition to the reactivity of the primary functional groups, the trans–cis isomerization of the peptidyl–prolyl amide bond and its impact on the protein structure and function are of major interest. A variety of proline ring substitutions occur in nature, and more substitutions have been generated via chemical synthesis. Particularly promising is the 19F-labelling of proline, which offers a relatively new research application area. For example, it circumvents the lack of common NH-NMR reporters in peptidyl–prolyl fragments. Obtaining structural information from selectively fluorine-labelled peptides, proteins, and non-peptidic structures requires the analysis of the physicochemical features of the 19F-carrying proline analogues. To better understand and ultimately predict the potential perturbations (e.g., in protein stability and dynamics) introduced by fluorine labels, we conducted a comprehensive survey of the physicochemical effects of CF3 substitutions at each ring position by comparing the behavior of CF3-substituted residues with the CH3-substituted analogues. The parameters analyzed include the acid–base properties of the main chain functional groups, carbonyl-group interaction around the residue, and the thermodynamics and kinetics of trans–cis isomerization. The results reveal significant factors to consider with the use of CF3-substituted prolines in NMR labeling and other applications. Furthermore, lipophilicity measurements demonstrate that CF3-substituted proline shows comparable hydrophobicity to valine, suggesting the potential application of these residues for enhancing interactions at nonpolar interfaces.
Introduction
Complex biological processes such as protein translation and folding are fundamentally influenced by the unique features of amino acid proline (Pro, Fig. 1).1–7 The secondary amino group of Pro creates a tertiary amide-based backbone structure that is prone to cis–trans isomerization issues, which is sometimes responsible for the special role of Pro in protein folding.4–8 Additionally, the cyclic nature of the Pro residue restricts the molecular conformation to certain envelope-type states of the pyrrolidine ring.9–12 As the only coded amino acid with a restricted ϕ torsion, Pro is typically positioned in specific structural contexts in biological systems relative to other amino acid residues. | ||
| Fig. 1 Principle of the study design. | ||
Several unique biological phenomena associated with the presence of Pro in polypeptide structures have been investigated through the substitution with structural analogues of Pro (ProAs). ProAs were shown to impact translation yields13,14 and velocities,15 folding kinetics,16–18 protein structural stability,19–25 aggregation properties,17,26 biological potency of peptides27,28 and more. In each individual case, changes resulting from the substitution of a Pro with an analogue were attributed to structural differences or are rationalized using data obtained from experimental molecular models.29–32 Parallel to these efforts, significant computational data have been generated for select ProAs in recent literature.33–38
Curiously, the majority of the characterized substitution effects have involved very closely related ProAs and typically these substitutions occupied position 4 of the ring. A more general summary characterizing the effect of substitutions at each ring position is lacking. Therefore, we decided to summarize the physicochemical data of molecular models based on ProAs bearing methyl- (CH3–) and trifluoromethyl-group (CF3–) substitutions (Fig. 1). We started from a basic assumption that a methyl group would mimic the presence of an aliphatic substituent, whereas a trifluoromethyl group would exhibit an electron-withdrawing effect in addition to larger steric demands. The resulting physicochemical outcomes are presented in this work. While we sought to locate general trends related to the movement of substituents along the heterocyclic ring, our primary goal was to discern effects resulting from the presence of the CF3 group due to its utility in 19F labelling of polypeptides for NMR studies. The latter application is useful in peptide and protein labelling studies,39,40 although other applications are also possible. This study is limited to a select set of regioisomeric ProAs collected from synthetic and commercial sources.
Results
Available regioisomers
All isomeric methylprolines have been well described in the literature. In contrast, all trifluoromethylated prolines are relatively recent constructs, and these are all products of the XXI century chemistry. In 2002, the synthesis of the first trifluoromethylated proline, 4CF3Pro, was reported independently by two groups.41–43 The synthesis of this regioisomer was later addressed by others.44,45 In 2006, the first synthesis of 2CF3Pro was reported,46 and additional syntheses of this amino acid and its analogues were established later.47–50 The synthesis of 5CF3Pro was first reported in 2012,51 and since then additional synthetic approaches to this compound and its diastereomers have been reported.52,53 Synthesis of 3CF3Pro has not yet been provided in the literature; nonetheless, this regioisomer is available from commercial sources.Thus, we located the set of regioisomeric trifluoromethylated prolines shown in Fig. 2, while the methylproline analogues were collected from commercial and synthetic sources (see ESI†). For example, a very convenient synthesis of 5CH3Pro from a glutamic acid derivative and Meldrum's acid was recently reported by Mohite and Bhat.54
 | ||
| Fig. 2 Regioisomers of trifluoromethyl- and methyl-proline selected for this study. Indices ‘t’ and ‘c’ indicate the configuration of the substituent relative to the carboxyl group. | ||
Amino group
Physicochemical examinations were then performed on the identified set of ProAs and their derivatives. For example, for α-trifluoromethyl amino acids, it has been established that the ammonium group is severely deactivated in nucleophilic reactions due to the electron-withdrawing effect of the fluorinated moiety. As a result, special synthetic strategies towards the incorporation of these amino acids into peptides have been developed.55–59 In order to quantify the amino group deactivation, we examined the acidity of the ammonium group in the set of selected amino acids (Table 1 and Fig. 3).| AA | pKa | ΔpKa | K trans/cis | ΔpKa* | ||||
|---|---|---|---|---|---|---|---|---|
| AA | AA-OMe | Ac-AA | Ac-AA-X | |||||
| trans Amide | cis Amide | X = O− | X = OH | |||||
| a ΔpKa and ΔpKa* values are defined in eqn (1) and (2). | ||||||||
| Pro | 10.68 | 8.47 | 3.55 | 2.85 | 0.70 | 0.81 ± 0.02 | 3.77 ± 0.05 | 0.67 |
| Trifluoromethylated | ||||||||
| 2tCF3Pro | 6.07 | 3.25 | 2.53 | 1.37 | 1.16 | 5.85 ± 0.11 | 100 ± 3 | 1.23 |
| 3tCF3Pro | 8.56 | 6.21 | 2.81 | 2.17 | 0.64 | 1.27 ± 0.02 | 5.61 ± 0.06 | 0.65 |
| 4cCF3Pro | 8.46 | 6.15 | 3.21 | 2.57 | 0.64 | 0.90 ± 0.02 | 3.59 ± 0.09 | 0.60 |
| 5cCF3Pro | 5.80 | 2.91 | 3.66 | 2.96 | 0.70 | 0.42 ± 0.01 | 2.27 ± 0.03 | 0.67 |
| Methylated | ||||||||
| 2tCH3Pro | 10.94 | 8.42 | 4.06 | 2.98 | 1.08 | 2.56 ± 0.09 | 28.3 ± 1.0 | 1.04 |
| 3tCH3Pro | 10.68 | 8.37 | 3.58 | 2.83 | 0.75 | 0.90 ± 0.01 | 5.28 ± 0.06 | 0.77 |
| 4cCH3Pro | 10.73 | 8.49 | 3.54 | 2.79 | 0.75 | 1.03 ± 0.01 | 6.24 ± 0.11 | 0.78 |
| 5cCH3Pro | 10.54 | 8.26 | 3.83 | 3.03 | 0.80 | 0.65 ± 0.02 | 3.73 ± 0.06 | 0.76 |
 | ||
| Fig. 3 pKa values for the ammonium group in trifluoromethyl- (A) and methyl- (B) group-containing proline (solid circles) and their methyl esters (open circles). The values were determined in aqueous solution at 295–298 K. Henceforth, the curve fits in figures represent the most rational fits for optimal presentation of the data. | ||
While the pKa of the ammonium group in methylproline remains nearly identical to that of Pro, the trifluoromethyl-group imposes a severe reduction of the value. These reductions are 4.6–4.9 pKa units at positions 2 and 5 and 2.2 units at positions 3 and 4. The magnitudes of the effects are similar to those previously described for trifluoromethylated pyrrolidines60 and morpholines.61
Notably, for the free amino acid, the C-terminal carboxyl-group remains ionized over the entire pH range of the ammonium protonation transition. In this case, the presence of the C-terminal charge stabilizes the ammonium group in the protonated state.62 We mimicked the removal of the compensatory charge by esterification into a methyl ester. This reduces the pKa by 2.3 units in all cases, with the exception of three derivatives in which the difference was larger. These cases were observed for 2CH3Pro (Δ 2.5 units) and 2CF3Pro and 5CF3Pro (both Δ 2.9 units). The larger differences in this case can be attributed to the interference of the ammonium group with the proximal substituents due to the CO2− ↔ NH2+ hydrogen bond. The effect is analogous to that of the C5 hydrogen bond in protein structures, which was described recently.63
Carboxyl-group
We then examined the acidity of the carboxyl group in the N-acetyl derivatives, which is comparable to the C-terminal residue in a polypeptide. The N-acetylation has a dual effect by both removing the N-terminal charge and simultaneously generating two new conformational states of the formally single N–C(![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O) bond: s-trans and s-cis amides, where prefix ‘s-’ refers to the single bond (Scheme 1). This transition between the two conformations is slow on the NMR time scale, thereby allowing the determination of the pKa for each form separately.64 We determined the acidity of the N-acetyl derivatives. The results are shown in Fig. 4 (Table 1). The analyses revealed similar acidities among the 5CF3Pro, methyl-bearing derivatives, and Pro, while re-positioning of the CF3-group (from 4 to 3 to 2) gradually increased the acidity over the range of 1.6 pKa units.
O) bond: s-trans and s-cis amides, where prefix ‘s-’ refers to the single bond (Scheme 1). This transition between the two conformations is slow on the NMR time scale, thereby allowing the determination of the pKa for each form separately.64 We determined the acidity of the N-acetyl derivatives. The results are shown in Fig. 4 (Table 1). The analyses revealed similar acidities among the 5CF3Pro, methyl-bearing derivatives, and Pro, while re-positioning of the CF3-group (from 4 to 3 to 2) gradually increased the acidity over the range of 1.6 pKa units.
 | ||
| Scheme 1 Amide isomerism in N-acetyl proline derivatives. | ||
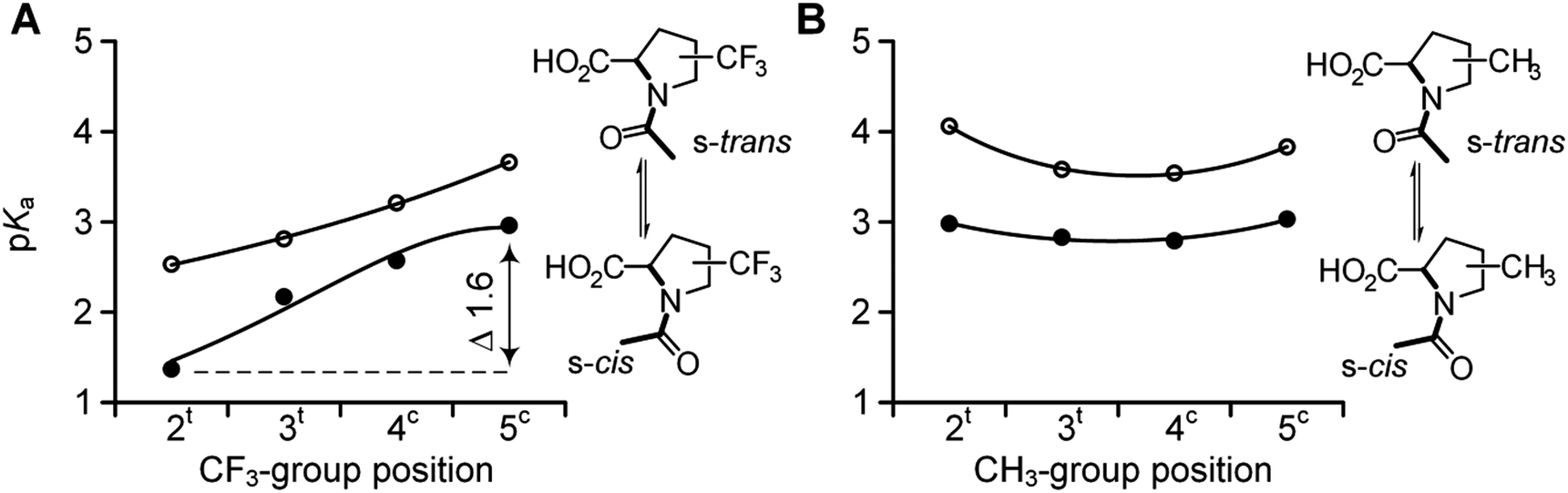 | ||
| Fig. 4 pKa values of the two rotameric forms in N-acetyl trifluoromethyl- (A) and methyl- (B) proline derivatives: s-cis (full circles) and s-trans (empty circles). | ||
Inter-carbonyl alignment
The difference between the pKa of the s-trans and s-cis amides is always positive (eqn (1)–(3)). We have previously demonstrated that this difference is indicative of the inter-carbonyl interaction, which plays an important role in the folding propensities of the amino acids.64 The ΔpKa is between 0.67 and 0.70 for N-acetyl proline, and this increase indicates an attraction between the carbonyl groups. In ProAs, this effect can be related to the stabilization of the C4-exo side chain envelope conformation.65 The inter-carbonyl alignment is an important contributor to the stability of polypeptide structures containing ProAs. This phenomenon has been attributed previously to the n→π* orbital interaction between the adjacent carbonyl groups.66Previous studies enable us to speculate that examined diastereomers of 3-, 4- and 5-methylprolines should exhibit elevated ΔpKa values due to the larger contribution of the side chain C4-exo conformation and more favourable angles between the interacting carbonyl groups.67–70 Indeed, we observed that the experimental ΔpKa values are higher for methylprolines compared to Pro (Fig. 5 and Table 1). Interestingly, for corresponding trifluoromethylprolines, the inter-carbonyl alignment is systematically lowered. Supporting this observation, we previously reported that the trans-amide stability in a model 4CF3Pro derivative was not enhanced despite the C4-exo conformation dominant in solution and crystal structure.45 Thus, fluorination of the molecules slightly reduces the energy of the inter-carbonyl interaction, as can be clearly deduced from the presented data.
| ΔpKa = pKa(trans) − pKa(cis) | (1) |
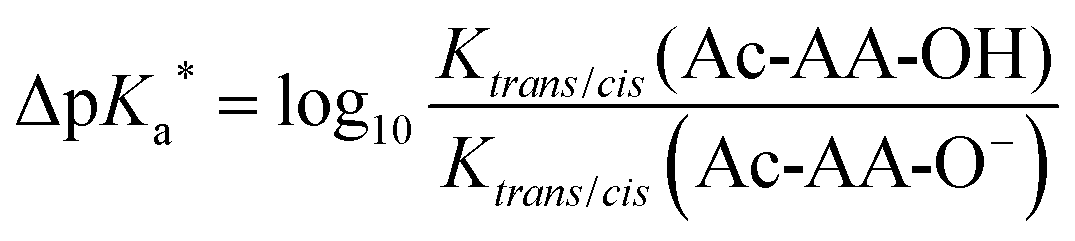 | (2) |
| ΔpKa = ΔpKa* > 0 | (3) |
 | ||
| Fig. 5 Experimental ΔpKa for N-acetyl trifluoromethyl- (full circles) and methyl- (empty circles) prolines as derived from the experimental pKa values and equilibrium populations according to eqn (1) and (2). Dashed lines indicate reference values for N-acetyl proline. | ||
In contrast to these tendencies, the 2-substituted Pro derivatives exhibited a remarkably higher interaction of carbonyl groups relative to the other cases. A previously reported crystal structure of a 2CH3Pro derivative illustrates that elevated alignment energy may result from the increased proximity of interacting groups due to steric effects of the α-substituent.71 The larger steric effect of the CF3-group can explain the best alignment of the carbonyl groups featured by this amino acid. These results highlight potency of the α-substituents to stabilize the trans-amide bond according to stereoelectronic effect in addition to the steric one.
Amide rotameric preference
We then determined the trans/cis amide rotameric preference in N-acetyl derivatives bearing carboxylate, carboxylic acid and methyl ester groups at the C-terminus. Increasing the polarity of the carbonyl group leads to an increase in the inter-carbonyl alignment and increases the trans/cis amide ratio in the order of ester ≥ acid > salt (Scheme 1). The amide isomerism is impacted by steric factors due to the presence of bulky substituents around the amide bond for substitutions at the 2- and 5-positions in the ring. Fluorination evidently increases the original steric effect in both 2- and 5-substituted structures (Table 2). However, additional contribution of the inter-carbonyl alignment leads to additional stabilization of the trans-amide but not cis-amide structure. Thus, the substituent position confers an asymmetric effect on the trans/cis amide ratio (Fig. 6). Although, another reason for the asymmetric shape of the curves can be non-additivity of the steric sizes of the substituents.| Ac-AA-OMe | K trans/cis (at 298 K) | Kinetics | ||||
|---|---|---|---|---|---|---|
| T, K | k, s−1 | E ≠, kJ mol−1 | ||||
| cis → trans | trans → cis | cis → trans | trans → cis | |||

|
4.95 ± 0.05 | 310 | 0.033 ± 0.002 | 0.0070 ± 0.0005 | 84.8 ± 0.2 | 88.8 ± 0.2 |
| Trifluoromethylated | ||||||

|
199 ± 4 | 298 | 1.19 ± 0.22 | 0.007 ± 0.001 | 72.6 ± 0.5 | 85.3 ± 0.4 |

|
6.60 ± 0.15 | 298 | 0.032 ± 0.011 | 0.0038 ± 0.0004 | 81.5 ± 1.1 | 86.8 ± 0.3 |
|
4.36 ± 0.18 | 298 | 0.034 ± 0.003 | 0.008 ± 0.001 | 81.4 ± 0.2 | 84.9 ± 0.3 |

|
2.78 ± 0.18 | 298 | 0.508 ± 0.015 | 0.180 ± 0.009 | 74.7 ± 0.1 | 77.2 ± 0.2 |
| Methylated | ||||||
|
39 ± 1 | 310 | 0.067 ± 0.004 | 0.0020 ± 0.0005 | 83.0 ± 0.1 | 92.0 ± 0.8 |
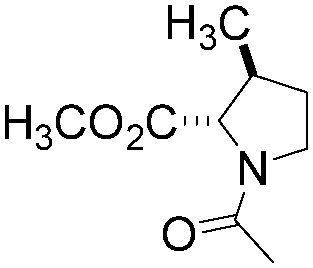
|
6.51 ± 0.10 | 310 | 0.027 ± 0.001 | 0.005 ± 0.001 | 85.3 ± 0.1 | 89.7 ± 0.6 |

|
8.05 ± 0.13 | 310 | 0.041 ± 0.011 | 0.004 ± 0.001 | 84.3 ± 0.7 | 90.3 ± 0.7 |

|
4.64 ± 0.02 | 310 | 0.238 ± 0.011 | 0.055 ± 0.002 | 79.7 ± 0.1 | 83.5 ± 0.1 |
 | ||
| Fig. 6 trans/cis Amide ratios in N-acetyl derivatives of trifluoromethyl- (A) and methyl- (B) prolines bearing C-terminal carboxylates (empty blue circles), carboxylic acid (orange triangles) and methyl esters (full red circles). Determined in deuterium oxide at 298 K. | ||
Amide rotation kinetics
The rotational velocity of the amide bond around Pro is an important contributor to protein folding kinetics. Peptidyl–prolyl cis–trans isomerases are a class of enzymes that accelerate the amide rotation around prolyl residues, and the catalytic centres of these enzymes are attractive targets for the development of pharmaceutical peptide-based inhibitors.72–76ProAs often alter the amide rotational velocities in peptidyl-prolyl fragments, although the overall effect is complex. In order to clarify the effects of positional substitutions in the Pro ring, we summarized the potential contributing forces in Scheme 2. Effects A, B and C occur in the ground state. Effect A is invariant in the ground state and correlates with the basicity of the nitrogen atom, while effects B and C vary for the trans- and cis-amides and are reflected in amide thermodynamics. Since the rotation proceeds via the syn/exo transition state,69,77 a substituent may sterically interfere with the oxygen atom, which shifts below the ring and opposite to the carboxyl group of Pro, thus creating effect D.
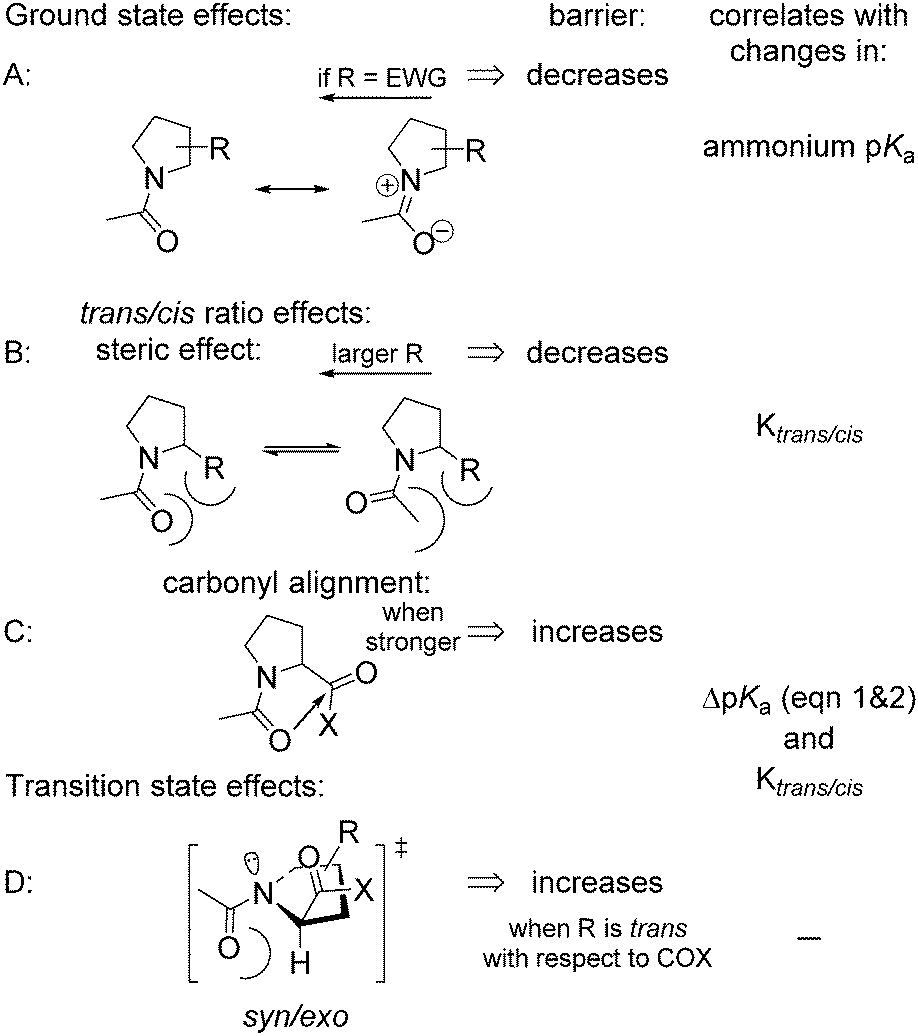 | ||
| Scheme 2 Potential effects contributing to the amide rotation barrier in N-acetyl prolyl derivatives. EWG = electron-withdrawing group. | ||
For example, an electron-withdrawing substituent usually reduces the barrier by destabilizing the ground state resonance (effect A, Scheme 2). However, the same substituent can have an opposite and compensatory effect on the trans → cis barrier by increasing the energy of the inter-carbonyl alignment. For example, this occurs in the case of 4-hydroxy78 and 4-fluoroprolines.79
Experimentally determined velocities of the cis → trans and trans → cis amide rotations in methyl esters of N-acetyl amino acids (water) are shown in Fig. 7 (see also Table S1 in the ESI† for the data on C-terminal carboxylates). These values can be rationalized based on the considerations outlined in Scheme 2. For example, compared to Pro, the 2CH3Pro derivative exhibits a decreased cis → trans barrier due to effect B, whereas the trans → cis barrier increases due to the additional contribution from effects C and D. In the derivative analogues of 2CF3Pro, both rotational barriers are decreased primarily due to the additional contribution of effect A, which is not present in methylprolines. Overall, the rotational barrier is the parameter that is most complex among all presented so far.
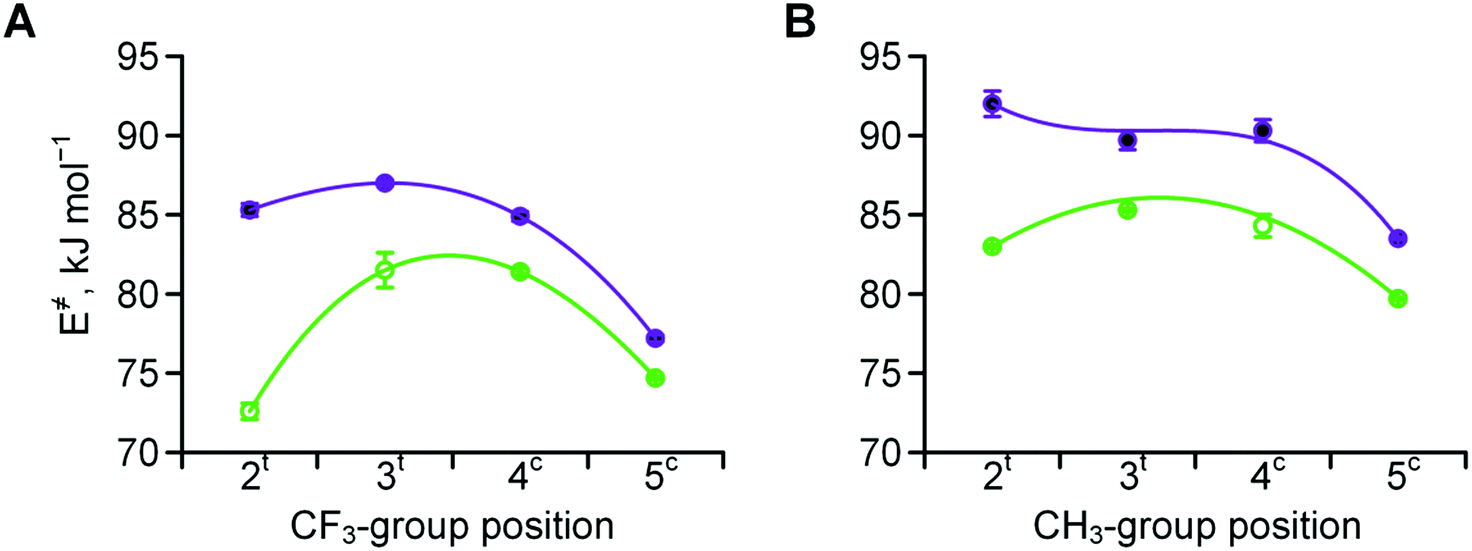 | ||
| Fig. 7 Amide rotation barriers determined for methyl esters of N-acetyl trifluoromethyl- (A) and methyl- (B) prolines: trans → cis (full purple circles) and cis → trans (empty green circles). Measured in aqueous medium at 298 or 310 K (see Table 2 for details). | ||
Lipophilicity
The attempts to classify Pro in the paradigm of hydrophobic/hydrophilic dualism create an ambiguity in the literature, as evidence exists supporting both classifications of Pro in polypeptides. In natural proteins, Pro can be present in both hydrophobic interiors and exposed, highly solvated stretches.80 For example, Pro can accommodate very lipophilic prosthetic groups such as retinal in a hydrophobic core of proteins.81 Pro interactions with aromatic residues are also thought to be guided by the hydrophobic contacts between the aromatic ring and the ring structure of proline.82 In many other instances, Pro occupies solvated and polar stretches primarily due to the extended nature of the secondary structures it forms.80 Noteworthy, we recently demonstrated that the addition of Pro systematically decreases the lipophilicity in an oligoproline peptide series.83Both trifluoromethyl- and methyl-groups are expected to increase the lipophilicity of an organic molecule,84,85 and the same effect should potentially be seen in the amino acid derivatives. A recent study of aliphatic fluorine-containing amino acids demonstrated that the main effect of fluorination on the overall polarity occurs due to the alteration of the backbone contacts with water,86 although we presume that for ProAs, the backbone-solvation considerations are less relevant because the backbone-forming tertiary amide is less accessible to the solvent.
In order to characterize the outcome of CF3-/CH3-group substitutions, we examined experimental octan-1-ol/water partitioning (log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P) values for the methyl esters of the N-acetyl amino acids (Fig. 8 and Table S2 in the ESI†). Our results demonstrate that the presence of a methyl group increased the lipophilicity by approximately 0.4logP units, whereas the trifluoromethyl moiety exhibits a larger effect of approximately 0.7logP units. The lipophilic contribution of the CF3 group is slightly higher in the 2CF3Pro derivative, presumably due to the partial intermolecular compensation of dipoles from the trifluoromethyl- and carboxymethyl-moieties. Overall, these data suggest that trifluoromethylprolines can be considered as hydrophobic amino acids, with the logP close to that of valine (Fig. 9).
P) values for the methyl esters of the N-acetyl amino acids (Fig. 8 and Table S2 in the ESI†). Our results demonstrate that the presence of a methyl group increased the lipophilicity by approximately 0.4logP units, whereas the trifluoromethyl moiety exhibits a larger effect of approximately 0.7logP units. The lipophilic contribution of the CF3 group is slightly higher in the 2CF3Pro derivative, presumably due to the partial intermolecular compensation of dipoles from the trifluoromethyl- and carboxymethyl-moieties. Overall, these data suggest that trifluoromethylprolines can be considered as hydrophobic amino acids, with the logP close to that of valine (Fig. 9).
 | ||
| Fig. 8 logP values for the methyl esters of N-acetylated trifluoromethyl- (empty circles) and methyl- (full circles) prolines. The dashed line represents the reference value for proline. Values are for octan-1-ol/water partitioning at 295 ± 2 K. | ||
 | ||
| Fig. 9 The position of trifluoromethyl- (CF3Pros) and methyl- (CH3Pros) prolines on the lipophilicity scale based on logPoctan-1-ol/water values previously reported for methyl esters of N-acetyl amino acids.80 | ||
The hydrophobicity of amino acid analogues is an important consideration when designing peptide sequences, and some trifluoromethylated ProAs have recently been applied in attempts to improve the membrane permeability of therapeutic peptide constructs.87 We have demonstrated that the local polarity differences introduced by the incorporation of 4-methyl- and 4-fluoroprolines impact structural stability at a level nearly equal to the preorganization from the inter-carbonyl alignment.88
A notable fact is that 4-methylprolines are abundant in natural products,89,90 and these are the simplest alkylproline derivatives among others occurring in nature.91 Another common modification is hydroxylation in position 4; finally extensive experimental data have addressed 4-fluoroprolines.19,20 For this reason, we compared the lipophilicity of the 4-substituted proline derivatives in Fig. 10. The data illustrate that the 4-hydroxyl and 4-methyl moieties have opposite effects, with the former enhancing the molecular hydrophilicity and the latter enhancing the molecular lipophilicity. Furthermore, 4-fluorination increases polarity, though less so than hydroxylation.
 | ||
| Fig. 10 Comparison of the experimental logPoctan-1-ol/water values for 4-substituted prolines. Molecular conformation is sketched taking into account the preferred side-chain conformation.29 | ||
Discussion
A number of chemical routes have been developed for the convenient synthesis of amino acids bearing CF3-groups over the last two decades. Further application of these compounds in engineering and/or in peptides and proteins requires a comprehensive understanding of the physicochemical effects of trifluoromethylation. Interestingly, since the synthesis of the first CF3-bearing proline in 2002,41–43 only one of these compounds has undergone physiochemical characterization through investigation of trans/cis amide properties and pKa (in 2015).45In an attempt to provide the missing data we have systematically characterized all of the trifluoromethylproline regioisomers and compared obtained values with those of analogous methylproline counterparts (Fig. 2). The results of the N-terminal basicity and C-terminal acidity provide a numerical description for the electron-withdrawing effect of the CF3-group (Fig. 3 and 4). Subsequently, we noticed that the inter-carbonyl alignment (represented by the ΔpKa and ΔpKa* values and trans/cis equilibrium constants) is reduced in the trifluoromethyl-derivatives compared to the methyl derivatives in the 3-, 4-, and 5-substituted amino acids (Fig. 5 and 6). For the 2-position, however, substitutions the quaternization of the residue significantly enhances the original interaction between the carbonyl groups due to the steric reasons.
The kinetics of the amide bond rotation are reflected in the trans → cis and cis → trans activation barriers (Fig. 7). The latter parameters are quite complex, as they are impacted by the amide thermodynamics as well as the N-terminal basicity. For instance, the curves in Fig. 7A (trifluoromethyl derivatives) can be considered as composite results of the barriers for the corresponding methylated amino acids (Fig. 7B) and the basicity trends (Fig. 3A). Transition state effects should be considered when the substituents interfere with the upstream carbonyl oxygen atom moving below the pyrrolidine ring. Another important observation is that trifluoromethyl substituents cause the amino acid to exhibit lipophilicity levels equivalent to those of valine, as demonstrated by the experimental examination of the octan-1-ol/water partitioning constants (Fig. 8 and 9).
Our data will enable researchers to rationally and more accurately predict and plan experiments that involve Pro substitutions, in applications such as in the design of peptidyl–prolyl cis–trans isomerase inhibitors,72–76 expansion of the genetic amino acid repertoire13–15 as well as in protein engineering21–26 and development of other peptide therapeutics.87 However, the most outstanding application is the use of fluorine-labelled ProAs in 19F NMR studies of polypeptides. A few works have suggested the use of fluoroprolines for this purpose (Fig. 11).92,93 Recently vicinal difluoroprolines were proposed for use in 19F NMR labelling, as these derivatives have a reduced conformational bias in the side chain and the amide bond.94
 | ||
| Fig. 11 19F NMR labelling schemes for proline residues in polypeptides. Ticks indicate the reported examples of 19F NMR labelling of polypeptides. | ||
To a great dissatisfaction, studies of the polarity changes introduced in ProAs are quite underrepresented in the literature. Even for fluoroprolines, which are most well studied, there is a very little awareness of the polarity changes introduced by these residues in the polypeptide structures. In this context, we recently demonstrated that local polarity changes introduced by ProAs may have quite a significant effect when placed on a periphery of a 9 kDa foldon trimeric propeller structure.88 As seen from further analysis of the logP values, 4-fluorination increases the polarity (Fig. 10), which may reduce the folding stability when the Pro residue is located at a buried, interior position. Geminal difluoroprolines95 may potentially reduce the polarity of the proline residue following partial C–F dipole compensation84,85 while maintaining conformational bias at minimum.96 However, these would also have poor NMR properties due to a large coupling between the fluorine atoms.94
Alternatively, Tressler and Zondlo adapted the use of perfluoro-tert-butytoxy 19F probes97,98 and proposed the use of O-perfluoro-tert-butyl-4-hydroxyprolines in NMR applications,99 although these bulky lipophilic compounds also demonstrated a significant conformational bias in trans/cis amide equilibrium. Other proposed Pro substitutes are trifluoromethyl-4,5-methanoprolines, which were originally introduced as 19F NMR labels for solid-state NMR studies in lipid membranes.100 Later studies demonstrated that 4,5-methanolprolines also impose a notable conformational alteration depending on the stereochemistry of the cyclopropane unit.101 A recent 19F NMR study fully supported this conclusion.102 However, their use in NMR labeling can justified given that the conformational bias is taken into account when interpreting the data.
A single trifluoromethyl group represents another alternative to the above-mentioned 19F NMR labelling strategies. The utility of this labelling scheme follows in particular from the good relaxation profile of the axially rotating trifluoromethyl-group.103 Our study together with our previous report45 shows that the trans/cis ratios are minimally perturbed for the 3- and 4-position substitutions, as these positions are distant from the backbone. Thus, the hydrophobic residues of CF3-proline are good candidates for the design and engineering of non-polar interaction interfaces. These insights have enabled the recent application of 4CF3Pro for the labelling and detection of the first transmembrane polyproline helix by means of solid state 19F NMR.104 Also, some other ProAs, 4-trifluoromethyl-3,4-dehydroproline105 and difluoro-4,5-methanoprolines106 can be considered promising fluorine-bearing proline substitutes. However, these compounds exhibit severely compromised stability, in particular, in basic media.
Finally, we believe that the physicochemical data reported in this study will further support the use of CF3-containing prolines for purposes ranging from customizable design of small molecules to the reprogramming of complex biological processes and structures.
Conflicts of interest
The authors declare the following competing financial interest. SP is affiliated to a commercial company, which sells some of the discussed amino acids. VK and NB declare no conflict of interests.Acknowledgements
VK acknowledges the DFG research group FOR 1805 for a postdoctoral position. Enamine Ltd is acknowledged for provision of some amino acids.Notes and references
- P. Hunter, S. Arenz, L. V. Bock, M. Graf, J. O. Frister, A. Heuer, L. Peil, A. L. Starosta, I. Wohlgemuth, F. Peske, J. Nováček, O. Berninghausen, H. Grubmüller, T. Tenson, R. Beckman, M. V. Rodnina, A. C. Vaiana and D. N. Wilson, Mol. Cell, 2017, 68, 515–527, DOI:10.1016/j.molcel.2017.10.014.
- L. Zhang and D. F. Becker, Front. Plant Sci., 2015, 6, 552, DOI:10.3389/fpls.2015.00552.
- S.-J. Chen, X. Wu, B. Wadas, J.-H. Oh and A. Varshavsky, Science, 2017, 355, eaal3655, DOI:10.1126/science.aal3655.
- T. R. Anderson, J. H. Lee, C. Charlier, J. Ying and A. Bax, ChemBioChem, 2018, 19, 37–42, DOI:10.1002/cbic.201700548.
- P. Cortes-Hernandez and L. Domínguez-Ramírez, PLoS One, 2017, 12, e0188935, DOI:10.1371/journal.pone.0188935.
- A. Taler-Verčič, S. Hasanbašić, S. Berbić, V. Stoka, D. Turk and E. Žerovnik, Int. J. Mol. Sci., 2017, 18, 549, DOI:10.3390/ijms18030549.
- P. Ahuja, F.-X. Cantrelle, I. Huvent, X. Hanoulle, J. Lopez, C. Smet, J.-M. Wieruszeski, I. Landrieu and G. Lippens, J. Mol. Biol., 2016, 428, 79–91, DOI:10.1016/j.jmb.2015.11.023.
- cis–trans-Isomerization in Biochemistry, ed. C. Dugave, Wiley, Weinheim, 2006 Search PubMed.
- Y. K. Kang, J. Phys. Chem. B, 2004, 108, 5463–5465, DOI:10.1021/jp049658f.
- Y. K. Kang and H. Y. Choi, Biophys. Chem., 2004, 111, 135–142, DOI:10.1016/j.bpc.2004.05.006.
- Y. K. Kang, J. Phys. Chem. B, 2007, 111, 10550–10556, DOI:10.1021/jp073411b.
- C. B. Braga, L. C. Ducati, C. F. Tormena and R. Rittner, J. Phys. Chem. A, 2014, 118, 1748–1758, DOI:10.1021/jp5007632.
- T. Kawakami, T. Ishizawa and H. Murakami, J. Am. Chem. Soc., 2013, 135, 12297–12304, DOI:10.1021/ja405044k.
- B.-S. Shin, T. Katoh, E. Gutierrez, J.-R. Kim, H. Suga and T. E. Dever, Nucleic Acids Res., 2017, 45, 8392–8402, DOI:10.1093/nar/gkx532.
- L. K. Doerfel, I. Wohlgemuth, V. Kubyshkin, A. L. Starosta, D. N. Wilson, N. Budisa and M. V. Rodnina, J. Am. Chem. Soc., 2015, 137, 12997–13006, DOI:10.1021/jacs.5b07427.
- T. Steiner, P. Hess, J. H. Bae, B. Wiltschi, L. Moroder and N. Budisa, PLoS ONE, 2008, 3, e1680, DOI:10.1371/journal.pone.0001680.
- V. Y. Torbeev and D. Hilvert, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 20051–20056, DOI:10.1073/pnas.1310414110.
- D. Roderer, R. Glockshuber and M. Rubini, ChemBioChem, 2015, 16, 2162–2166, DOI:10.1002/cbic.201500342.
- S. P. Nadarajan, K. Deepankumar, J.-H. Seo and H. Yun, Biotechnol. Bioprocess Eng., 2017, 22, 504–511, DOI:10.1007/s12257-017-0257-2.
- R. W. Newberry and R. T. Raines, Top. Heterocycl. Chem., 2017, 48, 1–25, DOI:10.1007/7081_2015_196.
- D. Naduthambi and N. J. Zondlo, J. Am. Chem. Soc., 2006, 128, 12430–12431, DOI:10.1021/ja0648458.
- M. D. Shoulders, K. A. Satyshur, K. T. Forest and R. T. Raines, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 559–564, DOI:10.1073/pnas.0909592107.
- B. Holzberger and A. Marx, J. Am. Chem. Soc., 2010, 132, 15708–15713, DOI:10.1021/ja106525y.
- T.-Y. Zheng, Y.-J. Lin and J.-C. Horng, Biochemistry, 2010, 49, 4255–4263, DOI:10.1021/bi100323v.
- U. Arnold and R. T. Raines, Org. Biomol. Chem., 2016, 14, 6780–6785, 10.1039/c6ob00980h.
- V. Torbeev, M.-O. Ebert, J. Dolenc and D. Hilvert, J. Am. Chem. Soc., 2015, 137, 2524–2535, DOI:10.1021/ja510109p.
- M. A. El-Maghraby, Indian J. Chem., Sect. B: Org. Chem. Incl. Med. Chem., 1976, 14, 988–990 Search PubMed.
- E. Katz, W. K. Williams, K. T. Mason and A. B. Mauger, Antimicrob. Agents Chemother., 1977, 11, 1056–1063, DOI:10.1128/AAC.11.6.1056.
- M. D. Shoulders and R. T. Raines, Annu. Rev. Biochem., 2009, 78, 929–958, DOI:10.1146/annurev.biochem.77.032207.120833.
- S. A. Cadamuro, R. Reichold, U. Kusebauch, H.-J. Musiol, C. Renner, P. Tavan and L. Moroder, Angew. Chem., Int. Ed., 2008, 47, 2143–2146, DOI:10.1002/anie.200704310.
- M. Umashankara, M. V. Sonar, N. D. Bansode and K. N. Ganesh, J. Org. Chem., 2015, 80, 8552–8560, DOI:10.1021/acs.joc.5b01032.
- C. R. Forbes, A. K. Pandey, H. K. Ganguly, G. P. A. Yap and N. J. Zondlo, Org. Biomol. Chem., 2016, 14, 2327–2346, 10.1039/C5OB02473K.
- A. Flores-Ortega, J. Casanovas, R. Nussinov and C. Alemán, J. Phys. Chem. B, 2008, 112, 14045–14055, DOI:10.1021/jp807638p.
- A. Flores-Ortega, A. I. Jiménez, C. Cativiela, R. Nussinov, C. Alemán and J. Casanovas, J. Org. Chem., 2008, 73, 3418–3427, DOI:10.1021/jo702710x.
- Y. K. Kang, H. S. Park and B. J. Byun, Biopolymers, 2009, 91, 444–455, DOI:10.1002/bip.21150.
- J. G. Warren, G. Revilla-López, C. Alemán, A. I. Jiménez, C. Cativiela and J. Torras, J. Phys. Chem. B, 2010, 114, 11761–11770, DOI:10.1021/jp105456r.
- Y. K. Kang, B. J. Byun and H. S. Park, Biopolymers, 2011, 95, 51–61, DOI:10.1002/bip.21534.
- H. S. Park, B. J. Byun, D. Motooka, K. Kawahara, M. Doi, T. Nakazawa, Y. Kobayashi and Y. K. Kang, Biopolymers, 2012, 97, 629–641, DOI:10.1002/bip.22054.
- S. L. Cobb and C. D. Murphy, J. Fluorine Chem., 2009, 130, 132–143, DOI:10.1016/j.jfluchem.2008.11.003.
- E. N. G. Marsh and Y. Suzuki, ACS Chem. Biol., 2014, 9, 1242–1250, DOI:10.1021/cb500111u.
- J. Del Valle and M. Goodman, Angew. Chem., Int. Ed., 2002, 41, 1600–1602, DOI:10.1002/1521-3773(20020503)41:9<1600::AID-ANIE1600>3.0.CO;2-V.
- X-l. Qiu and F.-l. Qing, J. Chem. Soc., Perkin Trans. 1, 2002, 2052–2057, 10.1039/B206719F.
- X.-l. Qiu and F.-l. Qing, J. Org. Chem., 2002, 67, 7162–7164, DOI:10.1021/jo0257400.
- R. Nadano, Y. Iwai, T. Mori and J. Ichikawa, J. Org. Chem., 2006, 71, 8748–8754, DOI:10.1021/jo061421n.
- V. Kubyshkin, S. Afonin, S. Kara, N. Budisa, P. K. Mykhailiuk and A. S. Ulrich, Org. Biomol. Chem., 2015, 13, 3171–3181, 10.1039/C5OB00034C.
- G. Chaume, M.-C. Van Severen, S. Marinkovic and T. Brigaud, Org. Lett., 2006, 8, 6123–6126, DOI:10.1021/ol062593.
- C. Caupène, G. Chaume, L. Ricard and T. Brigaud, Org. Lett., 2009, 11, 209–212, DOI:10.1021/ol8024567.
- G. Huang, Z. Yin and X. Zhang, Chem. – Eur. J., 2013, 19, 11992–11998, DOI:10.1002/chem.201301479.
- N. Lensen, J. Marais and T. Brigaud, Org. Lett., 2015, 17, 342–345, DOI:10.1021/ol503448w.
- J. Hao, T. Milcent, P. Retailleau, V. A. Soloshonok, S. Ongeri and B. Crousse, Eur. J. Org. Chem., 2018 DOI:10.1002/ejoc.201800255.
- I. S. Kondratov, V. G. Dolovanyuk, N. A. Tolmachova, I. I. Gerus, K. Bergander, R. Fröhlich and G. Haufe, Org. Biomol. Chem., 2012, 10, 8778–8785, 10.1039/C2OB26176F.
- S. Ortial, R. Dave, Z. Benfodda, D. Bénimélis and P. Meffre, Synthesis, 2014, 569–573, DOI:10.1055/s-0033-1340553.
- H. Lubin, J. Pytkowicz, G. Chaume, G. Sizun-Thomé and T. Brigaud, J. Org. Chem., 2015, 80, 2700–2708, DOI:10.1021/jo502885v.
- A. R. Mohite and R. G. Bhat, J. Org. Chem., 2012, 77, 5423–5428, DOI:10.1021/jo300653u.
- B. Koksch, N. Sewald, H.-J. Hofmann, K. Burger and H.-D. Jakubke, J. Pept. Sci., 1997, 3, 157–167, DOI:10.1002/(SICI)1099-1387(199705)3:3<157::AID-PSC94>3.0.CO;2-W.
- G. Chaume, N. Lensen, C. Caupène and T. Brigaud, Eur. J. Org. Chem., 2009, 5717–5724, DOI:10.1002/ejoc.200900768.
- D. Maisch, P. Wadhwani, S. Afonin, C. Böttcher, B. Koksch and A. S. Ulrich, J. Am. Chem. Soc., 2009, 131, 15596–15597, DOI:10.1021/ja9067595.
- I. Jlalia, N. Lensen, G. Chaume, E. Dzhambazova, L. Astasidi, R. Hadjiolova, A. Bocheva and T. Brigaud, Eur. J. Med. Chem., 2013, 62, 122–129, DOI:10.1016/j.ejmech.2012.12.041.
- S. L. Grage, S. Kara, A. Bordessa, V. Doan, F. Rizzolo, M. Putzu, T. Kubař, A. M. Papini, G. Chaume, T. Brigaud, S. Afonin and A. S. Ulrich, Chem. – Eur. J., 2018, 24, 4328–4335, DOI:10.1002/chem.201704307.
- V. S. Yarmolchuk, O. V. Shishkin, V. S. Starova, O. A. Zaporozhets, O. Kravchuk, S. Zozulya, I. V. Komarov and P. K. Mykhailiuk, Eur. J. Org. Chem., 2013, 3086–3093, DOI:10.1002/ejoc.201300121.
- A. V. Shcherbatiuk, O. S. Shyshlyk, D. V. Yarmoliuk, O. V. Shishkin, S. V. Shishkina, V. S. Starova, O. A. Zaporozhets, S. Zozulya, R. Moriev, O. Kravchuk, O. Manoilenko, A. A. Tolmachev and P. K. Mykhailiuk, Tetrahedron, 2013, 69, 3796–3804, DOI:10.1016/j.tet.2013.03.067.
- M. Johansson, K.-W. Ieong, S. Trobro, P. Strazewski, J. Åqvist, M. Y. Pavlov and M. Ehrenberg, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 79–84, DOI:10.1073/pnas.1012612107.
- R. W. Newberry and R. T. Raines, Nat. Chem. Biol., 2016, 12, 1084–1088, DOI:10.1038/nchembio.2206.
- V. Kubyshkin, P. Durkin and N. Budisa, New J. Chem., 2016, 40, 5209–5220, 10.1039/C5NJ03611A.
- J. A. Hodges and R. T. Raines, J. Am. Chem. Soc., 2005, 127, 15923–15932, DOI:10.1021/ja054674r.
- R. W. Newberry and R. T. Raines, Acc. Chem. Res., 2017, 50, 1838–1846, DOI:10.1021/acs.accounts.7b00121.
- M. D. Shoulders, J. A. Hodges and R. T. Raines, J. Am. Chem. Soc., 2006, 128, 8112–8113, DOI:10.1021/ja061793d.
- G. Chaume, J. Simon, N. Lensen, J. Pytkowicz, T. Brigaud and E. Miclet, J. Org. Chem., 2017, 82, 13602–13608, DOI:10.1021/acs.joc.7b01944.
- V. Kubyshkin and N. Budisa, Org. Biomol. Chem., 2017, 15, 6764–6772, 10.1039/C7OB01421J.
- W. Kim, K. I. Hardcastle and V. P. Conticello, Angew. Chem., Int. Ed., 2006, 45, 8141–8145, DOI:10.1002/anie.200603227.
- P. K. Mykhailiuk, V. Kubyshkin, T. Bach and N. Budisa, J. Org. Chem., 2017, 82, 8831–8841, DOI:10.1021/acs.joc.7b00803.
- X. J. Wang and F. A. Etzkorn, Pept. Sci., 2006, 84, 125–146, DOI:10.1002/bip.20240.
- K. P. Lu and X. Z. Zhou, Nat. Rev. Mol. Cell Biol., 2007, 8, 904–916, DOI:10.1038/nrm2261.
- K. E. Duncan, B. R. Dempsey, L. E. Killip, J. Adams, M. L. Bailey, G. A. Lajoie, D. W. Litchfield, C. J. Brandl, G. S. Shaw and B. H. Shilton, J. Med. Chem., 2011, 54, 3854–3865, DOI:10.1021/jm200156c.
- Z. Lu and T. Hunter, Cell Res., 2014, 24, 1033–1049, DOI:10.1038/cr.2014.109.
- B. M. Dunyak and J. E. Gestwicki, J. Med. Chem., 2016, 59, 9622–9644, DOI:10.1021/acs.jmedchem.6b00411.
- S. Fischer, R. L. Dunbrack Jr. and M. Karplus, J. Am. Chem. Soc., 1994, 116, 11931–11937, DOI:10.1021/ja00105a036.
- N. W. Owens, C. Braun, J. D. O’Neil, K. Marat and F. Schweizer, J. Am. Chem. Soc., 2007, 129, 11670–11671, DOI:10.1021/ja073488d.
- C. Renner, S. Alefelder, J. H. Bae, N. Budisa, R. Huber and L. Moroder, Angew. Chem., Int. Ed., 2001, 40, 923–925, DOI:10.1002/1521-3773(20010302)40:5<923::AID-ANIE923>3.0.CO;2-#.
- B. K. Kay, M. P. Williamson and M. Sudol, FASEB J., 2000, 14, 231–241, DOI:10.1096/fasebj.14.2.231.
- I. Gushchin, V. Shevchenko, V. Polovinkin, K. Kovalev, A. Alekseev, E. Round, V. Borshchevskiy, T. Balandin, A. Popov, T. Gensch, C. Fahlke, C. Bamann, D. Willbold, G. Büldt, E. Bamberg and V. Gordeliy, Nat. Struct. Mol. Biol., 2015, 22, 390–395, DOI:10.1038/nsmb.3002.
- N. J. Zondlo, Acc. Chem. Res., 2013, 46, 1039–1049, DOI:10.1021/ar300087y.
- V. Kubyshkin and N. Budisa, J. Pept. Sci., 2018, 24, e3076, DOI:10.1002/psc.3076.
- Q. A. Huchet, B. Kuhn, B. Wagner, H. Fischer, M. Kansy, D. Zimmerli, E. M. Carreira and K. Müller, J. Fluorine Chem., 2013, 152, 119–128, DOI:10.1016/j.jfluchem.2013.02.023.
- V. Kubyshkin and N. Budisa, Beilstein J. Org. Chem., 2017, 13, 2442–2457, DOI:10.3762/bjoc.13.241.
- J. R. Robalo, S. Huhmann, B. Koksch and A. V. Verde, Chem, 2017, 3, 881–897, DOI:10.1016/j.chempr.2017.09.012.
- M. Oliver, C. Gadais, J. García-Pindado, M. Teixidó, N. Lensen, G. Chaume and T. Brigaud, RSC Adv., 2018, 8, 14597–14602, 10.1039/C8RA02511H.
- D. Dietz, V. Kubyshkin and N. Budisa, ChemBioChem, 2015, 16, 403–406, DOI:10.1002/cbic.201402654.
- A. C. Murphy, M. I. Mitova, J. W. Blunt and M. H. G. Munro, J. Nat. Prod., 2008, 71, 806–809, DOI:10.1021/np070673w.
- L. Liu, J. Jokela, L. Herfindal, M. Wahlstein, J. Sinkkonen, P. Permi, D. P. Fewer, S. O. Døskeland and K. Sivonen, ACS Chem. Biol., 2014, 9, 2646–2655, DOI:10.1021/cb500436p.
- J. Janata, Z. Kamenik, R. Gazak, S. Kadlcik and L. Najmanova, Nat. Prod. Rep., 2018, 35, 257–289, 10.1039/C7NP00047B.
- C. A. Thomas, E. R. Talaty and J. G. Bann, Chem. Commun., 2009, 3366–3368, 10.1039/B821952D.
- A. K. Pandey, D. Naduthambi, K. M. Thomas and N. J. Zondlo, J. Am. Chem. Soc., 2013, 135, 4333–4363, DOI:10.1021/ja3109664.
- G.-J. Hofman, E. Ottoy, M. E. Light, B. Kieffer, I. Kuprov, J. C. Martins, D. Sinnaeve and B. Linclau, Chem. Commun., 2018, 54, 5118–5121, 10.1039/C8CC01493K.
- C. Doebelin, Y. He and T. M. Kamenecka, Tetrahedron Lett., 2016, 57, 5658–5660, DOI:10.1016/j.tetlet.2016.11.012.
- M. D. Shoulders, K. J. Kamer and R. T. Raines, Bioorg. Med. Chem. Lett., 2009, 19, 3859–3862, DOI:10.1016/j.bmcl.2009.03.168.
- Z.-X. Jiang and Y. B. Yu, J. Org. Chem., 2007, 72, 1464–1467, DOI:10.1021/jo0616308.
- B. C. Buer, B. J. Levin and E. N. G. Marsh, J. Pept. Sci., 2013, 19, 308–314, DOI:10.1002/psc.2501.
- C. M. Tressler and N. J. Zondlo, J. Org. Chem., 2014, 79, 5880–5886, DOI:10.1021/jo5008674.
- P. K. Mykhailiuk, S. Afonin, G. V. Palamarchuk, O. V. Shishkin, A. S. Ulrich and I. V. Komarov, Angew. Chem., Int. Ed., 2008, 47, 5765–5767, DOI:10.1002/anie.200801022.
- G. Berger, M. Vilchis-Reyes and S. Hanessian, Angew. Chem., Int. Ed., 2015, 54, 13268–13272, DOI:10.1002/anie.201506208.
- S. Afonin, V. Kubyshkin, P. K. Mykhailiuk, I. V. Komarov and A. S. Ulrich, J. Phys. Chem. B, 2017, 121, 6479–6491, DOI:10.1021/acs.jpcb.7b02852.
- S. L. Grage, U. H. N. Dürr, S. Afonin, P. K. Mykhailiuk, I. V. Komarov and A. S. Ulrich, J. Magn. Reson., 2008, 191, 16–23, DOI:10.1016/j.jmr.2007.11.016.
- V. Kubyshkin, S. L. Grage, J. Bürck, A. S. Ulrich and N. Budisa, J. Phys. Chem. Lett., 2018, 9, 2170–2174, DOI:10.1021/acs.jpclett.8b00829.
- V. Kubyshkin and N. Budisa, Beilstein J. Org. Chem., 2016, 12, 589–593, DOI:10.3762/bjoc.12.57.
- V. S. Kubyshkin, P. K. Mykhailiuk, S. Afonin, S. L. Grage, I. V. Komarov and A. S. Ulrich, J. Fluorine Chem., 2013, 152, 136–143, DOI:10.1016/j.jfluchem.2013.03.002.
Footnote |
| † Electronic supplementary information (ESI) available: Description of experimental procedures and NMR data for compounds, and Tables S1 and S2. See DOI: 10.1039/c8nj02631a |
| This journal is © The Royal Society of Chemistry and the Centre National de la Recherche Scientifique 2018 |